
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 12 October 2022
Sec. Cellular Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.971745
This article is part of the Research TopicIntra and Inter Organ Cross-Talk and Cellular CommunicationView all 6 articles
 Emmanuel Somm1,2,3*
Emmanuel Somm1,2,3* François R. Jornayvaz1,2,3*
François R. Jornayvaz1,2,3*Interleukin-18 (IL-18) is a classical member of the IL-1 superfamily of cytokines. As IL-1β, IL-18 precursor is processed by inflammasome/caspase-1 into a mature and biologically active form. IL-18 binds to its specific receptor composed of two chains (IL-18Rα and IL-18Rβ) to trigger a similar intracellular signaling pathway as IL-1, ultimately leading to activation of NF-κB and inflammatory processes. Independently of this IL-1-like signaling, IL-18 also specifically induces IFN-γ production, driving the Th1 immune response. In circulation, IL-18 binds to the IL-18 binding protein (IL-18BP) with high affinity, letting only a small fraction of free IL-18 able to trigger receptor-mediated signaling. In contrast to other IL-1 family members, IL-18 is produced constitutively by different cell types, suggesting implications in normal physiology. If the roles of IL-18 in inflammatory processes and infectious diseases are well described, recent experimental studies in mice have highlighted the action of IL-18 signaling in the control of energy homeostasis, pancreatic islet immunity and liver integrity during nutritional stress. At the same time, clinical observations implicate IL-18 in various metabolic diseases including obesity, type 1 and 2 diabetes and nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH). In the present review, we summarize and discuss both the physiological actions of IL-18 in metabolism and its potential roles in pathophysiological mechanisms leading to the most common human metabolic disorders, such as obesity, diabetes and NAFLD/NASH.
Interleukin-18 (IL-18) is a member of the IL-1 superfamily of cytokines structurally similar to IL-1beta (IL-1β) (1, 2). Originally, Kupffer cell (liver-resident macrophage) was described as the main source of IL-18 (3). Nevertheless, many other cell types, including both hematopoietic cells and non-hematopoietic cells (such as intestinal epithelial cells, keratinocytes, endothelial cells) have also the potential to produce IL-18 in the basal state or under stimulation (3). IL-18 is regulated at the transcriptional, post-transcriptional and post-translational levels. The IL18 gene, which contains 7 exons, is located on chromosome 11 in humans and chromosome 9 in mice (4). The human IL18 promoter presents several single nucleotide polymorphisms (SNPs) at the 5′-end impacting gene transcription (5). IL18 gene expression could also be regulated by miRNAs (6). IL-18 is characterized by a specific mechanism of cellular production (partly shared with IL-1β). In contrast to cytokines expressed and subsequently secreted thank to their signal peptide, IL-18 is stored in the cytosol of its producing cells as a biologically inactive precursor (pro-IL-18) (4, 7). Pro-IL-18 requires post-translational processing to become biologically active and released extracellularly, meaning that constitutive or stimulated expression of IL-18 does not necessarily imply its secretion. Caspase-1 (initially named IL-1β-converting enzyme) is the major IL-18-processing enzyme, acting in large cytoplasmic multiprotein complexes named inflammasomes (8, 9). Inflammasomes are activated by specific stimuli and are composed of a sensor molecule, an adaptor protein, and an effector molecule responsible for precursor cleavage. Of note, caspase-1 requires autolysis to be cleaved into active caspase-1 which converts pro-IL-18 into mature IL-18. In addition, active caspase-1 cleaves gasdermin D and liberates a pore-forming domain allowing the release of mature IL-18 (10). The nucleotide-binding oligomerization domain leucine rich repeat and pyrin domain containing 3 (NLRP3) inflammasome was originally demonstrated to be a cytoplasmic platform necessary for caspase-1 activation (11). In some cells (such as blood-derived macrophages), NLRP3 inflammasome activation requires two signals: priming (generally mediated by Toll-like receptor [TLR]) and activation (triggered by various cellular responses including endogenous or exogenous particulates/microcrystals/metabolites/mitochondrial oxidized DNA). Bacterial or viral components are also sensed by inflammasome subunits, promoting inflammasome assembly and caspase-1-mediated IL-18 secretion (12). In Kupffer cells, LPS can trigger IL-18 secretion without priming (13). IL-18 maturation can also occur through the activation of other inflammasomes, such as NLRP6 or NLRP9. Other proteases, including caspase-8 (an apoptosis-initiating protease), proteinase 3, chymase or granzyme B are also involved in conversion of pro-IL-18 into mature IL-18 independently of NLRC4, NLRP3, or caspase-1 (3, 14). IL-18 signals through the IL-18 receptor (IL-18R), belonging to the IL-1R family. IL-18R is composed of the IL-18Rα chain (IL-18R1/IL-1Rrp) and the IL-18Rβ chain (IL-18R accessory protein/IL-1RAcPL) (4). Binding of IL-18 to IL-18Rα and IL-18Rβ chains forms a trimer. The intracellular region of IL-18R contains a TIR domain (analog to TLR) that binds MyD88 and initiates a signal transmission into the cell (15). MyD88 recruits IRAK1 and IRAK4 (16, 17), followed by binding to TRAF6, degradation of inhibitor of κB (IκB), phosphorylation and nuclear translocation of p65/p50 NF-κB (18). Other kinases, including the Mitogen-Activated Protein Kinase (MAPK) cascade of Extracellular Signal-regulated Kinase (ERK), c-jun N-terminal kinase (JNK), and p38 are also activated and implicated in IL-18 signaling (19). Together, these signals induce IFN-γ production or cell proliferation. IL-18 also induces the phosphorylation and activation of phosphatidylinositol-3 kinase (PI3K)/Akt/S6 and mammalian target of rapamycin (mTOR) (20). This signaling enhances the proliferation and survival of Natural Killer (NK) cells. IL-18R expression is stimulated by IL-12, IFN-α or Signal Transducer and Activator of Transcription (STAT)4 in T cells and NK cells (3). IL-18R is also expressed in non-immune cells (epithelial cells, nerve cells, etc…) in which IL-18 signaling is involved in cell survival and differentiation. Surprisingly, IL-18Rα also binds to IL-37 (IL-1F7), preventing IL-18 signaling. IL-18Rα/IL-37 complex binds to IL-1R8 (TIR8/SIGIRR), promoting an anti-inflammatory effect through STAT3 (21). In circulation, IL-18 binds to the IL-18 binding protein (IL-18BP) with high affinity (higher affinity than IL-18R), leaving only a small fraction of free IL-18 able to trigger receptor-mediated signaling. In consequence, IL-18BP can be considered as a negative regulator of the IL-18 signaling pathway (22, 23). Immunologic functions of IL-18 are pleiotropic and include stimulation of IFN-γ production, activation of basophils and mast cells, allergic inflammation, defense against extracellular pathogens, helminth infection and intracellular pathogens (bacteria, protozoan, virus) (3).
If the role of IL-18 in inflammatory and infectious diseases is well established, recent experimental studies in mice have involved IL-18 signaling in the control of energy homeostasis, pancreatic islet immunity and liver integrity during nutritional stress. At the same time, clinical observations have implicated IL-18 in various metabolic diseases including obesity, type 1 (T1D) and type 2 (T2D) diabetes, and nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH). In the present review, we summarize and discuss the physiological roles of IL-18 in metabolism and its potential involvement in pathophysiological mechanisms leading to the most common human metabolic disorders, such as obesity, diabetes, and NAFLD/NASH.
Due to behavioral, environmental, and sometimes genetic factors, the prevalence of obesity has risen to unacceptable levels worldwide (24). Obesity is associated with a shortened life span, predisposing to T2D, cardiovascular diseases, liver disorders, and cancers among others (24). In this pathophysiological context, a huge research effort has been brought to the study of cytokines (including IL-18) produced by the adipose tissue and upregulated during obesity. Beyond inflammation, these ‘‘adipokines’’ are also involved in the crosstalk between metabolic organs, acting on several physiological processes (such as energy homeostasis or glucose/fat metabolism) involved in obesity onset (25).
First evidence for a role of IL-18 in the regulation of body weight and fat mass comes from the phenotyping of mice globally deficient in IL-18. IL-18-/- mice displayed primary hyperphagia leading to obesity and insulin resistance in the liver, adipose tissue, and muscle (26). Similar hyperphagia was observed in mice deficient in IL-18R and in mice overexpressing IL-18BP, while central administration of recombinant IL-18 (rIL-18) inhibited food intake and reversed hyperglycemia in IL-18-/- mice (26). Both central and peripheral administration of IL-18 suppressed appetite and weight regain in food-deprived mice (27). In animal models of binge eating, a down-regulation of the IL-18/IL-18R system (but increased expression of IL-18BP) was specifically observed in the preoptic and anterior-tuberal region of the hypothalamus (28). In contrast, food restricted animals exhibited increased IL-18 expression (28). In humans, plasma IL-18 levels were significantly decreased in patients with anorexia nervosa and circulating IL-18 levels correlated to body mass index (BMI) in controls, but not in anorexic patients (29). Additional studies have attempted to elucidate the mechanisms of IL-18 action on food intake in the central nervous system (CNS).
Firstly, investigations concerning the distribution of IL-18Rα in the mouse brain allowed exploring the exact sites of central IL-18 action. In situ hybridization combined with immunohistochemistry revealed that IL-18Rα is expressed in neuronal cell bodies, as well as on their dendrites, throughout the brain, with particularly high levels in regions involved in metabolic control such as the hypothalamus and the thalamus but also in other regions of the limbic system such as the hippocampus, the amygdala, and the cerebral cortex (30).
Secondly, cellular mechanisms underlying the anorexigenic effects of IL-18 have also been deeply investigated. A high expression of both subunits of the IL-18R was detected in the bed nucleus of the stria terminalis (BST), a region of extended amygdala known to influence feeding via its projections to the lateral hypothalamus (LH) (31). Local injection of rIL-18 in this area significantly reduced c-fos activation and food intake (31). In BST brain slices, IL-18 reduced the excitatory input on neurons through a presynaptic mechanism (31). This effect was cell-specific, only observed in Type III GABAergic neurons located in the juxtacapsular BST. In consequence, IL-18 increases the firing of glutamatergic LH neurons through a mechanism of disinhibition (31).
In addition, to hyperphagia, adult IL-18-/- mice gained 2- to 3-fold more weight than wild-type mice per unit of food consumed (low- or high-fat diet) (27). This suggests that in addition to its anorexic action, IL-18 limits food efficiency. Accordingly, indirect calorimetry revealed a reduced energy expenditure in IL-18-/- mice, in association with an increased respiratory exchange ratio (RER), suggesting a preferential oxidation of carbohydrate at the expense of fat (27). The reduction in energy expenditure of IL-18-/- mice was seen across fasting/feeding conditions, low/high-fat diets, low/high levels of physical activity and times of day, suggesting an underlying action of IL-18 on basal metabolic rate (32). The circadian amplitude of energy expenditure, but not those of RER, food intake, or motor activity, was also blunted in IL-18-/-mice (32). In accordance, hepatic gene expression of circadian regulators [such as circadian locomotor output cycles kaput (CLOCK), brain and muscle Arnt-like protein (BMAL1), and period circadian clock 2 (PER2)] was also altered in IL-18-/- mice (33).
