 Raghuveer Kavarthapu
Raghuveer Kavarthapu Maria L. Dufau
Maria L. Dufau- Section on Molecular Endocrinology, Division of Developmental Biology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, United States
The prolactin receptor (PRLR) is a member of the lactogen/cytokine receptor family, which mediates multiple actions of prolactin (PRL). PRL is a major hormone in the proliferation/differentiation of breast epithelium that is essential for lactation. It is also involved in breast cancer development, tumor growth and chemoresistance. Human PRLR expression is controlled at the transcriptional level by multiple promoters. Each promoter directs transcription/expression of a specific non-coding exon 1, a common non-coding exon 2 and coding exons E3-11. The identification of exon 11 of PRLR led to finding of alternative spliced products and two novel short forms (SF) that can inhibit the long form (LF) of PRLR activity with relevance in physiological regulation and breast cancer. Homo and heterodimers of LF and SF are formed in the absence of PRL that acts as a conformational modifier. Heterodimerization of SF with LF is a major mechanism through which SF inhibits some signaling pathways originating at the LF. Biochemical/molecular modeling approaches demonstrated that the human PRLR conformation stabilized by extracellular intramolecular S−S bonds and several amino acids in the extracellular D1 domain of PRLR SF are required for its inhibitory actions on PRLR LF-mediated functions. Studies in breast cancer cells demonstrated that the transcription of PRLR was directed by the preferentially utilized PIII promoter, which lacks an estrogen responsive element. Complex formation of non-DNA bound ERα dimer with Sp1 and C/EBPβ dimers bound to their sites at the PRLR promoter is required for basal activity. Estradiol induces transcriptional activation/expression of the PRLR gene, and subsequent studies revealed the essential role of autocrine PRL released by breast cancer cells and CDK7 in estradiol-induced PRLR promoter activation and upregulation. Other studies revealed stimulation of the PRLR promoter activity and PRLR LF protein by PRL in the absence of estrogen via the STAT5/phospho-ERα activation loop. Additionally, EGF/ERBB1 can induce the transcription of PRLR independent of estrogen and prolactin. The various regulatory modalities contributing to the upregulation of PRLR provide options for the development of therapeutic approaches to mitigate its participation in breast cancer progression and resistance.
Introduction
Prolactin (PRL) is a multifaceted protein hormone produced and secreted by the anterior pituitary gland, and it is also found in extra-pituitary tissues, including the mammary gland, brain, decidua, gonads, pancreas, immune cells, liver and adipose tissue, where it exerts autocrine and paracrine functions [reviewed in (1–4)]. Prolactin directs several physiological functions, such as lactation, immunomodulatory actions, and glucose and lipid metabolism, and is involved in pathological modalities, such as prolactinoma, hypogonadism and several cancers [reviewed in (2–4)]. The highly diversified actions of PRL are mediated through its transmembrane prolactin receptor (PRLR), a member of the lactogen/cytokine receptor family, which is expressed ubiquitously and functions as a dimer activated by PRL. A short isoform of PRLR with 310 amino acids (aa) was initially cloned from rat liver (5). The long form (LF) of PRLR with 610 aa was isolated from human hepatoma and breast cancer cells (6) and from the rat ovary (7). The monomeric structure of human PRLR (hPRLR) was resolved using combined NMR and computational approaches (8). PRLR gene expression is controlled by multiple promoters that regulate sustained PRLR levels and function (9). In humans, there are several PRLR isoforms, including the LF, many short forms (SFs), an intermediate variant and a soluble isoform [ (10), reviewed in (11)]. The expression of these isoforms of PRLR widely varies in different tissues and is required for specific functions of the organ system at specific times. Certain short isoforms of hPRLR can interfere with the essential signaling of the long isoform, thereby exerting inhibitory action [reviewed in (11, 12)].
PRL is structurally similar to growth hormone and can act as a growth factor, immunomodulator, or neurotransmitter in an autocrine and paracrine manner [reviewed in (2)]. PRL mediates its actions through PRLR, resulting in the activation of the janus tyrosine kinase (JAK)/signal transducer and activator of transcription (STAT) and mitogen-activated protein kinase (MAPK) signaling pathways [reviewed in (2)]. PRL/PRLR has been implicated in the development of several cancers and tumor progression [reviewed in (13–20)]. Hence, PRLR signaling has emerged as a relevant target in breast cancer. PRL is normally secreted in a pulsatile fashion, and this is clinically important for diagnostics related to problems in lactation and female infertility. Additionally, elevated circulating or locally produced PRL levels are associated with the risk of breast cancer [reviewed in (17, 18)]. PRL, through its cognate receptor, stimulates various downstream signaling cascades involving STAT5, RAS and MAPK, and phosphatidylinositol 3-kinase (PI3K) have been implicated in mammary tumorigenesis [reviewed in (18, 19, 21)]. The gain-of-function mutations in PRLR reported in benign and malignant breast cancer patients may support the hypothesis that PRLR signaling cascades could participate in benign breast tumorigenesis (22). In this review, we provide an overview of the current understanding of the transcriptional regulation of PRLR and signal transduction in physiological and pathological modalities in mammary glands with special emphasis on the role of PRL/PRLR signaling in breast cancer.
