 Ye Lv
Ye Lv Yi-Yang Luo
Yi-Yang Luo Hui-Wen Ren
Hui-Wen Ren Cheng-Jie Li
Cheng-Jie Li Zhi-Xin Xiang
Zhi-Xin Xiang Zhi-Lin Luan
Zhi-Lin Luan- 1Advanced Institute for Medical Sciences, Dalian Medical University, Dalian, China
- 2Dalian Key Laboratory for Nuclear Receptors in Major Metabolic Diseases, Dalian Medical University, Dalian, China
As a member of the nuclear receptor (NR) superfamily, pregnane X receptor (PXR; NR1I2) is a ligand-activated transcription factor that plays a crucial role in the metabolism of xenobiotics and endobiotics in mammals. The tissue distribution of PXR is parallel to its function with high expression in the liver and small intestine and moderate expression in the kidney, stomach, skin, and blood-brain barrier, which are organs and tissues in frequent contact with xenobiotics. PXR was first recognized as an exogenous substance receptor regulating metabolizing enzymes and transporters and functioning in detoxification and drug metabolism in the liver. However, further research revealed that PXR acts as an equally important endogenous substance receptor in the metabolism and homeostasis of endogenous substances. In this review, we summarized the functions of PXR in metabolism of different substances such as glucose, lipid, bile acid, vitamin, minerals, and endocrines, and also included insights of the application of PXR ligands (drugs) in specific diseases.
Introduction
As a member of the nuclear receptor (NR) superfamily, pregnane X receptor (PXR; NR1I2) is a ligand-activated transcription factor first reported in 1998 and named based on its activation by endogenous pregnane 21-carbon steroids (1, 2). PXR is highly distributed in small intestine, liver, rectum, colon and bladder, while its expression in other organs and tissues is either moderate, low or undetectable (3), and the statistics from the GTEx and Tabula Muris databases also support this view (Figure 1) (4, 5). PXR can be activated by numerous chemical compounds. Besides pregnane, steroid hormones, bile acids and other endobiotic chemicals, various clinical drugs (e.g., statins, antidepressants, anticonvulsants) and environmental pollutants have been demonstrated as PXR ligands (Table 1) (35–39). Activated PXR, through direct binding to the genomic regions or indirect crosstalk with other transcriptional factors, controls various genes involved in biotransformation, transport, inflammation, oxidative stress and etc. (35).
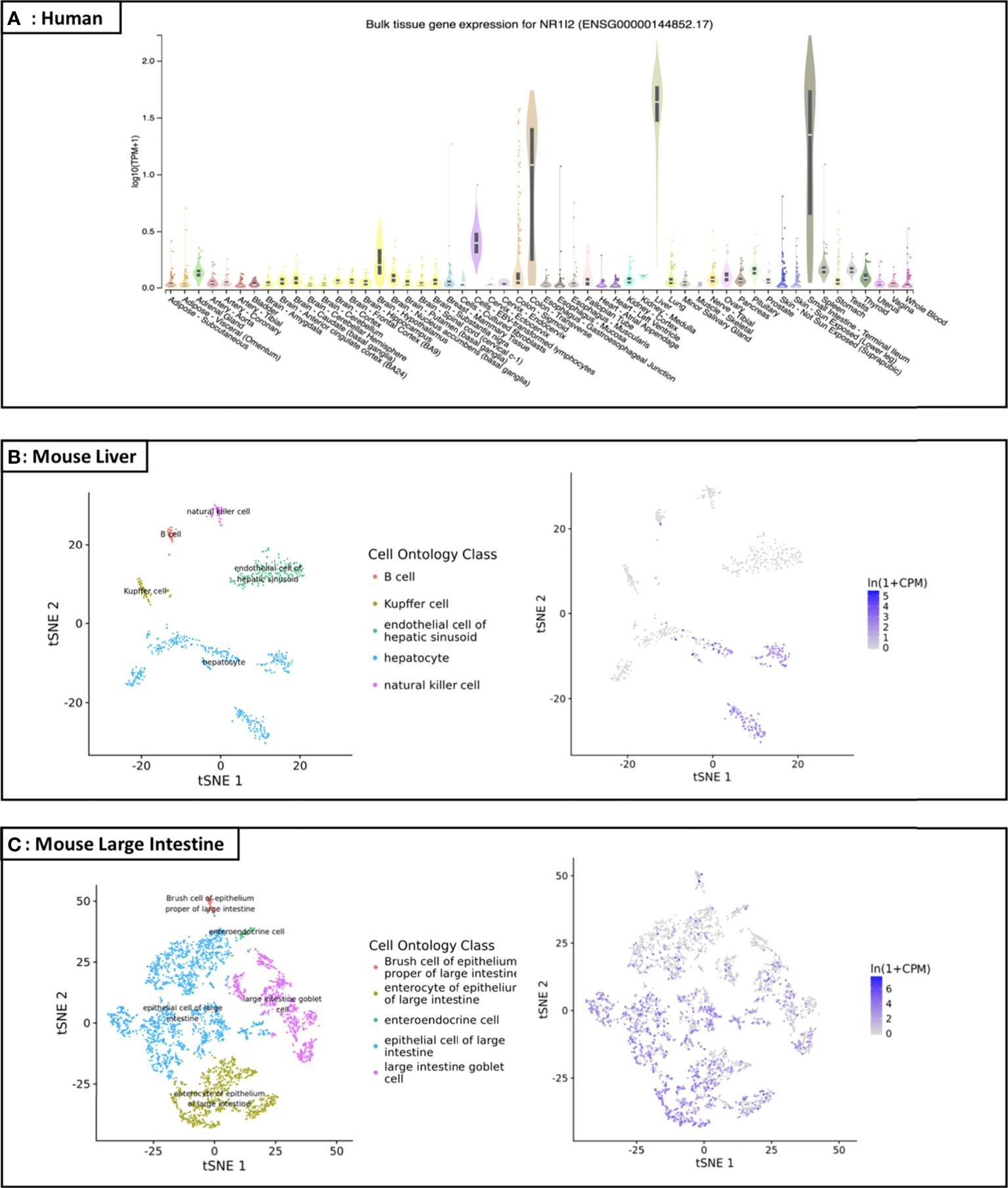
Figure 1 Distribution map of PXR in human and specific organs of mice (A) Expression profile of the NR1I2 (PXR) gene in human: According to the GTEx database (https://gtexportal.org/home/gene/NR1I2), the NR1I2 (PXR) gene is highly and specifically expressed in small intestine, liver, rectum, and colon, while its expression in other organs/tissues is either low or undetectable. TPM on the vertical axis represents the transcript quantification value, and the horizontal axis represents different tissues (TPM: transcripts per kilobase of exon model per Million mapped reads; tSNE: t-distributed stochastic neighbor embedding); (B) Liver cell scRNA-seq analysis demonstrating that mouse PXR mRNA is highly expressed in the liver, especially in hepatocyte; (C) Large intestine cell scRNA-seq analysis demonstrating that mouse PXR mRNA is expressed in the large intestine, especially in epithelial cell and enterocyte of epithelium.
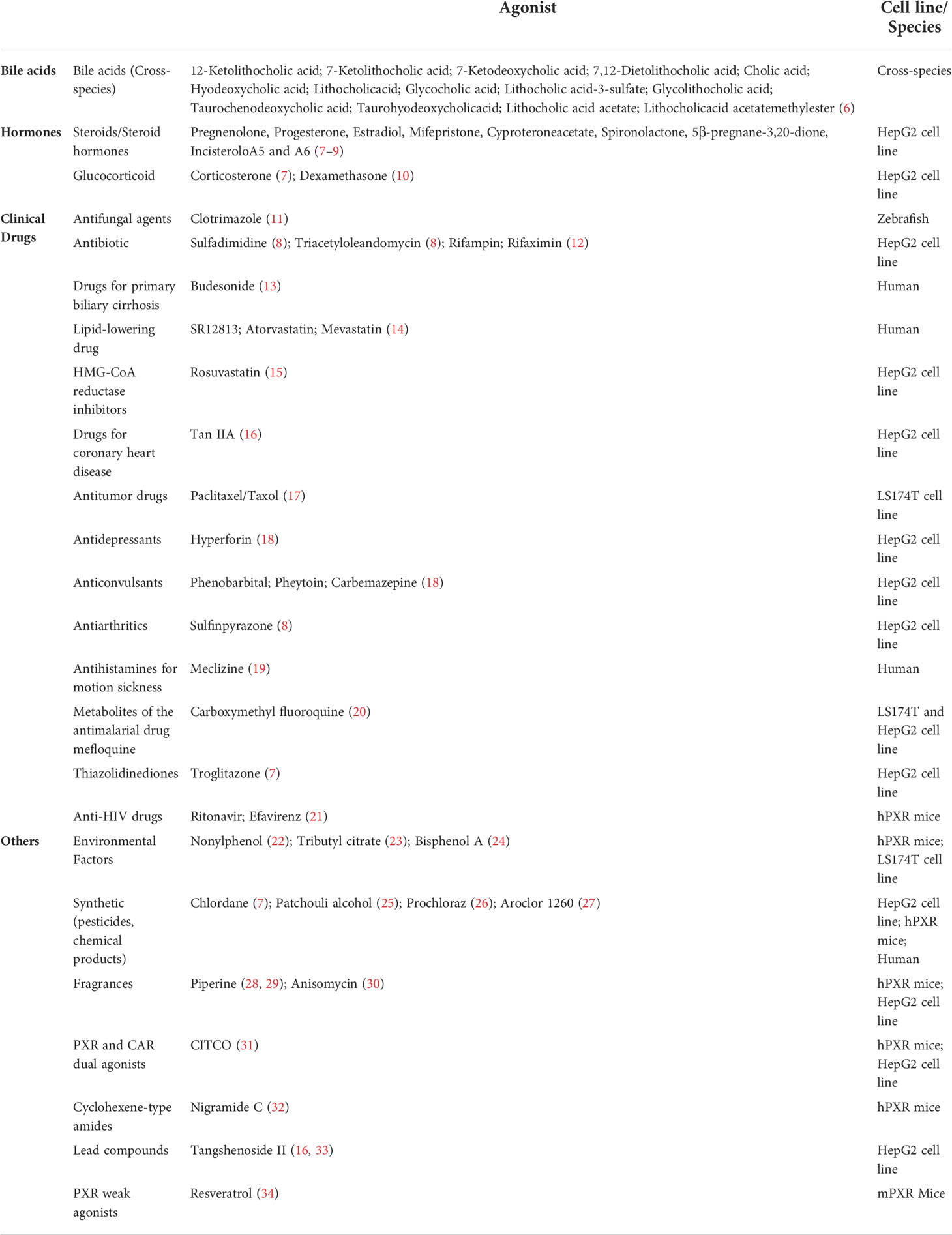
Table 1 The agonists of PXR ligand.
PXR is unique among NRs on account of its broad ligand spectrum, which make it a perfect tool for sensing changes in the external chemical environment. Although originally identified as a receptor for exogenous substances, PXR is now recognized as an equally important receptor for endogenous substances and plays a variety of functions in the metabolism of these substances. Many studies have shown that PXR is involved in a range of physiological and pathological processes through regulating metabolism of a large group of substances. In this review, we summarized the functions of PXR in substance metabolism in aspects of glucose and lipid metabolism, bile acid circulation, and endocrine homeostasis, and also included insights of the application of PXR ligands (drugs) in specific diseases.
The transcriptional regulatory characteristics of PXR
PXR share a common protein structure with most NRs which consists of a typical N-terminal non-ligand-dependent activation function 1 (AF-1), a highly conserved DNA-binding domain (DBD), a less conserved hinge region, a C-terminal ligand-binding domain (LBD) and an activation function 2 (AF-2) (Figure 2A) (2, 3, 40). It has been reported that PXR can be modified by acetylation, phosphorylation, ubiquitination, and SUMOylation through protein-protein interactions (Figure 2A), indicating that PXR is implicated in posttranslational modifications which may ultimately affect its transcriptional regulation and metabolic detoxification process. The interaction centered by PXR will illustrated the multifunctional property of it in different signaling pathways (41). Being part of a chaperone protein complex consisting of heat shock protein 90 (Hsp90) and CAR cytoplasmic retention protein (CCRP), PXR is predominantly localized in the cytoplasm (42). After activation by ligand binding, PXR is transferred from the cytoplasm to the nucleus and forms a heterodimer with retinoid X receptor (RXR). All in all, molecular analysis based on both in vivo and in vitro models have systematically revealed the mechanism of PXR activation (Figure 2B) (43, 44). After recruiting a large number of co-activators, the DBD domain of PXR promotes the DNA binding specificity of PXR through two highly conserved zinc finger motifs as well as the P- and D-box motifs. PXR binds as heterodimers with RXR to repeats of the nucleotide hexamer AGG/TTCA with variable spacing (45) (Figure 3). PXR functions as a trans-factor and regulates its downstream target genes by binding to specific promoter DNA reaction elements. Initial studies suggested that the PXR/RXR co-activation complex binds only to direct repeat sequences in the enhancer regions of target genes, such as DR3 (directed repeat 3) (46).
