 Xinyi Fang
Xinyi Fang Haoran Wu
Haoran Wu Jiahua Wei
Jiahua Wei Runyu Miao
Runyu Miao Yanjiao Zhang1
Yanjiao Zhang1 Jiaxing Tian
Jiaxing Tian- 1Institute of Metabolic Diseases, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 2Graduate College, Beijing University of Chinese Medicine, Beijing, China
- 3Graduate College, Changchun University of Chinese Medicine, Changchun, China
Berberine is a natural active ingredient extracted from the rhizome of Rhizoma Coptidis, which interacts with multiple intracellular targets and exhibits a wide range of pharmacological activities. Previous studies have preliminarily confirmed that the regulation of mitochondrial activity is related to various pharmacological actions of berberine, such as regulating blood sugar and lipid and inhibiting tumor progression. However, the mechanism of berberine’s regulation of mitochondrial activity remains to be further studied. This paper summarizes the molecular mechanism of the mitochondrial quality control system and briefly reviews the targets of berberine in regulating mitochondrial activity. It is proposed that berberine mainly regulates glycolipid metabolism by regulating mitochondrial respiratory chain function, promotes tumor cell apoptosis by regulating mitochondrial apoptosis pathway, and protects cardiac function by promoting mitophagy to alleviate mitochondrial dysfunction. It reveals the mechanism of berberine’s pharmacological effects from the perspective of mitochondria and provides a scientific basis for the application of berberine in the clinical treatment of diseases.
Introduction
Recent studies have found that berberine, an active ingredient from traditional Chinese medicine, has various medicinal effects. Berberine is not only irreplaceable for its dominance of antibacterial, anti-inflammatory and antiviral in traditional pharmacological research, but also has become a current research hotspot because of its remarkable curative effect in the treatment of tumors, diabetes, cardiovascular diseases, and digestive system disease. Although the pharmacological effects of berberine have been confirmed in clinical patients and animal models in the past decade, its specific targets and mechanism of action on the body are still unclear, which limits its further wide application and promotion in clinical practice. Mitochondria are organelles that exist in most cells and are involved in maintaining the stability of the internal environment by producing reactive oxygen species (ROS) and regulating cellular metabolism. A large number of studies have shown that mitochondria are one of the important targets for berberine to exert its pharmacological effects, but its specific mechanism needs to be further studied. Based on this, this paper intends to systematically review the research progress of berberine in improving glycolipid metabolism disorder, anti-tumor, and treating cardiovascular disease by regulating mitochondria, in order to provide a reference for elucidating the pharmacological mechanism of action of berberine.
The pharmacokinetics and toxicology of berberine
Berberine is a natural isoquinoline alkaloid with yellow needle-like crystals in appearance, mostly in stable forms of hydrochloride and sulfate (1). Modern pharmacological research showed that berberine has inhibitory effects on a variety of gram-positive and gram-negative bacteria, and its mechanism of action may be to selectively inhibit bacterial RNA transcription, protein/lipid biosynthesis, and glycolysis. Berberine is mainly used to treat bacterial infectious diseases such as bacillary dysentery, gastroenteritis and carbuncle (2). With in-depth research, berberine has been found to have other extensive pharmacological actions, such as anti-oxidation, blood sugar and blood lipid regulation, anti-tumor, treatment of cardiovascular disease, etc. These new findings expand the range of applications of berberine (3, 4).
The source of berberine
Berberine mainly exists in Ranunculaceae, Berberaceae, Papaveraceae, Rutaceae, Fangchiaceae, Rhamnaceae and other plants, with the highest content in Rhizoma Coptidis and Phellodendron. It can be extracted by acid water extraction, alkaline water extraction, ethanol extraction, liquid membrane method, two-phase extraction, supercritical fluid extraction, etc. Plant extraction and isolation, tissue culture, biological fermentation and chemical synthesis are the main ways to obtain berberine. Among them, chemical synthesis is the most important and direct source. At present, the generally accepted mainstream pathway of berberine biosynthesis is L-tyrosine as the starting material, which is converted into dopamine and 4-hydroxyphenylacetaldehyde, respectively, and then the skeleton is synthesized by biological enzymes such as synthase and transferase, and finally converted into natural berberine after skeleton modification.
Pharmacokinetic study of berberine
The bioavailability of berberine is closely related to the route of administration. According to the experimental data, the bioavailability of berberine decreases in the following order of administration routes: intravenous injection, intraperitoneal injection, intragastric oral administration (5). Oral administration of berberine has the characteristics of difficult absorption, rapid metabolism and low blood concentration. Berberine has poor intestinal absorption and low bioavailability after oral administration. The study (6) found that the peak plasma concentration (Cmax) of berberine after oral administration of 40mg•kg-1 in rats was only 10μg•L-1. There are two main reasons for the low bioavailability of berberine. One is that berberine is a quaternary ammonium base with a lipid-water partition coefficient of -1.5. Drugs containing quaternary ammonium groups in the structure have strong hydrophilicity and low ability to penetrate cell membranes, which limit the transmembrane transport and intestinal absorption of drugs (7). Second, berberine is a substrate of P-glycoprotein (P-gp) (8). The P-glycoprotein is located on the cell membrane and acts as a drug efflux pump, and its transport leads to the secretion of absorbed berberine back into the intestine. In terms of clinical medication, although intravenous injection can increase the drug concentration in the blood, clinical trials found that it will cause serious side effects (e.g., drop in blood pressure and respiratory arrest). Thus, oral administration is the main clinical berberine administration method (9).
The uptake and distribution of berberine in tissues are very important for its biological activity. Studies have shown that berberine is widely distributed in the body after rapid absorption, and the liver is the main organ for the distribution of berberine. Tan et al. (10) deeply studied the organ distribution of berberine in rats, and the results showed that berberine (200mg•kg-1, orally) was rapidly distributed in major organs, such as the liver, kidney, muscle, lung, brain, heart, pancreas and fat. Ma et al. (11) determined the dose-related tissue concentration of the Rhizoma Coptidis alkaloids in mice using high-performance liquid chromatography with ultraviolet detection. The research indicated that four Rhizoma Coptidis alkaloids were detected in the brain, heart, and lung tissues of mice that received the oral total extract of Rhizoma Coptidis. Berberine can also penetrate the blood-brain barrier (12). Liu et al. (13) administered berberine to rats by gavage and found that the distribution of berberine in the liver was 70 times the exposure in the blood. Berberine and its metabolites are widely distributed in tissues and remain relatively stable, which may be the reason they are still active in the body even at low blood concentrations, and can still exert good antibacterial, anti-inflammatory, and hypoglycemic effects.
The main site of berberine metabolism in the body is the liver, which can be rapidly metabolized by a variety of P450 enzymes in the liver. The main metabolic pathway of phase I is methylation and demethylation, and the phase II metabolic pathway is glucuronidation and sulfation. At the same time, after berberine enters the body via oral administration, it will inevitably come in contact with gut microbiota. Gut microbiota can produce a variety of enzymes, such as glycosidase, nitroreductase, etc. Biotransformation of gut microbiota regulates berberine absorption and metabolism in vivo. Studies have shown that nitroreductase produced by gut microbiota is an important biological factor that positively regulates the intestinal absorption and metabolism of berberine. Berberine, which is not easily absorbed by the gut, can be converted into dihydroberberine by the nitroreductase produced by gut microbiota, and then the dihydroberberine enters the intestinal wall, where it is oxidized to berberine in the intestinal wall tissue, finally enters the blood circulation (14). In addition, the gut microbiota also plays an important role in the enterohepatic circulation of berberine metabolites. Therefore, both liver and gut microbiota are involved in the metabolism of berberine in vivo.
Berberine is mainly excreted in the urine and bile in animals, with less excretion in the stool. There is still a lack of human sample studies on the excretion kinetics of berberine. MA et al (15) studied the excretion kinetics of berberine after oral administration in rats and found that berberine was mainly excreted in the form of phase I metabolites in bile and urine.
Toxicology study of berberine
The toxic effects of berberine are usually expressed as median lethal dose (LD50), which varies with the biological species and route of administration. Berberine acute toxicity test showed that the dose of dead mice after intragastric administration was 83.2 g•kg-1, while the LD50 after intravenous administration was 9.04 mg•kg-1. It can be seen that the bioavailability of oral administration is significantly different from that of intravenous administration. It is worth noting that no matter how different the doses of berberine administered by different routes are, the blood concentration of berberine at the time of mouse death is not different, about 0.5-0.7 mg•L-1. It is suggested that the toxicity of berberine is affected by its blood concentration (5). Emerging studies have shown that berberine is almost safe at conventional doses, with a relatively low incidence of adverse reactions, such as gastrointestinal discomfort, and transient increases in plasma bilirubin levels (16). Although berberine is relatively safe, it should be used with caution in specific circumstances to avoid adverse reactions. For example, A mouse study found that administration of berberine (5 mg/kg) could induce skeletal muscle atrophy via increasing atrogin-1 expression (17), whether this effect extends to humans is not currently known. Berberine can be toxic to nerve cells by affecting the mitochondrial respiratory chain and N-methyl-D-aspartic acid receptors (18). Berberine replaces bilirubin in binding to albumin, suggesting that the use of the herb and other traditional Chinese medicines containing a high proportion of berberine is best avoided in jaundiced neonates and pregnant women (19). Berberine interacts with macrolides to inhibit human ether-a-go-go-related gene (hERG) channels, leading to arrhythmias (20). Berberine in combination with statins increases cardiotoxicity by inhibiting CYP3A4 and hERG potassium channels (21).
Research progress on the regulation mechanism of mitochondrial activity
Mitochondria are an important organelle ubiquitous in eukaryotic cells. They are mainly divided into the outer membrane, intermembrane space, inner membrane and matrix. Different enzymes and biological factors are distributed in different spaces. For example, there are anti-apoptotic Bcl-2 family proteins and ion channel proteins on the outer membrane, cytochrome C, apoptosis-inducing factor and procaspase 2, 3, and 9 are distributed in the intermembrane space, the inner membrane is the aggregation site of the complexes that make up the mitochondrial respiratory chain complex enzymes, the tricarboxylic acid cycle-related enzymes and mitochondrial genome are distributed in the matrix. The difference in the permeability of the inner and outer mitochondrial membranes creates a trans-mitochondrial membrane potential that maintains mitochondrial integrity and performs its normal function. Mitochondria are important places for cells to perform aerobic respiration and oxidative phosphorylation to synthesize ATP. Furthermore, mitochondria are also involved in various physiological processes such as cell proliferation, differentiation, signal transduction, innate immune regulation, autophagy, and apoptosis (22). As a highly dynamic organelle, mitochondria are precisely regulated by a variety of proteins for mitochondrial fusion, fission, biosynthesis and clearance (mainly mitophagy) to ensure that mitochondria function effectively, adapt to the energy metabolism of cells, and maintain cells in various physiological activity (Figure 1).
