
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 20 October 2022
Sec. Molecular and Structural Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.972890
This article is part of the Research Topic Pharmacology of Endocrine Related GPCRs View all 5 articles
 Valentina Spigoni1
Valentina Spigoni1 Gloria Cinquegrani1Nicolas Thomas Iannozzi1Giulia Frigeri2Giulia Maggiolo2Marta Maggi2Vanessa Parello1Alessandra Dei Cas1,2*
Gloria Cinquegrani1Nicolas Thomas Iannozzi1Giulia Frigeri2Giulia Maggiolo2Marta Maggi2Vanessa Parello1Alessandra Dei Cas1,2*Ketogenesis takes place in hepatocyte mitochondria where acetyl-CoA derived from fatty acid catabolism is converted to ketone bodies (KB), namely β-hydroxybutyrate (β-OHB), acetoacetate and acetone. KB represent important alternative energy sources under metabolic stress conditions. Ketogenic diets (KDs) are low-carbohydrate, fat-rich eating strategies which have been widely proposed as valid nutritional interventions in several metabolic disorders due to its substantial efficacy in weight loss achievement. Carbohydrate restriction during KD forces the use of FFA, which are subsequently transformed into KB in hepatocytes to provide energy, leading to a significant increase in ketone levels known as “nutritional ketosis”. The recent discovery of KB as ligands of G protein-coupled receptors (GPCR) - cellular transducers implicated in a wide range of body functions - has aroused a great interest in understanding whether some of the clinical effects associated to KD consumption might be mediated by the ketone/GPCR axis. Specifically, anti-inflammatory effects associated to KD regimen are presumably due to GPR109A-mediated inhibition of NLRP3 inflammasome by β-OHB, whilst lipid profile amelioration by KDs could be ascribed to the actions of acetoacetate via GPR43 and of β-OHB via GPR109A on lipolysis. Thus, this review will focus on the effects of KD-induced nutritional ketosis potentially mediated by specific GPCRs in metabolic and endocrinological disorders. To discriminate the effects of ketone bodies per se, independently of weight loss, only studies comparing ketogenic vs isocaloric non-ketogenic diets will be considered as well as short-term tolerability and safety of KDs.
Ketone bodies (KB) - β-hydroxybutyrate (β-OHB), acetoacetate and the less abundant acetone - are produced by ketogenesis in the hepatocyte mitochondria and converted into energy in the mitochondria of several extrahepatic organs (i.e. brain, heart, kidney cortex and skeletal muscle) by ketolysis (Figure 1) (1).
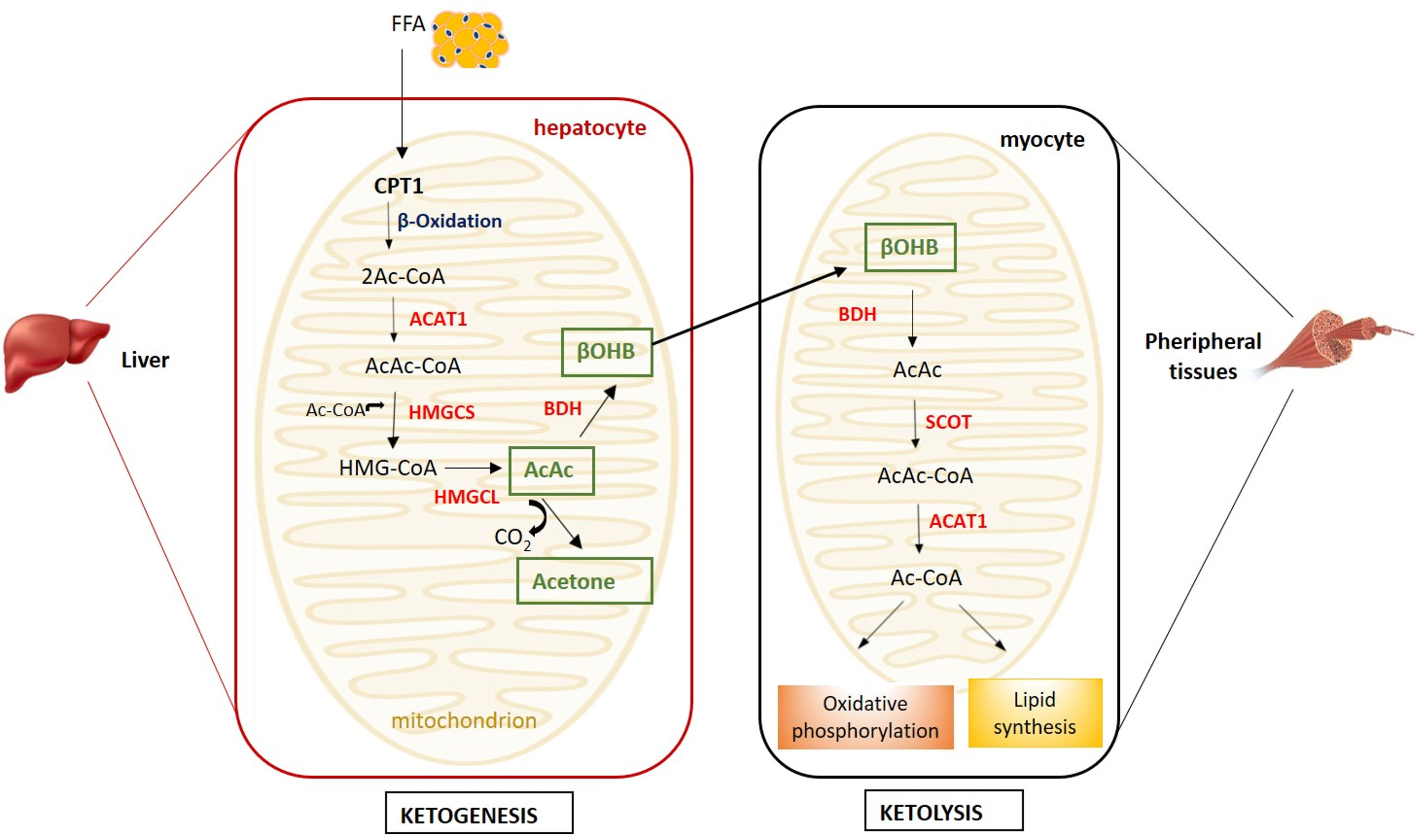
Figure 1 Metabolism of ketone bodies. Ketogenesis takes place in hepatocyte mitochondria, while ketolysis in the mitochondria of peripheral tissues (i.e. muscle, heart, kidney and brain). (ACAT1, acetyl coenzyme A acetyltransferase; AcAc, acetoacetate; Ac-CoA, acetyl-CoA; AcAc-CoA, acetoacetyl-CoA; BDH, β-hydroxybutyrate dehydrogenase; βOHB, β-hydroxybutyrate; CPT1, carnitine palmitoyltransferase 1; FFA, free fatty acids; HMGCL, HMG-CoA lyase; HMG-CoA, β-hydroxy-β-methylglutaryl-CoA; HMGCS, HMG-CoA synthase; SCOT, succinyl-CoA-3-ketoacid-CoA transferase).
Briefly, in the ketogenesis process, free fatty acids (FFA) mobilised from the adipose tissue are transported into the liver mitochondria by carnitine palmitoyltransferase (CPT1) and undergo β-oxidation resulting into two acetyl-CoA molecules subsequently condensed to acetoacetyl-CoA by acetyl coenzyme A acetyltransferase (ACAT1) (2). The condensation of acetoacetyl-CoA with a third molecule of acetyl-CoA, by β-hydroxy-β-methylglutaryl-CoA (HMG-CoA) synthase, leads to the formation of HMG-CoA, an intermediate in the mevalonate and ketogenesis pathways and a precursor of cholesterol biosynthesis (3). The HMG-CoA lyase converts the HMG-CoA to the β-keto acid acetoacetate, which, in turn, can be converted to acetone and CO2 by non-enzymatic decarboxylation or to β-OHB by β-OHB-dehydrogenase. The liver releases β-OHB, which, in a process named ketolysis, is re-converted to acetoacetate and subsequently to acetyl-CoA by the succinyl-CoA-3-ketoacid-CoA transferase (SCOT), which is expressed in all mammalian cells with mitochondria, with the exception of hepatocytes (2) (Figure 1).
Acetyl-CoA enters the Krebs cycle for ATP production through oxidative phosphorylation.
KB utilization as metabolic substrate is energetically more efficient in terms of ATP synthesis per molecule of oxygen invested than glucose or FFA and allows the transfer of lipid-derived energy from liver to extrahepatic organs as an alternative fuel source (4). Alternatively, acetyl-CoA deviates from the Krebs cycle to lipid synthesis. Acetone does not convert backwards to acetyl-CoA and it is excreted through urine or exhaled (5).
Several conditions are associated to physiological plasma ketosis. KB plasma levels are normally < 0.5mM, but during fasting, a decrease in insulin/glucagon ratio is paralleled by a concomitant increase in plasma FFA and KB, which, in the presence of a prolonged starvation, may excessively increase up to 5-fold (1.3 mM) and 20-fold (7-10 mM), respectively (6–8). Prolonged exercise may also induce a physiological increase in plasma KB up to 1-2 mM (1). Other biological conditions, such as pregnancy or the neonatal period, are associated to a mild increase in ketosis (1).
The nutritional ketosis following a ketogenic diet (KD) regimen also represents a physiological mechanism, in which KB concentrations are < 3.0 mM and blood pH remains within the normal range (1, 9). Conversely, diabetic ketoacidosis, a severe acute complication of diabetes, is a pathological condition in which KB reach extremely high plasma concentrations (≥ 3.0 mM (10)) with a concomitant blood pH fall below the normal range (11). In diabetic conditions, insulin deficiency prompts the body to metabolize triglycerides and amino acids, instead of glucose, for energy. Serum levels of glycerol and FFA consequently rise due to exacerbated lipolysis (1). Moreover, glycerol provides the substrates for hepatic gluconeogenesis, which is further stimulated by the excess glucagon that accompanies insulin deficiency (1). In non-diabetic individuals, ketoacidosis is prevented as the preserved insulin secretion reduces the release of FFA from the adipose tissue, leading to a decreased ketogenesis in hepatocytes, accompanied by urinary KB excretion (9).
Thus, KB can be either a necessary nutrient or the reflection of a pathological status depending on their plasmatic concentrations.
