 Rong Tang
Rong Tang Wallace Y. Langdon
Wallace Y. Langdon Jian Zhang3*
Jian Zhang3*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 28 July 2022
Sec. Molecular and Structural Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.971162
This article is part of the Research TopicRegulation of Hormone and Growth factor Signalling by Ubiquitin and Ubiquitin-like Protein ModificationsView all 5 articles
Receptor tyrosine kinases (RTKs) serve as transmembrane receptors that participate in a broad spectrum of cellular processes including cellular growth, motility, differentiation, proliferation, and metabolism. Hence, elucidating the regulatory mechanisms of RTKs involved in an assortment of diseases such as cancers attracts increasing interest from researchers. Members of the Cbl family ubiquitin ligases (c-Cbl, Cbl-b and Cbl-c in mammals) have emerged as negative regulators of activated RTKs. Upon activation of RTKs by growth factors, Cbl binds to RTKs via its tyrosine kinase binding (TKB) domain and targets them for ubiquitination, thus facilitating their degradation and negative regulation of RTK signaling. RTKs such as epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGF), fibroblast growth factor receptor (FGFR) and hepatocyte growth factor receptor (HGFR) undergo ubiquitination upon interaction with Cbl family members. In this review, we summarize the current knowledge related to the negative regulation of RTKs by Cbl family proteins.
Receptor tyrosine kinases (RTKs) are a family of cell surface receptors that participate in morphogenesis, cellular fate processes and pathogenesis. Aberrantly activated RTKs are involved in various diseases such as malignancies and immunological disorders (1, 2). RTKs are tightly regulated by interacting proteins such as ubiquitin ligases. Ubiquitylation of RTKs promotes their trafficking and targeted lysosomal degradation (3, 4). The Casitas B-lineage lymphoma (Cbl) family of proteins (c-Cbl, Cbl-b and Cbl-3) are well-known negative regulators of RTK signaling through their E3 ubiquitin ligase activity (5, 6). Cbl proteins can directly interact with activated RTKs via the binding of its Src homology 2 (SH2)-like tyrosine kinase binding (TKB) domain to specific phosphotyrosine peptide motifs of RTKs, leading to the ubiquitination and degradation of RTKs (7). Here, we summarize current knowledge on the negative regulation of RTKs by Cbl family ubiquitin ligases.
RTKs are transmembrane proteins that bind extracellular growth factors to control a wide range of cellular processes such as cell growth, motility, proliferation, differentiation and metabolism (1). All RTKs have a similar protein structure consisting of an extracellular ligand binding region, a single transmembrane α-helix, and a cytoplasmic kinase domain that includes a protein tyrosine kinase domain (TKD), a carboxyl (C-) terminal tail plus a juxtamembrane regulatory region (8). Humans possess 58 known RTKs, which are subdivided into 20 different subfamilies based on their variable extracellular ligand binding domain (9, 10). Typical members of this family contain growth factor receptors such as epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), Nerve growth factor receptor (NGFR), hepatocyte growth factor receptor (HGFR) and colony-stimulating factor 1 receptor (CSF-1R) (11). Besides the insulin receptor (IR) family, other known RTKs are monomers in the membrane of cells (2). Dysregulation or mutation of RTKs and the resultant aberrant activation of downstream signaling pathways have been involved in the development and progression of diseases, such as cancers, immunological disorders and diabetes (12–14).
RTKs are routinely activated by binding receptor-specific ligands to their extracellular regions. Growth factor ligands induce the dimerization and/or oligomerization of RTKs, which leads to autophosphorylation resulting in the recruitment and activation of various downstream signaling proteins and signaling cascades (15, 16). RTKs signaling is negatively regulated by protein-tyrosine phosphatases and ubiquitin ligases. Of the negative regulators, the Cbl family emerges as the most widely studied ubiquitin ligase that associates with RTKs (6, 7) (Figure 1).
