 Megan Cairns
Megan Cairns Danzil Joseph
Danzil Joseph M. Faadiel Essop
M. Faadiel Essop- 1Centre for Cardio-Metabolic Research in Africa, Division of Medical Physiology, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
- 2Centre for Cardio-Metabolic Research in Africa, Department of Physiological Sciences, Faculty of Science, Stellenbosch University, Stellenbosch, South Africa
The heart is a highly metabolic organ with extensive energy demands and hence relies on numerous fuel substrates including fatty acids and glucose. However, oxidative stress is a natural by-product of metabolism that, in excess, can contribute towards DNA damage and poly-ADP-ribose polymerase activation. This activation inhibits key glycolytic enzymes, subsequently shunting glycolytic intermediates into non-oxidative glucose pathways such as the hexosamine biosynthetic pathway (HBP). In this review we provide evidence supporting the dual role of the HBP, i.e. playing a unique role in cardiac physiology and pathophysiology where acute upregulation confers cardioprotection while chronic activation contributes to the onset and progression of cardio-metabolic diseases such as diabetes, hypertrophy, ischemic heart disease, and heart failure. Thus although the HBP has emerged as a novel therapeutic target for such conditions, proposed interventions need to be applied in a context- and pathology-specific manner to avoid any potential drawbacks of relatively low cardiac HBP activity.
Introduction
Next to the brain, the heart is the most energy-intensive organ in the body (1). Fuel substrate metabolism plays a key role to ensure optimal cardiac function. However, a fine balance exists between over- and under-utilization and a shift in either direction contributes towards cardiac dysfunction. Oxidation of a number of fuel substrates ensures a constant supply of ATP for cardiac contractility and ion gradient regulation (2). These energy substrates can be crudely divided into “preferred” (free fatty acids and glucose), or “alternative” (branched-chain amino acids [BCAAs] and ketones) which contribute 90% and 10%, respectively, towards cardiac ATP production (2). The dynamics between, and within, such substrate groups underlie the development of various pathologies ranging from diabetes to cardiac hypertrophy and heart failure (2, 3).
The switch from fatty acids to glucose as the main fuel substrate for energy production is commonly observed in heart failure, myocardial ischemia, cardiac hypertrophy, and increased cardiac workload (4–6). However, elevated glucose uptake and glycolysis rates in the hypertrophied heart do not necessarily correlate with increased pyruvate oxidation (7). This suggests that glucose enters glycolysis but that the rate of acetyl-CoA turnover in the Krebs cycle does not change – thus providing evidence for attenuated pyruvate dehydrogenase (PDH) activity. The delinking of glycolysis from mitochondrial oxidative phosphorylation promotes proton accumulation and a pro-oxidative milieu (8, 9).
Oxidative stress is a natural by-product of metabolism that can trigger intracellular damage ranging from DNA strand breaks to lipid and protein peroxidation (10). The damage mediated in this fashion leads to inhibition of glycolytic enzymes, including hexokinase, glyceraldehyde-3-phosphate dehydrogenase, and phosphofructokinase (PFK), through a combination of poly-ADP-ribose polymerase-dependent and independent mechanisms (11–16). This results in upstream glycolytic intermediate accumulation (11–15). Such intermediates are subsequently shunted into various non-oxidative glucose pathways (NOGPs) such as the polyol pathway, advanced glycation end-product (AGE) pathway, pentose phosphate pathway, and the hexosamine biosynthetic pathway (HBP). Although the NOGPs usually function at relatively low basal levels of activity, enhanced flux can occur with increased oxidative stress, or during hyperglycemic, diabetic, and hypertrophic conditions (13–15).
The links between elevated protein O-GlcNAcylation due to increased HBP flux and cardiac pathology are well established, particularly in the context of diabetes (17–19). The goal of this review is therefore not to reconsider the well-understood pathological mechanisms linked to diabetes due to chronic HBP activation. Rather, we aim to provide a contextualized understanding of the HBP in terms of cardiac function – from its acute and beneficial upregulation, to dysfunction induced by chronic O-GlcNAcylation across numerous cardiac pathologies. This includes a summary of available literature highlighting the significant findings of clinical trials, in vivo, in vivo transgenic, and in vitro studies regarding the impact of HBP regulation on the heart’s function.
Hexosamine biosynthetic pathway and the heart
Inhibition of PFK increases intracellular fructose-6-phosphate which can be shunted into the HBP. Glutamine fructose-6-phosphate amidotransferase (GFAT) isoforms are location-specific with GFAT1 expressed in skeletal muscle and the heart, while GFAT2 is predominant in the heart, placenta, and spinal cord (20, 21). Nabeebaccus et al. (2021) recently found GFAT1 as the primary isoform in cardiomyocytes while GFAT2 is predominantly expressed in fibroblasts (22). Upon shunting into the HBP, GFAT1/2 converts fructose-6-phosphate into glucosamine-6-phosphate, which is metabolized to uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) (23). O-GlcNAc transferase (OGT) is the primary enzyme responsible for catalyzing the reaction whereby O-GlcNAc binds to protein serine or threonine residues often in direct competition with phosphorylation (24). Binding to these sites may directly alter protein function, or indirectly alter alternative activation sites through steric or electrosteric actions (25). O-GlcNAcase (OGA) exerts opposing effects on OGT by hydrolyzing O-GlcNAc residues from proteins (24). The dynamics between OGT and OGA (in addition to GFAT1/2 activity) underlie the shift away from cardioprotection and towards cardiac pathology.
Detection of O-GlcNAc moieties on proteins can be crudely divided into two categories: overall and site-specific. The use of antibodies (particularly CTD110.6) designed to detect O-GlcNAcylated serine and threonine residues are most commonly utilized in Western blotting to determine the overall change in specific (via immunoprecipitation) or total protein modifications (26–29). The primary drawback of this method is the antibodies themselves as they lack specificity and may skew results based on protein abundance (30). Proteomic techniques such as β-Elimination followed by Michael addition (BEMAD) enable the identification of site-specific O-GlcNAcylation, thus allowing for greater understanding regarding the effects of O-GlcNAcylation in terms of protein function (31). However, this technique is most often applied at the peptide (versus full protein) level.
The HBP displays tissue and organ-specific expression profiles that fluctuate according to circadian rhythm, substrate availability, as well as age (32). Studies demonstrated the pivotal role of the HBP and downstream O-GlcNAcylation on pre- and post-natal cardiac development (33, 34). Complete OGT deficient animals display overt cardiac failure with severe cardiac fibrosis, apoptosis, and hypertrophy compared to wild-type littermates (33). Although O-GlcNAcylation is essential for normal cardiac development, Dupas et al. (2021) highlighted the time-dependent decrease in cardiac O-GlcNAc from birth (34). Such a decrease in O-GlcNAcylation suggests a shift away from non-oxidative glucose metabolism in the heart. Conversely, excessive protein O-GlcNAcylation is observed in multiple organs with increasing age in rat and mouse models, contributing to adverse clinical outcomes (35, 36). For example, young mice subjected to cardiac arrest and resuscitation displayed increased unfolded protein response pathways and post-translational modifications including O-GlcNAcylation, which corresponded with improved recovery not observed in aged animals (36, 37). The most accepted rationale behind this increase is the gradual shift into cellular senescence during the aging process and the subsequent decline of proteostasis (35, 36).
The influence of the HBP on cardiac function is gaining increasing attention and understanding (refer to Tables 1–4 for a summary of currently available literature on the role of the HBP in the cardiovascular system). Here a comprehensive literature search was conducted using the search engines Scopus® and PubMed, with keywords including “hexosamine biosynthetic pathway”, “O-GlcNAc”, and “heart” with the focus on original and clinical research article titles between the years 2000 and 2022. The most researched areas of O-GlcNAc pathology include the influence of altered glucose levels, ischemia-reperfusion injury (IR/I), hypertrophy, hypertension, and heart failure. These data reveal that the HBP is intricately involved in cardiac function and pathology and that O-GlcNAcylation affects various pathways and proteins (Tables 1–4). Such alterations may be protective or detrimental depending on the experimental and/or clinical contexts, and this often depends on the dynamic cycling, extent, and duration of protein O-GlcNAcylation.
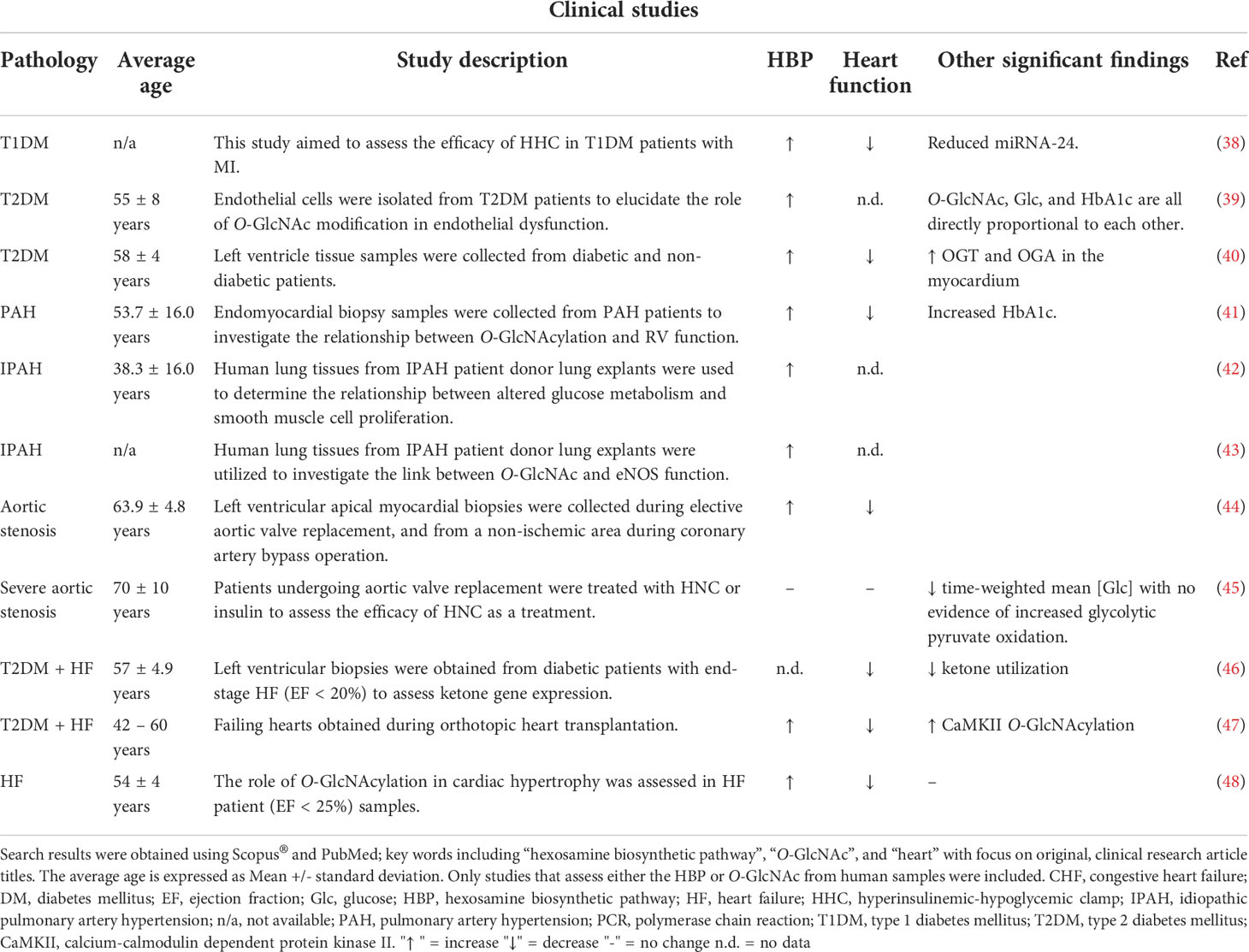
Table 1 Summary of clinical studies (2000 – 2022) investigating the role of the HBP or O-GlcNAc in various pathological conditions.

Table 2 Summary of available literature (2000 – 2022) utilizing non-transgenic animal models to investigate the role of the HBP or O-GlcNAc in various pathologies.