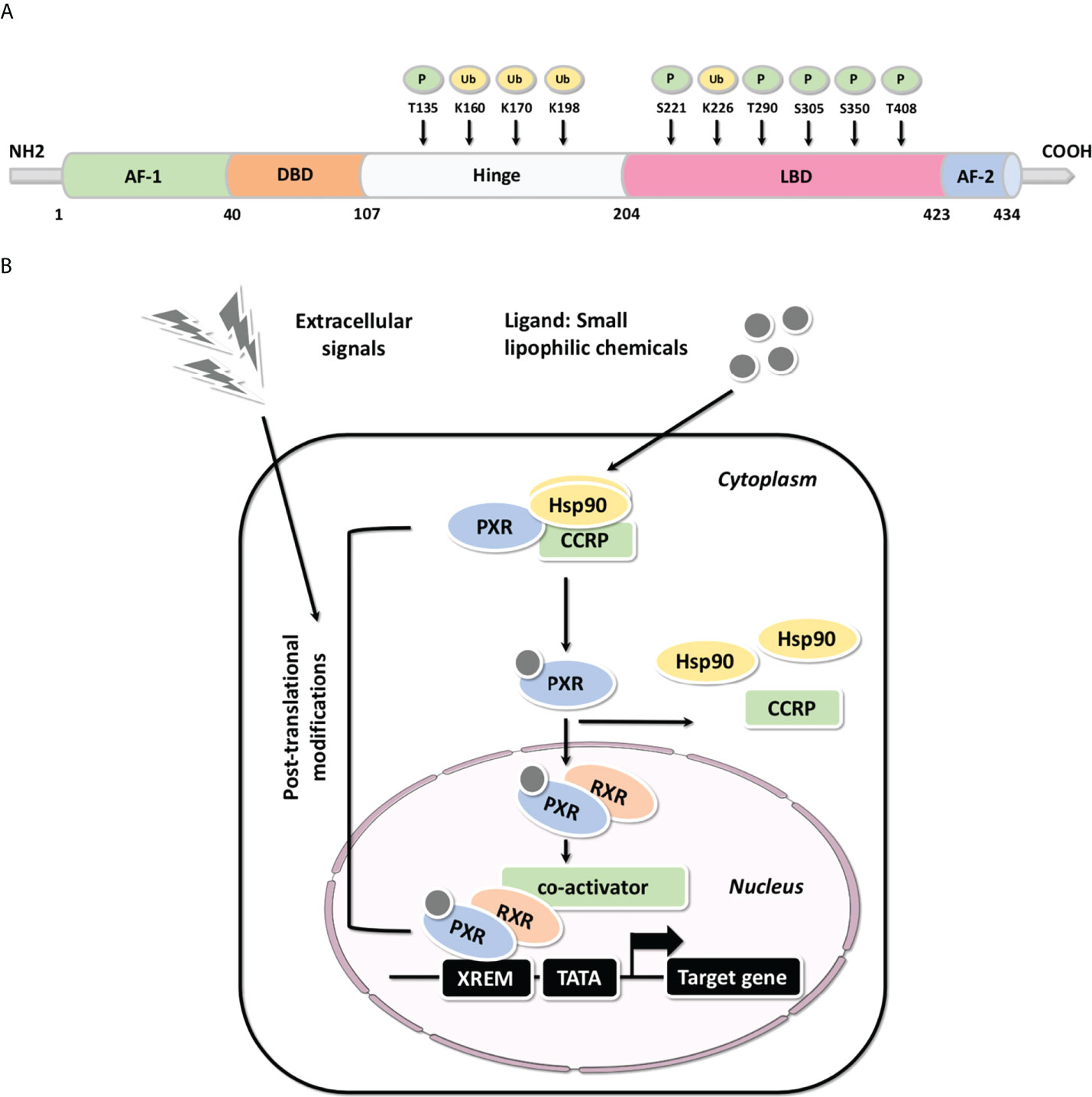
Figure 2 The structure and molecular mechanisms associated with PXR (A) Common structure of metabolic nuclear receptor and the post-translational modifications of PXR protein structure. The domain structure of metabolic nuclear receptor is presented, including the typical N-terminal non-ligand-dependent AF-1, a highly conserved DBD, a less conserved hinge region, a C-terminal LBD and AF-2; PXR may be modified by phosphorylation and ubiquitination through protein-protein interactions, thus, reported phosphorylation and ubiquitination are highlighted (P: Phosphorylation; Ub: Ubiquitination). (B) The molecular mechanisms of PXR-mediated gene activation: Molecular analysis based on both in vivo and in vitro models have systematically illustrated the mechanism of PXR activation.
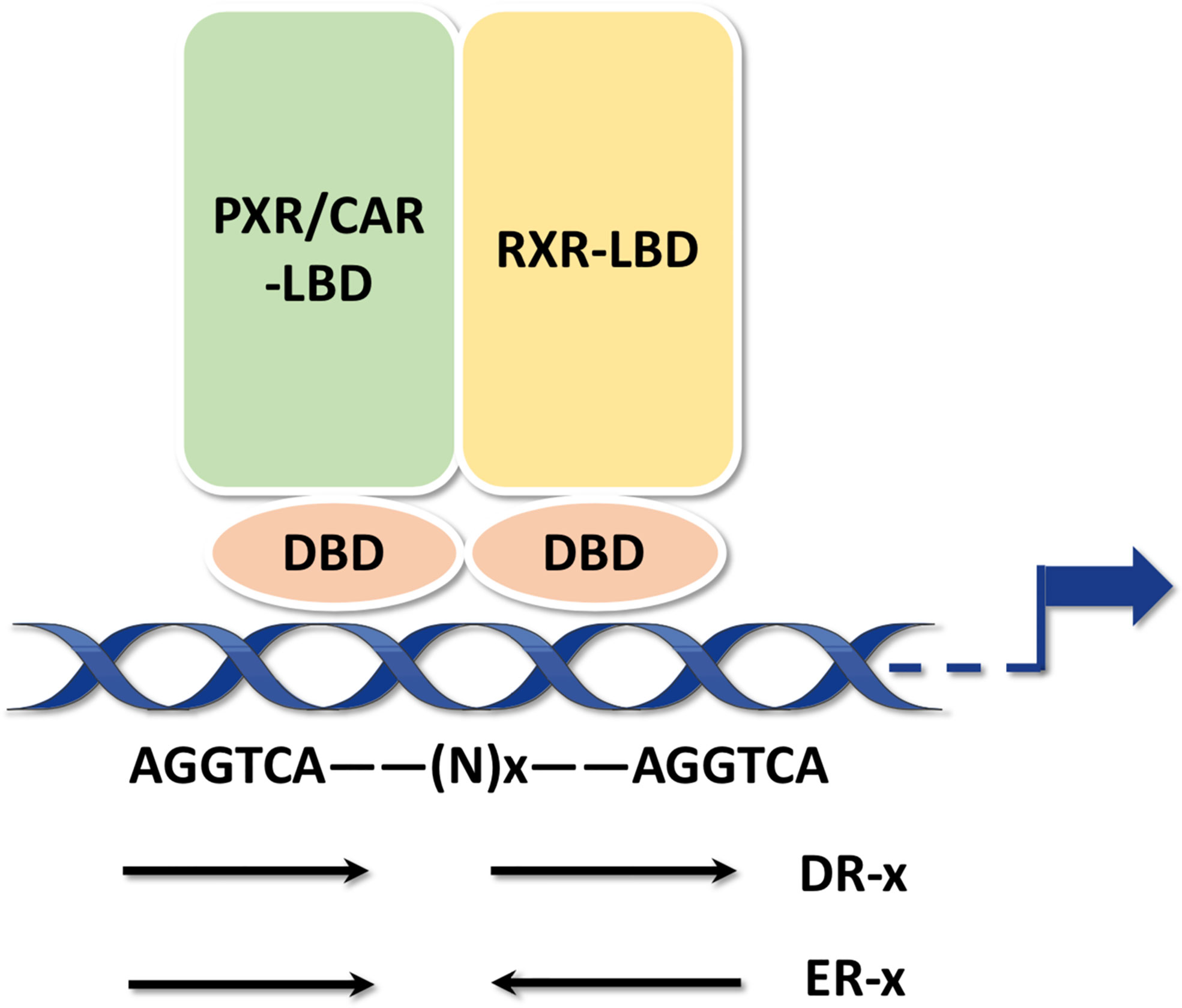
Figure 3 PXR response elements PXR binds as heterodimers with RXR to repeats of the nucleotide hexamer AGGTCA with variable spacing. The hexamers can be arranged either as DRs or ERs.
However, Drocourt et al. found that PXR/RXR heterodimer not only binds DR3 i found that PXR/RXR heterodimer not only binds DR3 in the enhancer region of the human CYP3A4 gene but also acts on the ER6 (everted repeat 6) element. The PXR-bound DR3 and ER6 are highly conserved and generally contain AG(G/T)TCA or TGA(A/C)CT sequences (47, 48). By binding to DR3 and ER6, activated PXR/RXR heterodimer promotes transcriptional regulation of many genes in the cytochrome P450 3A (CYP3A) family, the most abundant, clinically significant group of cytochrome P-450 isoenzymes, such as CYP3A1, CYP3A2, CYP3A23, CYP3A4, CYP3A6, CYP3A7. CYP3A4 is a major target gene for PXR and involved in 60% of drug transport in vivo. Although, mouse genes (e.g. Cyp3a23, Cyp3a1) are absence in humans, but they are considered as clinically significant. Recent studies have revealed that PXR can bind not only to DR3 and ER6 in the promoter region of its target genes, but also to other response elements. Geick et al. reported that PXR/RXR heterodimers can bind to three types of DR4, with DR4(I) and DR4(III) having the highest affinity. The binding of PXR to DR4 is essential for transcription of certain downstream target genes, such as the multi-drug resistance gene 1 (MDR1) and CYP2B3 families (49). Jeske et al. found that PXR can directly bind DR4 and ER8 on the first intron at the 5’ end of sphingomyelin phosphodiesterase acid-like (SMPDL) 3A, and in the presence of non-ligands can then bind (50). As a newly discovered hepatic phospholipase, SMPDL can activate the carbamate precursor drug CS-917 and serves as a promising candidate for the treatment of type 2 diabetes (51). It has also been found that PXR/RXR can bind to ER8 in the promoter region of multidrug resistance protein 2 (MRP2) and promote MRP2 protein transcriptional expression. In summary, the binding elements of PXR/RXR on DNA are divided into: direct repeats (DR4, DR5, DR9, DR9, DR14, DR19), everted repeats (ER6 and ER8), and inverted repeats (IRs) (52).
In addition to xenobiotic receptors above, PXR and CAR can also collaboratively exhibit promiscuous xenobiotic activation. They govern the transcription of a broad spectrum of distinct and overlapping genes encoding phase I, phase II drug-metabolizing enzymes (DMEs), as well as uptake and efflux transporters (53–55). Notably, CAR and PXR share significant cross-talk in both target gene recognition by binding to the similar xenobiotic responsive elements in their target gene promoters, and in accommodating a diverse array of xenobiotic activators (56, 57). Coordinately, CAR and PXR regulate a largely overlapping set of xenobiotic metabolizing genes. These target genes include several CYPs (i.e. CYP3A4, CYP2B6, CYP2Cs, and CYP2A6) (58, 59), UGTs (i.e. UGT1A1, UGT1A6, and UGT1A9) (60, 61), GSTs, and SULTs; as well as drug transporters such as MRPs, MDR1 and OATPs (62). On the other hand, CAR displays unique activation mechanisms compared with PXR and other orphan receptors, involving both direct ligand binding and indirect ligand-independent pathways (Figure 4) (63).
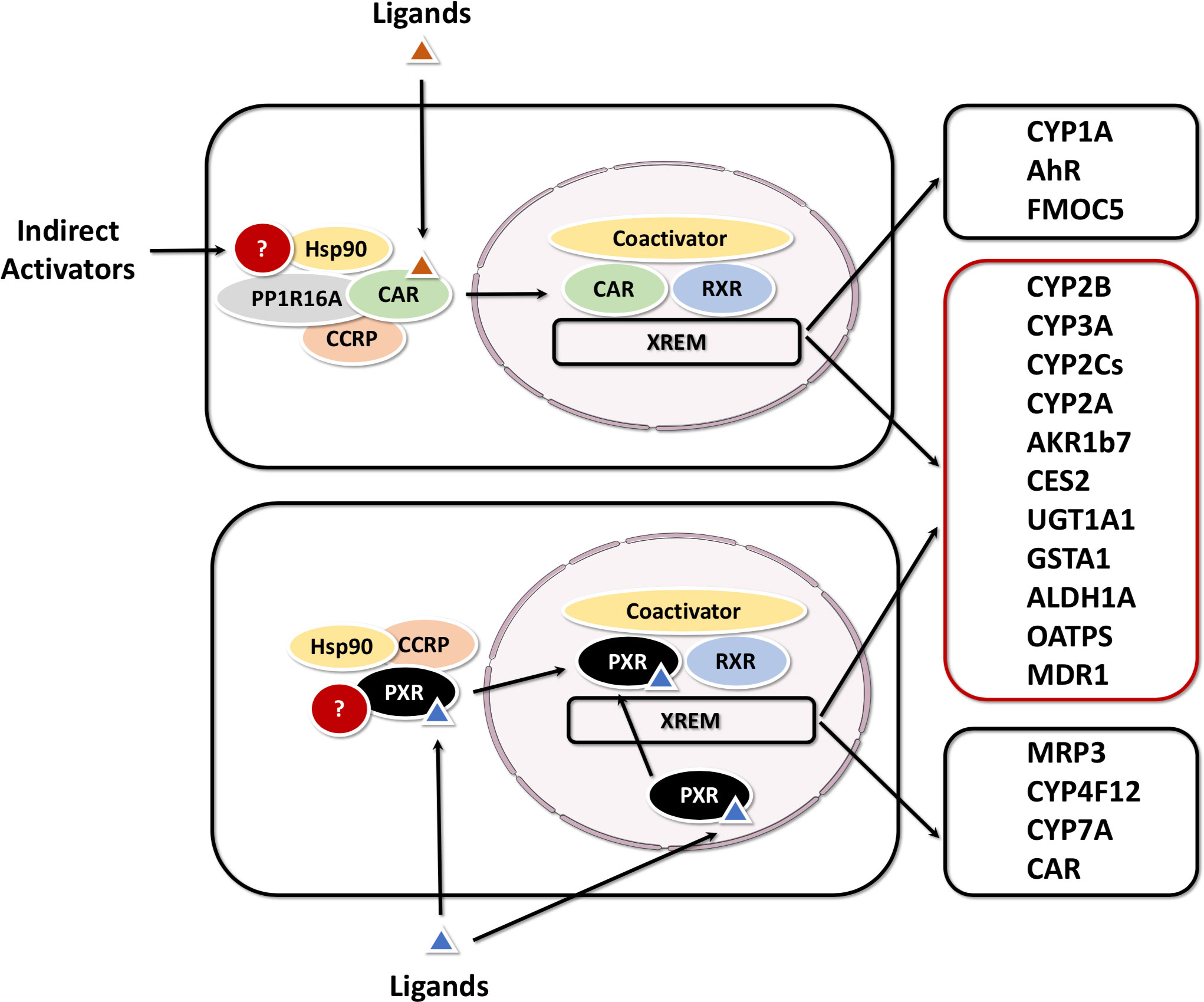
Figure 4 Activation mechanisms and target genes of CAR and PXR The activation of PXR is purely ligand dependent, while CAR can be activated by either direct (ligand binding) or indirect mechanisms. CAR and PXR shared target genes are grouped in a red box, CAR or PXR-specific targets in a black box.
The development of structural genomics has provided insight into the structural basis of NR-regulated transcription. Watkins et al. completed the first X-ray crystallographic analysis of PXR LBD (64). Similar to the LBDs of other members in the NR superfamily, the PXR LBD contains a triple helix sandwich (H1/H3, H4/H5/H8, H7/H10) (65, 66). A fragment containing 45 amino acids is inserted between helix 1 (H1) and helix 3 (H3) as a β-sheet in the PXR LBD and forms one of the five chains of β-sheet in the PXR structure. However, other NRs contain only two or three β-sheet chains. Part of helix 2 (H2) in the PXR LBD is replaced by a β-fold, forming a spherical ligand-binding cavity with H1 and H3, also known as the “ligand-binding pocket”. With the potential for flexible expansion and contraction, a ring-like structure in the PXR LBD is formed by β2, β3 and β4-expanded helix 6 (H6) which graphically serves as a “ligand-binding pocket”. The “ligand binding pocket” has a uniform distribution of hydrophobic amino acid residues on its surface, allowing the ligand to maintain equilibrium in any orientation through hydrogen bonding and van der Waals forces (67). The above molecular features allow PXR to recognize a wide range of xenobiotics.