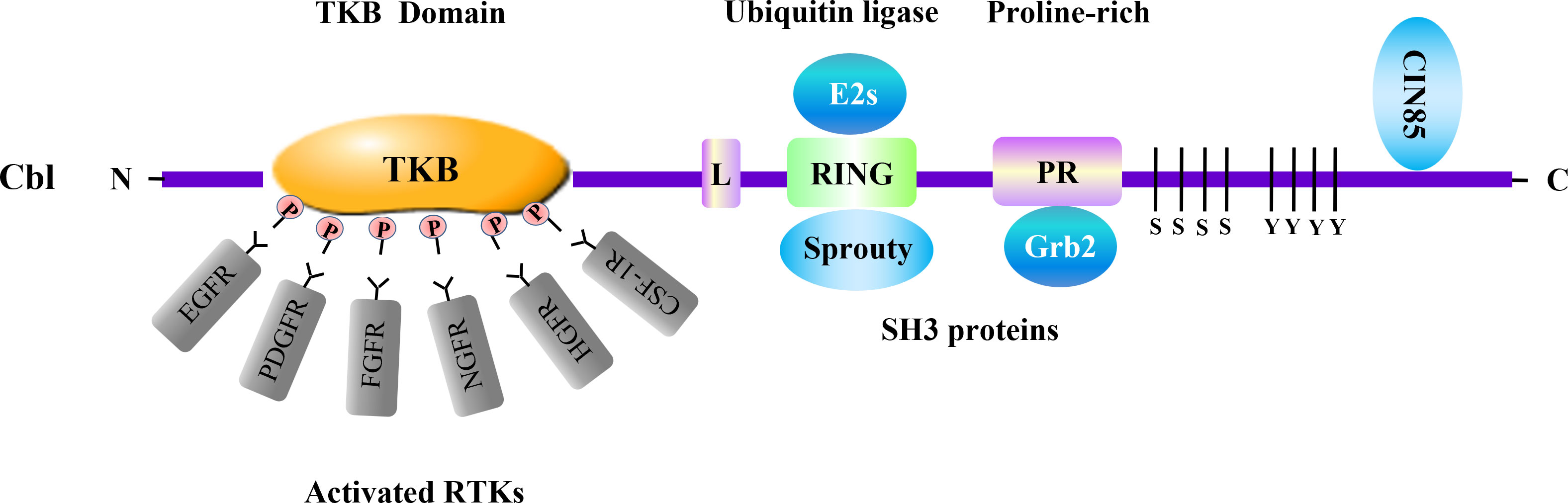
Figure 1 Cbl and adaptor proteins in negative regulation of RTKs. Cbl acts as a ubiquitin ligase involved in negative regulation of multiple RTKs. The Cbl protein consists of several domains which associate with distinct signaling transducers. Among them, the tyrosine kinase binding (TKB) domain binds to various activated RTKs. The RING-finger (RING) domain, which is crucial for the enzymatic activity of Cbl, interacts with adaptor protein Sprouty. Cbl RING finger domain can associate with E2 ubiquitin-conjugating enzymes (E2s) including Ubc4 and UbcH7. The proline-rich region (PR) serves as binding site for SH3-containing proteins such as Grb2. The SH3 domain of CIN85 can interact with the distal carboxyl terminus of Cbl. These molecules are involved in RTKs regulation conducted by Cbl. L, linker.
As an adaptor molecule and a RING-type E3 ubiquitin ligase, the Cbl family contains three distinct mammalian members, c-Cbl, Cbl-b and Cbl-3 (17). The Cbl family proteins have a conserved N-terminus composed of a tyrosine kinase binding (TKB) domain, an alpha helical linker region and a catalytic RING finger domain. In addition, c-Cbl and Cbl-b contain a less conserved C-terminal region including proline-rich (PR) regions, tyrosine phosphorylation sites and a ubiquitin-associated (UBA)/Leucine zipper (LZ) domain (18). Numerous studies have confirmed that Cbl proteins function as negative regulators in signaling pathways that include RTKs, T cell receptors (TCRs), B cell receptors (BCRs) and C-type lectin receptors (CLRs) that regulate innate and adaptive immune responses (19, 20). Cbl proteins are recruited to activated RTKs through the binding its TKB domain to phosphopeptide motifs produced by receptor autophosphorylation, targeting RTKs for ubiquitylation and degradation by E2 ubiquitin-conjugating (Ubc) enzyme (21–23). The RING domain of Cbl protein recruits the E2 Ubc enzymes including Ubc4, UbcH7, UbcH5B/C, mediating the transfer of ubiquitin from the E2 to the target RTK, synergistically support the ligand-induced ubiquitinationaa (24–26). Hence, Cbl proteins serve as negative regulator of RTKs (Table 1).

Table 1 Negative Regulation of RTKs by Cbls.
The EGFR (also recognized as ErbB1 or HER1) belongs to the ERBB family of RTKs that consists of three additional members including ErbB2/HER-2, ErbB3/HER-3, and ErbB4/HER-4 (37, 38). The EGFR emerges as a typical transmembrane receptor that triggers signaling cascades through ligand-elicited dimerization and tyrosine kinase activation (39). The EGFR possesses diverse ligands such as amphiregulin (AR), betacellulin, epidermal growth factor (EGF), transforming growth factor (TGF) -α and heparin-binding EGF. Aberrant EGFR activation stimulates multiple signaling pathways that mediate cellular dysfunctions and pathologies (40, 41).