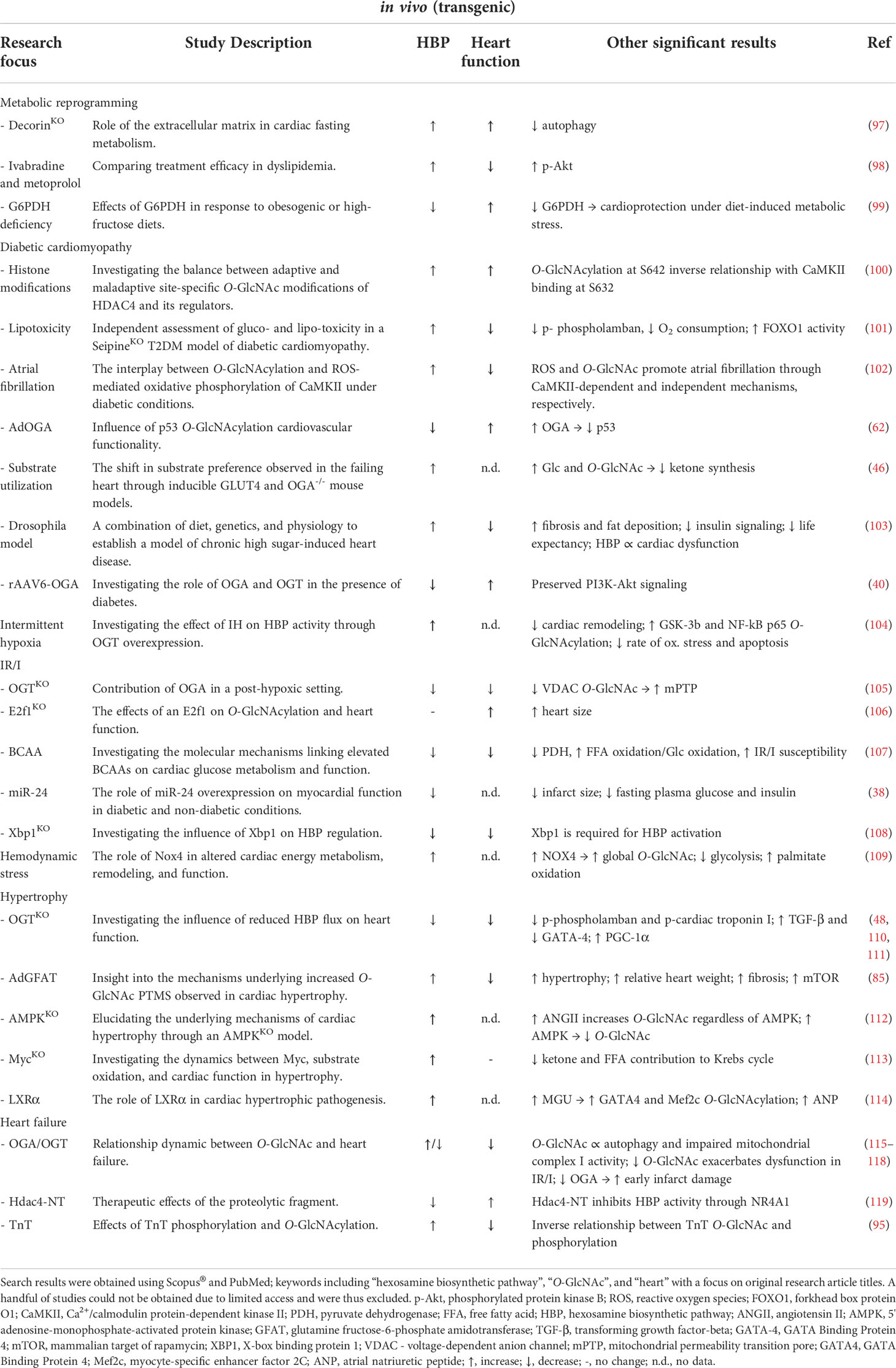
Table 3 Summary of available literature (2000 – 2022) utilizing transgenic rat and mouse models to investigate the role of the HBP or O-GlcNAc in various pathologies.
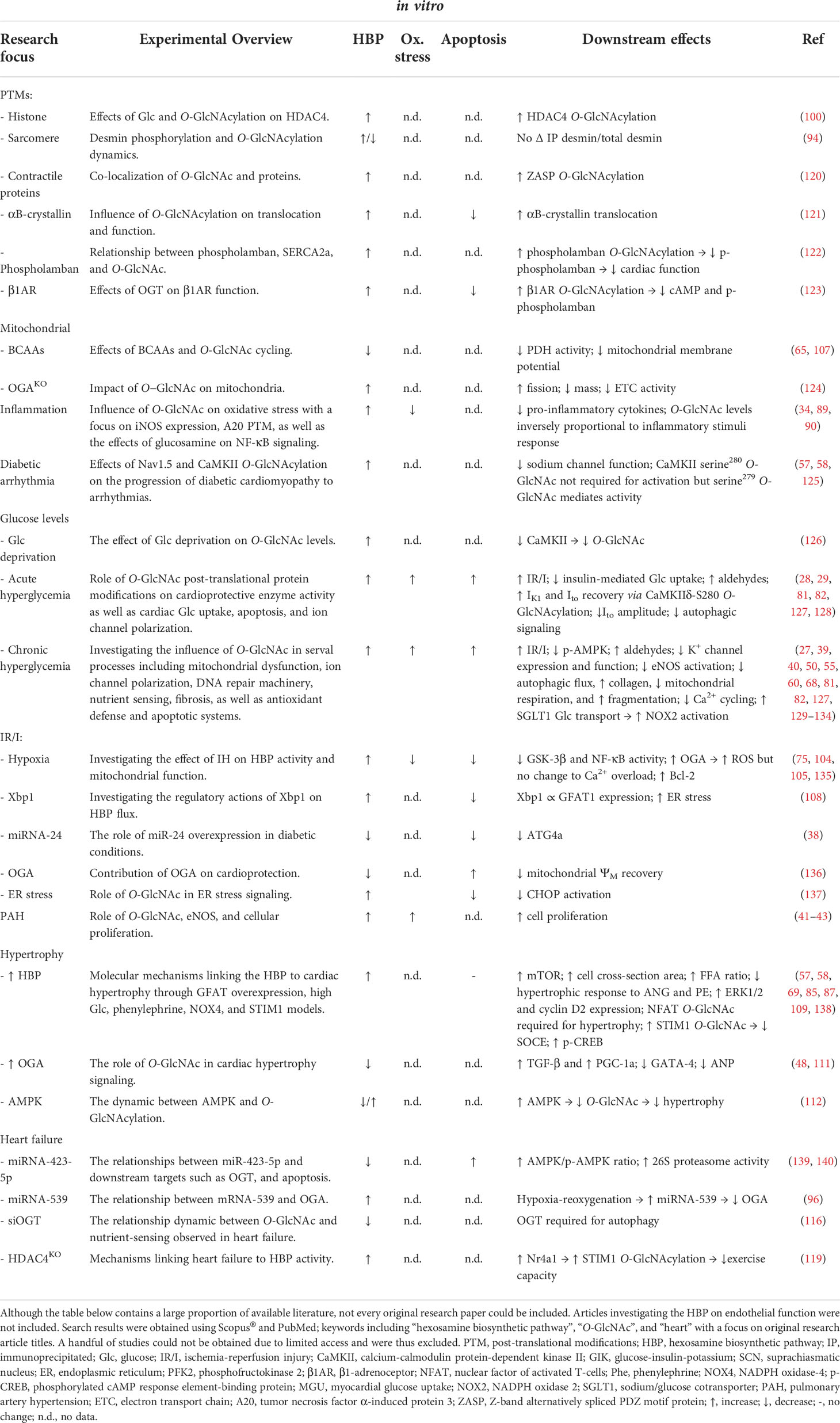
Table 4 Summary of available literature (2000 – 2022) utilizing in vitro models to investigate the underlying mechanisms between the HBP or O-GlcNAc in various pathologies.
The following sections aim to highlight the potential benefits and harms of altered HBP and O-GlcNAcylation in both pre-clinical and clinical studies (Tables 1–4). The benefits of acute HBP flux on the heart will also be discussed, followed by the effects of chronic activation.
HBP flux: Benefits of acute upregulation
While the majority of literature highlights the detrimental effects of excessive HBP activation, increased flux can be beneficial within an acute context. O-GlcNAcylation appears to promote cellular survival, for e.g. exposure to bacterial lipopolysaccharide or trauma events increases HBP activity. Here, lipopolysaccharide increases O-GlcNAcylation of calcium transporters (e.g. L-type subunits of the sarcoplasmic reticulum Ca2+-ATPases (LTCC)) and enhances cardiac contractility (88, 141), whereas attenuated inflammation and increased O-GlcNAcylation occur as a result of trauma-hemorrhage events (89–93). Moreover, increased HBP activity is indirectly proportional to both inflammatory and oxidative markers in an acute context, with the opposite observed in a chronic setting (142–144). O-GlcNAcylation also influences cardiomyocyte apoptosis. αB-crystallin is an intracellular chaperone that prevents the toxic accumulation of misfolded proteins and inhibits apoptosis when active. While the majority of post-translational modifications (PTMs) decrease αB-crystallin’s function as a molecular chaperone, O-GlcNAcylation on threonine170 attenuates cardiomyocyte apoptosis through increased αB-crystallin activity and translocation (121). While this is beneficial in an acute context, chronic HBP activation is associated with increased apoptosis and diminished autophagy (17, 74, 145, 146). Findings from our laboratory showed higher apoptosis under simulated hyperglycemic conditions that were linked to O-GlcNAcylation of the pro-apoptotic protein Bad, with a concomitant decrease in its phosphorylation (27). This further highlights the context-specific, dual nature of HBP activity in the heart.
With diminished autophagic and apoptotic signaling, cardiac cells should face increased hypertrophic stimuli. In contrast, enhanced O-GlcNAcylation of histone deacetylase 4 (HDAC4) serine642 elevates its activity and hinders gene transcription, ultimately lowering transforming growth factor-beta (TGF-β) myoblast differentiation and hypertrophy (100). While this may appear to prevent cardiomyocyte differentiation and hypertrophy in an acute context, O-GlcNAc also binds to the theonine63 residue of mothers against decapentaplegic homolog 4 (SMAD4), stabilizing it and preventing its binding to glycogen synthase kinase 3β, and ultimately augmenting downstream TGF-β signaling (147). Thus a combination of SMAD-independent (i.e. mammalian target of rapamycin) and SMAD-dependent signaling may compensate for reduced TGF-β/glycogen synthase kinase 3β pathway activity in the long term and thereby contribute to cardiac hypertrophy.
Acute HBP upregulation can therefore confer cardioprotection against hypertrophy and apoptosis, as well as improved functionality. Although various proteins require O-GlcNAcylation for activation or repression under normal physiological conditions, constant O-GlcNAcylation can potentiate protein dysfunction and cardiac pathology, for example in the context of ischemia-reperfusion.
Dual role of O-GlcNAcylation in ischemia-reperfusion
Depending on the duration and frequency of the ischemic event, O-GlcNAcylation is either protective or detrimental to cardiac function and health (Figure 1). For example, increased HBP flux is often observed during ischemic preconditioning, intermittent hypoxia, and acute IR/I episodes where it confers cardioprotection (77, 78). Here, increased myocardial glucose uptake elevates myocardial O-GlcNAc levels during ischemic preconditioning and improves mitochondrial function through increased calcium sensitivity and reduced mitochondrial transition pore opening (mPTP) (148). Subsequently, this will minimize myocardial infarct damage following reperfusion under experimentally induced diabetic and non-diabetic conditions (77, 78, 148, 149). Acute upregulation of X-box binding protein 1 (Xbp1) due to endoplasmic reticulum stress during IR/I increases HBP flux and confers cardio-protection by increasing autophagy of misfolded proteins (108).