KDs were first used since the 1920s to treat refractory epilepsy in children (12) and increasing evidence shows encouraging results in pathological conditions such as Alzheimer disease (13), cancer (14), and metabolic diseases (15). KDs are defined as low-carbohydrate (usually to < 50 g/day) and adequate protein intakes to induce ketosis without restricting fat intake (16). Similar to fasting conditions, during a carbohydrate-restricted diet, the body undergoes metabolic changes to provide an energy source relying on gluconeogenesis and ketogenesis. Following depletion of glycogen stores, the body is forced to use FFA, which are subsequently transformed into KB in hepatocytes, to provide energy (1). KB become the primary energy source by cells with mitochondria and, most importantly, by the brain. This significant rise in KB in course of KD it is known as “nutritional ketosis” (17), a condition in which plasma KB concentration physiologically increases (at least > 0.5 mM (18)), resulting in a rather safe nutritional approach (19).
Different variants of the KD have been defined in the last few years, although a commonly accepted classification is still lacking.
Types of KDs includes: the classic long-chain triglycerides (LCT) KD, which is the most used KD, and it contains four times as many grams of fat for every 1 g of protein and carbohydrates (4:1 ratio) (20, 21). The medium-chain triglyceride (MCT, C6–C12) KD is more ketogenic than (LCT KD); MCTs are readily absorbed into the portal vein -as they do not require micelle formation with bile acids- and are directly transformed into KB. The MCT KD allows the intake of a higher ratio of carbohydrates and protein compared to the high-fat KD with consequent increase in adherence and prevention of muscle catabolism (22). Differently from high fat KD, the MCT KD is built only on the percentage of calorie from MCT oil (23). MCT KD is commonly used for the treatment of refractory childhood epilepsy (24, 25) and other neurological disorders such as cognitive dysfunction in Alzheimer’s disease and mild cognitive impairment (26).
The very low carbohydrate ketogenic diet (VLCKD) represents the most widespread KD type and is traditionally characterized by carbohydrate limitation (<50 g per day) with unrestricted fat consumption and a moderate increase in protein intake (0.8-1.2 g per day) (27). The total amount of calories to be provided for a single individual is based on anthropometric measurements, dietary intake and physical activity (28).
The very low calorie KDs have an extremely low caloric intake (400–800 kcal/day) and contain an amount of protein of 1.2–1.4 g/kg of ideal body weight, a very limited carbohydrate content (< 30–50 g/day, < 5–10%) and a determined amount of fats (∼20 g/day) (29).
Beyond calorie restriction, the increase in KB levels is associated with a specific anorexic effect, by preventing the increase in ghrelin secretion in response to weight loss, decreasing hunger (30). However, the exact threshold of ketosis needed to induce appetite suppression, as well as the exact mechanisms mediating such an effect, have yet to be elucidated (31). The suppression/blunting of hunger sensation and consequent reduction in caloric intake has a fundamental role in diet compliance and short-term weigh loss outcomes (32–34).
Generally, KDs are recommended for a limited time and the shift to a standard diet should be gradual and supervised (35). KDs may be fraught with side effects which usually disappear within a few days to a few weeks like nausea, vomiting, headache, tiredness, insomnia and exercise intolerance. Long-term adverse effects may include hepatic steatosis, hypoproteinemia and kidney stones (35). The main contraindications to implement a KD are pancreatitis, liver failure, primary carnitine deficiency, carnitine palmitoyltransferase deficiency, carnitine translocase deficiency, porphyrias, or pyruvate kinase deficiency (36).
It has recently emerged that KDs may hold additional beneficial effects on metabolism and cardiovascular risk reduction, which may be partially independent of weight loss. The increase in KB following a keto diet regimen would potentially produce tissue specific effects by the activation of cell membrane G protein-coupled receptors (GPCRs) which have been recently discovered to be engaged by β-OHB and acetoacetate.
GPCRs - which represent the largest protein family in the human proteome - are signal transducers located in the plasma membrane of eukaryotic cells. GPCRs share a common structure consisting of seven 25-amino acid α-helical segments crossing through the plasma membrane, with the amino-terminus and carboxy-terminus outside the cell and in the cytoplasm, respectively (37). The variety of ligands capable of binding to GPCRs is notable, ranging from a single photon to large proteins and including ions, odorants, nucleotides, peptides, amines, lipids and metabolic intermediates. Following ligand engagement, GPCRs interact with G proteins in the plasma membrane leading to the production of cAMP and of other second messengers. A subsequent cascade of phosphorylation, involving extracellular regulated protein kinases 1/2 (ERK1/2), transduces cellular responses by modulating cell metabolism and function (38).
Over time, the number of GPCRs discovered has increased as well as that of their binding energy substrates such as fatty acids and sucrose, and key metabolic intermediates including acetate and lactate (39–41). The recent discovery of KB as GPCR ligands has aroused a great interest in understanding whether some of the clinical effects associated to KDs might be mediated by ketone/GPCR axis; however, data are scarce in the literature.
Thus, this review will focus on the effects of KD-induced nutritional ketosis potentially mediated by specific GPCRs in metabolic and endocrinological disorders.
β-OHB is the most represented KB in humans and its concentrations typically reach ∼0.2–0.4 mM after an overnight fast, ∼1–2 mM following 2–3 days of fasting, and plateaus at ∼6–8 mM upon prolonged starvation (1, 42–44).
Most of β-OHB actions seem to be mediated by the activation of a specific GPCR, namely GPR109A, also known as Hydroxycarboxylic Acid Receptor 2 (HCAR2). GPR109A is classically recognized as the receptor for butyrate and nicotinic acid and is abundantly expressed in adipocytes, macrophages, and neutrophils (45). The plasma concentrations of β-OHB able to engage GPR109A (EC50 = 0.77 ± 0.06 mM) (46) are those achievable after ∼2–3 days of starvation (1), or following a ketogenic diet regimen (47–49).
The binding of β-OHB to GPR109A is implicated in the reduction of both lipid metabolism and inflammation (Table 1). In adipocytes, GPR109A activation results in Gi/G0 protein-mediated inhibition of adenylate cyclase, leading to a mitigated cAMP response with a consequent reduction in hormone sensitive lipase activity, resulting in lipolysis inhibition (57). The consequent reduced FFA release and hepatic delivery limit substrate availability for hepatic triglyceride and has been proposed as a negative feedback mechanism in KB synthesis (46, 58). Accordingly, a calorie-restricted diet or β-OHB administration resulted in the suppression of hepatic lipid accumulation through GPR109A/AMP-activated protein kinase (AMPK) pathway in aged rats (51). If the results on lipolysis inhibition are robust, less consistent are the findings on the resulting modifications in plasma lipoprotein profile (59).
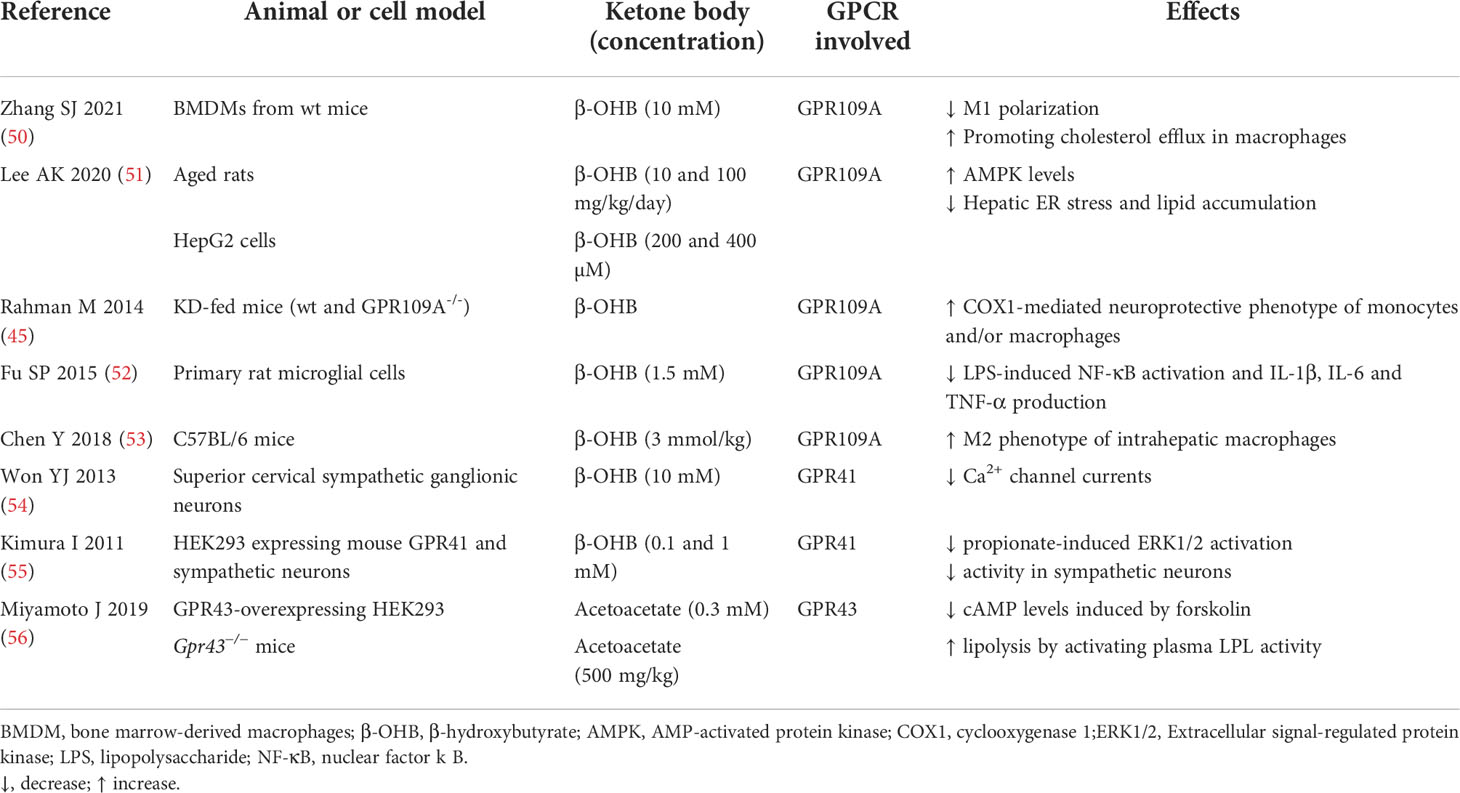
Table 1 Summary of studies on GPCR-mediated actions of ketone bodies.
