
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 18 August 2022
Sec. Pediatric Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.951331
This article is part of the Research Topic Endocrine Aspects of Noonan Syndrome and Related Syndromes View all 6 articles
 Stefano Stagi1*
Stefano Stagi1* Vittorio Ferrari1Marta Ferrari1Manuela Priolo2
Vittorio Ferrari1Marta Ferrari1Manuela Priolo2 Marco Tartaglia3 on behalf of the Meyer University Hospital Noonan Study group
Marco Tartaglia3 on behalf of the Meyer University Hospital Noonan Study groupNoonan syndrome (NS) is a disorder characterized by a typical facial gestalt, congenital heart defects, variable cognitive deficits, skeletal defects, and short stature. NS is caused by germline pathogenic variants in genes coding proteins with a role in the RAS/mitogen-activated protein kinase signaling pathway, and it is typically associated with substantial genetic and clinical complexity and variability. Short stature is a cardinal feature in NS, with evidence indicating that growth hormone (GH) deficiency, partial GH insensitivity, and altered response to insulin-like growth factor I (IGF-1) are contributing events for growth failure in these patients. Decreased IGF-I, together with low/normal responses to GH pharmacological provocation tests, indicating a variable presence of GH deficiency/resistance, in particular in subjects with pathogenic PTPN11 variants, are frequently reported. Nonetheless, short- and long-term studies have demonstrated a consistent and significant increase in height velocity (HV) in NS children and adolescents treated with recombinant human GH (rhGH). While the overall experience with rhGH treatment in NS patients with short stature is reassuring, it is difficult to systematically compare published data due to heterogeneous protocols, potential enrolment bias, the small size of cohorts in many studies, different cohort selection criteria and varying durations of therapy. Furthermore, in most studies, the genetic information is lacking. NS is associated with a higher risk of benign and malignant proliferative disorders and hypertrophic cardiomyopathy, and rhGH treatment may further increase risk in these patients, especially as dosages vary widely. Herein we provide an updated review of aspects related to growth, altered function of the GH/IGF axis and cell response to GH/IGF stimulation, rhGH treatment and its possible adverse events. Given the clinical variability and genetic heterogeneity of NS, treatment with rhGH should be personalized and a conservative approach with judicious surveillance is recommended. Depending on the genotype, an individualized follow-up and close monitoring during rhGH treatments, also focusing on screening for neoplasms, should be considered.
Noonan syndrome (NS, OMIM PS163950) is one of the most common non-chromosomal disorders affecting development and growth (1). It is largely transmitted as a dominant trait, even though two recessive forms have recently been identified (2, 3). NS was first described by the cardiologist Jaqueline Noonan in 1968 (4), who reported a previously unrecognized phenotype with pulmonary valve stenosis (PVS), short stature, variable cognitive deficits, and facial dysmorphism as recurrent major features. NS is a syndromic condition characterized by a distinctive facial gestalt (e.g., relative macrocephaly, hypertelorism, ptosis, and low-set/posteriorly rotated ears), failure to thrive in the first years of life, reduced postnatal growth, a wide spectrum of congenital and acquired cardiac defects (most commonly PVS and hypertrophic cardiomyopathy [HCM)]), varying degrees of developmental delay (DD)/intellectual disability (ID), typical chest deformities (superior pectus carinatum, inferior pectus excavatum), tendency to bleed and cryptorchidism in males (5–7).
The variable clinical phenotype of NS overlaps with those of other genetic syndromes originally categorized as NS-spectrum disorders (NSSD) (Figure 1). Among these, NS with multiple lentigines (NSML, previously known as LEOPARD syndrome; OMIM 151100), Mazzanti syndrome (also known as NS-like disorder with loose anagen hair) (NS-LAH; OMIM 607721 and 617506), Legius syndrome (LGSS; OMIM 611431), and a phenotype originally denominated neurofibromatosis-NS (OMIM 601321), a condition representing a distinct form of neurofibromatosis 1 (NF1, OMIM 162200) are the most closely related conditions. There are also significant clinical overlaps with cardiofaciocutaneous syndrome (CFCS; OMIM 115150), and Costello syndrome (CS; OMIM 218040) (8–10), and other recently recognized diseases (11, 12). Unsurprisingly, the molecular mechanisms underlying the pathogenesis of these disorders are closely related. These disorders are caused by germline mutations in genes encoding components of the RAS-mitogen-activated protein kinase (MAPK) pathway, a signal transduction cascade controlling key cellular processes (e.g., proliferation, differentiation, migration and metabolism) in response to a wide array of growth factors, hormones and cytokines (Figure 1) (13). Because of these shared mechanisms, these disorders are collectively termed as RASopathies (7–10). Multiple key players in this pathway are mutated in NSSD, with an overall gain-of-function effect on signal flow through the RAS-MAPK pathway (9).
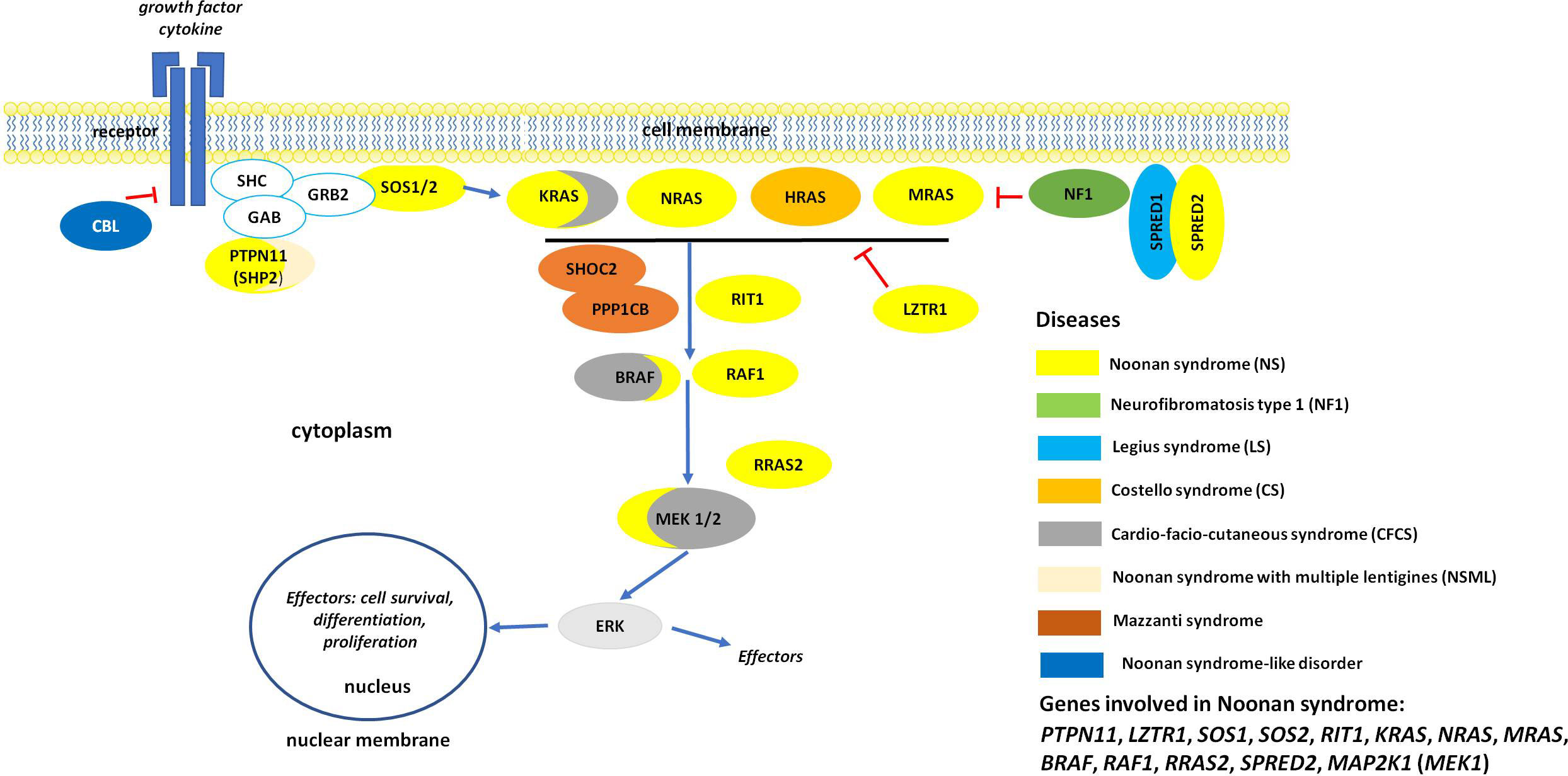
Figure 1 The RAS/MAPK signaling pathway and proteins involved in RASopaties. Overview of the RAS-MAPK signal transduction pathway. As shown, an extracellular stimulus triggers the activation of cell surface receptors (here a tyrosine kinase receptor, RTK), whose activation promotes the translocation of signal transducers positively controlling the function of RAS proteins (e.g., SOS1/2). These small monomeric GTPases, when activated, mediate the activation of the RAF kinases (BRAF and RAF1), which in turn phosphorylate and activate the dual-specificity kinases MEK1/2. Activated ERK1/2 is the last tier of the cascade and controls the function of a number of cytoplasmic and nuclear proteins. MAPK signaling is switched off by multiple circuits involving other proteins mutated in NS and related disorders (e.g., neurofibromin, CBL, LZTR1, SPRED1 and SPRED2).
The wide clinical variability characterizing NS results from a particularly marked genetic heterogeneity. Heterozygous germline mutations in 12 genes have been reported to be associated with NS (PTPN11, SOS1, SOS2, KRAS, NRAS, RIT1, MRAS, RAF1, BRAF, MAP2K1, LZTR1, and RRAS2) (1, 6, 8, 10, 14). Moreover, at least two genes have been reported as causing recessive forms of NS (LZTR1 and SPRED2) (2, 3). Mutations in the PTPN11 gene account for approximately 50% of NS cases, and mutations in LZTR1, RIT1, SOS1 and RAF1 for the majority of the remaining cases (1, 6–8, 10). Genetic mutations have not been identified in a small proportion of subjects with a clinical diagnosis of NS, supporting the existence of additional genes implicated in the disorder (14). Most NSSD causative pathogenic variants act through a gain of function mechanism on the RAS/MAPK signaling cascade that destabilizes the autoinhibitory mechanisms that maintain these proteins in their catalytically inactive conformation. Inactivating pathogenic variants affecting regulatory proteins that negatively control the RAS/MAPK cascade may also cause NSSD, and may act as either loss-of-function (LoF) (e.g., SPRED1, SPRED2, neurofibromin and LZTR1 in the recessive form of NS) or dominant-negative (DN) (LZTR1 in the dominant form of NS) mutations (3, 11, 15, 16).
From an auxological point of view, NS is one of the most prevalent non-chromosomal disorders affecting development and growth (17, 18). In affected subjects, birth weight and length are generally normal, although postnatal growth failure is observed (19, 20). Commonly, height tends to follow the third centile from ages two to four years until puberty, when below-average height velocity and an attenuated pubertal growth spurt tend to occur with a near adult height in the lower limits of normal values (21–23).
In NS, decreased insulin-like growth factor I (IGF-I) and IGF binding protein 3 (IGFBP-3), together with low responses to GH pharmacological provocation tests (GHST), may suggest, for some patients, an impaired growth hormone (GH) release or disturbance of the GH/IGF-I axis. Variable GH resistance, particularly in subjects with PTPN11 pathogenic variants, has also been reported (21). Short stature may be in part related to other factors (24, 25), such as congenital heart disease (CHD) requiring surgery (25); in fact, cardiac involvement is a main clinical feature (70-80%) of NS and cardiac anomalies are mostly represented by congenital heart diseases (in particular PVS) and HCM (20%) (25). Nearly 50% of NS patients present with some electrocardiographic abnormalities, even in the absence of structural cardiac abnormalities (25).
Emerging evidence shows that the molecular cause underlying the disease has a specific impact on stature, as demonstrated by a more severely impaired growth in patients carrying PTPN11, RAF1, and KRAS pathogenic variants compared to those with SOS1 variants (24).
Although neurocutaneous manifestations have been considered hallmark features in NS and other RASopathies, other organs and systems, including the musculoskeletal system, may be affected (26). Musculoskeletal anomalies include scoliosis, kyphosis, anterior chest wall anomalies, pes planus, osteopenia, and hand anomalies (26, 27). The central nervous system may also be affected by congenital malformations; a few cases of Arnold-Chiari I malformation have been described, though the true incidence in NS is not known (28, 29).
Finally, NS is associated with a higher risk for benign and malignant proliferative disorders, such as juvenile myelomonocytic leukemia (JMML) and/or other hematological malignancies, as well as solid tumors, specifically neuroblastoma, solid brain tumors, and embryonal rhabdomyosarcoma (30–33).
NS diagnostic criteria were originally published by van der Burgt in 2007 (34). They are of particular value in research, and they were the basis for the more recently developed guidelines by Dyscerne (35), which set out recommended baseline investigations and age-specific management of patients. Similar recommendations are provided by Romano et al. (36), Roberts et al. (1) and, after GH treatment approvals in Europe, by Carcavilla et al. (8).