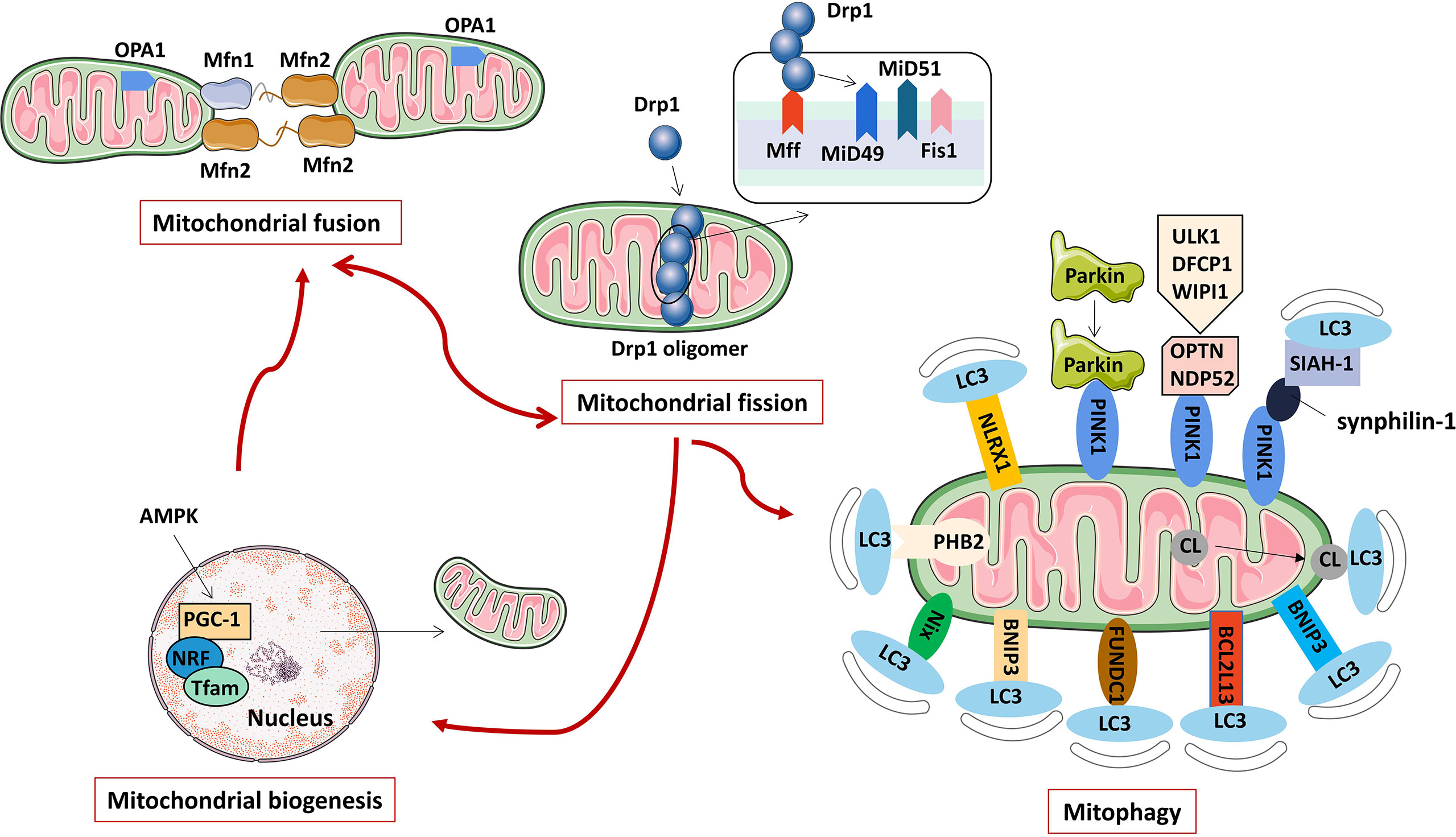
Figure 1 The regulation mechanism of mitochondrial activity (including mitochondrial fusion, fission, biogenesis, and mitophagy).
Regulatory mechanisms of mitochondrial dynamics
Mitochondrial dynamics refers to the continuous fission and fusion of mitochondria to control mitochondrial morphology, provide energy for cells, and regulate processes such as autophagy, calcium homeostasis, innate immunity, signal transduction, and apoptosis (23). Mitochondrial fission is a proliferative process that produces one or more daughter mitochondria, which are required for the formation of new mitochondria. Mitochondrial fission not only increases the number of mitochondria, but also separates damaged mitochondria from the mitochondrial network and promotes autophagy of damaged mitochondria (24). Mitochondrial fusion is a phenomenon in which two adjacent mitochondrial outer and inner membranes fuse to form a mitochondrion with a fibrous extension and a network-like structure. Mitochondrial fusion is important for maintaining normal mitochondrial function. When mitochondria are slightly damaged, mitochondria fuse, which can maintain the stability of the mitochondrial structure, allow material exchange between mitochondria, and repair their functional defects. The dynamic balance of mitochondrial fission and fusion is crucial to maintaining the quality and function of mitochondria, and is an important basis for ensuring the normal activities of cells. Enhanced or weakened mitochondrial fusion/fission protein functions can disrupt the mitochondrial dynamic balance, which may lead to oxidative stress, mitochondrial dysfunction and metabolic changes, and ultimately promote the occurrence of related diseases.
Mitochondrial fission is mainly regulated by mitochondrial dynamin-related protein1 (Drp1) and Drp1 receptor proteins located in the inner or outer mitochondrial membrane, such as mitochondrial fission factor (Mff), mitochondrial fission protein 1 (Fis1), mitochondrial dynamics proteins of 49 kDa (MiD49) and mitochondrial dynamics proteins of 51 kDa (MiD51) (25). Drp1 is a protein necessary for mitochondrial fission. It is generally located in the cytoplasm under physiological conditions. It is mainly composed of an N-terminal GTPase domain, a helical middle domain and a C-terminal GTPase effector domain (26). Under the stimulation of various mitochondrial fission factors, Drp1 is recruited to mitochondria by its receptor protein, oligomerization occurs, and multiple Drp1 assemble into cyclic polymers around mitochondria. Under the action of GTPase hydrolysis, Drp1 cyclic polymers shrink, which ruptures the inner and outer mitochondrial membranes, eventually leading to mitochondrial fission (27). After mitochondrial fission, Drp1 can revert to monomers and re-enter the cytoplasm for reuse (28).
Mitochondrial fusion includes three steps, mitochondrial tethering, mitochondrial outer membrane fusion, and mitochondrial inner membrane fusion (29). In mammals, it is mainly regulated by mitofusin 1 (Mfn1), mitofusin 2 (Mfn2) and optic atrophy 1 (OPA1) protein (30). Among them, Mfn1 and Mfn2 mainly mediate mitochondrial outer membrane fusion, and OPA1 mainly participates in the mitochondrial inner membrane fusion process. Mfn is widely expressed on the outer mitochondrial membrane, and the interaction between Mfn1 and Mfn2 undergoes cis dimerization to form Mfn1/Mfn2 homodimers or Mfn1-Mfn2 heterodimers, which promote the occurrence of adjacent mitochondrial outer membranes tethering and subsequent mitochondrial outer membrane fusion (31, 32). Next, OPA1, which localizes to the inner mitochondrial membrane, mediates mitochondrial inner membrane fusion and cristae remodeling (33). Studies have shown that OPA1 induces mitochondrial inner membrane fusion in a manner dependent on Mfn1, but not Mfn2, suggesting that there may be information transmission between the inner and outer membranes during mitochondrial fusion, and there may be an interaction between the outer membrane protein Mfn1 and the inner membrane protein OPA1 (34). However, the mechanism of OPA1-induced mitochondrial inner membrane fusion has not been fully revealed, and more in-depth studies are still needed.
Regulatory mechanism of mitophagy
Mitophagy refers to the depolarization of mitochondria in cells under stress such as ROS, nutrient deficiency, and cell aging. The damaged mitochondria are specifically encapsulated into autophagosomes and fused with lysosomes to complete the process of degradation of damaged mitochondria, thereby maintaining a stable intracellular environment (35). Mitophagy not only removes damaged mitochondria, but also degrades normal mitochondria for survival when cells are in a harsh environment (36). Mitophagy is a type of selective autophagy that controls the quantity and quality of mitochondria and maintains the normal function of the mitochondrial network. Abnormal mitophagy can cause many pathological changes, which can lead to Alzheimer’s disease, Parkinson’s disease, heart failure, tumors and other diseases.
Mitophagy in mammalian cells mainly includes the ubiquitination degradation pathway mediated by PTEN-induced putative kinase protein 1 (PINK1) and the receptor recognition pathway mediated by different proteins such as Nip3-like protein X (NIX), Bcl-2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3), FUN14 domain-con-taining protein 1 (FUNDC1), FK506 binding protein 8 (FKBP8), Bcl-2-like protein 13 (Bcl2L13), nucleotide-binding domain and leucine-rich-repeat-containing proteins X1 (NLRX1), prohibitin-2 (PHB2) and cardiolipin (CL). Parkin is an E3 ubiquitin ligase present in the cytoplasm (37), and PINK1 is a mitochondrial serine/threonine kinase. Studies have confirmed that PINK1 is an upstream regulatory protein of Parkin, and the two synergistically mediate the polyubiquitination process of damaged mitochondrial surface structures or functional proteins, and play a key role in depolarizing mitophagy degradation (38). Recent studies have shown that PINK1 can also induce mitophagy in a Parkin-independent manner. PINK1 recruits LC3 adaptor proteins OPTN and NDP52 to mitochondria, which in turn recruits autophagy initiator UNC-51-like kinase 1 (ULK1), double FYVE containing protein 1 (DFCP1), and WD repeat domain phosphoinositide-interacting protein 1 (WIPI1) to induce mitophagy (39). Additionally, synphilin-1 overexpression recruits PINK1, and the PINK-synphilin-1 complex can further recruit the E3 ubiquitin ligase SIAH-1 and promote the ubiquitination of mitochondrial proteins to induce mitophagy (40). NIX and BNIP3 are localized in the outer mitochondrial membrane (OMM) and belong to the BH3-only subfamily of B cell lymphoma-2 (Bcl-2) family proteins. NIX and BNIP3 can directly bind to LC3 through their BH3 domains to induce mitophagy (41, 42). Under hypoxic conditions, the mitochondrial outer membrane protein FUNDC1 induces mitophagy in a Parkin-independent manner by directly binding to LC3 (43). The activity of FUNDC1-induced mitophagy is regulated by phosphorylation and ubiquitination modifications (44, 45). BCL2L13 in the mammalian cell is a homologue of Atg32 in the yeast cell. Atg32 is the only mitophagy receptor molecule in yeast cells. BCL2L13 induces mitophagy through a Parkin-independent mechanism, and its mitophagy-inducing activity is regulated by phosphorylation modifications (46). FKBP8 is localized to the mitochondrial outer membrane and can bind to LC3A through its LIR amino acid sequence to mediate mitophagy, and this effect is independent of the PINK1/Parkin pathway (47). NLRX1 is a member of the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family, and its N-terminus contains a mitochondrial localization sequence, making it the only NLR family mitochondrial protein (48). NLRX1 has been identified as a new mitophagy receptor involved in mitophagy (49), but its specific activation pathway and molecular mechanism have not been fully clarified. So far, the roles of many OMM proteins in mediating mitophagy have been confirmed, but the regulatory roles of inner mitochondrial membrane (IMM) proteins in mitophagy have not been fully elucidated. Recent studies have observed that PHB2 localized to the IMM is also a receptor protein that mediates mitophagy during stress and embryonic development (50). In addition, as a characteristic phospholipid of IMM, CL is involved in lipid-protein interactions and is one of the necessary raw materials for maintaining mitochondrial functions (eg, cristae formation, membrane fusion, etc.) (51). The study found that CL is a mitophagy receptor, and mitochondrial damage or membrane depolarization can cause CL to translocate from IMM to OMM, which is the initiation signal of CL-mediated mitophagy (52, 53).