In addition to the effects on lipolysis inhibition, emerging evidence points to GPR109A as the molecular transducer of β-OHB-mediated effects also on the mitigation of the inflammatory burden, through the reduction in NLR family pyrin domain containing 3 (NLRP3) inflammasome activity (50). Briefly, the inflammasome is a multiprotein intracellular complex which can be activated by diverse signals such as endoplasmic reticulum (ER) stress, reactive oxygen species (ROS), and excessive Ca2+ leading to caspase 1 activation which in turn activates proinflammatory interleukin (IL)-1β and IL-18. Specifically, in macrophages, inflammasome activation triggers pro-atherogenic M1 polarization -in contrast to anti-inflammatory M2- contributing to the pro-inflammatory e pro-atherogenic environment (60).
A recent work demonstrated that the exogenous administration of 200 mg/kg/day of β-OHB resulted in a strong fat reduction in apoE −/− atherosclerotic mice even in the presence of a high‐fat diet. β-OHB decreased pro-inflammatory M1 polarization and cholesterol accumulation in bone marrow‐derived macrophages (BMDMs) (50). These protective effects were no longer evident in GPR109A −/− and NLRP3 −/− BMDMs, indicating that β-OHB attenuates atherosclerosis via GPR109A-NLRP3 dependent pathway. Mechanistically, β-OHB - via GPR109A - promoted the influx of Ca2+ by reducing Ca2+ release from ER thereby maintaining Ca2+ storage and inhibiting its depletion induced by NLRP3 inflammasome activation. Overall, this study suggests that the treatment with β-OHB significantly blunts systemic inflammation and atherogenesis mostly through GPR109A-expressing macrophages (50).
Most of the knowledge concerning the anti-inflammatory action of β-OHB mediated by GPR109A comes primarily from studied conducted in neurodegenerative diseases. Nicotinic acid, a GPR109A agonist, induced a neuroprotective phenotype of monocytes/macrophages in a mouse model of stroke resulting in the reduction of ischemic brain damage (45). GPR109A activation in macrophages led to Prostaglandin D2 (PGD2) production by Cyclooxygenase (COX)-1 and PGD2 synthase, underscoring the key role of PGD2 in the regulation of inflammation and innate immune response (61).
Another study demonstrated that β-OHB, inhibited pro-inflammatory enzyme and cytokine [tumor necrosis factor (TNF)-α, IL-1β, and IL-6] production in microglia via GPR109A, through the reduction of nuclear factor-κB (NF-κB) activation (52). This anti-inflammatory action of β-OHB has been recently proposed as a tool to prevent (or slow down) also the progression of Parkinson’s disease.
GPR109A-mediated anti-inflammatory action of β-OHB was shown to be also protective in alcohol hepatitis, characterized by liver inflammation which may progress into systemic inflammatory response and high neutrophil infiltration in the liver (62). The β-OHB intake in mice has an anti-inflammatory and hepatoprotective role through a GPR109A-dependent pathway. Specifically, β-OHB supplementation (i.p. 3 mmol/kg) induced anti-inflammatory IL-10 transcripts and promoted the switch of mouse intrahepatic macrophages towards an anti-inflammatory M2 phenotype (53). This effect was mediated by the activation of protein kinase A (PKA), which reduces mitochondrial membrane potential and was no longer evident in GPR109A-knockout mice (63). GPR109A-mediated effects of β-OHB in modulating macrophage polarization (shown in Figure 2) support a key role of GPR109A signalling as anti-inflammatory regulator of the low-grade inflammation associated with insulin resistance and obesity (64).
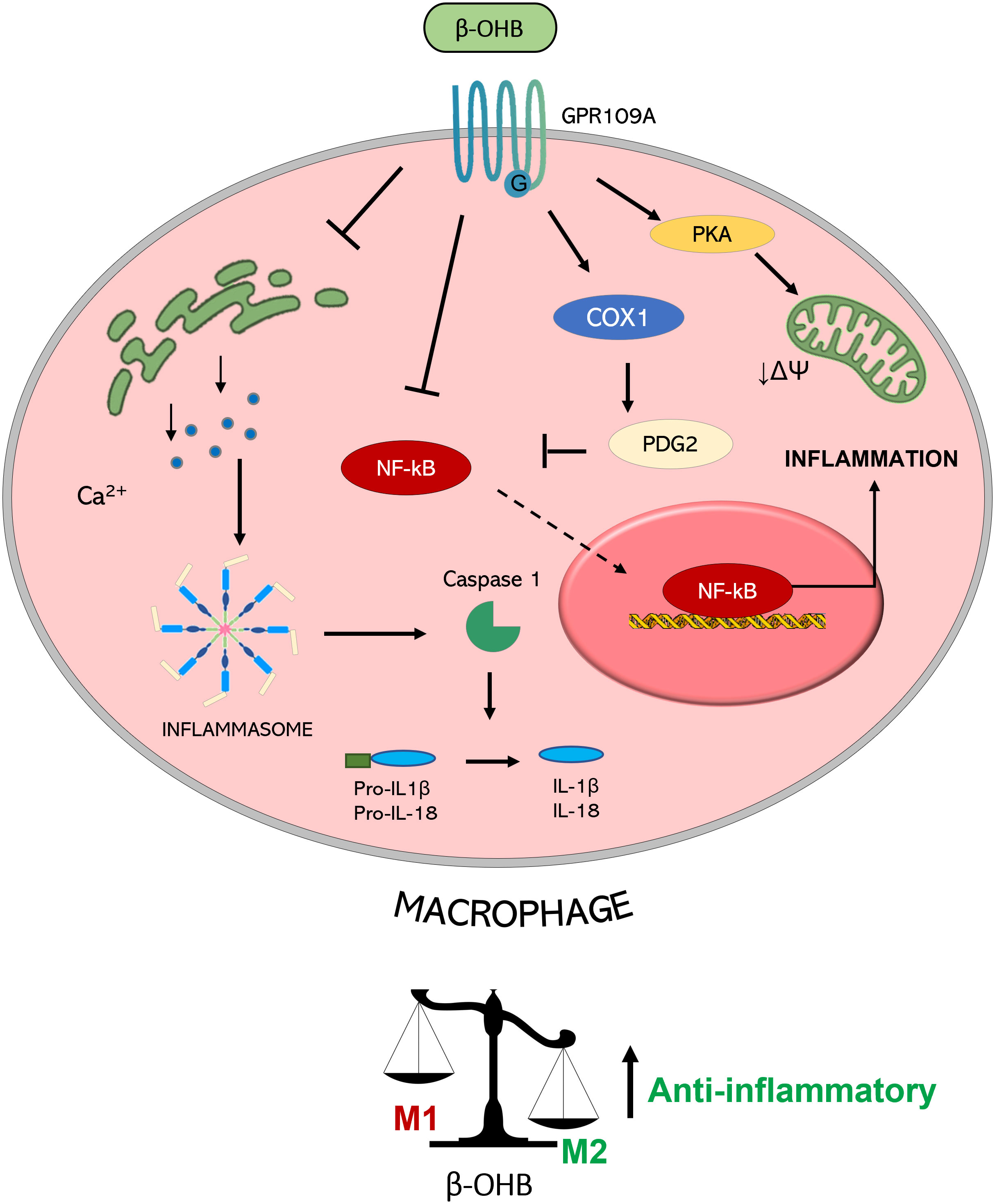
Figure 2 Molecular mechanisms triggered by GPR109A-β-OHB axis in macrophages. GPR109A engagement by β-OHB leads to a reduced inflammasome activation mediated by a reduction of Ca2+ release from endoplasmic reticulum. This inhibition of inflammasome activity causes a lower IL-1β and IL-18 release from macrophages. A reduction of inflammatory mediator release is also achieved by the inhibition of the transcription factor NF-κB which can be mediated by the COX-1-mediated synthesis of PDG2, a known inhibitor of NF-κB activity. All together these effects converge in the induction of an anti-inflammatory M2 phenotype of macrophages. This switch is also obtained by a PKA-mediated decrease in the mitochondrial membrane potential which guides M2 polarization. (PGD2: Prostaglandin D2; COX-1: Cyclooxygenase 1; PKA: Protein kinase A; NF-κB: nuclear factor-κB; IL: interleukin; βOHB: β-hydroxybutyrate; ΔΨ: mitochondrial membrane potential).
Some evidence (Table 1) points to β-OHB as a novel ligand also for GPR41, a GPCR expressed in human adipocytes (65), sympathetic neurons (54) colon epithelial, endothelial (66) and mononu clear cells, and classically activated by short-chain fatty acids (SCFAs) such as acetate, propionate, and butyrate (67). Studies on knockout mice implicate GPR41 in chronic inflammatory disorders such as obesity, colitis, asthma and arthritis, although its protective or causative role is inconsistent among studies (68). Propionate potently activated sympathetic nervous system (SNS) and β-OHB - through unclear molecular mechanisms - antagonized SCFA-GPR41 signaling thereby suppressing SNS activity (55), which is known to be finely regulated for the balance of energy intake, dietary excess and starvation. This β-OHB inhibitory effect on SNS was demonstrated both in cell culture stimulated with β-OHB 10-100 mM and in streptozotocin-induced diabetic mice, in which β-OHB concentration reached 2.6 mM (55). Although other studies consider β-OHB a GPR41 agonist, rather than antagonist (54), it is established that β-OHB can control energy balance by directly regulating GPR41-mediated sympathetic activation (55). Further studies are necessary to confirm these findings and to unravel the precise molecular mechanism underlying the involvement of GPR41 in the modulation of cell metabolism.
GPR43 is a SCFA receptor which reduces cAMP levels, activates the ERK1/2 pathway, and increases Ca2+ concentrations (69). Although highly expressed in adipose tissues, it is also involved in the modulation of inflammation and body metabolism through gut microbiota (70, 71).
Recent evidence reported that the KB acetoacetate functions as an endogenous activator of GPR43 with binding affinity comparable to that of SCFA (i.e. propionate and acetate) under ketogenic conditions (Table 1). As already stated, during fasting KB plasma concentrations were increased (>0.5 mM) and SCFA levels were highly decreased (from 0.5 to 0.1 mM), suggesting that the primary ligand for GPR43 in plasma switches from SCFAs to KB under fasting conditions.