After 2007, when the Food and Drug Administration (FDA) approved recombinant human GH (rhGH) treatment for short stature in NS, the treatment was subsequently approved in Brazil, Israel, Japan, South Korea, Switzerland and some European countries (8, 17). For this reason, many studies conducted in the past evaluating the response and safety of rhGH treatment in NS patients were performed in subjects with concomitant GH deficiency (GHD) who were therefore treated with standard doses for this diagnosis. To date, most studies on rhGH treatment for NS have been retrospective and observational, involving small numbers of patients, with variable ages at the start of treatment and treated with different doses. Unfortunately, molecular characterization is available for only a small number of these cohorts (8, 17).
Here, we review and critically assess data from the literature on the growth pattern characterizing NS, altered GH/IGF-1 axis, and efficacy and safety of rhGH treatment in NS. PubMed and Google scholar tools were used to retrieve relevant publications by using the following terms: Noonan syndrome, PTPN11, KRAS, SOS1, RAF1, NRAS, BRAF, MEK1, RIT1, SOS2, LZTR1, MRAS, CBL, RRAS, RRAS2, RASA2, SPRED1, growth, growth hormone, GH, GH treatment, puberty, scoliosis, cancer, tumor, brain tumor, MRI, Chiari malformation, dysembryoplastic, pilocytic, medulloblastoma, oligodendroglioma, glioneuronal, astrocytoma, glioma, ependymoma,pulmonary stenosis, and hypertrophic cardiopathy. We included reviews, case reports, case series and case report abstracts.
Up to 70% of NS subjects present with postnatal short stature (17, 37), even though the majority has a normal birth weight and length (20); the presence of edema may result in an overestimation of weight which should be carefully evaluated (20). Some authors have reported a significantly higher frequency of NS subjects who are small for gestational age (SGA) compared with the general population (24%), particularly for length (24), and in specific genotypes (24, 38). Attention should be paid to SGA patients whose phenotypic characteristics are suggestive of a RASopathy, and to children who do not respond to GH treatment (39). The concomitant diagnosis of SGA and NS may affect the responsiveness of a child to the growth-promoting effects of rhGH treatment (39). Prematurity has also been reported in a significant proportion of patients and should be taken into account when evaluating NS individuals (24, 40).
Feeding difficulties in NS subjects are extremely frequent (19) and may cause transient failure to thrive and poor weight gain during the neonatal period and infancy in 55 to 100% of cases, depending on the molecular subtype (20, 41).
Birth length is, however, usually normal, although postnatal growth failure is commonly observed starting during the first years of life (20); in fact, mean height tends to follow the third centile from ages two to four years until puberty, when below-average height velocity and an attenuated pubertal growth spurt tend to occur (20).
As bone age is usually delayed, prolonged growth into the 20s may occur, attenuating the growth deficit in some subjects (20). Near adult height approaches the lower limits of normal values: 161-167 cm in males and 150-155 cm in females, with a -1.5 SDS compared to the normal population (20, 21). On the contrary, more than 50% of females and nearly 40% of males have an adult height below the third centile (22).
Growth charts, not genotype-specific, have been developed, although evidence for the occurrence of short stature is represented by pooled data expressed in standard deviation scores (SDS) from patients at different ages, thus precluding a longitudinal description of growth (20–22). Currently available NS-specific growth charts should be used with caution, as they are not genotype-specific and often refer to patients for whom the clinical diagnosis had not been molecularly confirmed.
As reported, in patients with NSSD, the underlying molecular cause of the disease has a specific impact on stature (24). In fact, growth retardation appears to be significantly less severe and less frequent in patients with NSML and NS associated with SOS1 mutations compared to patients with NSSD associated with pathogenic variants in other RASopathy genes, such as patients with PTPN11, RAF1, and KRAS mutations (24).
NS individuals generally present with a normal BMI during the first years of life (24, 42), followed by relative ‘thinness’ with a BMI in the lower normal range; it is rare for NS patients to be overweight or obese, suggesting that NS-causing mutations could impact energy metabolism regulation (36, 43, 44). The greatest decrease in BMI has been associated with pathogenic SHOC2, KRAS and HRAS variants, occurring in NS-LAH, CFCS and CS. These patients present with marked failure to thrive substantially due to muscle tissue wasting rather a decrease in adipose tissue (44). A possible correlation between metabolism and control of energy storage has been hypothesized (45, 46), which may involve two important hormones involved in unsatiety signals (insulin and leptin) and the RAS/MAPK pathway. As proof, patients with CS display an increased resting energy expenditure and a high calorie intake compared with the recommended levels of energy intake (47).
There may be other metabolic alterations in RASopathies. In the NSML mouse model, reduced fat mass and resistance to diet-induced obesity with increased carbohydrate metabolism/insulin sensitivity has been reported (48). On the other hand, the NS mouse model shows an insulin resistant phenotype associated with inflammation of tissues involved in the regulation of glucose metabolism likely due to increased activation of macrophage and triggered monocyte infiltration through SHP2 induced RAS/MAPK hyperactivation (49).
Puberty is often delayed in NS subjects (5), in both females (22%) and males (34%) (50). Those with delayed puberty, frequently resembling a constitutional delay of growth and puberty, are usually shorter and thinner than NS individuals with normal puberty (50). Unfortunately, the molecular available data do not allow us to make accurate genotype-phenotype correlations (50).
As NS is a condition characterized by clinical variability and genetic heterogeneity, different mechanisms implicated with altered GH secretory dynamics or response have been reported, including GHD, neurosecretory dysfunction, and GH insensitivity (GHI) (17, 21). Decreases in IGF-I and IGF-binding protein 3, together with low responses to provocation tests, suggest impaired GH release, or disturbance of the GH/IGF-I axis, at least in a proportion of affected individuals (21).
Recent data suggest that germline activating BRAF mutations may lead to an abnormal differentiation of pituitary hormone-producing cells in the progenitors of the pituitary gland with postnatal hypopituitarism, suggesting a biological role of the MAPK pathway in the etiology of pituitary hormone deficiencies, and a biological link between congenital forms of human hypopituitarism and RASopathies (51). Data on hypopituitarism in NS are poor (52) and there remain many questions about the presence of GHD in subjects with NS.
In NS with growth impairment, the results of provocatory tests should also be taken into account as data indicate that approximately 40% of NS subjects have a GH peak below 10 ng/mL after GHST (53, 54) and some individuals may also present with a range of disturbances in GH secretion, such as low levels of mean overnight GH concentration and unusual GH pulsatility with high trough GH concentration, indicating a concurrent presence of a neurosecretory dysfunction (55). On the other hand, a recent paper studying 24hGH profiles in patients with NS or Turner syndrome (TS), and unaffected prepubertal children showed that GH-baseline, GH mean values, GHmax and other parameters were significantly higher in NS patients, particularly in those with PTPN11 variants, compared to healthy children (56). In fact, a mild GHI, related to a post-receptor signaling defect due to upregulation of SHP2, the protein encoded by PTPN11, has been reported in NS individuals with pathogenic PTPN11 variants (21). SHP2 acts as a negative regulator of the GH receptor signaling pathway and its anomalous activation could be partially compensated via a more elevated GH secretion, as occasionally observed in NS subjects treated with rhGH who showed a mild resistance to rhGH treatment (21). NS individuals carrying PTPN11 variants also have lower levels of insulin-like growth factor1 (IGF-1) than those without a PTPN11 variant (37) although it is not completely clear whether this indicates GHD or GHI in these individuals. We believe that it is important to assess whether sensitivity to GH is normal or decreased before introducing rhGH treatment (57). In a short and/or slowly growing NS child, serum IGF-I levels could be useful in distinguishing between GHD or GHI (57). If serum IGF-I is low (or in the lower half of the reference range) for age, sex, and pubertal stage, or height velocity is reduced, assessment of the endogenous GH reserve by GHST could be informative (58). “Classical” GHD can be excluded in the presence of a normal stimulated GH peak (57). If a diagnosis of GHD is established, we suggest that magnetic resonance imaging (MRI) of the hypothalamic/pituitary region is also carried out (58).
After 2007, when the use of rhGH received approval for treatment of short stature in NS by FDA, many countries (e.g., Brazil, Israel, Japan, South Korea, and some European countries), began to treat NS patients (59–64). In addition to cases treated for documented GHD, rhGH treatment has also been initiated in NS individuals in the presence of markedly short stature, defective function of the GH-IGF-I axis, and/or a documented response to rhGH treatment (59–62). No standard dose has been established, however, based on available data from Phase III clinical trials (65, 66), another long-term interventional study (61) and long-term real world data from the NordiNET IOS and ANSWER international registries (67), the recommended dose is 0.066 mg/kg/day (0.46 mg/kg/week); however, we suggest an initial dose regimen of 0.033 mg/kg/day which may be increased up to 0.066 mg/kg/day in cases of poor response, as reported on the medication label and also stated by other authors (8).
Unfortunately, it is difficult to systematically compare published data on rhGH treatment in NS due to heterogeneous protocols, potential enrolment bias, the small size of the cohorts studied, different cohort selection criteria and varying durations of therapy (Table 1) (18, 54, 85). Most data on response to rhGH treatment derive from uncontrolled observational studies, frequently involving small numbers of patients, with different ages at onset of therapy, different rhGH doses, and varying durations of treatment (18). Noticeably, most individuals treated with rhGH were not genetically characterized, which is a drawback due to the marked clinical variability characterizing the disorder and reported genotype-phenotype correlations.
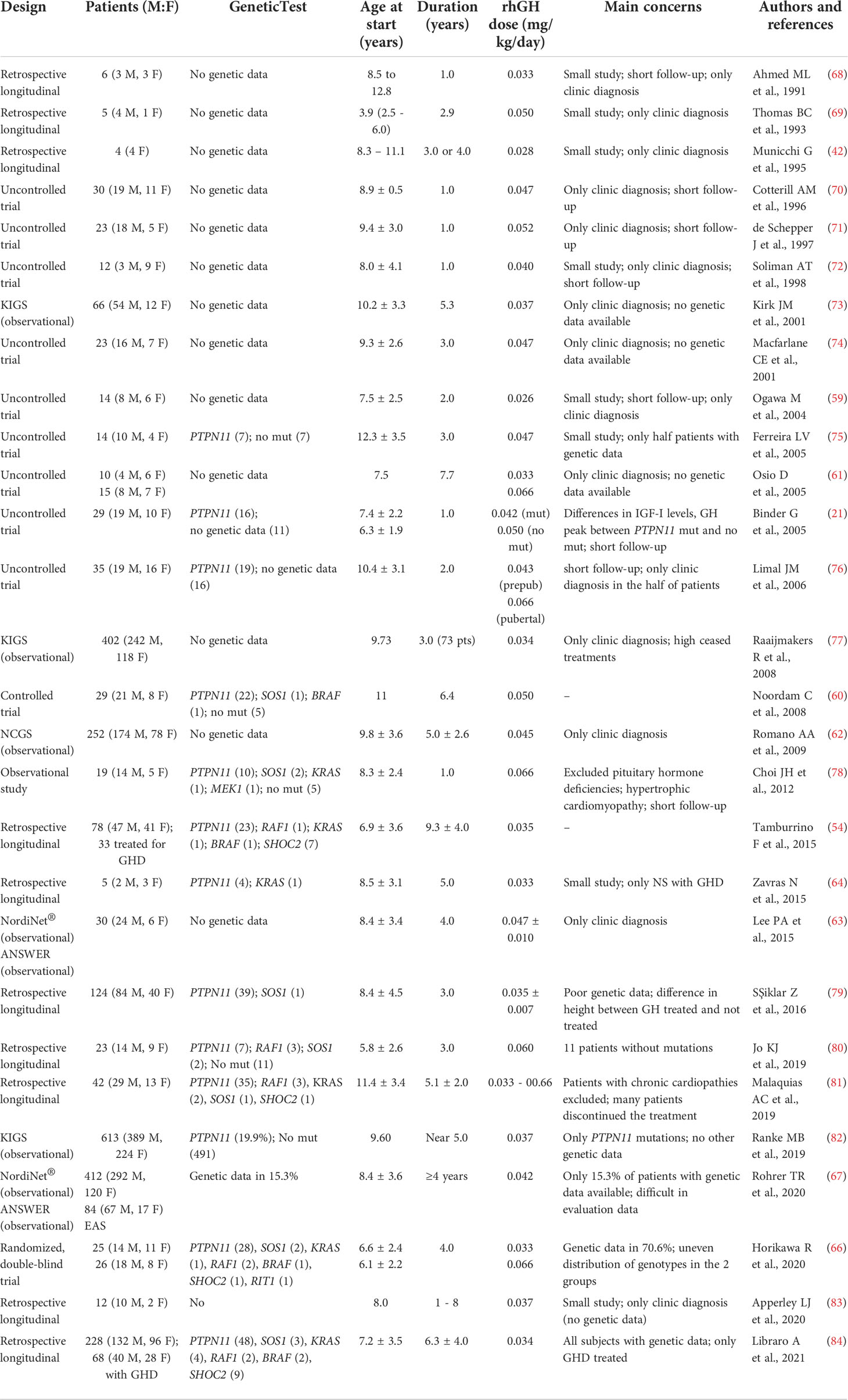
Table 1 Studies assessing the use of rhGH in Noonan syndrome.
Short and long term studies have demonstrated a consistent and significant increase in HV in NS children and adolescents treated with rhGH (57, 59–61, 67, 86). Increases in height SDS vary from 0.6 to 1.8 and may depend on age at start of treatment and duration of treatment, age at onset of puberty and/or GH sensitivity (59, 60, 86).