PRL and PRLR
PRL and PRLR distribution and biological functions
Prolactin is a 23 kDa protein hormone containing 199 aa that is produced in the lactotroph cells of the anterior pituitary gland. Prolactin is also secreted in an extra-pituitary and autocrine/paracrine manner from different tissues, such as mammary glands, brain, decidua, gonads, pancreas, immune cells, liver, and fat [reviewed in (1–3)]. The gene encoding human PRL is ∼10 kb with five exons, and four introns is present on chromosome 6. In addition to its normal expression in the anterior pituitary, it is also expressed in mammary epithelial cells, the decidua, brain, myometrium, endometrium, lacrimal gland, thymus, spleen, skin fibroblasts, sweat glands and immune system cells. PRL secretion is regulated by several factors. Ovarian steroids, specifically estrogens, modulate PRL synthesis and prolactin release while suppressing dopamine synthesis [reviewed in (1–3)]. Increased PRL secretion (prolactinomas) directly suppresses the secretion of GnRH and indirectly suppresses follicle-stimulating hormone and luteinizing hormone, thereby disrupting the HPG axis and the ovulatory cycle (reviewed in 1-2). PRL performs innumerable physiological functions in the body, including mammary gland development, lactation, gonadal functions (luteal cycle, uterine actions, Leydig cell development), parental behavior, preadipocyte differentiation, osmoregulation, angiogenesis, immunomodulatory function, islet cell proliferation, adrenal steroidogenesis, bone and calcium homeostasis, anovulation, reduced stress responses, oxytocin secretion and lipid metabolism. Prolactin also promotes neurogenesis in maternal and fetal brains [reviewed in (2, 3)].
PRLR has been identified in numerous cells and tissues of adult mammals. Cellular proliferation is also an important function of PRL in mammals. The expression of receptors (short and long forms) tends to vary with the stage of the estrous cycle, pregnancy, and lactation (23). The expression of PRLR in the late gestational fetal rat using in situ hybridization and immunocytochemistry showed that the long and short isoforms of PRLR were expressed during late fetal development (days 17.5 to 20.5). These studies showed PRLR transcripts were widely expressed in tissues from all three germ layers, in addition to the classic target organs of PRL (24). The expression of PRLR mRNA in the fetal adrenal cortex, gastrointestinal and bronchial mucosae, renal tubular epithelia, choroid plexus, thymus, liver, pancreas, and epidermis was higher than that in other tissues (23).
PRLR structure and isoforms
PRLRs belong to the lactogen/cytokine receptor superfamily, which mediates the various cellular actions of PRL in different target tissues. PRL binds to preexisting PRLR dimers and acts as a conformational modifier, which results in the activation of the JAK/STAT pathway and MAPK and SRC kinases, thus leading to the induction of PRL responsive genes [reviewed in (4, 18)].
PRLR structure
PRLR has three main domains: extracellular, transmembrane and intracellular. The extracellular domain is divided into two fibronectin domains, D1 and D2, and the WS motif in D2 acts as a molecular switch during ligand-bound activation of PRLR (25). PRLR has a single-pass transmembrane chain, and the receptor chain does not possess kinase activity. The receptor chain is dependent on the associated kinases to transduce phosphorylation-based signal cascades. The intracellular domain includes proline-rich sequence-mediated JAK2 association to the prolactin receptor is required but not sufficient for signal transduction Box-1/2 sub-domains. Box-1 is known to interact with JAK2 and SRC family kinases such as FYN [reviewed in (4)]. The intracellular domain of PRLR-LF is intrinsically highly unstructured/disordered and binds to negatively charged lipids of the inner plasma membrane through conserved motifs resembling immuno receptor tyrosine-based activation motifs. However, this lipid association of the PRLR intracellular domain is not accompanied by induced folding and is independent of specific tyrosine phosphorylation. These attributes may contribute to regulating intracellular signaling (26). There are two short isoforms of hPRLR generated by alternative splicing to exon 11 (10).
PRLR gene and isoforms
The genomic organization of the hPRLR gene (>200 kb) is complex and subject to alternative splicing, which results in several isoforms of the receptor. The gene resides on chromosome 5p14-13. The hPRLR gene contains eleven exons, where exon 1 consists of six non-coding sequences (hE13, hE1N1-5) that are alternatively spliced to a common non-coding exon 2, and only exons 3 to 11 are coding exons [ (9); Figure 1]. The LF of hPRLR is encoded by exons 3-10 (Figure 2). Additionally, an intermediate form (412 aa) with partial deletion of 198 aa within the cytoplasmic domain in exon 10 of the LF was isolated from the rat Nb2 lymphoma cell line (28) and human breast cancer cells (29). In addition, a soluble PRLR lacking transmembrane and cytoplasmic regions was isolated from the rat ovary (7). The transmembrane domain is encoded by Exon 8, while most of the intracellular domain is encoded by Exon 10 (Figure 2). Alternative splicing of exons 10 and 11 with a truncated intracellular domain resulted in two novel SFs of hPRLR, S1a and S1b, which inhibit the LF signal induced by PRL [ (10); Figures 2, 3]. In addition, a unique spliced variant designated S1c, which completely lacks exon 10, has been identified in human spermatozoa (30). These short forms S1a and S1b are expressed as cell surface transmembrane receptors with a reduced cytoplasmic domain and unique C-termini S1b was far more effective in inhibiting the PRL-induced activation of the β-casein gene promoter mediated by LF (31, 32). A subsequent study demonstrated a naturally occurring ΔS2 deletion variant of SF in normal and cancerous human cells. These studies have also demonstrated that removal of the S2 extracellular subdomain can alter the conformation of the intracellular signaling region of the LF and both SFs (S1a and S1b), thereby supporting the concept that the conformation of the ECD can affect the conformation of the intracellular domain (33).
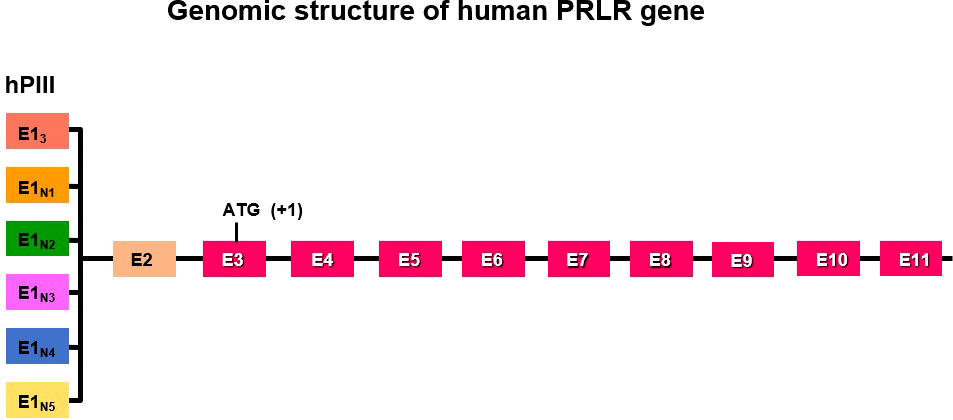
Figure 1 Schematic representation of multiple exons 1 (hE13, hE1N1-5) driven by individual promoters and alternative splicing to common exon 2 of human PRLR. Exon 3 has initiation codon. PIII/hEI3 is the predominantly utilized generic promoter in addition to five specific exon 1/promoters (hE1N1 to hE1N5).