Under ketogenic conditions acetoacetate increased plasma lipid utilization by controlling the activation of lipoprotein lipase (LPL) via adipose GPR43 (56). Furthermore, plasma levels of glucose and triglycerides were significantly reduced, and cholesterol levels increased, in wild-type compared to Gpr43−/− mice. Similarly, the decrease in body weight was significantly blunted in the Gpr43−/− mice compared with the wild-type mice during fasting. Thus, the acetoacetate-GPR43 axis plays a key role on metabolic homeostasis by controlling body weight and lipid levels.
The following paragraphs will discuss the results of clinical trials on the effects of KD in different metabolic and endocrinological disorders [obesity, diabetes, non-alcoholic fatty liver disease (NAFLD) and polycystic ovary syndrome (PCOS)]. The rationale for the use of KDs is the substantial efficacy in short-term weight loss; however, it is still unclear whether KDs may have additional benefits vs isocaloric non-ketogenic diets. In this context we will explore the hypothesis that KB, by binding to GPCR family receptors, may mediate some of these additional beneficial effects (i.e lipid metabolism and anti-inflammatory actions). To discriminate the effects of KB per se, independently of weight loss, only studies comparing ketogenic vs isocaloric non-ketogenic diets have been herein considered, unless otherwise specified. Short-term tolerability and safety of KDs are reported in Tables 2 and 3.
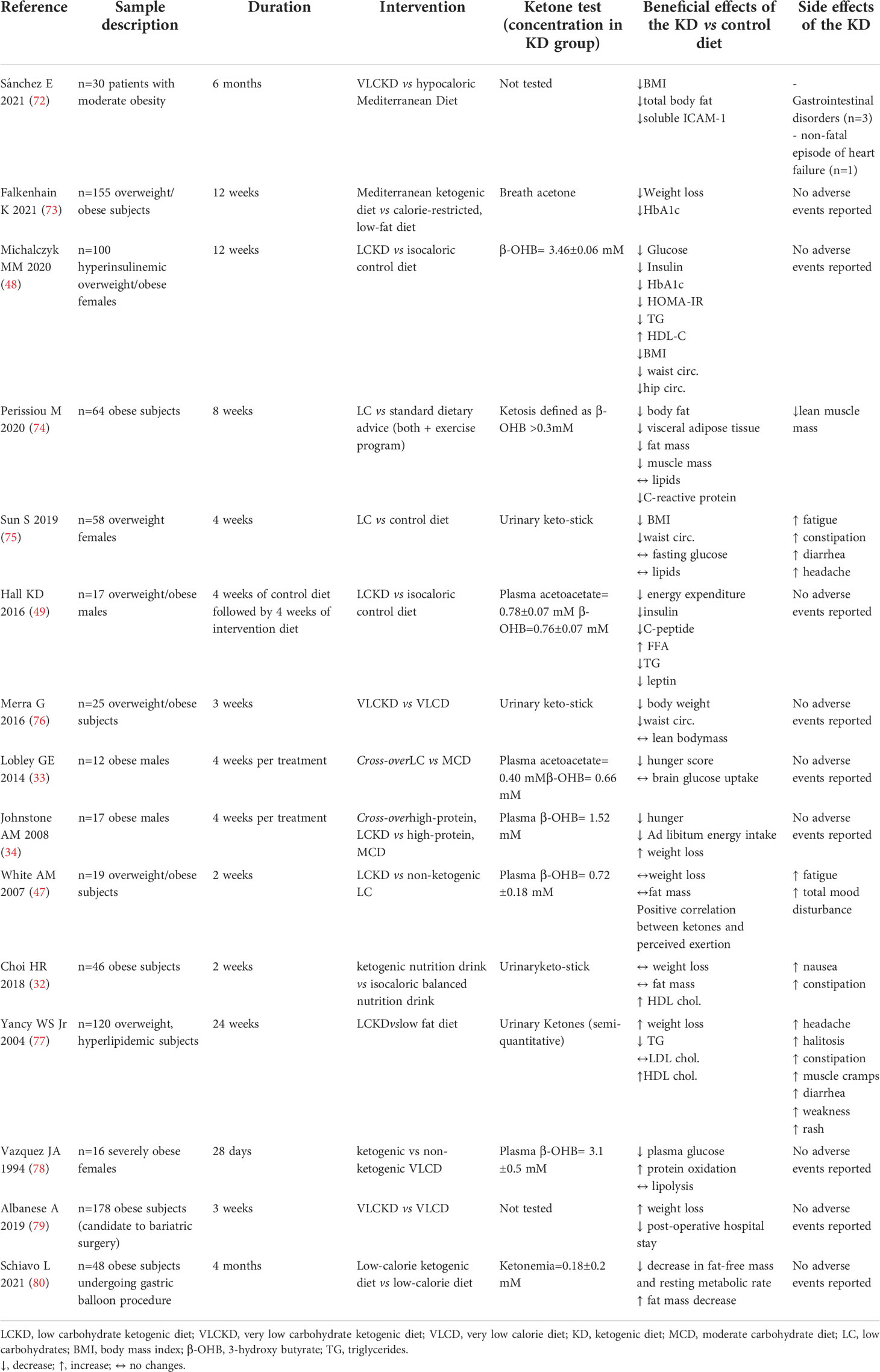
Table 2 Summary of trials evaluating KD effects vs comparative diet in obese subjects.

Table 3 Summary of trials evaluating KD effects vs comparative diet in subjects with type 2 diabetes.
We searched in Medline (PubMed) using the terms “clinical trials” in adults using the query “ketogenic diet” OR “very low carbohydrate ketogenic diet” AND “obesity”, “diabetes”, “NAFLD”, “PCOS”.
Obesity is recognized as one of the major public health hazards worldwide as it is associated with cardiometabolic and cerebrovascular diseases leading to reduced life quality and increased mortality (85). Over the past decades, there has been a rising prevalence of obesity and its prevention and treatment are mainly based on lifestyle changes, including physical activity and healthy eating habits (86).
Several studies provided robust evidence of the beneficial effect of KDs in reducing body weight in obesity. Importantly, KD-induced weight loss is generally higher than that achieved with an isocaloric non-ketogenic control diet (Table 2), as shown in several populations including hyperinsulinemic (48) overweight females (75), overweight/obese subjects (34, 72–74, 76, 77) and in morbidly obese subjects candidate to bariatric surgery (79). In addition, weight reduction following a KD is demonstrated to be accompanied by favourable changes in body composition with a higher decrease in waist circumference (48, 75, 76) and fat mass loss (72, 74), also in subjects who underwent gastric balloon procedure (80), vs control diets.
Results on weight management with KDs are generally obtained in animal models (87–90). Mice maintained on a KD exhibited higher weight reduction compared to chow-fed animals both at 6 and 12 weeks. Nevertheless, a few studies in obese mice (ob/ob) reported no differences in body weight loss between KD- and chow-fed animals (91, 92).
Favourable data of KD on metabolism have been confirmed also in human studies. A 12-week low-carbohydrate KD intervention led to a more marked improvement in glucose (48, 74, 78) and insulin (48, 49) levels compared to an isocaloric control diet in overweight/obese females (48). The amelioration in metabolic profile associated to a KD in obese subjects, has likely to be ascribed to the very low carbohydrate intake and the consequent decrease in plasma glucose/insulin levels.
KDs might display additional weight loss-independent benefits, particularly on lipid metabolism and inflammatory processes via GPCR signaling activation.
Briefly, as already described, GPR109A engagement by β-OHB induces a decrease in adipocyte lipolysis, by blunting hormone sensitive lipase activation (46, 51, 57). This antilipolytic effect in hepatocytes decreases FFA serum levels with a consequent shortage in substrate availability for triglyceride synthesis. Moreover, GPR43 engagement by acetoacetate promotes lipid utilization by increasing plasma LPL activation (56), thus contributing to lipid profile amelioration. In addition to the above-described mechanisms, insulin reduction secondary to carbohydrate restriction might further contribute to a more favourable anti-atherogenic lipid profile by inhibiting HMG-CoA reductase enzyme and consequently endogenous cholesterol biosynthesis (93).
Accordingly, some clinical evidence showed an increase in HDL-cholesterol and a reduction in triglyceride levels in overweight/obese women (48) and in subjects with obesity and dyslipidaemia (77) randomised to a VLCKD compared to those in the isocaloric control diet. In the latter study, although the KD did not affect total low-density lipoprotein (LDL)-cholesterol, it led to a shift from small, dense LDL to large, buoyant LDL, associated to an anti-atherogenic lipid profile (94). In line, a recent study showed that a 2-week low-calorie ketogenic nutrition drink in obese adults was more effective in ameliorating lipid profile compared to an isocaloric balanced nutrition drink (32). Although these favourable mechanistical premises, contrasting literature is present as neutral findings on KD effects on lipid profile were also reported in human studies (74, 75). A very recent systematic review of trials comparing KDs vs balanced diets in obese subjects reported no statistically significant differences in total, high-density lipoprotein (HDL) and LDL-cholesterol or triglycerides (95).
These findings may be reconciled taking into consideration the high (saturated) fat intake of these diets which might blunt/abolish the above-mentioned beneficial effects on lipid profile.
Another major point is the possible GPCR-mediated anti-inflammatory effect in course of KD.
A recent study conducted in obese individuals randomised to a 8-week VLCKD vs standard diet (both associated to an exercise program) showed a correlation between circulating β-OHB levels and the reduction of visceral fat and inflammatory markers (74) which define the adiposity state, pointing to a direct role of ketones in the amelioration of metaflammation (72). These effects should be ascribed to the activation of the β-OHB-GPR109A axis which triggers an anti-inflammatory macrophage phenotype in several animal models (50, 52, 53), ultimately reducing cardiovascular risk.
Thus, KDs might be beneficial in blunting chronic low-grade inflammation which plays a pivotal role in the pathogenesis of cardiometabolic disorders, such as obesity. Nevertheless, future studies are warranted to better explore the impact of this anti-inflammatory mechanism in mediating the beneficial effects of KDs in obesity. Clinical trials on the effects of KDs in subjects with overweight/obesity are reported in Table 2, including adverse events.
Type 2 diabetes mellitus (T2D) is a major public health global emergency (96). The dietary approach, combined with physical exercise, represents the first-line treatment in T2D. KDs have been taken into consideration, also in recent guidelines (97), among the strategies to adopt in order to obtain weight reduction, ameliorate glucose control and limit the need for anti-hyperglycemic medications in T2D (98).