Many retrospective, observational studies on rhGH treatment in NS patients with and without GHD as well as clinical trials in NS patients with short stature have been carried out over the past 20 years (42, 54, 59, 60, 64, 68–72, 74, 75, 78–80, 83). In some studies, auxological and safety data were consequent to rhGH dosages established on the whole cohort of NS patients with no distinction being made between subjects with GHD and subjects without GHD, while other studies included only NS subjects with GHD (54, 59, 60, 64, 69, 72, 75, 78–80, 83). In other studies, doses of rhGH varied according to different parameters, for example the pubertal stage of patients (21, 61, 66, 76, 81). International Registers (e.g., the Kabi International Growth Study [KIGS] database or the US National Cooperative Growth Study [NCGS]), report data for NS cohorts with and without GHD who were treated with rhGH with different dosages (62, 63, 67, 73, 77, 82).
Some short-term studies have demonstrated an increase in HV and an increase in mean height SDS (63, 68, 70, 71, 74), particularly in the first year of treatment (63, 68, 70, 71, 74), suggesting that short-term use of rhGH for managing short stature is effective in NS. In a study involving 30 patients with a clinical diagnosis of NS without genetic characterization, nearly 80% of patients (aged from 4.5 to 14.0 years and treated with 0.045 mg/kg/day of rhGH for 1 year) showed a significant increase in mean height SDS (-3.01 ± 0.1 to -2.36 ± 0.1) and HV (4.9 ± 0.2 to 8.1 ± 0.4 cm/yr) (70), highlighting the effectiveness of rhGH treatment in both prepubertal and pubertal patients (70, 76, 78). However, other studies indicate that high HV typically and gradually decreases every year after the first year of treatment (74, 78) despite adherence to therapy (63, 74). This waning effect is likely to be due to many co-occurring factors, such as age at the beginning of therapy, GH secretory status, and variable GHI. Indeed, a recent randomized, double-blind, multi-center trial investigating the effect of dose on the growth-promoting effect of rhGH in prepubertal children with NS demonstrated a significant increase in height gain with a dose of 0.066 versus 0.033 mg/kg/day (66). In addition to dose, factors associated with improved outcome include earlier initiation of rhGH therapy and longer prepubertal duration of therapy (61, 62, 82). The presence of genetic heterogeneity in NS also raises the possibility that different responses to rhGH treatment may be genotype-related (78). Unfortunately, genetic data are lacking in the majority of papers. Some data show that prepubertal NS children with GHD present an increased growth rate during rhGH treatment at a rate equivalent to girls with Turner syndrome but at a lower rate than in idiopathic GHD (63, 64). Nonetheless, these studies used very different rhGH dosages (63, 64).
Long-term data on the effect of rhGH treatment on height outcomes are poor and limited to a small number of patients. Data on adult height (AH) or near-adult height (NAH) in NS patients treated with rhGH are also available (42, 54, 61–63, 76, 77, 82, 87, 88), but refer to small cohorts whose results may be biased because of a number of factors, such as age at start of treatment, duration of treatment, and definitions of NAH (60–62, 81, 82).
Patients who were treated with rhGH for more than 3 years (median, 6.4 years) showed an increase in height of 1.3 SDS (26), with the majority of patients achieving an AH within the normal range and 30% remaining below -2 SDS (37, 84). Again, these differences might be due to several confounding factors (37), as well as the dose of rhGH (84). After one year of rhGH treatment, the mean difference between chronological age and bone age decreased (68), and this was particularly evident in subjects having a significantly delayed bone age at the start of rhGH therapy (62), reflecting a normalization rather than excessive acceleration of bone development in these individuals (37).
The bone age of rhGH-treated NS individuals with a significant increase in AH did not excessively advance during rhGH treatment (60, 74). Some data also show that, after rhGH treatment, most NS patients present with significant gains in AH, despite the pubertal growth spurt occurring much later than normal (37).
However, the possible relationship between a “reduced” advancement in bone age with later pubertal development and the effect on final stature is not clear.
Some studies report a more significant increment in height SDS after one to three years of rhGH treatment in patients without PTPN11 mutations (75, 76, 87). However, other data do not confirm these findings (78).
There are contrasting data about the influence of pre-treatment values of IGF-1 and IGFBP-3 on the effectiveness of rhGH treatment. While some studies suggest that basal IGF-I and IGFBP-3 levels before rhGH treatment are significantly related to final response (60), other data indicate that these levels cannot predict changes in height SDS (63, 67). Other studies reported significantly lower IGF-1 and IGFBP-3 levels at the start of rhGH therapy in some NS individuals carrying PTPN11 mutations (37). The choice of reference population is important when interpreting the magnitude of rhGH response. This is clearly illustrated by the differences in mean adjusted ΔHSDS at 5 years (national reference, 1.17, Ranke 1.46), though a similar effectiveness of treatment was observed irrespective of the reference used (88).
In summary, the available data confirm that rhGH treatment is associated with an increase in HSDS in NS individuals during childhood with a final increase in AH. It seems that the earlier rhGH treatment is started, the more likely an optimal height is reached due to height normalization and the delayed pubertal onset frequently observed in NS subjects. However, the scarcity of data on genetically characterized cohorts does not allow us to accurately determine whether response to rhGH treatment also depends on genotype.
Tables 2 and (3A, 3B and 3C) show the results of the major studies on rhGH therapy in NS. Side effects in children were infrequently reported. Based on the data, rhGH treatment does not seem to influence cardiac physiology and function (38). The decision to use rhGH in patients with NS should, however, be made on a case-by-case basis (8). The accumulated safety data on rhGH treatment in NS are reassuring (60) and include no significant evidence of adverse cardiac effects or increased occurrence of malignancies (60–62).
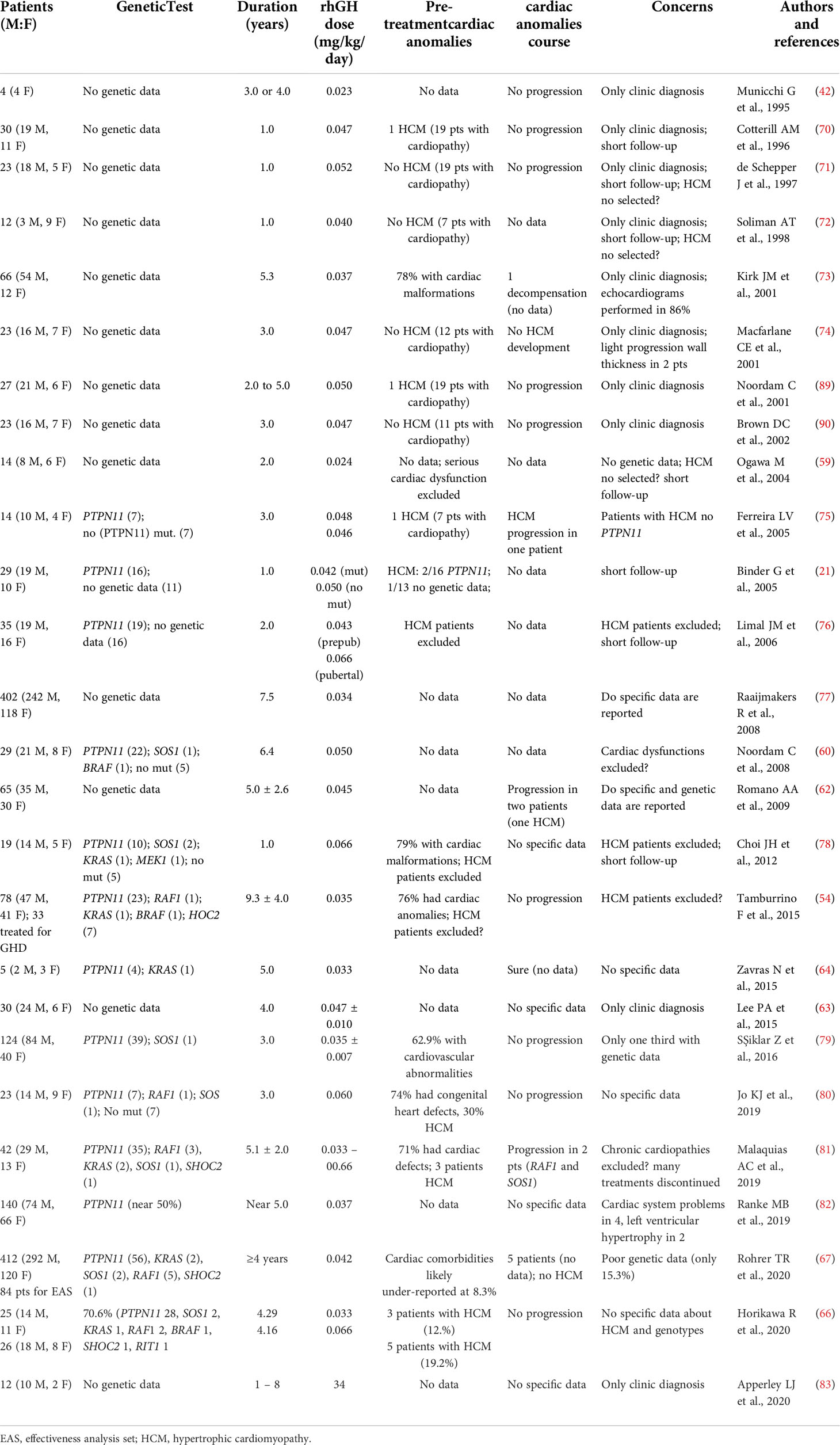
Table 2 Studies evaluating the course of cardiac anomalies during rhGH treatment in rasopathies.
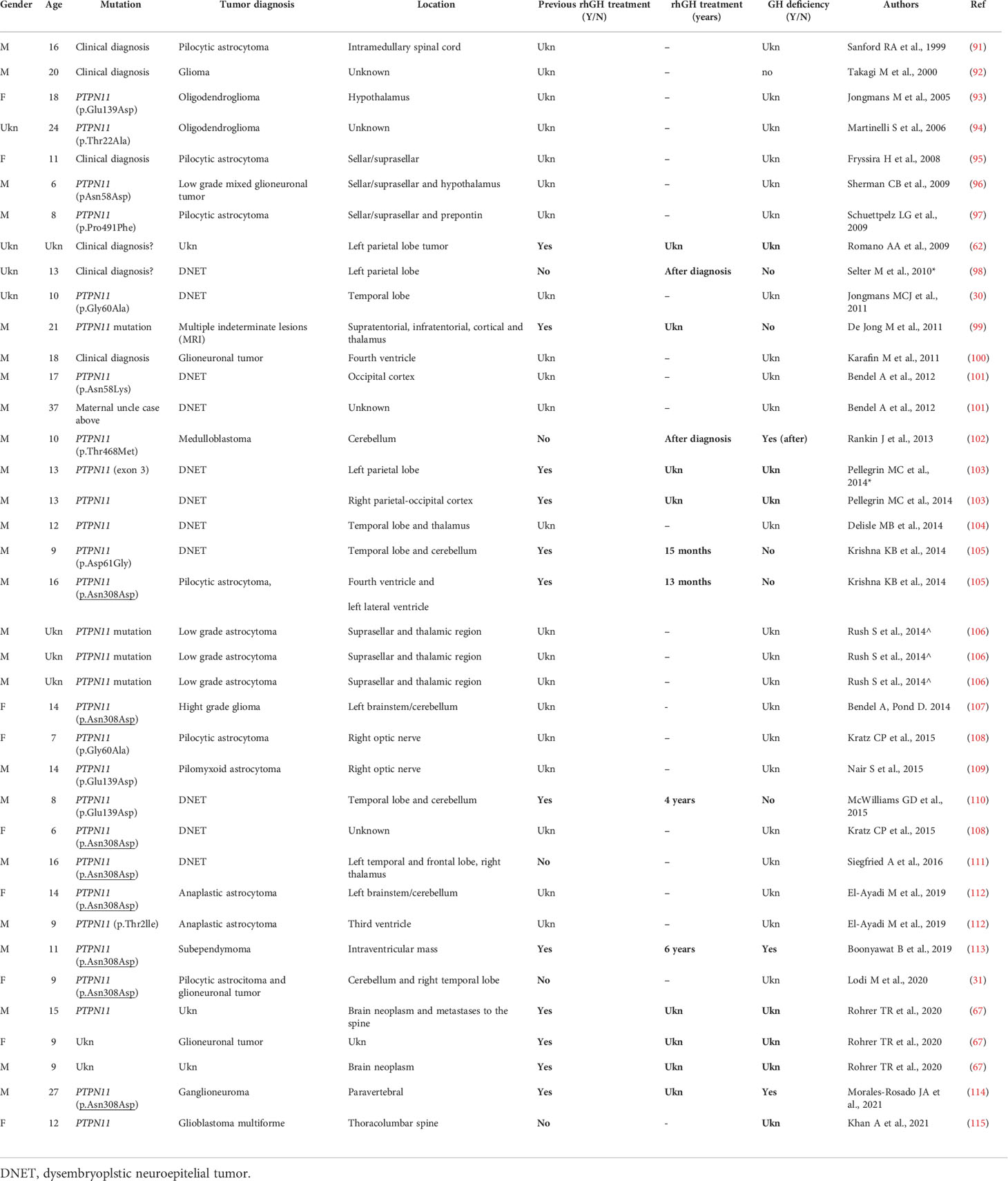
Table 3A Primary brain tumors in Noonan syndrome with PTPN11 mutations.