Taken together, these data demonstrate that endogenous IL-18 not only suppresses appetite but also promotes energy expenditure and lipid oxidation (as illustrated in Figure 1).
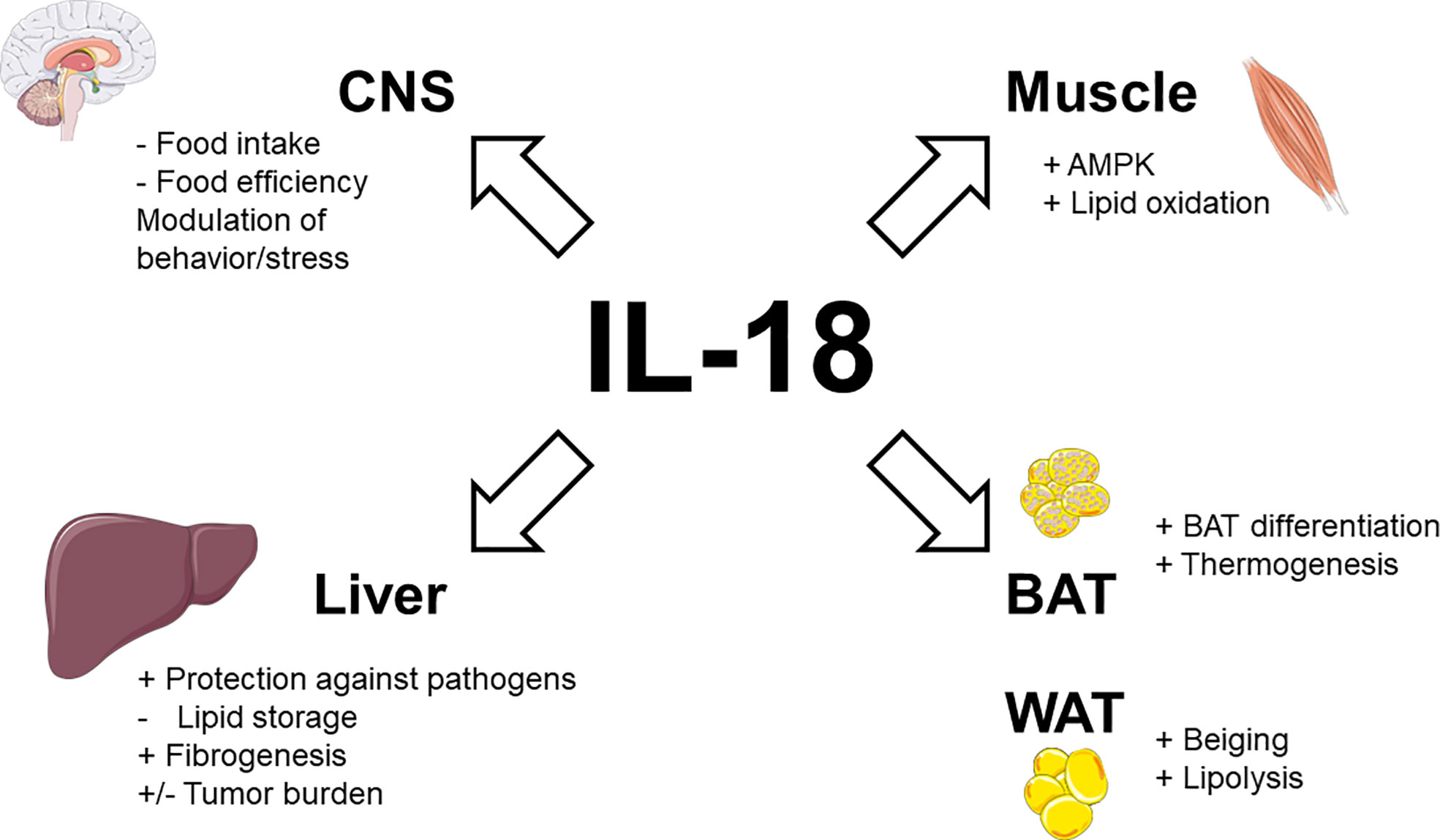
Figure 1 Physiological actions of IL-18 signaling in metabolic tissues. Stimulatory effects are indicated by plus signs (+), inhibitory effects by minus signs (−). Figure produced using illustrations from Servier Medical Art (smart.servier.com) under Creative Commons Attribution 3.0 unported license.
Outside the metabolic field, IL-18-/- mice also exhibited a reduction in depressive-like behavior and a decreased expression of neuroendocrine genes in the amygdala (34). In humans, association between polymorphisms in the IL-18 gene (resulting in higher IL-18 production) and depressive behavior have been reported (35, 36). In addition, other observations also involved IL-18 in stress responses and the hypothalamic-pituitary axis (37, 38–43).
In addition to its role in the inhibition of food intake, some evidence has implicated peripherally-produced and -acting IL-18 in the regulation of body weight homeostasis and fat mass accretion.
In vitro, brown adipocyte precursors isolated from IL-18-/- mice showed an impaired differentiation featured by increased lipid accumulation and decreased gene expression of type 2 iodothyronine deiodinase when compared to those of control mice (44). Similar findings were observed in vivo, since the BAT of IL-18-/- mice spontaneously accumulated fat and overexpressed Apoc3 and Insig1 (44). Treatment with rIL-18 reduced the size and number of fat droplets in BAT of IL-18-/- mice (44). The surrounding perivascular adipose tissue (PVAT) of IL-18-/- mice exhibited a conversion from brown adipose tissue-like features to white adipose tissue-like features, impacting the aorta physiology (45). In line with these observations, IL-18-/- mice failed to develop diet-induced thermogenesis as shown by non-induction of uncoupling protein-1 (UCP-1) in BAT and inguinal white adipose tissue (iWAT) (46). This defect could result from non-activation of some IL-18R expressing subpopulations of group 2 innate lymphoid cells (ILC2s) (47), an immune cell type known to promote adipocyte beiging (48). Surprisingly, IL-18R-/- mice were overweighted on standard chow diet but appeared to resist to high-fat diet (HFD)-induced obesity or cold exposure-induced hypothermia (46), suggesting the absence of BAT dysfunction. This discrepancy between IL-18-/- and IL-18R-/- mice suggests that a more complex signaling system than IL-18/IL-18R interaction could be involved in the adipose thermogenic action of IL-18 (46). In this way, the anti-inflammatory IL-37 which binds IL-18Rα/IL-1R8 heterodimer has not yet been identified in mice (49).
IL-18 is produced in WAT. In mice and humans, obesity was associated with increased IL-18 levels in WAT, which contribute to systemic concentrations (50–55). Expression of the IL-18R and the IL-18BP was also observed in human WAT, mirroring that of IL-18 (56). IL-18 gene expression was evident in human subcutaneous and visceral adipose tissues (56), but a higher secretion levels of IL-18 was observed in explant from visceral WAT, possibly due to increased inflammasome and caspase-1 activation in this intraperitoneal depot (57). The cellular source of IL-18 in WAT was a matter of debate since several studies reported that expression of IL-18 occurs in both mature adipocytes and the stromal vascular fraction (56), while other stated that most of the IL-18 released by explants of human adipose tissue are derived from the non-fat cells, not from the adipocytes (58). In this way, IL-18 expression is similar in mesothelial cells and the stromal vascular fraction (59). Other observations showed that IL-18 was immunolocalized in WAT neutrophils and mast cells, but not in macrophages or in adipocytes (59). In humans, IL-18 expression was increased in adipose tissue from HIV-associated lipodystrophic patients (60) and a haplotype associated with lower IL-18 levels was associated with a higher body mass index (61). Weight loss after bariatric surgery (62–64), or after lifestyle changes (53, 65), resulted in decreased IL-18 concentrations in the blood.
Paracrine effects of IL-18 in WAT could contribute to regulation of adiposity. Through an activating mutation in the inflammasome NLRP1, it has been shown that IL-18 triggers lipolysis in WAT (66). Interestingly, this action could explain susceptibility to obesity/diabetes of C57BL/6 mice compared to the healthy metabolic status of Balb/c mice. In fact, while C57BL/6 mice harbor the NLRP1b2 allelic inflammasome variant, Balb/c mice harbor the NLRP1b1 inflammasome which produces more important IL-18 levels, resulting in higher lipolysis, reduced WAT inflammation and improved insulin sensitivity (67). Mechanistically, IL-18 promoted adipose hormone-sensitive lipase (HSL) phosphorylation and activation (67). IL-18 also enhanced insulin-mediated glucose uptake in adipocytes and could counteract the suppression of glucose uptake caused by Tumor Necrosis Factor (TNF)-α in 3T3-L1 adipocytes (68). Underlying mechanisms involved phosphorylation of protein kinase B (Akt) and downregulation of phosphorylated p38 MAPK (68).
Obesity-related upregulation of IL-18, which experimentally limits food intake and food efficiency while promoting energy expenditure and fat oxidation could appear surprising. However, as for leptin, obesity could reflect a state of IL-18 resistance. In this way, it is interesting to note that phosphorylation and then activation of STAT3 is triggered both following IL-18 signaling (27) and leptin signaling (69, 70), suggesting a crosstalk/convergence of these signaling pathways. In accordance with this perspective, monocytes from obese patients (leptin resistant) were also desensitized to IL-18 (71). The stimulatory activity of IL-18 signaling on brown adipocyte differentiation and thermogenesis, as well as on white adipocyte transdifferentiation (into brown-like adipocyte-beiging) and lipolysis are summarized in Figure 1.
Immunohistochemistry demonstrated that IL-18 is solely expressed by type II (fast-twitch) fibers in different human skeletal muscles (72). This suggests that the level of daily muscle activity does not influence basal IL-18 expression, which is rather implicated in normal physiology (72). A basic study in mice deficient in IL-18 signaling confirmed that the action of IL-18 in skeletal muscle could contribute to its anti-obesity effect. In addition to increased weight gain, IL-18R-/- mice display ectopic lipid deposition, inflammation, and reduced AMP-activated protein kinase (AMPK) signaling in skeletal muscle (73). Conversely, electroporation of IL-18 into normal skeletal muscle activated AMPK and concomitantly inhibited HFD-induced weight gain (73). In vitro, treatment of myotubes and skeletal muscle strips with IL-18 also activated AMPK and increased fat oxidation (73). Patients with human immunodeficiency virus (HIV)-lipodystrophy had lower levels of muscular IL-18 and IL-18R gene expression (74). These low levels of IL-18 correlated to high muscular levels of deleterious lipid species as ceramides and sphingosine-1P and increased levels of triglycerides in circulation (74). The combined activations of AMPK and lipid oxidation by IL-18 signaling in skeletal muscle are shown in Figure 1.