In KD-fed diabetic rodents, KD strongly reduced blood glucose levels (99–103), improving glucose tolerance (102, 104) and insulin sensitivity (100, 102–104) compared to those fed with a control diet, even at weight loss equipoise (99, 104).
Accordingly, several studies demonstrated KD-associated beneficial effects on weight loss, lipid and glucose parameters, and insulin sensitivity in individuals with T2D (28, 81–84).
Subjects with T2D randomized to a very low-calorie KD for 4 months showed a significant reduction in weight, waist circumference, HbA1c and homeostatic model assessment for insulin resistance (HOMA-IR) compared to those allocated to a standard low-calorie diet (82). Similarly, T2D subjects following a VLCKD showed a decrease in HbA1c levels (81, 83) and an amelioration in lipid profile (81) compared to those on a control diet. A KD-induced decrease in glucose parameters was also confirmed in a short-term (4 days) clinical study (84). Of note, VLCKD consumption was also associated to a reduction in diabetes medication use compared to non-ketogenic control diet (83).
Mechanistically, lipid profile amelioration in T2D by VLCKD could be ascribed to the above-described actions of acetoacetate via GPR43 (56) and of β-OHB via GPR109A on lipolysis (46, 51) and to the insulin resistance amelioration.
Conversely, the improvement in glucose profile associated to the consumption of a KD in T2D is unlikely to be mediated by GPCR activation, but rather by the limited carbohydrate intake.
Results from controlled clinical trials on the effects of KD in subjects with T2D are summarized in Table 3.
Despite these limited but encouraging results, it should be taken into consideration that carbohydrate restriction associated to KDs may increase the risk of hypoglycemia in these patients, especially in those treated with insulin and/or insulin secretagogues. Thus, KDs should be initiated under strict medical supervision, particularly in course of diuretic therapy, in order to avoid hypoglycemia and dehydration (105, 106). Importantly, KDs should not be prescribed to patients treated with sodium-glucose cotransporter (SGLT)-2 inhibitors for their significantly higher risk of developing euglycemic diabetic ketoacidosis (28, 107). Indeed, this drug class enhances the production of KB in the liver, by increasing glucagon levels and reducing the insulin:glucagon ratio per se (108–110).
Clinical trials investigating KD effects in individuals with type 1 diabetes (T1D) are very limited. The randomization to a VLCKD in a cross-over study reduced glucose variability compared to an isocaloric high-carbohydrate diet (111). Nevertheless, in an uncontrolled study the consumption of a VLCKD was associated to a high risk of dyslipidaemia and to a high frequency of hypoglycaemic episodes in T1D subjects (112). Interestingly, a case report showed that a healthy young woman, in course of a KD for weight reduction, developed diabetic ketoacidosis which was considered the “precipitating factor” leading to T1D diagnosis (113).
Thus, a KD might be appropriate for selected T1D patients, but only following a careful evaluation of the burden of risks (dyslipidemia, diabetic ketoacidosis and hypoglycemia) and benefits (weight loss and better glycemic control) (114). Nonetheless, the latest Standards of Medical Care by American Diabetes Association do not recommend KDs in the medical nutrition therapy for T1D (115).
NAFLD is a metabolic liver disease strongly associated with obesity and T2D; its prevalence is increasing - it affects a quarter of global population- along with that of other metabolic disorders. NAFLD is the commonest cause of chronic liver disease and it is defined as an increase in liver fat content, in the absence of secondary cause of steatosis (116). In fact, the clinical onset of the disease is a much more complex process, closely related to insulin resistance and to the limited expandability and dysfunctionality of adipose tissue (117).
In obese mice, a 3-week VLCKD regimen increased pro-inflammatory M1/M2 macrophage ratio in adipose tissue without ameliorating NAFLD, in contrast to the low-fat control diet (118). In line, long-term administration of a KD in mice induced systemic glucose intolerance, hepatic ER stress, steatosis, cellular injury and macrophage accumulation (90). Though, data from the literature are discordant as a very recent paper demonstrated that KD prevented steatosis and insulin resistance by reducing lipogenesis, diacylglycerol accumulation and protein kinase C activity in rat liver (119).
In humans, a very recent randomized 6-week intervention trial showed that the consumption of a KD or an isocaloric low-fat diet similarly and safely reduced liver fat in individuals with NAFLD (120). Nevertheless, randomized trials on subjects with NAFLD are deficient, and the efficacy of KD in NAFLD is still a matter of debate.
KDs may display a dual effect in NAFLD treatment. If on the one hand KD-mediated reduction in insulinemia and lipogenesis and increase in FFA oxidation (121) are beneficial; on the other, the high fat content of the diet might worsen liver fat accumulation.
Therefore, long-term randomized clinical trials are needed to assess safety and efficacy of KDs in NAFLD (122).
PCOS is an endocrine disorder characterized by anovulation, polycystic ovaries and hirsutism. PCOS is often associated to insulin resistance, dyslipidaemia, NAFLD, obesity, and ultimately to an augmented risk for the development of cardiometabolic diseases. To the best of our knowledge, no studies to date have compared the effects of a KD versus a non-ketogenic control diet in women with PCOS. Nevertheless, some pilot short-term uncontrolled studies in overweight (123) and obese (124, 125) women with PCOS showed beneficial effects of KDs in reducing body weight, fat mass (123, 124), visceral adipose tissue (123) and cholesterol levels, accompanied by an improvement in insulin sensitivity (reduced HOMA-IR) (123, 124) and to a reduction in the luteinizing hormone (LH)/follicle-stimulating hormone (FSH) ratio and testosterone levels (123–125).
In a very recent study conducted in 18 obese women with PCOS and liver dysfunction the intake of a KD for 12 weeks was more effective compared to a conventional pharmacological treatment (polyene phosphatidylcholine) in reducing fasting glucose, body weight and liver enzymes; both interventions equally decreased plasma estradiol and progesterone levels (126). In conclusion, comparative studies are needed to ascertain whether KDs may display additional endocrinological and metabolic benefits in PCOS, independently of weight loss.
Nutritional ketosis induced through a ketogenic diet leads to higher, but controlled, plasma ketone concentrations (>0.5-1.0 mM). Most human studies provide evidence of a higher effect in short-term weight loss of the KDs compared to isocaloric balanced diets in T2D and obese/overweight subjects, nevertheless controversies remain about the use of KDs in other metabolic disease such as T1D and NAFLD. Scarce evidence exists regarding the effects of KDs in PCOS.
The important weight loss effect with KDs compared to standard diets is largely attributable to higher plasma ketones (31, 127); however, the exact threshold of ketosis needed to control appetite, as well as the exact mechanisms underlying this effect have yet to be established.
KDs might also display additional cardiometabolic benefits, beyond (and in addition to) weight loss, which may rely on different mechanisms. The very limited carbohydrate assumption mainly accounts for the improvement in glucose levels and insulin resistance, often reported in subjects with obesity/diabetes.
Recent evidence supports a different mechanism for KD effects on the amelioration in lipid profile (in particular triglycerides) and inflammation which might be of particular relevance in insulin resistance-related disorders. Ketones can bind specific GPCRs - i.e. GPR109A (50), GPR41 (55) and GPR43 (56) - through which they are able to directly modulate lipid metabolism and inflammation. The activation of acetoacetate-GPR43 axis promotes lipid utilization by plasma LPL activation (56), whilst that of GPR109A by β-OHB decreased adipocytic lipase activity and consequent hepatic triglyceride synthesis (46, 51). In addition, the activation of GPR41 by β-OHB may probably negatively regulate energy intake and metabolism through the suppression of sympathetic activation (55). Comparably, GPR109A-mediated inhibition of NLRP3 inflammasome by β-OHB presumably accounts for the anti-inflammatory effects associated to a KD regimen, leading to an anti-atherogenic polarization of macrophages in M2 phenotype (1, 45, 50, 52).
However, despite a strong preclinical and animal evidence, (45, 50, 52) points to a role of ketones in the reduction of inflammation - which is a common trait of metabolic diseases - a robust clinical confirmation is still lacking. Mechanistic human studies are warranted to ascertain whether GPCR signalling activation by ketones might be of clinical relevance in favourably affecting lipid and inflammatory profiles, independently or in addition to the important weight reduction.
Human intervention studies with KDs are fraught with important limitations since high-quality and long-term evidence is currently scant.
Most interventions span over a period of a few weeks and KD adherence is limited by the very low carbohydrate content (<50g/day) (16). In addition, many studies report only partial data on calories/macronutrients composition of the diets and on the achieved plasma ketone levels, which may account for result heterogeneity among different studies. Ultimately, data on long-term beneficial effects and safety of KD on metabolic/endocrinological disorders are still needed.
In conclusion, although KDs hold a strong potential in the treatment of endocrinological and metabolic disorders due to a broader spectrum of short-term beneficial therapeutic effects in reducing weight and appetite, in ameliorating lipid and glucose profile and the inflammatory milieu, definitive conclusions are difficult to be drawn.
VS, GC and ADC concepted and designed the review and wrote the manuscript. NTI, GF, GM, MM and VP contributed to the review and interpretation of the literature and drafted the manuscript. ADC critically revised the manuscript. All authors contributed to the article and approved the submitted version.
This paper has been supported by “FIL funds for research” from University of Parma to ADC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev (1999) 15(6):412–26. doi: 10.1002/(sici)1520-7560(199911/12)15:6
2. Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab (2017) 25(2):262–84. doi: 10.1016/j.cmet.2016.12.022
4. Cahill GF Jr, Veech RL. Ketoacids? good medicine? Trans Am Clin Climatol Assoc (2003) 114:149–61.
5. Dhillon KK, Gupta S. Biochemistry, ketogenesis. In: StatPearls. Treasure Island (FL: StatPearls Publishing (2021). Available at: https://www.ncbi.nlm.nih.gov/books/NBK493179/.
6. Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes (2011) 60(10):2441–9. doi: 10.2337/db11-0425
7. Pinckaers PJ, Churchward-Venne TA, Bailey D, van Loon LJ. Ketone bodies and exercise performance: The next magic bullet or merely hype? Sports Med (2017) 47(3):383–91. doi: 10.1007/s40279-016-0577-y
8. Barnett CR, Barnett YA. Ketone bodies. In: Caballero B, editor. Encyclopedia of food sciences and nutrition, 2. (San Diego, California, USA: Academic Press) (2003). pp. 3421–5.
