
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 27 July 2022
Sec. Developmental Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.951534
This article is part of the Research Topic Advances and Perspectives in Livestock Reproductive and Developmental Endocrinology View all 6 articles
 Yuanmei Wang†
Yuanmei Wang† Jingwei Yuan†
Jingwei Yuan† Yanyan SunYunlei LiPanlin WangLei Shi
Yanyan SunYunlei LiPanlin WangLei Shi Aixin NiYunhe ZongJinmeng ZhaoShixiong BianHui Ma
Aixin NiYunhe ZongJinmeng ZhaoShixiong BianHui Ma Jilan Chen*
Jilan Chen*Sexual maturation is fundamental to the reproduction and production performance, heterosis of which has been widely used in animal crossbreeding. However, the underlying mechanism have long remained elusive, despite its profound biological and agricultural significance. In the current study, the reciprocal crossing between White Leghorns and Beijing You chickens were performed to measure the sexual maturation heterosis, and the ovary lncRNAs and mRNAs of purebreds and crossbreeds were profiled to illustrate molecular mechanism of heterosis. Heterosis larger than 20% was found for pubic space and oviduct length, whereas age at first egg showed negative heterosis in both crossbreeds. We identified 1170 known lncRNAs and 1994 putative lncRNAs in chicken ovary using a stringent pipeline. Gene expression pattern showed that nonadditivity was predominant, and the proportion of nonadditive lncRNAs and genes was similar between two crossbreeds, ranging from 44.24% to 49.15%. A total of 200 lncRNAs and 682 genes were shared by two crossbreeds, respectively. GO and KEGG analysis showed that the common genes were significantly enriched in the cell cycle, animal organ development, gonad development, ECM-receptor interaction, calcium signaling pathway and GnRH signaling pathway. Weighted gene co-expression network analysis (WGCNA) identified that 7 out of 20 co-expressed lncRNA-mRNA modules significantly correlated with oviduct length and pubic space. Interestingly, genes harbored in seven modules were also enriched in the similar biological process and pathways, in which nonadditive lncRNAs, such as MSTRG.17017.1 and MSTRG.6475.20, were strongly associated with nonadditive genes, such as CACNA1C and TGFB1 to affect gonad development and GnRH signaling pathway, respectively. Moreover, the results of real-time quantitative PCR (RT-qPCR) correlated well with the transcriptome data. Integrated with positive heterosis of serum GnRH and melatonin content detected in crossbreeds, we speculated that nonadditive genes involved in the GnRH signaling pathway elevated the gonad development, leading to the sexual maturation heterosis. We characterized a systematic landscape of ovary lncRNAs and mRNAs related to sexual maturation heterosis in chicken. The quantitative exploration of hybrid transcriptome changes lays foundation for genetic improvement of sexual maturation traits and provides insights into endocrine control of sexual maturation.
Sexual maturation is a pivotal development stage and indispensable for achieving successful reproduction (1). The initiation of sexual maturation is complicated, which is contingent upon integrating different stimuli into neuroendocrine and endocrine signals along the hypothalamic-pituitary-gonadal (HPG) axis. HPG axis is shown to be under a dual control system with a stimulatory and an inhibitory branch (2), in which hypothalamic gonadotropin-releasing hormone (GnRH) played a central and positive role in activation of reproductive function (3, 4), whereas gonadotropin-inhibitory hormone (GnIH) have inhibitory effect on GnRH release and then delayed onset of sexual maturation. The release and secretion of GnIH are primarily under the influence of melatonin (MT). Heterosis refers to the superior performance of hybrid compared with their parent in production, fertility and adaptability (5, 6). It has been comprehensively exploited to improve yield and quality in plant and animal. The underlying mechanism has been studied for over century, but still remains elusive (7). As an importantly economic trait in chicken, sex maturation is controlled by HPG signals. Attaining sexual maturation at an early age was shown to increase total egg production (8, 9), which has been extensively utilized in crossbreeding in poultry industry. Therefore, it is of great value to unravel the molecular mechanism that regulates the heterosis of sexual maturation.
The superiority phenotype depends on the alteration of gene expression. In recent decades, genome-wide gene expression changes of hybrids relative to their parents have been illustrated in different species, such as microorganisms, plants and animals. The transcriptional changes in hybrids generally associated with processes involved in the energy production, metabolic rates, stress response and hormone signaling (10). In chickens, multiple studies illustrated the heterosis mechanism of important traits at different stages through genome-wide expression analysis of brain, liver and muscle (11–13). Long non-coding RNAs (lncRNAs) are pivotal epigenomic factors, a class of endogenous more than 200 bases in length, and could affect gene expression by acting as cis- or trans- regulators (14). It was reported that lncRNA could involve in sexual maturation through regulating oogenesis and folliculogenesis (15). Recent study in C. elegans found the lncRNAs let 7 could schedule sexual maturation through regulating their target genes (16). Moreover, Shu et al. (17) profiled lncRNA expression of pepper leaves and concluded that lncRNAs participated in seedling and flowering heterosis. Taken together, the integrated analysis of lncRNA and mRNA may provide novel clues for exploring the mechanism of sexual maturation heterosis.
Chicken meat and egg are great protein sources for human. Heterosis utilization is routine for economic traits improvement in poultry breeding (18). It was documented that negative heterosis for age at first egg (AFE) was observed in hybrids of different chicken breeds (19, 20). Moreover, the expression of ER and FSHR was reported to be correlated with AFE heterosis in chicken (21). Nevertheless, previous study mostly concentrated on exploring heterosis mechanism through characterizing several candidate genes. The genome-wide gene expression of sexual maturation heterosis was rarely reported and investigated. In this study, White Leghorn and Beijing You chicken were employed as parents to generate purebreds and reciprocal crossbreeds, respectively. The White Leghorn is a dominant commercial layer breed around the world. The Beijing You chicken is a traditional Chinese indigenous breed with head crest, beard and fuzzy shank, which produced much less egg mass compared to the White Leghorn. These two breeds have previously been reported genetically distant (22), and this would contribute the heterosis of traits. The transcriptome analysis of the ovary in purebreds and crossbreeds was implemented to reveal the key lncRNAs and genes regulating the formation of sexual maturation heterosis. Our study would provide new insights into the molecular basis of sexual maturation heterosis in chicken, and lay theoretical foundation for effectively utilization of heterosis.
All experimental procedures involving the use of animals were conducted according to the Guidelines for Experiment established by the Science Research Department of the Institute of Animal Sciences, Chinese Academy of Agriculture Sciences (IAS-CAAS), and ethical approval was received from the Animal Ethics Committee of the IAS-CAAS (no. IAS 2022-35).
In the present study, White Leghorn and Beijing You chicken were employed as parents to generate purebreds (WW and YY) and reciprocal crossbreeds (WY and YW). Briefly, 30 males and 150 unrelated females from White Leghorn, and 30 males and 150 unrelated females from Beijing You chicken were selected for mating to generate WW and YY and for reciprocal mating to generate WY and YW based on laying consistency, semen quality and body weight. Chickens from a single hatch were raised under the same environment with free access to feed and water in the experimental farm of IAS-CAAS.
Egg was collected for each individual daily from the age at first egg (AFE). When the egg laying ratio of population reached 5%, thirty chickens from each group were randomly selected to measure pubic space. Meanwhile, six individuals from WW, YY, WY and YW were selected based on the average of pubic space of each group, respectively. Subsequently, all visible follicles were carefully excluded when ovary tissue was collected as previously reported (23). Then, the ovary was frozen immediately in liquid nitrogen for RNA sequencing. In addition, the oviduct was captured for length measurement by image J (24). Heterosis of phenotype was calculated according to the following equation:
Where , and are the mean phenotype of reciprocal crossbreeds, maternal and purebreds, respectively. In addition, Student’s t-value was estimated to evaluate the significance of H% based on the formula of Wu and Zhang (25):
where F1i is the phenotype of individual i in crossbreeds; N is the number of chickens in WY or YW. P value was obtained according to the student’s t-test and degrees of freedom. H% was considered statistically significant if P < 0.05.
Total RNA was extracted using TRIzol® Reagent (Invitrogen, USA) following the manufacturer’s instructions. The RNA integrity and quality were determined by agarose gel electrophoresis and NanoPhotometer® spectrophotometer (IMPL EN, CA, USA). Three micrograms of total RNA were used for the construction of lncRNA library, and then the ribosomal RNA was removed from total RNA with TruSeq Stranded Total RNA Library Prep Gold Kits (Illumina, United States). Finally, 24 RNA sequencing libraries were constructed for paired-end sequencing according to the instructions of NEB Next® Ultra Directional RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA). RNA-Seq were performed using Novaseq6000 (Illumina, San Diego, United States) to generate 150bp paired-end reads.
The reads, including adapter contamination, low-quality reads (Phred Quality Score < 5%), reads with poly-N > 5% and reads matched rRNA were filtered out using in-house perl scripts to generate clean reads, which were aligned to the chicken reference genome (http://ftp.ensembl.org/pub/release-106/fasta/gallus_gallus/) using HiSAT2 (v2.1.0) (26) with default parameters, and then the mapped reads was assembled by StringTie (v2.1.5) (26) with gene transfer format (GTF) file of Ensembl gene annotation (http://ftp.ensembl.org/pub/release-106/gtf/gallus_gallus/). According to the chicken gene annotation, the number and proportion of three lncRNA functional elements (exon, intron and intergenic) were calculated based on the unique mapping sequences.
The potential lncRNAs were winnowed with the following criteria. Firstly, the transcripts less than 200 bp and without strand information were removed. Secondly, transcripts with class code “i”, “u” and “x” were retained. Subsequently, the sequence of known lncRNAs were download from two lncRNA databases, Domestic-Animal LncRNA Database (ALDB) (12) and NONCODE (27). We then used BLAST to align the unannotated transcripts to known lncRNAs. The transcripts perfectly aligned with sequences in either ALDB or NONCODE were regarded as known lncRNAs. Then, the protein coding potential of the remaining transcripts was predicted using CNCI (28), CPC (29), PLEK (30) and Pfam (31) software. Only transcripts with CNCI score < 0, CPC score < 0, PLEK score < 0 and Pfam E-value < 1e-5 were established as putative lncRNAs. Finally, the cis- and trans- acting relationship between lncRNAs and potential protein-coding genes was predicted according to their distances and expression correlations (32).
Differentially expressed lncRNAs (DELs) and differentially expressed genes (DEGs) between two different groups were analyzed based on gene counts by DESeq2 (v1.16.1) (33) in R project. Meanwhile, |Log2(fold change) | >1 and adjusted P value < 0.05 were taken as criteria to identify the DELs and DEGs in the corresponding comparison. These genes were further classified into three inheritance patterns: additivity, dominance and overdominance, based on the gene expression level of purebreds and crossbreeds as previously reported (34). Briefly, additivity (IV and X) occurred when gene expression was significantly different between two purebreds, and gene expression of crossbreeds was higher than one purebred but lower than the another. Gene expression within crossbreeds that was not significantly different from one purebred but significantly higher (or lower) than the another was regarded as dominance (II, IV, IX, and XI). Gene expression within crossbreeds that was significantly higher (or lower) than both purebreds was considered as overdominance (V, VI, VIII, III, VII, and X).