Type 1 diabetes mellitus (T1D) is an endocrine disorder in which pancreatic β-cells stop producing insulin, typically due to autoimmune destruction (75). Incidence peaks in puberty/early adulthood, but onset can occur at any age (75). Type 2 diabetes mellitus (T2D) is one of the most common illnesses encountered by internists (76). Featured by insulin resistance, T2D is increasing worldwide due to populations aging and obesity pandemic (76). Although diabetes care is improving by many measures, complications are still common, including visual loss, amputation, atherosclerotic disorders or end-stage renal disease (76). Management of both T1D and T2D should focus on optimizing glucose control to reduce acute consequences (such as diabetic ketoacidosis or hyperosmolarity) and long-term complications (including microvascular and macrovascular diseases) (75). Experimental and clinical evidence has involved IL-18 in the onset/progression of both T1D and T2D.
Innate immunity contributes to the induction and amplification of the immune response inducing β-cells loss in T1D (77). In addition, a crosstalk between immune cells and stressed β-cells is mediated by cytokines and others immunogenic signals delivered by stressed β-cells (77). IL-18 has been detected in rodent pancreatic β-cells (78, 79), exhibiting only a minor stimulatory effect on insulin secretion (80). If a protective role has been early conferred to IL-18 in diabetes onset in Non Obese Diabetic (NOD) mice [in link with impaired progression from Th2- to Th1-dependent insulitis (81)], further works have globally attributed a deleterious role of IL-18 in T1D. IL-18 was detected in NOD mouse pancreatic islets during early stages of insulitis (82), mediating islet injury (83). In response to the alkylating agent cyclophosphamide, macrophages from NOD mice presented an increase in IL-18 gene expression closely associated with diabetes development, while macrophages from Balb/c mice did not (82, 84). IL-18 has been implicated in the expansion of islet-destructive T-cells during pre-diabetes. In fact, IL-18 expanded pathogenic T-cells in the periphery of NOD mice (85), while IL-18-/- mice exhibited a reduced T-cell turnover, an increased prevalence of naïve/quiescent T-cells and less effector T-cells, resulting in disease protection (85). In addition, islet-reactive T-cells failed to become activated and expanded in the lymphoid organs of IL-18-/- mice (85). Systemic administration of IL-18 also promoted diabetes development in young NOD mice (86), while endogenous IL-18 was required to observe the full diabetogenic effect of streptozotocin in C57BL/6 mice (87, 88).
Clinical evidence also confirmed a role for IL-18 in T1D. From a genetic perspective, the genomic loci idd2, associated with T1D, maps in close proximity to the IL-18 gene (82). Positive association between T1D and polymorphisms in the promoter of IL-18 gene (leading to increased IL-18 gene expression) were found in some studies (89, 90), but not in others (91–93). IL-18 serum levels and IL-18 protein expression in pancreatic islets were increased selectively in T1D patients (94–96). IL-18 was also detected in islets infected with enterovirus in pancreas of patients presenting a fulminant T1D (97).
Several properties of IL-18 previously described, including stimulation of insulin-mediated glucose uptake (68), activation of AMPK (73) or phosphorylation of STAT3 suggest a beneficial role for IL-18 in glucose homeostasis. Nevertheless, numerous clinical studies have described an upregulation of IL-18 circulating levels in T2D. Notably, patients with prediabetes have higher levels of IL-18 compared to obese normoglycemic controls (98). Circulating IL-18 was increased in patients with T2D (99–101), independently of a generalized pro-inflammatory state (102). This association was independent of usual risk factors, including BMI and other adipokines levels (103, 104). In addition, circulating IL-18 levels were positively correlated with the Homeostasis Model Assessment of Insulin Resistance (HOMA-IR) index (105) and glucose intolerance, independently of BMI or age (104, 106, 107). Conversely, a decrease in IL-18 was an independent factor for the improvement of β-cell function in T2D (108). A polymorphism of the IL-18 gene associated with increased circulating levels of IL-18 has been linked to impaired insulin sensitivity (109). Similarly to insulin, high blood IL-18 levels observed in T2D could reflect a state of IL-18 resistance.
Obesity and T2D are frequent causes of NAFLD, the most common hepatic disease in industrialized countries (110, 111). A significant proportion of patients with NAFLD develop a state of hepatic inflammation (NASH), which can lead to fibrosis and cirrhosis, potentially resulting in hepatocellular carcinoma (HCC) (112). Whilst changes in eating habits, weight loss or physical activity have beneficial effects on liver steatosis, no efficient pharmacologic treatment are available to limit the progression of NAFLD, NASH and fibrosis (113). Some clinical observations as well as basic studies in rodents suggested a role for IL-18 in the onset/progression of NAFLD/NASH. The next sections will summarize how IL-18 can directly or indirectly act on the liver status before reviewing its implication in hepatic carcinogenesis.
IL-18 is mostly produced by macrophages in the liver (114, 115), mediating hepatic defense against bacteria, parasites, virus, and drug-induced injuries (116–119). Ability of IL-18 to activate NK cells, inducing apoptosis of infected/damaged hepatocytes through the Fas ligand (FasL) pathway (115, 120), is central in this context. Nevertheless, IL-18 signaling required a fine-tuning. Uncontrolled IL-18 signaling due to loss-of-function in IL-18BP could result in massive death of human hepatocytes in vitro and in patients (115). Metabolically, toxic lipids activate the NLRP3 inflammasome and IL-18 production in different NAFLD/NASH murine models (121–123). Several experimental evidence suggest that IL-18 is involved in both steatotic and fibrotic processes.
Observations in rodents suggest that IL-18 could suppress hepatic lipid deposition. NLRP1-/- mice (with low IL-18 levels) spontaneously developed hepatic steatosis, a situation aggravated on HFD (66). In contrast, mice with constitutively activated NLRP1 (with high IL-18 levels) were devoid of lipid vacuoles in the liver, and depletion in IL-18 reversed this protective effect (66, 124). Obese C57BL/6 mice (naturally harboring the NLRP1b2 allele-resulting in low IL-18 levels) have increased hepatic steatosis when compared to obese NLRP1b1 transgenic C57BL/6 mice or Balb/c mice (naturally harboring the NLRP1b1 allele-resulting in high IL-18 levels) (67). High levels of IL-18 also mediated the reduction in hepatic steatosis observed in mice with a conditional deficiency in Src homology-2 domain-containing protein tyrosine phosphatase-2 (SHP2) in macrophages (125). In accordance, administration of exogenous IL-18 counteracted steatohepatitis in mice on HFD (66) while IL-18-/- mice exhibited hepatic steatosis, insulin resistance, increased expression of gluconeogenic genes and defective phosphorylation of STAT3 (26). Several explanations have been argued to explain this NAFLD/NASH phenotype of IL-18-/- mice. The initial study suggested that primary hyperphagia and resulting obesity were the cause of steatosis in IL-18-/- mice (26). Later work showed that IL-18-/- mice developed hypercholesterolemia and hypertriglyceridemia before the manifestation of obesity (33) suggesting a primary effect of IL-18 on the liver. In accordance, hepatic transcriptional changes were observed in the liver of IL-18-/- mice before obesity onset (33). Finally, a role for altered gut microbiota composition has been advanced to explain steatosis in IL-18-/- mice (126). Hepatic insulin-resistance resulting from IL-18 deficiency seems to be sex-hormone-dependent (127). In addition, IL-18R-/- mice (but not IL-1R-/- mice) were protected from precocious dietary liver damage, possibly due to silencing of early pro-inflammatory genes initiating NASH (128).
Other basic studies suggest that IL-18 could enhance hepatic fibrosis. In fact, IL-18 induce multiple functional changes in Hepatic Stellate Cells (HSCs), the resident perisinusoidal cells orchestrating the deposition of extracellular matrix (ECM) in normal and fibrotic liver (129). In vitro, monosodium urate-induced inflammasome activation led to overexpression of TGF-β and collagen 1 in primary mouse HSC and HSC line (130). In vivo, NLRP3-/- mice had reduced chemically-induced liver fibrosis (130) while conditional NLRP3 knock-in mice expressing an hyperactive NLRP3 present HSC activation with increased collagen deposition in the liver (131). These changes were only partially attenuated by treatment with an interleukin-1 receptor antagonist suggesting that beyond IL-1β, IL-18 is also involved in fibrogenesis (131). In this way, adoptive transfer of CD4+ T cells from IL-18 transgenic mice (but not from wild-type littermates) in SCID mice resulted in massive periportal fibrosis (132). The direct consequences of IL-18 signaling on liver physiology, including protection against pathogens, inhibition of lipid storage and stimulation of fibrogenesis are detailed in Figure 1.
Clinically, circulating IL-18 levels were associated with increased liver injury markers and portal fibrosis in obese subjects with NAFLD (133), as well as with plasma concentrations of liver injury markers in healthy subjects (134). Patients with NAFLD had significantly higher IL-18 and IL-18/IL-18BP ratio compared with healthy controls (135). Plasma levels of IL-18 and IL-18BP were elevated in chronic liver diseases such as cirrhosis, correlating with inflammation, liver injury and severity of the disease (136). However, IL-18BP levels may not be sufficient to counteract the overwhelming pro-inflammatory response in end stage liver disease (136). Genetically, IL-18 variants, resulting in higher IL-18 levels, were significantly associated with chronic liver diseases (including cirrhosis) in the overall population (137).
Taken together, these results suggest that a physiological/limited amount of IL-18 exhibits interesting anti-steatotic properties while IL-18 excess could be deleterious for liver integrity.
The intestinal mucosal and portal vein enables transport of gut-derived products directly to the liver and in turn, the liver secretes bile and other compounds in the intestine. This direct interaction, concomitantly allowing nutrients to directly reach the liver and limiting the dissemination of microbes and toxins to the systemic circulation is called the ‘‘gut-liver axis’’ (138). Dysbiosis and gut leakiness play a critical role in the development of NAFLD/NASH (139, 140). IL-18 controls the gut-liver axis at multiple interconnected levels, including the maintenance of intestinal epithelial barrier, the production of intestinal mucus and the production of intestinal Anti-Microbial Peptide (AMP), all impacting gut microbiota composition.
Several evidence confer a deleterious role to IL-18 in gut epithelium integrity. Systemic administration of IL-12 and IL-18 to wild-type mice induced intestinal mucosal inflammation (141) while administration of IL-18BP reduced intestinal inflammation and ulceration (142). In addition, IL-18 overproduction in the mucosa exacerbated infiltration of macrophages and colitis (143) in mouse models of gut inflammation. In line, mice with a genetic deletion of IL-18 or its receptor IL-18R1 in intestinal epithelial cells were protected from chemically-induced mucosal damage (144). In humans, IL-18 is produced by gut epithelial cells and macrophages and this production was increased during inflammatory bowel diseases (145–148). Nevertheless, other studies have suggested a protective role for physiologic amount of IL-18 on intestinal epithelium, in particular through its crosstalk with IL-22. In fact, IL-18 could increase the ratio of IL-22/IL-22BP, which exerts protective properties during the peak of gut epithelial damage (149). In turn, a study in ileum organoids showed that IL-22 transcriptionally activates epithelial IL-18 (150). In colitis mouse models, inflammasome activation led to an increase in both IL-18 production and mucosal barrier integrity, resulting in a decreased hepatic bacterial load (151). Epithelium-derived IL-18 seems to contribute to epithelial proliferation through induction of stem cell genes (150). In this way, it has been suggested that IL-18 polymorphisms known to reduce IL-18 mRNA and protein levels may be involved in the susceptibility to Crohn’s disease (152).