9. Caprio M, Infante M, Moriconi E, Armani A, Fabbri A, Mantovani G, et al. Cardiovascular endocrinology club of the Italian society of endocrinology. very-low-calorie ketogenic diet (VLCKD) in the management of metabolic diseases: systematic review and consensus statement from the Italian society of endocrinology (SIE). J Endocrinol Invest. (2019) 42(11):1365–86. doi: 10.1007/s40618-019-01061-2
10. Sheikh-Ali M, Karon BS, Basu A, Kudva YC, Muller LA, Xu J, et al. Can serum beta-hydroxybutyrate be used to diagnose diabetic ketoacidosis? Diabetes Care (2008) 31(4):643–7. doi: 10.2337/dc07-1683
11. Ford W, Self WH, Slovis C, McNaughton CD. Diabetes in the emergency department and hospital: Acute care of diabetes patients. Curr Emerg Hosp Med Rep (2013) 1(1):1–9. doi: 10.1007/s40138-012-0007-x
13. Augustin K, Khabbush A, Williams S, Eaton S, Orford M, Cross JH, et al. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol (2018) 17(1):84–93. doi: 10.1016/S1474-4422(17)30408-8
14. Klement RJ, Champ CE, Otto C, Kämmerer U. Anti-tumor effects of ketogenic diets in mice: a meta-analysis. PloS One (2016) 11(5):e0155050. doi: 10.1371/journal.pone.0155050
15. Geng S, Zhu W, Xie C, Li X, Wu J, Liang Z, et al. Medium-chain triglyceride ameliorates insulin resistance and inflammation in high fat diet-induced obese mice. Eur J Nutr (2016) 55(3):931–40. doi: 10.1007/s00394-015-0907-0
16. Kirkpatrick CF, Bolick JP, Kris-Etherton PM, Sikand G, Aspry KE, Soffer DE, et al. Review of current evidence and clinical recommendations on the effects of low-carbohydrate and very-low-carbohydrate (including ketogenic) diets for the management of body weight and other cardiometabolic risk factors: A scientific statement from the national lipid association nutrition and lifestyle task force. J Clin Lipidol. (2019) 13(5):689–711:e1. doi: 10.1016/j.jacl.2019.08.003
17. Prabhakar A, Quach A, Zhang H, Terrera M, Jackemeyer D, Xian X, et al. Acetone as biomarker for ketosis buildup capability–a study in healthy individuals under combined high fat and starvation diets. Nutr J (2015) 14:41. doi: 10.1186/s12937-015-0028-x
18. Anderson JC, Mattar SG, Greenway FL, Lindquist RJ. Measuring ketone bodies for the monitoring of pathologic and therapeutic ketosis. Obes Sci Pract (2021) 7(5):646–56. doi: 10.1002/osp4.516
19. Kolb H, Kempf K, Röhling M, Lenzen-Schulte M, Schloot NC, Martin S. Ketone bodies: from enemy to friend and guardian angel. BMC Med (2021) 19(1):313. doi: 10.1186/s12916-021-02185-0
20. Hassan AM, Keene DL, Whiting SE, Jacob PJ, Champagne JR, Humphreys P. Ketogenic diet in the treatment of refractory epilepsy in childhood. Pediatr Neurol (1999) 21(2):548–52. doi: 10.1016/s0887-8994(99)00045-4
21. Coppola G, Veggiotti P, Cusmai R, Bertoli S, Cardinali S, Dionisi-Vici C, et al. The ketogenic diet in children, adolescents and young adults with refractory epilepsy: an Italian multicentric experience. Epilepsy Res (2002) 48(3):221–7. doi: 10.1016/s0920-1211(01)00315-1
22. Huttenlocher PR, Wilbourn AJ, Signore JM. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology (1971) 21(11):1097–103. doi: 10.1212/wnl.21.11.1097
23. Zupec-Kania BA, Spellman E. An overview of the ketogenic diet for pediatric epilepsy. Nutr Clin Pract (2008) 23(6):589–96. doi: 10.1177/0884533608326138
24. Chang P, Terbach N, Plant N, Chen PE, Walker MC, Williams RS. Seizure control by ketogenic diet-associated medium chain fatty acids. Neuropharmacology (2013) 69:105–14. doi: 10.1016/j.neuropharm.2012.11.004
25. Liu YM. Medium-chain triglyceride (MCT) ketogenic therapy. Epilepsia (2008) 49 Suppl8::33–6. doi: 10.1111/j.1528-1167.2008.01830.x
26. Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond). (2009) 6:31. doi: 10.1186/1743-7075-6-31
27. Gibson AA, Seimon RV, Lee CM, Ayre J, Franklin J, Markovic TP, et al. Do ketogenic diets really suppress appetite? a systematic review and meta-analysis. Obes Rev (2015) 16(1):64–76. doi: 10.1111/obr.12230
28. Gupta L, Khandelwal D, Kalra S, Gupta P, Dutta D, Aggarwal S. Ketogenic diet in endocrine disorders: Current perspectives. J Postgrad Med (2017) 63(4):242–51. doi: 10.4103/jpgm.JPGM_16_17
29. Terzikhan N, Doets EL, Vonk Noordegraaf-Schouten M. Extensive literature search and review as preparatory work for the evaluation of the essential composition of total diet replacement products for weight control. EFSA Supporting Publications. (2015) 12:590E. doi: 10.2903/sp.efsa.2015.EN-590
30. Sumithran P, Prendergast LA, Delbridge E, Purcell K, Shulkes A, Kriketos A, et al. Ketosis and appetite-mediating nutrients and hormones after weight loss. Eur J Clin Nutr (2013) 67(7):759–64. doi: 10.1038/ejcn.2013.90
31. Deemer SE, Plaisance EP, Martins C. Impact of ketosis on appetite regulation-a review. Nutr Res (2020) 77:1–11. doi: 10.1016/j.nutres.2020.02.010
32. Choi HR, Kim J, Lim H, Park YK. Two-week exclusive supplementation of modified ketogenic nutrition drink reserves lean body mass and improves blood lipid profile in obese adults: A randomized clinical trial. Nutrients (2018) 10(12):1895. doi: 10.3390/nu10121895
33. Lobley GE, Johnstone AM, Fyfe C, Horgan GW, Holtrop G, Bremner DM, et al. Glucose uptake by the brain on chronic high-protein weight-loss diets with either moderate or low amounts of carbohydrate. Br J Nutr (2014) 111(4):586–97. doi: 10.1017/S0007114513002900
34. Johnstone AM, Horgan GW, Murison SD, Bremner DM, Lobley GE. Effects of a high-protein ketogenic diet on hunger, appetite, and weight loss in obese men feeding ad libitum. Am J Clin Nutr (2008) 87(1):44–55. doi: 10.1093/ajcn/87.1.44
35. Masood W, Annamaraju P, Uppaluri KR. Ketogenic diet. In: StatPearls. Treasure Island (FL: StatPearls Publishing (2021).
36. Batch JT, Lamsal SP, Adkins M, Sultan S, Ramirez MN. Advantages and Disadvantages of the Ketogenic Diet: A Review Article. Cureus (2020) 12(8):e9639. doi: 10.7759/cureus.9639
37. Yang D, Zhou Q, Labroska V, Qin S, Darbalaei S, Wu Y, et al. G Protein-coupled receptors: structure- and function-based drug discovery. Signal Transduct Target Ther (2021) 6(1):7. doi: 10.1038/s41392-020-00435-w
38. Stevens RC, Cherezov V, Katritch V, Abagyan R, Kuhn P, Rosen H, et al. The GPCR network: a large-scale collaboration to determine human GPCR structure and function. Nat Rev Drug Discovery (2013) 12(1):25–34. doi: 10.1038/nrd3859
39. Priyadarshini M, Villa SR, Fuller M, Wicksteed B, Mackay CR, Alquier T, et al. FFAR2, regulates insulin secretion. Mol Endocrinol (2015) 29(7):1055–66. doi: 10.1210/me.2015-1007
40. Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J, et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem (2009) 284(5):2811–22. doi: 10.1074/jbc.M806409200
41. Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-mediated signaling of metabolites. Cell Metab (2017) 25(4):777–96. doi: 10.1016/j.cmet.2017.03.008
42. Senior B, Loridan L. Direct regulatory effect of ketones on lipolysis and on glucose concentrations in man. Nature (1968) 219(5149):83–4. doi: 10.1038/219083a0
43. Cahill GF Jr. Starvation in man. N Engl J Med (1970) 282(12):668–75. doi: 10.1056/NEJM197003192821209
44. Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids (2004) 70(3):243–51. doi: 10.1016/j.plefa.2003.11.001
45. Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Müller-Fielitz H, et al. The β-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun (2014) 5:3944. doi: 10.1038/ncomms4944
46. Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem (2005) 280(29):26649–52. doi: 10.1074/jbc.C500213200
47. White AM, Johnston CS, Swan PD, Tjonn SL, Sears B. Blood ketones are directly related to fatigue and perceived effort during exercise in overweight adults adhering to low-carbohydrate diets for weight loss: a pilot study. J Am Diet Assoc (2007) 107(10):1792–6. doi: 10.1016/j.jada.2007.07.009
48. Michalczyk MM, Klonek G, Maszczyk A, Zajac A. The effects of a low calorie ketogenic diet on glycaemic control variables in hyperinsulinemic Overweight/Obese females. Nutrients (2020) 12(6):1854. doi: 10.3390/nu12061854
49. Hall KD, Chen KY, Guo J, Lam YY, Leibel RL, Mayer LE, et al. Energy expenditure and body composition changes after an isocaloric ketogenic diet in overweight and obese men. Am J Clin Nutr (2016) 104(2):324–33. doi: 10.3945/ajcn.116.133561
50. Zhang SJ, Li ZH, Zhang YD, Chen J, Li Y, Wu FQ, et al. Ketone body 3-hydroxybutyrate ameliorates atherosclerosis via receptor Gpr109a-mediated calcium influx. Adv Sci (Weinh). (2021) 8(9):2003410. doi: 10.1002/advs.202003410
51. Lee AK, Kim DH, Bang E, Choi YJ, Chung HY. β-hydroxybutyrate suppresses lipid accumulation in aged liver through GPR109A-mediated signaling. Aging Dis (2020) 11(4):777–90. doi: 10.14336/AD.2019.0926
