 Liu Yang
Liu Yang Xuejiao Zhang
Xuejiao Zhang Qing Wang
Qing Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 15 December 2022
Sec. Cardiovascular Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.992937
This article is part of the Research TopicAdvances in Research of the Cardiovascular Disease Continuum: Endocrine Aspects of Disease Pathophysiology, Risk Predictors, Therapeutics, and Management of Diabetes and Hypertension Volume IIView all 9 articles
Atherosclerosis is a lipid-driven chronic inflammatory disease that is widespread in the walls of large and medium-sized arteries. Its pathogenesis is not fully understood. The currently known pathogenesis includes activation of pro-inflammatory signaling pathways in the body, increased oxidative stress, and increased expression of cytokines/chemokines. In the innate immune response, inflammatory vesicles are an important component with the ability to promote the expression and maturation of inflammatory factors, release large amounts of inflammatory cytokines, trigger a cascade of inflammatory responses, and clear pathogens and damaged cells. Studies in the last few years have demonstrated that NLRP3 inflammatory vesicles play a crucial role in the development of atherosclerosis as well as its complications. Several studies have shown that NLRP3 binding to ligands promotes inflammasome formation, activates caspase-1, and ultimately promotes its maturation and the maturation and production of IL-1β and IL-18. IL-1β and IL-18 are considered to be the two most prominent inflammatory cytokines in the inflammasome that promote the development of atherosclerosis. SGLT2 inhibitors are novel hypoglycemic agents that also have significant antiatherosclerotic effects. However, their exact mechanism is not yet clear. This article is a review of the literature on the effects and mechanisms of SGLT2 inhibitors on the NLRP3 inflammasome, focusing on their role in antiatherosclerosis.
Atherosclerosis is a lipid-driven chronic inflammatory disease that is widespread in the walls of large and medium-sized arteries (1, 2). In recent years, fatal vascular diseases caused by atherosclerosis have included stroke, acute myocardial infarction (MI), and severe peripheral vascular disease (3, 4). Pathological changes in atherosclerosis include endothelial damage, lipid deposition, macrophages, the formation of foam cells and the proliferation and migration of smooth muscle cells (SMCs) (5). Previous research has shown that the complex pathogenesis of atherosclerosis includes activation of proinflammatory signaling pathways in the body, increased oxidative stress, and increased expression of cytokines/chemokines (6). Its main causes include lipid accumulation in the arterial wall and chronic inflammation (2, 7, 8). Diabetes has been shown to be an independent risk contributor to the accelerated progression of atherosclerosis (9–11). Research has shown that the rate of vascular disorders in T2DM patients is two to four times higher than that in nondiabetic patients (12). Diabetic patients are prone to atherosclerosis, which is influenced by hyperglycemia, advanced glycosylation end product production, dyslipidemia, inflammation, insulin resistance, endothelial dysfunction and oxidative stress (4, 9, 13).
The results of several recent studies have shown that inflammation is crucial for the onset and progression of atherosclerosis and its complications (1, 14). The existence of inflammasomes was first demonstrated by Martinon et al. in 2002 (15). It is a protein complex that is composed of pattern recognition receptors (PRRs) activated by various physiological or causative stimuli (16). Pattern recognition receptors (PRRs) target pathogenicity in the innate immune response. The inflammasome is an essential part of the innate immune system and has the capacity to promote the expression and maturation of inflammatory factors, release large amounts of inflammatory cytokines, trigger a cascade of inflammatory responses and clear pathogens and damaged cells (17, 18). The most typically characterized and widely studied inflammasomes are the nucleotide binding domain and leucine-rich repeat containing family pyrin domain containing 3 (NLRP3) (19). NLRP3 is an innate immune cell sensor (20). It identifies nonmicrobiological red flags and causes and promotes a bacterial inflammatory reaction in different disease conditions (17, 21, 22). The NLRP3 inflammasome consists of three parts (8, 23–26): (1) The sensor molecule NLRP3 (a toll-like receptor), which includes three structural domains: a pyrin structural domain (PYD), a nucleoside oligomerization structural domain (NACHT) and a leucine-rich repeat sequence structural domain (LRR). (2) Apoptotic spot-like protein (ASC), containing an N-terminal pyrin domain (PYD) and a C-terminal PYD recruitment domain (CARD), also called Pycard. (3) Procaspase-1 contains a CARD and a catalytic structural domain (Caspase-1). ASC plays a critical linkage role between receptor NLRP3 and effector caspase-1 due to the lack of pyrin domain in caspase-1 (27). The NLRP3 inflammasome is a concentric circle-shaped tissue. The NLRP3 protein is located in the middle of the circle, and the ASC layer surrounds the NLRP3 protein. Caspase-1 is located in the outermost layer and is attached to the ASC layer (17, 28). The body can regulate the activation of the NLRP3 inflammasome at multiple levels, and in macrophages, two independent signals are usually required (29). First, inflammatory factors induce NLRP3 and pro-IL-1β expression through stimulation of nuclear factor-κβ (NF-κβ) (30, 31). When pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) are activated, this leads to the interaction of PYD of NLRP3 inflammatory vesicle proteins with PYD of ASC. The accumulation of these structural domains eventually leads to the release of active caspase-1, converting inactive procaspase-1 to active caspase-1 through the action of NLRP3 inflammatory vesicles, thereby upregulating multiple inflammatory cytokines and initiating defense mechanisms (29, 32). Inactive pro-interleukin-1β (Pro-IL-1β) can be converted to active IL-1β by caspase-1 and released from cells to mediate inflammatory responses in tissues (25, 30, 33–35). Active caspase-1 is able to cleave gas protein D (GSDMD), causing the N-terminal structural domain of GSDMD to form a pore in the plasma membrane, which triggers cell lysis until death, also known as pyroptosis (36–39). Pyroptosis is a form of cell death that is distinct from apoptosis. It forces the exposure of intracellular pathogens to other immune factors and triggers the release of cytokines and the production of DAMPs to enhance the immune system’s response to infection (29, 38, 40). Activation of NLRP3 inflammatory vesicles is directly associated with the pathophysiology of chronic inflammatory diseases, such as diabetes, and its associated complications (29, 41–43). Wan et al. found that NLRP3 expression levels and plasma IL-1β levels are dramatically higher in PBMCs from diabetic individuals than in those from healthy controls (44). In addition, aberrant activation of the NLRP3 inflammasome is related to several types of inflammatory diseases, including obesity (45), diabetes (46, 47), atherosclerosis (8, 48, 49), nonalcoholic steatohepatitis (50), gout (51–53), and Alzheimer’s disease (54, 55).
The NLRP3 inflammasome is mainly expressed in monocytes, macrophages, smooth muscle cells, endothelial cells, and dendritic cells (24, 56, 57). Nevertheless, studies of atherosclerosis have concentrated on the activation of inflammatory vesicles in monocytes, macrophages and vascular smooth muscle cells (24). Vascular smooth muscle cells (VSMCs) are the mesangial cells of coronary arteries (58). In the early stage of atherosclerosis, activated vascular smooth muscle cells have a good ability to proliferate and migrate. These cells can migrate from the arterial mesothelium to the intima, forming fibrous cap-like structures. The fibrous cap secretes extracellular matrix, which buries lipids in deeper layers and prevents plaque degradation. Massive lipid deposition in plaques leads to activation of the NLRP3 inflammatory vesicle response and exacerbation of the inflammatory response, ultimately leading to plaque necrosis (59). It has been shown that in foam cells and macrophages, NLRP3 inflammatory vesicles are mainly localized in the cytoplasm and are associated with intracellular and extracellular crystallization of cholesterol crystals (60). In macrophages, activation of NLRP3 inflammatory vesicles can stimulate the formation of cholesterol crystals, which are necessary for the development of atherosclerosis (49); activation is an important mechanism driving the development of atherosclerotic inflammation (61).
The NLRP3 inflammasome plays an important role in the molecular etiology of atherosclerosis (48, 49, 62, 63). Multiple studies in atherosclerotic patients and animal models have shown that the NLRP3 inflammasome can increase IL-1β and IL-18 production, leading to the progression and instability of atherosclerotic plaques (64). Wan et al. (44) applied NLRP3 knockout technology to inhibit NLRP3 inflammasome activation and the expression of adhesion molecules ICAM-1 and vascular cell adhesion molecule-1 (VCAM-1) in the intima, reducing atherosclerosis and stabilizing atherosclerotic plaques in a diabetic atherosclerosis mouse model. Zheng et al. (65) showed that silencing of the NLRP3 gene delayed the progression of atherosclerosis in mice by administering NLRP3-RNA lentiviral suspension to ApoE-/- mice fed a high-fat diet, mainly by decreasing the plaque content of macrophages and increasing the plaque content of smooth muscle cells. The mRNA levels of NLRP3 inflammasome-related genes are significantly increased in human atherosclerotic plaques compared to nonatherosclerotic vessels, and particularly high expression is observed in patients with symptomatic lesions (66). NLRP3 can be overexpressed in the aorta of patients with atherosclerosis, making it an important risk factor for the development of and correlation with the severity of coronary artery disease (67). NLRP3, ASC, caspase-1, IL-1β, and IL-18 levels are differentially elevated in unstable carotid atherosclerotic plaques in patients undergoing carotid endarterectomy (60, 66, 68). NLRP3 inflammasomes have been shown to be activated by a variety of stimuli, including ion flux, mitochondrial dysfunction, reactive oxygen species production, and lysosomal damage (16, 69, 70). The mechanisms of NLRP3 inflammasome activation are quite complex, and how NLRP3 responds to these signaling events and initiates the assembly of the NLRP3 inflammasome is not fully understood (69). A variety of targets have been used to develop therapeutic strategies. For example, they inhibit the activity of upstream signaling pathways, block the assembly and activation of the NLRP3 inflammasome, inhibit caspase-1 activation and secretion of IL-1 and IL-18 factors (5).
