 Toshimi Michigami
Toshimi Michigami- Department of Bone and Mineral Research, Research Institute, Osaka Women’s and Children’s Hospital, Osaka Prefectural Hospital Organization, Izumi, Japan
Osteocytes are dendritic cells in the mineralized bone matrix that descend from osteoblasts. They play critical roles in controlling bone mass through the production of sclerostin, an inhibitor of bone formation, and receptor activator of nuclear factor κ B ligand, an inducer of osteoblastic bone resorption. Osteocytes also govern phosphate homeostasis through the production of fibroblast growth factor 23 (FGF23), which lowers serum phosphate levels by increasing renal phosphate excretion and reducing the synthesis of 1,25-dihydroxyvitamin D (1,25(OH)2D), an active metabolite of vitamin D. The production of FGF23 in osteocytes is regulated by various local and systemic factors. Phosphate-regulating gene homologous to endopeptidase on X chromosome (PHEX), dentin matrix protein 1 (DMP1), and family with sequence similarity 20, member C function as local negative regulators of FGF23 production in osteocytes, and their inactivation causes the overproduction of FGF23 and hypophosphatemia. Sclerostin has been suggested to regulate the production of FGF23, which may link the two functions of osteocytes, namely, the control of bone mass and regulation of phosphate homeostasis. Systemic regulators of FGF23 production include 1,25(OH)2D, phosphate, parathyroid hormone, insulin, iron, and inflammation. Therefore, the regulation of FGF23 in osteocytes is complex and multifactorial. Recent mouse studies have suggested that decreases in serum phosphate levels from youth to adulthood are caused by growth-related increases in FGF23 production by osteocytes, which are associated with the down-regulation of Phex and Dmp1.
Introduction
Osteocytes, which are terminally differentiated cells of the osteoblast lineage, are dendritic cells embedded within the mineralized bone matrix (1–3). Although osteocytes are the most abundant among all cells in bone, their location and inaccessibility has delayed our understanding of their function at the molecular level. In the past few decades, mounting evidence has indicated that osteocytes play important roles in bone homeostasis. They produce sclerostin, a secreted potent suppressor of bone formation (4, 5). Furthermore, critical roles for osteocyte-derived receptor activator of nuclear factor κ B ligand (RANKL) in the control of postnatal bone resorption have been demonstrated in mouse models in which its expression was specifically deleted from osteocytes (6).
Phosphate is an essential nutrient that mediates the majority of biological processes (7). Fibroblast growth factor 23 (FGF23), which functions as a central regulator of phosphate metabolism in mammals, is mainly produced by osteocytes (8). In addition to FGF23, several other molecules responsible for phosphate homeostasis are highly expressed in osteocytes, which include phosphate-regulating gene homologous to endopeptidase on X chromosome (PHEX), dentin matrix protein 1 (DMP1), and family with sequence similarity 20, member C (FAM20C), the genes responsible for hereditary hypophosphatemia (8–12). Current concepts on the molecular mechanisms by which osteocytes regulate phosphate metabolism are discussed herein.
Osteocyte differentiation from osteoblasts
Osteocytes account for 90-95% of all bone cells in adult bone and have the longest lifespan (1–3). In the process of osteocytogenesis, a subpopulation of matrix-producing osteoblasts on the bone surface become embedded within the matrix proteins they produce and differentiate into osteocytes with a decrease in the production of the bone matrix, marked changes in morphology, and the expression of genes that constitute the signature of osteocytes (1–3). Approximately 5 to 20% of osteoblasts mature into osteocytes, while the remainder die by apoptosis or become bone lining cells (3). During the maturation of osteoblasts into osteocytes, cell morphology changes to a stellate shape with long processes. Osteocytes reside in lacunae within the mineralized bone matrix, and interconnect with each other and with osteoblasts on the bone surface by their long cytoplasmic processes running through canaliculi. As osteoblasts differentiate into osteocytes, they acquire the expression of molecules that regulate bone homeostasis and phosphate metabolism (1–3).
Control of bone mass by osteocytes
Osteocytes embedded in the bone matrix sense mechanical signals and regulate bone formation and resorption. A previous study reported that the genetic ablation of osteocytes in mice led to osteoporotic bone loss and the suppression of mechanically-induced new bone formation (13). Sclerostin encoded by the SOST gene is secreted by osteocytes and suppresses bone formation (5). The inactivation and reduced expression of SOST in humans have been shown to be responsible for rare bone sclerosing diseases, such as sclerosteosis 1 and van Buchem diseases (14). Sclerostin binds to low-density lipoprotein receptor-related protein 5 (LRP5) and LRP6 to inhibit Wnt/ß-catenin signaling (15). Wnt/ß-catenin signaling plays a critical role in controlling bone mass by promoting the commitment of mesenchymal progenitor cells into osteoblasts as well as the proliferation and differentiation of osteoblasts. Furthermore, Wnt/ß-catenin signaling has been shown to inhibit the differentiation and activation of osteoclasts (16, 17). Mechanical signaling was found to suppress the expression of sclerostin in osteocytes, which promoted bone formation by enhancing Wnt/β-catenin signaling (4). The mechanosensor channel Piezo1 has recently been suggested to be involved in the suppression of Sost expression by mechanical force (18). The bone anabolic effects of parathyroid hormone (PTH) are also mediated by the down-regulation of Sost (19).