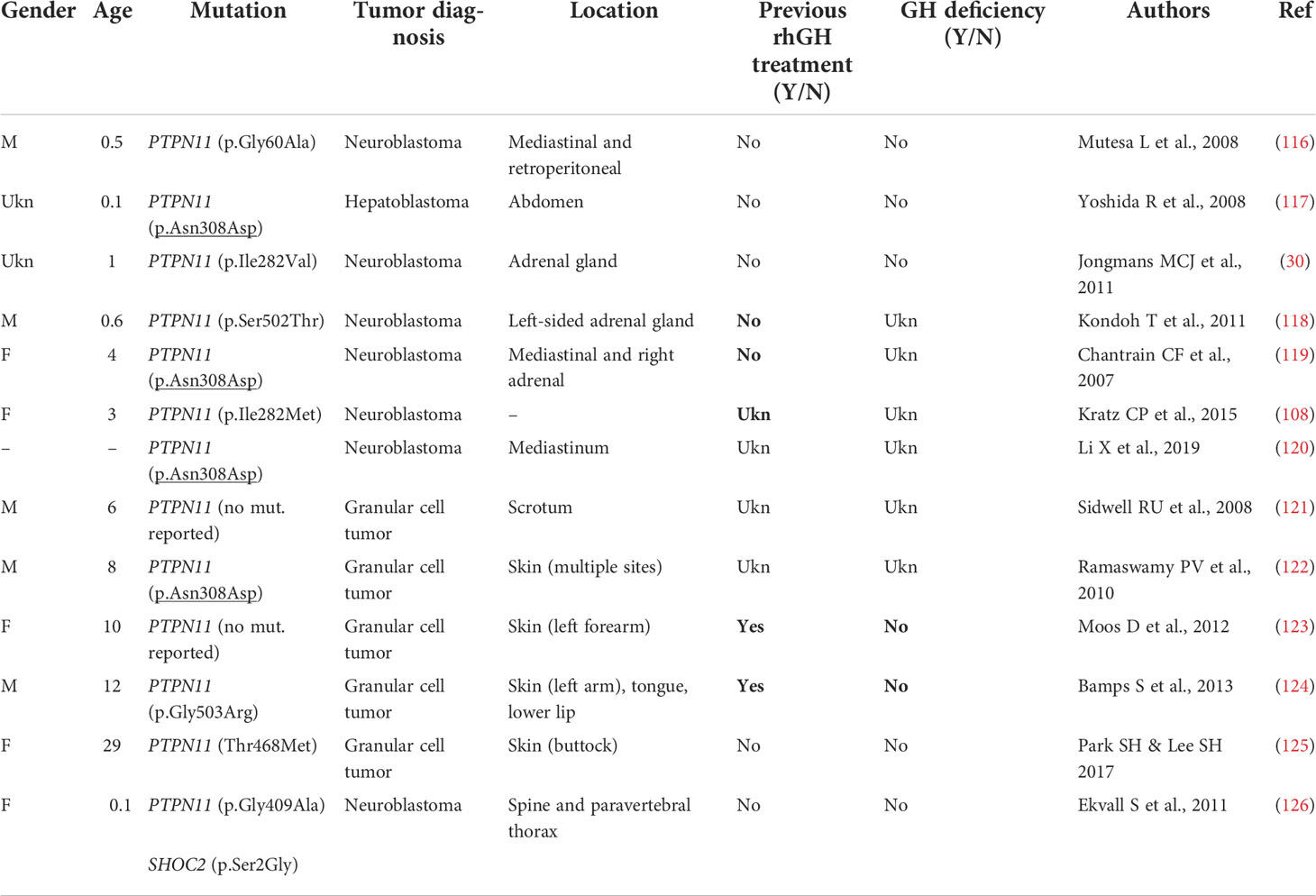
Table 3B Other (no hematological) primary tumors in Noonan syndrome with PTPN11 mutations.
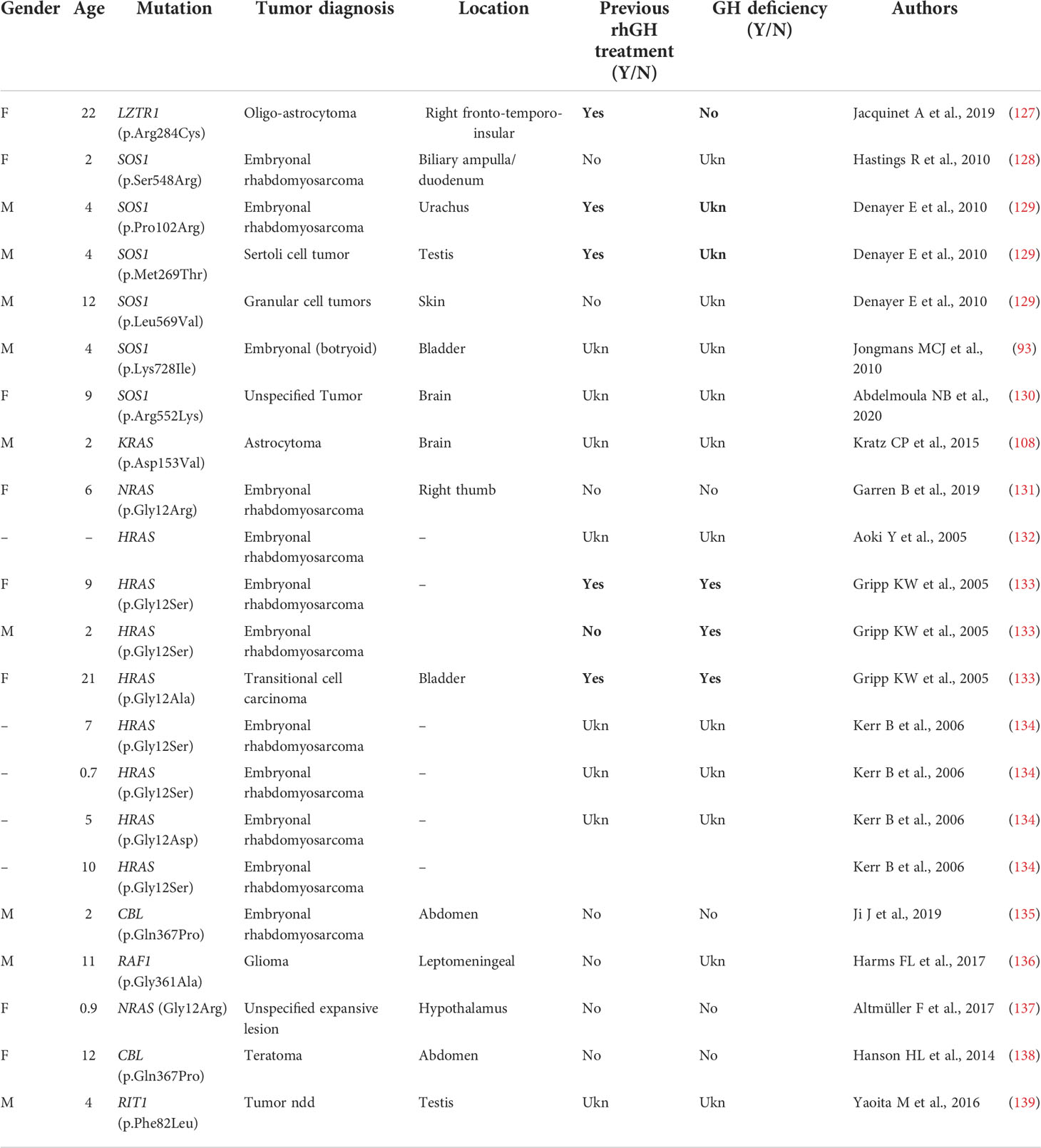
Table 3C Primary brain and other tumors in Noonan syndrome and other rasopathies with no PTPN11 mutations.
We review some of the major concerns and side effects during rhGH treatment in NS individuals.
Several studies show normal blood glucose levels during rhGH treatment (37). Recently, the metabolic impact of SHP2 hyperactivation has been investigated in 21 NS children carrying PTPN11 pathogenic variants. Although they presented with a lower BMI compared to normal weight healthy control subjects, they showed persistent increased glycemia and insulinemia levels after oral glucose tolerance testing (OGTT) (140). Such insulin resistance with reduced adiposity occurs without obvious signs of ectopic lipid deposits or lipotoxicity. This anomalous response is probably induced by a proinflammatory phenotype triggered by SHP2 hyperactivation which may alter hepatic macrophage homeostasis and promote insulin resistance. Larger studies are needed to further confirm this data (140).
Additional factors to be taken into account include height, age (early initiation maximizes prepubertal linear growth) and the presence of comorbidities. Nutrition should be assessed, and energy intake deficits resolved before initiation of treatment, and in cases of clinical features compatible with GHD, evaluation of the somatotropic axis should be considered.
Special attention should be paid to IGF-1 levels, carbohydrate metabolism and other possible adverse events. To the best of our knowledge, no specific studies on GH therapy at different dosages in patients with altered metabolic profiles have been performed.
If the patient exhibits a poor response despite 1-2 years of treatment at high doses, discontinuation of treatment should be considered, as the peak response is expected to occur in the early years of the treatment (8).
Patients with a clinical NS diagnosis without an identified molecular cause in known RASopathy genes should be considered for treatment with extreme caution; in these cases, assessment by an experienced clinical geneticist is recommended, as well as careful monitoring of the patient throughout treatment (8).
Cardiac involvement is one of the main clinical features of NS, occurring in at least 70-80% of individuals. The most common manifestations are congenital heart diseases (in particular PVS, 60-70% of patients) and HCM (nearly 20% of patients) (25). However, a wide spectrum of other abnormalities has been reported, including atrial and/or ventricular septal defects, pulmonary artery branch stenosis, and mitral valve or coronary artery anomalies (25).
Electrocardiographic abnormalities, such as multifocal atrial tachycardia, wide QRS intervals with a predominantly negative pattern in the left precordial leads and left axis deviation with giant Q waves, have also been reported in 50% of NS patients, even in the absence of structural cardiac abnormalities (25).
RAS signaling has a central role in both pathologic and physiologic cardiac hypertrophy as demonstrated in multiple in vitro and in vivo settings (141, 142). Expression of the dominant negative Raf-1 variant in mice has no effect on cardiac function at baseline but promotes cardiomyocyte apoptosis and increases mortality in settings of pressure overload (143).
Nonetheless, many issues, including the specific pathways activated by RAS GTPases which eventually lead to cardiac hyperplasia or hypertrophy, have not yet been elucidated (144). Among the RASopathies, the frequency of HCM is strictly correlated with the genotype, being particularly frequent in NS patients with RAF1, LZTR1, RIT1 and MRAS, NSML (narrow spectrum of PTPN11 mutations), and CS (narrow spectrum of HRAS mutations) (145–151).
Despite the good safety profile of rhGH treatment in NS patients presenting with HCM, a few adverse effects have been reported (Table 2). In a retrospective analysis (152), one case of HCM and one case of worsening HCM were reported (81, 152), but the genotype of the affected individuals is not known.
A progression of HCM associated with rhGH treatment has been reported in CS patients; nearly 20% of patients presented an increased severity of HCM during rhGH treatment (153). There is evidence of mild progression of PVS in NS individuals, but this does not appear to be related to rhGH treatment (60) (Table 2).
Two prospective studies specifically designed to evaluate cardiac anatomy and function after 1 and 4 years of rhGH therapy at different dosages, did not identify any change in myocardial function, or in ventricular wall thickness. Unfortunately, no genotype information on the enrolled patients were available (74, 89, 90).
An electrophysiologic phenotype has been also described in NSSD, with an increased incidence of multifocal atrial tachycardia and ectopic atrial tachycardia that occurs independently of HCM or PVS in 36% of patients (154–156). Calcium dysregulation may result in triggered activity giving rise to the atrial tachycardia, as well as contributing to the cardiomyopathy phenotype (154).
Unrelated arrhythmias have been described during rhGH treatment in NS (146, 147) some of which were evident also after interruption of the treatment (65, 157). This finding may be not strictly related to NS.
In conclusion, rhGH should be introduced only after a thorough cardiologic evaluation, particularly in patients carrying variants of specific genes (see previous paragraphs). NS patients undergoing rhGH therapy should always be closely monitored. At the first signs of HCM, discontinuing rhGH should be considered (150, 153). An assessment of the relative risks and benefits of rhGH treatment should be made for individual patients. Unfortunately, genotype-phenotype correlations are lacking from the vast majority of studies; we stress the need for collecting more complete data and longer follow-up
Dysregulation of the RAS/MAPK signaling pathway may increase risks for cancer and contribute to oncogenesis (9, 30, 31, 158, 159). NS is associated with a higher risk for benign and malignant proliferative disorders, such as juvenile myelomonocytic leukemia (JMML) and other hematological malignancies, as well as solid tumors, specifically neuroblastoma, brain tumors, and embryonal rhabdomyosarcoma (30, 31, 105, 108, 110, 112, 127, 158–165). JMML is occasionally observed in NS carrying specific SHP2 pathogenic missense substitutions, (e.g., Y62D and T73I). In these patients, JMML often presents with a benign course which commonly regresses spontaneously even though a severe course has also been described (160). Transient benign myeloproliferative disorder (MPD) is also estimated to occur in up to 10% of all NS children. This disorder generally resolves spontaneously over months or years, although an estimated 10% of cases of NS/MPD may progress to JMML. Thus, MPD in NS should be closely monitored. Nearly all patients with NS and MPD carry mutations in PTPN11 (162). The gain-of-function effect of these mutations is predicted to be intermediate, between that for NS without MPD (milder gain of function) and somatic mutations in JMML (stronger gain of function).