Several studies have shown that NLRP3 binding to ligands promotes inflammasome formation, activates caspase-1, and ultimately promotes its maturation and the maturation and secretion of IL-1β and IL-18 (66, 71, 72). IL-1β and IL-18 have important roles in the pathogenesis of atherosclerosis (73).
The interleukin 1 (IL-1) family acts on almost all tissues and cells throughout the body. It plays a key role in innate immunity and inflammatory responses (74). It is a key mediator of inflammatory, autoimmune, infectious and degenerative responses (75). Among all IL-1 family cytokines, IL-1α, IL-1β, IL-18 and IL-1Ra are the most extensively studied members (48). In recent years, interleukin-1β (IL-1β) and interleukin-18 (IL-18) have been identified as the two most important inflammatory cytokines that promote the development of atherosclerosis (76).
IL-1β is known to be a key cytokine in atherogenesis (77, 78). It is an inducible cytokine that is mainly produced by monocytes and macrophages as well as neutrophils (74). IL-1β acts mainly as a soluble mediator outside the cell and can act on tissues and organs at a distance. IL-1β gene expression is low or absent in blood mononuclear cells in healthy populations but significantly increased in disease states (79). Knockout of the IL-1β gene in ApoE-/- mice significantly reduces atherosclerotic plaque development (80). Ablation of the IL-1 receptor (IL-1R) attenuates plaque progression in atherosclerosis-prone mice (81). Active IL-1β has the following main effects: a. Significantly increases the lifespan and activity of neutrophils and macrophages (82) and induces lytic enzymes and fibroblast proliferation (83). b. Induces and controls the expression of genes related to fever, pain threshold, and vasodilation, leading to vascular endothelial cell responses and promoting immune cell responses to infected or injured tissues (84). c. Binding to IL-1R stimulates the activation of the NF-κβ and mitogen-activated protein kinase (MAPK) pathways (85). d. Stimulates the secretion of a range of other cytokines (86) and induces the production of endothelin-1 and adherence molecules in the endothelium, facilitating leukocyte migration and maintaining the cycle of inflammation (87).
A growing number of studies have demonstrated the critical role of IL-18 in atherosclerosis. IL-18 is related to IL-1β both biologically and structurally. Similar to IL-1β, it is generated as an inactive precursor that requires cleavage by caspase-1 to mature into a biologically active cytokine (88). In contrast, unlike IL-1β, IL-18 is constitutively expressed (89). IL-18 binding protein (BP) is an IL-18-specific inhibitor. It is a unique soluble protein that is mainly derived from endothelial cells and monocytes/macrophages. Structurally, IL-18BP has an immunoglobulin (Ig) structural domain (90). IL-18BP binds mature IL-18 (but not pro-IL-18) with high affinity and blocks its interaction with cell surface receptors, thus acting as a natural inhibitor (91). Circulating IL-18BP levels in healthy individuals range from 0.5-7 ng/ml, while elevated IL-18BP levels have been described in a number of autoimmune or inflammatory diseases (92–94). It has been shown that IL-18BP-expressing plasmid DNA prevents the development of fatty streaks in the thoracic aortas of apoE knockout mice and slows the progression of atherosclerotic plaques. IL-18BP has a high binding affinity for IL-18, and IL-18BP is an important regulator of immune and inflammatory responses in IL-18-related diseases (72). In ApoE-/- mouse models, IL-18 has been shown to promote atherogenesis via an interferon-γ (IFN-γ)-dependent pathway (95). In contrast, IL-18-deficient ApoE-/- mice show reduced atherosclerotic plaque extension (96). In addition, IL-18 gene variants affect clinical outcomes in patients with coronary artery disease (97). Atherosclerotic lesions are smaller in IL-18 gene-deficient mice (98). Overexpression of IL-18BP and IL-18 deletion in ApoE-/- mice blocked IL-18 activity in vivo, leading to impaired development of atherosclerotic injury (48). Studies have shown that IL-18R-/- mice exhibit increased body weight, ectopic lipid deposition, increased inflammation and diminished AMPK signaling pathways in skeletal muscle (99). Elevated IL-18 levels were found in obese and type 2 diabetic patients (100, 101). IL-18 is a costimulatory cytokine that mediates adaptive immunity and is required for interferon-gamma (IFN-γ) production (84).
The NLRP3 inflammasome plays an important role in the pathogenesis of atherosclerosis and may therefore be a promising target for therapeutic approaches. Drugs known to date to have targeted inhibitory effects on NLRP3 inflammasomes include statins, metformin (102), colchicine (74), plant compounds (artemisinin, curcumin, rosmarinic acid) (73), and the specific small molecule NLRP3 inhibitor MCC950 (103).
SGLTs are membrane proteins on renal tubular cells whose main role is to transport certain ions and small molecules that mediate glucose reabsorption in the kidney. There are two types of human SGLTs, SGLT1 and SGLT2. SGLT2 is a low-affinity, high-volume cotransporter that is mainly located in the S1 part of the renal proximal tubule and completes approximately 90% of glucose reabsorption. SGLT1 is mainly expressed in the S2 and S3 segments of the brush border of the small intestine and renal proximal tubule and is responsible for glucose transport in the intestinal lumen and reabsorption of the 10% of glucose not reabsorbed by SGLT2 in the renal proximal tubule (104, 105). The site of action of SGLT2 inhibitors is highly specific for the inhibition of renal glucose reabsorption. Sodium-glucose cotransporter-2 inhibitor (SGLT2i) is a blood glucose control drug for the treatment of diabetes. It targets renal glucose reabsorption in an insulin-independent manner to exert its unique hypoglycemic effect (106). The selectivity of SGLT2i for SGLT2 correlated well between in vivo and in vitro studies (r = 0.985; P < 0.05) (107). SGLT2i regulate glycemic control by enhancing glycosuria, osmotic diuresis and urinary sodium excretion and exert additional positive effects, such as weight loss (108). SGLT2 inhibitors are antiatherosclerotic mainly through several aspects: improving endothelial dysfunction, improving vascular smooth muscle dysfunction, reducing macrophage inflammation and foam cell formation, reducing oxidative stress, reducing inflammation, promoting autophagy, increasing ketone bodies, reducing body weight, lowering blood pressure, and lowering uric acid levels (10, 109–111). Four SGLT2i have been approved for marketing: canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin. A network meta-analysis of randomized controlled trials (RCTs) suggests that engramine may be superior to other SGLT2 inhibitors and has a lower risk of all-cause mortality and cardiovascular events in patients with T2DM (112). However, the main drugs known to have anti-inflammatory effects are dapagliflozin (113) and empagliflozin (114). How SGLT2 inhibitors alter the inflammatory process is not fully understood. Current studies on the effects of SGLT2i on NLRP3 inflammatory vesicles have focused on diabetic nephropathy (17, 115, 116), atherosclerosis (25, 110), steatohepatitis (117), and cardiomyopathy (118).
A study showed that SGLT2i treatment in diabetic mice improved atherosclerotic plaque regression while lowering blood glucose levels (119). After 30 days of SGLT2i treatment in patients with T2D combined with high cardiovascular risk, NLRP3 inflammasome activity in macrophages was dramatically attenuated, while IL-1β secretion was markedly reduced (120). This may be related to its ability to inhibit atherosclerosis by improving poor glucose tolerance associated with suppression of inflammation (121). SGLT2i can significantly inhibit the formation and development of aortic lesions in diabetic ApoE−/− mice and delay the formation of diabetic atherosclerosis (25). It has been shown that SGLT2 inhibitors can act directly on inflammatory pathways independent of hypoglycemic pathways (111). SGLT2 inhibitors are safe and well tolerated in adults who are overweight and obese but do not have diabetes (122). Under normal glucose conditions, SGLT2i inhibits SMC migration and proliferation by targeting il-17a-mediated oxidative stress, NLRP3 expression and inflammatory responses without inducing cell death (123). Synthesizing the current literature as well as research results, we summarized the effects and mechanisms of SGLT2i on the NLRP3 inflammasome in atherosclerosis treatment (Figure 1).