Osteocytes also regulate osteoclastic bone resorption by producing RANKL. Mice with the conditional deletion of RANKL from osteocytes and some mature osteoblasts exhibited markedly impaired osteoclastic bone resorption after birth, leading to the osteopetrotic phenotype (6). Therefore, osteocytes play critical roles in bone homeostasis by controlling the formation and resorption of bone in postnatal life.
Production and effects of FGF23
As osteoblasts mature into osteocytes, they acquire the expression of various molecules involved in phosphate homeostasis, which include the genes responsible for hereditary hypophosphatemic diseases (1–3, 8). The high expression of these molecules indicates that osteocytes play essential roles in the regulation of phosphate metabolism as well as the control of bone mass.
FGF23, the key regulator in phosphate metabolism, consists of 251 amino acids including an amino-terminal signal sequence of 24 amino acids (20). It is mainly produced by osteocytes and exerts its effects on distant target organs, such as the kidneys. Its endocrine function is suggested to be conferred by its low binding affinity to heparin/heparan sulfate (21). In the kidneys, the main target for FGF23, it increases phosphate excretion by reducing the expression of type IIa and IIc sodium/phosphate (Na+/Pi) co-transporters (designated as NaPi-IIa and NaPi-IIc, respectively) (20). In addition, FGF23 reduces the production of 1,25-dihydroxyvitamin D [1,25(OH)2D], an active metabolite of vitamin D, by suppressing the expression of 25-hydroxyvitamin D 1α-hydroxylase and inducing that of 24-hydroxylase, which leads to the decreased absorption of phosphate in the intestines (20). At physiological concentrations, FGF23 requires a single-pass transmembrane protein, αKlotho as a co-receptor for its signal transduction through the FGF receptor (FGFR) (22, 23). FGF23 is inactivated by proteolytic cleavage between Arg179 and Ser180. This cleavage is prevented by the O-glycosylation of FGF23 at Thr178, a process that is mediated by UDP-N-acetyl-α-D-galacosamine:polypeptide N-acetylgalactosaminyltransferase 3 (GalNAc-T3) (24).
Figure 1 summarizes the roles of osteocytes in the control of bone mass and the regulation of phosphate metabolism.
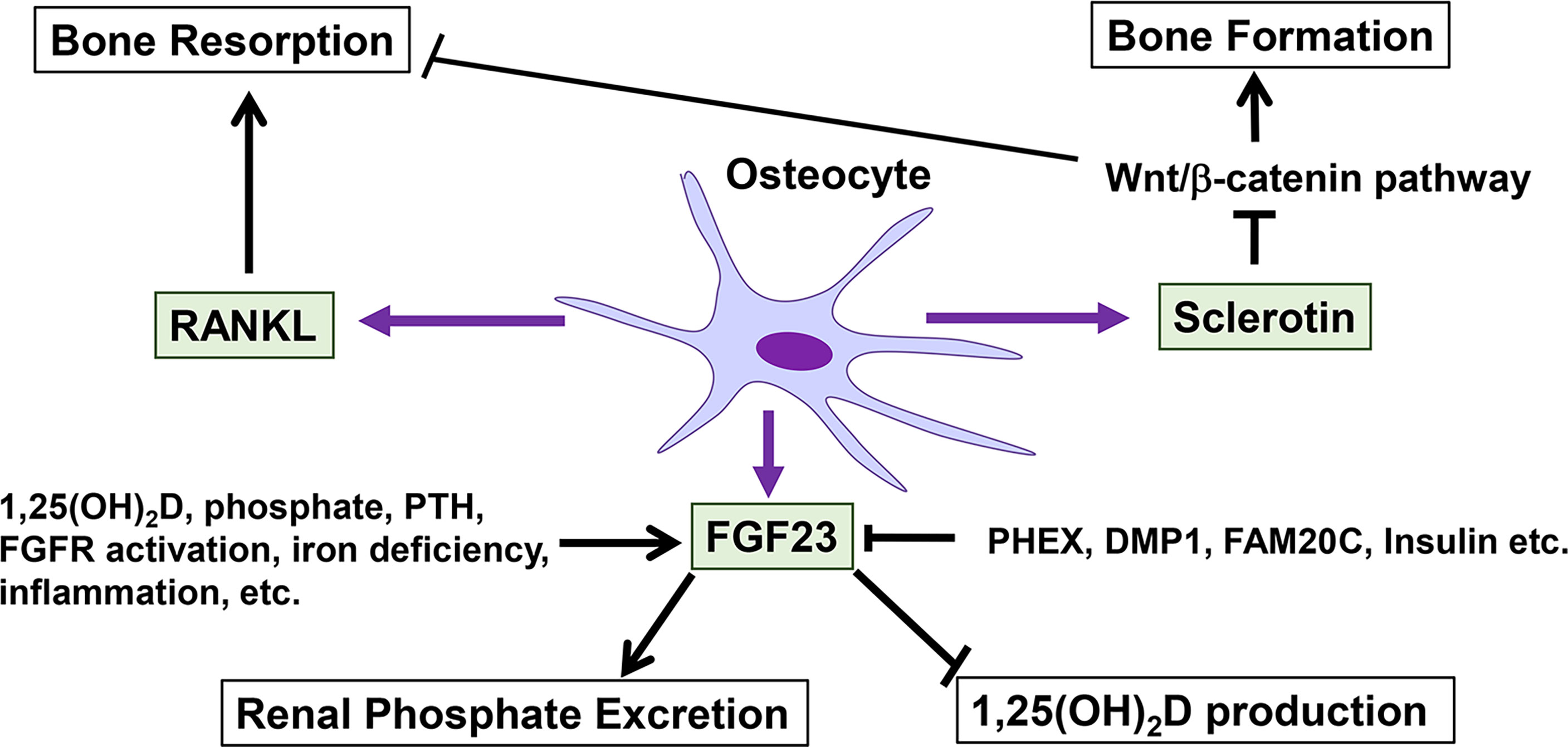
Figure 1 Roles of osteocytes in the control of bone mass and the regulation of phosphate metabolism. Osteocytes regulate bone formation and resorption through the production of sclerostin and RANKL. Sclerostin suppresses bone formation by inhibiting Wnt/β-catenin signaling. FGF23 produced by osteocytes plays central roles in phosphate metabolism by increasing renal phosphate excretion and decreasing the production of 1,25(OH)2D. The production of FGF23 in osteocytes is influenced by various positive and negative regulators.