Excessive IL-18 signaling through genetic deletion of IL-18BP resulted in loss of mature mucus-producing goblet cells associated with colitis (144). Goblet cells defect observed in IL-18BP-/- mice was rescued by concomitant deficiency for IL-18R1 in intestinal epithelial cells, demonstrating the autocrine/paracrine deleterious action of uncontrolled IL-18 production (144). Mechanistically, it seems that IL-18 excess inhibited the transcriptional program of goblet cells development (144). However, it has been recently observed that IL-18 could stimulate mucin secretion from goblet cells during Escherichia coli infection (150).
RNA-sequencing of colon from IL-18-/- mice, as well as administration of IL-18 to germ-free colon explants or mice, revealed that non-hematopoietic IL-18 induced AMP, in particular intelectin 1 (ITLN1), resistin-like molecule β/FIZZ2 (RELMβ) and angiogenin-4 (ANG4) in a NF-κB-dependent manner (151). This was confirmed by another study showing that IL-18 induces both Paneth cell-related AMP (ITLN1, ANG4) and Paneth cell-specific AMP (lysozyme, cryptdin) in a STAT-dependent manner (150). In contrast, IL-18-/- mice exhibited reduced mRNA levels of AMP and lysozyme-containing Paneth cells (150). Defect in IL-18 and AMPs due to intestinal NLRP6 inflammasome-deficiency resulted in a specific gut microbiota (151). Similarly, mice deficient in the AIM2 inflammasome had few colonic levels of IL-18, low expression of AMP, and were highly susceptible to colitis and microbiota dysbiosis (in particular Escherichia coli enrichment) (153). Recently, confocal microscopy studies revealed that intestinal neurons produce IL-18 (154). Deletion of IL-18 from the enteric neurons, but not from immune or epithelial cells, made mice susceptible to invasive Salmonella infection (154). Mechanistic approaches showed that enteric neuronal IL-18 was specifically required for homeostatic goblet cells AMP production (154). If IL-18 can modulate AMPs and impact gut microbiota, the reciprocal action is also true. In fact, both germ free mice and wild-type mice transplanted with IL-18-/- mice microbiota exhibited a suppression of colonic IL-18 levels (151). Metabolomic screening revealed that microbiota-derived taurine enhanced (while histamine and spermine suppressed) NLRP6 inflammasome-induced IL-18 secretion (151).
Several studies have shown that IL-18 modulates gut microbiota composition. NLRP1 and downstream IL-18 reduced the amount of beneficial butyrate-producing Clostridiales in the gut, aggravating experimentally-induced colitis (155). In NLRP3-/- mice, potentially pathogenic members of Enterobacteriaceae (including Citrobacter, Proteus or Shigella) were over-represented in the gut microbiota (156). Microbiota from NLRP6-/- and IL-18-/- mice on methionine-choline deficient (MCD) diet was characterized by increased proportion of Prevotellaceae and TM7 phylum (126). A significantly increased levels of Akkermansia muciniphila (a bacterial strain with ability to degrade mucus) has also been observed in IL-18-/- mice (157). This central role of IL-18 in microbiota maintenance impacts, in consequence, liver metabolism. In fact, increased proportion of Prevotellaceae and TM7 phylum in the microbiota from IL-18-/- mice led to an exacerbated influx of TLR4 and TLR9 agonists into the portal circulation, leading to hepatic overexpression of TNF-α that drives NASH progression (126).
The physiological functions of epithelial/neuronal-derived IL-18 in the gut/microbiota interface, including its roles in the control of intestinal barrier integrity and the production of mucus and AMPs (all required to maintain a normal microbiota in the digestive tract) are illustrated in the upper panel of Figure 2. In contrast, the deleterious action of immune cells-derived IL-18 (consecutive to gut microbial invasion) leading to altered gut microbiota composition, increased TLR agonists in the portal circulation and NASH progression is shown in the lower panel of Figure 2.
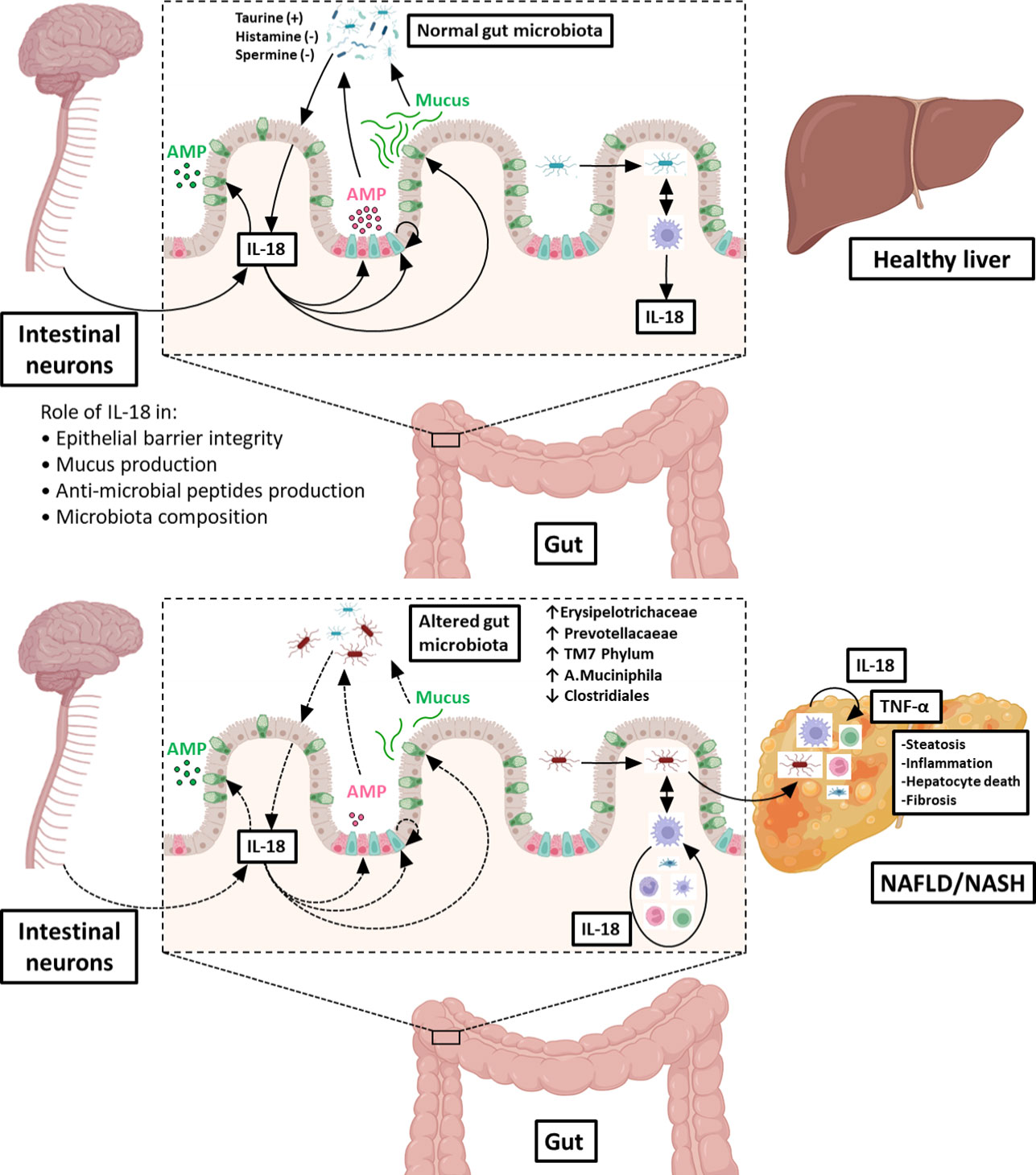
Figure 2 Role of IL‐18 in the gut microbiota balance and consequences on the gut-liver axis. Under physiological conditions (upper illustration), IL‐18 is produced by intestinal epithelial cells (in light pink) and is regulated by microbiota-derived metabolites (such as taurine, histamine or spermine). IL‐18 maintains intestinal barrier integrity through stimulation of production of anti‐microbial peptides (AMP) by Paneth cells (in dark pink), mucus synthesis by goblet cells (in green) and epithelial proliferation through induction of epithelial stem cells (in blue). In addition, IL-18 is also produced by intestinal neurons controlling homeostatic goblet cells AMP production. In this way, IL‐18 contribute to maintain a normal microbiota in the digestive tract. When commensal bacteria enter the mucosa, macrophages from the lamina propria secrete IL‐18, which participate to control infection. In pathological conditions (lower illustration), the intestinal epithelial barrier is disrupted, and microbes can enter in the lamina propria where they stimulate resident macrophages to produce IL‐18. This excess of IL-18 is deleterious to intestinal epithelial barrier integrity, leading to leukocytes recruitment from the blood and inhibition of mucus production by goblet cells destruction. Altogether, these injuries shift gut microbiota favoring dysbiosis. Some of the enriched bacterial populations lead to an increased influx of Toll-Like Receptor agonists into the portal circulation. Reaching the liver, theses noxious molecules lead to hepatic overexpression of TNF-α and others pro-inflammatory cytokines that drives NASH progression. Figure produced using illustrations from BioRender.com.
As a potent activator of NK cells, IL-18 could have a potential anti-cancer activity. This hypothesis has been validated by experimental observations in mice. Treatment with a combination of IL-18/IL-12 decreased tumor burden in mice with established HCC (158). In colorectal cancer with hepatic metastases, burden is exacerbated in NLRP3-deficient mice (159). Downstream NLRP3, IL-18 promoted the maturation of hepatic NK cells and triggered FasL mediated cytotoxicity (159, 160). Downregulated expression of NLRP3 inflammasome in HCC correlated with the aggravation of carcinoma, while reconstitution of NLRP3 inflammasome reversed the malignant phenotype of HCC (161). Nevertheless, in contrast to this protective role of NLRP3/IL-18 axis against hepatic tumor growth, other results showed that an inhibition of IL-18 signaling could protect TLR2-/- mice from diethylnitrosamine (DEN)-induced carcinogenesis (162). The underlying mechanism seems to involve a limitation of IL-18-induced accumulation of myeloid-derived suppressor cells (162).
From a clinical point of view, several observations have corroborated this role for IL-18 in liver cancer progression. Expressions of IL-18 and IL-18R were upregulated in HCC tissue specimens (163, 164). IL-18 suppressed the apoptosis of human HCC cells (163) and promoted hepatoma cells metastasis and migration (164). The underlying mechanism could be partially attributable to the increased activities of ECM metalloproteinase (MMP)-2/3/9 by IL-18 (164). Circulating levels of IL-18 were elevated in patients with HCC compared to controls and they significantly correlated with the presence of venous invasion and advanced tumor stages (165). Finally, mutations in IL-18 alleles contributed to susceptibility to HCC and severity of the disease in general populations and in patients infected with hepatitis virus (166–171).