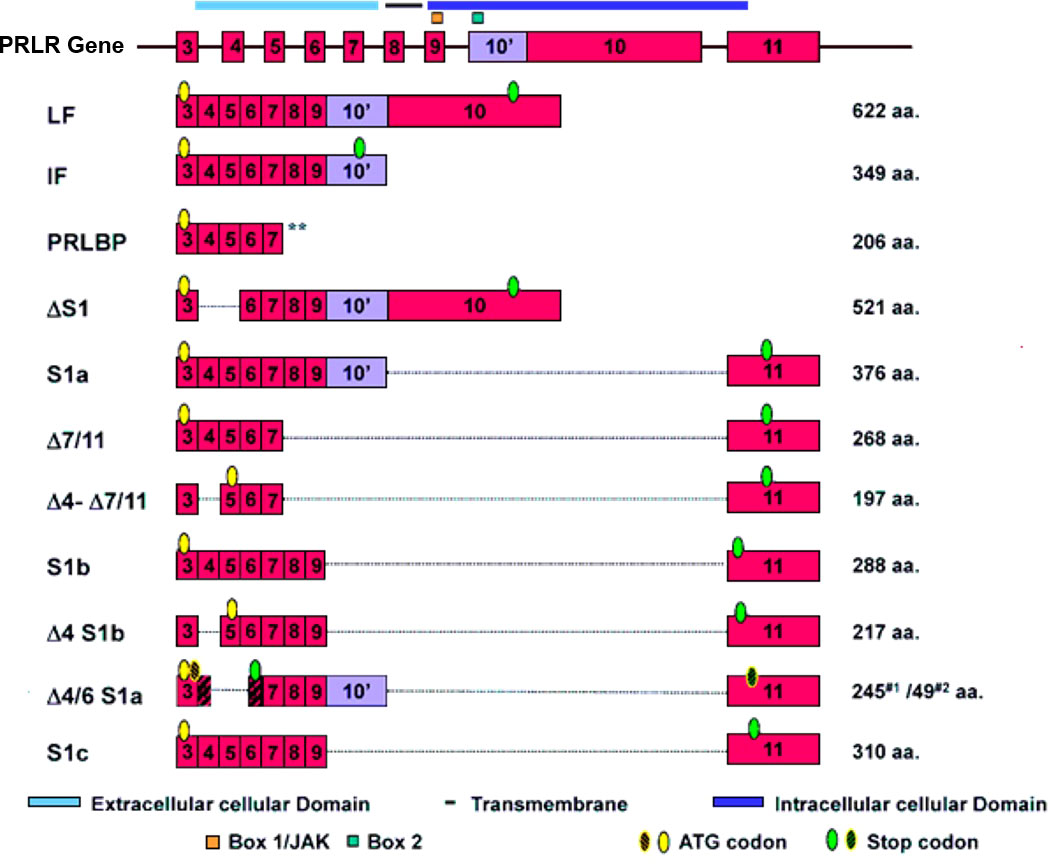
Figure 2 Schematic representation of human PRLR isoforms generated by alternative splicing [adapted from reference (27)] PRLR (prolactin receptor) Atlas Genet Cytogenet. ** indicates soluble form of PRLR.
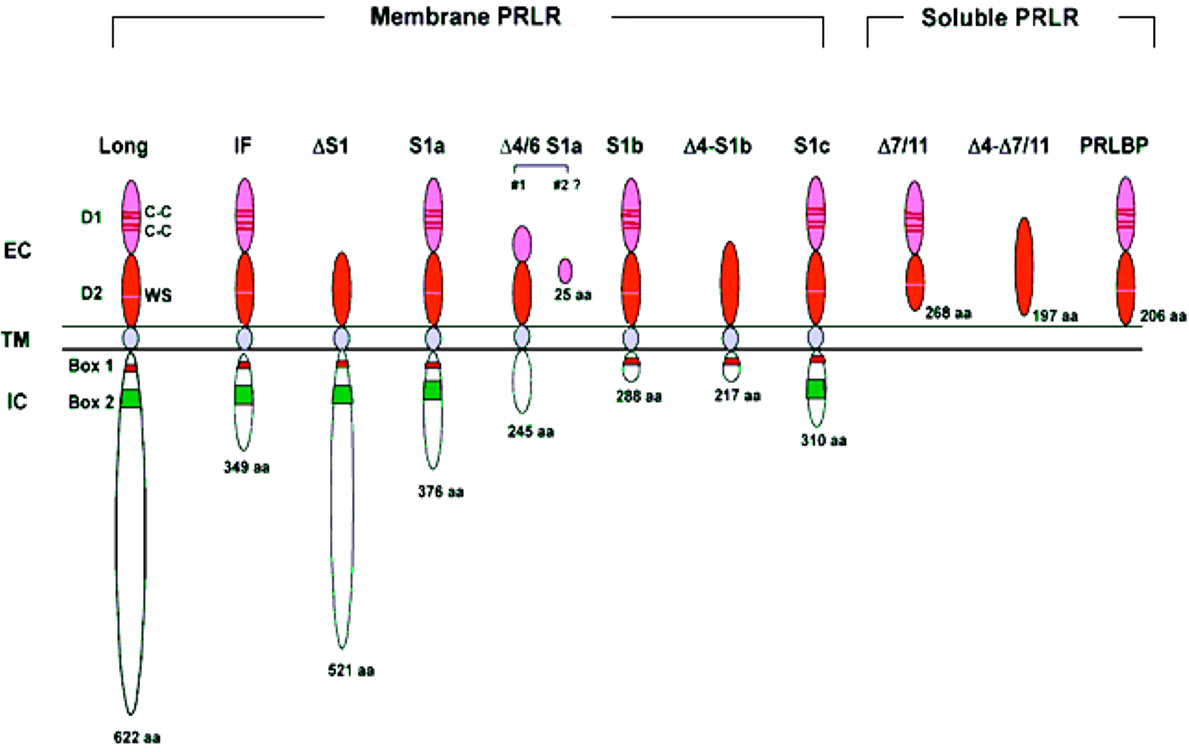
Figure 3 Structure of human PRLR variants. LF, long form; IF, intermediate form; S1a, S1b & S1c, short forms; PRLBP, prolactin binding protein; LFΔS1, long form lacking D1 domain; LFΔS2, long form lacking D2 domain; Δ, deleted exon. D1, D2, N-terminal subdomain; WS, WSXWS motif; C, cysteine; Y, tyrosine; EC, extracellular domain; TM, transmembrane domain; IC, intracellular domain [adapted from reference (27)].