“Benign” proliferative conditions include multiple giant cell lesions (MGCL) and granular cell tumors (30). To date, few NS individuals with MGCL and mutations in PTPN11 or SOS1 have been identified (32), whereas MGCL is more frequently reported in patients with other RASopathies, such as in CFCS subjects carrying pathogenic variants of BRAF and MEK1 (33).
In a cohort of 297 individuals carrying pathogenic PTPN11 variants, cancer risk was estimated as 3.5-fold higher than in the general population. When considering all the RASopathies, (in a cohort of 632 individuals with molecularly defined NSSD), a 8.0-fold higher risk than controls has been found, although for CS a 42.4-fold increased risk was present (22, 31, 159). These data, again, stress the importance of genetic characterization in individuals affected with NS and other RASopathies.
Dedicated guidelines for cancer surveillance in patients with NS have yet to be developed (166). Tables 3A, 3B and 3C show the correlation between genotype and oncological risk in NS, which should be taken into consideration before and during rhGH treatment as different radiological follow-up is likely to be necessary for the various genotypes.
The more common NS-associated solid and soft tissue tumors include glioneuronal tumor and astrocytoma (33, 110, 112). While specific associations between a subset of PTPN11 variants and pediatric hematological malignancies have been reported, the apparently higher incidence of PTPN11 variants in NS individuals with tumors is likely to reflect the higher frequency of variants in this gene with respect to other genes. A significantly higher cancer risk is observed in CS, with typical association with bladder cancer and embryonal rhabdomyosarcoma.
The available data on rhGH and cancer in NS are reassuring (67), but underlying susceptibility to tumor growth should be considered when rhGH therapy is started (105, 127). Follow-up must be based on clinical symptoms, regular physical examinations and complete blood counts. Recent recommendations advise obtaining a brain MRI prior to initiating rhGH treatment in patients with NS, particularly in those with PTPN11 mutations, as they appear to have a slightly increased risk for cranial neoplasms (105, 127, 166, 167).
The paucity of data on the long-term safety of rhGH therapy in patients with NS, especially regarding the risk of tumor development and tumor recurrence does not allow us to report a definitive consensus.
Data on IGF-I variations during rhGH treatment and variations in neoplastic risk in the medium and long term are largely lacking because rhGH treatment in non GHD patients was only introduced in 2007 after FDA approval.
Genotype characterization appears to be important in understanding the neoplastic risk for NS patients, and we stress the need to gather more data on different rhGH doses and differences between GHD Noonan and non-GHD Noonan.
Besides short stature, skeletal findings in NS include kyphosis, lordosis, scoliosis, anterior chest wall anomalies and hand anomalies, such as syndactyly, brachydactyly, and cubitus valgus (168, 169). Chest wall anomalies are also extremely frequent, mostly represented by a superior pectus carinatum with an inferior pectus excavatum. Osteopenia/osteoporosis has not been frequently reported but was observed in a 2/26 individuals in one cohort (170).
Although an increased risk for development or progression of scoliosis is not apparent in NS patients treated with rhGH, this feature has not systematically been studied and continued surveillance is necessary (67). Romano et al. (62) reported 6 cases of scoliosis in 370 patients over 5.6 years of rhGH treatment, whereas Kirk et al. (73) reported 1 case of worsening kyphoscoliosis among 66 patients who were treated with rhGH for up to 6 years. In an observational study including a large number of NS subjects treated with rhGH (only 15% with a genetic diagnosis), three patients presented with scoliosis and three experienced arthralgia episodes. One patient needed spinal fusion surgery at 16.5 years of age, whereas the other 2 cases were considered non-serious and possibly unrelated to rhGH treatment; none of these patients had a diagnosis of scoliosis before rhGH treatment started (67). On the other hand, the condition of one patient affected with scoliosis prior to starting rhGH treatment, did not worsen during the treatment (67).
The data suggest that scoliosis-related outcomes are better than for TS during rhGH treatment (171). For example, out of 49 girls with Turner syndrome followed by Ricotti et al. (172), 29 exhibited scoliosis at baseline, and 9 additional individuals developed minor scoliosis during the 4-year follow-up, suggesting that these problems may be related to a worsening of pre-existing scoliosis (172).
Arnold-Chiari malformation is commonly seen in RASopathies, and several cases have been reported in the medical literature in NS patients although the incidence is not known (28, 173). Arnold-Chiari is also observed in other medical conditions, including GHD (5–20%) (174), due to the underdevelopment of certain cranial bone structures.
NS is a highly heterogeneous disorder, with variable clinical features and genetic complexity (1, 6–8, 13, 14), which must be taken into consideration when evaluating rhGH treatment in these patients. In addition to auxological and safety data, genetic data, often lacking, is essential for identifying patients at risk for specific side effects and complications during rhGH treatment. In many papers, diagnosis of NS is based only on clinical assessment (75), which makes it difficult to analyze the results and side effects for specific genotypes.
The presence of varying degrees of GHI in subjects with NS should not prevent clinicians from evaluating rhGH treatment, which should be individualized. Given the emerging data about neoplasms in NS patients, we recommend a conservative approach and judicious surveillance (8).
As in the general population, it is important to rule out GHD. The correct starting standard dose of rhGH has not been established in NS and high doses of rhGH are not recommended (175). In non-NS patients, some data suggest an increased risk for cardiovascular events (176) and increased incidence of secondary tumors (177) in children with a primary tumor who had been treated with rhGH during childhood and adolescence. Other authors have not found a significant increase in overall mortality in low-risk patients, such as those with isolated GHD (177). Evaluation of the GH-IGF-I axis could help determine the most appropriate starting dosage of rhGH. Even though most studies have not shown an increase in the incidence of neoplasms in NS patients treated with rhGH, there are no long-term studies specifically designed to address this issue. Since malignancies for patients with NS tend to involve multiple sites and develop throughout life, a routine tumor surveillance program should be implemented. In line with the authors of previously published studies (105, 110), we recommend that when rhGH therapy is initiated in NS patients, the possibility of performing a brain MRI is considered, particularly in subjects with PTPN11 mutations who appear to have higher risk for CNS neoplasms.
Published data do not show any changes in myocardial function, or in ventricular wall thickness during rhGH treatment in NS patients but the lack of genetic data means that a definite conclusion cannot be reached. In children with a diagnosis of HCM a cautious approach and careful follow-up are necessary.
In conclusion, NS is a genetic disorder with substantial clinical variability, which in part is associated with the specific genes and mutations involved. Given the genetic and clinical complexity of the disorder and high prevalence of cardiac defects and malignancies, NS requires a multidisciplinary approach and follow-up.
The overall experience with rhGH treatment in most NS patients with short stature is reassuring; the data reveal few serious adverse effects. Therapy with rhGH increases HV in patients with NS, but firm conclusions regarding the effects of this therapy on near adult height and long-term health are not available. A better understanding of the causes of short stature, as well as response to rhGH treatment in NS, is needed and must be based on genetic characterization. Reliable large-scale and case-control studies are crucial in elucidating the long-term effects of rhGH treatment and defining the examinations necessary prior to treatment and in follow-up. Until we have more complete data, an individualized follow-up and close monitoring, also related to the cardiac, neoplastic and orthopedic risks, during rhGH treatment should be considered.
Silvia Favilli, Giovanni Battista Calabri, Giulio Porcedda, Gaia Spaziani, Luciano De Simone, Elena Andreucci, Giovanna Traficante, Giorgia Mancano, Angelica Pagliazzi, Sara Bargiacchi, Giulia Gori.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
MT and SS have received speakers’ bureau honoraria from Novo Nordisk. The other authors do not have any financial or non-financial competing interests in relation to this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet (2013) 381:333–42. doi: 10.1016/S0140-6736(12)61023-X
2. Johnston JJ, van der Smagt JJ, Rosenfeld JA, Pagnamenta AT, Alswaid A, Baker EH, et al. Autosomal recessive noonan syndrome associated with biallelic LZTR1 variants. Genet Med (2018) 20:1175–85. doi: 10.1038/gim.2017.249
3. Motta M, Fasano G, Gredy S, Brinkmann J, Bonnard AA, Simsek-Kiper PO, et al. SPRED2 loss-of-function causes a recessive noonan syndrome-like phenotype. Am J Hum Genet (2021) 108:2112–29. doi: 10.1016/j.ajhg.2021.09.007
4. Noonan JA. Hypertelorism with turner phenotype. a new syndrome with associated congenital heart disease. Am J Dis Child (1968) 116:373–80. doi: 10.1001/archpedi.1968.02100020377005
5. Noonan JA. Noonan syndrome and related disorders: alterations in growth and puberty. Rev Endocr Metab Disord (2006) 7:251–5. doi: 10.1007/s11154-006-9021-1
6. Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab (2011) 25:161–79. doi: 10.1016/j.beem.2010.09.002
7. Giugliano T, Santoro C, Torella A, Del Vecchio Blanco F, Grandone A, Onore ME, et al. Clinical and genetic findings in children with neurofibromatosis type 1, legius syndrome, and other related neurocutaneous disorders. Genes (Basel) (2019) 10:580. doi: 10.3390/genes10080580
8. Carcavilla A, Suárez-Ortega L, Rodríguez Sánchez A, Gonzalez-Casado I, Ramón-Krauel M, Labarta JI, et al. [Noonan syndrome: genetic and clinical update and treatment options]. Pediatr (2020) 93:61.e1–61.e14. doi: 10.1016/j.anpedi.2020.04.008
9. Riller Q, Rieux-Laucat F. RASopathies: from germline mutations to somatic and multigenic diseases. BioMed J (2021) 44:422–32. doi: 10.1016/j.bj.2021.06.004. S2319-4170(21)00070-6.
10. Pudewell S, Wittich C, Kazemein Jasemi NS, Bazgir F, Ahmadian MR. Accessory proteins of the RAS-MAPK pathway: moving from the side line to the front line. Commun Biol (2021) 4:696. doi: 10.1038/s42003-021-02149-3
11. Martinelli S, De Luca A, Stellacci E, Rossi C, Checquolo S, Lepri F, et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a noonan syndrome-like phenotype. Am J Hum Genet (2010) 87:250–7. doi: 10.1016/j.ajhg.2010.06.015
12. Motta M, Pannone L, Pantaleoni F, Bocchinfuso G, Radio FC, Cecchetti S, et al. Enhanced MAPK1 function causes a neurodevelopmental disorder within the RASopathy clinical spectrum. Am J Hum Genet (2020) 107:499–513. doi: 10.1016/j.ajhg.2020.06.018
13. Tartaglia M, Gelb BD. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann N Y Acad Sci (2010) 1214:99–121. doi: 10.1111/j.1749-6632.2010.05790.x
14. Tajan M, Paccoud R, Branka S, Edouard T, Yart A. The RASopathy family: Consequences of germline activation of the RAS/MAPK pathway. Endocr Rev (2018) 39:676–700. doi: 10.1210/er.2017-00232
15. Motta M, Fidan M, Bellacchio E, Pantaleoni F, Schneider-Heieck K, Coppola S, et al. Dominant noonan syndrome-causing LZTR1 mutations specifically affect the kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum Mol Genet (2019) 28:1007–22. doi: 10.1093/hmg/ddy412
16. Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, et al. Revised diagnostic criteria for neurofibromatosis type 1 and legius syndrome: an international consensus recommendation. Genet Med (2021) 23:1506–13. doi: 10.1038/s41436-021-01170-5
17. Shin YL. Correlation between genetic heterogeneity and variability for response to growth hormone in noonan syndrome. Korean J Pediatr (2019) 62:412–3. doi: 10.3345/kjp.2019.00220
18. Rodríguez F, Gaete X, Cassorla F. Etiology and treatment of growth delay in noonan syndrome. Front Endocrinol (Lausanne) (2021) 12:691240. doi: 10.3389/fendo.2021.691240
19. Tiemens DK, van Haaften L, Leenders E, van Wegberg AMJ, Gunther Moor B, Geelen J, et al. Feeding problems in patients with noonan syndrome: A narrative review. J Clin Med (2022) 11(3):754. doi: 10.3390/jcm11030754
20. Otten BJ, Noordam C. Growth in noonan syndrome. Horm Res (2009) 72 Suppl 2:31–5. doi: 10.1159/000243776
21. Binder G, Neuer K, Ranke MB, Wittekindt NE. PTPN11 mutations are associated with mild growth hormone resistance in individuals with noonan syndrome. J Clin Endocrinol Metab (2005) 90:5377–81. doi: 10.1210/jc.2005-0995
22. Noonan JA, Raaijmakers R, Hall BD. Adult height in noonan syndrome. Am J Med Genet A (2003) 123A:68–71. doi: 10.1002/ajmg.a.20502
23. Croonen EA, Draaisma JMT, van der Burgt I, Roeleveld N, Noordam C. First-year growth in children with noonan syndrome: Associated with feeding problems? Am J Med Genet A (2018) 176(4):951–8. doi: 10.1002/ajmg.a.38649
24. Cessans C, Ehlinger V, Arnaud C, Yart A, Capri Y, Barat P, et al. Growth patterns of patients with noonan syndrome: correlation with age and genotype. Eur J Endocrinol (2016) 174(5):641–50. doi: 10.1530/EJE-15-0922
25. Gelb BD, Roberts AE, Tartaglia M. Cardiomyopathies in noonan syndrome and the other RASopathies. Prog Pediatr Cardiol (2015) 39:13–9. doi: 10.1016/j.ppedcard.2015.01.002
26. Allanson JE, Roberts AE. Noonan syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2001). p. 1993–2021.
27. Stagi S, Iurato C, Lapi E, Cavalli L, Brandi ML, de Martino M. Bone status in genetic syndromes: a review. Hormones (Athens) (2015) 14:19–31. doi: 10.1007/BF03401378
28. Ejarque I, Millán-Salvador JM, Oltra S, Pesudo-Martínez JV, Beneyto M, Pérez-Aytés A. Malformacion de Arnold-chiari en el sindrome de noonan y otros sindromes de la via RAS/MAPK [Arnold-chiari malformation in noonan syndrome and other syndromes of the RAS/MAPK pathway]. Rev Neurol (2015) 60:408–12.