To conclude, research over the past 20 years has provided an increasingly complex view of the pleiotropic functions of IL-18. Beyond being a classic cytokine allowing fine-tuning of immune cells communication, this protein has emerged as a key regulator in the control of metabolism, in normal physiology, but also in pathological conditions such as obesity, diabetes and associated liver disorders.
Important metabolic functions resulting from physiological levels of IL-18 should clearly be dissociated from deleterious effects resulting from supra-physiological levels of IL-18 reached through immune stimulation (in a global pro-inflammatory context) or pharmacological administration (in an experimental context). Despite the significant advances in the understanding of physiological functions of IL-18 and its role in the occurrence and progression of metabolic diseases, several questions remain unanswered: What is the trigger that leads to IL-18 production by inflammasomes in metabolic diseases? Could different stimuli activate different inflammasomes, resulting in different production of IL-1 and IL-18? What is the respective contribution of different metabolic tissues (such as WAT and skeletal muscle) to circulating levels of IL-18? What are the molecular mechanisms underlying IL-18 resistance? Is there some link between leptin, insulin and IL-18 resistance? How circulating levels of IL-18 are related to IL-18 tissular actions? Can different cytokines (as IL-37 in humans) modulate IL-18/IL-18R interaction, and if so, how?
Answering these important questions will reinforce our understanding of IL-18 metabolic roles. In addition, more pre-clinical studies involving IL-18 regulatory steps, such as maturation by inflammasome, retention by IL-18BP or antagonism by anti-IL-18 or anti-IL-18R antibodies could open new therapeutic options for patients with metabolic diseases such as obesity, diabetes, and NAFLD/NASH.
ES contributed to the literature search and original draft preparation. ES and FJ contributed to text review and editing. All authors have read and agreed to the published version of the manuscript.
This work was funded by the Swiss National Science Foundation (SNSF grants 189003), the Swisslife Foundation, the Fondation pour l’innovation sur le cancer et la biologie and the Vontobel Foundation. Open access funding was provided by the University of Geneva.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev (2008) 223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x
2. Kaplanski G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol Rev (2018) 281:138–53. doi: 10.1111/imr.12616
3. Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in health and disease. Int J Mol Sci 20 (2019) 20. doi: 10.3390/ijms20030649
4. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol (2001) 19:423–74. doi: 10.1146/annurev.immunol.19.1.423
5. Giedraitis V, He B, Huang WX, Hillert J. Cloning and mutation analysis of the human IL-18 promoter: A possible role of polymorphisms in expression regulation. J Neuroimmunol (2001) 112:146–52. doi: 10.1016/S0165-5728(00)00407-0
6. Marques-Rocha JL, Samblas M, Milagro FI, Bressan J, Martinez JA, Marti A. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J (2015) 29:3595–611. doi: 10.1096/fj.14-260323
7. Bazan JF, Timans JC, Kastelein RA. A newly defined interleukin-1? Nature (1996) 379:591. doi: 10.1038/379591a0
8. Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science (1997) 275:206–9. doi: 10.1126/science.275.5297.206
9. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature (1997) 386:619–23. doi: 10.1038/386619a0
10. Feng S, Fox D, Man SM. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J Mol Biol (2018) 430:3068–80. doi: 10.1016/j.jmb.2018.07.002
11. Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. And small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature (2006) 440:233–6. doi: 10.1038/nature04517
12. Duncan JA, Canna SW. The NLRC4 inflammasome. Immunol Rev (2018) 281:115–23. doi: 10.1111/imr.12607
13. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature (1995) 378:88–91. doi: 10.1038/378088a0
14. Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol (2012) 189:5508–12. doi: 10.4049/jimmunol.1202121
15. Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity (1998) 9:143–50. doi: 10.1016/S1074-7613(00)80596-8
16. Kanakaraj P, Ngo K, Wu Y, Angulo A, Ghazal P, Harris CA, et al. Defective interleukin (IL)-18-mediated natural killer and T helper cell type 1 responses in IL-1 receptor-associated kinase (IRAK)-deficient mice. J Exp Med (1999) 189:1129–38. doi: 10.1084/jem.189.7.1129
17. Suzuki N, Chen NJ, Millar DG, Suzuki S, Horacek T, Hara H, et al. IL-1 receptor-associated kinase 4 is essential for IL-18-mediated NK and Th1 cell responses. J Immunol (2003) 170:4031–5. doi: 10.4049/jimmunol.170.8.4031
18. Robinson D, Shibuya K, Mui A, Zonin F, Murphy E, Sana T, et al. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity (1997) 7:571–81. doi: 10.1016/S1074-7613(00)80378-7
19. Kalina U, Kauschat D, Koyama N, Nuernberger H, Ballas K, Koschmieder S, et al. IL-18 activates STAT3 in the natural killer cell line 92, augments cytotoxic activity, and mediates IFN-gamma production by the stress kinase p38 and by the extracellular regulated kinases p44erk-1 and p42erk-21. J Immunol (2000) 165:1307–13. doi: 10.4049/jimmunol.165.3.1307
20. Deason K, Troutman TD, Jain A, Challa DK, Mandraju R, Brewer T, et al. BCAP links IL-1R to the PI3K-mTOR pathway and regulates pathogenic Th17 cell differentiation. J Exp Med (2018) 215:2413–28. doi: 10.1084/jem.20171810
21. Nold-Petry CA, Lo CY, Rudloff I, Elgass KD, Li S, Gantier MP, et al. IL-37 requires the receptors IL-18Ralpha and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat Immunol (2015) 16:354–65. doi: 10.1038/ni.3103
22. Dinarello CA. Targeting interleukin 18 with interleukin 18 binding protein. Ann Rheum Dis (2000) 59 Suppl 1:i17–20. doi: 10.1136/ard.59.suppl_1.i17
23. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol (2013) 4:289. doi: 10.3389/fimmu.2013.00289
24. Bray GA, Heisel WE, Afshin A, Jensen MD, Dietz WH, Long M, et al. The science of obesity management: An endocrine society scientific statement. Endocr Rev (2018) 39:79–132. doi: 10.1210/er.2017-00253
25. Schaffler A, Muller-Ladner U, Scholmerich J, Buchler C. Role of adipose tissue as an inflammatory organ in human diseases. Endocr Rev (2006) 27:449–67. doi: 10.1210/er.2005-0022
26. Netea MG, Joosten LA, Lewis E, Jensen DR, Voshol PJ, Kullberg BJ, et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med (2006) 12:650–6. doi: 10.1038/nm1415
27. Zorrilla EP, Sanchez-Alavez M, Sugama S, Brennan M, Fernandez R, Bartfai T, et al. Interleukin-18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc Natl Acad Sci USA (2007) 104:11097–102. doi: 10.1073/pnas.0611523104
28. Alboni S, Micioni Di Bonaventura MV, Benatti C, Giusepponi ME, Brunello N, Cifani C. Hypothalamic expression of inflammatory mediators in an animal model of binge eating. Behav Brain Res (2017) 320:420–30. doi: 10.1016/j.bbr.2016.10.044
29. Tanaka S, Oya-Ito T, Murakami Y, Saito K, Furuta S, Yu Y, et al. Decline of plasma concentrations of interleukin-18 in severely malnourished patients with anorexia nervosa: Exploratory analysis. Nutrients 11 (2019) 11. doi: 10.3390/nu11030540
30. Alboni S, Cervia D, Ross B, Montanari C, Gonzalez AS, Sanchez-Alavez M, et al. Mapping of the full length and the truncated interleukin-18 receptor alpha in the mouse brain. J Neuroimmunol (2009) 214:43–54. doi: 10.1016/j.jneuroim.2009.06.016
31. Francesconi W, Sanchez-Alavez M, Berton F, Alboni S, Benatti C, Mori S, et al. The proinflammatory cytokine interleukin 18 regulates feeding by acting on the bed nucleus of the stria terminalis. J Neurosci (2016) 36:5170–80. doi: 10.1523/JNEUROSCI.3919-15.2016
32. Zorrilla EP, Conti B. Interleukin-18 null mutation increases weight and food intake and reduces energy expenditure and lipid substrate utilization in high-fat diet fed mice. Brain Behav Immun (2014) 37:45–53. doi: 10.1016/j.bbi.2013.12.001
33. Yamanishi K, Maeda S, Kuwahara-Otani S, Watanabe Y, Yoshida M, Ikubo K, et al. Interleukin-18-deficient mice develop dyslipidemia resulting in nonalcoholic fatty liver disease and steatohepatitis. Transl Res (2016) 173:101–114.e7. doi: 10.1016/j.trsl.2016.03.010
34. Yamamoto Y, Tanahashi T, Katsuura S, Kurokawa K, Nishida K, Kuwano Y, et al. Interleukin-18 deficiency reduces neuropeptide gene expressions in the mouse amygdala related with behavioral change. J Neuroimmunol (2010) 229:129–39. doi: 10.1016/j.jneuroim.2010.07.024
35. Haastrup E, Bukh JD, Bock C, Vinberg M, Thorner LW, Hansen T, et al. Promoter variants in IL18 are associated with onset of depression in patients previously exposed to stressful-life events. J Affect Disord (2012) 136:134–8. doi: 10.1016/j.jad.2011.08.025
36. Swartz JR, Prather AA, Di Iorio CR, Bogdan R, Hariri AR. A functional interleukin-18 haplotype predicts depression and anxiety through increased threat-related amygdala reactivity in women but not men. Neuropsychopharmacology (2017) 42:419–26. doi: 10.1038/npp.2016.129
37. Yaguchi T, Nagata T, Yang D, Nishizaki T. Interleukin-18 regulates motor activity, anxiety and spatial learning without affecting synaptic plasticity. Behav Brain Res (2010) 206:47–51. doi: 10.1016/j.bbr.2009.08.033