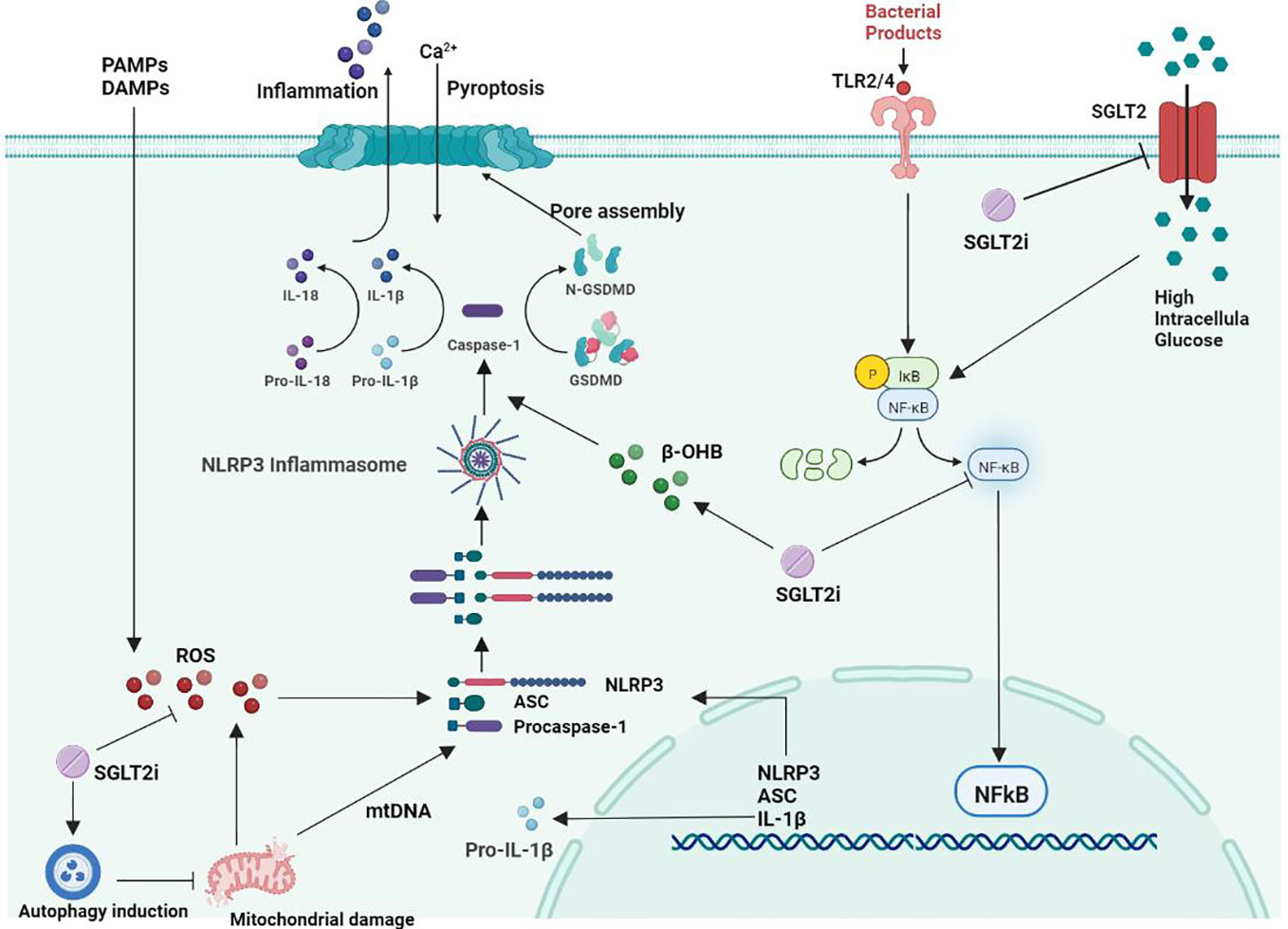
Figure 1 Mechanism of the effect of SGLT2 inhibitors on NLRP3 inflammatory vesicles in atherosclerosis.
Oxidative stress is a state of imbalance in the body that favors increased production of reactive oxygen species (ROS) and reduced antioxidant defense systems, resulting in abnormal cell signaling and dysfunction (124). As research progresses, there is increasing evidence that ROS play a more important role in atherogenesis than natural LDL. ROS play an essential role in the inflammatory response, apoptosis, cell growth, alterations in vascular tone, and oxidation of LDL cholesterol (125). Oxidative stress has emerged as a key factor in the pathogenesis of atherosclerosis (6). Studies have demonstrated that ROS generation in the vessel wall is increased in people with risk elements for atherosclerotic cardiovascular disease (CVD) (126). The importance of ROS for macrophage-mediated immune responses is unquestionable (127). Obesity can cause inflammation and insulin resistance, the key cause of which is the recruitment and polarization of macrophages (128). ROS play a key role in NLRP3 inflammasome activation (129, 130), and excessive ROS production is associated with vascular injury. All NLRP3 agonists trigger the production of ROS, which activate NLRP3 inflammatory vesicles through interaction with ROS-sensitive thioredoxin reductase (TXNIP) (25).
DiMarco et al. demonstrated that accelerated atherosclerosis in diabetes is associated with elevated ROS (131). SMC proliferation and migration play a key role in the pathogenesis of atherosclerosis. Persistent inflammation and oxidative stress are also involved in the development of vasoproliferative diseases. In a study by Sukhanov et al., human aortic smooth muscle cells (SMCs) were found to express SGLT2 mRNA and protein, and the application of SGLT2i treatment under normal glucose conditions reduced oxidative stress, NLRP3 expression, SMC migration and proliferation and did not induce cell death (123). Leng et al. showed that SGLT2i reduces the development of atherosclerosis in the aortic root, mainly by lowering blood glucose and lipids to inhibit the ROS-NLRP3-caspaspase-1 pathway in macrophages and reduce IL-1β and IL-18 production (25). In another study, Leng et al. effectively prevented hepatocyte inflammation by treating double HFD/STZ-fed ApoE-/- mice with SGLT2i, suggesting that SGLT2i may be involved in the inhibitory activity of ROS-NLRP3 inflammatory vesicles (117).
Sodium-glucose cotransporter 2 (SGLT2) is a transmembrane protein that transports large amounts of glucose from the extracellular to the intracellular compartment in the diabetic setting. Excess intracellular glucose can induce the activation of NF-kB, ultimately leading to increased expression of the proinflammatory molecule HMGB1 (132). HMGB1 is a DNA-binding protein in the nucleus, and when cells are activated to release HMGB1, it can act as a strong mediator of inflammation by inducing activation of its surface receptors RAGE and TLR-4, thereby increasing the activity of the NF-kB signaling pathway. SGLT2 inhibition may improve ROS, lipid peroxidation and NLRP3-related pathways by attenuating glucose accumulation in renal tubular cells and attenuating HMGB1 and RAGE/TLR-4 expression, thereby reducing NF-κB signaling pathway activity (133–135). Activation of NLRP3 inflammatory vesicles involves multiple signaling pathways, and the pathways interact with each other, of which the NF-κB signaling pathway is an essential part of the NLRP3 activation process (136). In atherosclerotic pathology, NF-κB regulates the expression of several genes, including cytokines (TNF-α, IL-1 and IL-6), monocyte chemotactic proteins and adhesion protein molecules (137). NF-κB is involved in the inflammatory response by binding to NF-κB inhibitor (IκB) retained in the cytoplasm, which leads to the formation of atherosclerotic plaques as well as plaque instability and rupture (138). Activation of NF-κB can induce the production of pro-IL-1β and increase the synthesis of NLRP3 (19). The effect of SGLT2 inhibitors on the NLRP3 inflammasome may be related to its inhibition of the NF-κB signaling pathway. Abdollahi et al. showed that SGLT2 inhibitors can exert direct anti-inflammatory effects independent of glucose concentration, at least in part through inhibition of TLR-4 expression and NF-κB activation and the secretion of proinflammatory mediators (139). Xu et al. showed that SGLT2i ameliorated inflammatory changes induced by NF-κB pathway inhibition in diabetic proximal renal tubular cells with mature IL-1β, IL-6 and TNF-α expression (115). By treating ApoE -/- mice that were induced with atherosclerosis with SGLT2 inhibitors, Liu et al. found that SGLT2 inhibitors significantly reduced inflammation levels in vivo, mainly by modulating NF-κB signaling to inhibit IL-1β expression in oxLDL-treated macrophages (140). The above studies suggest that SGLT2 inhibitors can exert their anti-inflammatory effects by inhibiting the NF-κB signaling pathway, and this anti-inflammatory effect is associated with restricted activation of the NLRP3 inflammasome.