ErbB-1 undergoes tyrosine phosphorylation in response to ligand binding, whereas overexpression of c-Cbl can mitigate this effect (27, 28, 42). c-Cbl causes the ubiquitin-dependent degradation and down-regulation of ErbB-1, but not ErbB-3. Through c-Cbl’s direct binding to phosphotyrosine 1045 (pY1045) in ErbB-1’s cytosplasmic domain, c-Cbl and ErbB-1 co-localize in endosomes, targeting ErbB-1 for lysosomal degradation through a ubiquitin-dependent process (43). Similarly, overexpression of Cbl-b or Cbl-3 in Chinese hamster ovary (CHO) cells accelerates removal of overexpressed EGFR from the cell surface, leading to its endocytosis and ubiquitination. Interestingly, alternative splicing of a short peptide of Cbl-3 (Cbl-3S), with a defective SH2 domain, does not influence ubiquitination of the EGFR (43). Thus, all three mammalian members of the Cbl family are implicated in desensitization of the EGFR. Furthermore, an endophilin-CIN85-Cbl complex induces ligand-dependent endocytosis and downregulation of the EGFR (44, 45).
The PDGF family consists of four polypeptide chains that form five biologically active isoforms: PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC and PDGF-DD (46). The PDGF ligands exhibit cellular effects by binding to two tyrosine kinase receptors: PDGFR-α and PDGFR-β. PDGFR-α and PDGFR-β bind to different ligands with diverse affinities, and they have similar but different activities (47). Ligand binding promotes dimerization, autophosphorylation and activation of the tyrosine kinase domain in PDGFRs (48, 49).
Stimulation of PDGFRs facilitates phosphorylation of c-Cbl, as well as their physical interaction (29). In turn, c-Cbl overexpression accelerates ligand-induced ubiquitination and subsequent degradation of PDGFR-α and PDGFR-β, as well as inhibiting proliferation and survival dependence by PDGF (50, 51). c-Cbl is able to negatively regulate PDGFR-dependent biological functions, which requires the intact tyrosine kinase binding domain of c-Cbl (30, 31, 50). Both c-Cbl and Cbl-b impact PDGFRβ polyubiquitination and internalization of the PDGFR stimulated by its ligand. Cbl-b together with c-Cbl form a complex which also interacts with PDGFRβ after PDGF-BB stimulation. c-Cbl is unable to bind directly to PDGFRβ, indicating that Cbl-b is essential for the interaction of c-Cbl with PDGFRβ (52).
The mammalian FGF family comprises 22 members which have homologous central protein sequences and structure. FGFs interact with four high affinity cell-surface RTKs designated FGFR1-4 (53–55). Binding of FGFs to FGFRs initiates intrinsic tyrosine kinase activity and multiple downstream signaling cascades, such as RAS-MAP and PI3K-AKT, mediating a wide range of cellular responses (32, 56).
FGFR2 activation recruits c-Cbl binding to the receptor, allowing ubiquitination and proteasome degradation of FGFR2 (33). Thus, c-Cbl mediates down-regulation of FGFR2 and attenuation of FGFR2 signaling, which results in PI3K/Akt attenuation and decreased osteoblast survival triggered by FGFR2 activation. On the contrary, no direct interaction has been observed between c-Cbl and FGFR3 during ubiquitination of FGFR3 (57, 58). Interestingly, a correlation has been found between c-Cbl expression and FGFR3 activity. Overactive FGFR3 mutations yield c-Cbl overexpression, while c-Cbl does not influence FGFR3 expression and ubiquitylation (59).
NGF, a member of the neurotrophin family, is expressed in both the nervous system and peripheral organs (60). NGF/NGFR signaling contributes to the development of the nervous system, angiogenesis and inflammatory diseases. NGF exerts its effects through binding to one of its two receptors: the high-affinity receptor tropomyosin receptor kinase A (TrkA) and the low-affinity receptor p75 neurotrophin receptor (p75) (34, 61). Binding of NGF to its receptors causes receptor dimerization, autophosphorylation and activation, which enhances the phosphorylation of downstream cellular proteins and signal transduction (62).
In response to NGF stimulation, c-Cbl is capable of mediating the internalization, endosomal trafficking, ligand-induced ubiquitination and subsequent lysosomal degradation of TrkA. TrkA ubiquitination and degradation also require direct association between c-Cbl and phosphorylated TrkA (63).
HGF, also known as scatter factor (SF), is mainly expressed in is stromal cells and fibroblasts. Mesenchymal epithelial transition factor (Met) the receptor of HGF, is primarily produced by epithelial cells (64). Following stimulation with HGF, the receptor tyrosine kinase Met is auto-phosphorylated and activated in the cytoplasm, triggering intracellular signaling cascade activation (35, 65).
c-Cbl, but not its oncogenic mutants v-Cbl or 70Z/3 Cbl, targets Met for ubiquitylation and degradation to downregulate HGF/Met signaling (66). c-Cbl is recruited to Met though two mechanisms: direct interaction with the juxtamembrane domain of Met by its TKB domain, and indirect interaction through Grb2 via its proline-rich domain (67). After binding to ligand-activated Met, c-Cbl undergoes tyrosine phosphorylation, and ubiquitinates Met through recruiting the endophilin-CIN85 complex, resulting in suppression of signal transduction and biological responses. Inhibition of this complex formation is able to prevent downregulation of Met. Thereby, the endophilin-CIN85-Cbl complex is involved in ligand-dependent Met downregulation (68).