Although PXR can accept a wide variety of ligands, the degree of ligand binding is species-specific. For example, pregnenolone-16α-carbonitrile (PCN) activates the rodent PXR but not the human PXR, and SR12813 and rifampicin activate human PXR at high levels but not rodent PXR. In organisms, xenobiotic promotion of PXR activation causes more extensive metabolic changes involving downstream target gene transcription other than the direct involvement of activated PXR in xenobiotic metabolic processes. In addition, PXR activation has also been reported associated with a variety of diseases, therefore, clinical application of PXR ligands requires consideration not only of individual patient differences, but also of changes in drug efficacy in the body when administering different drugs to patients.
PXR in glucose metabolism
Increasing evidence indicates that PXR activation functions in glucose homeostasis. Blood glucose concentration maintains relatively constant by hormones (insulin, glucagon, glucocorticoids etc.) that regulate the activity of key enzymes involved in various pathways of glucose metabolism. In mouse primary hepatocytes, human hepatoma HepG2 and Huh7 cells, PXR activation inhibits the expression of glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK), two key enzymes inhibiting gluconeogenesis (68–70). Kodama et al. showed that PXR regulates gluconeogenesis by interacting with forkhead box protein O1 (FOXO1), cAMP-response element binding protein (CREB) and hepatic nuclear factor 4 (HNF4) (70). HNF4, together with the nuclear receptor co-activator PGC-1α, positively regulates gluconeogenesis. Bhalla et al. found that PXR competes with HNF4 for PGC-1α, thereby inhibiting gluconeogenesis (68). In vivo experiments also confirmed the plausibility of these results. FOXO1 functions as a G6Pase and PEPCK activator in insulin deficiency. Under normal conditions, after binding to the insulin response sequence (IRS), insulin repatriates FOXO1 from the nucleus via PI3K-Akt pathway, thereby inhibiting G6Pase and PEPCK expression and FOXO1-mediated transactivation of gluconeogenesis. Glucagon increases intracellular cAMP formation, which activates protein kinase A (PKA), which in turn activates CREB that binds and regulates G6Pase and PEPCK transcription. On the other hand, PXR inhibits CREB binding to homologous binding elements, thus preventing the transcription of glucagon-activated G6Pase and thereby inhibiting the gluconeogenic process (69). Nakamura et al. demonstrated that Pxr activation by PCN inhibited hepatic G6pase and Pepck expression in rats and mice (71). Similarly, the expression of G6pase and Pepck in the liver of VP-hPXR mice treated with rifampicin was also decreased (72). But these did not occur in Pxr-knockout mice. The downregulation of G6pase and Pepck expression may indicate that Pxr activation reduces hepatocyte glucose output, potentially improving glucose homeostasis in type 2 diabetes. Studies have also shown inconsistent gluconeogenic responses in the liver following Pxr activation (15, 73). As mentioned earlier, G6pase and Pepck expression was inhibited in mouse liver and human hepatoma cells following Pxr activation. However, rifampicin treatment of human primary hepatocytes for 6 h was able to induce the 2 times expression of G6Pase; whereas rifampicin treatment of human primary hepatocytes for 24 h resulted in a 30% reduction in G6Pase mRNA compared to the control group. Another study showed that simvastatin treatment of human hepatocytes for 24 h resulted in 7 times increase in PEPCK1 mRNA expression compared to the control group. Gotoh and Negishi et al. found that the PXR in human hepatocyte can bind directly to the promoters of G6Pase and PEPCK to regulate blood glucose. There are two distinct binding sites, one is the classical direct PXR binding site, and the other is the IRS site via protein-protein interactions. The activation of PXR binding to the promoter requires the involvement of serum/glucocorticoid regulated kinase 2 (SGK2) dephosphorylation co-activating transcription factors. Interestingly, PXR not only alters the phosphorylation status of SGK2, but also binds to the activated SGK2 gene promoter to induce SGK2 expression (15). The mechanism of PXR-mediated regulation of human hepatic gluconeogenesis still needs further investigation.
In addition to regulating gluconeogenesis, PXR activation is also involved in the oxidative absorption of glucose. The hepatic level of glucose transporter 2 (GLUT2) mRNA was downregulated by PCN. In rat and mouse hepatocytes, PCN-mediated activation of PXR downregulated the expression of glucose transporter 2 (GLUT2) and glucokinase (GCK) indicating a detrimental role of PXR activation on glucose tolerance (74). GCK drives the phosphorylation of glucose to glucose-6-phosphate, which is the first step in glycolysis. Mutations leading to reduced GCK activity have been reported as the cause of early-onset type 2 diabetes, and GCK activators are being investigated as potential agents for type 2 diabetes (75). As a major component of green tea (–),-Epigallocatechin-3-gallate (EGCG) activated PXR and constitutive androstane receptor (CAR), accompanied by up-regulating expressions of PXR/CAR-mediated phase 2etabolism enzymes (SULT1A1, UGT1A1 and SULT2B1b) in small intestine and liver (76). Thereby, this process can inhibit the starch digestion and improving glucose homeostasis. Therefore, EGCG has been considered as a promising PXR/CAR activator and therapeutic intervention in diabetes.
As for some oral hypoglycemic drugs, Shashi et al. showed that some oral antidiabetic agents, such as rosiglitazone and pioglitazone (thiazolidinediones, TZDs), can also activate PXR and upregulate its downstream expression of CYP3A4, UGT1A1, MDR1 and thereby may inflict undesirable consequences (77). As GLUT2 and GCK have important functions in postprandial glucose uptake, their abnormal regulation may be involved in PXR-induced postprandial hyperglycemia. Indeed, hepatic GLUT2 and GCK knockout rats developed mild hyperglycemia under normal feeding (78, 79). The regulation of GLUT2 and GCK by PXR was demonstrated in the HepG2 model. Atorvastatin reduced protein levels of GLUT2 and GCK and decreased glucose consumption and uptake in HepG2 cells. However, pravastatin had no effect on GLUT2 and GCK expression and no effect on glucose utilization. Based on in vitro studies, atorvastatin, simvastatin, lovastatin and fluvastatin are PXR activators, whereas pravastatin and rosuvastatin are not agonists of the PXR. At the same time, PXR knockdown or overexpression can up and down-regulate GLUT2 and GCK expression accordingly (80). Fatemeh et al. also verified these results in Pxr wild-type and Pxr knockout mice treated with PCN, where only in the wild type was the level of Glut2 protein down-regulated and glucose tolerance impaired after PXR activation (81). In a word, as a xenobiotic sensing regulator, PXR plays a crucial role in hepatic glucose metabolism (Figure 5A). These results indicate that the activation of PXR impairs glucose tolerance and thus PXR represents a novel diabetogenic pathway.
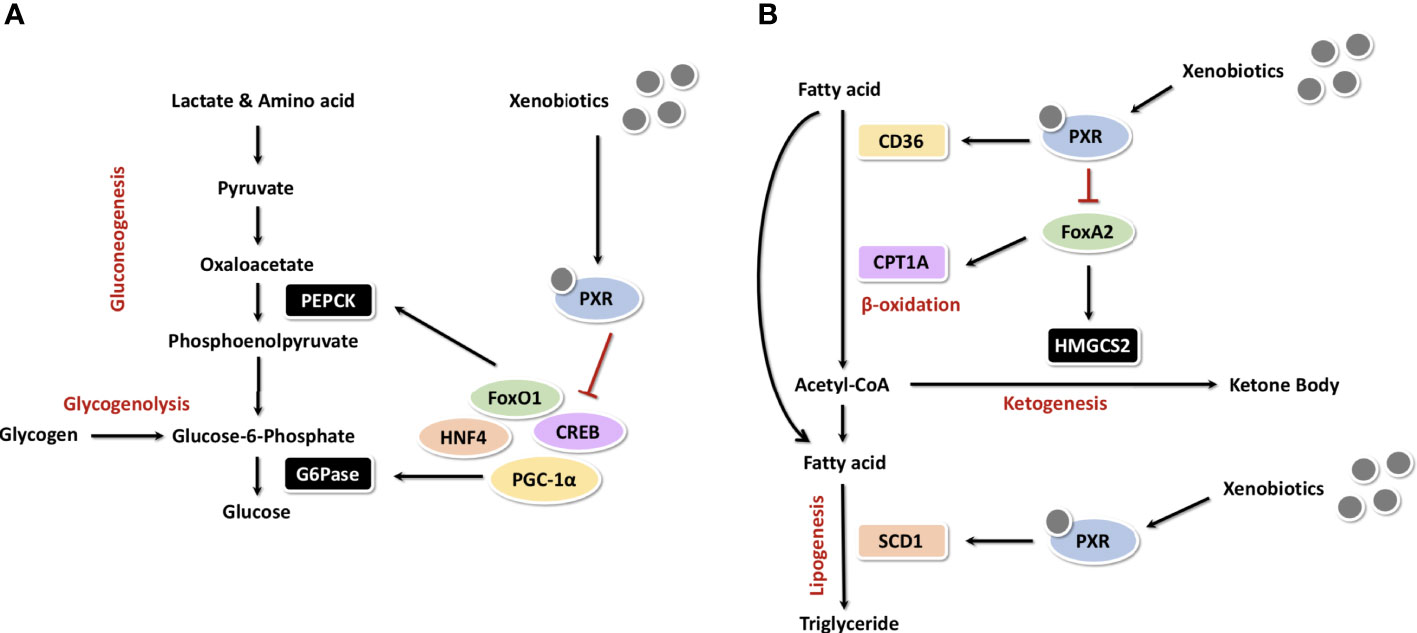
Figure 5 PXR in the regulation of hepatic glucose and cholesterol metabolism (A) As a xenobiotic sensing regulator, PXR plays a crucial role in hepatic glucose metabolism. (B) PXR in the regulation of hepatic cholesterol metabolism.
PXR in lipid metabolism
Triglycerides and fatty acids are important metabolic fuels. Lipid homeostasis balances lipid uptake and synthesis with lipid metabolism and secretion. When glucose and fatty acids exceed the body’s energy requirements, they are converted to triglycerides and stored in the liver. The expression of PXR and fatty acid binding protein 4 (FABP4) were increased by Valproate (valproic acid, VPA), a widely used drug in the therapy of epilepsy, in a dose-dependent manner. On the contrary, knockdown of PXR not only reduced lipid accumulation but also impaired the induction of FABP4 by VPA. While overexpression of PXR enhanced both lipid accumulation and FABP4 expression. These results suggest that PXR-mediated expression of FABP4 is responsible for lipid accumulation caused by VPA (82).
During fasting or exercise, fatty acid β-oxidation and ketogenesis increase in adipocytes, thereby promoting ketone body synthesis and energy production. The sterol regulatory element-binding protein-1c (SREBP-1c) is a major regulator of lipogenesis. Some NRs, such as liver X receptor (LXR), HNF4 and LRH-1, control lipid homeostasis by regulating the transcriptional activity of SREBP (83–85). VP-PXR transgenic mice develop intrahepatic triglyceride accumulation and are associated with upregulation of the fatty acid translocase CD36 and certain other lipogenic coenzymes, including SCD-1 and long-chain free fatty acid elongase. CD36 is a scavenger receptor with broad ligand specificity. The activation of CD36 promotes uptake of free fatty acids from the circulatory system and may be involved in hepatic steatosis (86). The correlation between CD36 levels and the storage and secretion of triglyceride suggests that CD36 plays an initiating role in hepatic steatosis. Moreover, PXR plays an essential role in CD36 transcription. Studies have shown that CD36 is a direct target gene of PXR transcriptional regulation (72). The expression of CD36 can also be positively regulated by LXR and peroxisome proliferator-activated receptor γ (PPARγ). Therefore, CD36 should be a common transcriptional target gene of LXR, PXR and PPARγ in the regulation of lipid homeostasis (87). Studies in the cardiovascular field have shown that higher Bisphenol A (BPA) exposure has been associated with an increased risk of atherosclerosis and cardiovascular disease (CVD). hPXR but ApoE knockout model mice were used by Sui et al. to study the teratogenic effects of BPA. It indicates that PXR epigenetically regulated CD36 expression by increasing H3K4me3 levels and decreasing H3K27me3 levels in the CD36 promoter in response to perinatal BPA exposure (88).
It has also been added that PXR can be activated by efavirenz, a drug commonly used in the treatment of HIV infection and proved as PXR-selective agonist. After efavirenz-mediated Pxr activation in mice, cholesterol biosynthetic cyclooxygenase (SQLE) can be regulated as a direct transcriptional target of Pxr in addition to CD36, leading to increased lipid uptake and cholesterol biosynthesis in hepatocytes. Considering that activation of PXR signaling may induce hypercholesterolemia and cirrhosis, the combination of this finding also suggests that PXR activation should be considered in patients on long-term PXR agonistic antiretroviral drugs (21). Similarly, Cobicistat (COBI) is the backbone of multiple regimens for antiretroviral therapy in AIDS patients. PXR (and CAR) modulate COBI hepatotoxicity through the CYP3A4-dependent pathways (89). The widely used anti-inflammatory drug hypocretin has also been reported as an agonist of PXR, the activation of PXR is followed by upregulation of the downstream proteins CYP3A11, CYP2B10, and organic anion transporter 2 (OATP2), which can also stimulate nuclear migration of YAP, leading to lipid accumulation (10). In addition to the accumulation of triglycerides in the liver of transgenic mice, PXR activation down-regulates hepatic PPARα activity and fibroblast growth factor 12 (FGF21) circulation, which could participate in the pleiotropic role of PXR in energy homeostasis (90).
Carnitine palmitoyltransferase 1A (CPT1A) and mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 (HMGCS2) are two important enzymes which involved in β-oxidation and ketogenesis. In the absence of insulin, the winged helix/forkhead transcription factor FoxA2 activates transcription of CPT1A and HMGCS2 (91). Insulin induces phosphorylation and exonucleation of FoxA2, which activates FoxA2 and suppresses transcription of CPT1A and HMGCS2 (92). Nakamura et al. showed that PCN down-regulates transcription of CPT1A and HMGCS2 in wild-type mice, but not in Pxr knockout mice. The mechanism may be that PXR directly binds to FoxA2, thus inhibits the activation of CPT1A and HMGCS2 genes (71). Figure 5B illustrates the overall mechanism of PXR in cholesterol metabolism.