52. Fu SP, Wang JF, Xue WJ, Liu HM, Liu BR, Zeng YL, et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro parkinson's disease models are mediated by GPR109A-dependent mechanisms. J Neuroinflammation. (2015) 12:9. doi: 10.1186/s12974-014-0230-3
53. Chen Y, Ouyang X, Hoque R, Garcia-Martinez I, Yousaf MN, Tonack S, et al. β-hydroxybutyrate protects from alcohol-induced liver injury via a Hcar2-cAMP dependent pathway. J Hepatol (2018) 69(3):687–96. doi: 10.1016/j.jhep.2018.04.004
54. Won YJ, Lu VB, Puhl HL 3rd, Ikeda SR. β-hydroxybutyrate modulates n-type calcium channels in rat sympathetic neurons by acting as an agonist for the G-protein-coupled receptor FFA3. J Neurosci (2013) 33(49):19314–25. doi: 10.1523/JNEUROSCI.3102-13.2013
55. Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci U S A. (2011) 108(19):8030–5. doi: 10.1073/pnas.1016088108
56. Miyamoto J, Ohue-Kitano R, Mukouyama H, Nishida A, Watanabe K, Igarashi M, et al. Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. Proc Natl Acad Sci U S A. (2019) 116(47):23813–21. doi: 10.1073/pnas.1912573116
57. Zhang Y, Schmidt RJ, Foxworthy P, Emkey R, Oler JK, Large TH, et al. Niacin mediates lipolysis in adipose tissue through its G-protein coupled receptor HM74A. Biochem Biophys Res Commun (2005) 334(2):729–32. doi: 10.1016/j.bbrc.2005.06.141
58. Chai JT, Digby JE, Choudhury RP. GPR109A and vascular inflammation. Curr Atheroscler Rep (2013) 15(5):325. doi: 10.1007/s11883-013-0325-9
59. Offermanns S. It ain't over 'til the fat lady sings. Sci Transl Med (2012) 4(148):148fs30. doi: 10.1126/scitranslmed.3004445
60. Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol (2013) 33(6):1120–6. doi: 10.1161/ATVBAHA.112.300173
61. Joo M, Sadikot RT. PGD synthase and PGD2 in immune response. Mediators Inflamm (2012) 2012:503128. doi: 10.1155/2012/503128
62. Mandrekar P, Bataller R, Tsukamoto H, Gao B. Alcoholic hepatitis: Translational approaches to develop targeted therapies. Hepatology (2016) 64(4):1343–55. doi: 10.1002/hep.28530
63. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell (2016) 167(2):457–70. doi: 10.1016/j.cell.2016.08.064
64. Ye L, Cao Z, Lai X, Wang W, Guo Z, Yan L, et al. Niacin fine-tunes energy homeostasis through canonical GPR109A signaling. FASEB J (2019) 33(4):4765–79. doi: 10.1096/fj.201801951R
65. Al-Lahham SH, Roelofsen H, Priebe M, Weening D, Dijkstra M, Hoek A, et al. Regulation of adipokine production in human adipose tissue by propionic acid. Eur J Clin Invest. (2010) 40(5):401–7. doi: 10.1111/j.1365-2362.2010.02278.x
66. Li M, van Esch BCAM, Henricks PAJ, Folkerts G, Garssen J. The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor α-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front Pharmacol (2018) 9:533. doi: 10.3389/fphar.2018.00533
67. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem (2003) 278(13):11312–9. doi: 10.1074/jbc.M211609200
68. Ang Z, Ding JL. GPR41 and GPR43 in obesity and inflammation - Protective or causative? Front Immunol (2016) 7:28. doi: 10.3389/fimmu.2016.00028
69. Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem (2003) 278(28):25481–9. doi: 10.1074/jbc.M301403200
70. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature (2009) 461(7268):1282–6. doi: 10.1038/nature08530
71. Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun (2013) 4:1829. doi: 10.1038/ncomms2852
72. Sánchez E, Santos MD, Nuñez-Garcia M, Bueno M, Sajoux I, Yeramian A, et al. Randomized clinical trial to evaluate the morphological changes in the adventitial vasa vasorum density and biological markers of endothelial dysfunction in subjects with moderate obesity undergoing a very low-calorie ketogenic diet. Nutrients (2021) 14(1):33. doi: 10.3390/nu14010033
73. Falkenhain K, Locke SR, Lowe DA, Reitsma NJ, Lee T, Singer J, et al. Keyto app and device versus WW app on weight loss and metabolic risk in adults with overweight or obesity: A randomized trial. Obes (Silver Spring). (2021) 29(10):1606–14. doi: 10.1002/oby.23242
74. Perissiou M, Borkoles E, Kobayashi K, Polman R. The effect of an 8 week prescribed exercise and low-carbohydrate diet on cardiorespiratory fitness, body composition and cardiometabolic risk factors in obese individuals: A randomised controlled trial. Nutrients (2020) 12(2):482. doi: 10.3390/nu12020482
75. Sun S, Kong Z, Shi Q, Hu M, Zhang H, Zhang D, et al. Non-Energy-Restricted low-carbohydrate diet combined with exercise intervention improved cardiometabolic health in overweight Chinese females. Nutrients (2019) 11(12):3051. doi: 10.3390/nu11123051
76. Merra G, Miranda R, Barrucco S, Gualtieri P, Mazza M, Moriconi E, et al. Very-low-calorie ketogenic diet with aminoacid supplement versus very low restricted-calorie diet for preserving muscle mass during weight loss: a pilot double-blind study. Eur Rev Med Pharmacol Sci (2016) 20(12):2613–21.
77. Yancy WS Jr, Olsen MK, Guyton JR, Bakst RP, Westman EC. A low-carbohydrate, ketogenic diet versus a low-fat diet to treat obesity and hyperlipidemia: a randomized, controlled trial. Ann Intern Med (2004) 140(10):769–77. doi: 10.7326/0003-4819-140-10-200405180-00006
78. Vazquez JA, Kazi U. Lipolysis and gluconeogenesis from glycerol during weight reduction with very-low-calorie diets. Metabolism (1994) 43(10):1293–9. doi: 10.1016/0026-0495(94)90225-9
79. Albanese A, Prevedello L, Markovich M, Busetto L, Vettor R, Foletto M. Pre-operative very low calorie ketogenic diet (VLCKD) vs. very low calorie diet (VLCD): Surgical impact. Obes Surg (2019) 29(1):292–6. doi: 10.1007/s11695-018-3523-2
80. Schiavo L, De Stefano G, Persico F, Gargiulo S, Di Spirito F, Griguolo G, et al. A randomized, controlled trial comparing the impact of a low-calorie ketogenic vs a standard low-calorie diet on fat-free mass in patients receiving an elipse™ intragastric balloon treatment. Obes Surg (2021) 31(4):1514–23. doi: 10.1007/s11695-020-05133-8
81. Hussain TA, Mathew TC, Dashti AA, Asfar S, Al-Zaid N, Dashti HM. Effect of low-calorie versus low-carbohydrate ketogenic diet in type 2 diabetes. Nutrition (2012) 28(10):1016–21. doi: 10.1016/j.nut.2012.01.016
82. Goday A, Bellido D, Sajoux I, Crujeiras AB, Burguera B, García-Luna PP, et al. Short-term safety, tolerability and efficacy of a very low-calorie-ketogenic diet interventional weight loss program versus hypocaloric diet in patients with type 2 diabetes mellitus. Nutr Diabetes. (2016) 6(9):e230. doi: 10.1038/nutd.2016.36
83. Saslow LR, Daubenmier JJ, Moskowitz JT, Kim S, Murphy EJ, Phinney SD, et al. Twelve-month outcomes of a randomized trial of a moderate-carbohydrate versus very low-carbohydrate diet in overweight adults with type 2 diabetes mellitus or prediabetes. Nutr Diabetes. (2017) 7(12):304. doi: 10.1038/s41387-017-0006-9
84. Myette-Côté É, Durrer C, Neudorf H, Bammert TD, Botezelli JD, Johnson JD, et al. The effect of a short-term low-carbohydrate, high-fat diet with or without postmeal walks on glycemic control and inflammation in type 2 diabetes: a randomized trial. Am J Physiol Regul Integr Comp Physiol (2018) 315(6):R1210–9. doi: 10.1152/ajpregu.00240.2018
85. Global BMI Mortality Collaboration, Di Angelantonio E, ShN B, Wormser D, Gao P, Kaptoge S, et al. Body-mass index and all-cause mortality: individual-participant-data meta-analysis of 239 prospective studies in four continents. Lancet (2016) 388(10046):776–86. doi: 10.1016/S0140-6736(16)30175-1
86. Bray GA, Heisel WE, Afshin A, Jensen MD, Dietz WH, Long M, et al. The science of obesity management: an endocrine society scientific statement. Endocr Rev (2018) 39(2):79–132. doi: 10.1210/er.2017-00253
87. Kennedy AR, Pissios P, Otu H, Xue B, Asakura K, Furukawa N, et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am J Physiol Endocrinol Metab (2007) 292(6):E1724–39. doi: 10.1152/ajpendo.00717.2006
88. Yamazaki T, Okawa S, Takahashi M. The effects on weight loss and gene expression in adipose and hepatic tissues of very-low carbohydrate and low-fat isoenergetic diets in diet-induced obese mice. Nutr Metab (Lond). (2016) 13:78. doi: 10.1186/s12986-016-0139-1
89. Bielohuby M, Menhofer D, Kirchner H, Stoehr BJM, Müller TD, Stock P, et al. Induction of ketosis in rats fed low-carbohydrate, high-fat diets depends on the relative abundance of dietary fat and protein. Am J Physiol Endocrinol Metab (2011) 300(1):E65–76. doi: 10.1152/ajpendo.00478.2010
90. Garbow JR, Doherty JM, Schugar RC, Travers S, Weber ML, Wentz AE, et al. Hepatic steatosis, inflammation, and ER stress in mice maintained long term on a very low-carbohydrate ketogenic diet. Am J Physiol Gastrointest Liver Physiol (2011) 300(6):G956–67. doi: 10.1152/ajpgi.00539.2010
91. Badman MK, Kennedy AR, Adams AC, Pissios P, Maratos-Flier E. A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss. Am J Physiol Endocrinol Metab (2009) 297(5):E1197–204. doi: 10.1152/ajpendo.00357.2009