Regulatory mechanisms of mitochondrial biosynthesis
Mitochondrial biogenesis refers to the process of mitochondrial proliferation and mitochondrial system synthesis and individual synthesis in the life cycle of a cell (54). Mitochondrial biosynthesis is a self-protection mechanism initiated after tissue cells are attacked. Under the synergistic effect of mitochondrial genes and nuclear genes, mitochondrial biosynthesis maintains the stability of its own structure, function and quantity by synthesizing and replacing abnormal mitochondria, ensuring the normal metabolic process of cell mitochondria (55). Under physiological conditions, mitochondrial biosynthesis increases with cell proliferation, while under stress conditions such as low temperature, exercise, oxidative stress, and inflammation, mitochondrial biosynthesis is more pronounced, which can reduce the cell damage caused by ROS while ensuring the body’s extra energy demand (56). Therefore, mitochondrial biosynthesis can not only ensure the internal balance of mitochondria, but also enhance the cells’ anti-infection and anti-oxidation capabilities.
Mitochondrial biosynthesis includes the synthesis of the mitochondrial inner membrane, outer membrane and encoded proteins, the synthesis and transfer of nuclear gene-encoded proteins, and the replication of mitochondrial DNA (mt DNA) (57). This process is regulated by a variety of factors, among which peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α)/nuclear respiratory factors (NRFs)/mitochondrial transcription factor A (TFAM) signaling pathway is the most important pathway known to regulate mitochondrial biosynthesis. Overexpression of PGC-1α can increase the steady-state level of mt DNA, NRF-1/2 as a downstream transcription factor target of PGC-1α, is a major transcriptional regulator linking nuclear-encoded genes and mitochondrial biosynthesis, NRF-1/2 and PGC-1α bind and co-activates, while NRF-1/2 is involved in the expression of mt DNA-encoded mitochondrial proteins (58). TFAM is required for mt DNA transcription and replication, and binds to mitochondrial promoter sequences (including heavy and light chain specific promoters) in a sequence-specific manner, thereby efficiently activating mt DNA transcription. In conclusion, once PGC-1α is activated by the upstream adenosine 5’monophosphate-activated protein kinase (AMPK) and deacetylase through phosphorylation and deacetylation, it can bind to and help activate the downstream NRF-1/2, and then bind to the TFAM promoter to activate TFAM, complete and promote the replication and transcription of mt DNA for mitochondrial biosynthesis (59).
Pharmacological effects of berberine targeting the mitochondria
The targeting mechanism of berberine exerting its pharmacological effects is complex. Previous studies have found that the regulation of mitochondrial activity is one of the ways that berberine exerts its pharmacological action (Figure 2, Table 1). Exploring the mechanism of berberine targeting the mitochondria has a certain guiding significance for promoting the clinical application of berberine.

Figure 2 Pharmacological effects of berberine targeting the mitochondria.
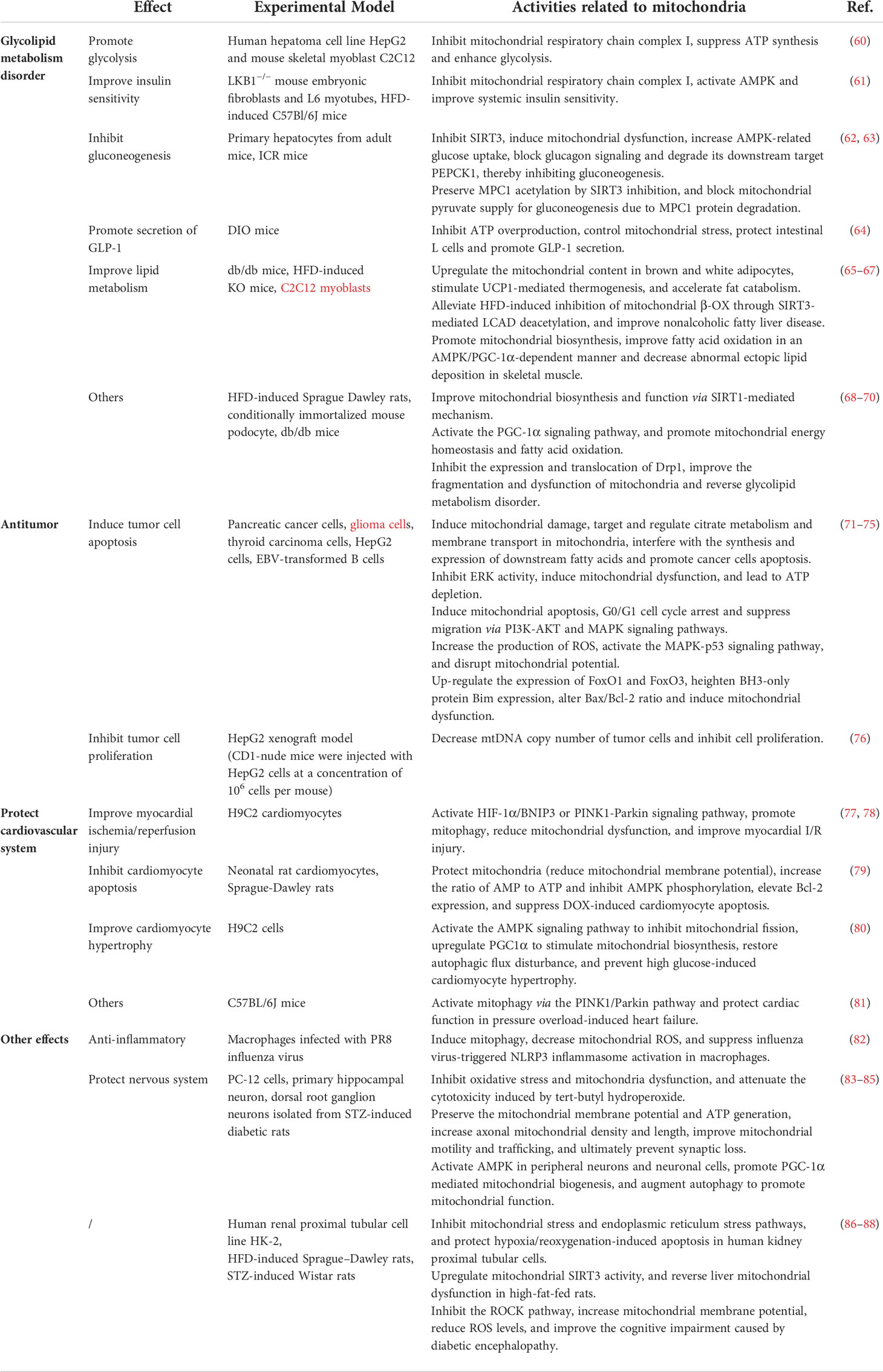
Table 1 Research progress on the pharmacological effects of berberine targeting mitochondria.
Regulate glycolipid metabolism
The relationship between mitochondria and the development of glycolipid metabolism disorder
Mitochondria, as the hub of body energy metabolism, are closely related to the occurrence and development of glycolipid metabolism disorder. Oxidative damage caused by mitochondrial ROS production plays a key role in the pathogenesis of glycolipid metabolism disorder, and is also an important pathophysiological feature of glycolipid metabolism disorder. In the respiratory electron transport chain, an appropriate amount of ROS can promote the secretion of insulin from islet β cells. However, patients with glycolipid metabolism disorder are exposed to a chronic state of hyperglycemia, causing oxidative stress damage to β cells and inhibiting β cell function and survival. Fluctuations in mitochondrial transmembrane potential can cause changes in intracellular adenosine triphosphate (ATP)/adenosine diphosphate (ADP) ratio and affect insulin secretion. Uncoupling protein 2 (UCP2), which is located in the inner mitochondrial membrane, is the master mitochondrial regulator of insulin secretion, and can cause proton leakage, decreased mitochondrial transmembrane potential, uncoupling of oxidative phosphorylation, and decreased ATP production, ultimately leading to a decrease in insulin secretion by β cells (89). Reduced mitochondrial biosynthesis is closely related to insulin resistance. Studies have shown that reduced mitochondria number and volume, and impaired mitochondrial respiration are found in muscle tissue in animal models of type 2 diabetes, obesity, and insulin resistance. The impaired mitochondrial respiration is mainly due to the downregulation of mitochondrial oxidative phosphorylation protein expression, resulting in mitochondrial biosynthesis reduced (90). The research of transgenic animal experiments indicated that overexpression of PGC-1α, which regulates mitochondrial biosynthesis, resulted in increased oxidative phosphorylation protein expression in rat muscle tissue and palmitic acid oxidation, enhanced insulin sensitivity, increased insulin-mediated glucose uptake, and decreased ROS production and inflammatory signaling (91). Activation of autophagy can induce islet β cells to be protected from oxidative stress, remove damaged organelle and their metabolites, promote mitochondrial and endoplasmic reticulum renewal, and maintain cellular homeostasis, whereas impaired autophagy will lead to mitochondrial dysfunction and endoplasmic reticulum stress, promote the occurrence and development of insulin resistance. Mitochondrial gene variation can also lead to glycolipid metabolism disorder. It may be related to insulin resistance, insufficient insulin secretion, oxidative phosphorylation damage, β cell apoptosis, and oxidative stress (92). The specific mechanism is not fully understood, further research and verification are needed. In addition, silent mating type information regulation 2 homolog 3 (SIRT3) located in the mitochondrial matrix plays a key role in energy metabolism, and its decreased activity can lead to impaired mitochondrial function and disturbance of body energy metabolism. SIRT3, which is considered to be a new potential target for regulating glycolipid metabolism disorder, can restore the activity of a series of metabolic enzymes in mitochondria through deacetylation, such as isocitrate, manganese superoxide dismutase and reduced coenzyme NADH, and increase the reduced glutathione content in mitochondria, reduce ROS generation, inhibit oxidative stress, and improve mitochondrial function (93, 94).
Mitochondrial damage plays an important role in the pathogenesis of glycolipid metabolism disorder, and mitochondrial dysfunction and glycolipid metabolism disorder have mutually causal relationships, forming a vicious circle. Mitochondrial membrane potential hyperpolarization, abnormal expression of uncoupling protein 2, reduced mitochondrial biosynthesis, and mitochondrial autophagy impairment lead to impaired mitochondrial function or structure, resulting in excessive production of ROS and aggravating insulin resistance. At the same time, mitochondrial gene variation is associated with the susceptibility to glycolipid metabolism disorder. Therefore, regulating mitochondrial biosynthesis and mitophagy, improving mitochondrial function, and mitochondrial gene therapy might be effective strategies to improve insulin resistance and prevent or delay glycolipid metabolism disorder.
Research progress of berberine in regulating glycolipid metabolism disorder by targeting the mitochondria
As the core mechanism of metabolic diseases such as diabetes, obesity and dyslipidemia, glycolipid metabolism disorder is quietly threatening human health. Previous studies have found that berberine can increase the expression of insulin receptors, promote glycolysis, enhance insulin sensitivity, promote the release and secretion of insulin, increase the consumption of glucose by liver cells, inhibit the differentiation of adipocytes, and regulate gut microbiota to regulate glycolipid metabolism, reduce triglyceride, cholesterol, low-density lipoprotein and blood glucose levels (95–97).