38. Sugama S, Fujita M, Hashimoto M, Conti B. Stress induced morphological microglial activation in the rodent brain: involvement of interleukin-18. Neuroscience (2007) 146:1388–99. doi: 10.1016/j.neuroscience.2007.02.043
39. Wang N, Sugama S, Conti B, Teramoto A, Shibasaki T. Interleukin-18 mRNA expression in the rat pituitary gland. J Neuroimmunol (2006) 173:117–25. doi: 10.1016/j.jneuroim.2005.12.009
40. Yang Y, Hahm E, Kim Y, Kang J, Lee W, Han I, et al. Regulation of IL-18 expression by CRH in mouse microglial cells. Immunol Lett (2005) 98:291–6. doi: 10.1016/j.imlet.2004.12.003
41. Tringali G, Pozzoli G, Vairano M, Mores N, Preziosi P, Navarra P. Interleukin-18 displays effects opposite to those of interleukin-1 in the regulation of neuroendocrine stress axis. J Neuroimmunol (2005) 160:61–7. doi: 10.1016/j.jneuroim.2004.10.028
42. Sugama S, Conti B. Interleukin-18 and stress. Brain Res Rev (2008) 58:85–95. doi: 10.1016/j.brainresrev.2007.11.003
43. Alboni S, Cervia D, Sugama S, Conti B. Interleukin 18 in the CNS. J Neuroinflamm (2010) 7:9. doi: 10.1186/1742-2094-7-9
44. Yamanishi K, Maeda S, Kuwahara-Otani S, Hashimoto T, Ikubo K, Mukai K, et al. Deficiency in interleukin-18 promotes differentiation of brown adipose tissue resulting in fat accumulation despite dyslipidemia. J Transl Med (2018) 16:314. doi: 10.1186/s12967-018-1684-3
45. Li W, Jin D, Takai S, Hayakawa T, Ogata J, Yamanishi K, et al. Impaired function of aorta and perivascular adipose tissue in IL-18-deficient mice. Am J Physiol Heart Circ Physiol 317 (2019) 317:H1142–56. doi: 10.1152/ajpheart.00813.2018
46. Pazos P, Lima L, Tovar S, Gonzalez-Touceda D, Dieguez C, Garcia MC. Divergent responses to thermogenic stimuli in BAT and subcutaneous adipose tissue from interleukin 18 and interleukin 18 receptor 1-deficient mice. Sci Rep (2015) 5:17977. doi: 10.1038/srep17977
47. Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol (2018) 19:1093–9. doi: 10.1038/s41590-018-0201-4
48. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature (2015) 519:242–6. doi: 10.1038/nature14115
49. Su Z, Tao X. Current understanding of IL-37 in human health and disease. Front Immunol (2021) 12:696605. doi: 10.3389/fimmu.2021.696605
50. Hung J, McQuillan BM, Chapman CM, Thompson PL, Beilby JP. Elevated interleukin-18 levels are associated with the metabolic syndrome independent of obesity and insulin resistance. Arterioscler Thromb Vasc Biol (2005) 25:1268–73. doi: 10.1161/01.ATV.0000163843.70369.12
51. Escobar-Morreale HF, Botella-Carretero JI, Villuendas G, Sancho J, San Millan JL. Serum interleukin-18 concentrations are increased in the polycystic ovary syndrome: relationship to insulin resistance and to obesity. J Clin Endocrinol Metab (2004) 89:806–11. doi: 10.1210/jc.2003-031365
52. Leick L, Lindegaard B, Stensvold D, Plomgaard P, Saltin B, Pilegaard H. Adipose tissue interleukin-18 mRNA and plasma interleukin-18: Effect of obesity and exercise. Obes (Silver Spring) (2007) 15:356–63. doi: 10.1038/oby.2007.528
53. Esposito K, Pontillo A, Ciotola M, Di Palo C, Grella E, Nicoletti G, et al. Weight loss reduces interleukin-18 levels in obese women. J Clin Endocrinol Metab (2002) 87:3864–6. doi: 10.1210/jcem.87.8.8781
54. Skurk T, Kolb H, Muller-Scholze S, Rohrig K, Hauner H, Herder C. The proatherogenic cytokine interleukin-18 is secreted by human adipocytes. Eur J Endocrinol (2005) 152:863–8. doi: 10.1530/eje.1.01897
55. Membrez M, Ammon-Zufferey C, Philippe D, Aprikian O, Monnard I, Mace K, et al. Interleukin-18 protein level is upregulated in adipose tissue of obese mice. Obes (Silver Spring) (2009) 17:393–5. doi: 10.1038/oby.2008.535
56. Wood IS, Wang B, Jenkins JR, Trayhurn P. The pro-inflammatory cytokine IL-18 is expressed in human adipose tissue and strongly upregulated by TNFalpha in human adipocytes. Biochem Biophys Res Commun (2005) 337:422–9. doi: 10.1016/j.bbrc.2005.09.068
57. Koenen TB, Stienstra R, van Tits LJ, Joosten LA, van Velzen JF, Hijmans A, et al. The inflammasome and caspase-1 activation: A new mechanism underlying increased inflammatory activity in human visceral adipose tissue. Endocrinology (2011) 152:3769–78. doi: 10.1210/en.2010-1480
58. Fain JN, Tichansky DS, Madan AK. Most of the interleukin 1 receptor antagonist, cathepsin s, macrophage migration inhibitory factor, nerve growth factor, and interleukin 18 release by explants of human adipose tissue is by the non-fat cells, not by the adipocytes. Metabolism (2006) 55:1113–21. doi: 10.1016/j.metabol.2006.04.008
59. Darimont C, Avanti O, Blancher F, Wagniere S, Mansourian R, Zbinden I, et al. Contribution of mesothelial cells in the expression of inflammatory-related factors in omental adipose tissue of obese subjects. Int J Obes (Lond) (2008) 32:112–20. doi: 10.1038/sj.ijo.0803688
60. Lindegaard B, Hansen AB, Pilegaard H, Keller P, Gerstoft J, Pedersen BK. Adipose tissue expression of IL-18 and HIV-associated lipodystrophy. AIDS (2004) 18:1956–8. doi: 10.1097/00002030-200409240-00013
61. Thompson SR, Sanders J, Stephens JW, Miller GJ, Humphries SE. A common interleukin 18 haplotype is associated with higher body mass index in subjects with diabetes and coronary heart disease. Metabolism (2007) 56:662–9. doi: 10.1016/j.metabol.2006.12.015
62. Vilarrasa N, Vendrell J, Sanchez-Santos R, Broch M, Megia A, Masdevall C, et al. Effect of weight loss induced by gastric bypass on proinflammatory interleukin-18, soluble tumour necrosis factor-alpha receptors, c-reactive protein and adiponectin in morbidly obese patients. Clin Endocrinol (Oxf) (2007) 67:679–86. doi: 10.1111/j.1365-2265.2007.02945.x
63. Tajik N, Keshavarz SA, Masoudkabir F, Djalali M, Sadrzadeh-Yeganeh HH, Eshraghian MR, et al. Effect of diet-induced weight loss on inflammatory cytokines in obese women. J Endocrinol Invest (2013) 36:211–5. doi: 10.3275/8465
64. Schernthaner GH, Kopp HP, Kriwanek S, Krzyzanowska K, Satler M, Koppensteiner R, et al. Effect of massive weight loss induced by bariatric surgery on serum levels of interleukin-18 and monocyte-chemoattractant-protein-1 in morbid obesity. Obes Surg (2006) 16:709–15. doi: 10.1381/096089206777346763
65. Esposito K, Pontillo A, Di Palo C, Giugliano G, Masella M, Marfella R, et al. Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA (2003) 289:1799–804. doi: 10.1001/jama.289.14.1799
66. Murphy AJ, Kraakman MJ, Kammoun HL, Dragoljevic D, Lee MK, Lawlor KE, et al. IL-18 production from the NLRP1 inflammasome prevents obesity and metabolic syndrome. Cell Metab (2016) 23:155–64. doi: 10.1016/j.cmet.2015.09.024
67. Salazar-Leon J, Valdez-Hernandez AL, Garcia-Jimenez S, Roman-Dominguez L, Huanosta-Murillo E, Bonifaz LC, et al. Nlrp1b1 negatively modulates obesity-induced inflammation by promoting IL-18 production. Sci Rep (2019) 9:13815. doi: 10.1038/s41598-019-49546-7
68. Yang YS, Li XY, Hong J, Gu WQ, Zhang YF, Yang J, et al. Interleukin-18 enhances glucose uptake in 3T3-L1 adipocytes. Endocrine (2007) 32:297–302. doi: 10.1007/s12020-008-9048-z
70. White DW, Kuropatwinski KK, Devos R, Baumann H, Tartaglia LA. Leptin receptor (OB-r) signaling. Cytoplasmic domain mutational analysis and evidence for receptor homo-oligomerization. J Biol Chem (1997) 272:4065–71. doi: 10.1074/jbc.272.7.4065
71. Zilverschoon GR, Tack CJ, Joosten LA, Kullberg BJ, van der Meer JW, Netea MG. Interleukin-18 resistance in patients with obesity and type 2 diabetes mellitus. Int J Obes (Lond) (2008) 32:1407–14. doi: 10.1038/ijo.2008.109
72. Plomgaard P, Penkowa M, Pedersen BK. Fiber type specific expression of TNF-alpha, IL-6 and IL-18 in human skeletal muscles. Exerc Immunol Rev (2005) 11:53–63.
73. Lindegaard B, Matthews VB, Brandt C, Hojman P, Allen TL, Estevez E, et al. Interleukin-18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes (2013) 62:3064–74. doi: 10.2337/db12-1095
74. Lindegaard B, Hvid T, Wolsk Mygind H, Mortensen OH, Grondal T, Abildgaard J, et al. Low expression of IL-18 and IL-18 receptor in human skeletal muscle is associated with systemic and intramuscular lipid metabolism-role of HIV lipodystrophy. PloS One (2018) 13:e0186755. doi: 10.1371/journal.pone.0186755
75. Syed FZ. Type 1 diabetes mellitus. Ann Intern Med (2022) 175:ITC33–48. doi: 10.7326/AITC202203150
77. Eizirik DL, Colli ML. Revisiting the role of inflammation in the loss of pancreatic beta-cells in T1DM. Nat Rev Endocrinol (2020) 16:611–2. doi: 10.1038/s41574-020-00409-6
78. Hong TP, Andersen NA, Nielsen K, Karlsen AE, Fantuzzi G, Eizirik DL, et al. Interleukin-18 mRNA, but not interleukin-18 receptor mRNA, is constitutively expressed in islet beta-cells and up-regulated by interferon-gamma. Eur Cytokine Netw (2000) 11:193–205.
79. Frigerio S, Hollander GA, Zumsteg U. Functional IL-18 is produced by primary pancreatic mouse islets and NIT-1 beta cells and participates in the progression towards destructive insulitis. Horm Res (2002) 57:94–104. doi: 10.1159/000057959
80. Krook H, Wallstrom J, Sandler S. Function of rat pancreatic islets exposed to interleukin-18 in vitro. Autoimmunity (1999) 29:263–7. doi: 10.3109/08916939908994745
81. Rothe H, Hausmann A, Casteels K, Okamura H, Kurimoto M, Burkart V, et al. IL-18 inhibits diabetes development in nonobese diabetic mice by counterregulation of Th1-dependent destructive insulitis. J Immunol (1999) 163:1230–6.