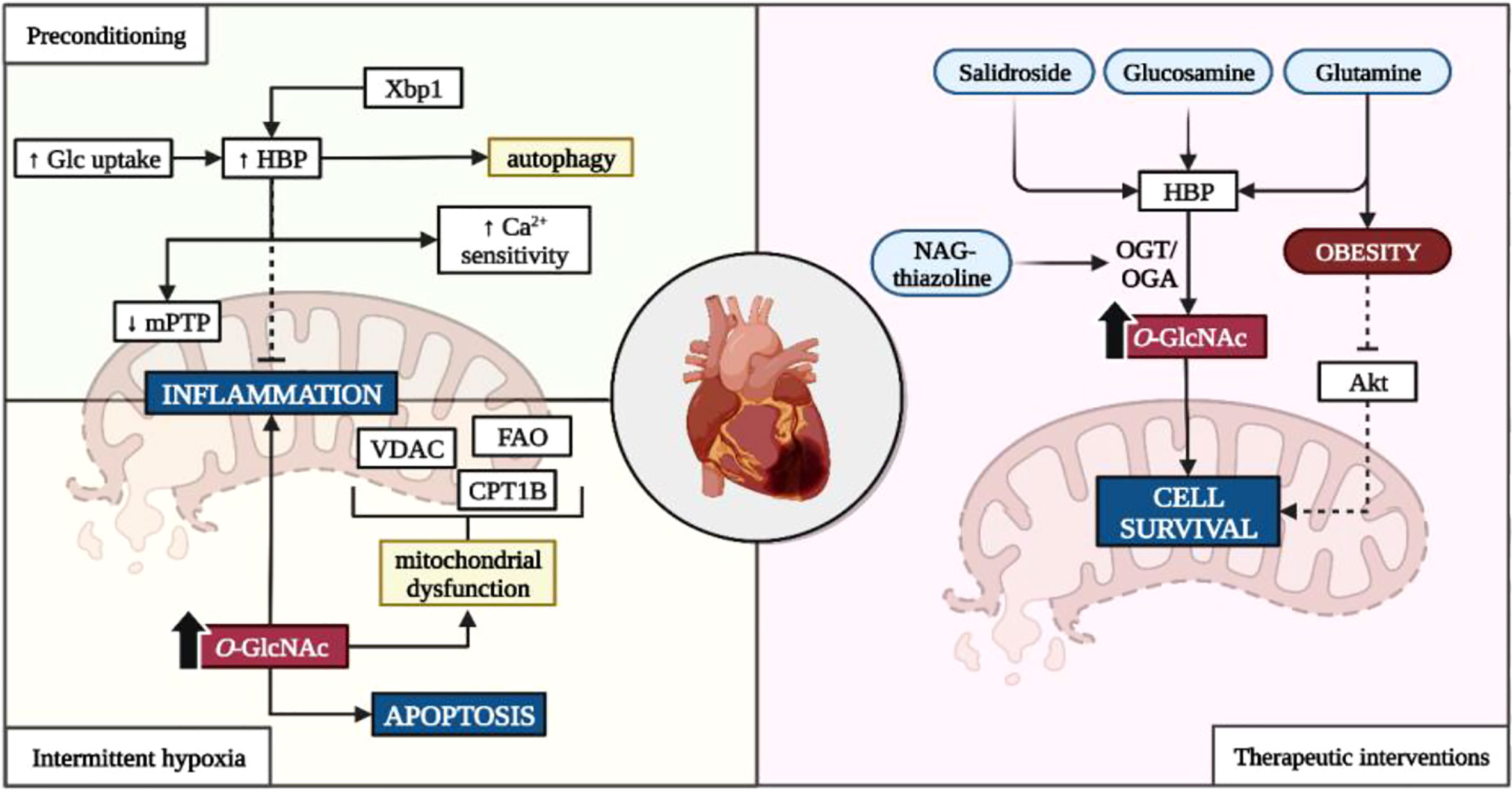
Figure 1 Influence of the HBP on ischemia-reperfusion injury. Glc, glucose; HBP, hexosamine biosynthetic pathway; Xbp1, X-box–binding protein-1; Ca2+, calcium; VDAC, voltage-dependent anion channels; CD36, cluster of differentiation 36; CPT1, carnitine palmitoyltransferase-1B; OGT, O-GlcNAc transferase; OGA, O-GlcNAcase; NAG, 1,2 dideoxy-2’-methyl-α-D-glycopyranoso-[2,1-d]-∆2’-thiazoline. Created with BioRender.com.
Exposure to intermittent hypoxia also upregulates the HBP which can either suppress inflammatory and hypertrophic genes (i.e. nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and nuclear factor of activated T cells (NFAT)), or promote apoptotic and inflammatory signaling depending on the duration of the ischemic event (74, 104). Consistent O-GlcNAcylation of mitochondrial proteins such as voltage-dependent anion channels (VDAC), carnitine-palmitate transferase 1B (CPT1B), and fatty acid oxidase (FAO) contributes to attenuated fatty acid β-oxidation, impaired calcium handling, increased mitochondrial fragmentation, and overall mitochondrial dysfunction (55, 56, 150). Moreover, elevated O-GlcNAcylation further hinders cardiac function through inflammatory and apoptotic signaling, and promotes hypertension (38, 54, 81, 82). This is further highlighted in diabetic studies, where chronically elevated glucose conditions exacerbate ischemic injury. Whilst the exact mechanisms remain unclear, Liu et al. (2017) suggested that the reduction in mitochondrial aldehyde dehydrogenase 2 (ALD2) activity due to its O-GlcNAcylation, impedes its protective effects and augments myocardial injury (82).
Although downregulating HBP activity should abate the negative consequences of chronic HBP activation, this is not always the case. Repression of OGT or OGA upregulation are primary intervention targets that are influenced by E2F transcription factor 1 (E2f1). E2f1 promotes entry into the S-phase of the cell cycle and alters metabolism by increasing pyruvate dehydrogenase kinase 4 activity which delinks glycolysis from mitochondrial oxidative phosphorylation (151). Increased E2f1 activity also potentiates phosphatase and tensin homolog-induced kinase 1 (PINK1) downregulation through miRNA-421 transcription, resulting in mitochondrial fragmentation and attenuated cardiomyocyte energetics (152). However, some like Dassanayaka et al. (2019) demonstrated that the lowering of E2f1 activity improved myocardial injury and cardiac remodeling without any effects on the HBP (106). This suggests that alternative NOGPs may utilize the accumulating glycolytic intermediates upstream of PDH, as opposed to the HBP.
Various interventions attempted to harness the potential benefits of an upregulated HBP in the context of ischemia (refer Figure 1). The most frequently reported compounds include salidroside, glucosamine, and glutamine as well as OGA inhibitors such as NAG-thiazoline due to their anti-hypoxia and pro-survival properties (135, 153–156). These treatments increase flux into the HBP through upregulated myocardial glucose uptake or as substrate intermediates within the HBP, as well as facilitating protein O-GlcNAcylation through OGT/OGA dynamics (135, 153–156). Although the exact mechanisms have not been fully elucidated, evidence suggests that protein O-GlcNAcylation influences Bcl-2 translocation (an intrinsic part of cell-death machinery) and cytoplasmic calcium concentration which mitigate injury following reperfusion (135, 155). However, the protective effects of glucosamine are diminished in obesity where increased HBP activity prevents insulin-dependent protein kinase B (Akt) signaling, thus hindering cell survival post-infarction (84).
Clinical and pre-clinical studies demonstrate the dual nature of the HBP during ischemic events. Increased HBP activity during mild ischemic or ischemic preconditioning protects the heart against ischemic injury. However, these effects do not extend to prolonged ischemic conditions that are associated with chronic HBP upregulation. Under such conditions, the HBP appears to be detrimental to cardiac health and function by disrupting normal cardiac metabolism and mitochondrial bioenergetics. Consistent O-GlcNAcylation of key proteins involved in these processes potentiate cardiac dysfunction and contribute towards increased IR/I.
Chronic HBP upregulation: Development of hypertension and cardiac hypertrophy
Clinical studies on idiopathic pulmonary artery hypertension observed increased HBP activity and circulating HbA1c levels with decreased heart function (41–43). In vivo experiments suggest increased HBP activation contributes to the lowered cardiac function observed in the clinical context (Tables 2, 3). Although the mechanisms underpinning such observations vary, elevated O-GlcNAcylation regulates adenosine monophosphate-activated protein kinase (AMPK) and endothelial nitric oxide synthase activity (eNOS) (43, 157). Here, O-GlcNAcylation of AMPKα and γ subunits increases its activity, which in turn results in AMPK phosphorylation of GFAT1/2 and OGT (threonine144), and creates a negative feedback loop in terms of HBP activity (112, 157). This bi-directional relationship improves heart function and limits cardiac hypertrophy in the event of increased AMPK activity and attenuated HBP flux (41, 73, 112). Interestingly, angiotensin II increases O-GlcNAcylation in the presence of active AMPK although the exact mechanism remains unclear (112).
Site-specific PTMs regulate eNOS synthase activity function, i.e. activated by serine1177,615,633 phosphorylation and inhibited by threonine495 phosphorylation (158). O-GlcNAc targets serine1177 and 615 without influencing other phosphorylation sites and lowering eNOS activity (43, 158). As a result, less nitric oxide is produced to subsequently lower vasodilatory capabilities and contributes to decreased vascular tone. Increased HBP flux promotes intracellular oxidative stress and cellular proliferation, further augmenting vascular dysfunction (41–43, 73). Nabeebaccus et al. (2017) observed that higher NAD(P)H oxidase 4 (NOX4) expression levels increased global O-GlcNAcylation through an activating transcription factor 4 (ATF4)-mediated increase in GFAT1 expression while shifting cardiac metabolism towards free fatty acid (FFA) β-oxidation versus glycolysis (109). Moreover, protein and mRNA analyses revealed a marked increase in cluster of differentiation 36 (CD36) O-GlcNAcylation (a major transporter of FFAs into the cytosol) which increases its activity and FFA influx into cardiomyocytes (109). Increased FFA β-oxidation can attenuate glycolysis via the Randle cycle enhancing HBP flux through upstream glycolytic substrate accumulation, and/or by higher GFAT1/2 expression. Although increased FFA β-oxidation is less efficient than glucose oxidative phosphorylation, mitochondrial bioenergetics and cardiac function were preserved in this case (109). This suggests that increased FFA β-oxidation may not be as detrimental to cardiac function in a hypertensive context, but that this may not hold when combined with other pathological complications such as ischemic events (reviewed by (159)).
Although cardiac hypertrophy is commonly observed under persistent hypertension, its development under diabetic and non-diabetic conditions remains a contentious topic. For example, under non-diabetic conditions, some indicated that the attenuated cardiac function observed in the hypertrophied heart (with increased HBP flux) is directly proportional to mammalian target of rapamycin (mTOR) activity (85). This resulted in increased protein synthesis and cellular growth, ultimately potentiating cardiac hypertrophy and fibrosis (85). However, others who characterized HBP flux in an ex vivo murine heart concluded that neither acute changes in glucose availability nor cardiac workload influenced its flux in a healthy heart (160). In vivo diabetic studies concluded that increased HBP activation blunted the hypertrophic response, although relatively high in vivo glucose levels and downregulated seipin increased extracellular signal-regulated kinase 1/2 (ERK1/2) mediated cyclin D2 expression (cell cycle progression), as well as repressor element 1-silencing transcription factor (REST) and forkhead box proteins of class O subgroup (FOXO) O-GlcNAcylation, ultimately promoting hypertrophic gene expression (49, 57, 58, 101). This suggests that although the stress-hypertrophy signaling may be blunted, the HBP can promote cardiac hypertrophy independent of stress signaling. The method by which diabetes is experimentally induced, and the condition of the heart (pre-intervention) may also be influencing factors. Genetically modified rodents such as the leptin receptor-deficient (db/db) mice and those injected with streptozotocin display distinct pathophysiological alterations that result in diabetes but may slightly alter their susceptibility to hypertrophy (161). Moreover, the duration of high glucose treatment (in vitro) or pressure overload intervention (in vivo/ex vivo) also contributes to the degree of dysfunction observed (Tables 2–4).
In summary, increased HBP activity and O-GlcNAcylation is observed in the context of cardiac hypertrophy and hypertension. Increased substrate availability combined with reduced antioxidant defense systems promotes flux into the HBP and O-GlcNAcylation of proteins such as AMPK and CD36. However, the effects of the HBP are not entirely clear under conditions where hypertension and cardiac hypertrophy exist in combination with diabetes. The degree by which the HBP influences all three pathologies centers around the duration and intensity of the stimulus (i.e. glucose overload, oxidative stress) as well as the experimental conditions.
Chronic HBP upregulation: Diabetic context
The greatest proportion of research work done thus far on the role of the HBP has focused on the diabetic context. In support, ~ 30% of articles cited in Tables 1–4 include either glycemic conditions, contributing factors (e.g., consumption of sugar-sweetened beverages), or diabetes-related pathology within a clinical/experimental context. For example, Wang et al. (2018) investigated the effects of a hyperinsulinemic hypoglycemic clamp in type 1 diabetes mellitus patients with myocardial infarction. Here, increased O-GlcNAc together with lowered miRNA-24 expression levels were found in patients with attenuated cardiac function (38). Increased miRNA-24 contributes to decreased apoptosis, lowered fasting glucose and insulin levels, and attenuated autophagy-related gene 4a (ATG4a – required for autophagy), as well as attenuated O-GlcNAc and OGT expression, thus conferring cardioprotective effects. Therefore, increasing miRNA-24 levels in circulation may prove a beneficial therapeutic target for both diabetic and ischemic heart conditions.
Experimental studies highlighted possible mechanisms for the blunted cardiac function observed in diabetic patients (38–40). Higher circulating glucose (indicated by elevated glycated hemoglobin levels) increases insulin-independent myocardial glucose uptake (39, 54). As the Krebs cycle possesses a limited capacity to convert pyruvate into reducing equivalents, glycolytic intermediates accumulate and are shunted into NOGPs such as the HBP (as discussed previously). Increased HBP flux promotes atrial fibrillation through calcium-calmodulin-dependent protein kinase II-dependent (CaMKII – cardiomyocyte calcium homeostasis) and independent mechanisms, respectively (59, 102). Elevated O-GlcNAcylation of key enzymes can increase oxidative stress, impair DNA repair machinery (8-oxoguanine DNA glycosylase (Ogg1)), increase apoptotic/autophagic stimuli, and promote mitochondrial stress. The combination of such PTMs contributes towards cardiac fibrosis, hypertrophy, and increased susceptibility to ischemic injury (26–29, 39, 46, 50, 55–65, 81, 82, 100, 127, 129–132, 134). Proteomic analysis suggests a potential role of O-GlcNAcylation in regulating myocardial actin (serine54) and troponin (serine150) functionality (162). While this may improve function in an acute context, chronic O-GlcNAcylation observed in conditions such as diabetes may result in decreased submaximal force development and reduced cardiac contractility (162). Increased HBP flux is also associated with metabolic derangements, lowered ketone utilization in the presence of enhanced production and availability, and increased aldehydes and FFAs (46, 50–53). Decreasing O-GlcNAcylation through OGA overexpression can lower p53 (“Guardian of the Genome”) expression thus inhibiting apoptotic signaling and improving cardiac function (62).