Local regulators of FGF23 production in osteocytes
PHEX, DMP1, and FAM20C are highly expressed in osteocytes (8). Since inactivating variants of these genes cause the overproduction of FGF23 in osteocytes (25), these molecules are considered to function as local negative regulators of FGF23 production in osteocytes.
The PHEX gene is responsible for X-linked hypophosphatemic rickets (XLH), the most common form of hereditary hypophosphatemia (9). Although the PHEX protein is suggested to function as a cell surface-bound, Zinc-dependent protease based on its structure (26), its physiological substrates remain elusive, and FGF23 does not serve as its substrate (27). DMP1 encodes an extracellular matrix protein belonging to the small integrin-binding ligand, N-linked glycoproteins (SIBLINGs) family, and its inactivating variants cause autosomal recessive hypophosphatemic rickets type 1 (ARHR1) (10, 11).
Studies using Phex-deficient hypophosphatemic Hyp mice and Dmp1-null mice have suggested that enhanced FGFR signaling in osteocytes contributes to the increased production of FGF23 in XLH and ARHR1 (8, 28, 29). In Hyp mice, the osteocytic expression of Fgf1, Fgf2, Fgfr1–3, and Egr-1, which is a target gene of activated FGFR signaling, was found to be markedly up-regulated (8), and the osteocyte-specific deletion of Fgfr1 partially restored the overproduction of FGF23 and attenuated hypophosphatemia and mineralization defects (29). In a culture of bone marrow stromal cells isolated from Dmp1-null mice, the inhibition of FGFR signaling using SU5402 prevented increases in FGF23 expression levels (28). Furthermore, mice with the transgenic overexpression of high-molecular-weight isoforms of FGF2 in osteoblast lineage cells exhibited elevated FGF23 levels and hypophosphatemic rickets (30). In humans, osteoglophonic dysplasia caused by activating variants in FGFR1 is often associated with hypophosphatemia due to elevated FGF23 levels (31). These findings support activated FGFR signaling in osteocytes increasing the production of FGF23.
The FAM20C gene encodes a secreted kinase that phosphorylates a broad range of substrates, including FGF23 and proteins of the SIBLINGs family, such as DMP1 (32, 33). Inactivating variants of FAM20C cause Raine syndrome (RNS), an autosomal recessive disease characterized by neonatal osteosclerotic bone dysplasia of an aggressive onset and poor prognosis. Patients with mild RNS may survive and manifest hypophosphatemic rickets due to elevated FGF23 levels as well as dental anomalies (12, 34). FAM20C has been shown to directly phosphorylate FGF23 on Ser180, which inhibits the O-glycosylation of FGF23 by GalNAc-T3. Therefore, the inactivation of FAM20C may increase the protein levels of intact FGF23 by reducing its cleavage (33).
Sclerostin may also function as a local regulator of FGF23. A recent study demonstrated that a treatment with an anti-sclerostin antibody reduced serum levels of intact FGF23 and increased serum phosphate levels in wild-type and Phex-deficient Hyp mice (35). While the serum intact FGF23 levels were reduced by anti-sclerostin antibody, the levels determined by C-terminal assay was found to be unchanged. Since the C-terminal FGF23 assay detects both the intact and cleaved C-terminal fragments of FGF23, the circulating C-terminal fragments of FGF23 were likely to be increased after the treatment with anti-sclerostin antibody (35). Considering a previous report demonstrating that the C-terminal fragments of FGF23 may antagonize the action of biologically active intact FGF23 (36), the elevation in serum phosphate levels following the treatment with anti-sclerostin antibody might be mediated by both the reduction in intact FGF23 levels and the increase in the C-terminal fragments. Although the regulation of FGF23 by sclerostin may be indirect and mediated by the control of bone turnover, a cell study using the osteocytic cell line IDG-SW3 suggested the direct stimulating effects of sclerostin on the synthesis of FGF23 (37). The regulation of FGF23 by sclerostin is interesting because it suggests a connection between the two important functions of osteocytes: the control of bone mass and the regulation of phosphate metabolism.
A clinical study previously demonstrated that the intravenous administration of pamidronate to patients with osteogenesis imperfecta rapidly decreased serum intact FGF23 levels (38), which suggested that bone turnover influences serum FGF23 levels. This concept was supported by a mouse study in which interleukin-1 (IL-1)-induced local bone resorption caused elevations in serum intact FGF23 levels without increasing its mRNA levels, and this elevation in FGF23 was prevented by a pre-treatment with a bisphosphonate pamidronate (39). Similarly, in nephrectomized rats with a high bone turnover renal osteodystrophy, a treatment with a bisphosphonate risedronate suppressed the elevation of serum intact FGF23 levels (40). These findings indicate that the release of FGF23 produced by osteocytes into the circulation is accelerated in association with bone resorption.