Autophagy refers to a mechanism by which intracellular macromolecules (such as organelles and protein aggregates) are broken down into their component parts and recycled within the lysosome (141). Defective or dysfunctional components of the cell are degraded. When mitochondrial autophagy removal is impaired, it can lead to reduced release of mitochondria-derived DAMPs and inhibit inflammasome activation (142). The autophagic status also influences the development and progression of atherosclerosis. The autophagy-deficient environment can exacerbate cholesterol crystal-mediated hyperactivation of macrophage inflammasomes and their atherogenic IL-1β response. Previous studies have shown that macrophage autophagy is impaired in atherosclerotic lesions in low-density lipoprotein receptor- or apolipoprotein E-knockout mice (143, 144).
Currently, an increasing number of studies are focusing on the relationship between NLRP3 inflammatory vesicles and autophagy. The mechanism of autophagy inhibition by NLRP3 inflammatory vesicles may lead to ASC reduction, NLRP3 phosphorylation and mitochondrial ROS clearance. Inhibition of cellular autophagy can lead to the accumulation of damaged mitochondria and ROS, which ultimately positively affects NLRP3 inflammasome activity, thereby mediating the inflammatory response (129). Autophagy has been suggested to be the ultimate cellular degradation system of the NLRP3 inflammasome (145). Numerous studies have further shown that autophagy can regulate inflammasome activation through multiple mechanisms, including the NLRP3 inflammasome (146–148). An SGLT2 inhibitor can restore phosphorylated AMPK and autophagy levels induced by high glucose in a dose-dependent manner, thereby inhibiting the activation of the NLRP3 inflammasome (115). Multiple studies have shown that SGLT2 inhibitor-mediated upregulation of autophagy can reduce NLRP3 inflammasome expression, thereby attenuating cardiomyocyte dysfunction and endothelial damage (33, 149).
Unlike other hypoglycemic agents, specific SGLT2i inhibit glucose reabsorption by proximal renal tubular cells, and the hypoglycemic effect can be independent of pancreatic cell function and insulin sensitivity. This insulin-independent decrease in blood glucose levels reduces the body’s need for insulin and induces an increase in the glucagon-to-insulin ratio (150). Systemic energy metabolism shifts to relative glucose deficiency and triggers increased lipolysis in adipocytes, fatty acid oxidation, and ketone body production in the liver (151, 152). It has been shown that SGLT2i can cause more than doubling of the rate of WAT lipolysis in rats (153), and WAT lipolysis can produce large amounts of β-hydroxybutyric acid, acetoacetic acid and acetone (154). SGLT2 inhibitors stimulate lipolysis and induce mild ketogenic effects in patients with type 2 diabetes (155, 156). β-hydroxybutyric acid (β-OHB) is an endogenous NLRP3 inflammasome inhibitor that can reduce the inflammatory response (157). β-OHB is an NLRP3 inflammasome inhibitor that can reduce NLRP3 inflammatory vesicle-mediated production of IL-1β and IL-18 in human monocytes (158). Bae et al. demonstrated that β-OHB can inhibit endoplasmic reticulum stress and NLRP3 inflammasome activity in rats by activating the AMPK pathway (159). Youm et al. found that β-OHB inhibited NLRP3 inflammasome activation and reduced macrophage and IL-1β production to stop the progression of atherosclerosis (160). However, supplementation, such as ketone supplements, can lead to the acute elevation of β-OHB in the blood, which can significantly increase the activation of caspase-1 and the secretion of the proinflammatory cytokine IL-1β in whole blood (62, 161–163). However, if supplementation with ketone supplements, for example, leads to an acute elevation of β-OHB in the blood, it can significantly increase activation of caspase-1 and increase secretion of the proinflammatory cytokine IL-1β in whole blood (161–163). Multiple studies (120, 164) have shown that in patients with T2D combined with CVD, SGLT2 inhibitors significantly inhibit NLRP3 inflammasome activation and IL-1β secretion in human macrophages by increasing serum β-OHB levels and decreasing serum insulin, glucose, and uric acid levels.
In addition, SGLT2i can affect atherosclerosis as well as cardiovascular events through multiple actions. Tighter glycemic control through SGLT2i treatment can reduce the number of monocytes, improve plasma lipoprotein profiles, and have a significant positive impact on the inhibition of atherosclerosis (165). SGLT2 inhibitors reduce the rate of major adverse cardiovascular events and heart failure hospitalizations in patients with T2DM, regardless of the presence of CVD (166, 167). SGLT2 inhibitors reduce the risk of new ventricular arrhythmic events in patients with T2DM combined with AMI (168). DAPA reduces intima-media thickening, eliminates cardiac hypertrophy and myocardial injury, reduces cardiac inflammation and fibrosis (169), and has an effect on myocardial remodeling (170). In addition, engramine may reduce debilitating conditions in patients with diabetes and hypertension (171). During treatment with SGLT2 inhibitors, a beneficial effect on the circadian rhythm of BP and sympathetic nerve activity (SNA) was demonstrated, resulting in a decrease in BP without a concomitant compensatory increase in HR (172). Treatment with SGLT2 inhibitors in T2DM patients treated with coronary artery bypass grafting (CABG) significantly reduced the amount of inflammatory factors such as IL-1, IL-6 and TNF-α (173).
In summary, NLPR3 inflammasome production has an important role in promoting the development of atherosclerosis. SGLT2 inhibitors can inhibit the development of atherosclerotic plaques by inhibiting NLRP3 inflammatory vesicles through multiple pathways. However, there may be crosstalk between the above mechanisms; for example, increased intracellular ROS levels can promote the activation of the NF-κB signaling pathway and the initiation of cellular autophagy, cellular autophagy can reduce intracellular ROS levels and inhibit the activation of the NF-κB signaling pathway, and β-OHB can promote myocardial autophagic flux and reduce the formation of mitochondrial ROS. A variety of SGLT2i have been shown to attenuate atherosclerotic lesions in animal models of diabetes (25, 174–176), and overall trial data estimated a relative reduction in the incidence of MACE (HR 0.89, 95% CI 0.82-0.96) and stroke (HR 0.92, 95% CI 0.79-1.08) of approximately 11% (177). In diabetic ApoE knockout (ApoE-/-) mice, SGLT2i (1 mg/kg/day) increased the aortic root atherosclerotic lesion area by 33% and reduced the atherosclerotic plaque size by 27% (178). Drugs specific for the treatment of atherosclerosis are still under further investigation. Indeed, the potential mechanism of SGLT2 inhibitors on the CV system is unclear (110). The results of the EMPA-REG study (179) suggest that SGLT2i activate a nonclassical RAAS pathway, namely, the angiotensin II type 2 receptor. Activation of angiotensin II type 2 receptors protects the cardiovascular system through multiple mechanisms, mainly including vasodilation, increased sodium excretion, anti-inflammation, and anti-arrhythmia. It has also been shown that SGLT2i may reduce cardiovascular (CV) risk in patients with type 2 diabetes mellitus (T2DM) by affecting the aldosterone/renin ratio (ARR) through diuretic and sympathetic depressant effects (104). An SGLT2 inhibitor, as a hypoglycemic agent, also inhibits atherosclerosis, but its mechanism and therapeutic effect still need more basic research and clinical observation to clarify. What is certain is that patients with type 2 diabetes mellitus combined with atherosclerosis benefit more from SGLT2 inhibitors.