Cholesterol is essential for the formation of cell membranes, bile acids and steroid hormones. Oxidized cholesterol metabolites are cytotoxic and are a risk factor for atherosclerosis. Cholesterol detoxification protects the body from producing excess cholesterol. In most tissues, the mitochondrial sterol 27-hydroxylase (CYP27A1) is an essential molecule for cholesterol shearing and hydroxylation. Li et al. found that PXR activated CYP27A1 and the cholesterol efflux transport proteins, ATP binding cassette (ABC) subfamily A member 1 (ABCA1) and subfamily G member 1 (ABCG1) in enterocytes (93).
Fibroblast growth factor (FGF) 15 plays a crucial role in the regulation of metabolism. Some findings suggest that PXR may negatively regulate FGF15 expression. In high fat diet (HFD)-fed Pxr knockout mice, intestinal FGF15 expression levels were significantly elevated and total lipids in feces were significantly increased compared with HFD-fed wild-type mice. These represent PXR as a potential therapeutic target for the treatment for metabolic disorders such as obesity (94).
Meng et al. experimented with quetiapine as a PXR agonist, a drug commonly used to treat psychiatric disorders. PXR activation stimulated the intestinal expression of cholesterol transporter Niemann-Pick C1-Like 1 (NPC1L1) and microsomal triglyceride transfer protein (MTP), leading to increased intestinal lipid absorption. Thus, NPC1L1 is a known PXR target gene, they identified a DR-1-type PXR response element in the MTP promoter and established MTP as a potentially novel transcriptional target of PXR (95).
High density lipoprotein (HDL) and its major component apolipoprotein A-I (ApoA-I) are involved in cholesterol reversal and associated with a reduced risk of atherosclerosis. ApoA-I and HDL cholesterol levels can be elevated by Pxr agonists in wild-type mice, but not in Pxr knockout mice. Bile acids mediated the downregulation of HDL cholesterol and lipid ApoA-I was completely absent in human Pxr transgenic mice (96). It has also been suggested that PXR has a pro-atherogenic effect. The expression of ABCA1 is reduced in hepatocytes after PXR activation (97). Clinical use of PXR-activating drugs can lead to hyperlipidemia and drug-induced hypercholesterolemia in some patients (98). Future studies will need to further elucidate the pathological role of PXR in hyperlipidemia.
PXR in bile acid circulation
Synthesized in the liver, bile acid is the end product of cholesterol catabolism and involved in the body’s removal of cholesterol (99). When bile acid excreted by the intestine, it promotes the absorption of cholesterol and fat-soluble vitamins. However, the excess of bile acid is cytotoxic and can lead to pathological cholestasis. Therefore, the level of bile acid needs to be strictly regulated to avoid toxic damage to the body. PXR plays a crucial role in the detoxification of bile acids. PCN reduced lithocholic acid (LCA)-induced toxicity in wild-type mice, but not in Pxr knockout mice, and Pxr transgenic mice were also tolerant to LCA toxicity. The protective effect of PXR can be explained by the regulation of genes involved in bile acid metabolism. The phase II metabolic enzyme SULT2A is a target gene of PXR and is involved in the detoxification of bile acid (100). In addition to regulating bile acid synthesis and metabolism, PXR also regulates the expression of bile acid transfer proteins, such as MRP2 and OATP2 (101, 102).
Drug-induced hepatotoxicity or acute liver failure remains a key issue in clinical medicine. PARP1-dependent poly(ADP-ribosyl)ation plays a key role in cellular stress responses and functions in multiple physiological and pathological processes. Wang et al. used a mouse model of Acetaminophen (APAP)-induced liver failure to investigate whether PARP1-dependent poly(ADP-ribosyl)ation was involved in the metabolic process. The result indicates that PARP1-dependent poly(ADP-ribosyl)ation of PXR in ligand-binding domain activates PXR competitively and solidly, facilitates its recruitment to target gene CYP3A11 promoter, and promotes CYP3A11 gene transcription, thus up-regulating APAP pro-toxic metabolism. Thus, the inhibition of PARP1-dependent poly(ADP-ribosyl)ation might represent a novel approach for the treatment of drug-induced hepatotoxicity (103). Zeng et al. ‘s experiment on palmitate (PA) treatment of HepG2 cells showed a significant reduction in mRNA levels of CYP3A, but the same results were observed in PXR knockout HepG2 cell lines. The above studies suggest that the transcriptional repression of CYP3A is not regulated by PXR. Although the results of the two experiments are controversial, they suggest that PXR interacts with CYP3A in some way (104).
Bilirubin is a degradation product of hemoglobin protein. UDP-glucuronosyltransferase (UGT) binds bilirubin and converts the neurotoxic unconjugated bilirubin into the non-toxic glucuronide bilirubin. Activation of PXR in mice suppresses hyperbilirubinemia. Oleanolic acid (OA) and ursolic acid (UA) activate the transcription of UGT1A1 and some important genes involved in bilirubin detoxification, such as OATP2 and MRP2 through PXR (60, 101, 105). OATP2 mediates the uptake of bilirubin from the blood into the liver and MRP2 facilitates the excretion of conjugated bilirubin into the bile ducts. Although PXR was initially characterized as a xenosensor, the discovery that certain bile acids such as LCA can serve as ligands for both human and mouse PXR provided a link between PXR and bile acid regulation (106). Below we will illustrate the role of PXR in the detoxification of bile acids and the implications in cholestatic disorders. It has been reported that PXR has some interactions with FXR in bile acid regulation. However neither conjugated LCA, nor any of the other conjugated bile acids activate PXR. In addition to direct activation by bile acids, PXR is a dependent transcriptional target of bile acid-activated FXR (107). PXR can mitigate the harmful effects of toxic bile acids (BA) such as LCA by activation of hepatic detoxification pathways. Activation of PXR induces the uptake of xenobiotics and endobiotics (phase 0), their modification by members of the cytochrome P450 subfamily (phase I), conjugation by glutathione S-transferases (GSTs), UDP-glucuronosyl-transferases (UGTs) and sulfotransferases (SULTs) (phase II) and elimination (phase III) by MRP2 (excretion of bilirubin and some bile acids), and the multidrug transporter MDR1 which excretion of a wide variety of xenobiotics and endobiotics. PXR can be directly activated by certain bile acids or indirectly via transcriptional regulation by FXR. Negative feedback on bile acid metabolism is mediated by inhibition of CYP7A1. During cholestasis bile acids can also be excreted back into the circulation via the sinusoidal ABC-transporters MRP3 and MRP4 (108) (Figure 6). Thus, PXR ligands may be potential agents for the treatment of hyperbilirubinemia.
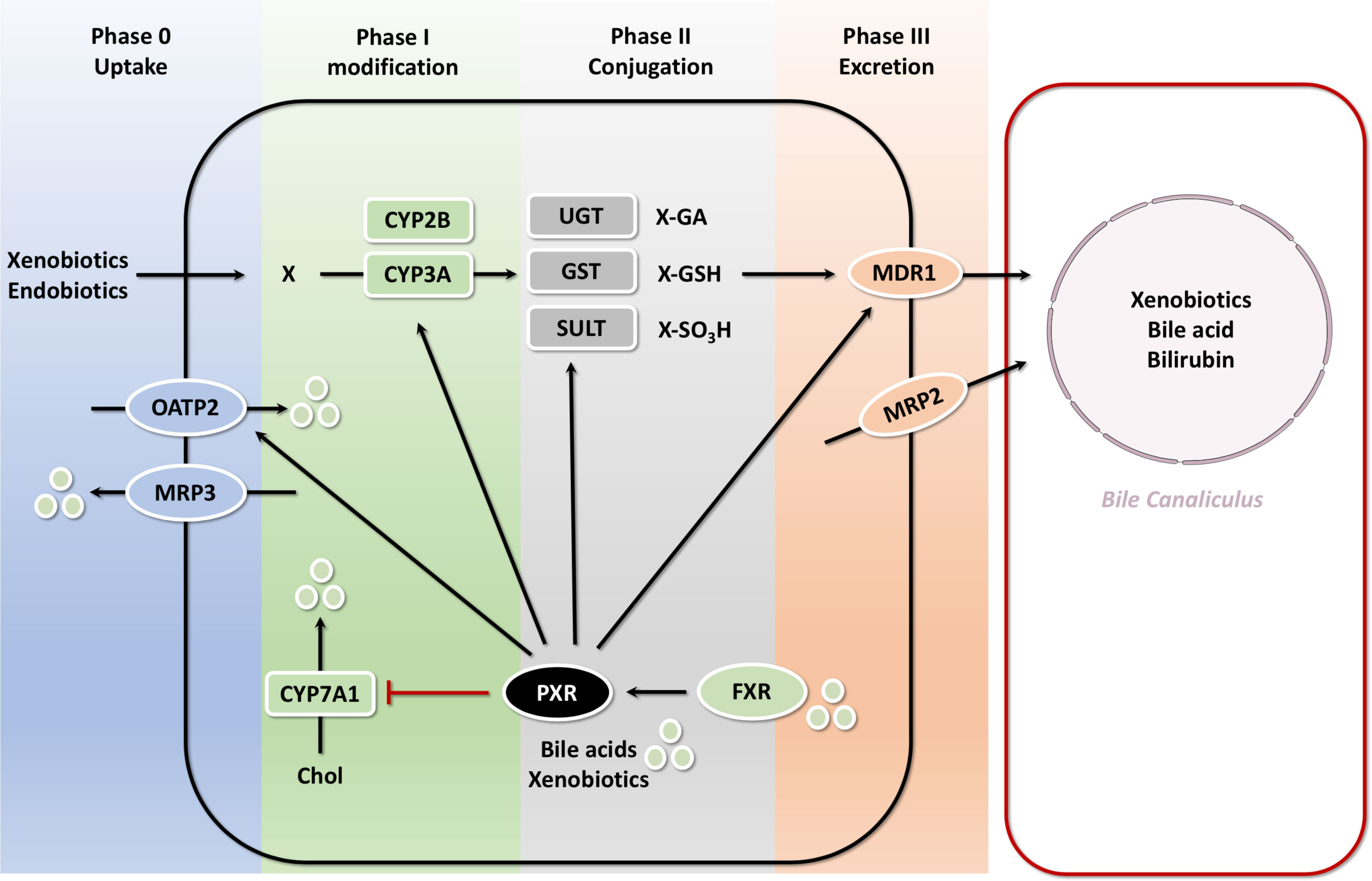
Figure 6 PXR-mediated bile acid transport and metabolism in the hepatocyte with FXR PXR is a dependent transcriptional target of bile acid-activated FXR. PXR can mitigate the harmful effects of toxic bile acids (BA) such as LCA by activation of hepatic detoxification pathways. Activation of PXR induces the uptake of xenobiotics and endobiotics (phase 0), their modification by members of the cytochrome P450 subfamily (phase I), conjugation by GSTs, UGTs and SULTs (phase II) and elimination (phase III) by MRP2, and the multidrug transporter MDR1 which excretion of a wide variety of xenobiotics and endobiotics. PXR can be directly activated by certain bile acids or indirectly via transcriptional regulation by FXR. Negative feedback on bile acid metabolism is mediated by inhibition of CYP7A1.
PXR in vitamin metabolism and bone metabolism
Vitamin K2 is essential for bone formation and is commonly used clinically in the treatment of osteoporosis. Vitamin K2 has been reported to activate PXR and promote the expression of PXR target genes. Treatment of osteosarcoma cells with vitamin K2 increased the expression of the osteoblast markers: bone alkaline phosphatase, osteoprotegerin, bone bridging protein and scaffold Gla protein (109). Vitamin K2 induced the expression of bone markers in primary osteoblasts of wild-type mice, but not in Pxr knockout mice. Ichikawa et al. found that certain PXR target genes are associated with bone function in osteoblasts (110). Besides, the results of Igarashi et al. showed that activation of PXR by vitamin K2 induced the expression of the osteoclastogenic transcription factor muscle segment homeobox 2 (MSX2), which is involved in osteoblast differentiation (111).
In addition, vitamin K2 prevents arterial calcification and atherosclerosis, and adequate intake of vitamin K2 reduces the risk of vascular damage. It is worth mentioning that calcification induced by menaquinone-4 (MK-4), the most common form of vitamin K2 present in animals, can be inhibited by inhibitors of PXR. It was also shown that MK-4 plays an accelerating role in the process of calcification in human aortic valve interstitial cells (HAVICs) through the PXR-BMP2-ALP pathway (112). MK-4 administration also altered mRNA levels of genes involved in drug metabolism (Abca3, Cyp2s1, Sult1b1), and mRNA levels of CYP7A1 and CYP8B1 are similarly changed in human hepatocarcinoma HepG2 cells (113). Besides, MK-4 along with other vitamin Ks, including vitamin K1, has the potential to induce MDR1 and CYP3A4 gene expression. But Pxr knockdown reversed MK-4-mediated stimulation of these genes, indicating the involvement of PXR in this effect. These results elucidate the importance of drug-nutrient interaction mediated via PXR (114).
Calcium is an important component of bone development and maintenance. Vitamin D regulates calcium absorption and excretion, and its activated metabolite 1,25(OH)2D3 binds to the vitamin D receptor (VDR). VDR activates 24-hydroxylation mediated by 25-hydroxyvitamin D (3)-24-hydroxylase (CYP24), which is essential in 1,25(OH)2D3 metabolism. Pascussi et al. reported that PXR activation upregulated CYP24 gene expression (115). However, Zhou et al. found that PXR activation inhibited CYP24 gene expression (116). Although the results of the two research are controversial, they suggest that PXR plays a potential function in bone homeostasis and further studies are needed to confirm. Ligand activation of PXR also inhibits the transcription of vitamin D3 25-hydroxylase (CYP2D25) which is an important hydroxylase in 1,25(OH)2D3 biosynthesis (117). Centuries ago, it was found that prolonged treatment with antitussive agents could lead to vitamin D deficiency or chondromalacia in patients. As many antitussive agents are PXR ligands, these results are significant for the prevention of drug-induced chondromalacia in patients.
The treatment and prevention of osteoclast-associated diseases also play a particularly crucial role in addressing problems related to bone metabolism. Guo et al. used common histamine H1 receptor antagonists to experiment in vivo and in vitro, meclizine reduced osteoclast formation and bone resorption in a dose-dependent manner, while knockdown of PXR with siRNA partially abrogated the osteoclastogenesis inhibition of meclizine (118). Besides, PXR also represses osteoblast differentiation through repression of the Hedgehog signaling pathway, it can repress the Hedgehog signaling-induced genes such as Gli1 and Hhip, and conversely induced the Hedgehog signaling-repressed genes such as Cdon, Boc, and Gas1 (119).