Our studies on human LF, S1a and S1b have revealed the existence of constitutive LF and SF homodimers and heterodimers (LF/S1a or LF/S1b) under non-reducing conditions in the absence of PRL that acts as conformational modifier (31). Both LF and SFs (S1a and S1b), as dimers, are capable of ligand binding and PRL-induced phosphorylation of JAK2, but only LF can activate downstream STAT5 signaling. S1a and S1b cannot induce the downstream activation of STAT5 due to the lack of an extended cytoplasmic domain. PRL signaling through the SF of PRLR in mouse ovaries actively regulates the expression of several genes and can profoundly affect follicular survival. SF can mediate the activation of MAPK and PI3K pathways [reviewed in (34)]. In breast cancer, the ratio of SFs (S1a and S1b) to LF is markedly reduced compared to that in adjacent tissue, which indicates that the loss of inhibitory regulation of LF could increase tumor cell proliferation (35). Two intramolecular disulfide bonds within the extracellular D1 domains are essential for the inhibitory function of S1b on LF. Additionally, the JAK2 association was disrupted. S−S bond disruption of S1b (S1bx) affects the dimerization interface, thus causing a significant decrease in LF heterodimerization with S1bx and an increase in homodimerization of S1bx. Therefore, stability of the PRLR structure by intramolecular S−S bonds is required for the inhibitory action of S1b on LF-mediated function (36). Additionally, mutations in E69 of the D1 domain of S1b and neighboring amino acid residues (R66, E67, E42) close to its surface binding domain cause a loss of its inhibitory effect, while those away from this region or mutants in the D2 domain have no effect. These findings underscore the significant role of extracellular D1 on the S1b conformation and its inhibitory action in PRL-induced LF function [ (12); Figure 4]. In addition, PRL signaling through SF of mouse PRLR can either stimulate or inhibit a substantial number of transcription factors in the decidua as well as ovary. Few transcription factors have been shown to be similarly regulated in both tissues, while most transcription factors are oppositely regulated by PRL (37). Additional studies are needed to better understand the role of alternatively spliced PRLR isoforms and the manner in which such splicing is regulated in breast cancer.
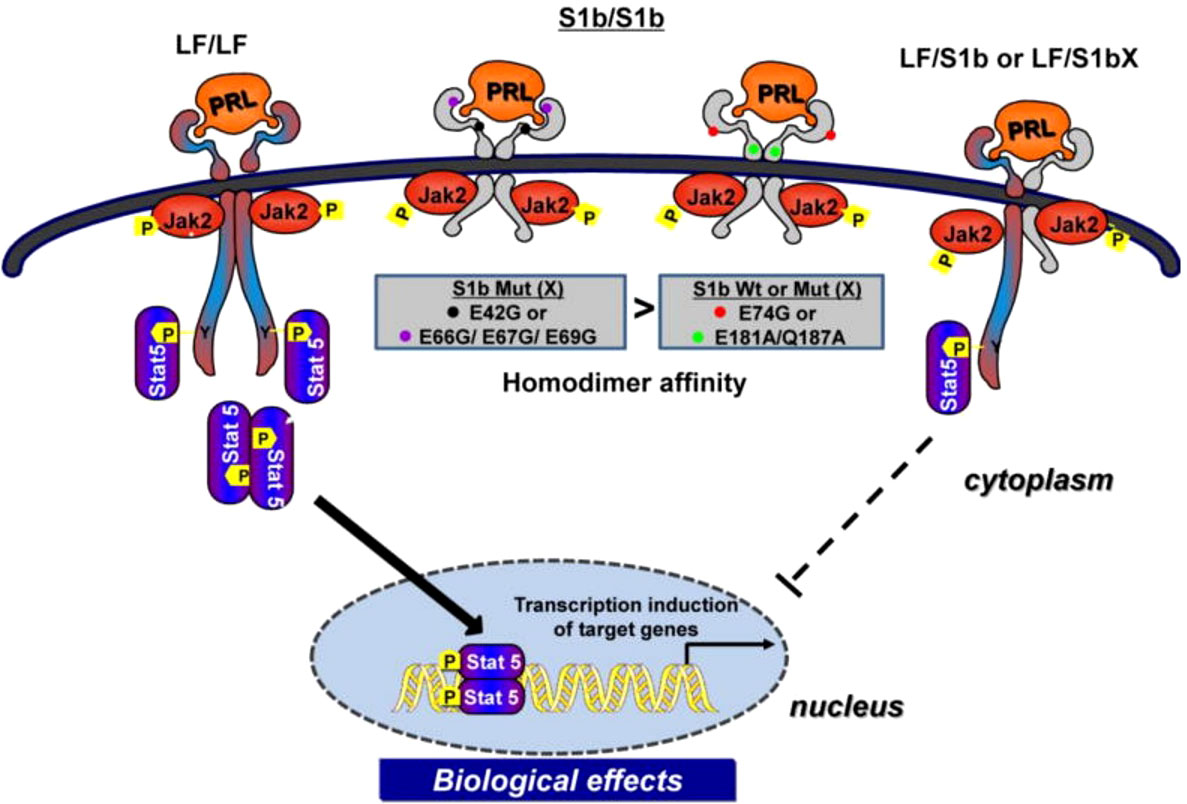
Figure 4 Mechanism of inhibitory action of short form S1b PRLR on PRL-induced long-from (LF) receptor signaling (reproduced from reference 12). Homodimer of the hPRLR long form (LF) mediates PRL stimulated JAK2/Stat5 signaling required for transcription/expression PRL/PRLR target genes which are essential for the various biological effects of the hormone. The inhibitory action of short form S1b on LF’s function induced by PRL results from LF/SF heterodimer formation and marked reduction of LF/LF homodimers which are required by Stat5 activation.