The weighted gene co-expression network analysis (WGCNA) was performed to identify co-expressed modules of highly correlated genes and summarized such modules based on the correlation between module eigengene and phenotypes (35). Briefly, an expression matrix of all lncRNAs and mRNAs was constructed. The top 40% most variant genes were used for subsequent analysis. After the weighted adjacency matrix established, an unsigned weighted correlation network was constructed by creating a matrix of Pearson correlation coefficients of each pair genes. Then, each topological matrix was used as input for linkage hierarchical clustering analysis, and primary gene modules was detected using a dynamic tree cutting algorithm (deepSplit = 2, minModuleSize = 50, mergeCutHeight = 0.25). Subsequently, module eigengenes were correlated with different traits and searched against the most significant correlations. Finally, the lncRNAs-mRNAs networks were visualized with Cytoscape (v.3.8.0), which was run with layout “attribute circle layout”.
To investigate biological function of nonadditive genes, we performed functional enrichment analysis, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways via KOBAS 3.0 platform (36). The GO terms and KEGG pathways with P value < 0.05 were considered significantly enriched.
GnRH and MT content was detected using the GnRH and MT assay kit (Beijing Laibotairui Technology Co. Ltd, Beijing, China) following the manufacture guidelines, respectively. Briefly, serum (40 μL) from each chicken was added to each well of 96-well plate. Then, the 96-well plate was incubated for one hour at 37°C. Following washed five times with washing liquid, color liquid A and B was added successively. After incubating for 10 min at 37°C, 50 μL of termination solution was added. Finally, the OD value was detected at 450 nm by Thermo Multiskan Ascent.
One microgram total RNA of each sample was reverse transcribed into cDNA using the PrimeScript RT Reagent Kit (TaKaRa, Shiga, Japan). The specific primer sequences were designed using Primer Premier5.0. The RT-qPCR reaction mixture (10 µL) consisted of cDNA 1.5 µL, forward primer 0.5 µL (10µM), reverse primer 0.5 µL (10µM), SYBR Green Master (Mix) 5 µL and ddH2O 2.3 µL. The amplification condition was as follows: 95°C for 3 min, followed by 40 cycles at 95°C for 3 s, 60 °C for 32s, finally, a single melt cycle was 95°C for 15 s and 65°C for 1 min. Triplicate was performed for each sample using PrimeScript One Step RT-PCR Kit (TaKaRa, Shiga, Japan). The results were normalized to GAPDH expression.
Fold changes in gene expression were calculated using the 2−ΔΔCt method. All data are presented as the mean ± standard deviation (SD). The correlation between results of RT-qPCR and RNA-seq was calculated using Pearson’s correlation method in R (v.4.0.2).
As shown in Figure 1A, the pubic space of crossbreeds was larger than the average value of purebreds, and significant positive heterosis (25.79%, 32.45%) was observed in both WY and YW (P < 0.05). Similarly, the larger oviduct length was observed in WY and YW compared with the average length of purebreds (Figure 1B), and heterosis of oviduct length was 30.55% in WY and 20.15% in YW, respectively. Moreover, AFE in crossbreeds was earlier than the average age of purebreds exhibiting negative heterosis (Figure 1C).
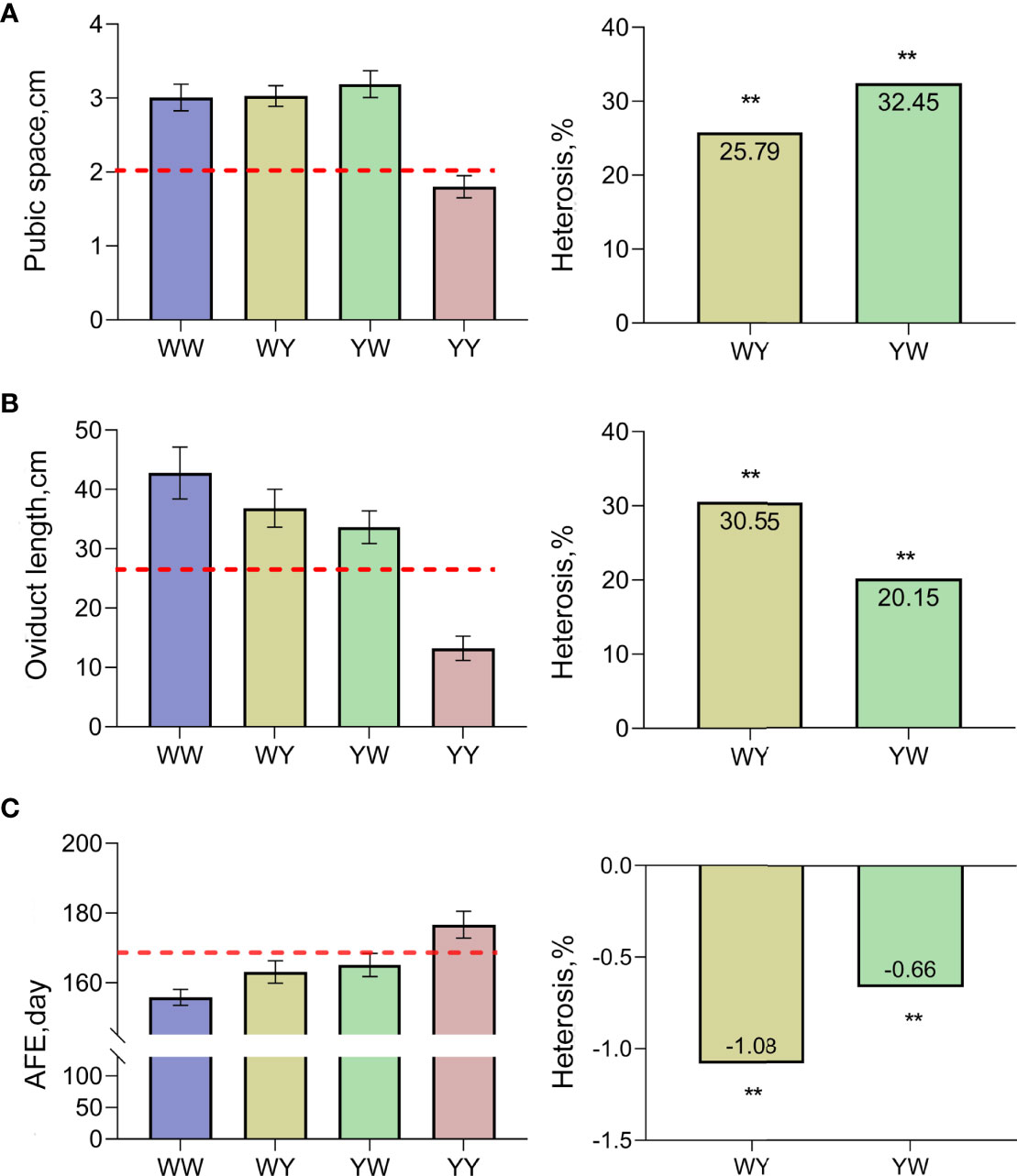
Figure 1 Heterosis of traits related to sexual maturation. (A) Heterosis of pubic space. (B) Heterosis of oviduct length. (C) Heterosis of AFE. The dashed line represents mid parent value. ** indicate that heterosis was highly significant (P < 0.01), respectively.
In the present study, an average of 36,579,837 raw reads was generated from each library, with average Q30 bases higher than 94.56% (Supplementary Table S1). The percentage of uniquely mapped reads and multi-mapped reads was 82.13% and 2.73%, respectively. In total, we characterized 3164 lncRNAs in chicken ovary (details in Supplementary Table S1). Among these lncRNAs, 1170 known lncRNA were identified through aligning lncRNAs sequence to the NONCODE or ALDB database. Besides known lncRNAs, a total of 1994 transcripts were identified as putative lncRNAs by CNCI, CPC, PLEK and Pfam software (Figure 2A). The majority of putative lncRNA were intergenic (50.82%), and 25.76% were intron, and 23.42% were antisense lncRNAs (Figure 2B). Furthermore, the length of the putative lncRNAs was mainly ranging from 2,800 to 3,000bp, similar to that of the known lncRNAs in ovary tissue (Figure 2C). The distributions of putative and known lncRNAs on the chromosomes were varied (Supplementary Figure S1). Moreover, the number of lncRNAs located on chromosome 1 was the largest, which accounted for 8.93% in putative lncRNAs and 13.08% in known lncRNAs. The most putative and known lncRNAs possessed two exons. The exon numbers of putative lncRNAs were greater than known lncRNAs (Figure 2D).
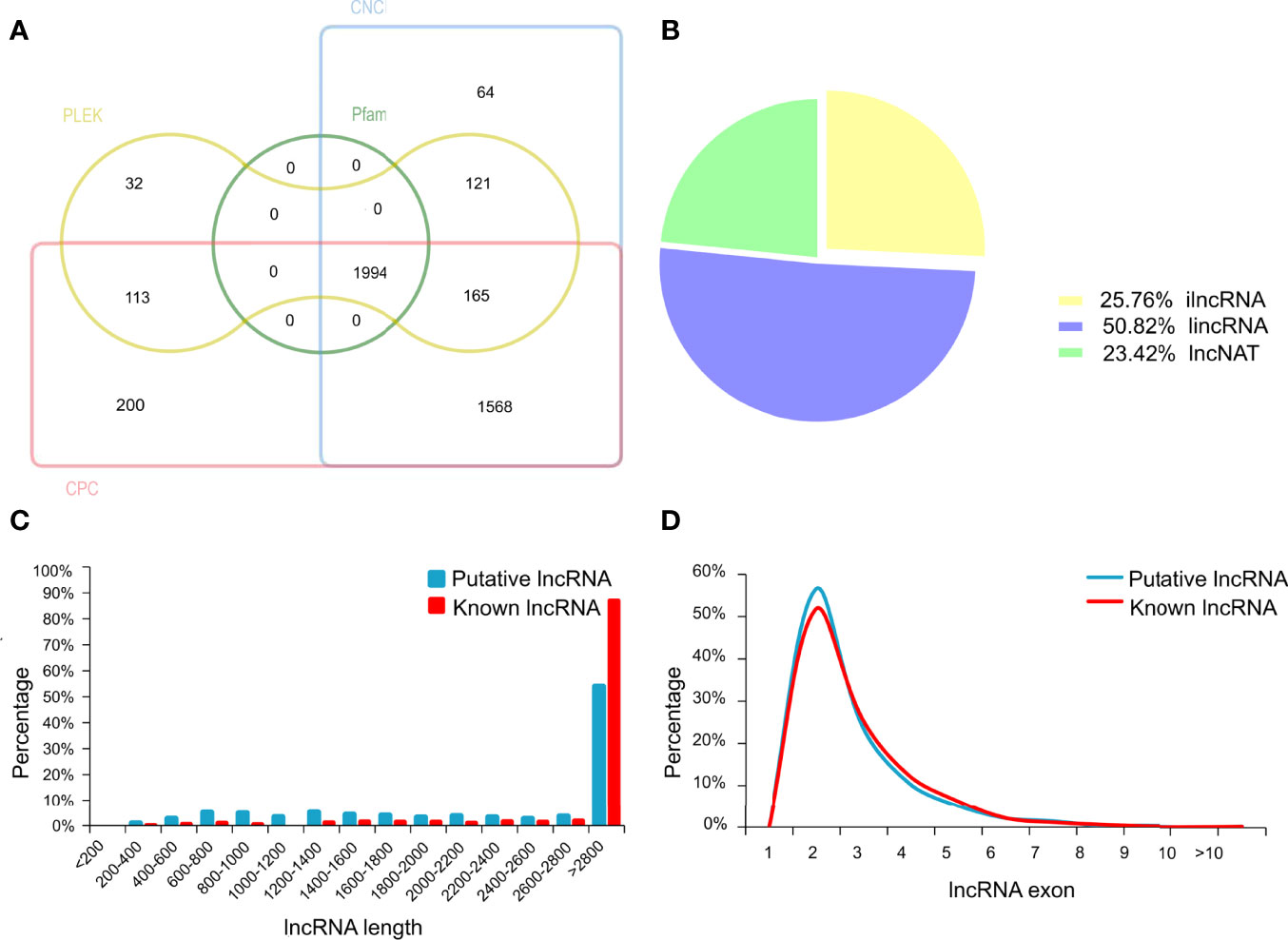
Figure 2 The identification of long non-coding RNAs (lncRNAs) in chicken ovary. (A) Venn diagram analysis showing the number of common and unique novel lncRNAs identified by CNCI, CPC, PLEK and Pfam database. (B) Classification of the uniquely mapped read locations including exon, intron and intergenic regions. ilncRNA, intergenic lncRNA; lincRNA, intron lncRNA; lncNAT, antisense lncRNA. (C) Length of known and novel lncRNAs. (D) The percentage of exon number for known and novel lncRNAs.