Systemic regulators of FGF23 production in osteocytes
In addition to the local regulators described above, FGF23 production by osteocytes is influenced by various systemic factors, some of which are described herein. Among the systemic regulators of FGF23 production, 1,25(OH)2D appears to be a principal regulator and increases the expression of FGF23 in osteoblast lineage cells through the vitamin D receptor (VDR)-mediated transactivation of its gene (41, 42). The importance of 1,25(OH)2D in the regulation of FGF23 is supported by clinical observations showing that patients with vitamin D deficiency have low levels of serum intact FGF23 (43).
PTH also stimulates the production of FGF23, as suggested by the elevated serum levels of FGF23 in patients and mouse models of hyperparathyroidism (44, 45). Elevated serum levels of intact FGF23 were also reported in patients with Jansen type metaphyseal chondrodysplasia, a skeletal dysplasia caused by activating variants in PTH receptor 1 (PTH1R) (46). The importance of PTH signaling in osteocytes for the regulation of FGF23 production has been shown in mouse studies demonstrating that the constitutive activation of PTH1R in osteocytes using a Dmp1 promoter increased the production of FGF23 (47).
Phosphate itself also regulates the production of FGF23 in osteocytes. Previous studies reported that dietary phosphate loading increased serum intact FGF23 levels in both humans and mice (48, 49). We recently showed that a 72-hour treatment of primary mouse osteocytes with high phosphate increased FGF23 production in vitro, and this increase occurred at the protein level rather than at the mRNA level (50). Furthermore, a treatment of osteoblast lineage cells with high phosphate up-regulated the expression of the Galnt3 gene, which prevented the cleavage-mediated inactivation of FGF23 (51).
Recent studies demonstrated that insulin signaling suppressed the osteocytic production of FGF23 through the activation of the AKT pathway. In clinical settings, negative correlations were reported between increases in plasma insulin levels after oral glucose loading and plasma intact FGF23 levels (52). A treatment of the cultured osteoblastic cell line UMR106 with insulin and insulin-like growth factor 1 (IGF-1) also suppressed the production of FGF23 through the activation of the AKT pathway and the inhibition of forkhead box protein 1 (FOXO1) (52). We recently reported that the osteocyte-specific deletion of phosphatase and tensin homolog deleted from chromosome 10 (PTEN), the molecule that antagonizes the insulin-induced activation of AKT, resulted in a decrease in the production of FGF23 in osteocytes, a reduction in renal phosphate excretion, and the attenuation of hyperphosphatemia (53). The knockdown of PTEN expression in UMR1-6 cells decreased the expression of Fgf23, which was partially restored by a treatment with rapamycin, suggesting the involvement of AKT/mechanistic target of rapamycin complex 1 (mTORC1) (53). These findings suggest that the insulin- and IGF1-induced activation of AKT in osteocytes inhibit the production of FGF23 through the FOXO1 and mTORC1 pathways.
Iron deficiency increases the production of FGF23 through the hypoxia-inducible factor 1α (HIF1α)-mediated transactivation of the gene (54, 55). The activation of the HIF pathway promotes the production of the hematopoietic hormone erythropoietin, which has been shown to increase the production of FGF23 (56).
Increased serum levels of intact FGF23 may be observed in patients with inflammatory diseases (57), and various proinflammatory cytokines, such as tumor necrosis factor α, IL-1β, and IL-6, have been reported to increase the expression of Fgf23 (58). Several mechanisms, including the activation of the HIF pathway, the involvement of NF-κB and signal transducer and activator of transcription 3, and lipocalin 2-mediated induction, have been suggested to contribute to inflammation-associated increases in the production of FGF23 (59–62).
Many other factors have also been suggested to regulate the production of FGF23, and there are several excellent review articles on this topic (57, 58). Therefore, the regulation of FGF23 is multifactorial and complex, and has not yet been elucidated in detail.
Growth-related changes in osteocytes and alterations in phosphate metabolism
Serum levels of phosphate are higher in children than in adults (63), which may be due to the high need for phosphate for the growth of the skeleton and soft tissues. However, the mechanisms underlying growth-related changes in phosphate metabolism remain unclear. Since osteocytes play central roles in phosphate homeostasis, we recently investigated the relationship between growth-related skeletal changes and alterations in phosphate metabolism from youth to adulthood using young (4-week-old) and adult (12-week-old) mice (50). Although serum phosphate levels were lower in young mice, serum intact FGF23 levels and the osteocytic production of FGF23 increased from youth to adulthood and were associated with the enhancement of the FGF23-mediated-bone-kidney axis (50). An analysis of osteocytes isolated from young and adult mice revealed that the mRNA and protein levels of Dmp1 and mRNA levels of Phex declined from youth to adulthood. Since they function in the negative regulation of FGF23 production, the down-regulation of Dmp1 and Phex may be one of the mechanisms contributing to growth-related increases in the production of FGF23 and decreases in serum phosphate levels (50). In isolated osteoblasts and osteocytes, gene responses to elevated extracellular phosphate levels were also markedly altered from youth to adulthood (50). These findings provide evidence for the critical roles of osteocytes in growth-related alterations in phosphate metabolism (Figure 2).