Mitochondria are essential for maintaining energy metabolism. A large amount of literature have reported that berberine inhibits hepatic gluconeogenesis, promotes the secretion of glucagon-like peptide-1 (GLP-1), upregulates glycolysis, enhances insulin sensitivity, and improves fatty acid oxidation by regulating mitochondrial activity, thereby regulating glycolipid metabolism. Xu et al. reported that elevated glycolysis may be a primary cause increased glucose consumption in hepatocytes and myotubes by berberine. In vitro experiment proved that berberine inhibited mitochondrial respiratory chain complex I, which led to the suppression of ATP synthesis, the enhancement of glycolysis, and the promotion of glucose metabolism, a process independent of AMPK activation (60). The results of animal experiments showed that berberine upregulates the mitochondrial content in brown and white adipocytes in db/db mice, and stimulates uncoupling protein 1 (UCP1)-mediated thermogenesis, thereby accelerating fat catabolism and improving obesity and other abnormal lipid metabolism diseases (65). Gomes et al. found that berberine improved mitochondrial biosynthesis and function via a silent mating type information regulation 2 homolog 1 (SIRT1)-mediated mechanism, thereby protecting against the deleterious effects of high-fat diet (HFD) feeding, hyperglycemia and fatty acids (68). Early studies found that berberine has direct inhibition of mitochondrial respiratory chain complex I, inhibiting mitochondrial oxidative respiration, increasing intracellular adenosine monophosphate (AMP)/ATP, thereby activating AMPK and improving systemic insulin sensitivity (61). Zhang et al. found that berberine inhibited mitochondrial function by inhibiting SIRT3. SIRT3 is a soluble protein deacetylase located in the mitochondrial matrix. It can interact with the subunits of the mitochondrial respiration Complex I. SIRT3 regulates the function of Complex I in the electron transport chain and maintains basal ATP yield. Inhibition of SIRT3 by berberine leads to mitochondrial dysfunction and increases the ratio of AMP/ATP, resulting in activating AMPK signaling, increasing AMPK-related glucose uptake, blocking glucagon signaling and degrading its downstream target PEPCK1, thereby inhibiting gluconeogenesis (62). Li et al. revealed that berberine reduced fasting hyperglycemia by reducing mitochondrial pyruvate import via inhibiting MPC1 function in the setting of lipid overload. Mitochondrial pyruvate import via mitochondrial pyruvate carrier (MPC) is a central step in hepatic gluconeogenesis. Berberine reduced hepatic acetyl CoA accumulation by limiting fatty acid oxidation and preserved MPC1 acetylation by SIRT3 inhibition, eventually blocking mitochondrial pyruvate supply for gluconeogenesis due to MPC1 protein degradation (63). However, recent studies identified that berberine can also regulate lipid metabolism by activating SIRT3. Inhibition of mitochondrial β-oxidation (β-OX) has been reported to be involved in the pathogenesis of the nonalcoholic fatty liver disease. SIRT3 regulates mitochondrial β-OX by deacetylating its substrate long-chain acyl-CoA dehydrogenase (LCAD). Xu et al. reported that berberine could alleviate HFD-induced inhibition of mitochondrial β-OX through SIRT3-mediated LCAD deacetylation, thereby improving nonalcoholic fatty liver disease in mice (66). Sun’s study showed that berberine controlled mitochondrial stress by inhibiting ATP overproduction in diet-induced obesity mice, thereby protecting intestinal L cells and promoting GLP-1 secretion (64). Yao et al. explored the effect of berberine on skeletal muscle lipid deposition and mitochondrial function from in vivo and in vitro experiments, and found that berberine could significantly reduce the content of triglycerides in the gastrocnemius muscle of obese mice, and it could promote mitochondrial biosynthesis and decrease abnormal ectopic lipid deposition in skeletal muscle by improving fatty acid oxidation in an AMPK/PGC-1α-dependent manner (67). Qin et al. found that berberine ameliorated fatty acid oxidation and related metabolic disorders in podocytes of diabetic kidney disease (DKD) mice by promoting PGC-1α to regulate mitochondrial energy homeostasis (69). Berberine activated the PGC-1α signaling pathway, which promoted mitochondrial energy homeostasis and fatty acid oxidation in podocytes. Lipid accumulation, excessive generation of mitochondrial ROS, mitochondrial dysfunction, and deficient fatty acid oxidation in DKD mouse models and cultured podocytes were suppressed by berberine. In addition, through inhibiting the expression and translocation of Drp1, berberine potently improved the fragmentation and dysfunction of mitochondria in podocytes, reduced mesangial matrix expansion, glomerular basement membrane thickening, podocyte damage, albuminuria and metabolic abnormalities of DKD (70).
Antitumor
The relationship between mitochondria and tumorigenesis
The occurrence and development of tumors is a complex biological process involving multiple genes and multiple signaling pathways. Compared with normal cells, tumor cells have the characteristics of infinite proliferation, abnormal energy metabolism, increased ROS, tissue infiltration and metastasis, and resistance to cell death (98). In recent years, people have gradually realized that mitochondrial energy metabolism, oxygen free radical homeostasis, calcium ion homeostasis, apoptosis and autophagy abnormalities, and impaired mitochondrial dynamics play important roles in tumorigenesis, and proposed a new strategy for tumor therapy targeting the mitochondria.
The energy metabolism of tumor cells is significantly different from that of normal cells, and their rapid proliferation requires a large amount of biological macromolecules such as carbohydrates, lipids, and proteins. Mitochondria are the most important organelles that regulate cell metabolism. Abnormal mitochondrial metabolism is closely related to the proliferation, metastasis and survival of tumor cells (99). Tumor cells rely mainly on aerobic glycolysis to generate ATP instead of more efficient mitochondrial oxidative phosphorylation (100). The main reason might be that the mitochondrial function of tumor cells is irreversibly damaged, and oxidative phosphorylation is inhibited. This is the famous Warburg effect, which is one of the unique metabolic characteristics of tumor cells. Glycolysis not only provides energy for the life activities of tumor cells, but its intermediate metabolites can also provide carbon precursors for the synthesis of biological macromolecules such as amino acids, fatty acids, and nucleotides, and provide raw materials for the anabolism of tumor cells. In addition, the lactic acid produced by the glycolysis of tumor cells is released to the outside of the cell, forming a local acidic microenvironment, destroying the components of the extracellular matrix, and further enhancing the ability of tumor invasion and metastasis (101). Damaged mitochondria can also lead to abnormally active lipid metabolism and amino acid metabolism in tumor cells, providing raw materials for tumor cell survival and rapid proliferation (102, 103). Significantly increased levels of ROS in tumor cells are another feature of tumor cell metabolism. ROS are involved in many processes such as tumor cell transformation, proliferation, survival, migration, invasion, metastasis, and angiogenesis, and are closely related to the biological characteristics of tumor cells (104, 105). Mitochondrial dysfunction can lead to abnormal mitochondrial ROS homeostasis, resulting in the instability of the cell genome, tumor suppressor gene mutations, finally causing tumorigenesis (106). Mitochondria are both the main site of intracellular ROS generation and the main target of ROS attack. In the process of tumorigenesis, because mt DNA is not protected by histones and lacks the corresponding damage repair system, it is easy to be damaged by ROS and cause gene mutation. Mutations in mt DNA encoding proteins related to the oxidative phosphorylation system directly lead to the abnormality of the mitochondrial oxidative phosphorylation system, generating more ROS, which in turn aggravates further damage to mitochondrial function and ultimately drives the transformation of tumor cells into cell types that rely primarily on glycolysis for energy (107, 108). Besides the endoplasmic reticulum, mitochondria are another important intracellular calcium reservoir. Mitochondria can regulate intracellular calcium homeostasis through multiple calcium transport systems present on their membranes. Abnormal mitochondrial calcium homeostasis is closely related to mitochondrial dysfunction and the occurrence and development of tumors, but its specific mechanism is still unclear and needs to be further explored (109, 110). Decreased apoptotic capacity is one of the hallmarks of tumors. Apoptosis mainly includes two representative pathways, the mitochondrial apoptosis pathway and the death receptor-mediated apoptosis pathway. Mitochondria act as the regulatory center of apoptotic activity, and when mitochondrial apoptosis is inhibited, it may lead to tumor cells acquiring the ability to resist cell death (111, 112). Therefore, targeting the mitochondria to induce tumor cell apoptosis can be one of the strategies for tumor therapy. Mitophagy is a key adaptive effect of tumor cell survival by eliminating damaged mitochondria, controlling ROS production, reducing the risk of cell carcinogenesis, and reducing apoptosis (113, 114). Autophagy has dual regulatory effects on cell survival and death, and abnormal mitophagy is closely related to tumorigenesis. In addition, the impaired mitochondrial dynamic can lead to mitochondrial dysfunction, resulting in cell energy metabolism, proliferation, apoptosis, and mitophagy abnormal. The fusion and fission of mitochondria can also lead to changes in mitochondrial localization, which in turn affects the migration and invasion of tumor cells. Studies have found that key molecules involved in the regulation of mitochondrial fission and fusion, such as Drp1, MFN1, and MFN2, are abnormally expressed in a variety of tumor tissues, and may be closely related to tumor progression (115). The ability of tumor cells to migrate and invade is closely related to their ability to form pseudopodia, and the dynamic changes of microfilaments and microtubules involved in pseudopodia formation require a large amount of ATP generation. Therefore, as the main organ of ATP production, mitochondrial function, quantity and distribution can affect the formation of pseudopodia, which in turn affects the migration and invasion ability of tumor cells (116).
Research progress of berberine in antitumor by targeting the mitochondria
Mitochondria play an important role in abnormal energy metabolism, changes in ROS homeostasis, tissue infiltration and metastasis, and resistance to cell death in tumors. In recent years, with the in-depth study of the pharmacological mechanism of berberine, it has been found that berberine mainly achieves antitumor effects by interfering with tumor cell proliferation, inhibiting tumor cell invasion and metastasis, and inducing tumor cell apoptosis (117–119). At present, extensive in vitro experiments revealed that berberine can induce tumor cell apoptosis and inhibit tumor cell proliferation by regulating mitochondrial activity, thereby exerting antitumor pharmacological effects. Liu et al. revealed through metabolomics and transcriptomic studies that berberine can induce mitochondrial damage in pancreatic cancer cells, and may directly or indirectly interfere with the synthesis and expression of downstream fatty acids by targeting and regulating citrate metabolism and membrane transport in mitochondria, thereby inhibiting the growth of pancreatic cancer cells, promoting their apoptosis and inhibiting their invasion and metastasis (71). Sun et al. found that berberine-induced oncosis of glioma cells was correlated with ATP depletion via the mitochondrial dysfunction induced by the inhibition of extracellular regulated protein kinases (ERK) activity. The down-regulation of mitochondrial phosphorylated extracellular regulated protein kinases (p-ERK) by berberine inhibited aerobic respiration and led to glycolysis, an inefficient energy production pathway (72). Prior studies have established that berberine could inhibit thyroid carcinoma cells by inducing mitochondrial apoptosis, G0/G1 cell cycle arrest and suppressing migration via phosphatidyl inositol 3-kinase (PI3K)-protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) signaling pathways (73). The mechanism of action of berberine in normal cells and tumor cells is similar such as regulating ROS. Oxidative stress plays an important role in the pathogenesis of many diseases. The beneficial effect of berberine is presumed to reside mostly in its antioxidant role (120). The mechanisms involved in the antioxidant role of berberine include scavenging ROS/RNS, removing free oxygen, reducing the destructiveness of superoxide ions and nitric oxide and so on. However, in tumor cells, berberine can increase the production of ROS and lead to the activation of the MAPK-p53 signaling pathway. The activated p53 and its downstream targets XAF1 and GADD45α interacted with PUMA, Bax and Bim in mitochondria, disrupted mitochondrial potential, and induced apoptosis in EBV+ B-lymphoma cells (74). Berberine can also induce mitochondrial dysfunction by significantly upregulating the expression of FoxO1 and FoxO3, thereby causing tumor cell apoptosis. Berberine significantly upregulated the mRNA expression of both FoxO1 and FoxO3a (75). FoxO transcription factors effectively heightened BH3-only protein Bim expression, and altered Bax/Bcl-2 ratio, culminating in mitochondrial dysfunction, caspases activation, and DNA fragmentation. Yan et al. found that berberine inhibited tumor growth and decreased mtDNA copy number of tumor cells in a dose-dependent manner in the HepG2 xenograft model, inhibiting cell proliferation (76).