A principal component analysis (PCA) was performed to visualize the group differences of gene expression. The purebreds and crossbreeds were obviously separated from each other (Supplementary Figure S2), indicating significant differences of gene expression between two purebreds and between purebreds and crossbreeds. With the criteria of |log2 (fold change) | > 1 and adjusted P value < 0.05, we identified 686 DELs between two purebreds and between purebreds and crossbreeds (Figure 3A), including 630 in YY vs. WW, 225 in WY vs. WW, 83 in WY vs. YY, 246 in YW vs. WW and 68 YW vs. YY (Supplementary Table S2). These DELs were clustered into 12 types (I, II, III, IV, V, VI, VII, VIII, IX, X, XI and XII, details in Supplementary Table S3) according to their expression in purebreds and crossbreeds. The 12 types were further clustered into three main inheritance patterns: additivity (IV, X), dominance (III, V, IX, XI) and overdominance (I, II, VI, VII, VIII, XII). The number of dominant lncRNAs was 288 in WY and 294 in YW, respectively. The number of overdominant lncRNAs was 2 and 2 in WY and YW, respectively. The nonadditive lncRNAs accounted for 44.24% in WY and 45.16% in YW, respectively (Figures 3B, C). Additionally, a total of 200 nonadditive lncRNAs were shared by WY and YW (Supplementary Figure S3A). The number of unique nonadditive lncRNAs was 90 in WY and 96 in YW, respectively.
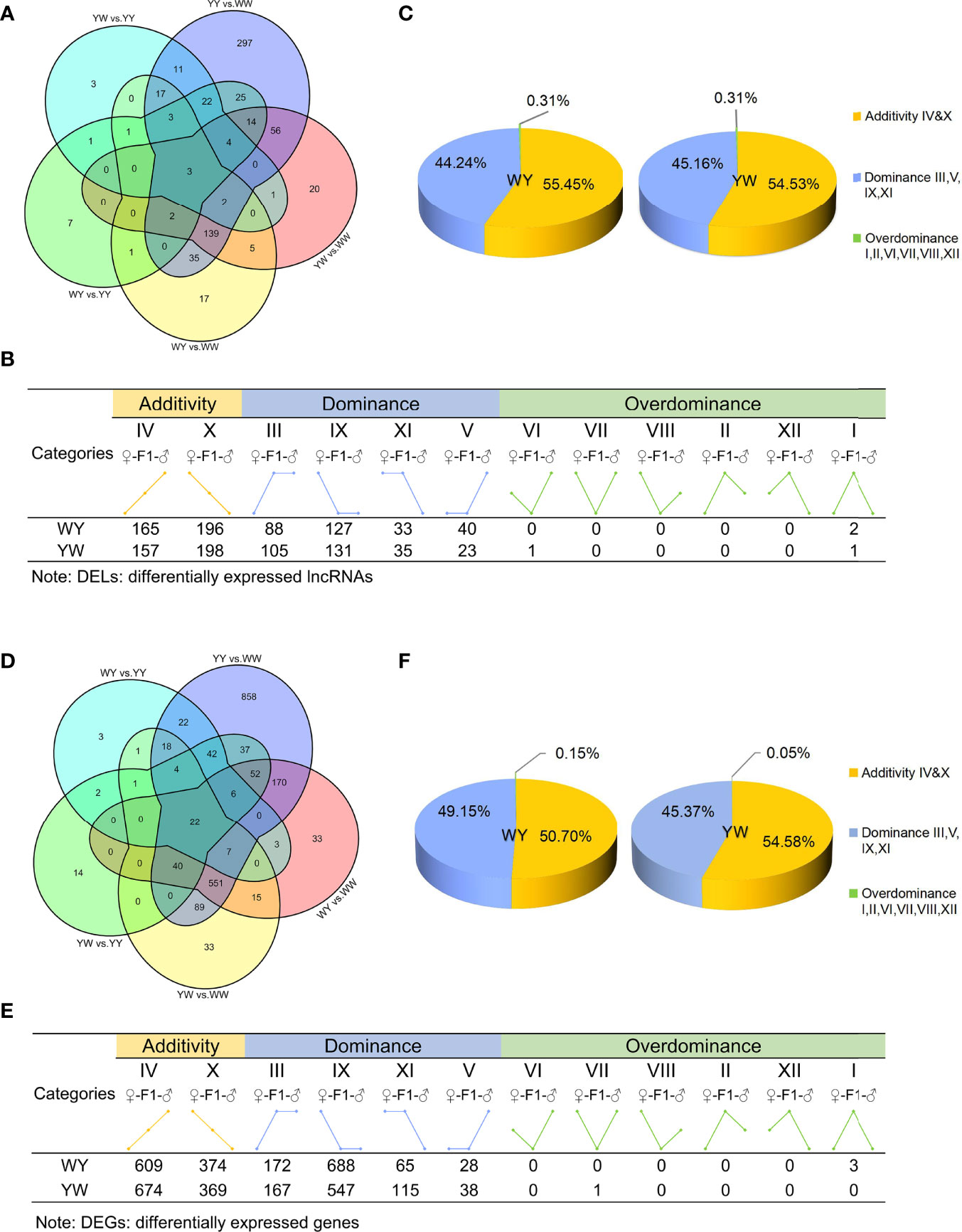
Figure 3 Analysis of lncRNAs and genes inheritance patterns. (A) The number of differentially expressed LncRNAs (DELs) among purebreds and crossbreeds. (B) Inheritance patterns of DELs between crossbreeds. DELs were divided into 12 types, e.g., class I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, and further classified into three inheritance patterns: additivity (class IV and X), dominance (class III, V, IX and XI) and overdominance (class I, II, VI, VII, VIII, XII), based on the level of gene expression exhibited by purebreds and crossbreeds. Additivity, dominance and overdominance are presented in orange, blue and green, respectively. (C) The proportion of DELs in additivity, dominance and overdominance pattern. (D) The number of DEGs among purebreds and crossbreeds. (E) Inheritance patterns of DEGs of crossbreeds. DEGs were divided into 12 types, e.g,. class I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, and further classified into three inheritance patterns: additivity (class IV and X), dominance (class III, V, IX and XI) and overdominance (class I, II, VI, VII, VIII, XII), based on the level of gene expression exhibited by crossbreeds and purebreds. Additivity, dominance and overdominance are presented in orange, blue and green, respectively. (F) The proportion of DEGs in additivity, dominance and overdominance pattern.
There were 2023 DEGs between two purebreds and between crossbreeds and purebreds identified (Figure 3D), including 1918 in YY vs. WW, 899 in WY vs. WW, 131 in WY vs. YY, 781 in YW vs. WW and 220 YW vs. YY (Supplementary Table S2). These DEGs were clustered into 12 types, and the gene number of different patterns in reciprocal crossbreeds was shown in Figure 3E. The dominant genes were 956 in WY and 868 in YW, respectively. The number of overdominant genes was 3 and 1 in WY and YW, respectively. The nonadditive genes, accounted for 49.30% in WY and 45.42% in YW (Figure 3F). Among the nonadditive genes, a total of 682 genes were shared by WY and YW, and the number of unique genes was 274 in WY and 186 in YW (Supplementary Figure S3B), respectively. As shown in Figure 4A, gene ontology (GO) analysis showed that the nonadditive genes were mainly enriched in three categories, including growth and development related GO terms, such as animal organ development, gland morphogenesis, female gonad development and female sex differentiation, ion transport related GO terms, such as calcium ion transport, calcium ion homeostasis and metal ion transport, and other GO terms, such as response to hormone, steroid binding, response to endogenous stimulus and sterol transport (details in Supplementary Table S4). Interestingly, the majority of GO terms were also enriched by shared nonadditive genes. Given that the nonadditive genes of WY and YW were also significantly enriched in the biological process of female gonad development, we further characterized the common nonadditive genes harbored in the GO terms shared by WY and YW. The four common nonadditive genes, including transforming growth factor beta 1 (TGFB1), collagen type VI alpha 1 chain (COL6A1), CCAAT/enhancer binding protein beta (CEBPB) and receptor tyrosine kinase (KIT) genes, were involved in the growth and development of ovarian cells (Supplementary Table S4).