CSF-1 serves as a growth factor that participates in the regulation of proliferation, differentiation and survival of mononuclear phagocytes (69). The biological activity of CSF-1 is mediated by its high-affinity cognate receptor CSF-1R encoded by the c-fms proto-oncogene. CSF-1 induces CSF-1R dimerization, resulting in the autophosphorylation of tyrosine residues in the cytoplasmic portion of the CSF-1R, which are the binding sites for Src homology 2 (SH2) containing proteins (36, 70).
c-Cbl down-regulates the CSF-1R by targeting it for polyubiquitination and subsequent enhancement in the endocytic rate, which inhibits macrophage proliferation in response to CSF-1 stimulation (71). Further study has confirmed that activated CSF-1R leads to autophosphorylation of multiple tyrosine residues, such as Tyr973 at its carboxy-terminus. The c-Cbl TKB domain binds to activated CSF-1R at the phosphorylated residue Tyr 973, resulting in CSF-1R signaling cessation (72).
CIN85, recognized as Cbl-interacting molecule of 85 kDa, is a modular-assembled adaptor protein (73). CIN85 consists of a proline-rich region, three SH3 domains and a coiled-coil region. The SH3 domains of CIN85 bind to the distal carboxyl termini of Cbl and Cbl-b, but not to the proline-rich region. There is no association between CIN85 and Cbl-3 in mammalian cells (74, 75).
Studies have indicated that CIN85 and Cbl family interactions are involved in the regulation of activated RTKs. These associations are further enhanced upon tyrosine phosphorylation of c-Cbl or Cbl-b stimulated after EGF and PDGF activation. CIN85 binding to Cbl-b is essential for internalization of EGFR, however it has no direct influence on receptor ubiquitination stimulated by Cbl-b (44). Further, c-Cbl has been found to mediate RTK endocytosis via an interaction with the CIN85-endophilin complex. The Cbl-CIN85-endophilin association induces ligand-stimulation degradation of the EGFR and c-Met. Suppression of the Cbl-CIN85-endophilin complex formation is enough to prevent RTK endocytosis and downregulation, without affecting the ubiquitination function of c-Cbl (45, 68).
Growth factor receptor bound protein 2 (Grb2) is a widely expressed adaptor molecule, which mediates various basic cellular functions and downstream signaling pathways (76). Grb2 consists of a Src homology2 (SH2) domain flanked by N- and C-terminal SH3 domains. The Grb2 SH2 domain associates directly with the tyrosine residues in autophosphorylated EGFR, and Grb2 also binds to other RTKs, such as HGFR (77).
Proline-rich sequences of c-Cbl interact with the SH3 domains of Grb2, which indirectly recruit c-Cbl to EGFRs or Met receptors (78, 79). Grb2 negatively regulates RTKs and downstream signaling, including the Ras pathway, through the recruitment of c-Cbl (80). Tyr1045 mutant EGFR, defective in the c-Cbl docking site, shows reduced ubiquitylation and endocytosis, even under the condition of c-Cbl overexpression. Unexpectedly, an EGFR mutant defective at Tyr1045 (Y1045F) still displays ubiquitination and downregulation, most notably in the presence of c-Cbl overexpression and Grb2, because the Grb2/c-Cbl complex is recruited to Grb2 docking sites on the EGFR (78). Additionally, other studies have shown that Grb2 exerts positive effects on RTK signaling and activates the Ras pathway via its interaction with guanine nucleotide exchange factor Sos (16). Hence, Grb2 acts as a double-edged sword in its regulation of RTKs signaling.
Sprouty was first discovered in Drosophila as a new antagonist of the FGF signaling pathway. There are four Sprouty isoforms in mammals, which include Sprouty1-4 ( (81). The Sprouty proteins are identified as an additional family of putative signaling regulators. The Sprouty family is shown to inhibit RTKs, specifically by suppressing downstream Ras/Raf/ERK signaling activation induced by growth factors such as FGF, PDGF, NGF and VEGF (82, 83).
Sprouty2 can directly bind to the RING finger domain of c-Cbl, and then remove c-Cbl from activated EGFR. As a result, Sprouty2 abrogates c-Cbl-mediated EGFR internalization and ubiquitylation, thus sustaining downstream receptor signaling (84–87). Other Sprouty homologs, such as Sprouty3 and Sprouty4, do not affect EGFR degradation, although they have the c-Cbl-binding motif (80). Moreover, Sprouty2 interacts with CIN85, and functions at the Cbl/CIN85 interface following EGF stimulation, consequently blocking EGFR downregulation (88).