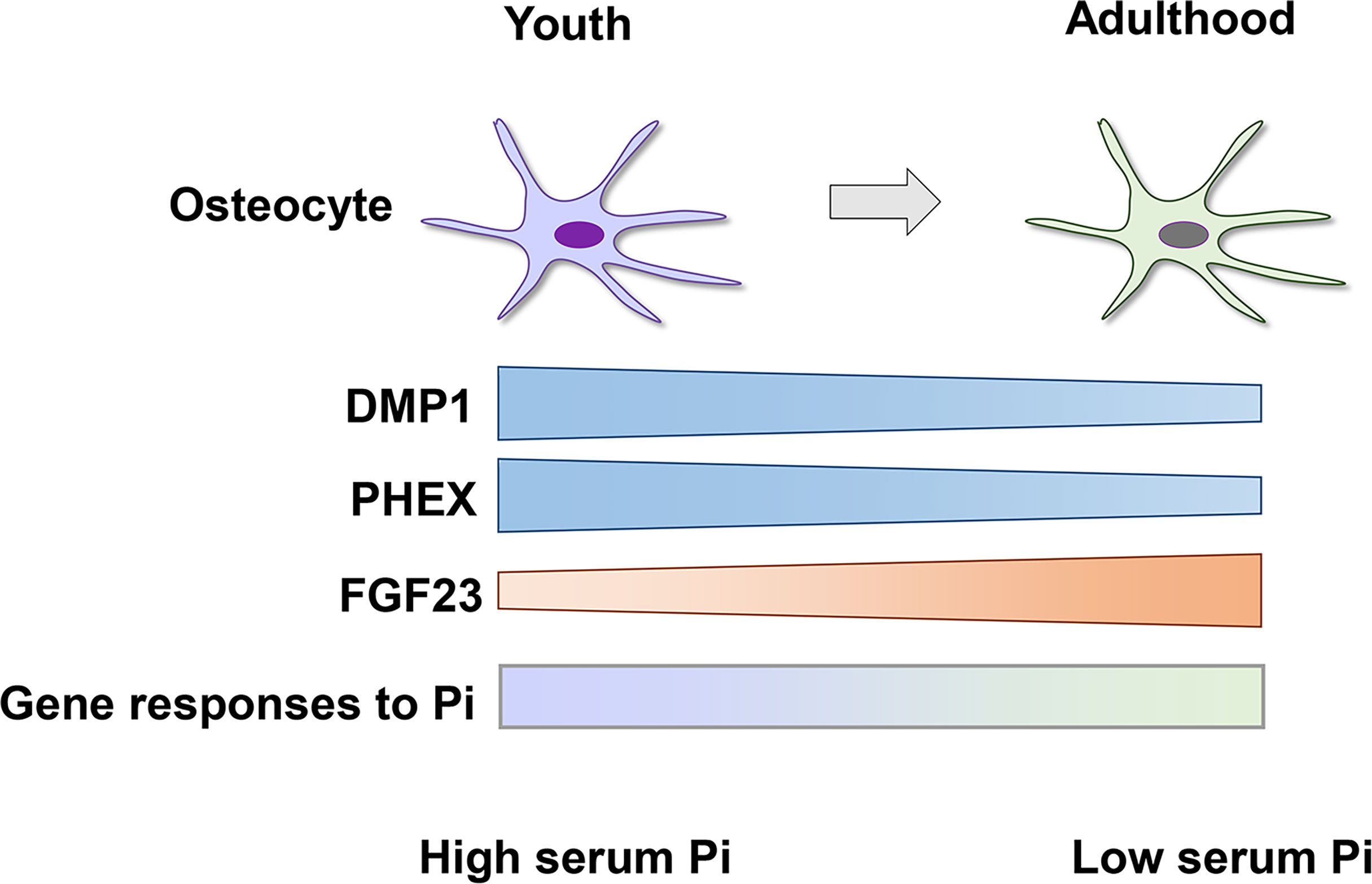
Figure 2 Growth-related changes in osteocytes cause a decrease in serum phosphate levels from youth to adulthood. The osteocytic expression of DMP1 and PHEX declines from youth to adulthood, which leads to an increase in the production of FGF23 and decrease in serum phosphate (Pi) levels. Gene responses of osteocytes to extracellular Pi are distinct between youth and adulthood (depicted as a shaded bar), suggesting growth-related differences in the sensitivity to the altered availability of Pi.
Phosphate sensing in osteocytes
To maintain phosphate homeostasis, organisms need to sense environmental and internal levels of phosphate and adapt to changes. Although the molecular mechanisms for phosphate sensing have been extensively investigated in unicellular organisms, such as bacteria and yeast, as well as in plants (64–66), the mechanisms by which mammals sense phosphate levels in individual cells or the whole body currently remain unknown. Previous studies, including ours, demonstrated that an elevation in extracellular phosphate directly exerted its effects on various cell types, including bone cells, through the activation of signaling pathways, such as the FGFR and Raf/MEK/ERK pathways (7, 67–70). The responsiveness of cells to elevated extracellular phosphate levels indicates that phosphate availability is detectable at the individual cell level. Since osteocytes play a central role in phosphate homeostasis, they may sense phosphate availability in the whole body. This concept appears to be supported by the close relationship between growth-related changes in osteocytic gene expression and their responses to phosphate and alterations in phosphate metabolism from youth to adulthood (50). A previous study reported that phosphate loading in mice up-regulated the skeletal expression of Galnt3 by activating FGFR and increased the production of FGF23, suggesting the involvement of FGFR in phosphate sensing in mammals (51).
The parathyroid glands also respond to altered levels of extracellular phosphate. The secretion of PTH is stimulated by phosphate, and a recent study has suggested that this process is mediated by a direct action of phosphate on the calcium-sensing receptor (71). Thus, calcium-sensing receptor may function as a phosphate sensor in the parathyroids.