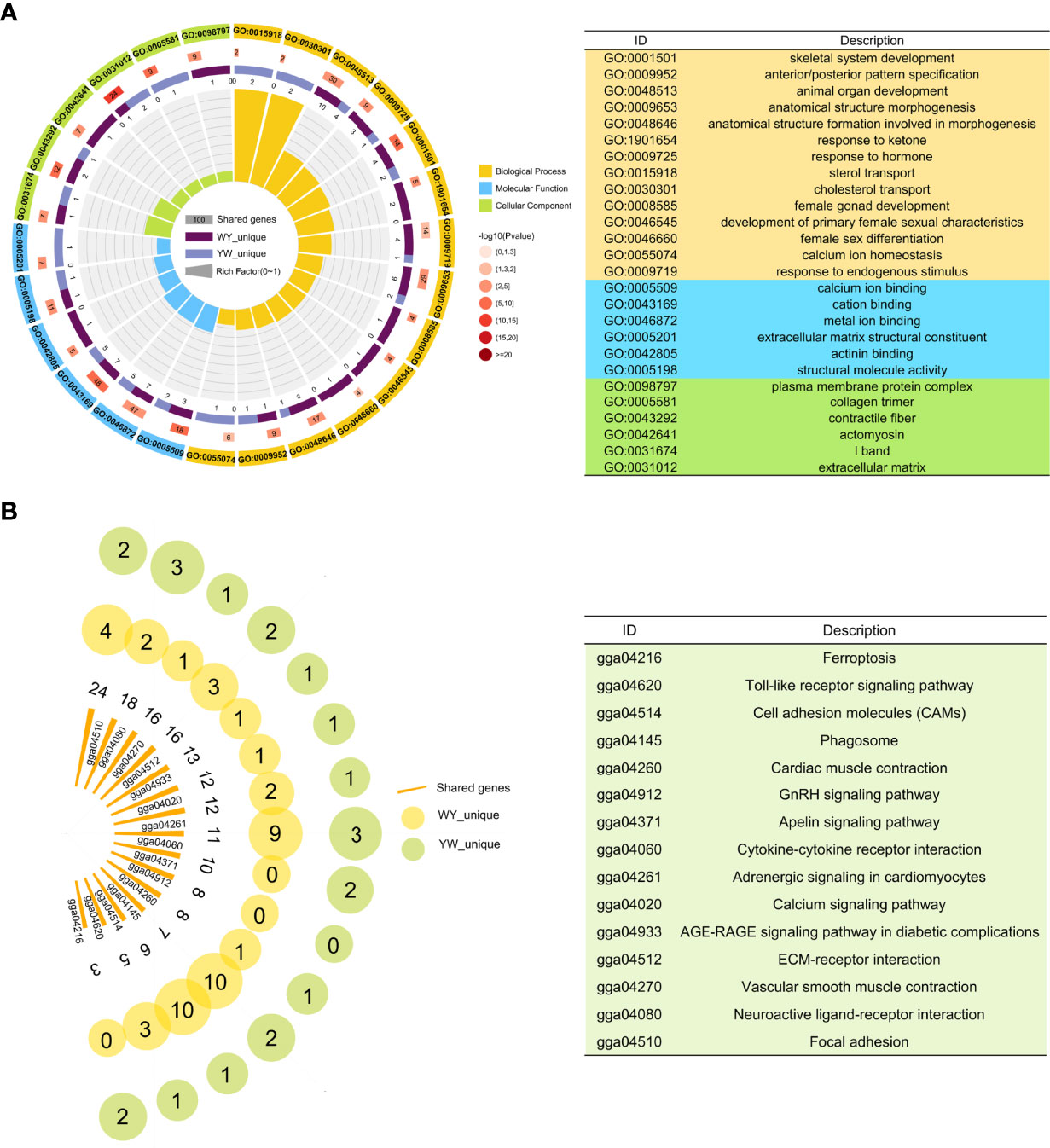
Figure 4 Function enrichment analysis of nonadditive genes. (A) Significant GO terms of nonadditive genes in crossbreeds. From outer to inner, the outermost circle represents the IDs of enriched GO terms. The names of GO ID in orange, blue and green represent biological process, molecular function and cell composition respectively. The second circle indicates shared genes enriched in GO terms. In the third circle, the piece in dark purple and light purple represents unique genes of WY and YW, respectively. In the innermost circle, each bar represents one GO term, and the size represents the rich factor. (B) KEGG pathway analysis for nonadditive genes. Each bar represents one pathway and the size represents the number of shared genes. The yellow and green circle represents the unique genes in WY and YW of each pathway, respectively.
KEGG pathway analysis showed that nonadditive genes were significantly enriched in the focal adhesion, ECM-receptor interaction, vascular smooth muscle contraction, GnRH signaling pathway, calcium signaling pathway and folate biosynthesis (Figure 4B). These pathways were also enriched by shared nonadditive genes. Regarding the GnRH signaling pathway, we found eight common nonadditive genes including G protein subunit alpha q (GNAQ), calcium voltage-gated channel subunit alpha1 C (CACNA1C), ENSGALG00000005884, adenylate cyclase 6 (ADCY6), calcium voltage-gated channel subunit alpha1 D (CACNA1D), ENSGALG00000008727, early growth response 1 (EGR1) and matrix metallopeptidase 2 (MMP2) were enriched in the pathway (Supplementary Table S4).
We constructed a co-expression network of lncRNAs and mRNAs to infer the underlying regulatory function and potential co-expressed genes and lncRNAs. Correlated lncRNAs and mRNAs were clustered into 20 modules by WGCNA, and each module contained at least 50 genes (Figure 5A). Based on the correlation between the clusters and phenotypes, seven modules were significantly correlated with pubic space or oviduct length (P < 0.05) (Figure 5B). Among these modules (ME), the midnightblue ME (r = -0.57, p = 0.01) and greenyellow ME (r = -0.56, p = 0.01) were negatively correlated with pubic space. The purple ME was negatively correlated with pubic space (r = -0.61, p = 0.005) and oviduct length (r = -0.57, p = 0.01). The green ME was positively correlated with the oviduct length (r = 0.53, p = 0.02). The turquoise ME (r = -0.59, p = 0.008), the brown ME (r = -0.54, p = 0.02) and the red ME (r = -0.64, p = 0.003) was negatively correlated with oviduct length. Additionally, for all lncRNAs and protein coding genes harbored in the co-expression network, the top 1% of highly correlated gene pairs were chosen to visualize the correlation of modules (Figure 5C). Function enrichment results of each cluster suggested that the highly correlated lncRNAs and genes were mainly enriched in the developmental related process. Notably, the annotated genes in cluster green ME, were significantly enriched in oocyte meiosis, ECM-receptor interaction, calcium ion binding and GnRH signaling pathway, suggesting that lncRNAs in this cluster is essential for those biological processes. In addition, genes in turquoise ME were found closely correlated with fatty acid biosynthetic process, cell cycle and peptide biosynthetic process (Figure 5D).
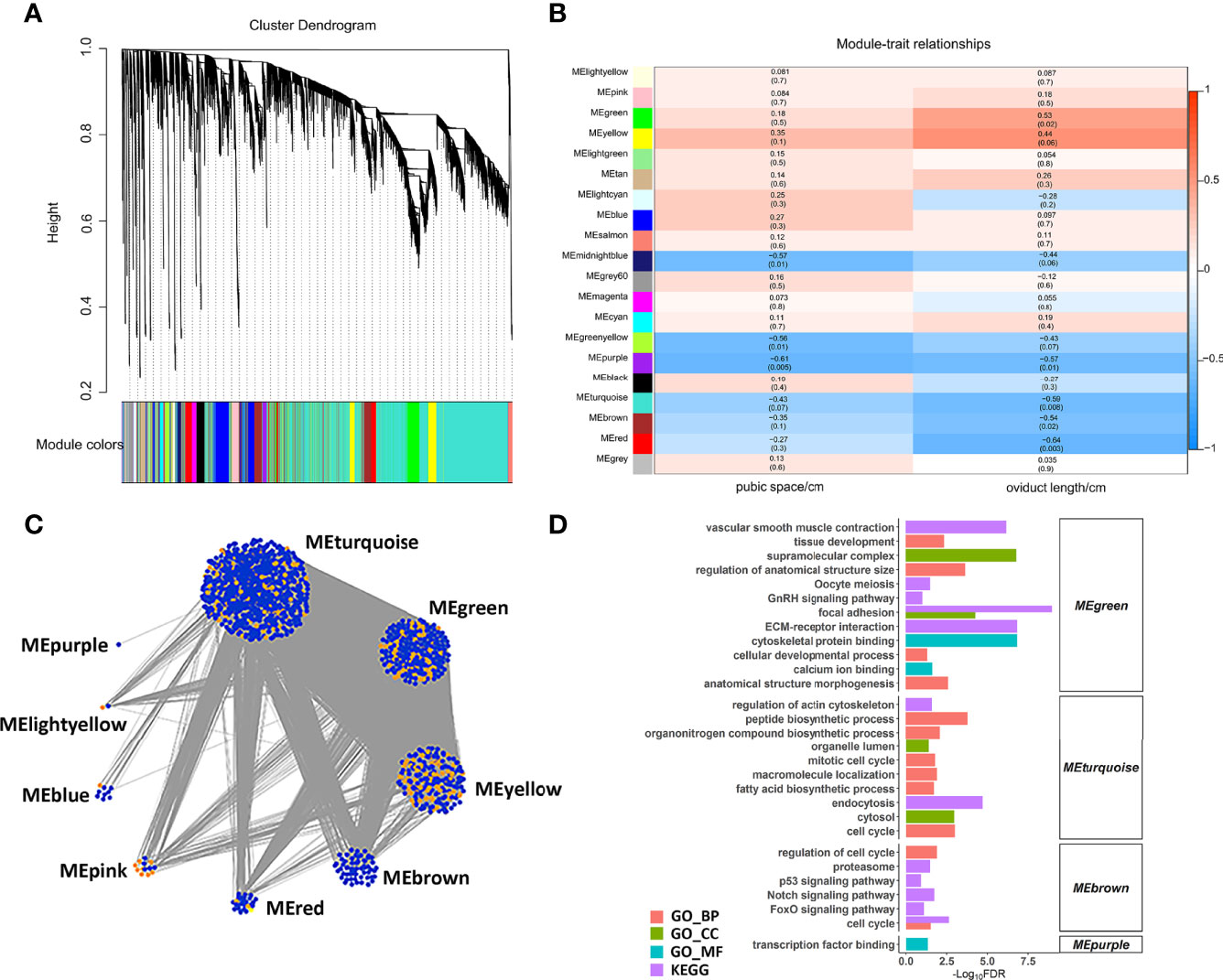
Figure 5 WGCNA analysis of lncRNAs and mRNAs. (A) Hierarchical cluster of 20 modules co-expressed mRNAs and lncRNAs. (B) Module-trait relationships. Each row represents a module eigengene, and each column present a trait. Each module includes the corresponding correlation and P value. (C) The networks of top 1% lncRNAs and mRNAs in nine selected modules. The orange and blue dots represent the hub lncRNAs and mRNAs, respectively. (D) The enriched function of each module.
We found 43 nonadditive genes were interactive and harbored in the seven associated modules, and these genes were regulated by 40 nonadditive lncRNAs in cis or trans (Figure 6A). Interestingly, function of these nonadditive genes were associated with reproductive structure development, gonad development, female gonad development, calcium signaling pathway and GnRH signaling pathway. The number of common nonadditive genes enriched in female gonad development and GnRH signaling pathway was four and eight in crossbreeds (Figure 6B), respectively. Moreover, CNCAN1C and TGFB1, which were regulated by MSTRG.17017.1 and MSTRG.6475.20 in trans, were enriched in GnRH signaling pathway and female gonad development, respectively. Accordingly, we found the expression of FSHR and ESR2 in crossbreeds was both higher than that of purebreds. But these two genes were not nonadditive genes (Supplementary Figure S4).