The majority of interventions in the context of diabetes are not preventative, and instead, focus on mitigating the effects of the HBP under established diabetic conditions. An interesting discrepancy is observed when comparing the findings of swim-exercised streptozotocin diabetic mice and treadmill-exercised db/db mice. Exercise significantly decreased cardiac O-GlcNAc in streptozotocin mice through increased OGA activity and expression, despite chronic hyperglycemia (163). However, db/db mice subjected to treadmill-exercise displayed increased cardiac protein O-GlcNAcylation and an upregulated hypertrophic response (125). Exercise was also investigated in the context of cardiac hypertrophy, where a single bout was sufficient to downregulate cytosolic protein O-GlcNAcylation and promote adaptive hypertrophic signaling (164). The discrepancy in findings may be in part due to the type of diabetic animal model utilized as well as the intensity and duration of the exercise protocol. Alternative interventions include vitamin D which lowers flux into NOGPs such as the HBP and AGE-RAGE pathways, mitigating protein O-GlcNAcylation and inflammation, respectively (66).
There is extensive evidence supporting increased HBP activity under diabetic conditions, regardless of the experimental induction. Increased O-GlcNAcylation of various proteins predisposes the heart to pathology through several mechanisms as highlighted in Tables 1–4. Combined with increased oxidative stress potentiating greater HBP activation, such dysfunctionality predisposes the heart to impaired contractility, hypertrophy, and fibrosis. These hallmarks of heart failure observed at a high frequency in diabetic patients, provide a mechanistic link between the two pathologies.
Heart failure: Role of the HBP
Clinical studies highlight increased O-GlcNAc levels in the myocardial tissue of patients with heart failure (ejection fraction < 20%) which provides a final clinical endpoint for enhanced HBP flux over an extended period (48). Moreover, reduced cardiac function under diabetic and non-diabetic conditions may be attributed to increased cardiac O-GlcNAcylation of target proteins (46–48). Diabetic patients diagnosed with heart failure also displayed attenuated ketone utilization, suggesting that alternative fuel substrates enter the heart, yet their potential energy is not harnessed (46).
Umapathi et al. (2021) highlighted the role of excessive O-GlcNAcylation in cardiac failure and sudden death (115). Transgenic mice overexpressing OGT developed significant cardiac pathologies including arrhythmias and dilation (115). Of note, crossing OGT transgenic mice with those overexpressing OGA, negated the detrimental effects of increased HBP activity. One key finding was that the O-GlcNAcylation of mitochondrial complex I impaired its function and activity (115). Mitochondrial complex I is considered the rate-limiting component of oxidative respiration and without sufficient activity, energy production is severely attenuated. Subsequently, cardiomyocytes are unable to contract efficiently, and the ejection fraction is ultimately reduced – thus returning to the diagnostic feature of heart failure.
A recently emerging biomarker of heart failure is miRNA-423-5p as congestive heart failure patients displayed increased plasma levels (139, 140). OGT is a downstream target with its expression significantly lowered in cardiomyocytes with increased expression of this miRNA. While this may appear beneficial in reducing O-GlcNAcylation, apoptotic stimuli and ubiquitin proteosome activity are elevated, thus contributing to cardiomyocyte death independently of chronic O-GlcNAcylation. Another diagnostic marker for heart failure is the increased serine phosphorylation of insoluble desmin (94). Desmin is a target of O-GlcNAcylation and is intricately involved in cardiac contractility (165, 166). Although site-specific O-GlcNAcylation can indirectly promote enzymatic activity by allosterically activating the phosphorylation site, this is not the case with desmin. Here, increased desmin O-GlcNAcylation does not alter its phosphorylation status but does confer improved cardiac function (94). These data indicate that this may not be solely attributed to desmin O-GlcNAcylation (no significant changes in expression) and instead likely be due to the acute upregulation of other O-GlcNAcylation targets during heart perfusion.
Autophagy plays a substantial role in the progression of heart failure and is also influenced by O-GlcNAcylation. Although pre-clinical work resulting in the downregulation of OGT expression indicates the role of O-GlcNAcylation in the initial phases of autophagy, chronic HBP upregulation hinders this process by modifying synaptosomal-associated protein 29 (SNAP29 – involved in the formation of the autophagolysosome) (60). Misfolded proteins and damaged organelles are subsequently unable to be degraded and interfere with intracellular homeostasis thus increasing the heart’s susceptibility to ischemic and hemodynamic injury (116). In support, therapeutic interventions aimed at increasing autophagy (e.g. spermidine) showed an improvement in cardiac health and function.
The role of O-GlcNAcylation in heart failure is somewhat controversial. Increased O-GlcNAcylation levels are observed in patients with heart failure and may contribute to autophagic dysregulation, impaired mitochondrial respiration, and altered ketone metabolism (Tables 1–4). However, diagnostic markers of heart failure (e.g. miRNA-423-5p) decrease O-GlcNAcylation which should provide protection; yet miRNA-423-5p promotes cardiomyocyte apoptosis independently of the HBP (139, 140). Thus, the HBP is not the sole contributor to a decreased ejection faction in the context of heart failure.
Regulating the HBP: Benefits and drawbacks
Substrate availability, circadian rhythm, and intracellular oxidative status all govern the activity of the HBP (32). Increased glycolytic flux with limited mitochondrial oxidative phosphorylation feeds glycolytic intermediates into the HBP, which results in a plethora of downstream PTMs. As the majority of such PTMs are detrimental to cardiovascular health in a chronic setting, there are emerging interventions to mitigate such detrimental effects.
Dietary supplementation with BCAAs influences cardiac glucose metabolism in a ratio-specific manner regarding its preferred substrates. A low BCAA/FFA ratio and glucose metabolism (enhanced catabolism) is beneficial while higher BCAA levels (lowered catabolism) can be damaging (107). Pre-clinical data highlight the effects of BCAA catabolic alterations (107, 167). Accumulation of BCAAs (lowered catabolism) inhibits the HBP and decreases O-GlcNAc PTMs, lowers PDH activity and myocardial glucose uptake, and promotes heart failure under conditions of pressure overload (107, 167). Decreased O-GlcNAc combined with lowered PDH activity suggest that glycolytic intermediates move into alternative NOGPs such as the AGE-RAGE or polyol pathway (Figure 2). Due to PDH inactivation, aerobic and anaerobic glycolysis are delinked, and the heart is therefore forced to rely on FFAs for mitochondrial ATP production. This results in increased injury during anoxic conditions due to reduced glucose catabolism, while excessive FFA utilization increases susceptibility to ischemic damage upon reperfusion – further compounding ischemic damage (107, 167). Promoting glucose metabolism by upregulating glucose transporter 1 (GLUT1) receptors rescues the heart from these damaging metabolic alterations and lowers ischemic damage (107). In contrast, enhanced BCAA catabolism is beneficial under certain circumstances, for e.g. it promotes survival through improving anti-oxidant defense systems and mitochondrial biogenesis in the heart following dietary supplementation (168). Therefore, the beneficial effects of BCAAs regarding the attenuation of HBP flux depends on various factors and the intracellular milieu at the time. This suggests that the downstream implications of proposed interventions to lower HBP activity need to be weighed against their potential benefits in a context-specific manner.
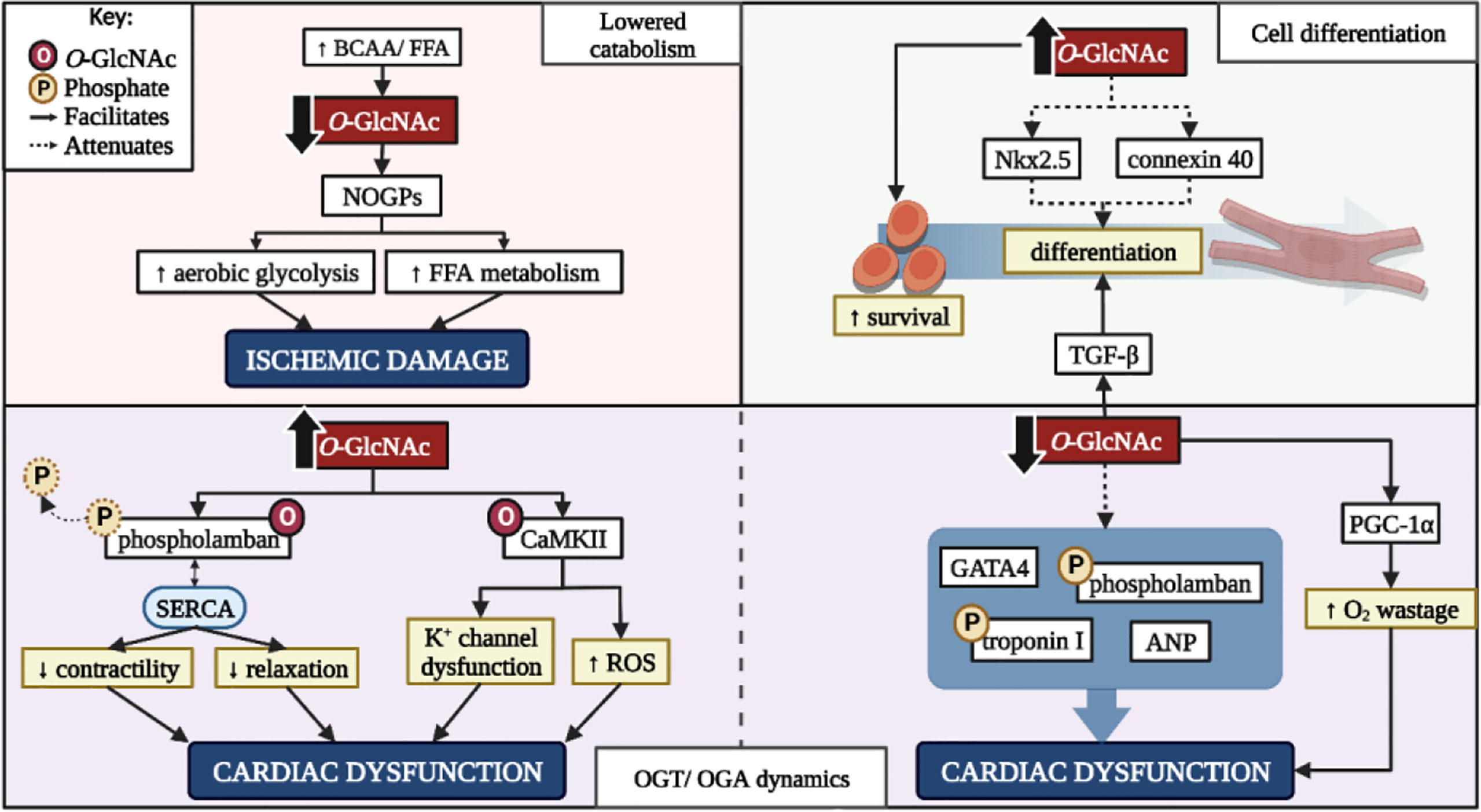
Figure 2 The benefits and drawbacks of altering cardiac O-GlcNAcylation. BCAA, branched-chain amino acid; FFA, free fatty acid; NOGPs, non-oxidative glucose pathways; K+, potassium; ROS, reactive oxygen species; CaMKII, calcium-calmodulin dependent protein kinase II; GATA4, GATA binding protein 4; ANP, atrial natriuretic peptide; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator-1α; OGT, O-GlcNAc transferase; OGA, O-GlcNAcase. Created with BioRender.com.
The impact of the HBP on in vitro cell differentiation is a lesser investigated area of research. The HBP is integral in mesenchymal stromal cell differentiation which extends to adult and embryonic cardiac stem cells (169–171). Evidence supports the shift away from oxidative metabolism and towards biosynthetic pathways to provide substrates for anabolic processes (170). However excessive O-GlcNAcylation hinders cardiac cell differentiation and may potentiate cardiomyopathy (171). Increased HBP activity decreases Nkx2.5 and connexin 40, whilst increasing smooth muscle actin expression (169). Nkx2.5 is heavily involved in cardiomyocyte cell fate during embryonic development and mutations within this gene are associated with congenital heart defects (172). Increased O-GlcNAcylation inhibits Nkx2.5 promotor activation thus attenuating its expression. Connexin 40 is a gap-junction protein intricately involved in the electrical conducting system of the heart (173) and lowered expression observed under conditions of increased HBP activity may occur as a direct or indirect result of O-GlcNAcylation, or Nkx2.5 expression (169). The combination of decreased Nkx2.5 and connexin 40 expression due to increased O-GlcNAcylation in adult cardiac mesenchymal cells limits their ability to differentiate and thus lower their efficacy as a cell-based therapeutic intervention (Figure 2). However, increased HBP activity in cardiac stem cells augments their regenerative capabilities and enhances cellular survival post-transplant (174). This further highlights the potential benefits and drawbacks of targeting increased HBP flux.