82. Rothe H, Jenkins NA, Copeland NG, Kolb H. Active stage of autoimmune diabetes is associated with the expression of a novel cytokine, IGIF, which is located near Idd2. J Clin Invest (1997) 99:469–74. doi: 10.1172/JCI119181
83. Lewis EC, Dinarello CA. Responses of IL-18- and IL-18 receptor-deficient pancreatic islets with convergence of positive and negative signals for the IL-18 receptor. Proc Natl Acad Sci USA (2006) 103:16852–7. doi: 10.1073/pnas.0607917103
84. Zaccone P, Phillips J, Conget I, Cooke A, Nicoletti F. IL-18 binding protein fusion construct delays the development of diabetes in adoptive transfer and cyclophosphamide-induced diabetes in NOD mouse. Clin Immunol (2005) 115:74–9. doi: 10.1016/j.clim.2004.11.007
85. Marleau AM, Sarvetnick NE. IL-18 is required for self-reactive T cell expansion in NOD mice. J Autoimmun (2011) 36:263–77. doi: 10.1016/j.jaut.2011.02.005
86. Oikawa Y, Shimada A, Kasuga A, Morimoto J, Osaki T, Tahara H, et al. Systemic administration of IL-18 promotes diabetes development in young nonobese diabetic mice. J Immunol (2003) 171:5865–75. doi: 10.4049/jimmunol.171.11.5865
87. Nicoletti F, Di Marco R, Papaccio G, Conget I, Gomis R, Bernardini R, et al. Essential pathogenic role of endogenous IL-18 in murine diabetes induced by multiple low doses of streptozotocin. prevention of hyperglycemia and insulitis by a recombinant IL-18-binding protein: Fc construct. Eur J Immunol (2003) 33:2278–86. doi: 10.1002/eji.200323864
88. Lukic ML, Mensah-Brown E, Wei X, Shahin A, Liew FY. Lack of the mediators of innate immunity attenuate the development of autoimmune diabetes in mice. J Autoimmun (2003) 21:239–46. doi: 10.1016/S0896-8411(03)00115-X
89. Kretowski A, Mironczuk K, Karpinska A, Bojaryn U, Kinalski M, Puchalski Z, et al. Interleukin-18 promoter polymorphisms in type 1 diabetes. Diabetes (2002) 51:3347–9. doi: 10.2337/diabetes.51.11.3347
90. Ide A, Kawasaki E, Abiru N, Sun F, Kobayashi M, Fukushima T, et al. Association between IL-18 gene promoter polymorphisms and CTLA-4 gene 49A/G polymorphism in Japanese patients with type 1 diabetes. J Autoimmun (2004) 22:73–8. doi: 10.1016/j.jaut.2003.10.001
91. Novota P, Kolostova K, Pinterova D, Novak J, Treslova L, Andel M, et al. Interleukin IL-18 gene promoter polymorphisms in adult patients with type 1 diabetes mellitus and latent autoimmune diabetes in adults. Immunol Lett (2005) 96:247–51. doi: 10.1016/j.imlet.2004.08.016
92. Martin RJ, Savage DA, Carson DJ, Maxwell AP, Patterson CC. Interleukin 18 promoter polymorphisms are not strongly associated with type I diabetes in a UK population. Genes Immun (2005) 6:171–4. doi: 10.1038/sj.gene.6364161
93. Szeszko JS, Howson JM, Cooper JD, Walker NM, Twells RC, Stevens HE, et al. Analysis of polymorphisms of the interleukin-18 gene in type 1 diabetes and hardy-Weinberg equilibrium testing. Diabetes (2006) 55:559–62. doi: 10.2337/diabetes.55.02.06.db05-0826
94. Nicoletti F, Conget I, Di Marco R, Speciale AM, Morinigo R, Bendtzen K, et al. Serum levels of the interferon-gamma-inducing cytokine interleukin-18 are increased in individuals at high risk of developing type I diabetes. Diabetologia (2001) 44:309–11. doi: 10.1007/s001250051619
95. Katakami N, Kaneto H, Matsuhisa M, Yoshiuchi K, Kato K, Yamamoto K, et al. Serum interleukin-18 levels are increased and closely associated with various soluble adhesion molecule levels in type 1 diabetic patients. Diabetes Care (2007) 30:159–61. doi: 10.2337/dc06-1768
96. Harms RZ, Yarde DN, Guinn Z, Lorenzo-Arteaga KM, Corley KP, Cabrera MS, et al. Increased expression of IL-18 in the serum and islets of type 1 diabetics. Mol Immunol (2015) 64:306–12. doi: 10.1016/j.molimm.2014.12.012
97. Aida K, Nishida Y, Tanaka S, Maruyama T, Shimada A, Awata T, et al. RIG-i- and MDA5-initiated innate immunity linked with adaptive immunity accelerates beta-cell death in fulminant type 1 diabetes. Diabetes (2011) 60:884–9. doi: 10.2337/db10-0795
98. Gateva A, Kamenov Z, Karamfilova V, Assyov Y, Velikova T, El-Darawish Y, et al. Higher levels of IL-18 in patients with prediabetes compared to obese normoglycaemic controls. Arch Physiol Biochem (2020) 126:449–52. doi: 10.1080/13813455.2018.1555667
99. Zhuang H, Han J, Cheng L, Liu SL. A positive causal influence of IL-18 levels on the risk of T2DM: A mendelian randomization study. Front Genet (2019) 10:295. doi: 10.3389/fgene.2019.00295
100. Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tsutsumi Z, Takahashi S, et al. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: Relationship with diabetic nephropathy. Metabolism (2003) 52:605–8. doi: 10.1053/meta.2003.50096
101. Zaharieva E, Kamenov Z, Velikova T, Tsakova A, El-Darawish Y, Okamura H. Interleukin-18 serum level is elevated in type 2 diabetes and latent autoimmune diabetes. Endocr Connect (2018) 7:179–85. doi: 10.1530/EC-17-0273
102. Thorand B, Kolb H, Baumert J, Koenig W, Chambless L, Meisinger C, et al. Elevated levels of interleukin-18 predict the development of type 2 diabetes: results from the MONICA/KORA augsburg study, 1984-2002. Diabetes (2005) 54:2932–8. doi: 10.2337/diabetes.54.10.2932
103. Hivert MF, Sun Q, Shrader P, Mantzoros CS, Meigs JB, Hu FB. Circulating IL-18 and the risk of type 2 diabetes in women. Diabetologia (2009) 52:2101–8. doi: 10.1007/s00125-009-1455-z
104. Bruun JM, Stallknecht B, Helge JW, Richelsen B. Interleukin-18 in plasma and adipose tissue: effects of obesity, insulin resistance, and weight loss. Eur J Endocrinol (2007) 157:465–71. doi: 10.1530/EJE-07-0206
105. Fischer CP, Perstrup LB, Berntsen A, Eskildsen P, Pedersen BK. Elevated plasma interleukin-18 is a marker of insulin-resistance in type 2 diabetic and non-diabetic humans. Clin Immunol (2005) 117:152–60. doi: 10.1016/j.clim.2005.07.008
106. Bosch M, Lopez-Bermejo A, Vendrell J, Musri M, Ricart W, Fernandez-Real JM. Circulating IL-18 concentration is associated with insulin sensitivity and glucose tolerance through increased fat-free mass. Diabetologia (2005) 48:1841–3. doi: 10.1007/s00125-005-1859-3
107. Olusi SO, Al-Awadhi A, Abraham M. Relations of serum interleukin 18 levels to serum lipid and glucose concentrations in an apparently healthy adult population. Horm Res (2003) 60:29–33. doi: 10.1159/000070824
108. Kim HJ, Kang ES, Kim DJ, Kim SH, Ahn CW, Cha BS, et al. Effects of rosiglitazone and metformin on inflammatory markers and adipokines: decrease in interleukin-18 is an independent factor for the improvement of homeostasis model assessment-beta in type 2 diabetes mellitus. Clin Endocrinol (Oxf) (2007) 66:282–9. doi: 10.1111/j.1365-2265.2006.02723.x
109. Presta I, Andreozzi F, Succurro E, Marini MA, Laratta E, Lauro R, et al. IL-18 gene polymorphism and metabolic syndrome. Nutr Metab Cardiovasc Dis (2009) 19:e5–6. doi: 10.1016/j.numecd.2008.10.002
110. Rinella ME. Nonalcoholic fatty liver disease: A systematic review. JAMA (2015) 313:2263–73. doi: 10.1001/jama.2015.5370
111. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
112. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol (2013) 10:656–65. doi: 10.1038/nrgastro.2013.183
113. Ratziu V, Francque S, Sanyal A. Breakthroughs in therapies for NASH and remaining challenges. J Hepatol (2022) 76:1263–78. doi: 10.1016/j.jhep.2022.04.002
114. Lebel-Binay S, Berger A, Zinzindohoue F, Cugnenc P, Thiounn N, Fridman WH, et al. Interleukin-18: biological properties and clinical implications. Eur Cytokine Netw (2000) 11:15–26.
115. Belkaya S, Michailidis E, Korol CB, Kabbani M, Cobat A, Bastard P, et al. Inherited IL-18BP deficiency in human fulminant viral hepatitis. J Exp Med (2019) 216:1777–90. doi: 10.1084/jem.20190669
116. Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Falcone EL, Holland SM, et al. Inflammasomes coordinate pyroptosis and natural killer cell cytotoxicity to clear infection by a ubiquitous environmental bacterium. Immunity (2015) 43:987–97. doi: 10.1016/j.immuni.2015.10.010
117. Zalinger ZB, Elliott R, Weiss SR. Role of the inflammasome-related cytokines il-1 and il-18 during infection with murine coronavirus. J Neurovirol (2017) 23:845–54. doi: 10.1007/s13365-017-0574-4
118. Serti E, Werner JM, Chattergoon M, Cox AL, Lohmann V, Rehermann B. Monocytes activate natural killer cells via inflammasome-induced interleukin 18 in response to hepatitis c virus replication. Gastroenterology (2014) 147:209–220.e3. doi: 10.1053/j.gastro.2014.03.046
119. Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest (2009) 119:305–14. doi: 10.1172/JCI35958
120. Tsutsui H, Matsui K, Okamura H, Nakanishi K. Pathophysiological roles of interleukin-18 in inflammatory liver diseases. Immunol Rev (2000) 174:192–209. doi: 10.1034/j.1600-0528.2002.017418.x
121. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
122. Camell CD, Nguyen KY, Jurczak MJ, Christian BE, Shulman GI, Shadel GS, et al. Macrophage-specific de Novo synthesis of ceramide is dispensable for inflammasome-driven inflammation and insulin resistance in obesity. J Biol Chem (2015) 290:29402–13. doi: 10.1074/jbc.M115.680199
123. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology (2011) 54:133–44. doi: 10.1002/hep.24341
124. Netea MG, Joosten LA. The NLRP1-IL18 connection: A stab in the back of obesity-induced inflammation. Cell Metab (2016) 23:6–7. doi: 10.1016/j.cmet.2015.12.014
125. Liu W, Yin Y, Wang M, Fan T, Zhu Y, Shen L, et al. Disrupting phosphatase SHP2 in macrophages protects mice from high-fat diet-induced hepatic steatosis and insulin resistance by elevating IL-18 levels. J Biol Chem (2020) 295:10842–56. doi: 10.1074/jbc.RA119.011840
126. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature (2012) 482:179–85. doi: 10.1038/nature10809
127. Lindegaard B, Abildgaard J, Heywood SE, Pedersen BK, Febbraio MA. Female sex hormones are necessary for the metabolic effects mediated by loss of interleukin 18 signaling. Mol Metab (2018) 12:89–97. doi: 10.1016/j.molmet.2018.04.005
128. Hohenester S, Kanitz V, Schiergens T, Einer C, Nagel J, Wimmer R, et al. IL-18 but not IL-1 signaling is pivotal for the initiation of liver injury in murine non-alcoholic fatty liver disease. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21228602
129. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun (2013) 4:2823. doi: 10.1038/ncomms3823
130. Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, et al. Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol (2009) 296:G1248–57. doi: 10.1152/ajpgi.90223.2008
131. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (2014) 59:898–910. doi: 10.1002/hep.26592
132. Finotto S, Siebler J, Hausding M, Schipp M, Wirtz S, Klein S, et al. Severe hepatic injury in interleukin 18 (IL-18) transgenic mice: A key role for IL-18 in regulating hepatocyte apoptosis in vivo. Gut (2004) 53:392–400. doi: 10.1136/gut.2003.018572
133. Mehta R, Neupane A, Wang L, Goodman Z, Baranova A, Younossi ZM. Expression of NALPs in adipose and the fibrotic progression of non-alcoholic fatty liver disease in obese subjects. BMC Gastroenterol (2014) 14:208. doi: 10.1186/s12876-014-0208-8
134. Lopez-Bermejo A, Bosch M, Recasens M, Biarnes J, Esteve E, Casamitjana R, et al. Potential role of interleukin-18 in liver disease associated with insulin resistance. Obes Res (2005) 13:1925–31. doi: 10.1038/oby.2005.237
135. Li Y, Li-Li Z, Qin L, Ying W. Plasma interleukin-18/interleukin-18 binding protein ratio in Chinese with NAFLD. Hepatogastroenterology (2010) 57:103–6.