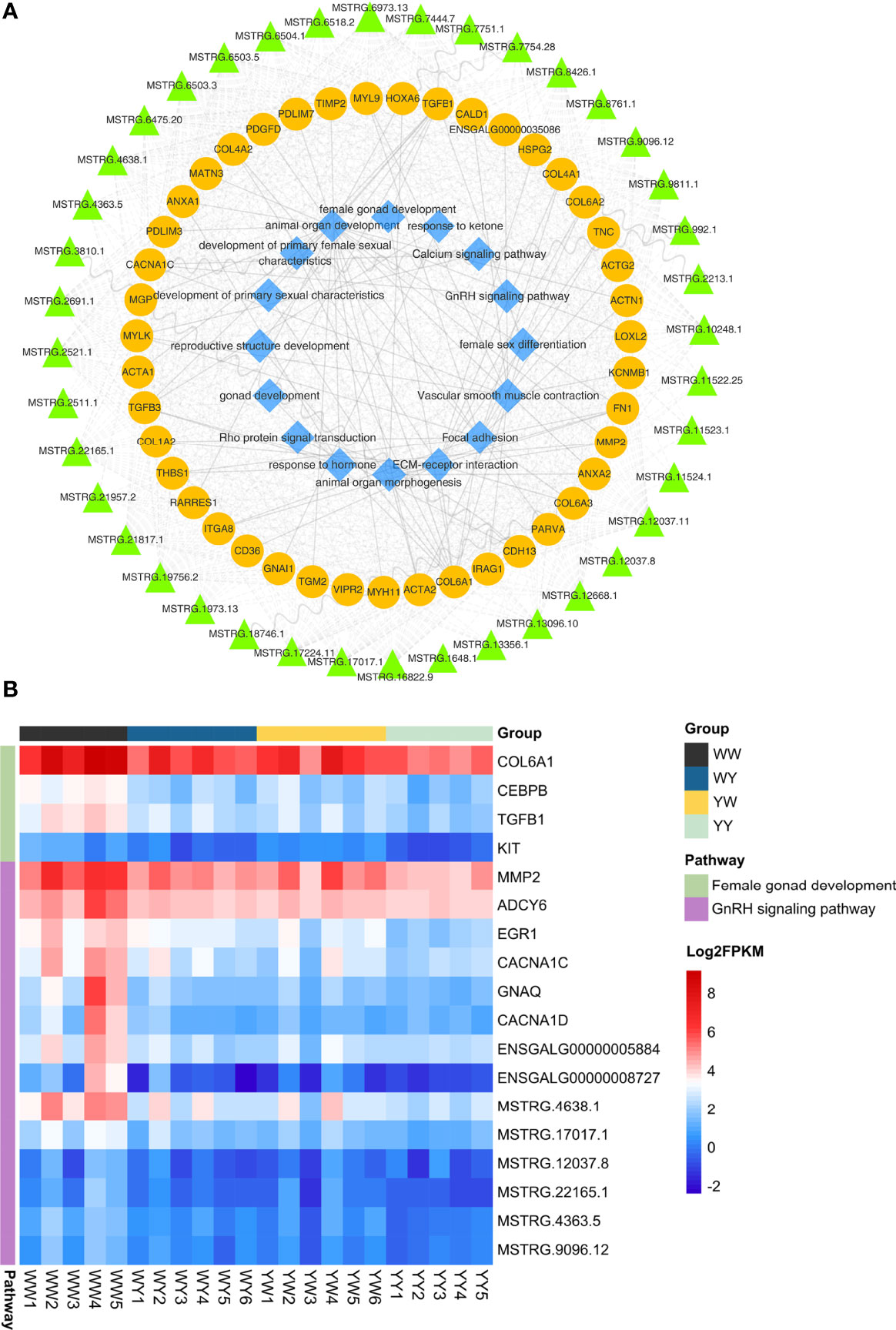
Figure 6 Co-expression network analysis of the paired lncRNA-mRNA involved in sexual maturation. (A) Network of lncRNAs, mRNAs and function. The green triangle, yellow circles and blue diamond represent lncRNAs, mRNAs and function, respectively. (B) The expression of nonadditive mRNAs and their lncRNAs enriched in GnRH signaling pathway and female gonad development.
The nonadditive genes were related to GnRH signaling pathway and female gonad development, implying that hormone secretion plays a vital role in positive heterosis of sexual maturation. Then, the serum GnRH and MT concentration were detected. GnRH content in crossbreeds was significantly higher than the average value of purebreds, and showed positive heterosis (P < 0.05) (Figure 7A). MT content was lower in reciprocal crossbreeds compared with the average value of purebreds exhibiting negative heterosis (Figure 7B).
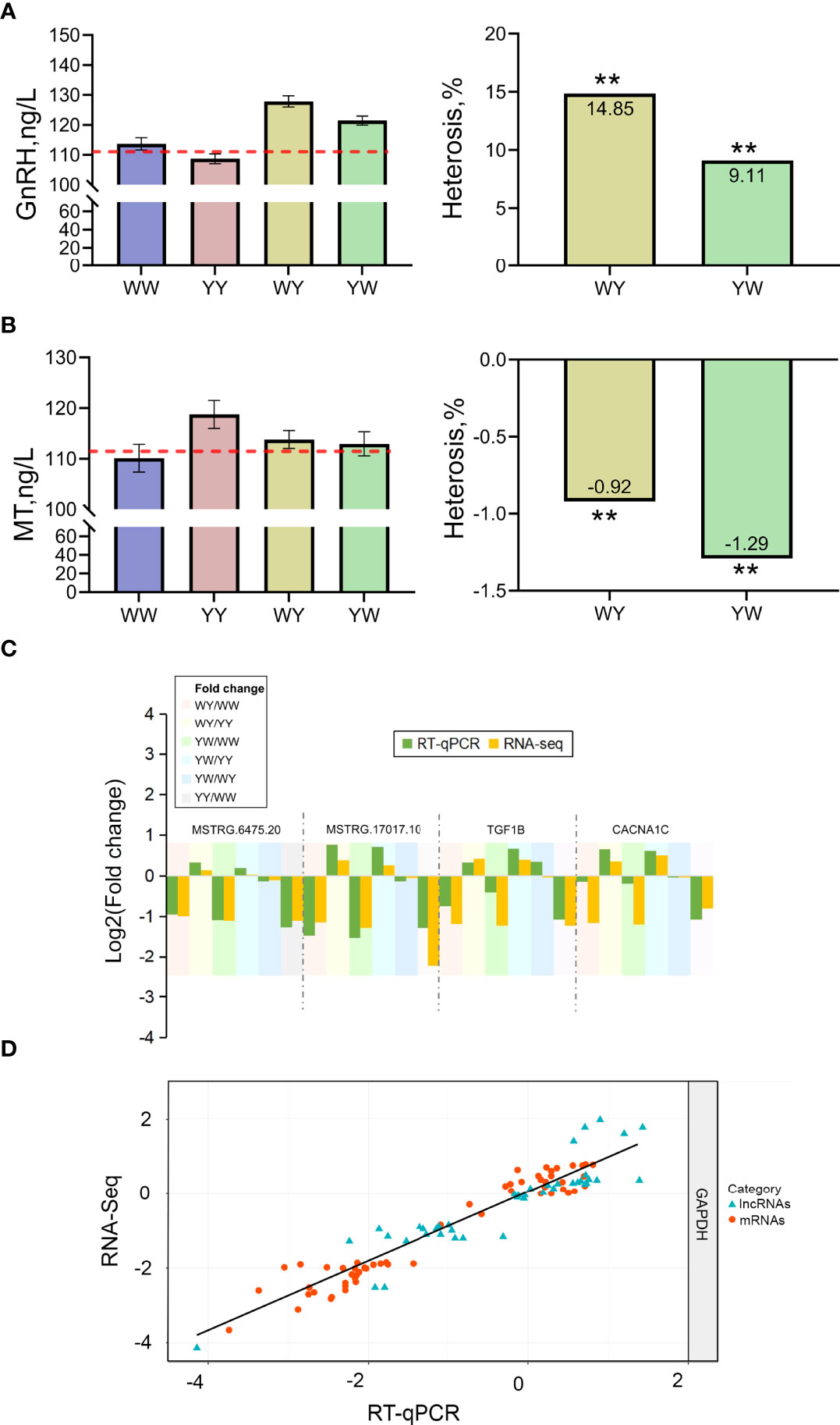
Figure 7 Validation of hormone concentration and gene expression. (A) The content of GnRH. (B) The content of MT. ** indicated adjusted P value less than 0.01. (C) The expression of candidate lncRNAs and mRNAs. (D) Correlation of gene expression level of 13 differentially expressed genes (DEGs) and six differentially expressed lncRNAs (DELs) using RNA-Seq and RT- qPCR. The x- and y- axis represents the log2 (fpkm) measured by RNA-Seq and RT-qPCR, respectively. GAPDH was used as reference gene. The blue and red dots represent the DEGs and DELs, respectively.
To validate the RNA sequencing results, the expression of 13 DEGs and 6 DELs was detected using RT-qPCR. The specific primers for genes and lncRNAs were listed in Table 1. The expression of DEGs and DELs between any two groups showed consistent trend with the results of RNA-seq. A high correlation coefficient of gene expression level was detected (R2 = 0.91), indicating that RNA-seq data were reliable (Figures 7C, D).