LY wrote the first draft of the manuscript; LY and XZ for the writing, review and editing; QW revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kobiyama K, Ley K. Atherosclerosis. Circ Res (2018) 123:1118–20. doi: 10.1161/CIRCRESAHA.118.313816
2. Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol (2006) 47(8 Suppl):C7–12. doi: 10.1016/j.jacc.2005.09.068
3. Saraiva JFK, Franco D. Oral GLP-1 analogue: perspectives and impact on atherosclerosis in type 2 diabetic patients. Cardiovasc Diabetol (2021) 20:235. doi: 10.1186/s12933-021-01417-0
4. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res (2016) 118:535–46. doi: 10.1161/CIRCRESAHA.115.307611
5. Jiang C, Xie S, Yang G, Wang N. Spotlight on NLRP3 inflammasome: Role in pathogenesis and therapies of atherosclerosis. J Inflamm Res (2021) 14:7143–72. doi: 10.2147/JIR.S344730
6. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep (2017) 19:42. doi: 10.1007/s11883-017-0678-6
7. Miname MH, Santos RD. Reducing cardiovascular risk in patients with familial hypercholesterolemia: Risk prediction and lipid management. Prog Cardiovasc Dis (2019) 62:414–22. doi: 10.1016/j.pcad.2019.10.003
8. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J Cell Physiol (2018) 233:2116–32. doi: 10.1002/jcp.25930
9. Poznyak A, Grechko AV, Poggio P, Myasoedova VA, Alfieri V, Orekhov AN. The diabetes mellitus-atherosclerosis connection: The role of lipid and glucose metabolism and chronic inflammation. Int J Mol Sci (2020) 21(5):1835. doi: 10.3390/ijms21051835
10. Salvatore T, Caturano A, Galiero R, Di Martino A, Albanese G, Vetrano E, et al. Cardiovascular benefits from gliflozins: Effects on endothelial function. Biomedicines (2021) 9(10):1356. doi: 10.3390/biomedicines9101356
11. McCormick LM, Kydd AC, Dutka DP. Cardiac protection via metabolic modulation: an emerging role for incretin-based therapies? Cardiovasc Hematol Agents Med Chem (2012) 10:319–24. doi: 10.2174/187152512803530360
12. Bertoluci MC, Rocha VZ. Cardiovascular risk assessment in patients with diabetes. Diabetol Metab Syndr (2017) 9:25. doi: 10.1186/s13098-017-0225-1
13. Gimbrone MA Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
14. Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci (Lond) (2018) 132:1243–52. doi: 10.1042/CS20180306
15. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
16. Prochnicki T, Mangan MS, Latz E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Res (2016) 5:F1000 Faculty Rev-1469. doi: 10.12688/f1000research.8614.1
17. Yaribeygi H, Katsiki N, Butler AE, Sahebkar A. Effects of antidiabetic drugs on NLRP3 inflammasome activity, with a focus on diabetic kidneys. Drug Discovery Today (2019) 24:256–62. doi: 10.1016/j.drudis.2018.08.005
18. Sharma D, Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol (2016) 213:617–29. doi: 10.1083/jcb.201602089
19. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
20. Hong P, Gu RN, Li FX, Xiong XX, Liang WB, You ZJ, et al. NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J Neuroinflamm (2019) 16:121. doi: 10.1186/s12974-019-1498-0
21. Amin J, Boche D, Rakic S. What do we know about the inflammasome in humans? Brain Pathol (2017) 27:192–204. doi: 10.1111/bpa.12479
22. Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science (2010) 327:296–300. doi: 10.1126/science.1184003
23. Silvis MJM, Demkes EJ, Fiolet ATL, Dekker M, Bosch L, van Hout GPJ, et al. Immunomodulation of the NLRP3 inflammasome in atherosclerosis, coronary artery disease, and acute myocardial infarction. J Cardiovasc Transl Res (2021) 14:23–34. doi: 10.1007/s12265-020-10049-w
24. Liaqat A, Asad M, Shoukat F, Khan AU. A spotlight on the underlying activation mechanisms of the NLRP3 inflammasome and its role in atherosclerosis: A review. Inflammation (2020) 43:2011–20. doi: 10.1007/s10753-020-01290-1
25. Leng W, Ouyang X, Lei X, Wu M, Chen L, Wu Q, et al. The SGLT-2 inhibitor dapagliflozin has a therapeutic effect on atherosclerosis in diabetic ApoE(-/-) mice. Mediators Inflamm (2016) 2016:6305735. doi: 10.1155/2016/6305735
26. Drenth JP, van der Meer JW. The inflammasome–a linebacker of innate defense. N Engl J Med (2006) 355:730–2. doi: 10.1056/NEJMcibr063500
27. Bryant C, Fitzgerald KA. Molecular mechanisms involved in inflammasome activation. Trends Cell Biol (2009) 19:455–64. doi: 10.1016/j.tcb.2009.06.002
28. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U.S.A. (2014) 111:7403–8. doi: 10.1073/pnas.1402911111
29. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi: 10.1038/nm.3806
30. Seok JK, Kang HC, Cho YY, Lee HS, Lee JY. Regulation of the NLRP3 inflammasome by post-translational modifications and small molecules. Front Immunol (2020) 11:618231. doi: 10.3389/fimmu.2020.618231
31. Qiao Y, Wang P, Qi J, Zhang L, Gao C. TLR-induced NF-kappaB activation regulates NLRP3 expression in murine macrophages. FEBS Lett (2012) 586:1022–6. doi: 10.1016/j.febslet.2012.02.045
32. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
33. Cao Z, Wang Y, Long Z, He G. Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim Biophys Sin (Shanghai) (2019) 51:1087–95. doi: 10.1093/abbs/gmz098
34. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis (2019) 10:128. doi: 10.1038/s41419-019-1413-8
35. Broz P, Dixit VM. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
36. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
37. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
38. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin d is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
39. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
40. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol (2010) 11:1136–42. doi: 10.1038/ni.1960
41. Dixit VD. Nlrp3 inflammasome activation in type 2 diabetes: is it clinically relevant? Diabetes (2013) 62:22–4. doi: 10.2337/db12-1115
42. De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol (2011) 32:373–9. doi: 10.1016/j.it.2011.05.004
43. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol (2010) 11:897–904. doi: 10.1038/ni.1935
44. Wan Z, Fan Y, Liu X, Xue J, Han Z, Zhu C, et al. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes Metab Syndr Obes (2019) 12:1931–42. doi: 10.2147/DMSO.S222053
45. Han JH, Shin H, Rho JG, Kim JE, Son DH, Yoon J, et al. Peripheral cannabinoid 1 receptor blockade mitigates adipose tissue inflammation via NLRP3 inflammasome in mouse models of obesity. Diabetes Obes Metab (2018) 20:2179–89. doi: 10.1111/dom.13350
46. Wu D, Yan ZB, Cheng YG, Zhong MW, Liu SZ, Zhang GY, et al. Deactivation of the NLRP3 inflammasome in infiltrating macrophages by duodenal-jejunal bypass surgery mediates improvement of beta cell function in type 2 diabetes. Metabolism (2018) 81:1–12. doi: 10.1016/j.metabol.2017.10.015
47. Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc Natl Acad Sci U.S.A. (2015) 112:11318–23. doi: 10.1073/pnas.1513509112
48. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res (2018) 122:1722–40. doi: 10.1161/CIRCRESAHA.118.311362
49. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464:1357–61. doi: 10.1038/nature08938
50. Kim SH, Kim G, Han DH, Lee M, Kim I, Kim B, et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy (2017) 13:1767–81. doi: 10.1080/15548627.2017.1356977
51. Dalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. Lancet (2021) 397:1843–55. doi: 10.1016/S0140-6736(21)00569-9
52. Dalbeth N, Choi HK, Joosten LAB, Khanna PP, Matsuo H, Perez-Ruiz F, et al. Gout. Nat Rev Dis Primers (2019) 5:69. doi: 10.1038/s41572-019-0115-y
53. So AK, Martinon F. Inflammation in gout: mechanisms and therapeutic targets. Nat Rev Rheumatol (2017) 13:639–47. doi: 10.1038/nrrheum.2017.155
54. Pawlos A, Broncel M, Wozniak E, Gorzelak-Pabis P. Neuroprotective effect of SGLT2 inhibitors. Molecules (2021) 26(23):7213. doi: 10.3390/molecules26237213
55. Wang BR, Shi JQ, Ge NN, Ou Z, Tian YY, Jiang T, et al. PM2.5 exposure aggravates oligomeric amyloid beta-induced neuronal injury and promotes NLRP3 inflammasome activation in an in vitro model of alzheimer's disease. J Neuroinflamm (2018) 15:132. doi: 10.1186/s12974-018-1178-5
56. Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol (2010) 10:210–5. doi: 10.1038/nri2725