Aside from HBP activity regulated by GFAT1/2, the dynamics between OGT and OGA also determine its influence on cardiac function. For example, an increased OGA/OGT ratio lowers overall O-GlcNAcylation together with increased TGF-β and peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) expression and activity, and decreased GATA binding protein 4 (GATA4), atrial natriuretic peptide (ANP), p-phospholamban, and p-cardiac troponin I levels (48, 110, 111). Increased TGF-β and PGC-1α expression and activity promote myoblast differentiation and mitochondrial biogenesis, respectively (111). While this appears beneficial, increased PGC-1α activity also enhances the heart’s reliance on FFAs for energy production, thus potentiating oxygen wastage, reactive oxygen species production, and NOGP flux. Decreased GATA4, ANP, p-phospholamban, and p-cardiac troponin I expression collectively promote cardiovascular inotropy and lusitropy, fibrosis, and hypertrophy (110). Thus, the combination of events following targeted OGT inactivation or OGA hyperactivation may promote more damage than relief, depending on the context of the intervention (Figure 2). This targeted therapeutic approach would be beneficial to the heart in the event of established HBP overactivation, and not as a preventative or preemptive measure.
Phospholamban plays an integral role in cardiac contractility through its interactions with the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) (175). Phosphorylation of phospholamban on serine16 renders it inactive and alleviates any inhibitory effects on SERCA, thus allowing for increased cardiac contractility and relaxation (175). However, O-GlcNAc targets the serine residues that alter phospholamban’s function, for e.g. Yokoe et al. (2010) demonstrated an increase in O-GlcNAcylated phospholamban with a concurrent decrease in phosphorylated phospholamban and cardiac function (122). Additionally, interactions of SERCA and phospholamban were increased in PUGNAc treated cells, suggesting that increased phospholamban O-GlcNAcylation promotes its negative regulation on SERCA. However, OGTKO models indicate reduced phosphorylated phospholamban with no effect of SERCA calcium handling (110). Further studies are required to understand this dynamic relationship of phospholamban O-GlcNAcylation and phosphorylation. CaMKII is another protein heavily involved in cardiac contractility and the addition of O-GlcNAc moieties regulate its activity in a residue-specific manner. For example, serine280 O-GlcNAcylation is not required for activation yet influences potassium channel activity, while O-GlcNAcylation of serine279 autonomously activates CaMKII (57, 58, 125). Increased O-GlcNAcylation of CaMKII thus augments its activity and facilitates numerous downstream effects ranging from altered ion channel function and inflammatory gene transcription, culminating in the development of cardiac arrhythmias and reduced heart function (47, 127, 176).
While an increased OGA/OGT activity ratio does influence cardiac O-GlcNAcylation, the localization of OGA and OGT also impacts cardiac function. Under normal conditions, OGT is primarily localized to the Z-line of myofilaments with OGA to the A-band (61). The delocalization of such enzymes correlates to increased cardiac O-GlcNAc levels and an impaired calcium response (61). This suggests that OGT and OGA localization, as well as their activity and expression, influence their respective roles in cardiac dysfunction. Without a sufficient response to physiological calcium levels, the heart struggles to meet the required ejection fraction for normal bodily function. Compensatory mechanisms to increase cardiac output include an increase in left ventricular wall mass which contributes to cardiac hypertrophy. Therefore, the potential effects of targeted therapies to lower O-GlcNAcylation need to be weighed against the HBP’s physiological role.
Conclusion
The HBP can be considered a double-edged sword in the context of the heart (Figure 3). Various proteins require O-GlcNAc moieties for activation or repression and can confer cardioprotection against hypertrophy and apoptosis. However, consistent HBP upregulation either due to intrinsic factors (i.e. circadian rhythm), substrate availability, or elevated intracellular oxidative status, contribute towards chronic protein O-GlcNAcylation and subsequent cardiac dysfunction (Figure 3). Clinical, in vivo, and in vitro studies highlighted excessive HBP activity in a number of cardiac pathologies ranging from diabetic cardiomyopathy to hypertrophy, hypertension, and heart failure (Figure 3). Thus, the HBP has a emerged as a promising therapeutic target, although the extent of such downregulation needs to be carefully considered as basal levels are required for optimal cardiac function.
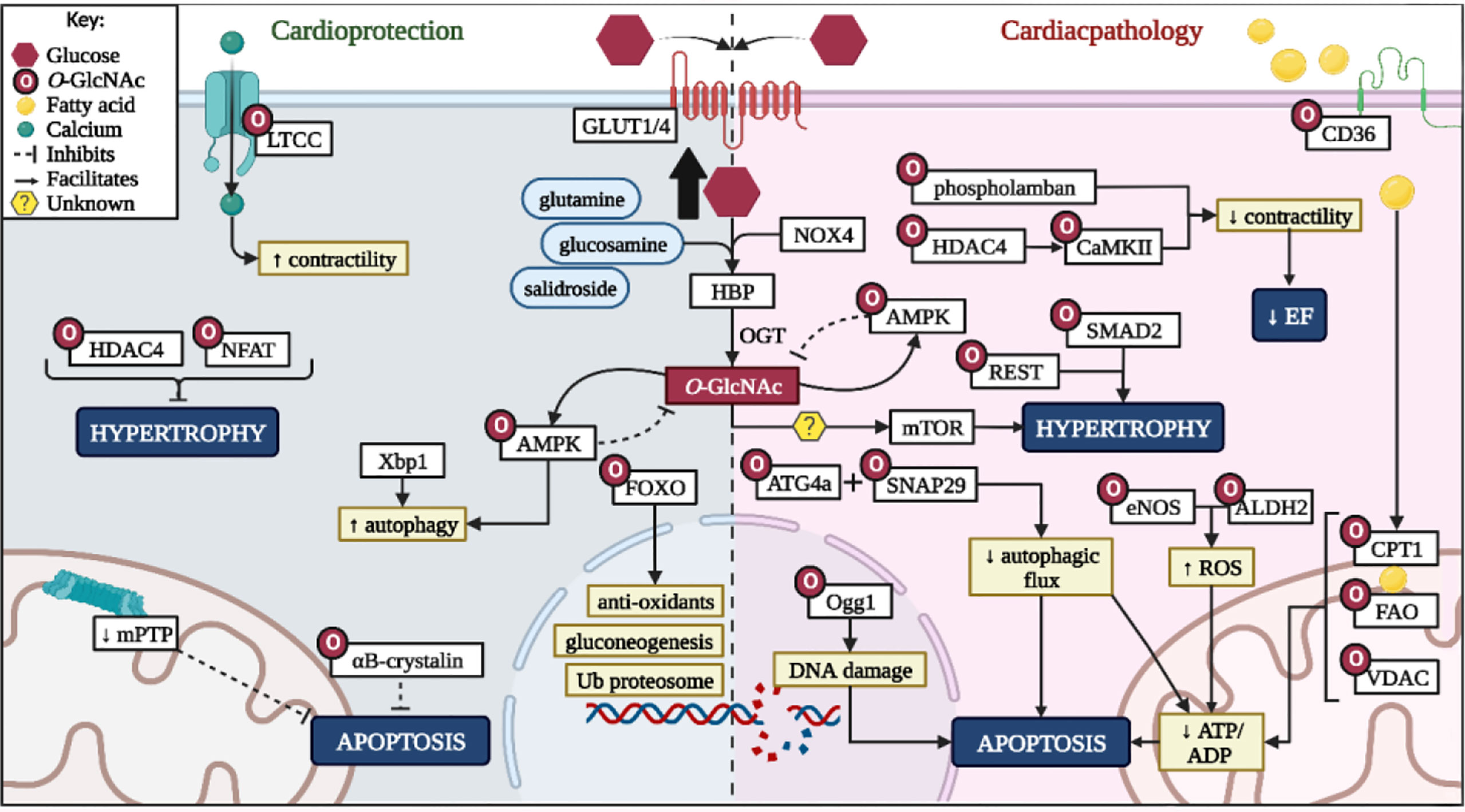
Figure 3 Summary of HBP role on cardioprotection and pathological effects. LTCC, L-type calcium channel; HDAC4, histone deacetylase 4; NFAT - nuclear factor of activated T-cells; Xbp1, X-box binding protein 1; GLUT1/4, glucose transporter 1/4; AMPK, 5ʹ-adenosine monophosphate-activated kinase; FOXO - forkhead box transcription factors; HBP, hexosamine biosynthetic pathway; OGT, O-GlcNAc transferase; NOX4, NADPH oxidase 4; ATG4a, autophagy related 4A cysteine peptidase; Ogg1 - 8-Oxoguanine glycosylase; SNAP29, synaptosomal-associated protein 29; mTOR, mammalian target of rapamycin; DNA, deoxyribonucleic acid; REST, repressor element 1-silencing transcription factor; SMAD2, mothers against decapentaplegic homolog 2; CaMKII, calcium-calmodulin dependent protein kinase II eNOS, endothelial nitric oxide; ALDH2, aldehyde dehydrogenase 2; ROS, reactive oxygen species; EF, ejection fraction; CD36, cluster of differentiation 36; CPT1, carnitine palmitoyltransferase-1/2; FAO, fatty acid β-oxidation enzymes; VDAC, voltage-dependent anion channels; ATP, adenosine triphosphate; ADP, adenosine diphosphate. Created with BioRender.com.
Author contributions
MC conceived the idea and wrote the first draft, edited and approved the final draft. DJ conceived the idea and wrote the first draft, edited and approved the final draft ME conceived the idea and wrote the first draft, edited and approved the final draft. All authors contributed to the article and approved the submitted version.
Funding
This work is based on the research supported wholly/in part by the National Research Foundation of South Africa (MND210428597749), National Research Foundation (129325 to MFE) and National Research Foundation (MND210428597749 to MC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: A question of balance. J Clin Invest (2005) 115(3):547–55.
2. Lopaschuk GD, Ussher JR. Evolving concepts of myocardial energy metabolism: More than just fats and carbohydrates. Circ Res (2016) 119(11):1173–6.
3. Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev (2010) 90(1):207–58.
4. Dávila-Román VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol (2002) 40(2):271–7.
5. Stuck BJ, Lenski M, Böhm M, Laufs U. Metabolic switch and hypertrophy of cardiomyocytes following treatment with angiotensin II are prevented by AMP-activated protein kinase. J Biol Chem (2008) 283(47):32562–9.
6. Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, et al. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ Res (2020) 126(2):182–96. doi: 10.1161/CIRCRESAHA.119.315483
7. Leong HS, Brownsey RW, Kulpa JE, Allard MF. Glycolysis and pyruvate oxidation in cardiac hypertrophy - why so unbalanced? Comp Biochem Physiol A Mol Integr Physiol (2003) 135(4):499–513.
8. Liu Q, Docherty JC, Rendell JC, Clanachan AS, Lopaschuk GD. High levels of fatty acids delay the recoveryof intracellular pH and cardiac efficiency inpost-ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol (2002) 39(4):718–25.
9. Dennis S. Protons in ischemia: Where do they come from; where do they go to? J Mol Cell Cardiol (1991) 23(9):1077–86.
10. Roy SG, De P, Mukherjee D, Chander V, Konar A, Bandyopadhyay D, et al. Excess of glucocorticoid induces cardiac dysfunction via activating angiotensin II pathway. Cell Physiol Biochem (2009) 24(1–2):1–10.
11. Kosova AA, Khodyreva SN, Lavrik OI. Role of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in DNA repair. Biochem (2017) 82(6):643–54. doi: 10.1134/S0006297917060013
12. Andrabi SA, Umanah GKE, Chang C, Stevens DA, Karuppagounder SS, Gagne J-P, et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci (2014) 111(28):10209–14. doi: 10.1073/pnas.1405158111
13. Song H, Yoon SP, Kim J. Poly(ADP-ribose) polymerase regulates glycolytic activity in kidney proximal tubule epithelial cells. Anat Cell Biol (2016) 49(2):79–87.
14. Pan L-Z, Ahn D-G, Sharif T, Clements D, Gujar S, Lee PWK. The NAD + synthesizing enzyme nicotinamide mononucleotide adenylyltransferase 2 (NMNAT-2) is a p53 downstream target. Cell Cycle (2014) 13(6):1041–8. doi: 10.4161/cc.28128
15. Kim J, Devalaraja-Narashimha K, Padanilam BJ. TIGAR regulates glycolysis in ischemic kidney proximal tubules. Am J Physiol Physiol (2015) 308(4):F298–F308. doi: 10.1152/ajprenal.00459.2014
16. Gibb AA, Lorkiewicz PK, Zheng YT, Zhang X, Bhatnagar A, Jones SP, et al. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem J (2017) 474(16):2785–801.
17. Chatham JC, Zhang J, Wende AR. Role of o-linked n-acetylglucosamine proteinmodification in cellular (Patho) physiology. Physiol Rev (2021) 101(2):427–93.
18. Ducheix S, Magré J, Cariou B, Prieur X. Chronic O-GlcNAcylation and diabetic cardiomyopathy: The bitterness of glucose. Front Endocrinol (2018) 9:1–11.