Although Cbl family proteins have been investigated widely in immune responses, extensive studies also indicate their crucial role in RTK signaling. Overall, Cbl family proteins interact with activated RTKs via binding its TKB domain to phosphopeptide motifs of activated RTKs, leading to RTK ubiquitylation and degradation. Importantly, Cbl family proteins can also recruit adaptor molecules (i.e. CIN85, Grb2 and Sprouty) to RTKs, which participate in the regulation of RTK signaling. The ability of Cbl proteins to interact with diverse RTKs, which are implicated in pathogenesis of varied diseases, indicate this family of proteins as attractive targets for therapeutic intervention. However, further research is still required to fully understand the underlying molecular mechanisms of the Cbl family in the regulation of RTKs, which may provide new clues to clinical applications in the future.
RT and JZ wrote and edited the manuscript. WL edited the manuscript.
This work is supported by the National Institutes of Health (NIH) grants R01 AI090901, AI121196, and 123253 (to JZ). JZ is a University of Iowa Health Care Distinguished Scholar.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
2. Choura M, Rebai A. Receptor tyrosine kinases: from biology to pathology. J Recept Signal Transduct Res (2011) 31:387–94. doi: 10.3109/10799893.2011.625425
3. Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene (2004) 23:2057–70. doi: 10.1038/sj.onc.1207390
4. Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol (2003) 5:461–6. doi: 10.1038/ncb983
5. Duan L, Reddi AL, Ghosh A, Dimri M, Band H. The cbl family and other ubiquitin ligases: destructive forces in control of antigen receptor signaling. Immunity (2004) 21:7–17. doi: 10.1016/j.immuni.2004.06.012
6. Keane MM, Rivero-Lezcano OM, Mitchell JA, Robbins KC, Lipkowitz S. Cloning and characterization of cbl-b: a SH3 binding protein with homology to the c-cbl proto-oncogene. Oncogene (1995) 10:2367–77.
7. Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC. The tyrosine kinase negative regulator c-cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science (1999) 286:309–12. doi: 10.1126/science.286.5438.309
8. Hubbard SR. Structural analysis of receptor tyrosine kinases. Prog Biophys Mol Biol (1999) 71:343–58. doi: 10.1016/s0079-6107(98)00047-9
9. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science (2002) 298:1912–34. doi: 10.1126/science.1075762
10. Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene (2000) 19:5548–57. doi: 10.1038/sj.onc.1203957
11. Trenker R, Jura N. Receptor tyrosine kinase activation: From the ligand perspective. Curr Opin Cell Biol (2020) 63:174–85. doi: 10.1016/j.ceb.2020.01.016
12. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer (2018) 17:58. doi: 10.1186/s12943-018-0782-4
13. LaVallee TM, Alvarado D, Garton AJ, Trombetta ES, Gedrich R, McMahon G. Emerging receptor tyrosine kinase drug targets: Implications for antibody-based therapies for oncology and immunologic disorders. Crit Rev Oncog (2015) 20:485–508. doi: 10.1615/CritRevOncog.v20.i5-6.170
14. Fountas A, Diamantopoulos LN, Tsatsoulis A. Tyrosine kinase inhibitors and diabetes: A novel treatment paradigm? Trends Endocrinol Metab (2015) 26:643–56. doi: 10.1016/j.tem.2015.09.003
15. Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol (2007) 19:117–23. doi: 10.1016/j.ceb.2007.02.010
16. Dikic I, Giordano S. Negative receptor signalling. Curr Opin Cell Biol (2003) 15:128–35. doi: 10.1016/s0955-0674(03)00004-8
17. Tsygankov AY, Teckchandani AM, Feshchenko EA, Swaminathan G. Beyond the RING: CBL proteins as multivalent adapters. Oncogene (2001) 20:6382–402. doi: 10.1038/sj.onc.1204781
18. Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Structure of the amino-terminal domain of cbl complexed to its binding site on ZAP-70 kinase. Nature (1999) 398:84–90. doi: 10.1038/18050
19. Tang R, Langdon WY, Zhang J. Regulation of immune responses by E3 ubiquitin ligase cbl-b. Cell Immunol (2019) 340:103878. doi: 10.1016/j.cellimm.2018.11.002
20. Swaminathan G, Tsygankov AY. The cbl family proteins: ring leaders in regulation of cell signaling. J Cell Physiol (2006) 209:21–43. doi: 10.1002/jcp.20694
21. Mohapatra B, Ahmad G, Nadeau S, Zutshi N, An W, Scheffe S, et al. Protein tyrosine kinase regulation by ubiquitination: critical roles of cbl-family ubiquitin ligases. Biochim Biophys Acta (2013) 1833:122–39. doi: 10.1016/j.bbamcr.2012.10.010