57. Centola M, Wood G, Frucht DM, Galon J, Aringer M, Farrell C, et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood (2000) 95:3223–31. doi: 10.1182/blood.V95.10.3223
58. Fang L, Wang KK, Zhang PF, Li T, Xiao ZL, Yang M, et al. Nucleolin promotes ang II-induced phenotypic transformation of vascular smooth muscle cells by regulating EGF and PDGF-BB. J Cell Mol Med (2020) 24:1917–33. doi: 10.1111/jcmm.14888
59. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
60. Shi X, Xie WL, Kong WW, Chen D, Qu P. Expression of the NLRP3 inflammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis (2015) 24:2455–66. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024
61. Jin Y, Fu J. Novel insights into the NLRP 3 inflammasome in atherosclerosis. J Am Heart Assoc (2019) 8:e012219. doi: 10.1161/JAHA.119.012219
62. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21:677–87. doi: 10.1038/nm.3893
63. Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin-1 in heart disease. Circulation (2013) 128:1910–23. doi: 10.1161/CIRCULATIONAHA.113.003199
64. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol (2015) 6:262. doi: 10.3389/fphar.2015.00262
65. Zheng F, Xing S, Gong Z, Mu W, Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein e-deficient mice. Mediators Inflammation (2014) 2014:507208. doi: 10.1155/2014/507208
66. Paramel Varghese G, Folkersen L, Strawbridge RJ, Halvorsen B, Yndestad A, Ranheim T, et al. NLRP3 inflammasome expression and activation in human atherosclerosis. J Am Heart Assoc (2016) 5(5):e003031. doi: 10.1161/JAHA.115.003031
67. Zheng F, Xing S, Gong Z, Xing Q. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ (2013) 22:746–50. doi: 10.1016/j.hlc.2013.01.012
68. Wang R, Wang Y, Mu N, Lou X, Li W, Chen Y, et al. Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab Invest (2017) 97:922–34. doi: 10.1038/labinvest.2017.30
69. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20(13):3328. doi: 10.3390/ijms20133328
70. Lee MS. Role of innate immunity in diabetes and metabolism: recent progress in the study of inflammasomes. Immune Netw (2011) 11:95–9. doi: 10.4110/in.2011.11.2.95
71. Cassel SL, Joly S, Sutterwala FS. The NLRP3 inflammasome: a sensor of immune danger signals. Semin Immunol (2009) 21:194–8. doi: 10.1016/j.smim.2009.05.002
72. Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev (2008) 223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x
73. Poznyak AV, Melnichenko AA, Wetzker R, Gerasimova EV, Orekhov AN. NLPR3 inflammasomes and their significance for atherosclerosis. Biomedicines (2020) 8(7):205. doi: 10.3390/biomedicines8070205
74. Martinez GJ, Celermajer DS, Patel S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis (2018) 269:262–71. doi: 10.1016/j.atherosclerosis.2017.12.027
75. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood (2011) 117:3720–32. doi: 10.1182/blood-2010-07-273417
76. Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res (2008) 79:360–76. doi: 10.1093/cvr/cvn120
77. Zimmer S, Grebe A, Latz E. Danger signaling in atherosclerosis. Circ Res (2015) 116:323–40. doi: 10.1161/CIRCRESAHA.116.301135
78. Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev (2006) 86:515–81. doi: 10.1152/physrev.00024.2005
79. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis of CANTOS and beyond. J Am Coll Cardiol (2017) 70:2278–89. doi: 10.1016/j.jacc.2017.09.028
80. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol (2003) 23:656–60. doi: 10.1161/01.ATV.0000064374.15232.C3
81. Chi H, Messas E, Levine RA, Graves DT, Amar S. Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein e heterozygote model: pharmacotherapeutic implications. Circulation (2004) 110:1678–85. doi: 10.1161/01.CIR.0000142085.39015.31
82. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11:519–31. doi: 10.1038/nri3024
83. Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol (2008) 130:147–58. doi: 10.1016/j.ijcard.2008.04.059
84. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
85. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol (2010) 10:89–102. doi: 10.1038/nri2691
86. Herman WH, Holcomb JM, Hricik DE, Simonson MS. Interleukin-1 beta induces endothelin-1 gene by multiple mechanisms. Transplant Proc (1999) 31:1412–3. doi: 10.1016/S0041-1345(98)02109-5
87. Wang X, Feuerstein GZ, Gu JL, Lysko PG, Yue TL. Interleukin-1 beta induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis (1995) 115(1):89–98. doi: 10.1016/0021-9150(94)05503-b
88. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res (2020) 126(9):1260–80. doi: 10.1161/CIRCRESAHA.120.315937
89. Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science (1997) 275:206–9. doi: 10.1126/science.275.5297.206
90. Kim SH, Azam T, Novick D, Yoon DY, Reznikov LL, Bufler P, et al. Identification of amino acid residues critical for biological activity in human interleukin-18. J Biol Chem (2002) 277:10998–1003. doi: 10.1074/jbc.M108311200
91. Kim SH, Eisenstein M, Reznikov L, Fantuzzi G, Novick D, Rubinstein M, et al. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc Natl Acad Sci U S A (2000) 97:1190–5. doi: 10.1161/JAHA.119.0122192.Maz9
92. Mazodier K, Marin V, Novick D, Farnarier C, Robitail S, Schleinitz N, et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood (2005) 106:3483–9. doi: 10.1182/blood-2005-05-1980
93. Ludwiczek O, Kaser A, Novick D, Dinarello CA, Rubinstein M, Vogel W, et al. Plasma levels of interleukin-18 and interleukin-18 binding protein are elevated in patients with chronic liver disease. J Clin Immunol (2002) 22:331–7. doi: 10.1023/A:1020600230977
94. Bresnihan B, Roux-Lombard P, Murphy E, Kane D, FitzGerald O, Dayer JM. Serum interleukin 18 and interleukin 18 binding protein in rheumatoid arthritis. Ann Rheum Dis (2002) 61:726–9. doi: 10.1136/ard.61.8.726
95. Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein e(-/-) mice through release of interferon-gamma. Circ Res (2002) 90:E34–8. doi: 10.1161/hh0202.105292
96. Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein e-knockout mice. Cardiovasc Res (2003) 59:234–40. doi: 10.1016/S0008-6363(03)00343-2
97. Tiret L, Godefroy T, Lubos E, Nicaud V, Tregouet DA, Barbaux S, et al. Genetic analysis of the interleukin-18 system highlights the role of the interleukin-18 gene in cardiovascular disease. Circulation (2005) 112:643–50. doi: 10.1161/CIRCULATIONAHA.104.519702
98. Tan HW, Liu X, Bi XP, Xing SS, Li L, Gong HP, et al. IL-18 overexpression promotes vascular inflammation and remodeling in a rat model of metabolic syndrome. Atherosclerosis (2010) 208:350–7. doi: 10.1016/j.atherosclerosis.2009.07.053
99. Lindegaard B, Matthews VB, Brandt C, Hojman P, Allen TL, Estevez E, et al. Interleukin-18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes (2013) 62:3064–74. doi: 10.2337/db12-1095
100. Opstad TB, Pettersen AA, Arnesen H, Seljeflot I. Circulating levels of IL-18 are significantly influenced by the IL-18 +183 A/G polymorphism in coronary artery disease patients with diabetes type 2 and the metabolic syndrome: an observational study. Cardiovasc Diabetol (2011) 10:110. doi: 10.1186/1475-2840-10-110
101. Jung C, Gerdes N, Fritzenwanger M, Figulla HR. Circulating levels of interleukin-1 family cytokines in overweight adolescents. Mediators Inflammation (2010) 2010:958403. doi: 10.1155/2010/958403
102. Yang F, Qin Y, Wang Y, Meng S, Xian H, Che H, et al. Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int J Biol Sci (2019) 15:1010–9. doi: 10.7150/ijbs.29680
103. Sharma A, Choi JSY, Stefanovic N, Al-Sharea A, Simpson DS, Mukhamedova N, et al. Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes (2021) 70:772–87. doi: 10.2337/db20-0357
104. Puglisi S, Rossini A, Poli R, Dughera F, Pia A, Terzolo M, et al. Effects of SGLT2 inhibitors and GLP-1 receptor agonists on renin-Angiotensin-Aldosterone system. Front Endocrinol (Lausanne) (2021) 12:738848. doi: 10.3389/fendo.2021.738848
105. Horiba N, Masuda S, Takeuchi A, Takeuchi D, Okuda M, Inui K. Cloning and characterization of a novel na+-dependent glucose transporter (NaGLT1) in rat kidney. J Biol Chem (2003) 278:14669–76. doi: 10.1074/jbc.M212240200
106. Tahrani AA, Barnett AH, Bailey CJ. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol (2013) 1:140–51. doi: 10.1016/S2213-8587(13)70050-0