Vitamin E is usually taken as an antioxidant in the daily diet. Vitamin E is metabolized by CYPs-mediated oxidative reactions and then excreted through β-oxidation and binding reactions including glucosylation and sulphation (120, 121). These processes are catalyzed and involved by enzymes and transfer proteins encoded by PXR target genes. Vitamin E activates PXR and may therefore regulate exogenous deleterious genes involved in its own metabolism. A study by Landes et al. using reporter gene analysis showed that PXR can be activated by some forms of vitamin E (122). Vitamin E metabolites were significantly decreased in the urine of wild-type mice following PCN treatment, but not in Pxr knockout mice, suggesting that this was caused by a PXR-mediated decrease in hepatic sterol carrier protein 2 expression and diminished β-oxidation (123). These results have led to widespread interest in investigating potential drug-drug interactions between vitamin E and PXR regulators.
PXR in endocrine homeostasis
The androgen receptor signaling pathway has an important role in the initiation and progression of prostate cancer. Therefore, androgen blockade is the most effective endocrine therapy for hormone-dependent prostate cancer. The two important PXR target genes, cytochrome P450 (CYP) 3As and hydroxysteroid sulfotransferase (SULT)2A1, function in androgen metabolism. CYP3As is an important enzyme that catalyzes the hydroxylation of testosterone and luteinizing hormone, producing the effects of hormone inactivation. Dehydroepiandrosterone-sulfotransferase 2Al (SULT2A1) is the main SULT isoform involved in androgen sulphonation (124). Zhang et al. reported a PXR-mediated metabolic blockade of androgens. This study revealed that PXR activation decreased androgenic activity and inhibited androgen-dependent prostate regeneration in castrated male rats which received daily testosterone injections to induce CYP3As and SULT2A1 expression (125).
In human prostate cancer cells (LAPC-4 and LA99 cells), treatment with rifampicin (RIF), the human PXR agonist, can inhibit the androgen-dependent proliferation of LAPC-4 cells but had essentially no effect on the growth of non-androgen-dependent homozygous LA99 cells. Downregulation of PXR or SULT2A1 by shRNA or siRNA in LAPC-4 cells abolished the effect of RIF, suggesting that the androgen inhibitory effect of RIF is PXR and SULT2A1 dependent. Thus, PXR may serve as a novel therapeutic target to reduce androgens for the treatment and inhibition of hormone-dependent prostate cancer (125).
Zhai et al. showed that PXR plays a crucial role in adrenal steroid homeostasis. The activation of PXR is accompanied by increased cytoplasmic levels of corticosterone and aldosterone and activation of adrenal steroidogenic enzymes such as CYP11a1, CYP11b1, CYP11b2 and 3β-hydroxysteroid dehydrogenase (3β-HSD) (126).
However, adrenocorticotropic hormone of pituitary secretion was normal in Pxr transgenic mice and cortisol was strongly inhibitory to dexamethasone, indicating that normal hypothalamic-pituitary-adrenal axis function even though adrenal steroid homeostasis was severely impaired. Consistent with these observations, some clinical studies have reported that RIF increases urinary steroid secretion and may also lead to misdiagnosis of Cushing’s syndrome (126). Thus, PXR is likely to affect endocrine homeostasis and to function in drug-hormone interactions.
Recently, some studies have linked endocrine disruption, chemical exposure, and cardiovascular disease risk in human, but the underlying mechanisms for this linkage are not clear. Many endocrine disorders involved the activation of the nuclear receptor PXR, and the PXR agonist tributyl citrate induces PXR target gene expression and activates PXR in the small intestine but has no effect on PXR activity in the liver. The mice exposure of tributyl citrate increased plasma total cholesterol and atherogenic LDL cholesterol levels in mice, but not in Pxr knockout mice (23).
Contribution of chemicals and drugs activating PXR in specific diseases
Recent studies have found that the detoxification system of PXR is a double-edged sword. Although detoxification is a beneficial protective mechanism against toxic compounds, it affects the absorption, distribution, metabolism and elimination of drugs in the body while making the half-life and tissue distribution of drugs in the body unpredictable. At the same time, it may lead to adverse drug reactions during clinical administration, such as reduced drug efficacy, induction of drug toxicity or drug resistance, thus affecting the clinical efficacy and safety of many drugs.
As for the discovery of PXR in aristolochic acid-induced kidney injury and other nephropathy. Atrazine is an herbicide, and environmental exposure to atrazine and its degradation products can cause nephrotoxicity. Atrazine exposure activates the PXR in mice, disrupting CYP450 homeostasis and exacerbating nephrotoxicity. Lycopene supplementation significantly prevented atrazine-induced nephrotoxicity and improved renal injury by modulating CYP450 homeostasis and PXR response (127). In addition, Ochratoxin A is present in food and decreases the survival of human proximal tubular cells and increases the expression of kidney injury molecule 1 (KIM-1).
Ochratoxin A may induce upregulation of PXR gene transcription and cause proximal tubular injury through PXR-related signaling pathways (128). Similarly, Ochratoxin A is also widely present in food and the environment and can cause chronic interstitial nephropathy. Studies have shown that Ochratoxin A does not activate PXR, but when combined with rifampicin, Ochratoxin A can down-regulate PXR gene expression, showing PXR antagonistic effects. In other words, Ochratoxin A is not due to the antagonism itself but due to the downregulation of PXR gene expression (129). In references 129 and 130, Ochratoxin A appear to have contradictory roles in relation to PXR. We suggest that PXR is involved in the regulation of renal drug metabolism and multiple other pathophysiological processes (not limited to the mechanisms explained in the two studies above). The regulation of drug metabolism by PXR in vivo is a double-edged sword, both in terms of accelerating toxicant metabolism and thus reducing nephrotoxicity, and in terms of accelerating drug metabolism and mediating drug-drug interactions. PXR is expected to be a therapeutic target in the pathogenesis of various kidney diseases, and to drive the process of clinical drug optimization and new drug development. PXR has a protective effect against acute toxicity induced by a high cholesterol diet. In PXR KO mice, high doses of cholestatic cholesterol feed lead to cholestasis and death due to severe liver and kidney failure. PXR signaling pathway protects the body from toxic dietary cholesterol metabolites, and activation of PXR improves acute renal failure associated with cholestatic liver disease (130).
Apart from renal disease, non-alcoholic fatty liver disease (NAFLD) which has a significant gender difference in the incidence during the whole population. In the process of NAFLD disease development, the expression of PXR and its target gene Cyp3a11 is progressively increased (131). Bile salts in human body may increase NAFLD risk by activating PXR receptor (132). However, polychlorinated biphenyls (PCBS), which can activate PXR, exist in the external environment. Wahlang et al. further found in animal experimental studies that Aroclor1260, a mixture of PCBS, aggravated NAFLD in diet-induced obese mice. Exposure to PCBS promotes the transition from hepatic steatosis to steatohepatitis, in part due to PXR activation. In vascular metabolic disease, Bisphenol A which is a basic chemical substance, is widely found in plastics and exposure to it is ubiquitous (133). In population-based studies, higher BPA exposure has been associated with an increased risk of atherosclerosis (88). In a similar way that BPA may increase the risk of atherosclerosis, some drugs in the clinic may increase the risk of cardiovascular disease by increasing circulating atherogenic lipids after PXR excitation. Karpale et al. conducted a serum metabolomic analysis in healthy volunteers, and found that administration of the PXR agonist rifampicin increased serum fractions of very low density lipoproteins and low density lipoproteins compared with placebo (134).
According to the latest studies, activation of PXR, the major regulator of drug metabolism and molecular mediator of clinically significant drug-drug interactions, has been shown to induce hypercholesterolemia. PXR may in part mediate hypercholesterolemic effects of drug treatment. In Table 2, we summarized the common drugs (as PXR ligand agonists) and their effects on lipid metabolism from five aspects, drug class, drug, mechanism and the influence on cholesterol and PXR.
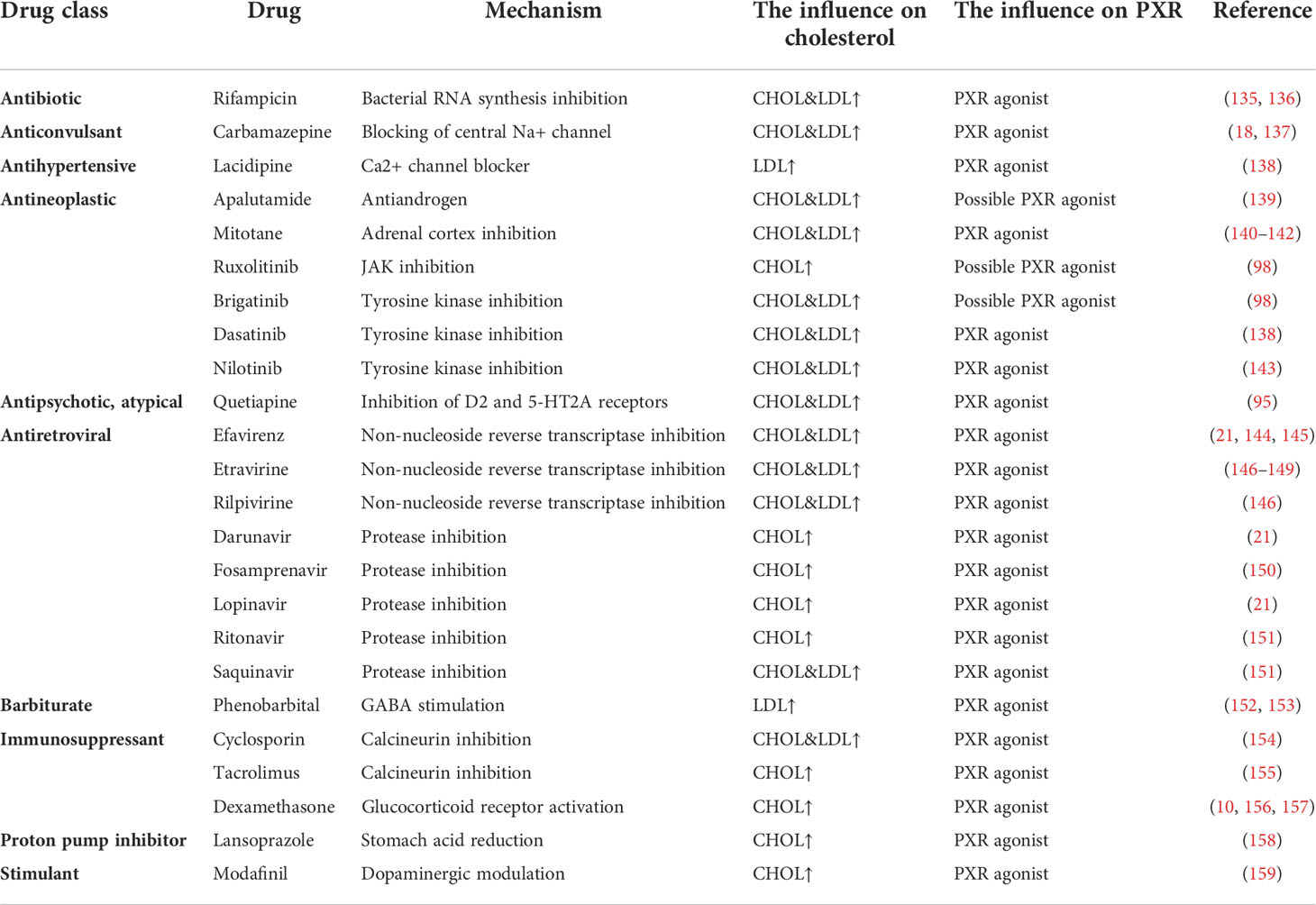
Table 2 Drugs which can increase the cholesterol and their potential to activate PXR.
Conclusion and perspective
According to accumulated evidence, PXR plays a significant role in substance metabolism (Figure 7), including but not limited to glucose metabolism, lipid metabolism and bile acid circulation. At the same time, some of the agonists that have been identified can also be involved in activating PXR during these processes, resulting in different effects. Not only PXR, but also other metabolic NRs may be also involved in the physiological and pathophysiological processes of substance metabolism. As the role of PXR in the regulation of substance metabolism becomes better understood, the use of PXR in the prevention and treatment of human diseases will gradually develop. Another challenge is that although the physiological functions of PXR have been discovered, the endogenous ligands or agonists remain largely elusive. Besides, targeted therapies for metabolic nuclear receptors will also become a new treatment in the future. Overall, PXR is still an attractive target, but the diversity of PXR biology and several pharmacological aspects of PXR modulation should be of concern for the rational therapeutic strategy and novel drug development.
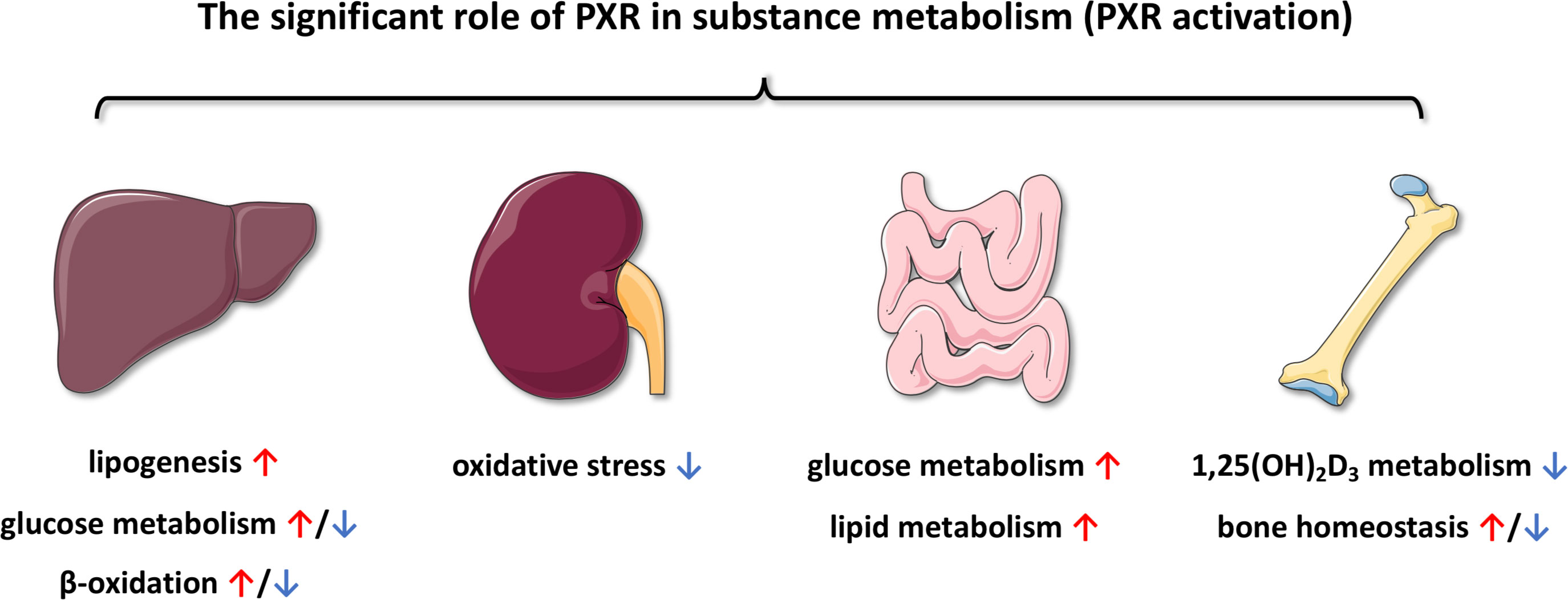
Figure 7 Schematic summary of the functions of PXR in substance metabolism among various organ.