92. Okuda T, Morita N. A very low carbohydrate ketogenic diet prevents the progression of hepatic steatosis caused by hyperglycemia in a juvenile obese mouse model. Nutr Diabetes. (2012) 2(11):e50. doi: 10.1038/nutd.2012.24
93. Zhu H, Bi D, Zhang Y, Kong C, Du J, Wu X, et al. Ketogenic diet for human diseases: the underlying mechanisms and potential for clinical implementations. Signal Transduct Target Ther (2022) 7(1):11. doi: 10.1038/s41392-021-00831-w
94. Westman EC, Yancy WS Jr, Olsen MK, Dudley T, Guyton JR. Effect of a low-carbohydrate, ketogenic diet program compared to a low-fat diet on fasting lipoprotein subclasses. Int J Cardiol (2006) 110(2):212–6. doi: 10.1016/j.ijcard.2005.08.034
95. López-Espinoza MÁ, Chacón-Moscoso S, Sanduvete-Chaves S, Ortega-Maureira MJ, Barrientos-Bravo T. Effect of a ketogenic diet on the nutritional parameters of obese patients: A systematic review and meta-analysis. Nutrients (2021) 13(9):2946. doi: 10.3390/nu13092946
96. Yuan X, Wang J, Yang S, Gao M, Cao L, Li X, et al. Effect of the ketogenic diet on glycemic control, insulin resistance, and lipid metabolism in patients with T2DM: a systematic review and meta-analysis. Nutr Diabetes. (2020) 10(1):38. doi: 10.1038/s41387-020-00142-z
97. Evert AB, Dennison M, Gardner CD, Garvey WT, Lau KHK, MacLeod J, et al. Nutrition therapy for adults with diabetes or prediabetes: A consensus report. Diabetes Care (2019) 42(5):731–54. doi: 10.2337/dci19-0014
98. Tay J, Luscombe-Marsh ND, Thompson CH, Noakes M, Buckley JD, Wittert GA, et al. Comparison of low- and high-carbohydrate diets for type 2 diabetes management: a randomized trial. Am J Clin Nutr (2015) 102(4):780–90. doi: 10.3945/ajcn.115.112581
99. Al-Khalifa A, Mathew TC, Al-Zaid NS, Mathew E, Dashti HM. Therapeutic role of low-carbohydrate ketogenic diet in diabetes. Nutrition (2009) 25(11-12):1177–85. doi: 10.1016/j.nut.2009.04.004
100. Zhang Q, Xu L, Xia J, Wang D, Qian M, Ding S. Treatment of diabetic mice with a combination of ketogenic diet and aerobic exercise via modulations of PPARs gene programs. PPAR Res (2018) 2018:4827643. doi: 10.1155/2018/4827643
101. Poplawski MM, Mastaitis JW, Isoda F, Grosjean F, Zheng F, Mobbs CV. Reversal of diabetic nephropathy by a ketogenic diet. PloS One (2011) 6(4):e18604. doi: 10.1371/journal.pone.0018604
102. Yang Z, Mi J, Wang Y, Xue L, Liu J, Fan M, et al. Effects of low-carbohydrate diet and ketogenic diet on glucose and lipid metabolism in type 2 diabetic mice. Nutrition (2021) 89:111230. doi: 10.1016/j.nut.2021.111230
103. Guo Y, Zhang C, Shang FF, Luo M, You Y, Zhai Q, et al. Ketogenic diet ameliorates cardiac dysfunction via balancing mitochondrial dynamics and inhibiting apoptosis in type 2 diabetic mice. Aging Dis (2020) 11(2):229–40. doi: 10.14336/AD.2019.0510
104. Zhang X, Qin J, Zhao Y, Shi J, Lan R, Gan Y, et al. Long-term ketogenic diet contributes to glycemic control but promotes lipid accumulation and hepatic steatosis in type 2 diabetic mice. Nutr Res (2016) 36(4):349–58. doi: 10.1016/j.nutres.2015.12.002
105. Yancy WS Jr, Foy M, Chalecki AM, Vernon MC, Westman EC. A low-carbohydrate, ketogenic diet to treat type 2 diabetes. Nutr Metab (Lond). (2005) 2:34. doi: 10.1186/1743-7075-2-34
106. Feinman RD, Pogozelski WK, Astrup A, Bernstein RK, Fine EJ, Westman EC, et al. Dietary carbohydrate restriction as the first approach in diabetes management: critical review and evidence base. Nutrition (2015) 31(1):1–13. doi: 10.1016/j.nut.2014.06.011
107. Kalra S, Jain A, Ved J, Unnikrishnan AG. Sodium-glucose cotransporter 2 inhibition and health benefits: The robin hood effect. Indian J Endocrinol Metab (2016) 20(5):725–9. doi: 10.4103/2230-8210.183826
108. Kalra S, Gupta Y, Patil S. Sodium-glucose cotransporter-2 inhibition and the insulin: Glucagon ratio: Unexplored dimensions. Indian J Endocrinol Metab (2015) 19(3):426–9. doi: 10.4103/2230-8210.152793
109. Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, et al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest. (2014) 124(2):499–508. doi: 10.1172/JCI72227
110. Kalra S, Sahay R, Gupta Y. Sodium glucose transporter 2 (SGLT2) inhibition and ketogenesis. Indian J Endocrinol Metab (2015) 19(4):524–8. doi: 10.4103/2230-8210.157859
111. Ranjan A, Schmidt S, Damm-Frydenberg C, Holst JJ, Madsbad S, Nørgaard K. Short-term effects of a low carbohydrate diet on glycaemic variables and cardiovascular risk markers in patients with type 1 diabetes: A randomized open-label crossover trial. Diabetes Obes Metab (2017) 19(10):1479–84. doi: 10.1111/dom.12953
112. Leow ZZX, Guelfi KJ, Davis EA, Jones TW, Fournier PA. The glycaemic benefits of a very-low-carbohydrate ketogenic diet in adults with type 1 diabetes mellitus may be opposed by increased hypoglycaemia risk and dyslipidaemia. Diabetes Med (2018). doi: 10.1111/dme.13663
113. Charoensri S, Sothornwit J, Trirattanapikul A, Pongchaiyakul C. Ketogenic diet-induced diabetic ketoacidosis in a young adult with unrecognized type 1 diabetes. Case Rep Endocrinol (2021) 2021:6620832. doi: 10.1155/2021/6620832
114. Buehler LA, Noe D, Knapp S, Isaacs D, Pantalone KM. Ketogenic diets in the management of type 1 diabetes: Safe or safety concern? Cleve Clin J Med (2021) 88(10):547–55. doi: 10.3949/ccjm.88a.20121
115. American Diabetes Association. 5. facilitating behavior change and well-being to improve health outcomes: Standards of medical care in diabetes–2021. Diabetes Care (2021) 44(Supplement_1):S53–72. doi: 10.2337/dc21-S005
116. Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol (2006) 40 Suppl 1:S5–10. doi: 10.1097/01.mcg.0000168638.84840.ff
117. Godoy-Matos AF, Silva Júnior WS, Valerio CM. NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol Metab Syndr (2020) 12:60. doi: 10.1186/s13098-020-00570-y
118. Yamazaki T, Okawa S, Takahashi M. The effects on weight loss and gene expression in adipose and hepatic tissues of very-low carbohydrate and low-fat isoenergetic diets in diet-induced obese mice. Nutr Metab (Lond). (2016) 13:78. doi: 10.1186/s12986-016-0139-1
119. Jani S, Da Eira D, Stefanovic M, Ceddia RB. The ketogenic diet prevents steatosis and insulin resistance by reducing lipogenesis, diacylglycerol accumulation and protein kinase c activity in male rat liver. J Physiol (2022) 600(18):4137–51. doi: 10.1113/JP283552
120. Crabtree CD, Kackley ML, Buga A, Fell B, LaFountain RA, Hyde PN, et al. Comparison of ketogenic diets with and without ketone salts versus a low-fat diet: Liver fat responses in overweight adults. Nutrients (2021) 13(3):966. doi: 10.3390/nu13030966
121. Pugliese N, Plaz Torres MC, Petta S, Valenti L, Giannini EG, Aghemo A. Is there an 'ideal' diet for patients with NAFLD? Eur J Clin Invest (2022) 52(3):e13659. doi: 10.1111/eci.13659
122. Parra-Vargas M, Rodriguez-Echevarria R, Jimenez-Chillaron JC. Nutritional approaches for the management of nonalcoholic fatty liver disease: An evidence-based review. Nutrients (2020) 12(12):3860. doi: 10.3390/nu12123860
123. Paoli A, Mancin L, Giacona MC, Bianco A, Caprio M. Effects of a ketogenic diet in overweight women with polycystic ovary syndrome. J Transl Med (2020) 18(1):104. doi: 10.1186/s12967-020-02277-0
124. Canciones RI, Losavio F, Ciolli F, Valenzano A, Cibelli G, Messina G, et al. Effects of mixed of a ketogenic diet in overweight and obese women with polycystic ovary syndrome. Int J Environ Res Public Health (2021) 18(23):12490. doi: 10.3390/ijerph182312490
125. Mavropoulos JC, Yancy WS, Hepburn J, Westman EC. The effects of a low-carbohydrate, ketogenic diet on the polycystic ovary syndrome: a pilot study. Nutr Metab (Lond). (2005) 2:35. doi: 10.1186/1743-7075-2-35
126. Li J, Bai WP, Jiang B, Bai LR, Gu B, Yan SX, et al. Ketogenic diet in women with polycystic ovary syndrome and liver dysfunction who are obese: A randomized, open-label, parallel-group, controlled pilot trial. J Obstet Gynaecol Res (2021) 47(3):1145–52. doi: 10.1111/jog.14650
Keywords: ketogenic diet, ketone bodies, GPCR (G protein coupled receptors), metabolic disorder, very low carbohydrate ketogenic diet
Citation: Spigoni V, Cinquegrani G, Iannozzi NT, Frigeri G, Maggiolo G, Maggi M, Parello V and Dei Cas A (2022) Activation of G protein-coupled receptors by ketone bodies: Clinical implication of the ketogenic diet in metabolic disorders. Front. Endocrinol. 13:972890. doi: 10.3389/fendo.2022.972890
Received: 19 June 2022; Accepted: 03 October 2022;
Published: 20 October 2022.
Edited by:
Aylin Carla Hanyaloglu, Imperial College London, United KingdomReviewed by:
Ruth Gutierrez-Aguilar, División de Investigación, Universidad Nacional Autónoma de México, MexicoCopyright © 2022 Spigoni, Cinquegrani, Iannozzi, Frigeri, Maggiolo, Maggi, Parello and Dei Cas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Dei Cas, YWxlc3NhbmRyYS5kZWljYXNAdW5pcHIuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.