Transcriptional regulation of PRLR
The expression of PRLR is controlled by multiple promoters using a complex regulatory transcriptional network. In the rat these promoters are PI (gonad specific and SF1 dependent), PII (liver specific induced by HNF4), and PIII a widely expressed in all tissues and requires CCAAT/enhancer binding protein-β (C/EBPβ) and specificity protein 1 (Sp1) for its activation. Promoter PI is inoperative in mice due to disruption of the SF1 motif. These multiple promoters have been shown to direct tissue-specific expression in the ovary, liver and mammary gland in rats [ (38), reviewed in (39)]. In humans, hPIII is a universal/generic promoter similar in structure and regulation to one of those found in rodents (PIII). This promoter in humans directs transcription in all PRL-responsive tissues as well as other promoter(s) of less known function (9). The promoters include the predominantly utilized generic promoter 1/exon 1 (PIII/hEI3), which is also present in rats and mice, and five human-specific exon 1/promoters (hE1N1 to hE1N5) [ (9, 39); Figure 1]. The PRLR promoters belong to the TATA-less/initiator class and are activated by estradiol 17β (E2). The preferentially utilized human promoter III (hPIII) promoter contains Sp1 and C/EBP elements that bind to Sp1/Sp3 and C/EBPβ, which are required for basal transcriptional activity (Figure 5). These promoters were found to be utilized in breast cancer tissue and cell lines, including MCF7 and T47D, and variably used in other tissues (40). Among these promoters, the generic hPIII (the human counterpart of rodent PIII), which drives the universal human E13 exon (the human counterpart of EIII in rodents), was functionally characterized in breast cancer cells, while the specific human exon hEN1 directed by promoter hPN1 is driven by domains containing an ETS element and a nuclear receptor NR half-site. The promoters for the specific human exons, i.e., hEN2-5, remain to be identified (39).
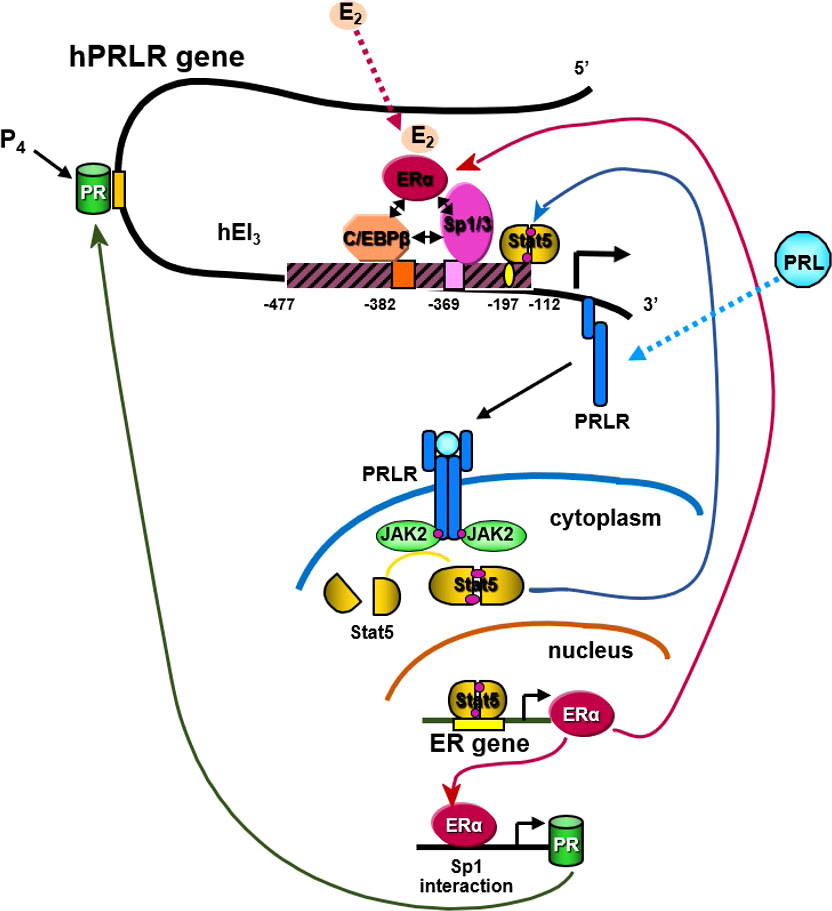
Figure 5 Transcriptional regulation of hPRLR induced by E2. E2 induces ligand mediated ERα-Sp1-C/EBPβ complex formation and binding to the hPIII promoter of the hPRLR gene (38, 39). This results in increased transcription of PRLR. PRL/PRLR activates JAK/STAT that can induce the transcription of ERα. P4 (progesterone)/Prog receptor can induce transcription of PRLR through association with Sp1and C/EBPβ through promoter hPIII.