Author contributions
YL retrieved literature and prepared the initial version of the manuscript and figures. Y-YL retrieved literature and prepared the figures. H-WR edited the initial version of the manuscript and provided suggestions to improve the manuscript. C-JL and Z-XX retrieved literature. Z-LL conceptualized the manuscript and prepared the final version of the manuscript and figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Education Department of Liaoning Province, China LZ2020023 (to Z-LL), Dalian Young Star of Science and Technology 2019RQ116 (to Z-LL) and Teaching Reform Project Foundation for Undergraduate Innovative Talent Cultivation of Dalian Medical University 111806010301 (to YL). We are also grateful for the support from Liaoning BaiQianWan Talents Program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Kliewer S, Moore J, Wade L, Staudinger J, Watson M, Jones S, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell (1998) 92(1):73–82. doi: 10.1016/S0092-8674(00)80900-9
2. Orans J, Teotico D, Redinbo M. The nuclear xenobiotic receptor pregnane X receptor: recent insights and new challenges. Mol Endocrinol (2005) 19(12):2891–900. doi: 10.1210/me.2005-0156
3. Zhang B, Xie W, Krasowski DM. PXR: a xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics (2008) 9(11):1695–709. doi: 10.2217/14622416.9.11.1695
4. Keen JC, Moore HM. The genotype-tissue expression (GTEx) project: Linking clinical data with molecular analysis to advance personalized medicine. J Pers Med (2015) 5(1):22–9. doi: 10.3390/jpm5010022
5. The Tabula Muris Consortium. Single-cell transcriptomics of 20 mouse organs creates a tabula muris. Nature (2018) 562(7727):367–72. doi: 10.1038/s41586-018-0590-4
6. Krasowski M, Yasuda K, Hagey L, Schuetz E. Evolution of the pregnane x receptor: adaptation to cross-species differences in biliary bile salts. Mol Endocrinol (Baltimore Md) (2005) 19(7):1720–39. doi: 10.1210/me.2004-0427
7. Jones S, Moore L, Shenk J, Wisely G, Hamilton G, McKee D, et al. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol (Baltimore Md) (2000) 14(1):27–39. doi: 10.1210/mend.14.1.0409
8. Luo G, Cunningham M, Kim S, Burn T, Lin J, Sinz M, et al. CYP3A4 induction by drugs: correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab Disposition: Biol Fate Chemicals (2002) 30(7):795–804. doi: 10.1124/dmd.30.7.795
9. Chianese G, Sepe V, Limongelli V, Renga B, D'Amore C, Zampella A, et al. Incisterols, highly degraded marine sterols, are a new chemotype of PXR agonists. Steroids (2014) 83:80–5. doi: 10.1016/j.steroids.2014.02.003
10. Jiao T, Yao X, Zhao Y, Zhou Y, Gao Y, Fan S, et al. Dexamethasone-induced liver enlargement is related to PXR/YAP activation and lipid accumulation but not hepatocyte proliferation. Drug Metab Disposition: Biol Fate Chemicals (2020) 48(9):830–9. doi: 10.1124/dmd.120.000061
11. Lille-Langøy R, Karlsen O, Myklebust L, Goldstone J, Mork-Jansson A, Male R, et al. Sequence variations in pxr (nr1i2) from zebrafish (Danio rerio) strains affect nuclear receptor function. Toxicol Sci an Off J Soc Toxicol (2019) 168(1):28–39. doi: 10.1093/toxsci/kfy269
12. Chang H, Chen C, Ma W, Cheng W, Lin Y, Lee Y, et al. Modulation of pregnane X receptor (PXR) and constitutive androstane receptor (CAR) activation by ursolic acid (UA) attenuates rifampin-isoniazid cytotoxicity. Phytomed Int J Phytother Phytopharmacol (2017) 36:37–49. doi: 10.1016/j.phymed.2017.09.016
13. Silveira M, Lindor K. Obeticholic acid and budesonide for the treatment of primary biliary cirrhosis. Expert Opin Pharmacother (2014) 15(3):365–72. doi: 10.1517/14656566.2014.873404
14. Flora G, Sahli K, Sasikumar P, Holbrook L, Stainer A, AlOuda S, et al. Non-genomic effects of the pregnane X receptor negatively regulate platelet functions, thrombosis and haemostasis. Sci Rep (2019) 9(1):17210. doi: 10.1038/s41598-019-53218-x
15. Gotoh S, Negishi M. Statin-activated nuclear receptor PXR promotes SGK2 dephosphorylation by scaffolding PP2C to induce hepatic gluconeogenesis. Sci Rep (2015) 5:14076. doi: 10.1038/srep14076
16. Zhang X, Ma Z, Liang Q, Tang X, Hu D, Liu C, et al. Tanshinone IIA exerts protective effects in a LCA-induced cholestatic liver model associated with participation of pregnane X receptor. J Ethnopharmacol (2015) 164:357–67. doi: 10.1016/j.jep.2015.01.047
17. Kim S, Hasanuzzaman Md, Cho M, Kim N, Choi H, Han J, et al. Role of 14-3-3 sigma in over-expression of p-gp by rifampin and paclitaxel stimulation through interaction with PXR. Cell Signalling (2017) 31:124–34. doi: 10.1016/j.cellsig.2017.01.001
18. Grewal G, Singh K, Kanojia N, Rawat C, Kukal S, Jajodia A, et al. Exploring the carbamazepine interaction with human pregnane X receptor and effect on ABCC2 using in vitro and in silico approach. Pharm Res (2017) 34(7):1444–58. doi: 10.1007/s11095-017-2161-z
19. Foo W, Tay H, Chan E, Lau A. Meclizine, a pregnane X receptor agonist, is a direct inhibitor and mechanism-based inactivator of human cytochrome P450 3A. Biochem Pharmacol (2015) 97(3):320–30. doi: 10.1016/j.bcp.2015.07.036
20. Piedade R, Traub S, Bitter A, Nüssler A, Gil J, Schwab M, et al. Carboxymefloquine, the major metabolite of the antimalarial drug mefloquine, induces drug-metabolizing enzyme and transporter expression by activation of pregnane X receptor. Antimicrobial Agents Chemother (2015) 59(1):96–104. doi: 10.1128/AAC.04140-14
21. Gwag T, Meng Z, Sui Y, Helsley R, Park S, Wang S, et al. Non-nucleoside reverse transcriptase inhibitor efavirenz activates PXR to induce hypercholesterolemia and hepatic steatosis. J Hepatol (2019) 70(5):930–40. doi: 10.1016/j.jhep.2018.12.038
22. Mota L, Barfield C, Hernandez J, Baldwin W. Nonylphenol-mediated CYP induction is PXR-dependent: The use of humanized mice and human hepatocytes suggests that hPXR is less sensitive than mouse PXR to nonylphenol treatment. Toxicol Appl Pharmacol (2011) 252(3):259–67. doi: 10.1016/j.taap.2011.02.017
23. Sui Y, Helsley R, Park S, Song X, Liu Z, Zhou C. Intestinal pregnane X receptor links xenobiotic exposure and hypercholesterolemia. Mol Endocrinol (2015) 29(5):765–76. doi: 10.1210/me.2014-1355
24. Sui Y, Park S, Helsley R, Sunkara M, Gonzalez F, Morris A, et al. Bisphenol a increases atherosclerosis in pregnane X receptor-humanized ApoE deficient mice. J Am Heart Assoc (2014) 3(2):e000492. doi: 10.1161/JAHA.113.000492
25. Zhang G, Liu M, Song M, Wang J, Cai J, Lin C, et al. Patchouli alcohol activates PXR and suppresses the NF-κB-mediated intestinal inflammatory. J Ethnopharmacol (2020) 248:112302. doi: 10.1016/j.jep.2019.112302
26. Marx-Stoelting P, Ganzenberg K, Knebel C, Schmidt F, Rieke S, Hammer H, et al. Hepatotoxic effects of cyproconazole and prochloraz in wild-type and hCAR/hPXR mice. Arch Toxicol (2017) 91(8):2895–907. doi: 10.1007/s00204-016-1925-2
27. Wahlang B, Falkner K, Clair H, Al-Eryani L, Prough R, States J, et al. Human receptor activation by aroclor 1260, a polychlorinated biphenyl mixture. Toxicol Sci an Off J Soc Toxicol (2014) 140(2):283–97. doi: 10.1093/toxsci/kfu083
28. Wang Y, Lin W, Chai S, Wu J, Ong S, Schuetz E, et al. Piperine activates human pregnane X receptor to induce the expression of cytochrome P450 3A4 and multidrug resistance protein 1. Toxicol Appl Pharmacol (2013) 272(1):96–107. doi: 10.1016/j.taap.2013.05.014
29. Hu D, Wang Y, Chen Z, Ma Z, You Q, Zhang X, et al. The protective effect of piperine on dextran sulfate sodium induced inflammatory bowel disease and its relation with pregnane X receptor activation. J Ethnopharmacol (2015) 169:109–23. doi: 10.1016/j.jep.2015.04.006
30. Saito K, Moore R, Negishi M. p38 mitogen-activated protein kinase regulates nuclear receptor CAR that activates the CYP2B6 gene. Drug Metab Disposition: Biol fate chemicals (2013) 41(6):1170–3. doi: 10.1124/dmd.113.051623
31. Lin W, Bwayi M, Wu J, Li Y, Chai S, Huber A, et al. CITCO directly binds to and activates human pregnane X receptor. Mol Pharmacol (2020) 97(3):180–90. doi: 10.1124/mol.119.118513
32. Kanno Y, Yatsu T, Li W, Koike K, Inouye Y. Nigramide c is a natural agonist of human pregnane x receptor. Drug Metab Disposition: Biol Fate Chemicals (2014) 42(6):1084–9. doi: 10.1124/dmd.114.057810
33. Chen K, Chen H, Chen K, Chen C. Treatment of cardiovascular disease by traditional Chinese medicine against pregnane X receptor. BioMed Res Int (2014) 2014:950191. doi: 10.1155/2014/950191
34. Dolezelova E, Prasnicka A, Cermanova J, Carazo A, Hyrsova L, Hroch M, et al. Resveratrol modifies biliary secretion of cholephilic compounds in sham-operated and cholestatic rats. World J Gastroenterol (2017) 23(43):7678–92. doi: 10.3748/wjg.v23.i43.7678
35. Oladimeji P, Chen T. PXR: More than just a master xenobiotic receptor. Mol Pharmacol (2018) 93(2):119–27. doi: 10.1124/mol.117.110155
36. Luo W, Xin Y, Zhao X, Zhang F, Liu C, Fan H, et al. Suppression of carboxylesterases by imatinib mediated by the down-regulation of pregnane X receptor. Br J Pharmacol (2017) 174(8):700–17. doi: 10.1111/bph.13731
37. Mo L, He J. Nuclear hormone receptors PXR and CAR and metabolic diseases. Hormone Mol Biol Clin Invest (2014) 19(2):129–40. doi: 10.1515/hmbci-2014-0006
38. He J, Nishida S, Xu M, Makishima M, Xie W. PXR prevents cholesterol gallstone disease by regulating biosynthesis and transport of bile salts. Gastroenterology (2011) 140(7):2095–106. doi: 10.1053/j.gastro.2011.02.055
39. Bwayi MN, Garcia-Maldonado E, Chai SC, Xie B, Chodankar S, Huber AD, et al. Molecular basis of crosstalk in nuclear receptors: Heterodimerization between PXR and CAR and the implication in gene regulation. Nucleic Acids Res (2022) 50(6):3254–75. doi: 10.1093/nar/gkac133
40. Luan ZL, Zhang C, Ming WH, Huang YZ, Guan YF, Zhang XY. Nuclear receptors in renal health and disease. EBioMedicine (2022) 76:103855. doi: 10.1016/j.ebiom.2022.103855
41. Niu X, Wu T, Li G, Gu X, Tian Y, Cui H. Insights into the critical role of the PXR in preventing carcinogenesis and chemotherapeutic drug resistance. Int J Biol Sci (2022) 18(2):742–59. doi: 10.7150/ijbs.68724
42. Zhang L, Yan J, Liu J, Meng C, Liu F, Xia C. Panaxytriol upregulates CYP3A4 expression based on the interaction of PXR, CAR, HSP90alpha, and RXRalpha. Phytomedicine (2022) 101:154097. doi: 10.1016/j.phymed.2022.154097
43. Umesono K, Evans R. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell (1989) 57(7):1139–46. doi: 10.1016/0092-8674(89)90051-2
44. Mangelsdorf D, Evans R. The RXR heterodimers and orphan receptors. Cell (1995) 83(6):841–50. doi: 10.1016/0092-8674(95)90200-7
45. Chai X, Zeng S, Xie W. Nuclear receptors PXR and CAR: Implications for drug metabolism regulation, pharmacogenomics and beyond. Expert Opin Drug Metab Toxicol (2013) 9(3):253–66. doi: 10.1517/17425255.2013.754010
46. Goodwin B, Hodgson E, Liddle C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol Pharmacol (1999) 56(6):1329–39. doi: 10.1124/mol.56.6.1329
47. Drocourt L, Ourlin J, Pascussi J, Maurel P, Vilarem M. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin d receptor pathway in primary human hepatocytes. J Biol Chem (2002) 277(28):25125–32. doi: 10.1074/jbc.M201323200
48. Parveen A, Alhusban M, Fantoukh OI, Ali Z, Chittiboyina AG, Khan IA, et al. Probing PXR activation and modulation of CYP3A4 by tinospora crispa and tinospora sinensis. J Ethnopharmacol (2022) 291:115159. doi: 10.1016/j.jep.2022.115159
49. Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem (2001) 276(18):14581–7. doi: 10.1074/jbc.M010173200
50. Jeske J, Bitter A, Thasler W, Weiss T, Schwab M, Burk O. Ligand-dependent and -independent regulation of human hepatic sphingomyelin phosphodiesterase acid-like 3A expression by pregnane X receptor and crosstalk with liver X receptor. Biochem Pharmacol (2017) 136:122–35. doi: 10.1016/j.bcp.2017.04.013
51. Kubota K, Inaba S, Nakano R, Watanabe M, Sakurai H, Fukushima Y, et al. Identification of activating enzymes of a novel FBPase inhibitor prodrug, CS-917. Pharmacol Res Perspectives (2015) 3(3):e00138. doi: 10.1002/prp2.138
52. Cui J, Gunewardena S, Rockwell C, Klaassen C. ChIPing the cistrome of PXR in mouse liver. Nucleic Acids Res (2010) 38(22):7943–63. doi: 10.1093/nar/gkq654
53. Klaassen CD, Slitt AL. Regulation of hepatic transporters by xenobiotic receptors. Curr Drug Metab (2005) 6(4):309–28. doi: 10.2174/1389200054633826
54. Handschin C, Meyer UA. Induction of drug metabolism: the role of nuclear receptors. Pharmacol Rev (2003) 55(4):649–73. doi: 10.1124/pr.55.4.2
55. Xie W, Evans RM. Orphan nuclear receptors: the exotics of xenobiotics. J Biol Chem (2001) 276(41):37739–42. doi: 10.1074/jbc.R100033200
56. Xie W, Barwick JL, Simon CM, Pierce AM, Safe S, Blumberg B, et al. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev (2000) 14(23):3014–23. doi: 10.1101/gad.846800
57. Wang H, LeCluyse EL. Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clin Pharmacokinet (2003) 42(15):1331–57. doi: 10.2165/00003088-200342150-00003
58. Ferguson SS, LeCluyse EL, Negishi M, Goldstein JA. Regulation of human CYP2C9 by the constitutive androstane receptor: Discovery of a new distal binding site. Mol Pharmacol (2002) 62(3):737–46. doi: 10.1124/mol.62.3.737
59. Goodwin B, Moore LB, Stoltz CM, McKee DD, Kliewer SA. Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol Pharmacol (2001) 60(3):427–31.