19. Qin CX, Sleaby R, Davidoff AJ, Bell JR, De Blasio MJ, Delbridge LM, et al. Insights into the role of maladaptive hexosamine biosynthesis and O-GlcNAcylation in development of diabetic cardiac complications. Pharmacol Res (2017) 116:45–56. doi: 10.1016/j.phrs.2016.12.016
20. UniProt Consortium. UniProtKB - O94808 (GFPT2_HUMAN). UniProt (2021). Available at: https://www.uniprot.org/uniprot/O94808.
21. NCBI. Gfpt2 glutamine-fructose-6-phosphate transaminase 2 [Rattus norvegicus (Norway rat)] (2021). Available at: https://www.ncbi.nlm.nih.gov/gene/360518.
22. Nabeebaccus A, Verma S, Zoccarato A, Emanuelli G, Santos CX, Streckfuss-Bömeke K, et al. Cardiomyocyte protein O-GlcNAcylation is regulated by GFAT1 not GFAT2. Biochem Biophys Res Commun (2021) 583:121–7.
23. Darley-Usmar VM, Ball LE, Chatham JC. Protein O-linked β-n-acetylglucosamine: A novel effector of cardiomyocyte metabolism and function. J Mol Cell Cardiol (2012) 52(3):538–49. doi: 10.1016/j.yjmcc.2011.08.009
24. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol (2017) 18(7):452–65. doi: 10.1038/nrm.2017.22
25. Liddy KA, White MY, Cordwell SJ. Functional decorations: Post-translational modifications and heart disease delineated by targeted proteomics. Genome Med (2013) 5(2):20. doi: 10.1186/gm424
26. Rajamani U, Joseph D, Roux S, Essop M. The hexosamine biosynthetic pathway can mediate myocardial apoptosis in a rat model of diet-induced insulin resistance. Acta Physiol (2011) 202(2):151–7. doi: 10.1111/j.1748-1716.2011.02275.x
27. Rajamani U, Essop M. Hyperglycemia-mediated activation of the hexosamine biosynthetic pathway results in myocardial apoptosis. Am J Physiol Physiol (2010) 299(1):C139–47. doi: 10.1152/ajpcell.00020.2010
28. Joseph D, Kimar C, Symington B, Milne R, Essop M. The detrimental effects of acute hyperglycemia on myocardial glucose uptake. Life Sci (2014) 105(1–2):31–42. doi: 10.1016/j.lfs.2014.04.009
29. Marsh SA, Powell PC, Dell’Italia LJ, Chatham JC. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci (2013) 92(11):648–56. doi: 10.1016/j.lfs.2012.06.011
30. Thompson JW, Griffin ME, Hsieh-Wilson LC. Methods for the detection, study, and dynamic profiling of O-GlcNAc glycosylation. Physiology and behavior (2018) 598:101–35. doi: 10.1016/bs.mie.2017.06.009
31. Banerjee PS, Hart GW, Won Cho J. Chemical approaches to study O-GlcNAcylation. Chem Soc Rev (2013) 42(10):4345–57.
32. Durgan DJ, Pat BM, Laczy B, Bradley JA, Tsai J-Y, Grenett MH, et al. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock*. J Biol Chem (2011) 286(52):44606–19. doi: 10.1074/jbc.M111.278903
33. Watson LJ, Long BW, DeMartino AM, Brittian KR, Readnower RD, Brainard RE, et al. Cardiomyocyte ogt is essential for postnatal viability. Am J Physiol Hear Circ Physiol (2014) 306(1):142–53.
34. Dupas T, Denis M, Dontaine J, Persello A, Bultot L, Erraud A, et al. Protein O-GlcNAcylation levels are regulated independently of dietary intake in a tissue and time-specific manner during rat postnatal development. Acta Physiol (2021) 231(3):1–15.
35. Fülöp N, Feng W, Xing D, He K, Nőt LG, Brocks CA, et al. Aging leads to increased levels of protein O-linked n-acetylglucosamine in heart, aorta, brain and skeletal muscle in brown-Norway rats. Biogerontology (2008) 9(3):139. doi: 10.1007/s10522-007-9123-5
36. Shen Y, Yan B, Zhao Q, Wang Z, Wu J, Ren J, et al. Aging is associated with impaired activation of protein homeostasis-related pathways after cardiac arrest in mice. J Am Heart Assoc (2018) 7(17):1–16.
37. Li R, Shen Y, Li X, Lu L, Wang Z, Sheng H, et al. Activation of the XBP1s/O-GlcNAcylation pathway improves functional outcome after cardiac arrest and resuscitation in young and aged mice. Shock (2021) 56(5):755–61. doi: 10.1097/SHK.0000000000001732
38. Wang D, Hu X, Lee SH, Chen F, Jiang K, Tu Z, et al. Diabetes exacerbates myocardial Ischemia/Reperfusion injury by down-regulation of MicroRNA and up-regulation of O-GlcNAcylation. JACC Basic Transl Sci (2018) 3(3):350–62.
39. Masaki N, Feng B, Bretón-Romero R, Inagaki E, Weisbrod RM, Fetterman JL, et al. O-GlcNAcylation mediates glucose-induced alterations in endothelial cell phenotype in human diabetes mellitus. J Am Heart Assoc (2020) 9(12):e014046.
40. Prakoso D, Lim SY, Erickson JR, Wallace RS, Lees JG, Tate M, et al. Fine-tuning the cardiac O-GlcNAcylation regulatory enzymes governs the functional and structural phenotype of the diabetic heart. Cardiovasc Res (2022) 118(1):212–25.
41. Prisco SZ, Rose L, Potus F, Tian L, Wu D, Hartweck L, et al. Excess protein o-glcnacylation links metabolic derangements to right ventricular dysfunction in pulmonary arterial hypertension. Int J Mol Sci (2020) 21(19):1–19.
42. Barnes JW, Tian L, Heresi GA, Farver CF, Asosingh K, Comhair SAA, et al. O-Linked β-n-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation (2015) 131(14):1260–8.
43. Aulak KS, Barnes JW, Tian L, Mellor NE, Haque MM, Willard B, et al. Specific O-GlcNAc modification at ser-615 modulates eNOS function. Redox Biol (2020) 36:101625. doi: 10.1016/j.redox.2020.101625
44. Lunde IG, Aronsen JM, Kvaløy H, Qvigstad E, Sjaastad I, Tønnessen T, et al. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol Genomics (2012) 44(2):162–72. doi: 10.1152/physiolgenomics.00016.2011
45. Duncan AE, Kashy BK, Sarwar S, Singh A, Stenina-Adognravi O, Christoffersen S, et al. Hyperinsulinemic normoglycemia does not meaningfully improve myocardial performance during cardiac surgery: A randomized trial. Anesthesiology (2015) 123(2):272–87.
46. Brahma MK, Ha CM, Pepin ME, Mia S, Sun Z, Chatham JC, et al. Increased glucose availability attenuates myocardial ketone body utilization. J Am Heart Assoc (2020) 9(15):e013039.
47. Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature (2013) 502(7471):372–6.
48. Dassanayaka S, Brainard RE, Watson LJ, Long BW, Brittian KR, DeMartino AM, et al. Cardiomyocyte ogt limits ventricular dysfunction in mice following pressure overload without affecting hypertrophy. Basic Res Cardiol (2017) 112(3):1–17.
49. Medford HM, Cox EJ, Miller LE, Marsh SA. Consuming a Western diet for two weeks suppresses fetal genes in mouse hearts. Am J Physiol Regul Integr Comp Physiol (2014) 306(8):519–26.
50. Cui Y-L, Xue R-Q, He X, Zhao M, Yu X-J, Liu L-Z, et al. Cholinergic drugs ameliorate endothelial dysfunction by decreasing O-GlcNAcylation via M3 AChR-AMPK-ER stress signaling. Life Sci (2019) 222(76):1–12.
51. Karimi M, Pavlov VI, Ziegler O, Sriram N, Yoon S-Y, Agbortoko V, et al. Robust effect of metabolic syndrome on major metabolic pathways in the myocardium. PloS One (2019) 14(12):e0225857. doi: 10.1371/journal.pone.0225857
52. Medford HM, Chatham JC, Marsh SA. Chronic ingestion of a Western diet increases O-linked-β-N-acetylglucosamine (O-GlcNAc) protein modification in the rat heart. Life Sci (2012) 90(23–24):883–8.
53. Tanaka T, Sohmiya K, Kono T, Terasaki F, Horie R, Ohkaru Y, et al. Thiamine attenuates the hypertension and metabolic abnormalities in CD36-defective SHR: Uncoupling of glucose oxidation from cellular entry accompanied with enhanced protein O-GlcNAcylation in CD36 deficiency. Mol Cell Biochem (2007) 299(1–2):23–35.
54. Driescher N, Joseph DE, Human VR, Ojuka E, Cour M, Hadebe N, et al. The impact of sugar-sweetened beverage intake on rat cardiac function. Heliyon (2019) 5(3):e01357.
55. Cividini F, Scott BT, Dai A, Han W, Suarez J, Diaz-Juarez J, et al. O-GlcNAcylation of 8-oxoguanine DNA glycosylase (Ogg1) impairs oxidative mitochondrial DNA lesion repair in diabetic hearts. J Biol Chem (2016) 291(51):26515–28.
56. Ma J, Banerjee P, Whelan SA, Liu T, Wei AC, Ramirez-Correa G, et al. Comparative proteomics reveals dysregulated mitochondrial O-GlcNAcylation in diabetic hearts. J Proteome Res (2016) 15(7):2254–64.
57. Marsh SA, Dell’Italia LJ, Chatham JC. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids (2011) 40(3):819–28.
58. Ding F, Yu L, Wang M, Xu S, Xia Q, Fu G. O-GlcNAcylation involvement in high glucose-induced cardiac hypertrophy via ERK1/2 and cyclin D2. Amino Acids (2013) 45(2):339–49.
59. Yu P, Hu L, Xie J, Chen S, Huang L, Xu Z, et al. O-GlcNAcylation of cardiac Nav1.5 contributes to the development of arrhythmias in diabetic hearts. Int J Cardiol (2018) 260:74–81. doi: 10.1016/j.ijcard.2018.02.099
60. Huang L, Yuan P, Yu P, Kong Q, Xu Z, Yan X, et al. O-GlcNAc-modified SNAP29 inhibits autophagy-mediated degradation via the disturbed SNAP29-STX17-VAMP8 complex and exacerbates myocardial injury in type I diabetic rats. Int J Mol Med (2018) 42(6):3278–90. doi: 10.3892/ijmm.2018.3866
61. Ramirez-Correa GA, Ma J, Slawson C, Zeidan Q, Lugo-Fagundo NS, Xu M, et al. Removal of abnormal myofilament O-GlcNAcylation restores Ca2+ sensitivity in diabetic cardiac muscle. Diabetes (2015) 64(10):3573–87.
62. Si R, Zhang Q, Tsuji-Hosokawa A, Watanabe M, Willson C, Lai N, et al. Overexpression of p53 due to excess protein O-GlcNAcylation is associated with coronary microvascular disease in type 2 diabetes. Cardiovasc Res (2020) 116(6):1186–98.
63. De Blasio MJ, Huynh N, Deo M, Dubrana LE, Walsh J, Willis A, et al. Defining the progression of diabetic cardiomyopathy in a mouse model of type 1 diabetes. Front Physiol (2020) 11:1–16.
64. Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, et al. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem (2003) 278(45):44230–7. doi: 10.1074/jbc.M303810200
65. Banerjee PS, Ma J, Hart GW. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci USA (2015) 112(19):6050–5.
66. Derakhshanian H, Djazayery A, Javanbakht MH, Eshraghian MR, Mirshafiey A, Jahanabadi S, et al. Vitamin d downregulates key genes of diabetes complications in cardiomyocyte. J Cell Physiol (2019) 234(11):21352–8.
67. Fülöp N, Mason MM, Dutta K, Wang P, Davidoff AJ, Marchase RB, et al. Impact of type 2 diabetes and aging on cardiomyocyte function and O-linked n-acetylglucosamine levels in the heart. Am J Physiol Cell Physiol (2007) 292(4):2006–7.
68. Fricovsky ES, Suarez J, Ihm SH, Scott BT, Suarez-Ramirez JA, Banerjee I, et al. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol (2012) 303(7):R689–R699.
69. Chen X, Zhang L, He H, Sun Y, Shen Q, Shi L. Increased O-GlcNAcylation induces myocardial hypertrophy. Vitr Cell Dev Biol Anim (2020) 56(9):735–43.
70. Greenman AC, Diffee GM, Power AS, Wilkins GT, Gold OMS, Erickson JR, et al. Increased myofilament calcium sensitivity is associated with decreased cardiac troponin I phosphorylation in the diabetic rat heart. Exp Physiol (2021) 106(11):2235–47.
71. Kim HS, Woo JS, Joo HJ, Moon WK. Cardiac transcription factor Nkx2.5 is downregulated under excessive O-GlcNAcylation condition. PloS One (2012) 7(6):3–8.
72. Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, et al. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem (2012) 287(35):30024–34.