136. Ludwiczek O, Kaser A, Novick D, Dinarello CA, Rubinstein M, Vogel W, et al. Plasma levels of interleukin-18 and interleukin-18 binding protein are elevated in patients with chronic liver disease. J Clin Immunol (2002) 22:331–7. doi: 10.1023/A:1020600230977
137. Zhang S, Yang X, Wang W. Associations of genetic polymorphisms in CTLA-4 and IL-18 with chronic liver diseases: Evidence from a meta-analysis. Genomics (2020) 112:1889–96. doi: 10.1016/j.ygeno.2019.11.001
138. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
139. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol (2018) 68:280–95. doi: 10.1016/j.jhep.2017.11.014
140. Milosevic I, Vujovic A, Barac A, Djelic M, Korac M, Radovanovic Spurnic A, et al. Gut-liver axis, gut microbiota, and its modulation in the management of liver diseases: A review of the literature. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20020395
141. Chikano S, Sawada K, Shimoyama T, Kashiwamura SI, Sugihara A, Sekikawa K, et al. IL-18 and IL-12 induce intestinal inflammation and fatty liver in mice in an IFN-gamma dependent manner. Gut (2000) 47:779–86. doi: 10.1136/gut.47.6.779
142. Ten Hove T, Corbaz A, Amitai H, Aloni S, Belzer I, Graber P, et al. Blockade of endogenous IL-18 ameliorates TNBS-induced colitis by decreasing local TNF-alpha production in mice. Gastroenterology (2001) 121:1372–9. doi: 10.1053/gast.2001.29579
143. Ishikura T, Kanai T, Uraushihara K, Iiyama R, Makita S, Totsuka T, et al. Interleukin-18 overproduction exacerbates the development of colitis with markedly infiltrated macrophages in interleukin-18 transgenic mice. J Gastroenterol Hepatol (2003) 18:960–9. doi: 10.1046/j.1440-1746.2003.03097.x
144. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell (2015) 163:1444–56. doi: 10.1016/j.cell.2015.10.072
145. Monteleone G, Trapasso F, Parrello T, Biancone L, Stella A, Iuliano R, et al. Bioactive IL-18 expression is up-regulated in crohn's disease. J Immunol (1999) 163:143–7.
146. Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF Jr., Foley E, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in crohn's disease: expression and localization in intestinal mucosal cells. J Immunol (1999) 162:6829–35.
147. Pages F, Berger A, Lebel-Binay S, Zinzindohoue F, Danel C, Piqueras B, et al. Proinflammatory and antitumor properties of interleukin-18 in the gastrointestinal tract. Immunol Lett (2000) 75:9–14. doi: 10.1016/S0165-2478(00)00285-6
148. Leach ST, Messina I, Lemberg DA, Novick D, Rubenstein M, Day AS. Local and systemic interleukin-18 and interleukin-18-binding protein in children with inflammatory bowel disease. Inflamm Bowel Dis (2008) 14:68–74. doi: 10.1002/ibd.20272
149. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491:259–63. doi: 10.1038/nature11535
150. Chiang HY, Lu HH, Sudhakar JN, Chen YW, Shih NS, Weng YT, et al. IL-22 initiates an IL-18-dependent epithelial response circuit to enforce intestinal host defence. Nat Commun (2022) 13:874. doi: 10.1038/s41467-022-28478-3
151. Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell (2015) 163:1428–43. doi: 10.1016/j.cell.2015.10.048
152. Gao SJ, Zhang L, Lu W, Wang L, Chen L, Zhu Z, et al. Interleukin-18 genetic polymorphisms contribute differentially to the susceptibility to crohn's disease. World J Gastroenterol (2015) 21:8711–22. doi: 10.3748/wjg.v21.i28.8711
153. Hu S, Peng L, Kwak YT, Tekippe EM, Pasare C, Malter JS, et al. The DNA sensor AIM2 maintains intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep (2015) 13:1922–36. doi: 10.1016/j.celrep.2015.10.040
154. Jarret A, Jackson R, Duizer C, Healy ME, Zhao J, Rone JM, et al. Enteric nervous system-derived IL-18 orchestrates mucosal barrier immunity. Cell (2020) 180:50–63.e12. doi: 10.1016/j.cell.2019.12.016
155. Tye H, Yu CH, Simms LA, de Zoete MR, Kim ML, Zakrzewski M, et al. NLRP1 restricts butyrate producing commensals to exacerbate inflammatory bowel disease. Nat Commun (2018) 9:3728. doi: 10.1038/s41467-018-06125-0
156. Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis (2011) 17:1359–72. doi: 10.1002/ibd.21478
157. Seregin SS, Golovchenko N, Schaf B, Chen J, Pudlo NA, Mitchell J, et al. NLRP6 protects Il10(-/-) mice from colitis by limiting colonization of akkermansia muciniphila. Cell Rep (2017) 19:733–45. doi: 10.1016/j.celrep.2017.03.080
158. Subleski JJ, Scarzello AJ, Alvord WG, Jiang Q, Stauffer JK, Kronfli A, et al. Serum-based tracking of de novo initiated liver cancer progression reveals early immunoregulation and response to therapy. J Hepatol (2015) 63:1181–9. doi: 10.1016/j.jhep.2015.06.021
159. Dupaul-Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A, et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity (2015) 43:751–63. doi: 10.1016/j.immuni.2015.08.013
160. Dagenais M, Saleh M. Linking cancer-induced Nlrp3 inflammasome activation to efficient NK cell-mediated immunosurveillance. Oncoimmunology (2016) 5:e1129484. doi: 10.1080/2162402X.2015.1129484
161. Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest (2014) 94:52–62. doi: 10.1038/labinvest.2013.126
162. Li S, Sun R, Chen Y, Wei H, Tian Z. TLR2 limits development of hepatocellular carcinoma by reducing IL18-mediated immunosuppression. Cancer Res (2015) 75:986–95. doi: 10.1158/0008-5472.CAN-14-2371
163. Asakawa M, Kono H, Amemiya H, Matsuda M, Suzuki T, Maki A, et al. Role of interleukin-18 and its receptor in hepatocellular carcinoma associated with hepatitis c virus infection. Int J Cancer (2006) 118:564–70. doi: 10.1002/ijc.21367
164. Zhang Y, Li Y, Ma Y, Liu S, She Y, Zhao P, et al. Dual effects of interleukin-18: inhibiting hepatitis b virus replication in HepG2.2.15 cells and promoting hepatoma cells metastasis. Am J Physiol Gastrointest Liver Physiol (2011) 301:G565–73. doi: 10.1152/ajpgi.00058.2011
165. Tangkijvanich P, Thong-Ngam D, Mahachai V, Theamboonlers A, Poovorawan Y. Role of serum interleukin-18 as a prognostic factor in patients with hepatocellular carcinoma. World J Gastroenterol (2007) 13:4345–9. doi: 10.3748/wjg.v13.i32.4345
166. Quan Y, Yang J, Qin T, Hu Y. Associations between twelve common gene polymorphisms and susceptibility to hepatocellular carcinoma: evidence from a meta-analysis. World J Surg Oncol (2019) 17:216. doi: 10.1186/s12957-019-1748-8
167. Teixeira AC, Mendes CT Jr., Marano LA, Deghaide NH, Secaf M, Elias J Jr., et al. Alleles and genotypes of polymorphisms of IL-18, TNF-alpha and IFN-gamma are associated with a higher risk and severity of hepatocellular carcinoma (HCC) in Brazil. Hum Immunol (2013) 74:1024–9. doi: 10.1016/j.humimm.2013.04.029
168. Lau HK, Hsieh MJ, Yang SF, Wang HL, Kuo WH, Lee HL, et al. Association between interleukin-18 polymorphisms and hepatocellular carcinoma occurrence and clinical progression. Int J Med Sci (2016) 13:556–61. doi: 10.7150/ijms.15853
169. Sharafelldin H, Morsy A, Elghobary H, Osman E, Rady N. Association between TNF-alpha, interleukin-18 polymorphisms and risk of hepatocellular carcinoma in Egyptian patients. Asian Pac J Cancer Prev (2021) 22:887–91. doi: 10.31557/APJCP.2021.22.3.887
170. Kim YS, Cheong JY, Cho SW, Lee KM, Hwang JC, Oh B, et al. A functional SNP of the interleukin-18 gene is associated with the presence of hepatocellular carcinoma in hepatitis b virus-infected patients. Dig Dis Sci (2009) 54:2722–8. doi: 10.1007/s10620-009-0970-6
Keywords: obesity, diabetes mellitus, NAFLD, NASH, gut microbiota, inflammation, interleukin-18
Citation: Somm E and Jornayvaz FR (2022) Interleukin-18 in metabolism: From mice physiology to human diseases. Front. Endocrinol. 13:971745. doi: 10.3389/fendo.2022.971745
Received: 17 June 2022; Accepted: 23 September 2022;
Published: 12 October 2022.
Edited by:
Nabil Rabhi, Boston University, United StatesReviewed by:
Adeline Bertola, Université Côte d’Azur, CNRS, FranceCopyright © 2022 Somm and Jornayvaz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emmanuel Somm, ZW1tYW51ZWwuc29tbUB1bmlnZS5jaA==; François R. Jornayvaz, ZnJhbmNvaXMuam9ybmF5dmF6QGhjdWdlLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.