60. Xie W, Yeuh M, Radominska-Pandya A, Saini S, Negishi Y, Bottroff B, et al. Control of steroid, heme, and carcinogen metabolism by nuclear pregnane X receptor and constitutive androstane receptor. Proc Natl Acad Sci USA (2003) 100(7):4150–5. doi: 10.1073/pnas.0438010100
61. Buckley DB, Klaassen CD. Induction of mouse UDP-glucuronosyltransferase mRNA expression in liver and intestine by activators of aryl-hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, peroxisome proliferator-activated receptor alpha, and nuclear factor erythroid 2-related factor 2. Drug Metab Dispos (2009) 37(4):847–56. doi: 10.1124/dmd.108.024190
62. Falkner KC, Prough RA. Regulation of the rat glutathione s-transferase A2 gene by glucocorticoids: crosstalk through C/EBPs. Drug Metab Rev (2007) 39(2-3):401–18. doi: 10.1080/03602530701511216
63. Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, et al. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J Biol Chem (2003) 278(19):17277–83. doi: 10.1074/jbc.M300138200
64. Watkins R, Wisely G, Moore L, Collins J, Lambert M, Williams S, et al. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science (2001) 292(5525):2329–33. doi: 10.1126/science.1060762
65. Wurtz J, Bourguet W, Renaud J, Vivat V, Chambon P, Moras D, et al. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol (1996) 3(1):87–94. doi: 10.1038/nsb0196-87
66. Buchman C, Chai S, Chen T. A current structural perspective on PXR and CAR in drug metabolism. Expert Opin Drug Metab toxicol (2018) 14(6):635–47. doi: 10.1080/17425255.2018.1476488
67. Willson T, Kliewer SPXR. CAR and drug metabolism. Nat Rev Drug discovery. (2002) 1(4):259–66. doi: 10.1038/nrd753
68. Bhalla S, Ozalp C, Fang S, Xiang L, Kemper J. Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1alpha. functional implications in hepatic cholesterol and glucose metabolism. J Biol Chem (2004) 279(43):45139–47. doi: 10.1074/jbc.M405423200
69. Kodama S, Moore R, Yamamoto Y, Negishi M. Human nuclear pregnane X receptor cross-talk with CREB to repress cAMP activation of the glucose-6-phosphatase gene. Biochem J (2007) 407(3):373–81. doi: 10.1042/BJ20070481
70. Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol (2004) 24(18):7931–40. doi: 10.1128/MCB.24.18.7931-7940.2004
71. Nakamura K, Moore R, Negishi M, Sueyoshi T. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J Biol Chem (2007) 282(13):9768–76. doi: 10.1074/jbc.M610072200
72. Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, et al. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem (2006) 281(21):15013–20. doi: 10.1074/jbc.M511116200
73. Gotoh S, Negishi M. Serum-and glucocorticoid-regulated kinase 2 determines drug-activated pregnane X receptor to induce gluconeogenesis in human liver cells. J Pharmacol Exp Ther (2014) 348(1):131–40. doi: 10.1124/jpet.113.209379
74. Rysä J, Buler M, Savolainen M, Ruskoaho H, Hakkola J, Hukkanen J. Pregnane X receptor agonists impair postprandial glucose tolerance. Clin Pharmacol Ther (2013) 93(6):556–63. doi: 10.1038/clpt.2013.48
75. Matschinsky F. GKAs for diabetes therapy: Why no clinically useful drug after two decades of trying? Trends Pharmacol Sci (2013) 34(2):90–9. doi: 10.1016/j.tips.2012.11.007
76. Li X, Li S, Chen M, Wang J, Xie B, Sun Z. (-)-Epigallocatechin-3-gallate (EGCG) inhibits starch digestion and improves glucose homeostasis through direct or indirect activation of PXR/CAR-mediated phase II metabolism in diabetic mice. Food Funct (2018) 9(9):4651–63. doi: 10.1039/C8FO01293H
77. Singh S, Yende A, Ponnusamy K, Tyagi R. A comprehensive evaluation of anti-diabetic drugs on nuclear receptor PXR platform. Toxicol vitro (2019) 60:347–58. doi: 10.1016/j.tiv.2019.06.015
78. Postic C, Shiota M, Niswender K, Jetton T, Chen Y, Moates J, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using cre recombinase. J Biol Chem (1999) 274(1):305–15. doi: 10.1074/jbc.274.1.305
79. Burcelin R, del Carmen Muñoz M, Guillam M, Thorens B. Liver hyperplasia and paradoxical regulation of glycogen metabolism and glucose-sensitive gene expression in GLUT2-null hepatocytes. further evidence for the existence of a membrane-based glucose release pathway. J Biol Chem (2000) 275(15):10930–6. doi: 10.1074/jbc.275.15.10930
80. Ling Z, Shu N, Xu P, Wang F, Zhong Z, Sun B, et al. Involvement of pregnane X receptor in the impaired glucose utilization induced by atorvastatin in hepatocytes. Biochem Pharmacol (2016) 100:98–111. doi: 10.1016/j.bcp.2015.11.023
81. Hassani-Nezhad-Gashti F, Rysä J, Kummu O, Näpänkangas J, Buler M, Karpale M, et al. Activation of nuclear receptor PXR impairs glucose tolerance and dysregulates GLUT2 expression and subcellular localization in liver. Biochem Pharmacol (2018) 148:253–64. doi: 10.1016/j.bcp.2018.01.001
82. Yan L, Yang K, Wang S, Xie Y, Zhang L, Tian X. PXR-mediated expression of FABP4 promotes valproate-induced lipid accumulation in HepG2 cells. Toxicol letters (2021) 346:47–56. doi: 10.1016/j.toxlet.2021.04.016
83. Yoshikawa T, Shimano H, Amemiya-Kudo M, Yahagi N, Hasty A, Matsuzaka T, et al. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol (2001) 21(9):2991–3000. doi: 10.1128/MCB.21.9.2991-3000.2001
84. Misawa K, Horiba T, Arimura N, Hirano Y, Inoue J, Emoto N, et al. Sterol regulatory element-binding protein-2 interacts with hepatocyte nuclear factor-4 to enhance sterol isomerase gene expression in hepatocytes. J Biol Chem (2003) 278(38):36176–82. doi: 10.1074/jbc.M302387200
85. Kanayama T, Arito M, So K, Hachimura S, Inoue J, Sato R. Interaction between sterol regulatory element-binding proteins and liver receptor homolog-1 reciprocally suppresses their transcriptional activities. J Biol Chem (2007) 282(14):10290–8. doi: 10.1074/jbc.M700270200
86. Febbraio M, Silverstein R. CD36: implications in cardiovascular disease. Int J Biochem Cell Biol (2007) 39(11):2012–30. doi: 10.1016/j.biocel.2007.03.012
87. Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology (2008) 134(2):556–67. doi: 10.1053/j.gastro.2007.11.037
88. Sui Y, Park S, Wang F, Zhou C. Perinatal bisphenol a exposure increases atherosclerosis in adult Male PXR-humanized mice. Endocrinology (2018) 159(4):1595–608. doi: 10.1210/en.2017-03250
89. Shehu A, Zhu J, Li J, Lu J, McMahon D, Xie W, et al. Targeting xenobiotic nuclear receptors PXR and CAR to prevent cobicistat hepatotoxicity. Toxicol Sci an Off J Soc Toxicol (2021) 181(1):58–67. doi: 10.1093/toxsci/kfab023
90. Barretto S, Lasserre F, Fougerat A, Smith L, Fougeray T, Lukowicz C, et al. Gene expression profiling reveals that PXR activation inhibits hepatic PPARα activity and decreases FGF21 secretion in Male C57Bl6/J mice. Int J Mol Sci (2019) 20(15):3767. doi: 10.3390/ijms20153767
91. Shizu R, Ezaki K, Sato T, Sugawara A, Hosaka T, Sasaki T, et al. PXR suppresses PPARalpha-dependent HMGCS2 gene transcription by inhibiting the interaction between PPARalpha and PGC1alpha. Cells (2021) 10(12):3550. doi: 10.3390/cells10123550
92. Wolfrum C, Asilmaz E, Luca E, Friedman J, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature (2004) 432(7020):1027–32. doi: 10.1038/nature03047
93. Li T, Chen W, Chiang J. PXR induces CYP27A1 and regulates cholesterol metabolism in the intestine. J Lipid Res (2007) 48(2):373–84. doi: 10.1194/jlr.M600282-JLR200
94. Zhao L, Xu J, Shi Z, Englert N, Zhang S. Pregnane X receptor (PXR) deficiency improves high fat diet-induced obesity via induction of fibroblast growth factor 15 (FGF15) expression. Biochem Pharmacol (2017) 142:194–203. doi: 10.1016/j.bcp.2017.07.019
95. Meng Z, Gwag T, Sui Y, Park S, Zhou X, Zhou C. The atypical antipsychotic quetiapine induces hyperlipidemia by activating intestinal PXR signaling. JCI Insight (2019) 4(3):e125657. doi: 10.1172/jci.insight.125657
96. Masson D, Lagrost L, Athias A, Gambert P, Brimer-Cline C, Lan L, et al. Expression of the pregnane X receptor in mice antagonizes the cholic acid-mediated changes in plasma lipoprotein profile. Arteriosclerosis thrombosis Vasc Biol (2005) 25(10):2164–9. doi: 10.1161/01.ATV.0000183674.88817.fb
97. Sporstøl M, Tapia G, Malerød L, Mousavi S, Berg T. Pregnane X receptor-agonists down-regulate hepatic ATP-binding cassette transporter A1 and scavenger receptor class b type I. Biochem Biophys Res Commun (2005) 331(4):1533–41. JB, communications br. doi: 10.1016/j.bbrc.2005.04.071
98. Karpale M, Hukkanen J, Hakkola J. Nuclear receptor PXR in drug-induced hypercholesterolemia. Cells (2022) 11(3):313. doi: 10.3390/cells11030313
99. Fiorucci S, Rizzo G, Donini A, Distrutti E, Santucci L. Targeting farnesoid X receptor for liver and metabolic disorders. Trends Mol Med (2007) 13(7):298–309. doi: 10.1016/j.molmed.2007.06.001
100. Sonoda J, Xie W, Rosenfeld J, Barwick J, Guzelian P, Evans RM. Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR). Proc Natl Acad Sci U S A (2002) 99(21):13801–6. doi: 10.1073/pnas.212494599
101. Staudinger J, Goodwin B, Jones S, Hawkins-Brown D, MacKenzie K, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA (2001) 98(6):3369–74. doi: 10.1073/pnas.051551698
102. Kast H, Goodwin B, Tarr P, Jones S, Anisfeld A, Stoltz C, et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem (2002) 277(4):2908–15. doi: 10.1074/jbc.M109326200
103. Wang C, Xu W, Zhang Y, Huang D, Huang K. Poly(ADP-ribosyl)ated PXR is a critical regulator of acetaminophen-induced hepatotoxicity. Cell Death Disease (2018) 9(8):819. doi: 10.1038/s41419-018-0875-4
104. Zeng H, Lin Y, Gong J, Lin S, Gao J, Li C, et al. CYP3A suppression during diet-induced nonalcoholic fatty liver disease is independent of PXR regulation. Chemico-Biolog Interactions (2019) 308:185–93. doi: 10.1016/j.cbi.2019.05.038
105. Yao N, Zeng C, Zhan T, He F, Liu M, Liu F, et al. Oleanolic acid and ursolic acid induce UGT1A1 expression in HepG2 cells by activating PXR rather than CAR. Front Pharmacol (2019) 10:1111. doi: 10.3389/fphar.2019.01111
106. Xie W, Radominska-Pandya A, Shi Y, Simon C, Nelson M, Ong E, et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci USA (2001) 98(6):3375–80. doi: 10.1073/pnas.051014398
107. Jung D, Mangelsdorf DJ, Meyer UA. Pregnane X receptor is a target of farnesoid X receptor. J Biol Chem (2006) 281(28):19081–91. doi: 10.1074/jbc.M600116200
108. Jonker JW, Liddle C, Downes M. FXR and PXR: potential therapeutic targets in cholestasis. J Steroid Biochem Mol Biol (2012) 130(3-5):147–58. doi: 10.1016/j.jsbmb.2011.06.012
109. Tabb M, Sun A, Zhou C, Grün F, Errandi J, Romero K, et al. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J Biol Chem (2003) 278(45):43919–27. doi: 10.1074/jbc.M303136200
110. Ichikawa T, Horie-Inoue K, Ikeda K, Blumberg B, Inoue S. Steroid and xenobiotic receptor SXR mediates vitamin K2-activated transcription of extracellular matrix-related genes and collagen accumulation in osteoblastic cells. J Biol Chem (2006) 281(25):16927–34. doi: 10.1074/jbc.M600896200
111. Igarashi M, Yogiashi Y, Mihara M, Takada I, Kitagawa H, Kato S. Vitamin K induces osteoblast differentiation through pregnane X receptor-mediated transcriptional control of the Msx2 gene. Mol Cell Biol (2007) 27(22):7947–54. doi: 10.1128/MCB.00813-07
112. Yang W, Yu Z, Chiyoya M, Liu X, Daitoku K, Motomura S, et al. Menaquinone-4 accelerates calcification of human aortic valve interstitial cells in high-phosphate medium through PXR. J Pharmacol Exp Ther (2020) 372(3):277–84. doi: 10.1124/jpet.119.263160