73. Lima VV, Giachini FRC, Choi H, Carneiro FS, Carneiro ZN, Fortes ZB, et al. Impaired vasodilator activity in deoxycorticosterone acetate-salt hypertension is associated with increased protein O-GlcNAcylation. Hypertension (2009) 53(2):166–74.
74. Guo X, Shang J, Deng Y, Yuan X, Zhu D, Liu H. Alterations in left ventricular function during intermittent hypoxia: Possible involvement of O-GlcNAc protein and MAPK signaling. Int J Mol Med (2015) 36(1):150–8.
75. Ngoh GA, Watson LJ, Facundo HT, Jones SP. Augmented O-GlcNAc signaling attenuates oxidative stress and calcium overload in cardiomyocytes. Amino Acids (2011) 40(3):895–911. doi: 10.1007/s00726-010-0728-7
76. Ou W, Liang Y, Qing Y, Wu W, Xie M, Zhang Y, et al. Hypoxic acclimation improves cardiac redox homeostasis and protects heart against ischemia-reperfusion injury through upregulation of O-GlcNAcylation. Redox Biol (2021) 43:101994. doi: 10.1016/j.redox.2021.101994
77. Kristiansen SB, Pælestik KB, Johnsen J, Jespersen NR, Pryds K, Hjortbak MV, et al. Impact of hyperglycemia on myocardial ischemia-reperfusion susceptibility and ischemic preconditioning in hearts from rats with type 2 diabetes. Cardiovasc Diabetol (2019) 18(1):1–10. doi: 10.1186/s12933-019-0872-7
78. Jensen RV, Johnsen J, Kristiansen SB, Zachara NE, Bøtker HE. Ischemic preconditioning increases myocardial O-GlcNAc glycosylation. Scand Cardiovasc J (2013) 47(3):168–74. doi: 10.3109/14017431.2012.756984
79. Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, et al. Cardioprotection by n-acetylglucosamine linkage to cellular proteins. Circulation (2008) 117(9):1172–82.
80. Pælestik KB, Jespersen NR, Jensen RV, Johnsen J, Bøtker HE, Kristiansen SB. Effects of hypoglycemia on myocardial susceptibility to ischemia–reperfusion injury and preconditioning in hearts from rats with and without type 2 diabetes. Cardiovasc Diabetol (2017) 16(1):148. doi: 10.1186/s12933-017-0628-1
81. Mapanga RF, Rajamani U, Dlamini N, Zungu-Edmondson M, Kelly-Laubscher R, Shafiullah M, et al. Oleanolic acid: A novel cardioprotective agent that blunts hyperglycemia-induced contractile dysfunction. PloS One (2012) 7(10):e47322. doi: 10.1371/journal.pone.0047322
82. Liu B, Wang J, Li M, Yuan Q, Xue M, Xu F, et al. Inhibition of ALDH2 by O-GlcNAcylation contributes to the hyperglycemic exacerbation of myocardial ischemia/reperfusion injury. Oncotarget (2017) 8(12):19413–26.
83. Liu J, Pang Y, Chang T, Bounelis P, Chatham J, Marchase R. Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J Mol Cell Cardiol (2006) 40(2):303–12.
84. Jin L, Gao F, Jiang T, Liu B, Li C, Qin X, et al. Hyper-O-GlcNAcylation impairs insulin response against reperfusion-induced myocardial injury and arrhythmias in obesity. Biochem Biophys Res Commun (2021) 558:126–33. doi: 10.1016/j.bbrc.2021.04.066
85. Tran DH, May HI, Li Q, Luo X, Huang J, Zhang G, et al. Chronic activation of hexosamine biosynthesis in the heart triggers pathological cardiac remodeling. Nat Commun (2020) 11(1):1771. doi: 10.1038/s41467-020-15640-y
86. Zhu WZ, Ledee D, Olson AK. Temporal regulation of protein O -GlcNAc levels during pressure-overload cardiac hypertrophy. Physiol Rep (2021) 9(15):1–17. doi: 10.14814/phy2.14965
87. Facundo HT, Brainard RE, Watson LJ, Ngoh GA, Hamid T, Prabhu SD, et al. O-GlcNAc signaling is essential for NFAT-mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am J Physiol Hear Circ Physiol (2012) 302(10):2122–31.
88. Ferron M, Cadiet J, Persello A, Prat V, Denis M, Erraud A, et al. O-GlcNAc stimulation: A new metabolic approach to treat septic shock. Sci Rep (2019) 9(1):18751.
89. Zou L, Yang S, Champattanachai V, Hu S, Chaudry IH, Marchase RB, et al. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-κB signaling. Am J Physiol Hear Circ Physiol (2009) 296(2):515–23.
90. Zou L, Yang S, Hu S, Chaudry IH, Marchase RB, Chatham JC. The protective effects of PUGNAc on cardiac function after trauma-hemorrhage are mediated via increased protein O-GlcNAc levels. Shock (2007) 27(4):402–8.
91. Nöt LG, Brocks CA, Vámhidy L, Marchase RB, Chatham JC. Increased O-linked β-n-acetylglucosamine levels on proteins improves survival, reduces inflammation and organ damage 24 hours after trauma-hemorrhage in rats. Crit Care Med (2010) 38(2):562–71.
92. Yang S, Zou L, Bounelis P, Chaudry I, Chatham JC, Marchase RB. Glucosamine administration during resuscitation improves organ function after trauma hemorrhage. SHOCK (2006) 25(6):600–7.
93. Nöt LG, Marchase RB, Fülöp N, Brocks CA, Chatham JC. Glucosamine administration improves survival rate after severe hemorrhagic shock combined with trauma in rats. Shock (2007) 28(3):345–52.
94. Mercier T, Bouvet M, Dubois-Deruy E, Dechaumes A, Beseme O, Richard V, et al. Interplay between phosphorylation and O-GlcNAcylation of sarcomeric proteins in ischemic heart failure. Front Endocrinol (2018) 9:1–10.
95. Dubois-Deruy E, Belliard A, Mulder P, Bouvet M, Smet-Nocca C, Janel S, et al. Interplay between troponin T phosphorylation and O-n-acetylglucosaminylation in ischaemic heart failure. Cardiovasc Res (2015) 107(1):56–65.
96. Muthusamy S, DeMartino AM, Watson LJ, Brittian KR, Zafir A, Dassanayaka S, et al. MicroRNA-539 is up-regulated in failing heart, and suppresses O-GlcNAcase expression. J Biol Chem (2014) 289(43):29665–76.
97. Gubbiotti MA, Seifert E, Rodeck U, Hoek JB, Iozzo RV. Metabolic reprogramming of murine cardiomyocytes during autophagy requires the extracellular nutrient sensor decorin. J Biol Chem (2019) 293(43):16940–50.
98. Vaillant F, Lauzier B, Ruiz M, Shi Y, Lachance D, Rivard ME, et al. Ivabradine and metoprolol differentially affect cardiac glucose metabolism despite similar heart rate reduction in a mouse model of dyslipidemia. Am J Physiol Hear Circ Physiol (2016) 311(4):H991–1003.
99. Hecker PA, Mapanga RF, Kimar CP, Ribeiro RF, Brown BH, O’Connell KA, et al. Effects of glucose-6-phosphate dehydrogenase deficiency on the metabolic and cardiac responses to obesogenic or high-fructose diets. Am J Physiol Endocrinol Metab (2012) 303(8):E298—E972.
100. Kronlage M, Dewenter M, Grosso J, Fleming T, Oehl U, Lehmann LH, et al. O-GlcNAcylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation (2019) 140(7):580–94.
101. Lugat A, Joubert M, Cariou B, Prieur X. Au cœur de la cardiomyopathie diabétique. Med Sci (2018) 34(6–7):563–70.
102. Mesubi OO, Rokita AG, Abrol N, Wu Y, Chen B, Wang Q, et al. Oxidized CaMKII and O-GlcNAcylation cause increased atrial fibrillation in diabetic mice by distinct mechanisms. J Clin Invest (2021) 131(2):1–14.
103. Na J, Musselman LP, Pendse J, Baranski TJ, Bodmer R, Ocorr K, et al. A drosophila model of high sugar diet-induced cardiomyopathy. PloS Genet (2013) 9(1):e1003175. doi: 10.1371/journal.pgen.1003175
104. Nakagawa T, Furukawa Y, Hayashi T, Nomura A, Yokoe S, Moriwaki K, et al. Augmented O-GlcNAcylation attenuates intermittent hypoxia-induced cardiac remodeling through the suppression of NFAT and NF-κB activities in mice. Hypertens Res (2019) 42:1858–71.
105. Ngoh GA, Watson LJ, Facundo HT, Dillmann W, Jones SP. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. J Mol Cell Cardiol (2008) 45(2):313–25.
106. Dassanayaka S, Brittian KR, Jurkovic A, Higgins LA, Audam TN, Long BW, et al. E2f1 deletion attenuates infarct-induced ventricular remodeling without affecting O-GlcNAcylation. Basic Res Cardiol (2019) 114(4):1–18. doi: 10.1007/s00395-019-0737-y
107. Li T, Zhang Z, Kolwicz SC, Abell L, Roe ND, Kim M, et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab (2017) 25(2):374–85. doi: 10.1016/j.cmet.2016.11.005
108. Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell (2014) 156(6):1179–92.
109. Nabeebaccus AA, Zoccarato A, Hafstad AD, Santos CXC, Aasum E, Brewer AC, et al. Nox4 reprograms cardiac substrate metabolism via protein O-GlcNAcylation to enhance stress adaptation. JCI Insight (2017) 2(24):1–16.
110. Zhu WZ, El-Nachef D, Yang X, Ledee D, Olson AK. O-GlcNAc transferase promotes compensated cardiac function and protein kinase a O-GlcNAcylation during early and established pathological hypertrophy from pressure overload. J Am Heart Assoc (2019) 8(11):1–21.
111. Brainard RE, Facundo HT. Cardiac hypertrophy drives PGC-1α suppression associated with enhanced O-glycosylation. Biochim Biophys Acta Mol Basis Dis (2021) 1867(5):166080.
112. Gélinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun (2018) 9(1):1–17. doi: 10.1038/s41467-017-02795-4
113. Ledee D, Smith L, Bruce M, Kajimoto M, Isern N, Portman MA, et al. C-myc alters substrate utilization and OGlcNAc protein posttranslational modifications without altering cardiac function during early aortic constriction. PloS One (2015) 10(8):1–16.
114. Cannon MV, Silljé HH, Sijbesma JW, Vreeswijk-Baudoin I, Ciapaite J, van der Sluis B, et al. Cardiac LXRα protects against pathological cardiac hypertrophy and dysfunction by enhancing glucose uptake and utilization. EMBO Mol Med (2015) 7(9):1229–43. doi: 10.15252/emmm.201404669
115. Umapathi P, Mesubi OO, Banerjee PS, Abrol N, Wang Q, Luczak ED, et al. Excessive O -GlcNAcylation causes heart failure and sudden death. Circulation (2021) 143(17):1687–703. doi: 10.1161/CIRCULATIONAHA.120.051911
116. Yu H, Wen L, Mu Y. O-GlcNAcylation is essential for autophagy in cardiomyocytes. Oxid Med Cell Longev (2020) 2020:1–9.
117. Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, et al. O-Linked β-n-acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci USA (2010) 107(41):17797–802.
118. Dassanayaka S, Brittian KR, Long BW, Higgins LA, Bradley JA, Audam TN, et al. Cardiomyocyte oga haploinsufficiency increases O-GlcNAcylation but hastens ventricular dysfunction following myocardial infarction. PloS One (2020) 15:1–20.
119. Lehmann LH, Jebessa ZH, Kreusser MM, Horsch A, He T, Kronlage M, et al. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat Med (2018) 24(1):62–72.
120. Leung MC, Hitchen PG, Ward DG, Messer AE, Marston SB. Z-band alternatively spliced PDZ motif protein (ZASP) is the major o-linked β-n-acetylglucosamine-substituted protein in human heart myofibrils. J Biol Chem (2013) 288(7):4891–8.
121. Krishnamoorthy V, Donofrio AJ, Martin JL. O-GlcNAcylation of αB-crystallin regulates its stress-induced translocation and cytoprotection. Mol Cell Biochem (2013) 379(1–2):59–68. doi: 10.1007/s11010-013-1627-5
122. Yokoe S, Asahi M, Takeda T, Otsu K, Taniguchi N, Miyoshi E, et al. Inhibition of phospholamban phosphorylation by O-GlcNAcylation: Implications for diabetic cardiomyopathy. Glycobiology (2010) 20(10):1217–26.
123. Cao H, Hu Y, Zhu X, Yao N, Gu J, Wang Y, et al. O-GlcNAc transferase affects the signal transduction of β1 adrenoceptor in adult rat cardiomyocytes by increasing the O-GlcNAcylation of β1 adrenoceptor. Biochem Biophys Res Commun (2020) 528(1):71–7. doi: 10.1016/j.bbrc.2020.05.010
124. Akinbiyi EO, Abramowitz LK, Bauer BL, Stoll MSK, Hoppel CL, Hsiao CP, et al. Blocked O-GlcNAc cycling alters mitochondrial morphology, function, and mass. Sci Rep (2021) 11(1):1–16. doi: 10.1038/s41598-021-01512-y
125. Cox EJ, Marsh SA. Exercise and diabetes have opposite effects on the assembly and O-GlcNAc modification of the mSin3A/HDAC1/2 complex in the heart. Cardiovasc Diabetol (2013) 12(1):1.