E2 can induce an increase in hPRLR mRNA transcripts directed by the hPIII promoter via a non-classical ERE independent mechanism in breast cancer cells [ (40); Figure 5]. The association of ERα with DNA-bound Sp1 and C/EBPβ is essential for E2-induced hPRLR gene transcription (Figure 5). The additional interaction between zinc fingers of Sp1 and leucine zipper of C/EBPβ stabilizes the ERα-Sp1-C/EBPβ complex. The enhanced complex formation of the ERα dimer (DNA binding domain) with Sp1 (zinc finger motifs) and C/EBPβ (basic region and leucine zipper) by E2 plays an essential role in the transcriptional activation of the PRLR gene [ (40, 41); Figure 5]. An autocrine/paracrine loop increases PRLR mRNA expression via its ligand PRL in breast cancer cells. Similarly, in MCF7 cells overexpressing PRL, upregulation of PRLR was observed in response to endogenous PRL but not exogenous PRL. Furthermore, there was an increase in ER levels and estrogen responsiveness in these MCF-7 cells. Owing to the positive effect of estrogen on PRLR transcription, this reciprocal regulation amplifies both ER and PRLR signaling in breast cancer (42). Another steroid hormone that can also regulate PRLR transcription is progesterone through its receptor (Figure 5). Progesterone participates in the menstrual cycle, pregnancy, and embryogenesis and can be involved in tumorigenesis as well as in normal growth. It has been reported that the progesterone receptor lacks the consensus sequence or half-sequence response element in the PRLR gene PIII promoter and demonstrated that progesterone induces an increase in PRLR mRNA in a non-classical manner by inducing the expression of PRLR through the cooperative activation of Sp1 and CEBPβ at the PIII promoter in mouse cells and T47D breast cancer cells (43).
PRLR Signal Transduction
JAK-STAT pathway
This is the most classical and well-studied downstream signaling pathway induced by the binding of PRL to PRLR. This pathway appears to mediate most of the PRL actions in lobuloalveolar development and lactation (Figure 6). The intracellular domain of PRLR is devoid of any intrinsic enzymatic activity; however, ligand-mediated activation of PRLR results in tyrosine phosphorylation of numerous cellular proteins, including the receptor itself [reviewed in (2, 4)]. Binding of PRL to PRLR results in conformational induction of preformed dimers and activation of JAK2 by transphosphorylation, which brings two JAK2 molecules close to each other [reviewed in (2, 4)]. JAK2 kinases are involved in the phosphorylation of Tyr residues of the PRLR itself, and the phosphotyrosines serve as potential docking sites for transducer molecules containing SH2 domains [ (45), reviewed (4)]. The phosphorylation of Tyr residues of PRLR occurs in all isoforms except short isoforms of PRLR [reviewed in (4)]. The LF of PRLR mediates several processes upon receptor activation due to the phosphorylation of several Tyr residues present in PRLR (45).
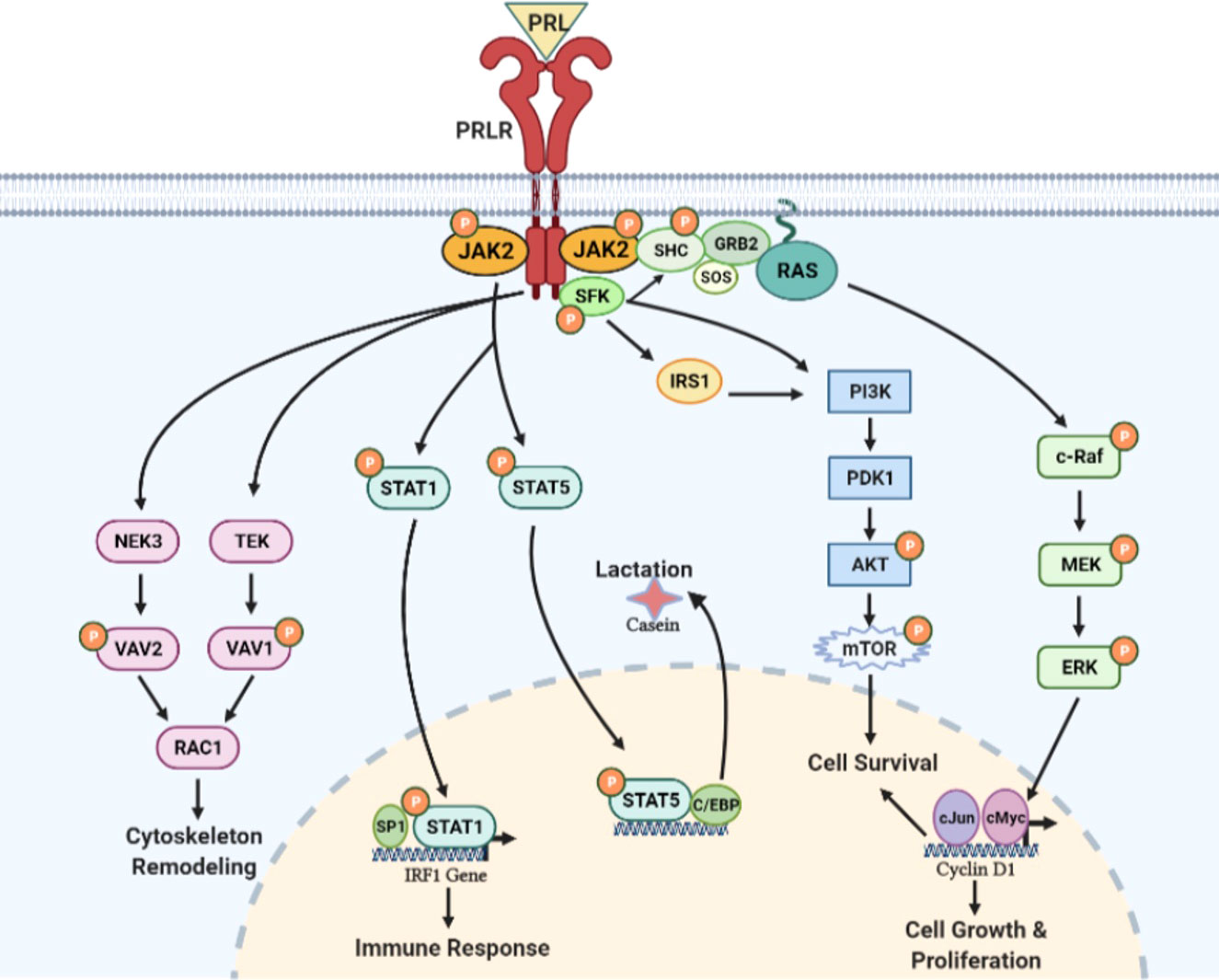
Figure 6 PRL/PRLR signaling pathways. PRL through its receptor PRLR induces multiple signaling cascades which include participation of JAK, SFK, PI3K/AKT, NEK3/VAV2, TEK/VAV1 kinases and STAT transcription factors. These signaling pathways are involved in lactation, immune response, cytoskeleton remodeling and cell growth, proliferation & survival [adapted from reference (44)].