22. Rubin C, Gur G, Yarden Y. Negative regulation of receptor tyrosine kinases: unexpected links to c-cbl and receptor ubiquitylation. Cell Res (2005) 15:66–71. doi: 10.1038/sj.cr.7290268
23. Thien CB, Langdon WY. Negative regulation of PTK signalling by cbl proteins. Growth Factors (2005) 23:161–7. doi: 10.1080/08977190500153763
24. Yokouchi M, Kondo T, Houghton A, Bartkiewicz M, Horne WC, Zhang H, et al. Ligand-induced ubiquitination of the epidermal growth factor receptor involves the interaction of the c-cbl RING finger and UbcH7. J Biol Chem (1999) 274:31707–12. doi: 10.1074/jbc.274.44.31707
25. Liyasova MS, Ma K, Voeller D, Ryan PE, Chen J, Klevit RE, et al. Cbl interacts with multiple E2s in vitro and in cells. PloS One (2019) 14:e0216967. doi: 10.1371/journal.pone.0216967
26. Thien CB, Langdon WY. C-cbl and cbl-b ubiquitin ligases: substrate diversity and the negative regulation of signalling responses. Biochem J (2005) 391:153–66. doi: 10.1042/BJ20050892
27. Waterman H, Levkowitz G, Alroy I, Yarden Y. The RING finger of c-cbl mediates desensitization of the epidermal growth factor receptor. J Biol Chem (1999) 274:22151–4. doi: 10.1074/jbc.274.32.22151
28. Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, et al. C-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev (1998) 12:3663–74. doi: 10.1101/gad.12.23.3663
29. Tang Z, Wang Y, Fan Y, Zhu Y, Chien S, Wang N. Suppression of c-cbl tyrosine phosphorylation inhibits neointimal formation in balloon-injured rat arteries. Circulation (2008) 118:764–72. doi: 10.1161/CIRCULATIONAHA.107.761932
30. Martinez-Martinez E, Tolle R, Donauer J, Gretzmeier C, Bruckner-Tuderman L, Dengjel J. Increased abundance of cbl E3 ligases alters PDGFR signaling in recessive dystrophic epidermolysis bullosa. Matrix Biol (2021) 103-104:58–73. doi: 10.1016/j.matbio.2021.10.004
31. Miyake S, Lupher ML Jr., Druker B, Band H. The tyrosine kinase regulator cbl enhances the ubiquitination and degradation of the platelet-derived growth factor receptor alpha. Proc Natl Acad Sci USA (1998) 95:7927–32. doi: 10.1073/pnas.95.14.7927
32. Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev (2005) 16:139–49. doi: 10.1016/j.cytogfr.2005.01.001
33. Xian W, Schwertfeger KL, Rosen JM. Distinct roles of fibroblast growth factor receptor 1 and 2 in regulating cell survival and epithelial-mesenchymal transition. Mol Endocrinol (2007) 21:987–1000. doi: 10.1210/me.2006-0518
34. Neet KE, Campenot RB. Receptor binding, internalization, and retrograde transport of neurotrophic factors. Cell Mol Life Sci (2001) 58:1021–35. doi: 10.1007/PL00000917
35. Maroun CR, Rowlands T. The met receptor tyrosine kinase: a key player in oncogenesis and drug resistance. Pharmacol Ther (2014) 142:316–38. doi: 10.1016/j.pharmthera.2013.12.014
36. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood (2002) 99:111–20. doi: 10.1182/blood.v99.1.111
37. Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell (2003) 11:507–17. doi: 10.1016/s1097-2765(03)00047-9
38. Burgess AW. EGFR family: structure physiology signalling and therapeutic targets. Growth Factors (2008) 26:263–74. doi: 10.1080/08977190802312844
39. Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res (2006) 12:5268–72. doi: 10.1158/1078-0432.CCR-05-1554
40. Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer (2001) 37(Suppl 4):S3–8. doi: 10.1016/s0959-8049(01)00230-1
41. Sabbah DA, Hajjo R, Sweidan K. Review on epidermal growth factor receptor (EGFR) structure, signaling pathways, interactions, and recent updates of EGFR inhibitors. Curr Top Med Chem (2020) 20:815–34. doi: 10.2174/1568026620666200303123102
42. Galisteo ML, Dikic I, Batzer AG, Langdon WY, Schlessinger J. Tyrosine phosphorylation of the c-cbl proto-oncogene protein product and association with epidermal growth factor (EGF) receptor upon EGF stimulation. J Biol Chem (1995) 270:20242–5. doi: 10.1074/jbc.270.35.20242
43. Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell (1999) 4:1029–40. doi: 10.1016/s1097-2765(00)80231-2
44. Haglund K, Shimokawa N, Szymkiewicz I, Dikic I. Cbl-directed monoubiquitination of CIN85 is involved in regulation of ligand-induced degradation of EGF receptors. Proc Natl Acad Sci USA (2002) 99:12191–6. doi: 10.1073/pnas.192462299
45. Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature (2002) 416:183–7. doi: 10.1038/416183a
47. Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev (2004) 15:197–204. doi: 10.1016/j.cytogfr.2004.03.007
48. Board R, Jayson GC. Platelet-derived growth factor receptor (PDGFR): a target for anticancer therapeutics. Drug Resist Updat (2005) 8:75–83. doi: 10.1016/j.drup.2005.03.004
49. Lewis NL. The platelet-derived growth factor receptor as a therapeutic target. Curr Oncol Rep (2007) 9:89–95. doi: 10.1007/s11912-007-0003-6
50. Miyake S, Mullane-Robinson KP, Lill NL, Douillard P, Band H. Cbl-mediated negative regulation of platelet-derived growth factor receptor-dependent cell proliferation. a critical role for cbl tyrosine kinase-binding domain. J Biol Chem (1999) 274:16619–28. doi: 10.1074/jbc.274.23.16619
51. Reddi AL, Ying G, Duan L, Chen G, Dimri M, Douillard P, et al. Binding of cbl to a phospholipase Cgamma1-docking site on platelet-derived growth factor receptor beta provides a dual mechanism of negative regulation. J Biol Chem (2007) 282:29336–47. doi: 10.1074/jbc.M701797200
52. Rorsman C, Tsioumpekou M, Heldin CH, Lennartsson J. The ubiquitin ligases c-cbl and cbl-b negatively regulate platelet-derived growth factor (PDGF) BB-induced chemotaxis by affecting PDGF receptor beta (PDGFRbeta) internalization and signaling. J Biol Chem (2016) 291:11608–18. doi: 10.1074/jbc.M115.705814
53. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem (2011) 149:121–30. doi: 10.1093/jb/mvq121
54. Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol (2001) 2:REVIEWS3005. doi: 10.1186/gb-2001-2-3-reviews3005
55. Jaye M, Schlessinger J, Dionne CA. Fibroblast growth factor receptor tyrosine kinases: molecular analysis and signal transduction. Biochim Biophys Acta (1992) 1135:185–99. doi: 10.1016/0167-4889(92)90136-y
56. Korsensky L, Ron D. Regulation of FGF signaling: Recent insights from studying positive and negative modulators. Semin Cell Dev Biol (2016) 53:101–14. doi: 10.1016/j.semcdb.2016.01.023
57. Dufour C, Guenou H, Kaabeche K, Bouvard D, Sanjay A, Marie PJ. FGFR2-cbl interaction in lipid rafts triggers attenuation of PI3K/Akt signaling and osteoblast survival. Bone (2008) 42:1032–9. doi: 10.1016/j.bone.2008.02.009
58. Kaabeche K, Lemonnier J, Le Mee S, Caverzasio J, Marie PJ. Cbl-mediated degradation of Lyn and fyn induced by constitutive fibroblast growth factor receptor-2 activation supports osteoblast differentiation. J Biol Chem (2004) 279:36259–67. doi: 10.1074/jbc.M402469200
59. Monsonego-Ornan E, Adar R, Rom E, Yayon A. FGF receptors ubiquitylation: dependence on tyrosine kinase activity and role in downregulation. FEBS Lett (2002) 528:83–9. doi: 10.1016/s0014-5793(02)03255-6
60. Rocco ML, Soligo M, Manni L, Aloe L. Nerve growth factor: Early studies and recent clinical trials. Curr Neuropharmacol (2018) 16:1455–65. doi: 10.2174/1570159X16666180412092859
61. Bracci-Laudiero L, De Stefano ME. NGF in early embryogenesis, differentiation, and pathology in the nervous and immune systems. Curr Top Behav Neurosci (2016) 29:125–52. doi: 10.1007/7854_2015_420
62. Hirose M, Kuroda Y, Murata E. NGF/TrkA signaling as a therapeutic target for pain. Pain Pract (2016) 16:175–82. doi: 10.1111/papr.12342
63. Takahashi Y, Shimokawa N, Esmaeili-Mahani S, Morita A, Masuda H, Iwasaki T, et al. Ligand-induced downregulation of TrkA is partly regulated through ubiquitination by cbl. FEBS Lett (2011) 585:1741–7. doi: 10.1016/j.febslet.2011.04.056
64. Fu J, Su X, Li Z, Deng L, Liu X, Feng X, et al. HGF/c-MET pathway in cancer: from molecular characterization to clinical evidence. Oncogene (2021) 40:4625–51. doi: 10.1038/s41388-021-01863-w
65. Hammond DE, Carter S, Clague MJ. Met receptor dynamics and signalling. Curr Top Microbiol Immunol (2004) 286:21–44. doi: 10.1007/978-3-540-69494-6_2
66. Taher TE, Tjin EP, Beuling EA, Borst J, Spaargaren M, Pals ST. C-cbl is involved in met signaling in b cells and mediates hepatocyte growth factor-induced receptor ubiquitination. J Immunol (2002) 169:3793–800. doi: 10.4049/jimmunol.169.7.3793
67. Gao CF, Vande Woude GF. HGF/SF-met signaling in tumor progression. Cell Res (2005) 15:49–51. doi: 10.1038/sj.cr.7290264
68. Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S. The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-met. Nature (2002) 416:187–90. doi: 10.1038/416187a
69. Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y, et al. Biology and action of colony–stimulating factor-1. Mol Reprod Dev (1997) 46:4–10. doi: 10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V
70. Hamilton JA. CSF-1 signal transduction. J Leukoc Biol (1997) 62:145–55. doi: 10.1002/jlb.62.2.145
71. Lee PS, Wang Y, Dominguez MG, Yeung YG, Murphy MA, Bowtell DD, et al. The cbl protooncoprotein stimulates CSF-1 receptor multiubiquitination and endocytosis, and attenuates macrophage proliferation. EMBO J (1999) 18:3616–28. doi: 10.1093/emboj/18.13.3616
72. Wilhelmsen K, Burkhalter S, van der Geer P. C-cbl binds the CSF-1 receptor at tyrosine 973, a novel phosphorylation site in the receptor's carboxy-terminus. Oncogene (2002) 21:1079–89. doi: 10.1038/sj.onc.1205166
73. Dikic I. CIN85/CMS family of adaptor molecules. FEBS Lett (2002) 529:110–5. doi: 10.1016/s0014-5793(02)03188-5
74. Take H, Watanabe S, Takeda K, Yu ZX, Iwata N, Kajigaya S. Cloning and characterization of a novel adaptor protein, CIN85, that interacts with c-cbl. Biochem Biophys Res Commun (2000) 268:321–8. doi: 10.1006/bbrc.2000.2147
75. Szymkiewicz I, Kowanetz K, Soubeyran P, Dinarina A, Lipkowitz S, Dikic I. CIN85 participates in cbl-b-mediated down-regulation of receptor tyrosine kinases. J Biol Chem (2002) 277:39666–72. doi: 10.1074/jbc.M205535200
76. Chardin P, Cussac D, Maignan S, Ducruix A. The Grb2 adaptor. FEBS Lett (1995) 369:47–51. doi: 10.1016/0014-5793(95)00578-w
77. Wada S, Sasaki Y, Horimoto M, Ito T, Ito Y, Tanaka Y, et al. Involvement of growth factor receptor-bound protein-2 in rat hepatocyte growth. J Gastroenterol Hepatol (1998) 13:635–42. doi: 10.1111/j.1440-1746.1998.tb00702.x
78. Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, Elson A, et al. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J (2002) 21:303–13. doi: 10.1093/emboj/21.3.303
79. Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, et al. Mutation of the c-cbl TKB domain binding site on the met receptor tyrosine kinase converts it into a transforming protein. Mol Cell (2001) 8:995–1004. doi: 10.1016/s1097-2765(01)00378-1
80. Belov AA, Mohammadi M. Grb2, a double-edged sword of receptor tyrosine kinase signaling. Sci Signal (2012) 5:pe49. doi: 10.1126/scisignal.2003576
81. Guy GR, Jackson RA, Yusoff P, Chow SY. Sprouty proteins: modified modulators, matchmakers or missing links? J Endocrinol (2009) 203:191–202. doi: 10.1677/JOE-09-0110
82. Casci T, Vinos J, Freeman M. Sprouty, an intracellular inhibitor of ras signaling. Cell (1999) 96:655–65. doi: 10.1016/s0092-8674(00)80576-0
83. Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol (2006) 16:45–54. doi: 10.1016/j.tcb.2005.11.004
84. Wong ES, Lim J, Low BC, Chen Q, Guy GR. Evidence for direct interaction between sprouty and cbl. J Biol Chem (2001) 276:5866–75. doi: 10.1074/jbc.M006945200
85. Wong ES, Fong CW, Lim J, Yusoff P, Low BC, Langdon WY, et al. Sprouty2 attenuates epidermal growth factor receptor ubiquitylation and endocytosis, and consequently enhances Ras/ERK signalling. EMBO J (2002) 21:4796–808. doi: 10.1093/emboj/cdf493
86. Fong CW, Leong HF, Wong ES, Lim J, Yusoff P, Guy GR. Tyrosine phosphorylation of Sprouty2 enhances its interaction with c-cbl and is crucial for its function. J Biol Chem (2003) 278:33456–64. doi: 10.1074/jbc.M301317200
87. Rubin C, Litvak V, Medvedovsky H, Zwang Y, Lev S, Yarden Y. Sprouty fine-tunes EGF signaling through interlinked positive and negative feedback loops. Curr Biol (2003) 13:297–307. doi: 10.1016/s0960-9822(03)00053-8
Keywords: receptor tyrosine kinases, cbl, ubiquitination, negative regulation, degradation
Citation: Tang R, Langdon WY and Zhang J (2022) Negative regulation of receptor tyrosine kinases by ubiquitination: Key roles of the Cbl family of E3 ubiquitin ligases. Front. Endocrinol. 13:971162. doi: 10.3389/fendo.2022.971162
Received: 16 June 2022; Accepted: 11 July 2022;
Published: 28 July 2022.
Edited by:
Sudha K. Shenoy, Duke University, United StatesReviewed by:
Birgit Leitinger, Imperial College London, United KingdomCopyright © 2022 Tang, Langdon and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Zhang, amlhbi16aGFuZ0B1aW93YS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.