Protect the cardiovascular system
The relationship between mitochondria and the development of cardiovascular disease
Cardiomyocyte mitochondria are the main site of cardiac energy metabolism, and their dysfunction can lead to a variety of cardiovascular diseases. Mitochondrial quality control mainly ensures the relative stability of mitochondrial morphology, quantity and quality by regulating mitochondrial biogenesis, fusion, fission and autophagy to maintain the integrity of its structure and function. Currently, numerous studies have confirmed that mitochondrial quality control plays an important role in cardiovascular diseases such as ischemic heart disease, heart failure, atherosclerosis, diabetic cardiomyopathy (DCM) and hypertension. Therefore, mitochondrial quality control in the early stage of disease and reducing mitochondrial dysfunction have become new targets for cardiovascular disease treatment.
Mitochondrial biogenesis directly affects mitochondrial quality control and further affects myocardial functional status. Mitochondrial biogenesis disorder is an early change in the development of heart failure. Cardiomyocytes are sites of high energy consumption, and improving mitochondrial biogenesis disorder has cardioprotective effects (121). Even though genetic and environmental factors are different, mitochondrial dynamics imbalance can still be observed in a variety of cardiovascular diseases, such as excessive mitochondrial fission or reduced mitochondrial fusion involved in the progression of myocardial ischemia/reperfusion (I/R) injury and atherosclerosis, suggesting the balance between mitochondrial fusion and fission plays a significant role in the normal heart development and function (122, 123). Therefore, mitochondrial fusion and fission proteins may provide new therapeutic targets for a variety of cardiovascular diseases. Under normal circumstances, mitophagy can maintain the stability of mitochondrial function in cells, thereby ensuring normal heart functioning, whereas abnormal mitophagy is closely related to a variety of cardiovascular diseases. Cardiomyocyte mitophagy is enhanced under various cardiovascular stresses, and whether mitophagy is a potential protective mechanism or a pathological mechanism leading to cell death or further disease development remains inconclusive. Some studies have reported that mitophagy has a protective effect on ischemic myocardium (124), but other studies have shown that mitophagy is one of the causes of myocardial I/R injury (125). Abnormality of the mitochondrial quality control system will lead to mitochondrial dysfunction. Growing research has proved that mitochondrial dysfunction is the cause of many cardiovascular diseases. Mitochondrial dysfunction plays an important role in the process of atherosclerosis by affecting normal lipid metabolism, increasing ROS production, stimulating inflammatory responses and vessel wall remodeling (126, 127). Mitochondrial energy metabolism disorder is an important factor in causing myocardial I/R injury. The main mechanisms include reduced mitochondrial ATP production and excessive ROS production, resulting in oxidative stress, Ca2+ overload and persistent mitochondrial PTP opening (128). Increased ROS, decreased ATP production, Ca2+ overload, and inhibition of mitochondrial electron transport chain caused by mitochondrial dysfunction play an important role in the occurrence and development of hypertension (129, 130). Mitochondrial energy metabolism disorder not only reduces mitochondrial ATP synthesis but also increases mitochondrial ROS generation, thus playing an important role in the occurrence and development of heart failure (131). The pathogenesis of DCM is complex. Hyperglycemia, lipid accumulation, oxidative stress, inflammation, myocardial fibrosis, cardiomyocyte apoptosis, and mitochondrial damage are all involved in the pathophysiological process of DCM. Among them, mitochondrial damage caused by hyperglycemia leads to abnormal mitochondrial energy metabolism, increased ROS production, initiation of mitochondria-dependent apoptosis pathways, mitochondrial fission and fusion disorders, and changes in the content and structure of cardiolipin play a key role in the occurrence and development of DCM (132–134).
Research progress of berberine in protecting the cardiovascular system by targeting the mitochondria
Cardiovascular disease has become one of the diseases with the highest morbidity and mortality, and has received much attention in the world health system. With the continuous advancement of traditional Chinese medicine, a growing body of research has shown that traditional Chinese medicine has unique advantages in cardiovascular prevention and treatment. Berberine, as an active ingredient of the traditional Chinese medicine Rhizoma Coptidis, can regulate cardiovascular diseases such as hypertension, atherosclerosis, heart failure, DCM, ischemic heart disease, and arrhythmia by protecting vascular endothelial function, inhibiting inflammatory response, anti-platelet aggregation, dilating blood vessels, regulating autophagy, and anti-oxidation (135, 136).
Mitochondria are important targets for the treatment of cardiovascular disease. In recent years, with the deepening of the research on the pharmacological mechanism of berberine, it has been noticed that berberine can improve myocardial I/R injury, inhibit myocardial cell apoptosis, and protect cardiac function by regulating mitochondrial activity. In vitro experiments show that berberine could not only upregulate PTEN-induced putative kinase 1 (PINK1) and Parkin levels, activate the PINK1-Parkin signaling pathway, promote mitophagy, induce autophagic flux, reduce mitochondrial dysfunction, and improve myocardial I/R injury, but also can promote mitophagy, reduce myocardial enzyme activity, induce cardiomyocyte proliferation, inhibit cardiomyocyte apoptosis, and protect the heart from myocardial I/R injury, possibly through the hypoxia-inducible factor 1α (HIF-1α)/BNIP3 pathway (77, 78). Studies have shown that doxorubicin (DOX) could rapidly induce mitochondrial damage and increase intracellular AMP/ATP ratio, which in turn activates AMPK phosphorylation and promotes cardiomyocyte apoptosis. Lv et al. reported that berberine suppressed DOX-induced cardiomyocyte apoptosis via protecting mitochondria (reducing DOX-induced loss of mitochondrial membrane potential), reducing the increased ratio of AMP to ATP and inhibiting AMPK phosphorylation as well as elevating Bcl-2 expression in the early stage of DOX treatment (79). Hang et al. proposed that berberine could prevent high glucose-induced cardiomyocyte hypertrophy by activating the AMPK signaling pathway to inhibit mitochondrial fission, upregulating proliferator-activated receptor-gamma coactivator 1α (PGC1α) to stimulate mitochondrial biosynthesis, and restoring autophagic flux disturbance (80). Animal experiments confirmed that berberine activated mitophagy via the PINK1/Parkin pathway and protected cardiac function in pressure overload-induced heart failure (81).
Other pharmacological effects
Berberine has a wide range of pharmacological effects and can be applied to various diseases. In addition to hypoglycemic, lipid-lowering, cardiovascular protection, and antitumor effects, berberine also has pharmacological activities such as anti-inflammatory, antibacterial, and protection of the central nervous system. In recent years, a large number of studies have shown that berberine targeting the mitochondria as therapeutic targets to regulate the structure and function of mitochondria, thereby exerting pharmacological effects. Yu et al. found that berberine pretreatment had a protective effect on hypoxia/reoxygenation-induced apoptosis in human kidney proximal tubular cells, and the mechanism was related to the inhibition of mitochondrial stress and endoplasmic reticulum stress pathways (86). Liu et al. reported that berberine suppresses influenza virus-triggered NOD-like receptor pyrin domain-containing protein 3 inflammasome activation in macrophages by inducing mitophagy and decreasing mitochondrial ROS (82). Li et al. found that berberine attenuated the cytotoxicity induced by tert-butyl hydroperoxide via inhibiting oxidative stress and mitochondria dysfunction in PC-12 cells (83). Berberine significantly suppressed cytochrome c expression, upregulated the ratio of Bcl-2/Bax, and ameliorated mitochondrial dysfunction by optimizing mitochondria membrane potential status and ATP production, playing a protective role in anti-apoptotic and anti-oxidative stress. Zhao et al. reported that berberine protected against Aβ-induced axonal mitochondrial abnormalities in primary cultured hippocampal neurons by preserving the mitochondrial membrane potential and ATP generation, increasing axonal mitochondrial density and length, and improving mitochondrial motility and trafficking, ultimately preventing synaptic loss (84). Previous research indicated that berberine activates AMPK in peripheral neurons and neuronal cells, which promotes PGC-1α mediated mitochondrial biogenesis, and augments autophagy to promote mitochondrial function (85). The improved mitochondrial function by berberine further strengthened redox homeostasis through stimulation of endogenous antioxidant systems and inhibition of neuroinflammation. These effects lead to increased nerve conduction velocity, improved nerve blood flow, and decreased hyperalgesia in diabetic neuropathy. Teodoro et al. found that mitochondria isolated from the livers of high-fat-fed rats exhibited decreased calcium accumulation capacity and impaired oxidative phosphorylation capacity, such as impaired mitochondrial membrane potential, oxygen consumption, and cellular ATP levels, whereas berberine could reverse liver mitochondrial dysfunction in high-fat-fed rats by upregulating mitochondrial SIRT3 activity (87). Tian et al. proposed that berberine improved the cognitive impairment caused by diabetic encephalopathy by inhibiting the Rho/Rho kinase (ROCK) pathway in diabetic encephalopathy rats, increasing mitochondrial membrane potential and reducing ROS levels (88).
Summary and outlook
Mitochondria are ubiquitous organelles that mainly consist of a bilayer-membrane structure and matrix. It is the ‘power station’ of cells and plays a core role in cell energy production, metabolism and signal transmission. Notably, mitochondria are critical for respiration, apoptosis, and mitochondrial DNA inheritance, and alterations in their structural and functional integrity may lead to ATP depletion, ROS overproduction, decreased mitochondrial membrane potential, impaired mt DNA and other mitochondrial dysfunction, and further mediate cell apoptosis and inflammatory responses (137). The mitochondrial quality control system mainly includes mitochondrial biosynthesis, dynamics and autophagy. As an important endogenous mechanism for maintaining mitochondrial homeostasis in cells, the mitochondrial quality control system has two opposites. It not only involves the regeneration of mitochondria, but also includes the removal of damaged or aging mitochondria, which is the core of maintaining the normal mitochondrial structure and exerting the normal physiological function of mitochondria (138). Recent studies have suggested that mitochondria are the target organelles for many drugs and poisons. Berberine is a common active ingredient in a variety of heat-clearing and detoxifying traditional Chinese medicines. Its functions such as heat-clearing and detoxifying and antibacterial have been used in clinical practice, and it also has various pharmacological activities such as antitumor, hypoglycemic, blood lipid regulation, hypotensive and antiarrhythmic effects. With more in-depth research, the mechanism of berberine’s pharmacological activity by targeting the mitochondria has been elucidated. Berberine can affect the quality control and function of mitochondria to a certain extent by regulating the expression of oxidative stress, mitochondrial fusion and fission, mitophagy, mitochondrial biosynthesis, and intracellular calcium regulation, thereby exerting pharmacological effects.