29. Keh YS, Abernethy L, Pettorini B. Association between noonan syndrome and chiari I malformation: a case-based update. Childs Nerv Syst (2013) 29(5):749–52. doi: 10.1007/s00381-012-2000-9
30. Jongmans MC, van der Burgt I, Hoogerbrugge PM, Noordam K, Yntema HG, Nillesen WM, et al. Cancer risk in patients with noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet (2011) 19:870–4. doi: 10.1038/ejhg.2011.37
31. Lodi M, Boccuto L, Carai A, Cacchione A, Miele E, Colafati GS, et al. Low-grade gliomas in patients with noonan syndrome: Case-based review of the literature. Diagnostics (Basel) (2020) 10:582. doi: 10.3390/diagnostics10080582
32. Roberts AE. Noonan syndrome. In:Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA: University of Washington, Seattle (2001). p. 1993–2022.
33. Neumann TE, Allanson J, Kavamura I, Kerr B, Neri G, Noonan J, et al. Multiple giant cell lesions in patients with noonan syndrome and cardio-facio-cutaneous syndrome. Eur J Hum Genet (2009) 17(4):420–5. doi: 10.1038/ejhg.2008.188
35. DYSCERNE. Noonan syndrome guideline development group. 2010. management of noonan syndrome. a clinical guideline. Available at: https://rasopathiesnet.org (Accessed 15 December 2019).
36. Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics (2010) 126:746–59. doi: 10.1542/peds.2009-3207
37. Seo GH, Yoo HW. Growth hormone therapy in patients with noonan syndrome. Ann Pediatr Endocrinol Metab (2018) 23:176–81. doi: 10.6065/apem.2018.23.4.176
38. Seok EM, Park HK, Rho JG, Kum CD, Lee HS, Hwang JS. Effectiveness of growth hormone therapy in children with noonan syndrome. Ann Pediatr Endocrinol Metab (2020) 25(3):182–6. doi: 10.6065/apem.1938154.077
39. Olivieri DJ, Massingham LJ, Schwab JL, Quintos JB. Lack of catch-up growth with growth hormone treatment in a child born small for gestational age leading to a diagnosis of noonan syndrome with a pathogenic PTPN11 variant. Case Rep Endocrinol (2021) 2021:5571524. doi: 10.1155/2021/5571524
40. Malaquias AC, Brasil AS, Pereira AC, Arnhold IJ, Mendonca BB, Bertola DR, et al. Growth standards of patients with noonan and noonan-like syndromes with mutations in the RAS/MAPK pathway. Am J Med Genet A (2012) 158A(11):2700–6. doi: 10.1002/ajmg.a.35519
41. Digilio MC, Lepri F, Baban A, Dentici ML, Versacci P, Capolino R, et al. RASopathies: Clinical diagnosis in the first year of life. Mol Syndromol (2011) 1(6):282–9. doi: 10.1159/000331266
42. Municchi G, Pasquino AM, Pucarelli I, Cianfarani S, Passeri F. Growth hormone treatment in noonan syndrome: report of four cases who reached final height. Horm Res (1995) 44(4):164–7. doi: 10.1159/000184618
43. Tiemens D, Wegberg AV, Druten DV, Draaisma J. High energy expenditure in a patient with feeding problems and noonan syndrome spectrum disorder. BMJ Case Rep (2022) 15(3):e247513. doi: 10.1136/bcr-2021-247513
44. da Silva FM, Jorge AA, Malaquias A, da Costa Pereira A, Yamamoto GL, Kim CA, et al. Nutritional aspects of noonan syndrome and noonan-related disorders. Am J Med Genet A (2016) 170(6):1525–31. doi: 10.1002/ajmg.a.37639
45. Carpentieri G, Leoni C, Pietraforte D, Cecchetti S, Iorio E, Belardo A, et al. Hyperactive HRAS dysregulates energetic metabolism in fibroblasts from patients with Costello syndrome via enhanced production of reactive oxidizing species. Hum Mol Genet (2022) 31(4):561–75. doi: 10.1093/hmg/ddab270
46. Leoni C, Massese M, Gervasoni J, Primiano A, Giorgio V, Onesimo R, et al. Metabolic profiling of Costello syndrome: Insights from a single-center cohort. Eur J Med Genet (2022) 65(3):104439. doi: 10.1016/j.ejmg.2022.104439
47. Leoni C, Onesimo R, Giorgio V, Diamanti A, Giorgio D, Martini L, et al. Understanding growth failure in Costello syndrome: Increased resting energy expenditure. J Pediatr (2016) 170:322–4. doi: 10.1016/j.jpeds.2015.11.076
48. Yart A, Edouard T. Noonan syndrome: an update on growth and development. Curr Opin Endocrinol Diabetes Obes (2018) 25(1):67–73. doi: 10.1097/MED.0000000000000380
49. Tajan M, Batut A, Cadoudal T, Deleruyelle S, Le Gonidec S, Saint Laurent C, et al. LEOPARD syndrome-associated SHP2 mutation confers leanness and protection from diet-induced obesity. Proc Natl Acad Sci USA (2014) 111(42):E4494–503. doi: 10.1073/pnas.1406107111
50. Malaquias AC, Noronha RM, Homma TK, Albuquerque EVA, Bertola DR, Jorge AAL. Evaluation of puberty in patients with noonan syndrome and mutations in the RAS/MAPK genes, in: 58th Annual ESPE Meeting, Horm Res Paediatr (2019) 91:91–92
51. Gualtieri A, Kyprianou N, Gregory LC, Vignola ML, Nicholson JG, Tan R, et al. Activating mutations in BRAF disrupt the hypothalamo-pituitary axis leading to hypopituitarism in mice and humans. Nat Commun (2021) 12(1):2028. doi: 10.1038/s41467-021-21712-4
52. Couser NL, Keelean-Fuller D, Davenport ML, Haverfield E, Masood MM, Henin M, et al. Cleft palate and hypopituitarism in a patient with noonan-like syndrome with loose anagen hair-1. Am J Med Genet A (2018) 176(9):2024–7. doi: 10.1002/ajmg.a.40432
53. Romano AA, Blethen SL, Dana K, Noto RA. Growth hormone treatment in noonan syndrome: the national cooperative growth study experience. J Pediatr (1996) 128(5 Pt 2):S18–21. doi: 10.1016/s0022-3476(96)70005-7
54. Tamburrino F, Gibertoni D, Rossi C, Scarano E, Perri A, Montanari F, et al. Response to long-term growth hormone therapy in patients affected by RASopathies and growth hormone deficiency: Patterns of growth, puberty and final height data. Am J Med Genet A (2015) 167A(11):2786–94. doi: 10.1002/ajmg.a.37260.
55. Noordam C, van der Burgt I, Sweep CG, Delemarre-van de Waal HA, Sengers RC, Otten BJ. Growth hormone (GH) secretion in children with noonan syndrome: frequently abnormal without consequences for growth or response to GH treatment. Clin Endocrinol (Oxf) (2001) 54:53–9. doi: 10.1046/j.1365-2265.2001.01188.x
56. Dahlgren J, Albertsson-Wikland K. GH responsiveness in children with noonan syndrome compared to turner syndrome. Front Endocrinol (Lausanne) (2021) 12:737893. doi: 10.3389/fendo.2021.737893
57. Wit JM, Joustra SD, Losekoot M, van Duyvenvoorde HA, de Bruin C. Differential diagnosis of the short IGF-I-Deficient child with apparently normal growth hormone secretion. Horm Res Paediatr (2021) 94:81–104. doi: 10.1159/000516407
58. Collett-Solberg PF, Ambler G, Backeljauw PF, Bidlingmaier M, Biller BMK, Boguszewski MCS, et al. Diagnosis, genetics, and therapy of short stature in children: A growth hormone research society international perspective. Horm Res Paediatr (2019) 92:1–14. doi: 10.1159/000502231
59. Ogawa M, Moriya N, Ikeda H, Tanae A, Tanaka T, Ohyama K, et al. Clinical evaluation of recombinant human growth hormone in noonan syndrome. Endocr J (2004) 51:61–8. doi: 10.1507/endocrj.51.61
60. Noordam C, Peer PG, Francois I, De Schepper J, van den Burgt I, Otten BJ. Long-term GH treatment improves adult height in children with noonan syndrome with and without mutations in protein tyrosine phosphatase, non-receptor-type 11. Eur J Endocrinol (2008) 159:203–8. doi: 10.1530/EJE-08-0413
61. Osio D, Dahlgren J, Wikland KA, Westphal O. Improved final height with long-term growth hormone treatment in noonan syndrome. Acta Paediatr (2005) 94:1232–7. doi: 10.1111/j.1651-2227.2005.tb02081.x
62. Romano AA, Dana K, Bakker B, Davis DA, Hunold JJ, Jacobs J, et al. Growth response, near-adult height, and patterns of growth and puberty in patients with noonan syndrome treated with growth hormone. J Clin Endocrinol Metab (2009) 94:2338–44. doi: 10.1210/jc.2008-2094
63. Lee PA, Ross J, Germak JA, Gut R. Effect of 4 years of growth hormone therapy in children with noonan syndrome in the American norditropin studies: Web-enabled research (ANSWER) program registry. Int J Pediatr Endocrinol (2012) 2012:15. doi: 10.1186/1687-9856-2012-15
64. Zavras N, Meazza C, Pilotta A, Gertosio C, Pagani S, Tinelli C, et al. Five-year response to growth hormone in children with noonan syndrome and growth hormone deficiency. Ital J Pediatr (2015) 41:71. doi: 10.1186/s13052-015-0183-x
65. Ozono K, Ogata T, Horikawa R, Matsubara Y, Ogawa Y, Nishijima K, et al. Efficacy and safety of two doses of norditropin® (somatropin) in short stature due to noonan syndrome: A 2-year randomized, double-blind, multicenter trial in Japanese patients. Endocr J (2018) 65(2):159–74. doi: 10.1507/endocrj.EJ17-0313
66. Horikawa R, Ogata T, Matsubara Y, Yokoya S, Ogawa Y, Nishijima K, et al. Long-term efficacy and safety of two doses of norditropin® (somatropin) in noonan syndrome: A 4-year randomized, double-blind, multicenter trial in Japanese patients. Endocr J (2020) 67(8):803–18. doi: 10.1507/endocrj.EJ19-0371
67. Rohrer TR, Abuzzahab J, Backeljauw P, Birkegård AC, Blair J, Dahlgren J, et al. Long-term effectiveness and safety of childhood growth hormone treatment in noonan syndrome. Horm Res Paediatr (2020) 93:380–95. doi: 10.1159/000512429
68. Ahmed ML, Foot AB, Edge JA, Lamkin VA, Savage MO, Dunger DB. Noonan's syndrome: Abnormalities of the growth hormone/IGF-I axis and the response to treatment with human biosynthetic growth hormone. Acta Paediatr Scand (1991) 80(4):446–50. doi: 10.1111/j.1651-2227.1991.tb11880.x
69. Thomas BC, Stanhope R. Long-term treatment with growth hormone in noonan's syndrome. Acta Paediatr (1993) 82(10):853–5. doi: 10.1111/j.1651-2227.1993.tb17626.x
70. Cotterill AM, McKenna WJ, Brady AF, Sharland M, Elsawi M, Yamada M, et al. The short-term effects of growth hormone therapy on height velocity and cardiac ventricular wall thickness in children with noonan's syndrome. J Clin Endocrinol Metab (1996) 81(6):2291–7. doi: 10.1210/jcem.81.6.8964866
71. De Schepper J, Otten BJ, François I, Bourguignon JP, Craen M, van der Burgt I, et al. Growth hormone therapy in pre-pubertal children with noonan syndrome: first year growth response and comparison with turner syndrome. Acta Paediatr (1997) 86(9):943–6. doi: 10.1111/j.1651-2227.1997.tb15175.x
72. Soliman AT, Rajab A, el Zalabany M, alSalmi I, Fattah MA. Defective growth hormone (GH) secretion and short-term treatment in noonan syndrome. Indian J Pediatr (1998) 65(5):741–9. doi: 10.1007/BF02731057
73. Kirk JM, Betts PR, Butler GE, Donaldson MD, Dunger DB, Johnston DI, et al. Short stature in noonan syndrome: response to growth hormone therapy. Arch Dis Child (2001) 84(5):440–3. doi: 10.1136/adc.84.5.440
74. MacFarlane CE, Brown DC, Johnston LB, Patton MA, Dunger DB, Savage MO, et al. Growth hormone therapy and growth in children with noonan's syndrome: results of 3 years' follow-up. J Clin Endocrinol Metab (2001) 86(5):1953–6. doi: 10.1210/jcem.86.5.7468
75. Ferreira LV, Souza SA, Arnhold IJ, Mendonca BB, Jorge AA. PTPN11 (protein tyrosine phosphatase, nonreceptor type 11) mutations and response to growth hormone therapy in children with noonan syndrome. J Clin Endocrinol Metab (2005) 90(9):5156–60. doi: 10.1210/jc.2004-2559
76. Limal JM, Parfait B, Cabrol S, Bonnet D, Leheup B, Lyonnet S, et al. Noonan syndrome: relationships between genotype, growth, and growth factors. J Clin Endocrinol Metab (2006) 91(1):300–6. doi: 10.1210/jc.2005-0983
77. Raaijmakers R, Noordam C, Karagiannis G, Gregory JW, Hertel NT, Sipilä I, et al. Response to growth hormone treatment and final height in noonan syndrome in a large cohort of patients in the KIGS database. J Pediatr Endocrinol Metab (2008) 21(3):267–73. doi: 10.1515/jpem.2008.21.3.267
78. Choi JH, Lee BH, Jung CW, Kim YM, Jin HY, Kim JM, et al. Response to growth hormone therapy in children with noonan syndrome: correlation with or without PTPN11 gene mutation. Horm Res Paediatr (2012) 77(6):388–93. doi: 10.1159/000339677
79. Şıklar Z, Genens M, Poyrazoğlu Ş, Baş F, Darendeliler F, Bundak R, et al. The growth characteristics of patients with noonan syndrome: Results of three years of growth hormone treatment: A nationwide multicenter study. J Clin Res Pediatr Endocrinol (2016) 8(3):305–12. doi: 10.4274/jcrpe.3013