Conclusion
Osteocytes embedded in the mineralized bone matrix play central roles in the regulation of phosphate metabolism as well as in the control of bone mass. They control bone formation and resorption by producing sclerostin and RANKL, respectively. FGF23 produced mainly by osteocytes functions as a key regulator of phosphate homeostasis, and it increases renal phosphate excretion and decreases the synthesis of 1,25(OH)2D. The production of FGF23 in osteocytes is influenced by multiple local and systemic regulators, and some of the local regulators, such as PHEX, DMP1, and FAM20C, were found to be responsible for hereditary hypophosphatemic diseases associated with the overproduction of FGF23. Serum phosphate levels are higher in children to meet the high needs for phosphate during growth. Mouse studies have suggested that decreases in serum phosphate levels from youth to adulthood are associated with growth-related increases in the production of FGF23 in osteocytes, which may be attributed to the down-regulation of PHEX and DMP1. Since osteocytes govern phosphate homeostasis, they may be responsible for sensing phosphate availability in the whole body. Although the mechanisms by which mammals sense phosphate levels remain largely unknown, FGFR appears to be involved in the process. Further clarification of the mechanisms by which osteocytes sense phosphate availability and regulate the production of FGF23 will contribute to a more detailed understanding of the pathogenesis of conditions with abnormal phosphate metabolism and the development of effective treatments.
Author contributions
TM developed the concept and prepared the manuscript.
Funding
Preparation of the manuscript was supported by a grant from Japan Society for the Promotion of Science (JSPS KAKENHI Grant Number 21K07835) to TM.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Dallas SL, Prideaux M, Bonewald LF. The osteocyte: an endocrine cell and more. Endocr Rev (2013) 34(5):658–90. doi: 10.1210/er.2012-1026
3. Delgado-Calle J, Bellido T. The osteocyte as a signaling cell. Physiol Rev (2022) 102(1):379–410. doi: 10.1152/physrev.00043.2020
4. Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of sost/sclerostin. J Biol Chem (2008) 283(9):5866–75. doi: 10.1074/jbc.M705092200
5. Delgado-Calle J, Sato AY, Bellido T. Role and mechanism of action of sclerostin in bone. Bone (2017) 96:29–37. doi: 10.1016/j.bone.2016.10.007
6. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med (2011) 17(10):1231–4. doi: 10.1038/nm.2452
7. Michigami T, Kawai M, Yamazaki M, Ozono K. Phosphate as a signaling molecule and its sensing mechanism. Physiol Rev (2018) 98(4):2317–48. doi: 10.1152/physrev.00022.2017
8. Miyagawa K, Yamazaki M, Kawai M, Nishino J, Koshimizu T, Ohata Y, et al. Dysregulated gene expression in the primary osteoblasts and osteocytes isolated from hypophosphatemic hyp mice. PloS One (2014) 9(4):e93840. doi: 10.1371/journal.pone.0093840
9. Francis SH F, Korn B, Reinhardt R, de Jong P, Poustka A, Lehrach H, et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. the HYP consortium. Nat Genet (1995) 11(2):130–6. doi: 10.1038/ng1095-130
10. Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet (2006) 38(11):1248–50. doi: 10.1038/ng1868
11. Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet (2006) 38(11):1310–5. doi: 10.1038/ng1905
12. Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, et al. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res (2013) 28(6):1378–85. doi: 10.1002/jbmr.1850
13. Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab (2007) 5(6):464–75. doi: 10.1016/j.cmet.2007.05.001
14. Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet (2001) 68(3):577–89. doi: 10.1086/318811
15. Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, et al. Sclerostin binds to LRP5/6 and antagonizes canonical wnt signaling. J Biol Chem (2005) 280(20):19883–7. doi: 10.1074/jbc.M413274200
16. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med (2013) 19(2):179–92. doi: 10.1038/nm.3074
17. Kubota T, Michigami T, Ozono K. Wnt signaling in bone metabolism. J Bone Miner Metab (2009) 27(3):265–71. doi: 10.1007/s00774-009-0064-8
18. Sasaki F, Hayashi M, Mouri Y, Nakamura S, Adachi T, Nakashima T. Mechanotransduction via the Piezo1-akt pathway underlies sost suppression in osteocytes. Biochem Biophys Res Commun (2020) 521(3):806–13. doi: 10.1016/j.bbrc.2019.10.174
19. Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, et al. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res (2007) 22(12):1957–67. doi: 10.1359/jbmr.070804
20. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A (2001) 98(11):6500–5. doi: 10.1073/pnas.101545198
21. Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol (2007) 27(9):3417–28. doi: 10.1128/MCB.02249-06
22. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem (2006) 281(10):6120–3. doi: 10.1074/jbc.C500457200
23. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature (2006) 444(7120):770–4. doi: 10.1038/nature05315
24. Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem (2006) 281(27):18370–7. doi: 10.1074/jbc.M602469200
25. Fukumoto S. FGF23-related hypophosphatemic rickets/osteomalacia: diagnosis and new treatment. J Mol Endocrinol (2021) 66(2):R57–65. doi: 10.1530/JME-20-0089
26. Drezner MK. PHEX gene and hypophosphatemia. Kidney Int (2000) 57(1):9–18. doi: 10.1046/j.1523-1755.2000.00807.x
27. Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone (2004) 35(2):455–62. doi: 10.1016/j.bone.2004.04.002
28. Martin A, Liu S, David V, Li H, Karydis A, Feng JQ, et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J (2011) 25(8):2551–62. doi: 10.1096/fj.10-177816
29. Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PloS One (2014) 9(8):e104154. doi: 10.1371/journal.pone.0104154
30. Xiao L, Naganawa T, Lorenzo J, Carpenter TO, Coffin JD, Hurley MM. Nuclear isoforms of fibroblast growth factor 2 are novel inducers of hypophosphatemia via modulation of FGF23 and KLOTHO. J Biol Chem (2010) 285(4):2834–46. doi: 10.1074/jbc.M109.030577
31. White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet (2005) 76(2):361–7. doi: 10.1086/427956
32. Tagliabracci VS, Engel JL, Wen J, Wiley SE, Worby CA, Kinch LN, et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science (2012) 336(6085):1150–3. doi: 10.1126/science.1217817
33. Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A (2014) 111(15):5520–5. doi: 10.1073/pnas.1402218111
34. Takeyari S, Yamamoto T, Kinoshita Y, Fukumoto S, Glorieux FH, Michigami T, et al. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of raine syndrome. Bone (2014) 67:56–62. doi: 10.1016/j.bone.2014.06.026
35. Carpenter KA, Ross RD. Sclerostin antibody treatment increases bone mass and normalizes circulating phosphate levels in growing hyp mice. J Bone Miner Res (2020) 35(3):596–607. doi: 10.1002/jbmr.3923
36. Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, et al. Isolated c-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A (2010) 107(1):407–12. doi: 10.1073/pnas.0902006107
37. Ito N, Prideaux M, Wijenayaka AR, Yang D, Ormsby RT, Bonewald LF, et al. Sclerostin directly stimulates osteocyte synthesis of fibroblast growth factor-23. Calcif Tissue Int (2021) 109(1):66–76. doi: 10.1007/s00223-021-00823-6
38. Kitaoka T, Namba N, Miura K, Kubota T, Ohata Y, Fujiwara M, et al. Decrease in serum FGF23 levels after intravenous infusion of pamidronate in patients with osteogenesis imperfecta. J Bone Miner Metab (2011) 29(5):598–605. doi: 10.1007/s00774-011-0262-z
39. Yamazaki M, Kawai M, Miyagawa K, Ohata Y, Tachikawa K, Kinoshita S, et al. Interleukin-1-induced acute bone resorption facilitates the secretion of fibroblast growth factor 23 into the circulation. J Bone Miner Metab (2015) 33(3):342–54. doi: 10.1007/s00774-014-0598-2
40. Ishida H, Komaba H, Hamano N, Yamato H, Sawada K, Wada T, et al. Skeletal and mineral metabolic effects of risedronate in a rat model of high-turnover renal osteodystrophy. J Bone Miner Metab (2020) 38(4):501–10. doi: 10.1007/s00774-020-01095-0
41. Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin d. J Am Soc Nephrol (2006) 17(5):1305–15. doi: 10.1681/ASN.2005111185
42. Haussler MR, Livingston S, Sabir ZL, Haussler CA, Jurutka PW. Vitamin d receptor mediates a myriad of biological actions dependent on its 1,25-dihydroxyvitamin d ligand: Distinct regulatory themes revealed by induction of klotho and fibroblast growth factor-23. JBMR Plus (2021) 5(1):e10432. doi: 10.1002/jbm4.10432
43. Endo I, Fukumoto S, Ozono K, Namba N, Tanaka H, Inoue D, et al. Clinical usefulness of measurement of fibroblast growth factor 23 (FGF23) in hypophosphatemic patients: proposal of diagnostic criteria using FGF23 measurement. Bone (2008) 42(6):1235–9. doi: 10.1016/j.bone.2008.02.014
44. Kobayashi K, Imanishi Y, Miyauchi A, Onoda N, Kawata T, Tahara H, et al. Regulation of plasma fibroblast growth factor 23 by calcium in primary hyperparathyroidism. Eur J Endocrinol (2006) 154(1):93–9. doi: 10.1530/eje.1.02053
45. Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, et al. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol (2007) 18(10):2683–8. doi: 10.1681/ASN.2006070783
46. Brown WW, Juppner H, Langman CB, Price H, Farrow EG, White KE, et al. Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with jansen's metaphyseal chondrodysplasia. J Clin Endocrinol Metab (2009) 94(1):17–20. doi: 10.1210/jc.2008-0220
47. Rhee Y, Bivi N, Farrow E, Lezcano V, Plotkin LI, White KE, et al. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone (2011) 49(4):636–43. doi: 10.1016/j.bone.2011.06.025
48. Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab (2005) 90(3):1519–24. doi: 10.1210/jc.2004-1039
49. Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin d metabolism in mice. Endocrinology (2005) 146(12):5358–64. doi: 10.1210/en.2005-0777