The STAT family of proteins are the major transducers of cytokine receptor signaling, which contains eight members. STAT1/3 and STAT5a/5b have been identified as transducer molecules of PRLR. STAT contains five conserved domains: DNA-binding, SH3-like, SH2-like, and NH2- and COOH-terminal transactivating domains [ (46), reviewed in 4)]. As per the consensus model of STAT activation, a phosphorylated Tyr of the activated receptor interacts with the SH2 domain of STAT. Then, STAT is phosphorylated by receptor-associated JAK kinase. The phosphorylated STAT dissociates from the receptor and undergoes homodimerization or heterodimerization through an interaction involving the phosphotyrosine of each monomer and the SH2 domain of another phosphorylated STAT molecule. The STAT dimer translocates to the nucleus and activates a STAT DNA-binding motif in the promoter of target genes such as β-casein, IRF1, c-Myc, and cyclin-D (46). The consensus DNA motif (TTCxxxGAA), termed GAS (γ-interferon activated sequence), is recognized by STAT1, STAT3, and STAT5 homo or heterodimers [reviewed in (47, 48)].
RAS/RAF/MAP kinase pathway
In addition to JAK/STAT signaling initiated by the activation of PRLR, several reports implicate PRLR in the activation of the mitogen-activated protein (MAP) kinase cascade (49, 50). Phosphotyrosine residues of PRLR can serve as docking sites for adapter proteins (Shc/Grb2/SOS) connecting the receptor to the RAS/RAF/MAPK cascade [ (49, 50); Figure 6]. Although the JAK/STAT and MAPK pathways were initially regarded as independent or parallel pathways, results suggest that these pathways are interconnected [reviewed in (51)].
Other kinases: c-SRC and FYN
Several reports indicate PRL-induced activation of members of the Src kinase family, c-SRC (reviewed in 4) and FYN (52). PRL-induced rapid Tyr phosphorylation of insulin receptor substrate-1 (IRS-1) and a subunit of PI3K have been described. Both IRS-1 and PI3K seem to be associated with the PRLR complex. PRL-induced activation of PI3K has been proposed to be mediated by FYN (52). PRL/PRLR can also induce TEC-VAV1 and NEK3-VAV-1/VAV-2 signaling cascades and function in the regulation of the cytoskeleton (Figure 6). NEK3 kinase has been shown to regulates PRL-mediated cytoskeletal reorganization and motility of breast cancer cells (53, 54).
Role of PRL/PRLR signaling in breast cancer
PRL/PRLR induced signaling cascades promote breast development and progression
PRL plays a crucial role in mammary gland development and in the etiology and progression of breast cancer. Considerable supporting data indicate that PRL/PRLR hyper signaling contributes to the initiation of breast cancer. A strong correlation between breast cancer with increased PRL and PRLR has been reported in several studies (17, 55–58). Higher levels of PRL in postmenopausal women may eventually lead to an increased risk of breast tumors and metastatic cancer (58–60). In premenopausal women with breast cancer, there are higher-than-average levels of serum PRL together with elevated PRLR expression. These are associated with an increased risk of tumor progression and invasion (60–62). An association is observed between invasive breast cancer risk in postmenopausal women with high circulating PRL, particularly for ER-positive disease. PRL/PRLR is expressed in 95% of mammary tumors and 60% of male breast carcinomas (63). These findings were replicated in transgenic mice overexpressing PRL that develop mammary tumors and in in vitro studies where PRL played a role in the proliferation of breast cancer cells (64, 65). A direct correlation is observed between single nucleotide polymorphisms (SNPs) in the PRLR gene and benign breast tumor incidence. The two SNPs PRLR-I76V and I146L demonstrate constitutive receptor activity, and one of the SNPs (PRLRI146L) correlates with benign breast disease in a patient cohort, but these patients did not have high levels of serum PRL (22, 66). However, in subsequent study these SNPs in the PRLR gene were found not to be associated with breast cancer and multiple fibroadenoma (67). In another study, SNPs were found in PRL, and PRLR genes were associated with breast cancer metastasis in Taiwanese women (68). Studies from our lab and others in T47D and MCF-7 breast cancer cells have shown that the PI3K/AKT and RAF/MEK/ERK pathways are activated in parallel following PRL treatment, which leads to profound cell proliferation and survival (69, 70). PRLR can also induce the MAPK/ERK signaling cascade via the PI3-kinase-dependent RAC/PAK/RAF/MEK pathway, which is in turn controlled by JAK2, SRC family kinases and focal adhesion kinase (FAK) (71). In addition to the role of the predominant LF of PRLR in breast cancer, a recent study showed that the human intermediate PRLR (alternatively spliced isoform) is a mammary proto-oncogene capable of stimulating cell survival and proliferation (29). Many breast tumors are characterized by reduced STAT5 and high levels of PRLR expression and MAPK signal components, including AP-1 and pro-invasive matrix metalloproteinases [reviewed in (56)]. MMPs are highly invasive agents and are associated with resistance to chemotherapy and anti-estrogen treatments (72). Extracellular matrix components in the breast tumor microenvironment can also influence PRL/PRLR signaling (73, 74). In invasive breast cancer, there is a shift in PRL signaling from STAT5-mediated pathways to focal-adhesion kinase and MAPK pathways, thereby favoring proliferation (73, 74). PRL signaling in high-density stiff collagen matrices increases MMP expression, thereby promoting cellular motility. Therefore, the