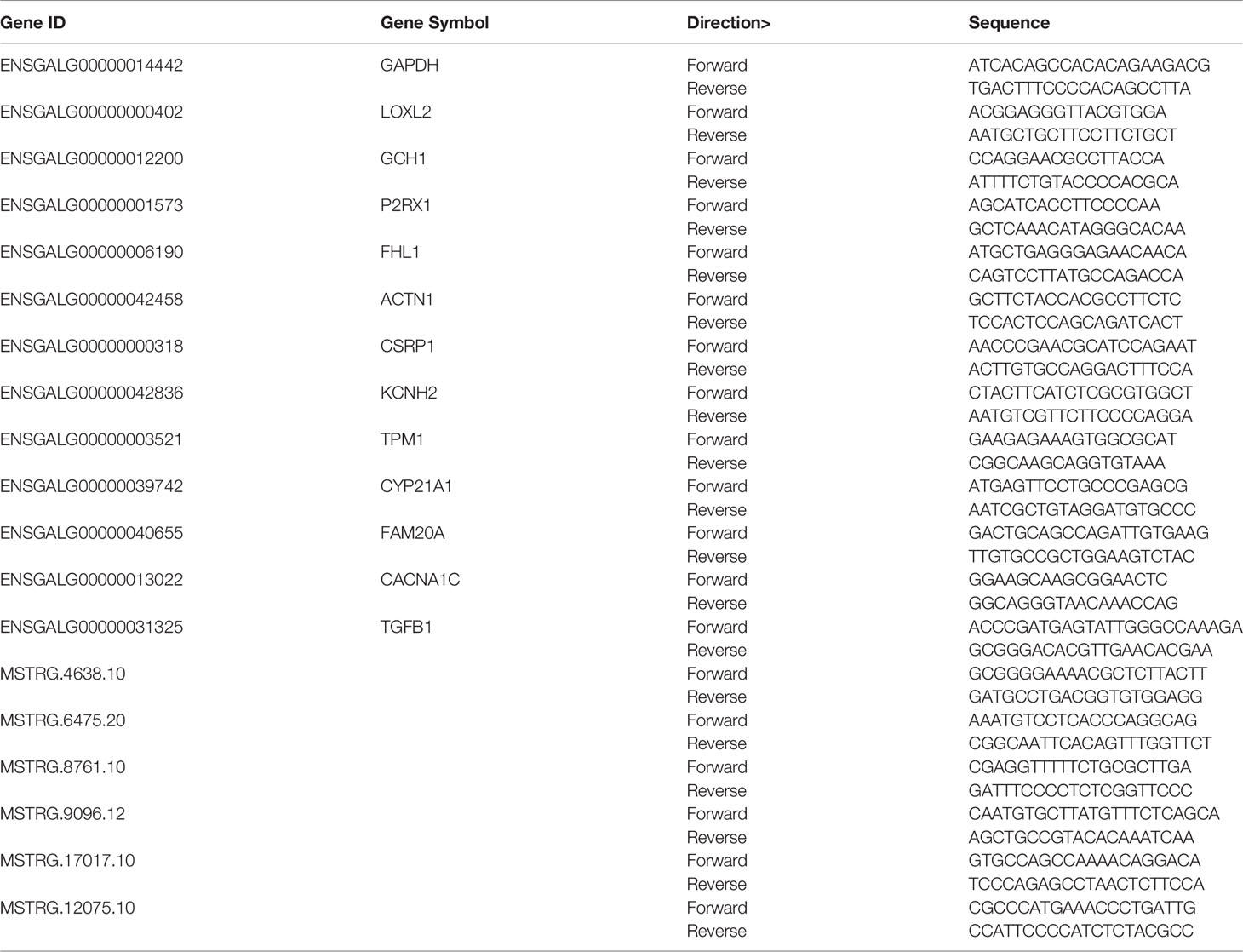
Table 1 Primers for differentially expressed mRNAs and lncRNAs used in RT-qPCR.
The utilization of sexual maturation heterosis has contributed to egg production improvement in chicken for decades. However, our understanding of its molecular basis is still rudimentary, constraining its flexible and accurate application. In the current study, we found negative heterosis of AFE in both crossbreeds. Similar results were also reported in hybrids of other chicken breeds (37, 38). Female sexual maturation is a complicated biological process accompanying with the growth and development of gonad and internal reproductive organs. Pubic space and oviduct length are important indicators of gonad development in chickens (39). Significantly positive heterosis for both traits were observed in crossbreeds, implying the superiority of sexual maturation in hybrids. Accumulating transcriptome analysis focused on heterosis of different traits were conducted, which facilitated the understanding of the molecular mechanism of hybrid vigor in different tissues of various species (10). Hence, the comprehensive gene expression patterns in purebreds and crossbreeds were profiled to reveal the potential mechanisms of sexual maturation heterosis. To our knowledge, the genome-wide expression of lncRNAs and mRNAs related to sexual maturation heterosis in chicken was firstly characterized in our study.
LncRNAs, an important epigenetic factor, could function in the reproductive process through cis and trans regulating gene activity. In the current study, a total of 3164 lncRNAs were identified. Fewer lncRNAs were identified than that in chicken brain (40), adipose tissue and liver (41), indicating tissue specific expression patterns and characteristics of lncRNAs. Additionally, the identified lncRNAs exhibited fewer exons, shorter transcripts, which is consistent with studies in other species (42). Consistent with previous research, intergenic lncRNAs was the most abundant class of lncRNAs in the genome (43, 44).
The number of DEGs and DELs between two parental lines was greater than that between crossbreeds and their purebreds, indicating the genetic difference between two purebreds was larger than any other groups. Similar results were also revealed by the previous study (45). Genome heterozygosity arising from genomics sequence variation between two purebreds is one fundamental driving force for the phenotype heterosis. As in the case of dominant or recessive allele pairs in classical genetics, the dominance expression of genes resulted in a nonlinear phenotypic effect of alleles at one locus (46). Therefore, nonadditive expression pattern is important for the formation of heterosis. Wu et al. (47) reported that dominant genes involved in carbohydrate metabolism were associated with heterosis for body weight in Drosophila melanogaster. It was also found the nonadditive genes may drive the formation of heat stress heterosis in cattle (48). In chicken, the nonadditive genes were illustrated to involved in oxidative phosphorylation, contributing to breast muscle mass heterosis (12). In the present study, most DEGs and DELs possessed nonadditivity pattern in hybrids, indicating nonadditivity was the critical gene expression pattern affecting traits heterosis. Functional enrichment analysis showed that nonadditive genes were associated with animal organ development, female gonad development, ECM-receptor interaction and GnRH signaling pathway, such as laminin subunit alpha 4 (VTN), CD36 molecule (CD36) and fibronectin 1 (FN1), which were revealed to function in ovary cell growth and proliferation (49–51) and specifically expressed in crossbreeds’ ovary. In addition, the nonadditive gene purinergic receptor P2X 1 (P2RX1) was identified, which was enriched in calcium signaling pathway. P2RX1 was demonstrated as potential candidate genes for egg production of ducks (52). Hence, we hypothesized these biological process and pathways might involve in the formation of sexual maturation heterosis through modulating gene expression.
The underlying mechanism of sexual maturation heterosis is complicated. WGCNA may be an effective method for mining valuable expression data to analyze the intricate genetic networks (35). By focusing on the association between co-expression modules and traits of interest, we identified seven modules were significantly correlated with pubic space and oviduct length. Genes harbored in 7 modules were enriched in GnRH signaling pathway, ECM-receptor interaction, calcium ion binding and tissue development, which were similar to the enrichment results of nonadditive genes. The overlapped biological processes may play more crucial role in the formation of sexual maturation heterosis.
The gonad development is part of prenatal development of the reproductive system and ultimately forms the ovary in the female (53). In this study, female gonad development was significantly enriched by nonadditive genes including CEBPB, COL6A1, TGFB1 and KIT. Previous studies demonstrated that CEBPB, COL6A1 and KIT may be essential for female reproduction through regulating the cell development, ovulation and luteinization of ovarian follicle (54–56). TGFB1 is implicated as a key regulator of the development and cyclic re-modelling characteristic of reproductive tissues (57). Altered plasma TGFB1 has been found closely associated with reproductive process in female (58). The up-regulated TGFB1 contributed follicles development and ovulation in sheep (44). In our study, the expression of TGFB1 was the lowest in YY, and significantly associated with oviduct length, suggesting TGFB1 may act an indicator to estimate the heterosis of sexual maturation. Additionally, previous studies found several ovarian lncRNAs could involve in follicular development by regulating target genes related to TGF-β in small-tailed sheep (59, 60). In the current study, TGFB1 were found significantly correlated with oviduct length, and regulated by MSTRG.17017.1 and MSTRG.6475.20 in trans. MSTRG.17017.1 and MSTRG.6475.20 were both novel lncRNAs in chicken. These indicated the two pairs of mRNAs and lncRNAs could be critical candidates for regulating the formation of sexual maturation heterosis. The putative function of candidate genes performed in the development and sexual maturation, supporting direct cue for nonadditive genes involved in sexual maturation heterosis.
GnRH signaling is regarded as the gonadotrope and endocrine control of fertility in mice (61) and goose (62). In chicken, GnRH signaling pathway could involve in ovarian function (49, 63). Here, the nonadditive genes, such as MMP2, ADCY6, EGR1, CACNA1C and GNAQ, were identified significantly enriched in GnRH signaling pathway. Importantly, GNAQ regulates estrus in sheep by controlling GnRH secretion and release through calcium signaling pathway (64). In ovary, GNAQ can also increase the size of ovarian follicles (65). Similarly, CACNA1C, associated with calcium channel function and crucial for onset of the reproductive maturation process (66). These two genes were involved in GnRH signaling pathway, implying they may act upstream factors to affect the downstream genes involved in gonad development. These results supported GnRH signaling pathway could involve the sexual maturation heterosis. Moreover, several lncRNAs were also evidenced involving in spermatogenesis and the time of puberty through regulating their target genes in bull (67). Herein, the expression differences of GNAQ and CACNA1C were regulated by MSTRG.19756.2 in trans. MSTRG.19756.2 was novel lncRNA. Our findings imply ovary MSTRG.19756.2 may be associated with reproductive impairment by controlling their target genes (GNAQ, CACNA1C) related with the GnRH signaling pathway. Admittedly, sexual maturation is initiated by the release of GnRH (68). The GnRH and MT content were further detected, and confirmed the results of gene expression. Upon light stimulation, decreasing levels of MT results in a decrease in GnIH synthesis (69), thus alleviating the inhibition on the HPG axis and allowing for the release of GnRH and subsequent activation of gonad development (70). Consistent with the previous study (71), we found that chickens with earlier AFE and larger pubic space had higher GnRH content but lower MT content, suggesting that chickens with higher GnRH could give impetus to development of reproductive tissue, contributing to the sexual maturation in crossbreeds. Interestingly, the crossbreeds shared same photoperiod stimulus with purebreds, but exhibited significant heterosis for sexual maturation. Hence, we speculated the disparity of light perception and the differences between crossbreeds and purebreds may drive the formation of sexual maturation heterosis (72). This may provide novel insights into understanding the neuroendocrine neurons controlling the onset of puberty and fertility in mammals including humans.
Our study focused on revealing the phenomenon of heterosis in chickens, and positive heterosis of sexual maturation was observed. Genome-wide gene expression pattern analysis showed that nonadditivity was the critical mode of mRNAs and lncRNAs action for sexual maturation heterosis. The nonadditive lncRNAs MSTRG.6475.20 and MSTRG.17017.1 and their target nonadditive genes (GNAQ, CACNA1C and TGFB1), which involved in GnRH signaling pathway and female gonad development, played a critical role in driving the formation of sexual maturation heterosis. Our findings would provide novel insight into the molecular basis of positive heterosis for sexual maturation in chicken and theoretical basis for improving egg production.
The transcriptome data are available in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) at NCBI, with the BioProject ID: PRJNA845127 and SRA Accession Number: SAMN28857461-28857484.
This study was carried out in line with the Guidelines for Experimental Animals established by the IAS-CAAS (IAS2022-35).
JC, YS and JY conceived and designed the project. JY and YW performed bioinformatics analyses, YW conducted the experiments and drafted the manuscript. YW, AN, YZ, JZ, SB and YL participated in animal manipulation and data collection. PW, LS, YL and HM discussed the manuscript. All authors contributed to sample collecting and approved the submitted version. All authors contributed to the article and approved the submitted version.