113. Sultana H, Watanabe K, Rana M, Takashima R, Ohashi A, Komai M, et al. Effects of vitamin K2; on the expression of genes involved in bile acid synthesis and glucose homeostasis in mice with humanized PXR. Nutrients (2018) 10(8):982. doi: 10.3390/nu10080982
114. Sultana H, Kato A, Ohashi A, Takashima R, Katsurai T, Sato S, et al. Effect of vitamin K-mediated PXR activation on drug-metabolizing gene expression in human intestinal carcinoma LS180 cell line. Nutrients (2021) 13(5):1709. doi: 10.3390/nu13051709
115. Pascussi J, Robert A, Nguyen M, Walrant-Debray O, Garabedian M, Martin P, et al. Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest (2005) 115(1):177–86. doi: 10.1172/JCI21867
116. Zhou C, Assem M, Tay J, Watkins P, Blumberg B, Schuetz E, et al. Steroid and xenobiotic receptor and vitamin d receptor crosstalk mediates CYP24 expression and drug-induced osteomalacia. J Clin Invest (2006) 116(6):1703–12. doi: 10.1172/JCI27793
117. Hosseinpour F, Ellfolk M, Norlin M, Wikvall K. Phenobarbital suppresses vitamin D3 25-hydroxylase expression: A potential new mechanism for drug-induced osteomalacia. Biochem Biophys Res Commun (2007) 357(3):603–7. doi: 10.1016/j.bbrc.2007.03.177
118. Guo J, Li W, Wu Y, Jing X, Huang J, Zhang J, et al. Meclizine prevents ovariectomy-induced bone loss and inhibits osteoclastogenesis partially by upregulating PXR. Front Pharmacol (2017) 8:693. doi: 10.3389/fphar.2017.00693
119. Saeki N, Itoh Y, Kanai R, Itoh S, Inububishi T, Akiyama S, et al. Pregnane X receptor (PXR) represses osteoblast differentiation through repression of the hedgehog signaling pathway. Exp Cell Res (2022) 416(1):113156. doi: 10.1016/j.yexcr.2022.113156
120. Birringer M, Drogan D, Brigelius-Flohe R. Tocopherols are metabolized in HepG2 cells by side chain omega-oxidation and consecutive beta-oxidation. Free Radical Biol Med (2001) 31(2):226–32. doi: 10.1016/S0891-5849(01)00574-3
121. Swanson J, Ben R, Burton G, Parker R. Urinary excretion of 2,7, 8-trimethyl-2-(beta-carboxyethyl)-6-hydroxychroman is a major route of elimination of gamma-tocopherol in humans. J Lipid Res (1999) 40(4):665–71. doi: 10.1016/S0022-2275(20)32145-3
122. Landes N, Pfluger P, Kluth D, Birringer M, Rühl R, Böl G, et al. Vitamin e activates gene expression via the pregnane X receptor. Biochem Pharmacol (2003) 65(2):269–73. doi: 10.1016/S0006-2952(02)01520-4
123. Cho J, Kang D, Ma X, Ahn S, Krausz K, Luecke H, et al. Metabolomics reveals a novel vitamin e metabolite and attenuated vitamin e metabolism upon PXR activation. J Lipid Res (2009) 50(5):924–37. doi: 10.1194/jlr.M800647-JLR200
124. Strott C. Sulfonation and molecular action. Endocrine Rev (2002) 23(5):703–32. doi: 10.1210/er.2001-0040
125. Zhang B, Cheng Q, Ou Z, Lee J, Xu M, Kochhar U, et al. Pregnane X receptor as a therapeutic target to inhibit androgen activity. Endocrinology (2010) 151(12):5721–9. doi: 10.1210/en.2010-0708
126. Zhai Y, Pai H, Zhou J, Amico J, Vollmer R, Xie W. Activation of pregnane X receptor disrupts glucocorticoid and mineralocorticoid homeostasis. Mol Endocrinol (2007) 21(1):138–47. doi: 10.1210/me.2006-0291
127. Xia J, Lin J, Li XN, Zhang C, Li N, Du ZH, et al. Atrazine-induced environmental nephrosis was mitigated by lycopene via modulating nuclear xenobiotic receptors-mediated response. J Nutr Biochem (2018) 51:80–90. doi: 10.1016/j.jnutbio.2017.09.006
128. Lee HJ, Pyo MC, Shin HS, Ryu D, Lee KW. Renal toxicity through AhR, PXR, and Nrf2 signaling pathway activation of ochratoxin a-induced oxidative stress in kidney cells. Food Chem Toxicol (2018) 122:59–68. doi: 10.1016/j.fct.2018.10.004
129. Doricakova A, Vrzal R. A food contaminant ochratoxin a suppresses pregnane X receptor (PXR)-mediated CYP3A4 induction in primary cultures of human hepatocytes. Toxicology (2015) 337:72–8. doi: 10.1016/j.tox.2015.08.012
130. Sonoda J, Chong LW, Downes M, Barish GD, Coulter S, Liddle C, et al. Pregnane X receptor prevents hepatorenal toxicity from cholesterol metabolites. Proc Natl Acad Sci USA (2005) 102(6):2198–203. doi: 10.1073/pnas.0409481102
131. Li X, Wang Z, Klaunig JE. Modulation of xenobiotic nuclear receptors in high-fat diet induced non-alcoholic fatty liver disease. Toxicology (2018) 410:199–213. doi: 10.1016/j.tox.2018.08.007
132. Chen J, Vitetta L. Gut microbiota metabolites in NAFLD pathogenesis and therapeutic implications. Int J Mol Sci (2020) 21(15):5214. doi: 10.3390/ijms21155214
133. Wahlang B, Alexander NC 2nd, Li X, Rouchka EC, Kirpich IA, Cave MC. Polychlorinated biphenyls altered gut microbiome in CAR and PXR knockout mice exhibiting toxicant-associated steatohepatitis. Toxicol Rep (2021) 8:536–47. doi: 10.1016/j.toxrep.2021.03.010
134. Karpale M, Karajamaki AJ, Kummu O, Gylling H, Hyotylainen T, Oresic M, et al. Activation of pregnane X receptor induces atherogenic lipids and PCSK9 by a SREBP2-mediated mechanism. Br J Pharmacol (2021) 178(12):2461–81. doi: 10.1111/bph.15433
135. Chen J, Raymond K. Roles of rifampicin in drug-drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann Clin Microbiol Antimicrob (2006) 5:3. doi: 10.1186/1476-0711-5-3
136. Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, et al. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci USA (1998) 95(21):12208–13. doi: 10.1073/pnas.95.21.12208
137. Crawford P, Chadwick DJ, Martin C, Tjia J, Back DJ, Orme M. The interaction of phenytoin and carbamazepine with combined oral contraceptive steroids. Br J Clin Pharmacol (1990) 30(6):892–6. doi: 10.1111/j.1365-2125.1990.tb05457.x
138. Lynch C, Sakamuru S, Huang R, Niebler J, Ferguson SS, Xia M. Characterization of human pregnane X receptor activators identified from a screening of the Tox21 compound library. Biochem Pharmacol (2021) 184:114368. doi: 10.1016/j.bcp.2020.114368
139. Duran I, Carles J, Bulat I, Hellemans P, Mitselos A, Ward P, et al. Pharmacokinetic drug-drug interaction of apalutamide, part 1: Clinical studies in healthy men and patients with castration-resistant prostate cancer. Clin Pharmacokinet (2020) 59(9):1135–48. doi: 10.1007/s40262-020-00882-2
140. Theile D, Haefeli WE, Weiss J. Effects of adrenolytic mitotane on drug elimination pathways assessed in vitro. Endocrine (2015) 49(3):842–53. doi: 10.1007/s12020-014-0517-2
141. Chortis V, Taylor AE, Schneider P, Tomlinson JW, Hughes BA, O'Neil DM, et al. Mitotane therapy in adrenocortical cancer induces CYP3A4 and inhibits 5alpha-reductase, explaining the need for personalized glucocorticoid and androgen replacement. J Clin Endocrinol Metab (2013) 98(1):161–71. doi: 10.1210/jc.2012-2851
142. van Erp NP, Guchelaar HJ, Ploeger BA, Romijn JA, Hartigh J, Gelderblom H. Mitotane has a strong and a durable inducing effect on CYP3A4 activity. Eur J Endocrinol (2011) 164(4):621–6. doi: 10.1530/EJE-10-0956
143. Harmsen S, Meijerman I, Maas-Bakker RF, Beijnen JH, Schellens JH. PXR-mediated p-glycoprotein induction by small molecule tyrosine kinase inhibitors. Eur J Pharm Sci (2013) 48(4-5):644–9. doi: 10.1016/j.ejps.2012.12.019
144. Fellay J, Marzolini C, Decosterd L, Golay KP, Baumann P, Buclin T, et al. Variations of CYP3A activity induced by antiretroviral treatment in HIV-1 infected patients. Eur J Clin Pharmacol (2005) 60(12):865–73. doi: 10.1007/s00228-004-0855-8
145. Hariparsad N, Nallani SC, Sane RS, Buckley DJ, Buckley AR, Desai PB. Induction of CYP3A4 by efavirenz in primary human hepatocytes: comparison with rifampin and phenobarbital. J Clin Pharmacol (2004) 44(11):1273–81. doi: 10.1177/0091270004269142
146. Sharma D, Lau AJ, Sherman MA, Chang TK. Agonism of human pregnane X receptor by rilpivirine and etravirine: comparison with first generation non-nucleoside reverse transcriptase inhibitors. Biochem Pharmacol (2013) 85(11):1700–11. doi: 10.1016/j.bcp.2013.04.002
147. Scholler-Gyure M, Kakuda TN, Raoof A, De Smedt G, Hoetelmans RM. Clinical pharmacokinetics and pharmacodynamics of etravirine. Clin Pharmacokinet (2009) 48(9):561–74. doi: 10.2165/10895940-000000000-00000
148. Havens JP, Podany AT, Scarsi KK, Fletcher CV. Clinical pharmacokinetics and pharmacodynamics of etravirine: An updated review. Clin Pharmacokinet (2020) 59(2):137–54. doi: 10.1007/s40262-019-00830-9
149. Kakuda TN, Van Solingen-Ristea RM, Onkelinx J, Stevens T, Aharchi F, De Smedt G, et al. The effect of single- and multiple-dose etravirine on a drug cocktail of representative cytochrome P450 probes and digoxin in healthy subjects. J Clin Pharmacol (2014) 54(4):422–31. doi: 10.1002/jcph.214
150. Svard J, Spiers JP, Mulcahy F, Hennessy M. Nuclear receptor-mediated induction of CYP450 by antiretrovirals: functional consequences of NR1I2 (PXR) polymorphisms and differential prevalence in whites and sub-Saharan africans. J Acquir Immune Defic Syndr (2010) 55(5):536–49. doi: 10.1097/QAI.0b013e3181f52f0c
151. Dussault I, Lin M, Hollister K, Wang EH, Synold TW, Forman BM. Peptide mimetic HIV protease inhibitors are ligands for the orphan receptor SXR. J Biol Chem (2001) 276(36):33309–12. doi: 10.1074/jbc.C100375200
152. Li L, Welch MA, Li Z, Mackowiak B, Heyward S, Swaan PW, et al. Mechanistic insights of phenobarbital-mediated activation of human but not mouse pregnane X receptor. Mol Pharmacol (2019) 96(3):345–54. doi: 10.1124/mol.119.116616
153. Burstein S, Klaiber EL. Phenobarbital-induced increase in 6-Beta-Hydroxycortisol excretion: Clue to its significance in human urine. J Clin Endocrinol Metab (1965) 25:293–6. doi: 10.1210/jcem-25-2-293
154. Wallace K, Cowie DE, Konstantinou DK, Hill SJ, Tjelle TE, Axon A, et al. The PXR is a drug target for chronic inflammatory liver disease. J Steroid Biochem Mol Biol (2010) 120(2-3):137–48. doi: 10.1016/j.jsbmb.2010.04.012
155. Shukla SJ, Sakamuru S, Huang R, Moeller TA, Shinn P, Vanleer D, et al. Identification of clinically used drugs that activate pregnane X receptors. Drug Metab Dispos (2011) 39(1):151–9. doi: 10.1124/dmd.110.035105
156. Pascussi JM, Drocourt L, Fabre JM, Maurel P, Vilarem MJ. Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: Synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol Pharmacol (2000) 58(2):361–72. doi: 10.1124/mol.58.2.361
157. Shi D, Yang D, Yan B. Dexamethasone transcriptionally increases the expression of the pregnane X receptor and synergistically enhances pyrethroid esfenvalerate in the induction of cytochrome P450 3A23. Biochem Pharmacol (2010) 80(8):1274–83. doi: 10.1016/j.bcp.2010.06.043
158. Novotna A, Dvorak Z. Omeprazole and lansoprazole enantiomers induce CYP3A4 in human hepatocytes and cell lines via glucocorticoid receptor and pregnane X receptor axis. PloS One (2014) 9(8):e105580. doi: 10.1371/journal.pone.0105580
Keywords: pregnane X receptor (PXR), nuclear receptor, metabolism, glycometabolism, lipid metabolism, bile acid, vitamin, endocrine homeostasis
Citation: Lv Y, Luo Y-Y, Ren H-W, Li C-J, Xiang Z-X and Luan Z-L (2022) The role of pregnane X receptor (PXR) in substance metabolism. Front. Endocrinol. 13:959902. doi: 10.3389/fendo.2022.959902
Received: 02 June 2022; Accepted: 28 July 2022;
Published: 16 August 2022.
Edited by:
Pierre De Meyts, Université Catholique de Louvain, BelgiumReviewed by:
Makoto Makishima, Nihon University, JapanRadim Vrzal, Palacký University Olomouc, Czechia
Copyright © 2022 Lv, Luo, Ren, Li, Xiang and Luan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-Lin Luan, emhpbGluX2x1YW5Ac2luYS5jb20=