126. Zou L, Zhu-Mauldin X, Marchase RB, Paterson AJ, Liu J, Yang Q, et al. Glucose deprivation-induced increase in protein O-GlcNAcylation in cardiomyocytes is calcium-dependent. J Biol Chem (2012) 287(41):34419–31. doi: 10.1074/jbc.M112.393207
127. Hegyi B, Borst JM, Bailey LRJ, Shen EY, Lucena AJ, Navedo MF, et al. Hyperglycemia regulates cardiac k+ channels via O-GlcNAc-CaMKII and NOX2-ROS-PKC pathways. Basic Res Cardiol (2020) 15(6):71. doi: 10.1007/s00395-020-00834-8
128. Lu S, Liao Z, Lu X, Katschinski DM, Mercola M, Chen J, et al. Hyperglycemia acutely increases cytosolic reactive oxygen species via O -linked GlcNAcylation and CaMKII activation in mouse ventricular myocytes. Circ Res (2020) 126(10)e80–e966. doi: 10.1161/CIRCRESAHA.119.316288
129. Aguilar H, Fricovsky E, Ihm S, Schimke M, Maya-Ramos L, Aroonsakool N, et al. Role for high-glucose-induced protein O-GlcNAcylation in stimulating cardiac fibroblast collagen synthesis. Am J Physiol Cell Physiol (2014) 306(9):794–804.
130. Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, et al. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem (2009) 284(1):547–55. doi: 10.1074/jbc.M808518200
131. Li SY, Sigmon VK, Babcock SA, Ren J. Advanced glycation endproduct induces ROS accumulation, apoptosis, MAP kinase activation and nuclear O-GlcNAcylation in human cardiac myocytes. Life Sci (2007) 80(11):1051–6.
132. Makino A, Suarez J, Gawlowski T, Han W, Wang H, Scott BT, et al. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am J Physiol Regul Integr Comp Physiol (2011) 300(6):1296–302.
133. Balteau M, Tajeddine N, De Meester C, Ginion A, Des Rosiers C, Brady NR, et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc Res (2011) 92(2):237–46.
134. Dassanayaka S, Readnower RD, Salabei JK, Long BW, Aird AL, Zheng Y-T, et al. High glucose induces mitochondrial dysfunction independently of protein O-GlcNAcylation. Biochem J (2015) 467(1):115–26.
135. Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial bcl-2. Am J Physiol Physiol (2008) 294(6):C1509–20.
136. Ngoh GA, Facundo HT, Hamid T, Dillmann W, Zachara NE, Jones SP. Unique hexosaminidase reduces metabolic survival signal and sensitizes cardiac myocytes to Hypoxia/Reoxygenation injury. Circ Res (2009) 104(1):41–9. doi: 10.1161/CIRCRESAHA.108.189431
137. Ngoh GA, Hamid T, Prabhu SD, Jones SP. O-GlcNAc signaling attenuates ER stress-induced cardiomyocyte death. Am J Physiol Hear Circ Physiol (2009) 297(5):H1711–H1719.
138. Zhu-Mauldin X, Marsh SA, Zou L, Marchase RB, Chatham JC. Modification of STIM1 by O-linked n-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes. J Biol Chem (2012) 287(46):39094–106.
139. Luo P, He T, Jiang R, Li G. MicroRNA-423-5p targets O-GlcNAc transferase to induce apoptosis in cardiomyocytes. Mol Med Rep (2015) 12(1):1163–8.
140. Luo P, Zhnag W. MicroRNA-423-5p mediates H2O2-induced apoptosis in cardiomyocytes through O-GlcNAc transferase. Mol Med Rep (2016) 14(1):857–64. doi: 10.3892/mmr.2016.5344
141. Ednie AR, Bennett ES. Intracellular O-linked glycosylation directly regulates cardiomyocyte l-type Ca2+ channel activity and excitation–contraction coupling. Basic Res Cardiol (2020) 115(6):59. doi: 10.1007/s00395-020-00820-0
142. Xu J, Zou M-H. Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation (2009) 120(13):1266–86.
143. Yao D, Xu L, Xu O, Li R, Chen M, Shen H, et al. O-Linked β-n-acetylglucosamine modification of A20 enhances the inhibition of NF-κB (nuclear factor-κB) activation and elicits vascular protection after acute endoluminal arterial injury. Arterioscler Thromb Vasc Biol (2018) 38(6):1309–20.
144. Hilgers RHP, Xing D, Gong K, Chen YF, Chatham JC, Oparil S. Acute O-GlcNAcylation prevents inflammation-induced vascular dysfunction. Am J Physiol Hear Circ Physiol (2012) 303(5):H513–H522.
145. Fülöp N, Zhang Z, Marchase RB, Chatham JC. Glucosamine cardioprotection in perfused rat hearts associated with increased O -linked n -acetylglucosamine protein modification and altered p38 activation. Am J Physiol Circ Physiol (2007) 292(5):H2227–36. doi: 10.1152/ajpheart.01091.2006
146. Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress: A survival response of mammalian cells. J Biol Chem (2004) 279(29):30133–42. doi: 10.1074/jbc.M403773200
147. Kim YJ, Kang MJ, Kim E, Kweon TH, Park YS, Ji S, et al. O-GlcNAc stabilizes SMAD4 by inhibiting GSK-3β-mediated proteasomal degradation. Sci Rep (2020) 10(1):19908.
148. Ma J, Liu T, Wei AC, Banerjee P, O’Rourke B, Hart GW. O-GlcNAcomic profiling identifies widespread O-linkedβ-N-acetylglucosamine modification (O-GlcNAcylation) in oxidative phosphorylation system regulating cardiac mitochondrial function. J Biol Chem (2015) 290(49):29141–53.
149. Chun WJ, Nah DY, Bae JH, Chung JW, Lee HS, Moon IS. Glucose-insulin-potassium solution protects ventricular myocytes of neonatal rat in an in vitro coverslip ischemia/reperfusion model. Korean Circ J (2015) 45(3):234–41.
150. Johnsen VL, Belke DD, Hughey CC, Hittel DS, Hepple RT, Koch LG, et al. Enhanced cardiac protein glycosylation (O-GlcNAc) of selected mitochondrial proteins in rats artificially selected for low running capacity. Physiol Genomics (2013) 45(1):17–25. doi: 10.1152/physiolgenomics.00111.2012
151. Zhang L, Mori J, Wagg C, Lopaschuk GD. Activating cardiac E2F1 induces up-regulation of pyruvate dehydrogenase kinase 4 in mice on a short term of high fat feeding. FEBS Lett (2012) 586(7):996–1003. doi: 10.1016/j.febslet.2012.02.027
152. Wang K, Zhou L-Y, Wang J-X, Wang Y, Sun T, Zhao B, et al. E2F1-dependent miR-421 regulates mitochondrial fragmentation and myocardial infarction by targeting Pink1. Nat Commun (2015) 6(1):7619.
153. Laczy B, Marsh SA, Brocks CA, Wittmann I, Chatham JC. Inhibition of O-GlcNAcase in perfused rat hearts by NAG-thiazolines at the time of reperfusion is cardioprotective in an O-GlcNAc-dependent manner. Am J Physiol Hear Circ Physiol (2010) 299(5):H1715–H1727.
154. Liu J, Marchase RB, Chatham JC. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J Mol Cell Cardiol (2007) 42(1):177–85.
155. Nagy T, Champattanachai V, Marchase RB, Chatham JC. Glucosamine inhibits angiotensin II-induced cytoplasmic Ca2+ elevation in neonatal cardiomyocytes via protein-associated O-linked n-acetylglucosamine. Am J Physiol Cell Physiol (2006) 290(1):57–65.
156. Wu T, Zhou H, Jin Z, Bi S, Yang X, Yi D, et al. Cardioprotection of salidroside from ischemia/reperfusion injury by increasing n-acetylglucosamine linkage to cellular proteins. Eur J Pharmacol (2009) 613(1–3):93–9. doi: 10.1016/j.ejphar.2009.04.012
157. Bullen JW, Balsbaugh JL, Chanda D, Shabanowitz J, Hunt DF, Neumann D, et al. Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc TRANSFERASE (OGT) AND AMP-ACTIVATED PROTEIN KINASE (AMPK). J Biol Chem (2014) 289(15):10592–606.
158. Musicki B, Kramer MF, Becker RE, Burnett AL. Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proc Natl Acad Sci (2005) 102(33):11870–5. doi: 10.1073/pnas.0502488102
159. Gao Q, Deng H, Li H, Sun C, Sun Y, Wei B, et al. Glycolysis and fatty acid β-oxidation, which one is the culprit of ischemic reperfusion injury? Int J Clin Exp Med (2018) 11(1):59–68.
160. Olson AK, Bouchard B, Zhu WZ, Chatham JC, Rosiers CD. Characterization of glucose flux through the hexosamine biosynthesis pathway (HBP) in ex vivo mouse heart. J Biol Chem (2020) 295(7):2018–33.
162. Ramirez-correa GA, Jin W, Wang Z, Zhong X, Gao WD, Dias WB, et al. Novel regulator of myocardial contractile function. Circ Res (2009) 103(12):1354–8.
163. Bennett CE, Johnsen VL, Shearer J, Belke DD. Exercise training mitigates aberrant cardiac protein O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci (2013) 92(11):657–63. doi: 10.1016/j.lfs.2012.09.007
164. Medford HM, Porter K, Marsh SA. Immediate effects of a single exercise bout on protein O-GlcNAcylation and chromatin regulation of cardiac hypertrophy. Am J Physiol Hear Circ Physiol (2013) 305(1):114–23.
165. Brodehl A, Gaertner-Rommel A, Milting H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys Rev (2018) 10(4):983–1006. doi: 10.1007/s12551-018-0429-0
166. Bennardini F, Wrzosek A, Chiesi M. Alpha b-crystallin in cardiac tissue. association with actin and desmin filaments. Circ Res (1992) 71(2):288–94. doi: 10.1161/01.RES.71.2.288
167. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation (2016) 133(21):2038–49. doi: 10.1161/CIRCULATIONAHA.115.020226
168. D’Antona G, Ragni M, Cardile A, Tedesco L, Dossena M, Bruttini F, et al. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab (2010) 12(4):362–72.
169. Zafir A, Bradley JA, Long BW, Muthusamy S, Li Q, Hill BG, et al. O-GlcNAcylation negatively regulates cardiomyogenic fate in adult mouse cardiac mesenchymal stromal cells. PloS One (2015) 10(11):1–17.
170. Abouleisa RRE, McNally L, Salama A bakr M, Hammad SK, Ou Q, Wells C, et al. Cell cycle induction in human cardiomyocytes is dependent on biosynthetic pathway activation. Redox Biol (2021) 46:102094. doi: 10.1016/j.redox.2021.102094
171. Kim HS, Park SY, Choi YR, Kang JG, Joo HJ, Moon WK, et al. Excessive O-GlcNAcylation of proteins suppresses spontaneous cardiogenesis in ES cells. FEBS Lett (2009) 583(15):2474–8. doi: 10.1016/j.febslet.2009.06.052
172. Anderson DJ, Kaplan DI, Bell KM, Koutsis K, Haynes JM, Mills RJ, et al. NKX2-5 regulates human cardiomyogenesis via a HEY2 dependent transcriptional network. Nat Commun (2018) 9(1):1373.
173. Delorme B, Dahl E, Jarry-Guichard T, Briand J-P, Willecke K, Gros D, et al. Expression pattern of connexin gene products at the early developmental stages of the mouse cardiovascular system. Circ Res (1997) 81(3):423–37. doi: 10.1161/01.RES.81.3.423
174. Zafir A, Readnower R, Long BW, McCracken J, Aird A, Alvarez A, et al. Protein O-GlcNAcylation is a novel cytoprotective signal in cardiac stem cells. Stem Cells (2013) 31(4):765–75.
Keywords: metabolism, heart, hexosamine biosynthetic pathway, oxidative stress, diabetes, cardiac hypertrophy, myocardial ischemia, heart failure
Citation: Cairns M, Joseph D and Essop MF (2022) The dual role of the hexosamine biosynthetic pathway in cardiac physiology and pathophysiology. Front. Endocrinol. 13:984342. doi: 10.3389/fendo.2022.984342
Received: 01 July 2022; Accepted: 10 October 2022;
Published: 24 October 2022.
Edited by:
Gaetano Santulli, Albert Einstein College of Medicine, United StatesReviewed by:
Julieta Palomeque, Universidad Nacional de La Plata, ArgentinaAnne-Sophie Vercoutter-Edouart, Centre National de la Recherche Scientifique (CNRS), France
Copyright © 2022 Cairns, Joseph and Essop. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Faadiel Essop, bWZlc3NvcEBzdW4uYWMuemE=