80. Jo KJ, Kim YM, Yoon JY, Lee YJ, Han YM, Yoo HW, et al. Comparison of effectiveness of growth hormone therapy according to disease-causing genes in children with noonan syndrome. Korean J Pediatr (2019) 62(7):274–80. doi: 10.3345/kjp.2018.06842
81. Malaquias AC, Noronha RM, Souza TTO, Homma TK, Funari MFA, Yamamoto GL, et al. Impact of growth hormone therapy on adult height in patients with PTPN11 mutations related to noonan syndrome. Horm Res Paediatr (2019) 91:252–61. doi: 10.1159/000500264
82. Ranke MB, Lindberg A, Carlsson M, Camacho-Hübner C, Rooman R. Treatment with growth hormone in noonan syndrome observed during 25 years of KIGS: Near adult height and outcome prediction. Horm Res Paediatr (2019) 91(1):46–55. doi: 10.1159/000498859
83. Apperley LJ, Ramakrishnan R, Dharmaraj P, Das U, Didi M, Blair J, et al. Effect of growth hormone therapy in patients with noonan syndrome: A retrospective study. Int J Endocrinol Metab (2020) 18(4):e107292. doi: 10.5812/ijem.107292
84. Libraro A, D'Ascanio V, Cappa M, Chiarito M, Digilio MC, Einaudi S, et al. Growth in children with noonan syndrome and effects of growth hormone treatment on adult height. Front Endocrinol (Lausanne) (2021) 12:761171. doi: 10.3389/fendo.2021.761171
85. Binder G. Noonan syndrome, the ras-MAPK signalling pathway and short stature. Horm Res (2009) 71 Suppl 2:64–70. doi: 10.1159/000192439
86. Dahlgren J. GH therapy in noonan syndrome: Review of final height data. Horm Res (2009) 72 Suppl 2:46–8. doi: 10.1159/000243779
87. Jeong I, Kang E, Cho JH, Kim GH, Lee BH, Choi JH, et al. Long-term efficacy of recombinant human growth hormone therapy in short-statured patients with noonan syndrome. Ann Pediatr Endocrinol Metab (2016) 21(1):26–30. doi: 10.6065/apem.2016.21.1.26
88. Ranke MB, Heidemann P, Knupfer C, Enders H, Schmaltz AA, Bierich JR. Noonan syndrome: Growth and clinical manifestations in 144 cases. Eur J Pediatr (1988) 148:220–7. doi: 10.1007/BF00441408
89. Noordam C, Draaisma JM, van den Nieuwenhof J, van der Burgt I, Otten BJ, Daniels O. Effects of growth hormone treatment on left ventricular dimensions in children with noonan's syndrome. Horm Res (2001) 56(3-4):110–3. doi: 10.1159/000048101
90. Brown DC, Macfarlane CE, McKenna WJ, Patton MA, Dunger DB, Savage MO, et al. Growth hormone therapy in noonan's syndrome: non-cardiomyopathic congenital heart disease does not adversely affect growth improvement. J Pediatr Endocrinol Metab (2002) 15(6):851–2. doi: 10.1515/jpem.2002.15.6.851
91. Sanford RA, Bowman R, Tomita T, De Leon G, Palka P. A 16-year-old male with noonan's syndrome develops progressive scoliosis and deteriorating gait. Pediatr Neurosurg (1999) 30(1):47–52. doi: 10.1159/000028761
92. Takagi M, Miyashita Y, Koga M, Ebara S, Arita N, Kasayama S. Estrogen deficiency is a potential cause for osteopenia in adult male patients with noonan's syndrome. Calcif Tissue Int (2000) 66(3):200–3. doi: 10.1007/s002230010040
93. Jongmans M, Sistermans EA, Rikken A, Nillesen WM, Tamminga R, Patton M, et al. Genotypic and phenotypic characterization of noonan syndrome: new data and review of the literature. Am J Med Genet A (2005) 134A(2):165–70. doi: 10.1002/ajmg.a.30598
94. Martinelli S, Carta C, Flex E, Binni F, Cordisco EL, Moretti S, et al. Activating PTPN11 mutations play a minor role in pediatric and adult solid tumors. Cancer Genet Cytogenet (2006) 166(2):124–9. doi: 10.1016/j.cancergencyto.2005.10.003
95. Fryssira H, Leventopoulos G, Psoni S, Kitsiou-Tzeli S, Stavrianeas N, Kanavakis E. Tumor development in three patients with noonan syndrome. Eur J Pediatr (2008) 167(9):1025–31. doi: 10.1007/s00431-007-0636-3
96. Sherman CB, Ali-Nazir A, Gonzales-Gomez I, Finlay JL, Dhall G. Primary mixed glioneuronal tumor of the central nervous system in a patient with noonan syndrome: a case report and review of the literature. J Pediatr Hematol Oncol (2009) 31(1):61–4. doi: 10.1097/MPH.0b013e31818ab2cf
97. Schuettpelz LG, McDonald S, Whitesell K, Desruisseau DM, Grange DK, Gurnett CA, et al. Pilocytic astrocytoma in a child with noonan syndrome. Pediatr Blood Cancer (2009) 53(6):1147–9. doi: 10.1002/pbc.22193
98. Selter M, Dresel R, Althaus J, Bartels MB, Dittrich S, Geb S, et al. Dysembryoplastic neuroepithelial tumor (DNET) in a patient with noonan syndrome. Neuropediatrics (2010) 41:P1356. doi: 10.1055/s-0030-1265602
99. de Jong M, Schieving J, Goraj B. Remarkable intra-cerebral lesions on MRI in a patient with noonan syndrome. Eur J Radiol Extra (2011) 78:e17–9. doi: 10.1016/j.ejrex.2011.01.005
100. Karafin M, Jallo GI, Ayars M, Eberhart CG, Rodriguez FJ. Rosette forming glioneuronal tumor in association with noonan syndrome: pathobiological implications. Clin Neuropathol (2011) 30(6):297–300. doi: 10.5414/np300374
101. Bendel A, Hansen M, Dugan S, Mendelsohn N. Dysembyoplastic neuroepithelial tumor in two relatives with noonan syndrome and a PTPN 11 mutation. Neuro Oncol (2012) 14:156. doi: 10.1093/neuonc/nos108
102. Rankin J, Short J, Turnpenny P, Castle B, Hanemann CO. Medulloblastoma in a patient with the PTPN11 p.Thr468Met mutation. Am J Med Genet A (2013) 161A(8):2027–9. doi: 10.1002/ajmg.a.36005
103. Pellegrin MC, Tornese G, Cattaruzz, Blank E, Kieslich M, Ventura A. A rare brain tumor in noonan syndrome: Report of two cases, in: 53rd ESPE Meeting. Horm Res Paediatr (2014) 82:94
104. Delisle M, Siegfried A, Tauber M, Cave H, Loukh N, Boetto S, et al. Dysembryoplastic neuroepithelial tumor (DNET) and Noonan syndrome. A case report. In: Abstracts of the XVIII international congress of neuropathology; Rio de Janeiro, brazil. 14–18 September 2014. Brain Pathol (2014) 24(Suppl. 1):73.
105. Bangalore Krishna K, Pagan P, Escobar O, Popovic J. Occurrence of cranial neoplasms in pediatric patients with noonan syndrome receiving growth hormone: Is screening with brain MRI prior to initiation of growth hormone indicated? Horm Res Paediatr (2017) 88(6):423–6. doi: 10.1159/000479107
106. Rush S, Madden J, Hemenway M, Foreman N. Mutations in PTPN11 gene may predispose to development of midline low grade gliomas. Abstracts from the 16th international symposium on pediatric neuro-oncology in conjunction with the 8th st. Jude-VIVA forum. Neuro-Oncology (2014) 16(1):i60–i70. doi: 10.1093/neuonc/nou073
107. Bendel A, Pond D. (2014). Abstracts from the 16th international symposium on pediatric neuro-oncology in conjunction with the 8th st. Jude-VIVA forum, June 28-July 2, 2014, Singapore, in: Central nervous system (CNS) glial neoplasms in three individuals with Noonan Syndrome (NS) and PTPN11 mutation, and a review of the literature, .
108. Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, et al. Cancer spectrum and frequency among children with noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer (2015) 112(8):1392–7. doi: 10.1038/bjc.2015.75
109. Nair S, Fort JA, Yachnis AT, Williams CA. Optic nerve pilomyxoid astrocytoma in a patient with noonan syndrome. Pediatr Blood Cancer (2015) 62(6):1084–6. doi: 10.1002/pbc.25382
110. McWilliams GD, SantaCruz K, Hart B, Clericuzio C. Occurrence of DNET and other brain tumors in noonan syndrome warrants caution with growth hormone therapy. Am J Med Genet A (2016) 170A(1):195–201. doi: 10.1002/ajmg.a.37379
111. Siegfried A, Cances C, Denuelle M, Loukh N, Tauber M, Cavé H, et al. Noonan syndrome, PTPN11 mutations, and brain tumors. A clinical report and review of the literature. Am J Med Genet A (2017) 173(4):1061–5. doi: 10.1002/ajmg.a.38108
112. El-Ayadi M, Ansari M, Kühnöl CD, Bendel A, Sturm D, Pietsch T, et al. Occurrence of high-grade glioma in noonan syndrome: Report of two cases. Pediatr Blood Cancer (2019) 66(5):e27625. doi: 10.1002/pbc.27625
113. Boonyawat B, Charoenpitakchai M, Suwanpakdee P. A first case report of subependymoma in PTPN11 mutation-associated noonan syndrome. Case Rep Neurol Med (2019) 2019:6091059. doi: 10.1155/2019/6091059
114. Morales-Rosado JA, Singh H, Olson RJ, Larsen BT, Hager MM, Klee EW, et al. Recurrent ganglioneuroma in PTPN11-associated noonan syndrome: A case report and literature review. Am J Med Genet A (2021) 185(6):1883–7. doi: 10.1002/ajmg.a.62178
115. Khan A, Soliman MAR, Ghannam MM, Jowdy PK, Hess R, Recker MJ, et al. Spinal cord glioblastoma multiforme in a patient with noonan syndrome: A clinical report. Clin Neurol Neurosurg (2021) 207:106725. doi: 10.1016/j.clineuro.2021.106725
116. Mutesa L, Pierquin G, Janin N, Segers K, Thomée C, Provenzi M, et al. Germline PTPN11 missense mutation in a case of noonan syndrome associated with mediastinal and retroperitoneal neuroblastic tumors. Cancer Genet Cytogenet (2008) 182(1):40–2. doi: 10.1016/j.cancergencyto.2007.12.005
117. Yoshida R, Ogata T, Masawa N, Nagai T. Hepatoblastoma in a noonan syndrome patient with a PTPN11 mutation. Pediatr Blood Cancer (2008) 50(6):1274–6. doi: 10.1002/pbc.21509
118. Kondoh T, Ishii E, Aoki Y, Shimizu T, Zaitsu M, Matsubara Y, et al. Noonan syndrome with leukaemoid reaction and overproduction of catecholamines: a case report. Eur J Pediatr (2003) 162(7-8):548–9. doi: 10.1007/s00431-003-1227-6
119. Chantrain CF, Jijon P, De Raedt T, Vermylen C, Poirel HA, Legius E, et al. Therapy-related acute myeloid leukemia in a child with noonan syndrome and clonal duplication of the germline PTPN11 mutation. Pediatr Blood Cancer (2007) 48(1):101–4. doi: 10.1002/pbc.20527
120. Li X, Yao R, Tan X, Li N, Ding Y, Li J, et al. Molecular and phenotypic spectrum of noonan syndrome in Chinese patients. Clin Genet (2019) 96(4):290–9. doi: 10.1111/cge.13588
121. Sidwell RU, Rouse P, Owen RA, Green JS. Granular cell tumor of the scrotum in a child with noonan syndrome. Pediatr Dermatol (2008) 25(3):341–3. doi: 10.1111/j.1525-1470.2008.00678.x
122. Ramaswamy PV, Storm CA, Filiano JJ, Dinulos JG. Multiple granular cell tumors in a child with noonan syndrome. Pediatr Dermatol (2010) 27(2):209–11. doi: 10.1111/j.1525-1470.2010.01111.x
123. Moos D, Droitcourt C, Rancherevince D, Marec Berard P, Skowron F. Atypical granular cell tumor occurring in an individual with noonan syndrome treated with growth hormone. Pediatr Dermatol (2012) 29(5):665–6. doi: 10.1111/j.1525-1470.2011.01641.x
124. Bamps S, Oyen T, Legius E, Vandenoord J, Stas M. Multiple granular cell tumors in a child with noonan syndrome. Eur J Pediatr Surg (2013) 23(3):257–9. doi: 10.1055/s-0032-1322537
125. Park SH, Lee SH. Noonan syndrome with multiple lentigines with PTPN11 (T468M) gene mutation accompanied with solitary granular cell tumor. J Dermatol (2017) 44(11):e280–1. doi: 10.1111/1346-8138.13960
126. Ekvall S, Hagenäs L, Allanson J, Annerén G, Bondeson ML. Co-Occurring SHOC2 and PTPN11 mutations in a patient with severe/complex noonan syndrome-like phenotype. Am J Med Genet A (2011) 155A(6):1217–24. doi: 10.1002/ajmg.a.33987
127. Jacquinet A, Bonnard A, Capri Y, Martin D, Sadzot B, Bianchi E, et al. Oligo-astrocytoma in LZTR1-related noonan syndrome. Eur J Med Genet (2020) 63(1):103617. doi: 10.1016/j.ejmg.2019.01.007
128. Hastings R, Newbury-Ecob R, Ng A, Taylor R. A further patient with noonan syndrome due to a SOS1 mutation and rhabdomyosarcoma. Genes Chromosomes Cancer (2010) 49(10):967–8. doi: 10.1002/gcc.20800
129. Denayer E, Devriendt K, de Ravel T, Van Buggenhout G, Smeets E, Francois I, et al. Tumor spectrum in children with noonan syndrome and SOS1 or RAF1 mutations. Genes Chromosomes Cancer (2010) 49(3):242–52. doi: 10.1002/gcc.20735
130. Abdelmoula NB, Louati R, Abdelmoula B, Aloulou S. TBIO-27. rasopathies and brain tumorogenesis: are SOS1 mutations concerned? Neuro-Oncology (2020) 22(3):iii471. doi: 10.1093/neuonc/noaa222.850
131. Garren B, Stephan M, Hogue JS. NRAS associated RASopathy and embryonal rhabdomyosarcoma. Am J Med Genet A (2020) 182(1):195–200. doi: 10.1002/ajmg.a.61395