This research was funded by the National Natural Science Foundation of China (32172721), China Agriculture Research System (CARS-40), the Fundamental Research Funds for Central Non-profit Scientific Institution (2021-YWF-ZYSQ-12), and the Agricultural Science and Technology Innovation Program (ASTIP-IAS04).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.951534/full#supplementary-material
Supplementary Figure 1 | Chromosomal distribution of putative and known lncRNAs.
Supplementary Figure 2 | Principal components analysis (PCA) of identified and genes in the crossbreeds (WY, YW) and purebreds (WW, YY).
Supplementary Figure 3 | The Venn analysis of nonadditive lncRNAs and genes in WY and YW. (A) The Venn analysis of nonadditive lncRNAs in WY and YW. (B) The Venn analysis of nonadditive genes in WY and YW.
Supplementary Figure 4 | The expression of FSHR and ESR2 in different groups.
Supplementary Table 1 | Summary statistics for transcriptome sequencing data, the identified lncRNAs.
Supplementary Table 2 | The results of differentially expressed lncRNAs and differentially expressed genes in different groups.
Supplementary Table 3 | Classification of different expression patterns of mRNAs and lncRNAs.
Supplementary Table 4 | The enriched GO terms and pathways of nonadditive genes and detailed information of the shared nonadditive genes enriched in key biological process.
1. Feibelmann TC, Silva AP, Resende DC, Resende EA, Scatena LM, Borges MF. Puberty in a Sample of Brazilian Schoolgirls: Timing and Anthropometric Characteristics. Arch Endocrinol Metab (2015) 59:105–11. doi: 10.1590/2359-3997000000021
2. Bédécarrats GY. Control of the Reproductive Axis: Balancing Act Between Stimulatory and Inhibitory Inputs. Poult Sci (2015) 94:810– 5. doi: 10.3382/ps/peu042
3. Rasier G, Toppari J, Parent AS, Bourguignon JP. Female Sexual Maturation and Reproduction After Prepubertal Exposure to Estrogens and Endocrine Disrupting Chemicals: A Review of Rodent and Human Data. Mol Cell Endocrinol (2006) 254– 255:187–201. doi: 10.1016/j.mce.2006.04.002
4. Roa J, Aguilar E, Dieguez C, Pinilla L, Tena– Sempere M. New Frontiers in Kisspeptin/GPR54 Physiology as Fundamental Gatekeepers of Reproductive Function. Front Neuroendocrinol (2008) 29:48–69. doi: 10.1016/j.yfrne.2007.07.002
5. Fujimoto R, Uezono K, Ishikura S, Osabe K, Peacock WJ, Dennis ES. Recent Research on the Mechanism of Heterosis is Important for Crop and Vegetable Breeding Systems. Breed Sci (2018) 68:145–58. doi: 10.1270/jsbbs.17155
6. Hochholdinger F, Baldauf JA. Heterosis in Plants. Curr Biol (2018) 28:R1089R1092. doi: 10.1016/j.cub.2018.06.041
7. Hochholdinger F, Hoecker N. Towards the Molecular Basis of Heterosis. Trends Plant Sci (2007) 12:42732. doi: 10.1016/j.tplants.2007.08.005
8. Cui Y, Wang J, Zhang H, Feng J, Wu S, Qi G. Effect of Photoperiod on Ovarian Morphology, Reproductive Hormone Secretion, and Hormone Receptor mRNA Expression in Layer Ducks During the Pullet Phase. Poult Sci (2019) 98:2439–47. doi: 10.3382/ps/pey601
9. Farghly M, Mahrose KM, Rehman ZU, Yu S, Abdelfattah MG, ElGarhy OH. Intermittent Lighting Regime as a Tool to Enhance Egg Production and Eggshell Thickness in Rhode Island Red Laying Hens. Poult Sci (2019) 98:2459–65. doi: 10.3382/ps/pez021
10. Liu W, Zhang Y, He H, He G, Deng X. From Hybrid Genomes to Heterotic Trait Output: Challenges and Opportunities. Curr Opin Plant Biol (2022) 66:102193. doi: 10.1016/j.pbi.2022.102193
11. Mai C, Wen C, Sun C, Xu Z, Chen S, Yang N. Implications of Gene Inheritance Patterns on the Heterosis of Abdominal Fat Deposition in Chickens. Genes (Basel) (2019) 10:824. doi: 10.3390/genes10100824
12. Mai C, Wen C, Xu Z, Xu G, Chen S, Zheng J, et al. Genetic Basis of Negative Heterosis for Growth Traits in Chickens Revealed by Genome–Wide Gene Expression Pattern Analysis. J Anim Sci Biotechnol (2021) 12:52. doi: 10.1186/s40104021005742
13. Zhuo Z, Lamont SJ, Abasht B. RNASeq Analyses Identify Additivity as the Predominant Gene Expression Pattern in F1 Chicken Embryonic Brain and Liver. Genes (Basel) (2019) 10:27. doi: 10.3390/genes10010027
14. Ponting CP, Oliver PL, Reik W. Evolution and Functions of Long Noncoding RNAs. Cell (2009) 136:629–41. doi: 10.1016/j.cell.2009.02.006
15. Gao X, Ye J, Yang C, Luo L, Liu Y, Ding J, et al. RNAseq Analysis of lncRNA–Controlled Developmental Gene Expression During Puberty in Goat & Rat. BMC Genet (2018) 19:19. doi: 10.1186/s1286301806089
16. Lawson H, Vuong E, Miller RM, Kiontke K, Fitch DH, Portman DS. The Makorin Lep–2 and the lncRNA Lep–5 Regulate Lin–28 to Schedule Sexual Maturation of the C. elegans nervous system. Elife (2019) 8:e43660. doi: 10.7554/eLife.43660
17. Shu H, Zhou H, Mu H, Wu S, Jiang Y, Yang Z, et al. Integrated Analysis of mRNA and Noncoding RNA Transcriptome in Pepper (Capsicum Chinense) Hybrid at Seedling and Flowering Stages. Front Genet (2021) 12:685788. doi: 10.3389/fgene.2021.685788
18. Haberfeld A, Dunnington EA, Siegel PB, Hillel J. Heterosis and DNA Fingerprinting in Chickens. Poult Sci (1996) 75:9513. doi: 10.3382/ps.0750951
19. Isa AM, Sun Y, Shi L, Jiang L, Li Y, Fan J, et al. Hybrids Generated by Crossing Elite Laying Chickens Exhibited Heterosis for Clutch and Egg Quality Traits. Poult Sci (2020) 99:6332–40. doi: 10.1016/j.psj.2020.08.056
20. Khawaja T, Khan SH, Mukhtar N, Ullah N, Parveen A. Production Performance, Egg Quality and Biochemical Parameters of Fayoumi, Rhode Island Red and Their Reciprocal Crossbred Chickens. J Appl Anim Res (2013) 41:20817. doi: 10.1080/09712119.2012.739969
21. Wang H, Zhang Y, Sun D, Yu Y. Relationship Between Differential Gene Expression Patterns in Chicken’s Ovary and Heterosis of Egg Number in a Chicken Diallel Cross. Chin J Anim Veterinar Sci (2005) 36:111–5. doi: 10.3321/j.issn:03666964.2005.02.002
22. Liu Z, Sun C, Yan Y, Li G, Li X, Wu G, et al. Design and Evaluation of a Custom 50K Infinium SNP Array for Egg–Type Chickens. Poult Sci (2021) 100(5):101044. doi: 10.1016/j.psj.2021.101044
23. Yin Z, Lian L, Zhu F, Zhang ZH, Hincke M, Yang N, et al. The Transcriptome Landscapes of Ovary and Three Oviduct Segments During Chicken (Gallus Gallus) Egg Formation. Genomics (2020) 112(1):24351. doi: 10.1016/j.ygeno.2019.02.003
24. Perkel JM. Ten Computer Codes That Transformed Science. Nature (2021) 589:344–48. doi: 10.1038/d41586021000752
25. Wu Z, Zhang W. Heterosis and Stastistical Tests. Heredit (Beijing) (1983) 5:2426. doi: CNKI:SUN:YCZZ.0.1983-01-008
26. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcriptlevel Expression Analysis of RNAseq Experiments With HISAT, StringTie and Ballgown. Nat Protoc (2016) 11:165067. doi: 10.1038/nprot.2016.095
27. Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, et al. NONCODE 2016: An Informative and Valuable Data Source of Long non–Coding RNAs. Nucleic Acids Res (2016) 44:D2038. doi: 10.1093/nar/gkv1252
28. Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C, et al. Utilizing Sequence Intrinsic Composition to Classify Protein–Coding and Long non–Coding Transcripts. Nucleic Acids Res (2013) 41:e166. doi: 10.1093/nar/gkt646
29. Kong L, Zhang Y, Ye Z, Liu X, Zhao S, Wei L, et al. CPC: Assess the Protein–Coding Potential of Transcripts Using Sequence Features and Support Vector Machine. Nucleic Acids Res (2007) 35:W3459. doi: 10.1093/nar/gkm391
30. Li A, Zhang J, Zhou Z. PLEK: A Tool for Predicting Long non–Coding RNAs and Messenger RNAs Based on an Improved K–Mer Scheme. BMC Bioinf (2014) 15:311. doi: 10.1186/1471210515311
31. ElGebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. The Pfam Protein Families Database in 2019. Nucleic Acids Res (2019) 47:D427D432. doi: 10.1093/nar/gky995
32. Xiang S, Li Z, Weng X. The Role of lncRNA RP11154D6 in Steroid–Induced Osteonecrosis of the Femoral Head Through BMSC Regulation. J Cell Biochem (2019) 120:18435–45. doi: 10.1002/jcb.29161
33. Love MI, Huber W, Anders S. Moderated Estimation of Fold Change and Dispersion for RNAseq Data With Deseq2. Genome Biol (2014) 15:550. doi: 10.1186/s1305901405508
34. SwansonWagner RA, Jia Y, DeCook R, Borsuk LA, Nettleton D, Schnable PS. All Possible Modes of Gene Action are Observed in a Global Comparison of Gene Expression in a Maize F1 Hybrid and its Inbred Parents. Proc Natl Acad Sci USA (2006) 103:6805–10. doi: 10.1073/pnas.0510430103
35. Langfelder P, Horvath S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinf (2008) 9:559. doi: 10.1186/147121059559
36. Bu D, Luo H, Huo P, Wang Z, Zhang S, He Z, et al. KOBASi: Intelligent Prioritization and Exploratory Visualization of Biological Functions for Gene Enrichment Analysis. Nucleic Acids Res (2021) 49:W317–25. doi: 10.1093/nar/gkab447