50. Michigami T, Tachikawa K, Yamazaki M, Nakanishi T, Kawai M, Ozono K. Growth-related skeletal changes and alterations in phosphate metabolism. Bone (2022) 161:116430. doi: 10.1016/j.bone.2022.116430
51. Takashi Y, Kosako H, Sawatsubashi S, Kinoshita Y, Ito N, Tsoumpra MK, et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc Natl Acad Sci USA (2019) 116(23):11418–27. doi: 10.1073/pnas.1815166116
52. Bar L, Feger M, Fajol A, Klotz LO, Zeng S, Lang F, et al. Insulin suppresses the production of fibroblast growth factor 23 (FGF23). Proc Natl Acad Sci USA (2018) 115(22):5804–9. doi: 10.1073/pnas.1800160115
53. Kawai M, Kinoshita S, Ozono K, Michigami T. Lack of PTEN in osteocytes increases circulating phosphate concentrations by decreasing intact fibroblast growth factor 23 levels. Sci Rep (2020) 10(1):21501. doi: 10.1038/s41598-020-78692-6
54. Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci USA (2011) 108(46):E1146–55. doi: 10.1073/pnas.1110905108
55. Wheeler JA, Clinkenbeard EL. Regulation of fibroblast growth factor 23 by iron, EPO, and HIF. Curr Mol Biol Rep (2019) 5(1):8–17. doi: 10.1007/s40610-019-0110-9
56. Daryadel A, Bettoni C, Haider T, Imenez Silva PH, Schnitzbauer U, Pastor-Arroyo EM, et al. Erythropoietin stimulates fibroblast growth factor 23 (FGF23) in mice and men. Pflugers Arch (2018) 470(10):1569–82. doi: 10.1007/s00424-018-2171-7
57. Rausch S, Foller M. The regulation of FGF23 under physiological and pathophysiological conditions. Pflugers Arch (2022) 474(3):281–92. doi: 10.1007/s00424-022-02668-w
58. Simic P, Babitt JL. Regulation of FGF23: Beyond bone. Curr Osteoporos Rep (2021) 19(6):563–73. doi: 10.1007/s11914-021-00703-w
59. Zhang B, Yan J, Umbach AT, Fakhri H, Fajol A, Schmidt S, et al. NFkappaB-sensitive Orai1 expression in the regulation of FGF23 release. J Mol Med (Berl) (2016) 94(5):557–66. doi: 10.1007/s00109-015-1370-3
60. Durlacher-Betzer K, Hassan A, Levi R, Axelrod J, Silver J, Naveh-Many T. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int (2018) 94(2):315–25. doi: 10.1016/j.kint.2018.02.026
61. Courbon G, Francis C, Gerber C, Neuburg S, Wang X, Lynch E, et al. Lipocalin 2 stimulates bone fibroblast growth factor 23 production in chronic kidney disease. Bone Res (2021) 9(1):35. doi: 10.1038/s41413-021-00154-0
62. David V, Francis C, Babitt JL. Ironing out the cross talk between FGF23 and inflammation. Am J Physiol Renal Physiol (2017) 312(1):F1–8. doi: 10.1152/ajprenal.00359.2016
63. Yamazaki Y, Imura A, Urakawa I, Shimada T, Murakami J, Aono Y, et al. Establishment of sandwich ELISA for soluble alpha-klotho measurement: Age-dependent change of soluble alpha-klotho levels in healthy subjects. Biochem Biophys Res Commun (2010) 398(3):513–8. doi: 10.1016/j.bbrc.2010.06.110
64. Silver J, Dranitzki-Elhalel M. Sensing phosphate across the kingdoms. Curr Opin Nephrol Hypertens (2003) 12(4):357–61. doi: 10.1097/00041552-200307000-00003
65. Mouillon JM, Persson BL. New aspects on phosphate sensing and signalling in saccharomyces cerevisiae. FEMS Yeast Res (2006) 6(2):171–6. doi: 10.1111/j.1567-1364.2006.00036.x
66. Chiou TJ, Lin SI. Signaling network in sensing phosphate availability in plants. Annu Rev Plant Biol (2011) 62:185–206. doi: 10.1146/annurev-arplant-042110-103849
67. Yamazaki M, Ozono K, Okada T, Tachikawa K, Kondou H, Ohata Y, et al. Both FGF23 and extracellular phosphate activate Raf/MEK/ERK pathway via FGF receptors in HEK293 cells. J Cell Biochem (2010) 111(5):1210–21. doi: 10.1002/jcb.22842
68. Kimata M, Michigami T, Tachikawa K, Okada T, Koshimizu T, Yamazaki M, et al. Signaling of extracellular inorganic phosphate up-regulates cyclin D1 expression in proliferating chondrocytes via the Na+/Pi cotransporter pit-1 and Raf/MEK/ERK pathway. Bone (2010) 47(5):938–47. doi: 10.1016/j.bone.2010.08.006
69. Nishino J, Yamazaki M, Kawai M, Tachikawa K, Yamamoto K, Miyagawa K, et al. Extracellular phosphate induces the expression of dentin matrix protein 1 through the FGF receptor in osteoblasts. J Cell Biochem (2017) 118(5):1151–63. doi: 10.1002/jcb.25742
70. Beck GR Jr., Knecht N. Osteopontin regulation by inorganic phosphate is ERK1/2-, protein kinase c-, and proteasome-dependent. J Biol Chem (2003) 278(43):41921–9. doi: 10.1074/jbc.M304470200
Keywords: phosphate, osteoblast, osteocyte, fibroblast growth factor 23, regulation
Citation: Michigami T (2022) Roles of osteocytes in phosphate metabolism. Front. Endocrinol. 13:967774. doi: 10.3389/fendo.2022.967774
Received: 13 June 2022; Accepted: 29 June 2022;
Published: 15 July 2022.
Edited by:
Mohammed S. Razzaque, Lake Erie College of Osteopathic Medicine, United StatesReviewed by:
Ryan Ross, Rush University Medical Center, United StatesHirotaka Komaba, Tokai University, Japan
Ronald Brown, University of Waterloo, Canada
Shuanhu Zhou, Brigham and Women’s Hospital and Harvard Medical School, United States
Copyright © 2022 Michigami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshimi Michigami, bWljaGlnYW1pQHdjaC5vcGhvLmpw