132. Aoki Y, Niihori H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet (2005) 37:1038–40.
133. Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet (2005) 137C(1):72–7. doi: 10.1002/ajmg.c.30065
134. Kerr B, Delrue MA, Sigaudy S, Perveen R, Marche M, Burgelin I, et al. Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet (2006) 43(5):401–5. doi: 10.1136/jmg.2005.040352
135. Ji J, Navid F, Hiemenz MC, Kaneko M, Zhou S, Saitta SC, et al. Embryonal rhabdomyosarcoma in a patient with a germline CBL pathogenic variant. Cancer Genet (2019) 231-232:62–6. doi: 10.1016/j.cancergen.2018.12.006
136. Harms FL, Alawi M, Amor DJ, Tan TY, Cuturilo G, Lissewski C, et al. The novel RAF1 mutation p.(Gly361Ala) located outside the kinase domain of the CR3 region in two patients with noonan syndrome, including one with a rare brain tumor. Am J Med Genet A (2018) 176(2):470–6. doi: 10.1002/ajmg.a.38569
137. Altmüller F, Lissewski C, Bertola D, Flex E, Stark Z, Spranger S, et al. Genotype and phenotype spectrum of NRAS germline variants. Eur J Hum Genet (2017) 25(7):823–31. doi: 10.1038/ejhg.2017.65
138. Hanson HL, Wilson MJ, Short JP, Chioza BA, Crosby AH, Nash RM, et al. Germline CBL mutation associated with a noonan-like syndrome with primary lymphedema and teratoma associated with acquired uniparental isodisomy of chromosome 11q23. Am J Med Genet A (2014) 164A(4):1003–9. doi: 10.1002/ajmg.a.36375
139. Yaoita M, Niihori T, Mizuno S, Okamoto N, Hayashi S, Watanabe A, et al. Spectrum of mutations and genotype-phenotype analysis in noonan syndrome patients with RIT1 mutations. Hum Genet (2016) 135(2):209–22. doi: 10.1007/s00439-015-1627-5
140. Paccoud R, Saint-Laurent C, Piccolo E, Tajan M, Dortignac A, Pereira O, et al. SHP2 drives inflammation-triggered insulin resistance by reshaping tissue macrophage populations. Sci Transl Med (2021) 13(591):eabe2587. doi: 10.1126/scitranslmed.abe2587
141. Ramos-Kuri M, Meka SH, Salamanca-Buentello F, Hajjar RJ, Lipskaia L, Chemaly ER. Molecules linked to ras signaling as therapeutic targets in cardiac pathologies. Biol Res (2021) 54(1):23. doi: 10.1186/s40659-021-00342-6
142. Force T, Hajjar R, Del Monte F, Rosenzweig A, Choukroun G. Signaling pathways mediating the response to hypertrophic stress in the heart. Gene Expr (1999) 7(4-6):337–48.
143. Harris IS, Zhang S, Treskov I, Kovacs A, Weinheimer C, Muslin AJ. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation (2004) 110:718–23. doi: 10.1161/01.CIR.0000138190.50127.6A
144. Sala V, Gallo S, Leo C, Gatti S, Gelb BD, Crepaldi T. Signaling to cardiac hypertrophy: insights from human and mouse RASopathies. Mol Med (2012) 18(1):938–47. doi: 10.2119/molmed.2011.00512
145. Motta M, Sagi-Dain L, Krumbach OHF, Hahn A, Peleg A, German A, et al. Activating MRAS mutations cause noonan syndrome associated with hypertrophic cardiomyopathy. Hum Mol Genet (2020) 29(11):1772–83. doi: 10.1093/hmg/ddz108
146. Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, et al. Gain-of-function RAF1 mutations cause noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet (2007) 39(8):1007–12. doi: 10.1038/ng2073
147. Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, et al. Germline gain-of-function mutations in RAF1 cause noonan syndrome. Nat Genet (2007) 39(8):1013–7. doi: 10.1038/ng2078
148. Kouz K, Lissewski C, Spranger S, Mitter D, Riess A, Lopez-Gonzalez V, et al. Genotype and phenotype in patients with noonan syndrome and a RIT1 mutation. Genet Med (2016) 18(12):1226–34. doi: 10.1038/gim.2016.32
149. Gelb BD, Tartaglia M. Noonan syndrome with multiple lentigines. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2007). p. 1993–2022.
150. Kerr B, Einaudi MA, Clayton P, Gladman G, Eden T, Saunier P, et al. Is growth hormone treatment beneficial or harmful in Costello syndrome? J Med Genet (2003) 40(6):e74. doi: 10.1136/jmg.40.6.e74
151. Pagnamenta AT, Kaisaki PJ, Bennett F, Burkitt-Wright E, Martin HC, Ferla MP, et al. Delineation of dominant and recessive forms of LZTR1-associated noonan syndrome. Clin Genet (2019) 95(6):693–703. doi: 10.1111/cge.13533
152. Romano A, Kaski JP, Dahlgren J, Kelepouris N, Pietropoli A, Rohrer TR, et al. Cardiovascular safety of growth hormone treatment in noonan syndrome: real-world evidence. Endocr Connect (2022) 11(1):e210549. doi: 10.1530/EC-21-0549
153. Kobayashi D, Cook AL, Williams DA. Progressively worsening hypertrophic cardiomyopathy in a child with newly diagnosed Costello syndrome while receiving growth hormone therapy. Cardiol Young (2010) 20(4):459–61. doi: 10.1017/S1047951110000260
154. Levin MD, Saitta SC, Gripp KW, Wenger TL, Ganesh J, Kalish JM, et al. Nonreentrant atrial tachycardia occurs independently of hypertrophic cardiomyopathy in RASopathy patients. Am J Med Genet A (2018) 176(8):1711–22. doi: 10.1002/ajmg.a.38854
155. Follansbee CW, Malloy-Walton L. Ventricular fibrillation due to a likely pathogenic SOS1 variant: An unrecognized etiology of infantile sudden death? HeartRhythm Case Rep (2021) 7(8):510–3. doi: 10.1016/j.hrcr.2021.06.010
156. Vos E, Leenders E, Werkman SR, Udink Ten Cate FEA, Draaisma JMT. The added value of the electrocardiogram in Noonan syndrome. Cardiol Young (2022) 32(6):936–43. doi: 10.1017/S1047951121003310.
157. Noordam C. Growth hormone and the heart in noonan syndrome. Horm Res (2009) 72 Suppl 2:49–51. doi: 10.1159/000243780
158. Smpokou P, Tworog-Dube E, Kucherlapati RS, Roberts AE. Medical complications, clinical findings, and educational outcomes in adults with noonan syndrome. Am J Med Genet A (2012) 158A(12):3106–11. doi: 10.1002/ajmg.a.35639
159. Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet (2011) 157C(2):83–9. doi: 10.1002/ajmg.c.30300
160. Niemeyer CM. RAS diseases in children. Haematologica (2014) 99(11):1653–62. doi: 10.3324/haematol.2014.114595
161. Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res (2004) 64(24):8816–20. doi: 10.1158/0008-5472.CAN-04-1923
162. Tartaglia M, Niemeyer CM, Shannon KM, Loh ML. SHP-2 and myeloid malignancies. Curr Opin Hematol (2004) 11(1):44–50. doi: 10.1097/00062752-200401000-00007
163. Rehman AU, Rahman MU, Khan MT, Saud S, Liu H, Song D, et al. The landscape of protein tyrosine phosphatase (Shp2) and cancer. Curr Pharm Des (2018) 24(32):3767–77. doi: 10.2174/1381612824666181106100837
164. Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood (2004) 103(6):2325–31. doi: 10.1182/blood-2003-09-3287
165. Mohi MG, Neel BG. The role of Shp2 (PTPN11) in cancer. Curr Opin Genet Dev (2007) 17(1):23–30. doi: 10.1016/j.gde.2006.12.011
166. Villani A, Greer MC, Kalish JM, Nakagawara A, Nathanson KL, Pajtler KW, et al. Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clin Cancer Res (2017) 23(12):e83–90. doi: 10.1158/1078-0432.CCR-17-0631
168. Sharland M, Burch M, McKenna WM, Paton MA. A clinical study of noonan syndrome. Arch Dis Child (1992) 67(2):178–83. doi: 10.1136/adc.67.2.178
169. Lee CK, Chang BS, Hong YM, Yang SW, Lee CS, Seo JB. Spinal deformities in noonan syndrome: a clinical review of sixty cases. J Bone Joint Surg Am (2001) 83(10):1495–502. doi: 10.2106/00004623-200110000-00006
170. Reinker KA, Stevenson DA, Tsung A. Orthopaedic conditions in Ras/MAPK related disorders. J Pediatr Orthop (2011) 31(5):599–605. doi: 10.1097/BPO.0b013e318220396e
171. Souza FM, Collett-Solberg PF. Adverse effects of growth hormone replacement therapy in children. Arq Bras Endocrinol Metabol (2011) 55(8):559–65. doi: 10.1590/s0004-27302011000800009
172. Ricotti S, Petrucci L, Carenzio G, Klersy C, Calcaterra V, Larizza D, et al. Prevalence and incidence of scoliosis in turner syndrome: a study in 49 girls followed-up for 4 years. Eur J Phys Rehabil Med (2011) 47(3):447–53.
173. Han Y, Chen M, Wang H. Chiari I malformation in patients with RASopathies. Childs Nerv Syst (2021) 37(6):1831–6. doi: 10.1007/s00381-020-05034-2
174. Loukas M, Shayota BJ, Oelhafen K, Miller JH, Chern JJ, Tubbs RS, et al. Associated disorders of chiari type I malformations: a review. Neurosurg Focus (2011) 31(3):E3. doi: 10.3171/2011.6.FOCUS11112
175. Xue Y, Gao Y, Wang S, Wang P. An examination of the effects of different doses of recombinant human growth hormone on children with growth hormone deficiency. Exp Ther Med (2016) 11(5):1647–52. doi: 10.3892/etm.2016.3091
176. Tidblad A, Bottai M, Kieler H, Albertsson-Wikland K, Sävendahl L. Association of childhood growth hormone treatment with long-term cardiovascular morbidity. JAMA Pediatr (2021) 175(2):e205199. doi: 10.1001/jamapediatrics.2020.5199
Keywords: Noonan syndrome, RASopathies, growth, puberty, growth hormone, cancer, hypertrophic cardiomyopathy, genotype-phenotype correlations
Citation: Stagi S, Ferrari V, Ferrari M, Priolo M and Tartaglia M (2022) Inside the Noonan “universe”: Literature review on growth, GH/IGF axis and rhGH treatment: Facts and concerns. Front. Endocrinol. 13:951331. doi: 10.3389/fendo.2022.951331
Received: 23 May 2022; Accepted: 18 July 2022;
Published: 18 August 2022.
Edited by:
Sandro Loche, Ospedale Microcitemico, ItalyReviewed by:
Alan David Rogol, University of Virginia, United StatesCopyright © 2022 Stagi, Ferrari, Ferrari, Priolo and Tartaglia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Stagi, c3RlZmFuby5zdGFnaUB1bmlmaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.