In improving glycolipid metabolism disorder, berberine can directly inhibit mitochondrial respiratory chain complex I or regulate the function of mitochondrial respiratory chain complex I by inhibiting SIRT3 in the mitochondrial matrix, thereby inhibiting ATP synthesis, upregulating the ratio of AMP/ATP, activating the AMPK signaling pathway to inhibit hepatic glycogen production, promote glycolysis, and improve insulin sensitivity. Berberine can also improve mitochondrial dysfunction, regulate mitochondrial energy homeostasis, and then regulate glycolipid metabolism disorder by promoting the expression of PGC-1α and upregulating mitochondrial biosynthesis, inhibiting the expression and mitochondrial translocation of Drp1 and downregulating mitochondrial fission. In antitumor progression, berberine can induce tumor cell apoptosis by activating PI3K-AKT and MAPK-p53 signaling pathways or promoting the expression of FoxO1 and FoxO3, and upregulating pro-apoptotic proteins such as Bax and Bim. Berberine also exerts antitumor effect by inhibiting cell proliferation by inhibiting ERK activity and inducing mitochondrial dysfunction or reducing mtDNA copy number. In the prevention and treatment of cardiovascular diseases, berberine mainly protects cardiac function by activating the PINK1-Parkin, HIF-1α/BNIP3 signaling pathways and promoting mitophagy. In addition, berberine can also improve mitochondrial function by protecting mitochondrial membrane potential, activating mitochondrial apoptosis signaling pathway, and promoting mitochondrial biogenesis, thereby exerting pharmacological effects such as anti-inflammatory and nerve protection. In summary, berberine mainly regulates the function of the mitochondrial respiratory chain, activates the important energy homeostasis regulator AMPK, regulates glycolipid metabolism, regulates mitochondrial apoptosis, promotes tumor cell apoptosis, promotes mitophagy to reduce mitochondrial dysfunction and protect heart function.
Mitochondria are important targets for berberine to exert its pharmacological effects, but the regulatory mechanism of berberine on mitochondria still needs to be further studied. Firstly, there are differences in mitochondria and their functions in different species. Different concentrations and dosages of berberine affect the regulation of mitochondria. Therefore, in order to prevent differences in research results, the research design should be more standardized. Secondly, the relevant research is still at an exploratory stage, and the research types mainly are animal experiments or cell experiments, and lack of in vitro mitochondrial experiments or clinical trials to evaluate the mechanism of action. Most importantly, there is currently a lack of multi-target, multi-path, multi-dimensional, and in-depth systematic research. Current research mostly focuses on the regulation of a specific pathway, the detection indicators are relatively limited, and pharmacokinetics is lacking. In addition, mitochondrial quality control is a continuous dynamic process, and static assessment of the roles and mechanisms of independent components of the mitochondrial quality control system at a single time point may generate research bias. Therefore, it is necessary to further explore the influence mechanism on different signaling pathways and their related factors in the process of applying berberine to intervene in mitochondria in the treatment of diseases. The normal maintenance of cell activity and physiological function relies on the cooperation of multiple intracellular and extracellular molecules and signaling pathways, and mitochondria are extensively involved in the regulation of physiological activities. Consequently, we need to comprehensively explore the mechanism of berberine regulating mitochondria to exert pharmacological effects through different technical means and different signal transduction pathways. The application of emerging omics technologies and methods such as mitochondrial proteomics and epigenetics is expected to bring light to screen and discover the targets of berberine.
Author contributions
JT developed the review question. The initial literature review was performed by XF. The first draft of the manuscript was written by XF with all authors commenting on subsequent versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81904187), Capital Health Development Research Project (CD2020-4-4155), CACMS Outstanding Young Scientific and Technological Talents Program (ZZ13-YQ-026), CACMS Innovation Fund (CI2021A01601), Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (ZYYCXTD-D-202001), Open Project of National Facility for Translational Medicine (TMSK-2021-407).
Acknowledgments
We would like to thank all the authors for their contribution to the realization of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Habtemariam S. Berberine pharmacology and the gut microbiota: A hidden therapeutic link. Pharmacol Res (2020) 155:104722. doi: 10.1016/j.phrs.2020.104722
2. Rabbani GH, Butler T, Knight J, Sanyal SC, Alam K. Randomized controlled trial of berberine sulfate therapy for diarrhea due to enterotoxigenic escherichia coli and vibrio cholerae. J Infect Dis (1987) 155(5):979–84. doi: 10.1093/infdis/155.5.979
3. Rajasekhar K, Samanta S, Bagoband V, Murugan NA, Govindaraju T. Antioxidant berberine-derivative inhibits multifaceted amyloid toxicity. iScience (2020) 23(4):101005. doi: 10.1016/j.isci.2020.101005
4. Meng FC, Wu ZF, Yin ZQ, Lin LG, Wang R, Zhang QW. Coptidis rhizoma and its main bioactive components: recent advances in chemical investigation, quality evaluation and pharmacological activity. Chin Med (2018) 13:13. doi: 10.1186/s13020-018-0171-3
5. Kheir MM, Wang Y, Hua L, Hu J, Li L, Lei F, et al. Acute toxicity of berberine and its correlation with the blood concentration in mice. Food Chem Toxicol (2010) 48(4):1105–10. doi: 10.1016/j.fct.2010.01.033
6. Zuo F, Nakamura N, Akao T, Hattori M. Pharmacokinetics of berberine and its main metabolites in conventional and pseudo germ-free rats determined by liquid chromatography/ion trap mass spectrometry. Drug Metab Dispos (2006) 34(12):2064–72. doi: 10.1124/dmd.106.011361
7. Battu SK, Repka MA, Maddineni S, Chittiboyina AG, Avery MA, Majumdar S. Physicochemical characterization of berberine chloride: a perspective in the development of a solution dosage form for oral delivery. AAPS PharmSciTech (2010) 11(3):1466–75. doi: 10.1208/s12249-010-9520-y
8. Zhang YT, Yu YQ, Yan XX, Wang WJ, Tian XT, Wang L, et al. Different structures of berberine and five other protoberberine alkaloids that affect p-glycoprotein-mediated efflux capacity. Acta Pharmacol Sin (2019) 40(1):133–42. doi: 10.1038/s41401-018-0183-7
9. Han Y, Xiang Y, Shi Y, Tang X, Pan L, Gao J, et al. Pharmacokinetics and pharmacological activities of berberine in diabetes mellitus treatment. Evid Based Complement Alternat Med (2021) 2021:9987097. doi: 10.1155/2021/9987097
10. Tan XS, Ma JY, Feng R, Ma C, Chen WJ, Sun YP, et al. Tissue distribution of berberine and its metabolites after oral administration in rats. PLoS One (2013) 8(10):e77969. doi: 10.1371/journal.pone.0077969
11. Ma BL, Ma YM, Shi R, Wang TM, Zhang N, Wang CH, et al. Identification of the toxic constituents in rhizoma coptidis. J Ethnopharmacol (2010) 128(2):357–64. doi: 10.1016/j.jep.2010.01.047
12. Zhang X, Zhang X, Wang C, Li Y, Dong L, Cui L, et al. Neuroprotection of early and short-time applying berberine in the acute phase of cerebral ischemia: up-regulated pAkt, pGSK and pCREB, down-regulated NF-κB expression, ameliorated BBB permeability. Brain Res (2012) 1459:61–70. doi: 10.1016/j.brainres.2012.03.065
13. Liu YT, Hao HP, Xie HG, Lai L, Wang Q, Liu CX, et al. Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats. Drug Metab Dispos (2010) 38(10):1779–84. doi: 10.1124/dmd.110.033936
14. Feng R, Shou JW, Zhao ZX, He CY, Ma C, Huang M, et al. Transforming berberine into its intestine-absorbable form by the gut microbiota. Sci Rep (2015) 5:12155. doi: 10.1038/srep12155
15. Ma JY, Feng R, Tan XS, Ma C, Shou JW, Fu J, et al. Excretion of berberine and its metabolites in oral administration in rats. J Pharm Sci (2013) 102(11):4181–92. doi: 10.1002/jps.23718
16. Feng X, Sureda A, Jafari S, Memariani Z, Tewari D, Annunziata G, et al. Berberine in cardiovascular and metabolic diseases: From mechanisms to therapeutics. Theranostics (2019) 9(7):1923–51. doi: 10.7150/thno.30787
17. Wang H, Liu D, Cao P, Lecker S, Hu Z. Atrogin-1 affects muscle protein synthesis and degradation when energy metabolism is impaired by the antidiabetes drug berberine. Diabetes (2010) 59(8):1879–89. doi: 10.2337/db10-0207
18. Kysenius K, Brunello CA, Huttunen HJ. Mitochondria and NMDA receptor-dependent toxicity of berberine sensitizes neurons to glutamate and rotenone injury. PLoS One (2014) 9(9):e107129. doi: 10.1371/journal.pone.0107129
19. Chan E. Displacement of bilirubin from albumin by berberine. Biol Neonate (1993) 63(4):201–8. doi: 10.1159/000243932
20. Zhi D, Feng PF, Sun JL, Guo F, Zhang R, Zhao X, et al. The enhancement of cardiac toxicity by concomitant administration of berberine and macrolides. Eur J Pharm Sci (2015) 76:149–55. doi: 10.1016/j.ejps.2015.05.009
21. Feng P, Zhao L, Guo F, Zhang B, Fang L, Zhan G, et al. The enhancement of cardiotoxicity that results from inhibiton of CYP 3A4 activity and hERG channel by berberine in combination with statins. Chem Biol Interact (2018) 293:115–23. doi: 10.1016/j.cbi.2018.07.022
22. Kiriyama Y, Nochi H. Intra- and intercellular quality control mechanisms of mitochondria. Cells (2017) 7(1):1. doi: 10.3390/cells7010001
23. Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev (2009) 89(3):799–845. doi: 10.1152/physrev.00030.2008
24. Yu SB, Pekkurnaz G. Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J Mol Biol (2018) 430(21):3922–41. doi: 10.1016/j.jmb.2018.07.027
25. Losón OC, Song Z, Chen H, Chan DC. Fis1, mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell (2013) 24(5):659–67. doi: 10.1091/mbc.E12-10-0721
26. Fröhlich C, Grabiger S, Schwefel D, Faelber K, Rosenbaum E, Mears J, et al. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J (2013) 32(9):1280–92. doi: 10.1038/emboj.2013.74
27. Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife (2015) 4:e11553. doi: 10.7554/eLife.11553
28. Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell (2001) 12(8):2245–56. doi: 10.1091/mbc.12.8.2245
29. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem (2018) 62(3):341–60. doi: 10.1042/EBC20170104
30. Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox homeostasis and mitochondrial dynamics. Cell Metab (2015) 22(2):207–18. doi: 10.1016/j.cmet.2015.06.006
31. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science (2004) 305(5685):858–62. doi: 10.1126/science.1099793
32. Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu B, et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature (2017) 542(7641):372–6. doi: 10.1038/nature21077
33. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell (2006) 126(1):177–89. doi: 10.1016/j.cell.2006.06.025
34. Mattie S, Riemer J, Wideman JG, McBride HM. A new mitofusin topology places the redox-regulated c terminus in the mitochondrial intermembrane space. J Cell Biol (2018) 217(2):507–15. doi: 10.1083/jcb.201611194
35. Green DR, Van Houten B. SnapShot: Mitochondrial quality control. Cell (2011) 147(4):950–50.e1. doi: 10.1016/j.cell.2011.10.036
36. Bhatia-Kiššová I, Camougrand N. Mitophagy: a process that adapts to the cell physiology. Int J Biochem Cell Biol (2013) 45(1):30–3. doi: 10.1016/j.biocel.2012.07.006
37. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol (2008) 183(5):795–803. doi: 10.1083/jcb.200809125
38. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent parkin for mitophagy. J Cell Biol (2010) 189(2):211–21. doi: 10.1083/jcb.200910140
39. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature (2015) 524(7565):309–14. doi: 10.1038/nature14893
40. Szargel R, Shani V, Abd Elghani F, Mekies LN, Liani E, Rott R, et al. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum Mol Genet (2016) 25(16):3476–90. doi: 10.1093/hmg/ddw189
41. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep (2010) 11(1):45–51. doi: 10.1038/embor.2009.256
42. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem (2012) 287(23):19094–104. doi: 10.1074/jbc.M111.322933
43. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol (2012) 14(2):177–85. doi: 10.1038/ncb2422
44. Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell (2014) 54(3):362–77. doi: 10.1016/j.molcel.2014.02.034
45. Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep (2017) 18(3):495–509. doi: 10.15252/embr.201643309
46. Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun (2015) 6:7527. doi: 10.1038/ncomms8527
47. Bhujabal Z, Birgisdottir ÅB, Sjøttem E, Brenne HB, Øvervatn A, Habisov S, et al. FKBP8 recruits LC3A to mediate parkin-independent mitophagy. EMBO Rep (2017) 18(6):947–61. doi: 10.15252/embr.201643147
48. Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. An n-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci (2009) 122(Pt 17):3161–8. doi: 10.1242/jcs.051193
49. Zhang Y, Yao Y, Qiu X, Wang G, Hu Z, Chen S, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol (2019) 20(4):433–46. doi: 10.1038/s41590-019-0324-2
50. Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell (2017) 168(1-2):224–38.e10. doi: 10.1016/j.cell.2016.11.042
51. Schlattner U, Tokarska-Schlattner M, Rousseau D, Boissan M, Mannella C, Epand R, et al. Mitochondrial cardiolipin/phospholipid trafficking: the role of membrane contact site complexes and lipid transfer proteins. Chem Phys Lipids (2014) 179:32–41. doi: 10.1016/j.chemphyslip.2013.12.008
52. Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol (2013) 15(10):1197–205. doi: 10.1038/ncb2837
53. Kagan VE, Jiang J, Huang Z, Tyurina YY, Desbourdes C, Cottet-Rousselle C, et al. NDPK-d (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ (2016) 23(7):1140–51. doi: 10.1038/cdd.2015.160
54. Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest (2000) 106(7):847–56. doi: 10.1172/JCI10268
55. Weinberg JM. Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol (2011) 22(3):431–6. doi: 10.1681/ASN.2010060643
56. Zhang Q, Wu Y, Zhang P, Sha H, Jia J, Hu Y, et al. Exercise induces mitochondrial biogenesis after brain ischemia in rats. Neuroscience (2012) 205:10–7. doi: 10.1016/j.neuroscience.2011.12.053
57. Li PA, Hou X, Hao S. Mitochondrial biogenesis in neurodegeneration. J Neurosci Res (2017) 95(10):2025–9. doi: 10.1002/jnr.24042
58. Islam H, Edgett BA, Gurd BJ. Coordination of mitochondrial biogenesis by PGC-1α in human skeletal muscle: A re-evaluation. Metabolism (2018) 79:42–51. doi: 10.1016/j.metabol.2017.11.001
59. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell (1999) 98(1):115–24. doi: 10.1016/S0092-8674(00)80611-X
60. Xu M, Xiao Y, Yin J, Hou W, Yu X, Shen L, et al. Berberine promotes glucose consumption independently of AMP-activated protein kinase activation. PLoS One (2014) 9(7):e103702. doi: 10.1371/journal.pone.0103702
61. Turner N, Li JY, Gosby A, To SW, Cheng Z, Miyoshi H, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes (2008) 57(5):1414–8. doi: 10.2337/db07-1552
62. Zhang B, Pan Y, Xu L, Tang D, Dorfman RG, Zhou Q, et al. Berberine promotes glucose uptake and inhibits gluconeogenesis by inhibiting deacetylase SIRT3. Endocrine (2018) 62(3):576–87. doi: 10.1007/s12020-018-1689-y
63. Li A, Liu Q, Li Q, Liu B, Yang Y, Zhang N. Berberine reduces pyruvate-driven hepatic glucose production by limiting mitochondrial import of pyruvate through mitochondrial pyruvate carrier 1. EBioMedicine (2018) 34:243–55. doi: 10.1016/j.ebiom.2018.07.039
64. Sun Y, Jin C, Zhang X, Jia W, Le J, Ye J. Restoration of GLP-1 secretion by berberine is associated with protection of colon enterocytes from mitochondrial overheating in diet-induced obese mice. Nutr Diabetes (2018) 8(1):53. doi: 10.1038/s41387-018-0061-x
65. Zhang Z, Zhang H, Li B, Meng X, Wang J, Zhang Y, et al. Berberine activates thermogenesis in white and brown adipose tissue. Nat Commun (2014) 5:5493. doi: 10.1038/ncomms6493
66. Xu X, Zhu XP, Bai JY, Xia P, Li Y, Lu Y, et al. Berberine alleviates nonalcoholic fatty liver induced by a high-fat diet in mice by activating SIRT3. FASEB J (2019) 33(6):7289–300. doi: 10.1096/fj.201802316R
67. Yao S, Yuan Y, Zhang H, Meng X, Jin L, Yang J, et al. Berberine attenuates the abnormal ectopic lipid deposition in skeletal muscle. Free Radic Biol Med (2020) 159:66–75. doi: 10.1016/j.freeradbiomed.2020.07.028
68. Gomes AP, Duarte FV, Nunes P, Hubbard BP, Teodoro JS, Varela AT, et al. Berberine protects against high fat diet-induced dysfunction in muscle mitochondria by inducing SIRT1-dependent mitochondrial biogenesis. Biochim Biophys Acta (2012) 1822(2):185–95. doi: 10.1016/j.bbadis.2011.10.008
69. Qin X, Jiang M, Zhao Y, Gong J, Su H, Yuan F, et al. Berberine protects against diabetic kidney disease via promoting PGC-1α-regulated mitochondrial energy homeostasis. Br J Pharmacol (2020) 177(16):3646–61. doi: 10.1111/bph.14935
70. Qin X, Zhao Y, Gong J, Huang W, Su H, Yuan F, et al. Berberine protects glomerular podocytes via inhibiting Drp1-mediated mitochondrial fission and dysfunction. Theranostics (2019) 9(6):1698–713. doi: 10.7150/thno.30640
71. Liu J, Luo X, Guo R, Jing W, Lu H. Cell metabolomics reveals berberine-inhibited pancreatic cancer cell viability and metastasis by regulating citrate metabolism. J Proteome Res (2020) 19(9):3825–36. doi: 10.1021/acs.jproteome.0c00394
72. Sun Y, Yu J, Liu X, Zhang C, Cao J, Li G, et al. Oncosis-like cell death is induced by berberine through ERK1/2-mediated impairment of mitochondrial aerobic respiration in gliomas. BioMed Pharmacother (2018) 102:699–710. doi: 10.1016/j.biopha.2018.03.132
73. Li L, Wang X, Sharvan R, Gao J, Qu S. Berberine could inhibit thyroid carcinoma cells by inducing mitochondrial apoptosis, G0/G1 cell cycle arrest and suppressing migration via PI3K-AKT and MAPK signaling pathways. BioMed Pharmacother (2017) 95:1225–31. doi: 10.1016/j.biopha.2017.09.010
74. Park GB, Park SH, Kim D, Kim YS, Yoon SH, Hur DY. Berberine induces mitochondrial apoptosis of EBV-transformed b cells through p53-mediated regulation of XAF1 and GADD45α. Int J Oncol (2016) 49(1):411–21. doi: 10.3892/ijo.2016.3502
75. Shukla S, Rizvi F, Raisuddin S, Kakkar P. FoxO proteins' nuclear retention and BH3-only protein bim induction evoke mitochondrial dysfunction-mediated apoptosis in berberine-treated HepG2 cells. Free Radic Biol Med (2014) 76:185–99. doi: 10.1016/j.freeradbiomed.2014.07.039
76. Yan XJ, Yu X, Wang XP, Jiang JF, Yuan ZY, Lu X, et al. Mitochondria play an important role in the cell proliferation suppressing activity of berberine. Sci Rep (2017) 7:41712. doi: 10.1038/srep41712
77. Zhu N, Cao X, Hao P, Zhang Y, Chen Y, Zhang J, et al. Berberine attenuates mitochondrial dysfunction by inducing autophagic flux in myocardial hypoxia/reoxygenation injury. Cell Stress Chaperones (2020) 25(3):417–26. doi: 10.1007/s12192-020-01081-5
78. Zhu N, Li J, Li Y, Zhang Y, Du Q, Hao P, et al. Berberine protects against simulated Ischemia/Reperfusion injury-induced H9C2 cardiomyocytes apoptosis In vitro and myocardial Ischemia/Reperfusion-induced apoptosis In vivo by regulating the mitophagy-mediated HIF-1α/BNIP3 pathway. Front Pharmacol (2020) 11:367. doi: 10.3389/fphar.2020.00367
79. Lv X, Yu X, Wang Y, Wang F, Li H, Wang Y, et al. Berberine inhibits doxorubicin-triggered cardiomyocyte apoptosis via attenuating mitochondrial dysfunction and increasing bcl-2 expression. PLoS One (2012) 7(10):e47351. doi: 10.1371/journal.pone.0047351
80. Hang W, He B, Chen J, Xia L, Wen B, Liang T, et al. Berberine ameliorates high glucose-induced cardiomyocyte injury via AMPK signaling activation to stimulate mitochondrial biogenesis and restore autophagic flux. Front Pharmacol (2018) 9:1121. doi: 10.3389/fphar.2018.01121
81. Abudureyimu M, Yu W, Cao RY, Zhang Y, Liu H, Zheng H. Berberine promotes cardiac function by upregulating PINK1/Parkin-mediated mitophagy in heart failure. Front Physiol (2020) 11:565751. doi: 10.3389/fphys.2020.565751
82. Liu H, You L, Wu J, Zhao M, Guo R, Zhang H, et al. Berberine suppresses influenza virus-triggered NLRP3 inflammasome activation in macrophages by inducing mitophagy and decreasing mitochondrial ROS. J Leukoc Biol (2020) 108(1):253–66. doi: 10.1002/JLB.3MA0320-358RR
83. Li Z, Jiang T, Lu Q, Xu K, He J, Xie L, et al. Berberine attenuated the cytotoxicity induced by t-BHP via inhibiting oxidative stress and mitochondria dysfunction in PC-12 cells. Cell Mol Neurobiol (2020) 40(4):587–602. doi: 10.1007/s10571-019-00756-7
84. Zhao C, Su P, Lv C, Guo L, Cao G, Qin C, et al. Berberine alleviates amyloid β-induced mitochondrial dysfunction and synaptic loss. Oxid Med Cell Longev (2019) 2019:7593608. doi: 10.1155/2019/7593608