107. Yamaguchi K, Kato M, Suzuki M, Hagita H, Takada M, Ayabe M, et al. In vitro-in vivo correlation of the inhibition potency of sodium-glucose cotransporter inhibitors in rat: a pharmacokinetic and pharmacodynamic modeling approach. J Pharmacol Exp Ther (2013) 345:52–61. doi: 10.1124/jpet.113.203125
108. Scheen AJ. Sodium-glucose cotransporter type 2 inhibitors for the treatment of type 2 diabetes mellitus. Nat Rev Endocrinol (2020) 16:556–77. doi: 10.1038/s41574-020-0392-2
109. Pahud de Mortanges A, Salvador D Jr., Laimer M, Muka T, Wilhelm M, Bano A. The role of SGLT2 inhibitors in atherosclerosis: A narrative mini-review. Front Pharmacol (2021) 12:751214. doi: 10.3389/fphar.2021.751214
110. Liu Z, Ma X, Ilyas I, Zheng X, Luo S, Little PJ, et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: from pharmacology to pre-clinical and clinical therapeutics. Theranostics (2021) 11:4502–15. doi: 10.7150/thno.54498
111. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose Co-transporter 2 (SGLT2) inhibitors: A state-of-the-Art review. JACC Basic Transl Sci (2020) 5:632–44. doi: 10.1016/j.jacbts.2020.02.004
112. Jiang Y, Yang P, Fu L, Sun L, Shen W, Wu Q. Comparative cardiovascular outcomes of SGLT2 inhibitors in type 2 diabetes mellitus: A network meta-analysis of randomized controlled trials. Front Endocrinol (Lausanne) (2022) 13:802992. doi: 10.3389/fendo.2022.802992
113. Rizvi SM, Shakil S, Biswas D, Shakil S, Shaikh S, Bagga P, et al. Invokana (Canagliflozin) as a dual inhibitor of acetylcholinesterase and sodium glucose co-transporter 2: advancement in alzheimer's disease- diabetes type 2 linkage via an enzoinformatics study. CNS Neurol Disord Drug Targets (2014) 13:447–51. doi: 10.2174/18715273113126660160
114. Behnammanesh G, Durante ZE, Peyton KJ, Martinez-Lemus LA, Brown SM, Bender SB, et al. Canagliflozin inhibits human endothelial cell proliferation and tube formation. Front Pharmacol (2019) 10:362. doi: 10.3389/fphar.2019.00362
115. Xu J, Kitada M, Ogura Y, Liu H, Koya D. Dapagliflozin restores impaired autophagy and suppresses inflammation in high glucose-treated HK-2 cells. Cells (2021) 10(6):1457. doi: 10.3390/cells10061457
116. Birnbaum Y, Bajaj M, Yang HC, Ye Y. Combined SGLT2 and DPP4 inhibition reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic nephropathy in mice with type 2 diabetes. Cardiovasc Drugs Ther (2018) 32:135–45. doi: 10.1007/s10557-018-6778-x
117. Leng W, Wu M, Pan H, Lei X, Chen L, Wu Q, et al. The SGLT2 inhibitor dapagliflozin attenuates the activity of ROS-NLRP3 inflammasome axis in steatohepatitis with diabetes mellitus. Ann Transl Med (2019) 7:429. doi: 10.21037/atm.2019.09.03
118. Ye Y, Bajaj M, Yang HC, Perez-Polo JR, Birnbaum Y. SGLT-2 inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther (2017) 31:119–32. doi: 10.1007/s10557-017-6725-2
119. Pennig J, Scherrer P, Gissler MC, Anto-Michel N, Hoppe N, Funer L, et al. Glucose lowering by SGLT2-inhibitor empagliflozin accelerates atherosclerosis regression in hyperglycemic STZ-diabetic mice. Sci Rep (2019) 9:17937. doi: 10.1038/s41598-019-54224-9
120. Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun (2020) 11:2127. doi: 10.1038/s41467-020-15983-6
121. Iwamoto M, Kubota T, Sakurai Y, Wada N, Shioda S, Yamauchi T, et al. The sodium-glucose co-transporter 2 inhibitor tofogliflozin suppresses atherosclerosis through glucose lowering in ApoE-deficient mice with streptozotocin-induced diabetes. Pharmacol Res Perspect (2022) 10:e00971. doi: 10.1002/prp2.971
122. Zheng H, Liu M, Li S, Shi Q, Zhang S, Zhou Y, et al. Sodium-glucose Co-Transporter-2 inhibitors in non-diabetic adults with overweight or obesity: A systematic review and meta-analysis. Front Endocrinol (Lausanne) (2021) 12:706914. doi: 10.3389/fendo.2021.706914
123. Sukhanov S, Higashi Y, Yoshida T, Mummidi S, Aroor AR, Jeffrey Russell J, et al. The SGLT2 inhibitor empagliflozin attenuates interleukin-17A-induced human aortic smooth muscle cell proliferation and migration by targeting TRAF3IP2/ROS/NLRP3/Caspase-1-dependent IL-1beta and IL-18 secretion. Cell Signal (2021) 77:109825. doi: 10.1016/j.cellsig.2020.109825
124. Peluso I, Morabito G, Urban L, Ioannone F, Serafini M. Oxidative stress in atherosclerosis development: the central role of LDL and oxidative burst. Endocr Metab Immune Disord Drug Targets (2012) 12:351–60. doi: 10.2174/187153012803832602
125. Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol 292 (2007) 292(5):H2023–31. doi: 10.1152/ajpheart.01283.2006
126. Forstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res (2017) 120:713–35. doi: 10.1161/CIRCRESAHA.116.309326
127. Herb M, Schramm M. Functions of ROS in macrophages and antimicrobial immunity. Antioxidants (Basel) (2021) 10(2):313. doi: 10.3390/antiox10020313
128. Yang Y, Zhao C, Ye Y, Yu M, Qu X. Prospect of sodium-glucose Co-transporter 2 inhibitors combined with insulin for the treatment of type 2 diabetes. Front Endocrinol (Lausanne) (2020) 11:190. doi: 10.3389/fendo.2020.00190
129. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
130. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem (2007) 282:2871–9. doi: 10.1074/jbc.M608083200
131. Di Marco E, Gray SP, Chew P, Koulis C, Ziegler A, Szyndralewiez C, et al. Pharmacological inhibition of NOX reduces atherosclerotic lesions, vascular ROS and immune-inflammatory responses in diabetic apoe(-/-) mice. Diabetologia (2014) 57:633–42. doi: 10.1007/s00125-013-3118-3
132. Ashrafi Jigheh Z, Ghorbani Haghjo A, Argani H, Sanajou D. Sodium-glucose co-transporters and diabetic nephropathy: Is there a link with toll-like receptors? Clin Exp Pharmacol Physiol (2020) 47:919–26. doi: 10.1111/1440-1681.13261
133. Quagliariello V, De Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A, et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol (2021) 20:150. doi: 10.1186/s12933-021-01346-y
134. Ashrafi Jigheh Z, Ghorbani Haghjo A, Argani H, Roshangar L, Rashtchizadeh N, Sanajou D, et al. Empagliflozin alleviates renal inflammation and oxidative stress in streptozotocin-induced diabetic rats partly by repressing HMGB1-TLR4 receptor axis. Iran J Basic Med Sci (2019) 22:384–90. doi: 10.22038/ijbms.2019.31788.7651
135. Wang XX, Levi J, Luo Y, Myakala K, Herman-Edelstein M, Qiu L, et al. SGLT2 protein expression is increased in human diabetic nephropathy: SGLT2 Protein inhibition decreases renal lipid accumulation, inflammation, and the development of nephropathy in diabetic mice. J Biol Chem (2017) 292:5335–48. doi: 10.1074/jbc.M117.779520
136. Williams JW, Huang LH, Randolph GJ. Cytokine circuits in cardiovascular disease. Immunity (2019) 50:941–54. doi: 10.1016/j.immuni.2019.03.007
137. Sun LF, An DQ, Niyazi GL, Ma WH, Xu ZW, Xie Y. Effects of tianxiangdan granule treatment on atherosclerosis via NFkappaB and p38 MAPK signaling pathways. Mol Med Rep (2018) 17:1642–50. doi: 10.3892/mmr.2017.8067
138. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat Immunol (2017) 18:861–9. doi: 10.1038/ni.3772
139. Abdollahi E, Keyhanfar F, Delbandi AA, Falak R, Hajimiresmaiel SJ, Shafiei M. Dapagliflozin exerts anti-inflammatory effects via inhibition of LPS-induced TLR-4 overexpression and NF-kappaB activation in human endothelial cells and differentiated macrophages. Eur J Pharmacol (2022) 918:174715. doi: 10.1016/j.ejphar.2021.174715
140. Liu Y, Wu M, Xu B, Kang L. Empagliflozin alleviates atherosclerosis progression by inhibiting inflammation and sympathetic activity in a normoglycemic mouse model. J Inflammation Res (2021) 14:2277–87. doi: 10.2147/JIR.S309427
141. Kim KH, Lee MS. Autophagy–a key player in cellular and body metabolism. Nat Rev Endocrinol (2014) 10:322–37. doi: 10.1038/nrendo.2014.35
142. Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
143. Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab (2012) 15:545–53. doi: 10.1016/j.cmet.2012.01.022
144. Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab (2012) 15:534–44. doi: 10.1016/j.cmet.2012.02.011
145. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13:132–41. doi: 10.1038/ncb2152
146. Zhou H, Feng L, Xu F, Sun Y, Ma Y, Zhang X, et al. Berberine inhibits palmitate-induced NLRP3 inflammasome activation by triggering autophagy in macrophages: A new mechanism linking berberine to insulin resistance improvement. BioMed Pharmacother (2017) 89:864–74. doi: 10.1016/j.biopha.2017.03.003
147. Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol (2016) 138:28–36. doi: 10.1016/j.jaci.2016.05.009
148. Chang YP, Ka SM, Hsu WH, Chen A, Chao LK, Lin CC, et al. Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J Cell Physiol (2015) 230:1567–79. doi: 10.1002/jcp.24903
149. Packer M. Autophagy stimulation and intracellular sodium reduction as mediators of the cardioprotective effect of sodium-glucose cotransporter 2 inhibitors. Eur J Heart Fail (2020) 22:618–28. doi: 10.1002/ejhf.1732
150. Fathi A, Vickneson K, Singh JS. SGLT2-inhibitors; more than just glycosuria and diuresis. Heart Fail Rev (2021) 26:623–42. doi: 10.1007/s10741-020-10038-w
151. Lauritsen KM, Voigt JH, Pedersen SB, Hansen TK, Moller N, Jessen N, et al. Effects of SGLT2 inhibition on lipid transport in adipose tissue in type 2 diabetes. Endocr Connect (2022) 11(4):e210558. doi: 10.1530/EC-21-0558
152. Kashiwagi A, Maegawa H. Metabolic and hemodynamic effects of sodium-dependent glucose cotransporter 2 inhibitors on cardio-renal protection in the treatment of patients with type 2 diabetes mellitus. J Diabetes Investig (2017) 8:416–27. doi: 10.1111/jdi.12644
153. Perry RJ, Rabin-Court A, Song JD, Cardone RL, Wang Y, Kibbey RG, et al. Dehydration and insulinopenia are necessary and sufficient for euglycemic ketoacidosis in SGLT2 inhibitor-treated rats. Nat Commun (2019) 10:548. doi: 10.1038/s41467-019-08466-w
154. Perry RJ, Shulman GI. Sodium-glucose cotransporter-2 inhibitors: Understanding the mechanisms for therapeutic promise and persisting risks. J Biol Chem (2020) 295:14379–90. doi: 10.1074/jbc.REV120.008387
155. Min SH, Oh TJ, Baek SI, Lee DH, Kim KM, Moon JH, et al. Degree of ketonaemia and its association with insulin resistance after dapagliflozin treatment in type 2 diabetes. Diabetes Metab (2018) 44:73–6. doi: 10.1016/j.diabet.2017.09.006
156. Ferrannini E. Sodium-glucose Co-transporters and their inhibition: Clinical physiology. Cell Metab (2017) 26:27–38. doi: 10.1016/j.cmet.2017.04.011
157. Yamanashi T, Iwata M, Kamiya N, Tsunetomi K, Kajitani N, Wada N, et al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci Rep (2017) 7:7677. doi: 10.1038/s41598-017-08055-1
158. Hattori Y. Beneficial effects on kidney during treatment with sodium-glucose cotransporter 2 inhibitors: proposed role of ketone utilization. Heart Fail Rev (2021) 26:947–52. doi: 10.1007/s10741-020-10065-7
159. Bae HR, Kim DH, Park MH, Lee B, Kim MJ, Lee EK, et al. Beta-hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget (2016) 7:66444–54. doi: 10.18632/oncotarget.12119
160. Katakami N, Mita T, Yoshii H, Shiraiwa T, Yasuda T, Okada Y, et al. Rationale, design, and baseline characteristics of the utopia trial for preventing diabetic atherosclerosis using an SGLT2 inhibitor: A prospective, randomized, open-label, parallel-group comparative study. Diabetes Ther (2017) 8:999–1013. doi: 10.1007/s13300-017-0292-1
161. Neudorf H, Durrer C, Myette-Cote E, Makins C, O'Malley T, Little JP. Oral ketone supplementation acutely increases markers of NLRP3 inflammasome activation in human monocytes. Mol Nutr Food Res (2019) 63:e1801171. doi: 10.1002/mnfr.201801171
162. Neudorf H, Myette-Cote E, Little JP. The impact of acute ingestion of a ketone monoester drink on LPS-stimulated NLRP3 activation in humans with obesity. Nutrients (2020) 12(3):854. doi: 10.3390/nu12030854
163. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
164. Gliozzi M, Macri R, Coppoletta AR, Musolino V, Carresi C, Scicchitano M, et al. From diabetes care to heart failure management: A potential therapeutic approach combining SGLT2 inhibitors and plant extracts. Nutrients (2022) 14(18):3737. doi: 10.3390/nu14183737
165. Al-Sharea A, Murphy AJ, Huggins LA, Hu Y, Goldberg IJ, Nagareddy PR. SGLT2 inhibition reduces atherosclerosis by enhancing lipoprotein clearance in ldlr(-/-) type 1 diabetic mice. Atherosclerosis (2018) 271:166–76. doi: 10.1016/j.atherosclerosis.2018.02.028
166. Xu J, Hirai T, Koya D, Kitada M. Effects of SGLT2 inhibitors on atherosclerosis: Lessons from cardiovascular clinical outcomes in type 2 diabetic patients and basic researches. J Clin Med (2021) 11(1):137. doi: 10.3390/jcm11010137
167. Juni RP, Kuster DWD, Goebel M, Helmes M, Musters RJP, van der Velden J, et al. Cardiac microvascular endothelial enhancement of cardiomyocyte function is impaired by inflammation and restored by empagliflozin. JACC Basic Transl Sci (2019) 4:575–91. doi: 10.1016/j.jacbts.2019.04.003
168. Cesaro A, Gragnano F, Paolisso P, Bergamaschi L, Gallinoro E, Sardu C, et al. In-hospital arrhythmic burden reduction in diabetic patients with acute myocardial infarction treated with SGLT2-inhibitors: Insights from the SGLT2-I AMI PROTECT study. Front Cardiovasc Med (2022) 9:1012220. doi: 10.3389/fcvm.2022.1012220
169. Hodrea J, Saeed A, Molnar A, Fintha A, Barczi A, Wagner LJ, et al. SGLT2 inhibitor dapagliflozin prevents atherosclerotic and cardiac complications in experimental type 1 diabetes. PloS One (2022) 17:e0263285. doi: 10.1371/journal.pone.0263285
170. Dhingra NK, Mistry N, Puar P, Verma R, Anker S, Mazer CD, et al. SGLT2 inhibitors and cardiac remodelling: a systematic review and meta-analysis of randomized cardiac magnetic resonance imaging trials. ESC Heart Fail (2021) 8:4693–700. doi: 10.1002/ehf2.13645
171. Mone P, Varzideh F, Jankauskas SS, Pansini A, Lombardi A, Frullone S, et al. SGLT2 inhibition via empagliflozin improves endothelial function and reduces mitochondrial oxidative stress: Insights from frail hypertensive and diabetic patients. Hypertension (2022) 79:1633–43. doi: 10.1161/HYPERTENSIONAHA.122.19586
172. Wan N, Rahman A, Hitomi H, Nishiyama A. The effects of sodium-glucose cotransporter 2 inhibitors on sympathetic nervous activity. Front Endocrinol (Lausanne) (2018) 9:421. doi: 10.3389/fendo.2018.00421
173. Sardu C, Massetti M, Testa N, Martino LD, Castellano G, Turriziani F, et al. Effects of sodium-glucose transporter 2 inhibitors (SGLT2-I) in patients with ischemic heart disease (IHD) treated by coronary artery bypass grafting via MiECC: Inflammatory burden, and clinical outcomes at 5 years of follow-up. Front Pharmacol (2021) 12:777083. doi: 10.3389/fphar.2021.777083
174. Dimitriadis GK, Nasiri-Ansari N, Agrogiannis G, Kostakis ID, Randeva MS, Nikiteas N, et al. Empagliflozin improves primary haemodynamic parameters and attenuates the development of atherosclerosis in high fat diet fed APOE knockout mice. Mol Cell Endocrinol (2019) 494:110487. doi: 10.1016/j.mce.2019.110487
175. Nasiri-Ansari N, Dimitriadis GK, Agrogiannis G, Perrea D, Kostakis ID, Kaltsas G, et al. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc Diabetol (2018) 17:106. doi: 10.1186/s12933-018-0749-1
176. Lee DM, Battson ML, Jarrell DK, Hou S, Ecton KE, Weir TL, et al. SGLT2 inhibition via dapagliflozin improves generalized vascular dysfunction and alters the gut microbiota in type 2 diabetic mice. Cardiovasc Diabetol (2018) 17:62. doi: 10.1186/s12933-018-0708-x
177. Nelson AJ, Harrington JL, Kolkailah AA, Pagidipati NJ, McGuire DK. Sodium-glucose cotransporter-2 inhibitors: Impact on atherosclerosis and atherosclerotic cardiovascular disease events. Heart Fail Clin (2022) 18:597–607. doi: 10.1016/j.hfc.2022.03.007
178. Terasaki M, Hiromura M, Mori Y, Kohashi K, Nagashima M, Kushima H, et al. Amelioration of hyperglycemia with a sodium-glucose cotransporter 2 inhibitor prevents macrophage-driven atherosclerosis through macrophage foam cell formation suppression in type 1 and type 2 diabetic mice. PLoS One (2015) 10:e0143396. doi: 10.1371/journal.pone.0143396
Keywords: atherosclerosis, SGLT2 inhibitor, NLRP3, inflammasome, diabetes mellitus
Citation: Yang L, Zhang X and Wang Q (2022) Effects and mechanisms of SGLT2 inhibitors on the NLRP3 inflammasome, with a focus on atherosclerosis. Front. Endocrinol. 13:992937. doi: 10.3389/fendo.2022.992937
Received: 13 July 2022; Accepted: 30 November 2022;
Published: 15 December 2022.
Edited by:
Gaetano Santulli, Albert Einstein College of Medicine, United StatesReviewed by:
Tianshu Zeng, Huazhong University of Science and Technology, ChinaCopyright © 2022 Yang, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Wang, d2FuZ19xaW5nQGpsdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.