37. Hristakieva P, Oblakova M, Lalev M, Mincheva N. Heterosis Effect in Hybrid Laying Hens. Biotechnol Anim Husbandr (2014) 30:303–11. doi: 10.3382/ps.0730001
38. Ahmed Soliman M, Hassan Khalil M, ElSabrout K, Kamel Shebl M. Crossing Effect for Improving Egg Production Traits in Chickens Involving Local and Commercial Strains. Veterinar World (2020) 13:407–12. doi: 10.14202/vetworld.2020.407412
39. Yang L, Mo C, Adetula AA, Elokil AA, Akbar BA, Huang T, et al. Bilateral Apex Pubis Distance: A Novel Index for Follicular Development and Egg Laying Status in Domestic Hens (Gallus Gallus Domesticus). Br Poult Sci (2020) 61:1951–99. doi: 10.1080/00071668.2019.1697429
40. Xu Z, Che T, Li F, Tian K, Zhu Q, Mishra SK, et al. The Temporal Expression Patterns of Brain Transcriptome During Chicken Development and Ageing. BMC Genomics (2018) 19:917. doi: 10.1186/s128640185301x
41. Jehl F, Muret K, Bernard M, Boutin M, Lagoutte L, Désert C, et al. An Integrative Atlas of Chicken Long non–Coding Genes and Their Annotations Across 25 Tissues. Sci Rep (2020) 10:20457. doi: 10.1038/s4159802077586x
42. Yuan J, Ni A, Li Y, Bian S, Liu Y, Wang P, et al. Transcriptome Analysis Revealed Potential Mechanisms of Resistance to Trichomoniasis Gallinae Infection in Pigeon (Columba Livia). Front Vet Sci (2021) 8:672270. doi: 10.3389/fvets.2021.672270
43. Zheng J, Wang Z, Yang H, Yao X, Yang P, Ren C, et al. Pituitary Transcriptomic Study Reveals the Differential Regulation of lncRNAs and mRNAs Related to Prolificacy in Different FecB Genotyping Sheep. Genes (Basel) (2019) 10:157. doi: 10.3390/genes10020157
44. Chen S, Guo X, He X, Di R, Zhang X, Zhang J, et al. Insight Into Pituitary lncRNA and mRNA at Two Estrous Stages in Small Tail Han Sheep With Different FecB Genotypes. Front Endocrinol (Lausan) (2021) 12:789564. doi: 10.3389/fendo.2021.789564
45. Wei G, Tao Y, Liu G, Chen C, Luo R, Xia H, et al. A Transcriptomic Analysis of Superhybrid Rice LYP9 and its Parents. Proc Natl Acad Sci USA (2009) 106:7695701. doi: 10.1073/pnas.0902340106
46. Seymour DK, Chae E, Grimm DG, Martín PC, HabringMüller A, Vasseur F, et al. Genetic Architecture of Nonadditive Inheritance in Arabidopsis Thaliana Hybrids. Proc Natl Acad Sci USA (2016) 113:E7317E7326. doi: 10.1073/pnas.1615268113
47. Wu X, Li R, Li Q, Bao H, Wu C. Comparative Transcriptome Analysis Among Parental Inbred and Crosses Reveals the Role of Dominance Gene Expression in Heterosis in Drosophila Melanogaster. Sci Rep (2016) 6:21124. doi: 10.1038/srep21124
48. Zhang G, Wang L, Huang D, Chen H, Li B, Wu Y, et al. Inheritance Patterns of Leukocyte Gene Expression Under Heat Stress in F1 Hybrid Cattle and Their Parents. J Dair Sci (2020) 103:10321–31. doi: 10.3168/jds.202018410
49. Ma Z, Jiang K, Wang D, Wang Z, Gu Z, Li G, et al. Comparative Analysis of Hypothalamus Transcriptome Between Laying Hens With Different Egg–Laying Rates. Poult Sci (2021) 100:101110. doi: 10.1016/j.psj.2021.101110
50. Sun T, Xiao C, Deng J, Yang Z, Zou L, Du W, et al. Transcriptome Analysis Reveals Key Genes and Pathways Associated With Egg Production in NandanYao Domestic Chicken. Comp Biochem Physiol Part D Genomics Proteomics (2021) 40:100889. doi: 10.1016/j.cbd.2021.100889
51. Karakaya C, Çil AP, Bilguvar K, Çakir T, Karalok MH, Karabacak RO, et al. Further Delineation of Familial Polycystic Ovary Syndrome (PCOS) via Whole–Exome Sequencing: PCOSrelated Rare FBN3 and FN1 Gene Variants are Identified. J Obstet Gynaecol Res (2022) 48:1202–11. doi: 10.1111/jog.15187
52. Bello SF, Xu H, Guo L, Li K, Zheng M, Xu Y, et al. Hypothalamic and Ovarian Transcriptome Profiling Reveals Potential Candidate Genes in Low and High Egg Production of White Muscovy Ducks (Cairina Moschata). Poultry Sci (2021) 100:101310. doi: 10.1016/j.psj.2021.101310
53. Zhang C, Kim AJ, RiveraPerez C, Noriega FG, Kim YJ. The Insect Somatostatin Pathway Gates Vitellogenesis Progression During Reproductive Maturation and the Post–Mating Response. Nat Commun (2022) 13:969. doi: 10.1038/s41467022285922
54. Fan HY, Liu Z, Johnson PF, Richards JS. CCAAT/enhancerbinding Proteins (C/EBP)–α and –β are Essential for Ovulation, Luteinization, and the Expression of Key Target Genes. Mol Endocrinol (2011) 25:25368. doi: 10.1210/me.20100318
55. Yenuganti VR, Ravinder, Singh D. Endotoxin Induced TLR4 Signaling Downregulates CYP19A1 Expression Through CEBPB in Buffalo Granulosa Cells. Toxicol In Vitro (2017) 42:93100. doi: 10.1016/j.tiv.2017.04.012
56. Merkwitz C, Lochhead P, Tsikolia N, Koch D, Sygnecka K, Sakurai M, et al. Expression of KIT in the Ovary, and the Role of Somatic Precursor Cells. Prog Histochem Cytochem (2011) 46:13184. doi: 10.1016/j.proghi.2011.09.001
57. Ingman WV, Robertson SA. The Essential Roles of TGFB1 in Reproduction. Cytokine Growth Factor Rev (2009) 20:233–9. doi: 10.1016/j.cytogfr.2009.05.003
58. Fornari MC, Sarto A, Berardi VE, Martínez MA, Rocha MG, Pasqualini S, et al. Effect of Ovaric Hyper–Stimulation on Blood Lymphocyte Subpopulations, Cytokines, Leptin and Nitrite Among Patients With Unexplained Infertility. Am J Reprod Immunol (2002) 48:394–403. doi: 10.1034/j.16000897.2002.01128.x
59. Miao X, Luo Q, Zhao H, Qin X. Ovarian Transcriptomic Study Reveals the Differential Regulation of miRNAs and lncRNAs Related to Fecundity in Different Sheep. Sci Rep (2016) 6:35299. doi: 10.1038/srep35299
60. Yang H, Ma J, Wang Z, Yao X, Zhao J, Zhao X, et al. Genomewide Analysis and Function Prediction of Long Noncoding RNAs in Sheep Pituitary Gland Associated With Sexual Maturation. Genes (Basel) (2020) 11:320. doi: 10.3390/genes11030320
61. Bliss SP, Navratil AM, Xie J, Roberson MS. GnRH Signaling, the Gonadotrope and Endocrine Control of Fertility. Front Neuroendocrinol (2010) 31:32240. doi: 10.1016/j.yfrne.2010.04.002
62. Luan X, Liu D, Cao Z, Luo L, Liu M, Gao M, et al. Transcriptome Profiling Identifies Differentially Expressed Genes in Huoyan Goose Ovaries Between the Laying Period and Ceased Period. PloS One (2014) 9:e113211. doi: 10.1371/journal.pone.0113211
63. Mu R, Yu Y, Gegen T, Wen D, Wang F, Chen Z, et al. Transcriptome Analysis of Ovary Tissues From Lowand High–Yielding Changshun Green–Shell Laying Hens. BMC Genomics (2021) 22:349. doi: 10.1186/s1286402107688x
64. Zhu M, Zhang H, Yang H, Zhao Z, Blair HT, Liang H, et al. Targeting GNAQ in Hypothalamic Nerve Cells to Regulate Seasonal Estrus in Sheep. Theriogenology (2022) 181:7988. doi: 10.1016/j.theriogenology.2022.01.005
65. Xie J, Kalwar Q, Yan P, Guo X. Effect of Concentrate Supplementation on the Expression Profile of miRNA in the Ovaries of Yak During non–Breeding Season. Anim (Basel) (2020) 10:1640. doi: 10.3390/ani10091640
66. Uengwetwanit T, Ponza P, Sangsrakru D, Wichadakul D, Ingsriswang S, Leelatanawit R, et al. Transcriptomebased Discovery of Pathways and Genes Related to Reproduction of the Black Tiger Shrimp (Penaeus Monodon). Mar Genomics (2018) 37:6973. doi: 10.1016/j.margen.2017.08.007
67. Liu H, Khan IM, Yin H, Zhou X, Rizwan M, Zhuang J, et al. Integrated Analysis of Long NonCoding RNA and mRNA Expression Profiles in Testes of Calves and Sexually Mature Wandong Bulls (Bos Taurus). Anim (Basel) (2021) 11:2006. doi: 10.3390/ani11072006
68. Mayer C, Boehm U. Female Reproductive Maturation in the Absence of Kisspeptin/GPR54 Signaling. Nat Neurosci (2011) 14:704–10. doi: 10.1038/nn.2818
69. Ubuka T, Bentley GE, Ukena K, Wingfield JC, Tsutsui K. Melatonin Induces the Expression of Gonadotropin–Inhibitory Hormone in the Avian Brain. Proc Natl Acad Sci USA (2005) 102:30527. doi: 10.1073/pnas.0403840102
70. Bédécarrats GY, McFarlane H, Maddineni SR, Ramachandran R. Gonadotropininhibitory Hormone Receptor Signaling and its Impact on Reproduction in Chickens. Gen Comp Endocrinol (2009) 163:711. doi: 10.1016/j.ygcen.2009.03.010
71. Bédécarrats GY, Hanlon C, Tsutsui K. Gonadotropin Inhibitory Hormone and its Receptor: Potential Key to the Integration and Coordination of Metabolic Status and Reproduction. Front Endocrinol (Lausan) (2021) 12:781543. doi: 10.3389/fendo.2021.781543
Keywords: Heterosis, chicken, sexual maturation, ovary, lncRNA, WGCNA
Citation: Wang Y, Yuan J, Sun Y, Li Y, Wang P, Shi L, Ni A, Zong Y, Zhao J, Bian S, Ma H and Chen J (2022) Genetic Basis of Sexual Maturation Heterosis: Insights From Ovary lncRNA and mRNA Repertoire in Chicken. Front. Endocrinol. 13:951534. doi: 10.3389/fendo.2022.951534
Received: 24 May 2022; Accepted: 13 June 2022;
Published: 27 July 2022.
Edited by:
Jun Wang, Jilin Agriculture University, ChinaCopyright © 2022 Wang, Yuan, Sun, Li, Wang, Shi, Ni, Zong, Zhao, Bian, Ma and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jilan Chen, Y2hlbi5qaWxhbkAxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.