85. Yerra VG, Kalvala AK, Sherkhane B, Areti A, Kumar A. Adenosine monophosphate-activated protein kinase modulation by berberine attenuates mitochondrial deficits and redox imbalance in experimental diabetic neuropathy. Neuropharmacology (2018) 131:256–70. doi: 10.1016/j.neuropharm.2017.12.029
86. Yu W, Sheng M, Xu R, Yu J, Cui K, Tong J, et al. Berberine protects human renal proximal tubular cells from hypoxia/reoxygenation injury via inhibiting endoplasmic reticulum and mitochondrial stress pathways. J Transl Med (2013) 11:24. doi: 10.1186/1479-5876-11-24
87. Teodoro JS, Duarte FV, Gomes AP, Varela AT, Peixoto FM, Rolo AP, et al. Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: a possible role for SirT3 activation. Mitochondrion (2013) 13(6):637–46. doi: 10.1016/j.mito.2013.09.002
88. Tian L, Ri H, Qi J, Fu P. Berberine elevates mitochondrial membrane potential and decreases reactive oxygen species by inhibiting the Rho/ROCK pathway in rats with diabetic encephalopathy. Mol Pain (2021) 17:1744806921996101. doi: 10.1177/1744806921996101
89. Affourtit C, Jastroch M, Brand MD. Uncoupling protein-2 attenuates glucose-stimulated insulin secretion in INS-1E insulinoma cells by lowering mitochondrial reactive oxygen species. Free Radic Biol Med (2011) 50(5):609–16. doi: 10.1016/j.freeradbiomed.2010.12.020
90. Franko A, von Kleist-Retzow JC, Neschen S, Wu M, Schommers P, Böse M, et al. Liver adapts mitochondrial function to insulin resistant and diabetic states in mice. J Hepatol (2014) 60(4):816–23. doi: 10.1016/j.jhep.2013.11.020
91. Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, et al. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab (2010) 12(6):633–42. doi: 10.1016/j.cmet.2010.11.008
92. Noda M, Yamashita S, Takahashi N, Eto K, Shen LM, Izumi K, et al. Switch to anaerobic glucose metabolism with NADH accumulation in the beta-cell model of mitochondrial diabetes. Characteristics betaHC9 Cells deficient mitochondrial DNA transcription J Biol Chem (2002) 277(44):41817–26. doi: 10.1074/jbc.M207690200
93. Choudhury M, Jonscher KR, Friedman JE. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging (Albany NY) (2011) 3(2):175–8. doi: 10.18632/aging.100289
94. Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell (2010) 40(6):893–904. doi: 10.1016/j.molcel.2010.12.013
95. Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med (2004) 10(12):1344–51. doi: 10.1038/nm1135
96. Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes (2006) 55(8):2256–64. doi: 10.2337/db06-0006
97. Xu X, Gao Z, Yang F, Yang Y, Chen L, Han L, et al. Antidiabetic effects of gegen qinlian decoction via the gut microbiota are attributable to its key ingredient berberine. Genomics Proteomics Bioinf (2020) 18(6):721–36. doi: 10.1016/j.gpb.2019.09.007
98. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
99. Finley LWS. Metabolic signal curbs cancer-cell migration. Nature (2019) 571(7763):39–40. doi: 10.1038/d41586-019-01934-9
100. Warburg OA. On the origin of cancer cell. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
101. Sun H, Chen L, Cao S, Liang Y, Xu Y. Warburg effects in cancer and normal proliferating cells: Two tales of the same name. Genomics Proteomics Bioinf (2019) 17(3):273–86. doi: 10.1016/j.gpb.2018.12.006
102. Burke AC, Huff MW. ATP-citrate lyase: genetics, molecular biology and therapeutic target for dyslipidemia. Curr Opin Lipidol (2017) 28(2):193–200. doi: 10.1097/MOL.0000000000000390
103. Li M, Chiang YL, Lyssiotis CA, Teater MR, Hong JY, Shen H, et al. Non-oncogene addiction to SIRT3 plays a critical role in lymphomagenesis. Cancer Cell (2019) 35(6):916–31.e9. doi: 10.1016/j.ccell.2019.05.002
104. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discovery (2009) 8(7):579–91. doi: 10.1038/nrd2803
105. Gupta SC, Hevia D, Patchva S, Park B, Koh W, Aggarwal BB. Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal (2012) 16(11):1295–322. doi: 10.1089/ars.2011.4414
106. Rottenberg H, Hoek JB. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell (2017) 16(5):943–55. doi: 10.1111/acel.12650
107. Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer (2002) 9:1–9. doi: 10.1186/1476-4598-1-9
108. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science (2008) 320(5876):661–4. doi: 10.1126/science.1156906
109. Ren T, Wang J, Zhang H, Yuan P, Zhu J, Wu Y, et al. MCUR1-mediated mitochondrial calcium signaling facilitates cell survival of hepatocellular carcinoma via reactive oxygen species-dependent P53 degradation. Antioxid Redox Signal (2018) 28(12):1120–36. doi: 10.1089/ars.2017.6990
110. Huang Q, Cao H, Zhan L, Sun X, Wang G, Li J, et al. Mitochondrial fission forms a positive feedback loop with cytosolic calcium signaling pathway to promote autophagy in hepatocellular carcinoma cells. Cancer Lett (2017) 403:108–18. doi: 10.1016/j.canlet.2017.05.034
111. Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ (2006) 13(8):1396–402. doi: 10.1038/sj.cdd.4401963
112. Davids MS, Letai A. Targeting the b-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol (2012) 30(25):3127–35. doi: 10.1200/JCO.2011.37.0981
113. Lu Z, Miao Y, Muhammad I, Tian E, Hu W, Wang J, et al. Colistin-induced autophagy and apoptosis involves the JNK-Bcl2-Bax signaling pathway and JNK-p53-ROS positive feedback loop in PC-12 cells. Chem Biol Interact (2017) 277:62–73. doi: 10.1016/j.cbi.2017.08.011
114. Lo AK, Lung RW, Dawson CW, Young LS, Ko CW, Yeung WW, et al. Activation of sterol regulatory element-binding protein 1 (SREBP1)-mediated lipogenesis by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) promotes cell proliferation and progression of nasopharyngeal carcinoma. J Pathol (2018) 246(2):180–90. doi: 10.1002/path.5130
115. Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy (2016) 12(6):999–1014. doi: 10.1080/15548627.2016.1166318
116. Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene (2013) 32(40):4814–24. doi: 10.1038/onc.2012.494
117. Iizuka N, Miyamoto K, Okita K, Tangoku A, Hayashi H, Yosino S, et al. Inhibitory effect of coptidis rhizoma and berberine on the proliferation of human esophageal cancer cell lines. Cancer Lett (2000) 148(1):19–25. doi: 10.1016/s0304-3835(99)00264-5
118. Ho YT, Yang JS, Li TC, Lin JJ, Lin JG, Lai KC, et al. Berberine suppresses in vitro migration and invasion of human SCC-4 tongue squamous cancer cells through the inhibitions of FAK, IKK, NF-kappaB, u-PA and MMP-2 and -9. Cancer Lett (2009) 279(2):155–62. doi: 10.1016/j.canlet.2009.01.033
119. Lin JP, Yang JS, Lee JH, Hsieh WT, Chung JG. Berberine induces cell cycle arrest and apoptosis in human gastric carcinoma SNU-5 cell line. World J Gastroenterol (2006) 12(1):21–8. doi: 10.3748/wjg.v12.i1.21
120. Neag MA, Mocan A, Echeverría J, Pop RM, Bocsan CI, Crişan G, et al. Berberine: Botanical occurrence, traditional uses, extraction methods, and relevance in cardiovascular, metabolic, hepatic, and renal disorders. Front Pharmacol (2018) 9:557. doi: 10.3389/fphar.2018.00557
121. Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, et al. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation (2005) 112(5):683–90. doi: 10.1161/CIRCULATIONAHA.104.524835
122. Anzell AR, Maizy R, Przyklenk K, Sanderson TH. Mitochondrial quality control and disease: Insights into ischemia-reperfusion injury. Mol Neurobiol (2018) 55(3):2547–64. doi: 10.1007/s12035-017-0503-9
123. Forte M, Schirone L, Ameri P, Basso C, Catalucci D, Modica J, et al. The role of mitochondrial dynamics in cardiovascular diseases. Br J Pharmacol (2021) 178(10):2060–76. doi: 10.1111/bph.15068
124. Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy (2012) 8(3):297–309. doi: 10.4161/auto.18658
125. Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation (2010) 121(18):2012–22. doi: 10.1161/CIRCULATIONAHA.109.906610
126. Yu E, Bennett MR. Mitochondrial dna damage and atherosclerosis. Trends Endocrinol Metab Tem (2014) 25(9):481–7. doi: 10.1016/j.tem.2014.06.008
127. Lee SR, Kim N, Noh YH, Xu Z, Ko KS, Rhee BD, et al. Mitochondrial dysfunction, and cardiac manifestations. Front Biosci (Landmark Ed) (2017) 22(7):1177–94. doi: 10.2741/4541
128. Penna C, Perrelli MG, Pagliaro P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxid Redox Signal (2013) 18(5):556–99. doi: 10.1089/ars.2011.4459
129. Chan SH, Wu CA, Wu KL, Ho YH, Chang AY, Chan JY. Transcriptional upregulation of mitochondrial uncoupling protein 2 protects against oxidative stress-associated neurogenic hypertension. Circ Res (2009) 105(9):886–96. doi: 10.1161/CIRCRESAHA.109.199018
130. Widder JD, Fraccarollo D, Galuppo P, Hansen JM, Jones DP, Ertl G, et al. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of thioredoxin 2. Hypertension (2009) 54(2):338–44. doi: 10.1161/HYPERTENSIONAHA.108.127928
131. Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res (2008) 80(1):30–9. doi: 10.1093/cvr/cvn184
132. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes (2007) 56(10):2457–66. doi: 10.2337/db07-0481
133. Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res (2008) 79(2):341–51. doi: 10.1093/cvr/cvn104
134. Li B, Zheng Z, Wei Y, Wang M, Peng J, Kang T, et al. Therapeutic effects of neuregulin-1 in diabetic cardiomyopathy rats. Cardiovasc Diabetol (2011) 10:69. doi: 10.1186/1475-2840-10-69
135. Tan HL, Chan KG, Pusparajah P, Duangjai A, Saokaew S, Mehmood Khan T, et al. Rhizoma coptidis: A potential cardiovascular protective agent. Front Pharmacol (2016) 7:362. doi: 10.3389/fphar.2016.00362
136. Fan X, Wang J, Hou J, Lin C, Bensoussan A, Chang D, et al. Berberine alleviates ox-LDL induced inflammatory factors by up-regulation of autophagy via AMPK/mTOR signaling pathway. J Transl Med (2015) 13:92. doi: 10.1186/s12967-015-0450-z
137. Colina-Tenorio L, Horten P, Pfanner N, Rampelt H. Shaping the mitochondrial inner membrane in health and disease. J Intern Med (2020) 287(6):645–64. doi: 10.1111/joim.13031
Keywords: berberine, mitochondria, pharmacological action, glycolipid metabolism disorder, tumor, cardiovascular diseases
Citation: Fang X, Wu H, Wei J, Miao R, Zhang Y and Tian J (2022) Research progress on the pharmacological effects of berberine targeting mitochondria. Front. Endocrinol. 13:982145. doi: 10.3389/fendo.2022.982145
Received: 30 June 2022; Accepted: 25 July 2022;
Published: 11 August 2022.
Edited by:
Yong Seek Park, Kyung Hee University, South KoreaReviewed by:
Daohua Xu, Guangdong Medical University, ChinaVictor M. Victor, University of Valencia, Spain
Copyright © 2022 Fang, Wu, Wei, Miao, Zhang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaxing Tian, dGluYV95YWlAMTI2LmNvbQ==