tumor microenvironment may be responsible for favoring one signaling pathway over another (73, 74). There is a complex interplay between PRLR and estrogen receptor (ERα), and there is an important role for the tumor microenvironment. The co-expression of PRLR and ERα in a non-compliant, rigid matrix is associated with increased tumor invasiveness and reduced responsiveness to estrogen antagonists (75). PRLR signaling can also induce motility and invasion of T47D breast cancer cells by activating downstream effectors such as TEC and NEK3 kinases, thus leading to cytoskeletal and focal adhesion reorganization (54, 76). Several studies using breast cancer cells have shown that PRL activates unliganded ERα through phosphorylation at the Ser118 and Ser167 residues. The activation of ERα promotes ligand-independent transcriptional initiation of ERE-dependent target genes, which seems to be an important factor in the proliferative and transcriptional actions of PRL in breast cancer cells (70, 77, 78). The most significant transcription factor in PRL/PRLR signaling is STAT, which regulates the growth, differentiation, and survival of mammary tissue. STAT3 and STAT5 are activated/overexpressed in several types of cancers, including breast cancer [reviewed in (79, 80)]. STAT5 can act as both a tumor suppressor and an oncogene in breast cancer under different circumstances. In ER-positive breast cancer, STAT5 expression enhanced the response to hormone therapy and increased the overall survival of patients [reviewed in (81)]. Recent studies have shown that phosphorylation of STAT5a serine residues (S726 and S780) may regulate its activity to promote cell proliferation in MCF-7 cells (82). Reports have indicated that STAT5 acts as a suppressor of breast cancer invasion and metastatic progression and can be used as a tumor marker of favorable prognosis [reviewed in (79)]. STAT5 is progressively inactivated with the progression to metastatic breast cancer due to enhanced regulation by tyrosine phosphatases (83). The activation of STAT5 in breast cancer cells promotes homotypic adhesion and inhibits the invasive characteristics of cells (84).
PRL and cyclin-dependent kinase 7 (CDK7) in estrogen-induced upregulation of PRLR in breast cancer cells
We demonstrated the essential role of endogenous PRL in the upregulation of the PRLR promoter, which involves the requisite participation of E2/ERα at the hPIII promoter along with STAT5a (Figure 5). Phosphorylated STAT5a, which associates with its functional element at hPIII, interacts with non-DNA-bound E2/ERα, which in turn associates in a complex with Sp1 and C/EBPβ bound to their cognate DNA sites at the PRLR hPIII promoter [ (85); Figure 5]. We have shown in MCF-7 cells that E2 induces ERα phosphorylation at S118 via CDK7 kinase and greatly increases the recruitment of E2/ERα to the hPIII promoter over basal unliganded ERα (86). Phosphorylation of ERα at S118 is necessary for its association with the Sp1-C/EBPβ complex and its interaction with STAT5a. Inhibition by the specific CDK7 inhibitor THZ1 markedly reduced E2-induced ERα phosphorylation at S118, while the JAK2 inhibitor AG490 or MEK inhibitor U0219, which inhibits downstream JAK2-induced pathways known to phosphorylate unliganded ERα at S118 and S167, had no effect. Targeting CDK7 kinase, which is known to regulate both transcription and the cell cycle, and ERα phosphorylation with the THZ1 inhibitor was found to effectively inhibit the transcription of PRLR and cell migration in breast cancer cells (85). Our studies may provide insights for therapeutic approaches that will mitigate the transcription/expression of PRLR and its participation in breast cancer progression fueled by E2 and PRL via their cognate receptors (Figure 5).
Crosstalk between PRLR and other receptors in breast cancer
Studies have indicated that PRLR signaling crosstalk with other receptors can influence signal transduction. PRLR interacts with integrin via the signal regulatory protein alpha transmembrane glycoprotein and SHP2 (87). PRL/PRLR and E2/ER synergistically can regulate the gene expression and proliferation of breast cancer cells (87). Furthermore, PRLR signaling tends to activate the unliganded ER (70, 77). PRL and estrogen cooperatively induce phosphorylation of ERK1/2 and enhance prolonged activation of AP-1 in breast cancer cells (88). This type of signaling pathway crosstalk can promote breast cancer progression and chemotherapeutic resistance. Crosstalk occurs between PRLR and EGFR/HER2 (Figure 7). PRLR can activate HER2 signaling via JAK2 (70, 85). We have demonstrated that PRL/PRLR induces HER2 phosphorylation at Tyr residues 1221 and 1222 through JAK2, thereby activating downstream PI3K/AKT pathways in both MCF-7 and T47D cells (70). This crosstalk between PRLR and HER2 signaling further facilitates the phosphorylation of ERα, its recruitment to the PRLR promoter and upregulation of PRLR transcription. Interestingly, we also found that EGF/EGFR in MCF-7 and T47D cells can induce PRLR transcription via downstream MAPK and PI3K signaling pathways (Figure 7). Crosstalk between the PRLR and progesterone receptor (PR) signaling pathways has been shown to be relevant to both breast development and progression. Both PR and STAT5a are key transcription factors in these pathways and have been shown to be mediators of breast cancer stem cell outgrowth (89). This evidence, coupled with their established function in the same transcriptional complexes at phospho-PR-target genes with high cancer relevance, supports the importance of PR-PRLR crosstalk [reviewed in (90)].
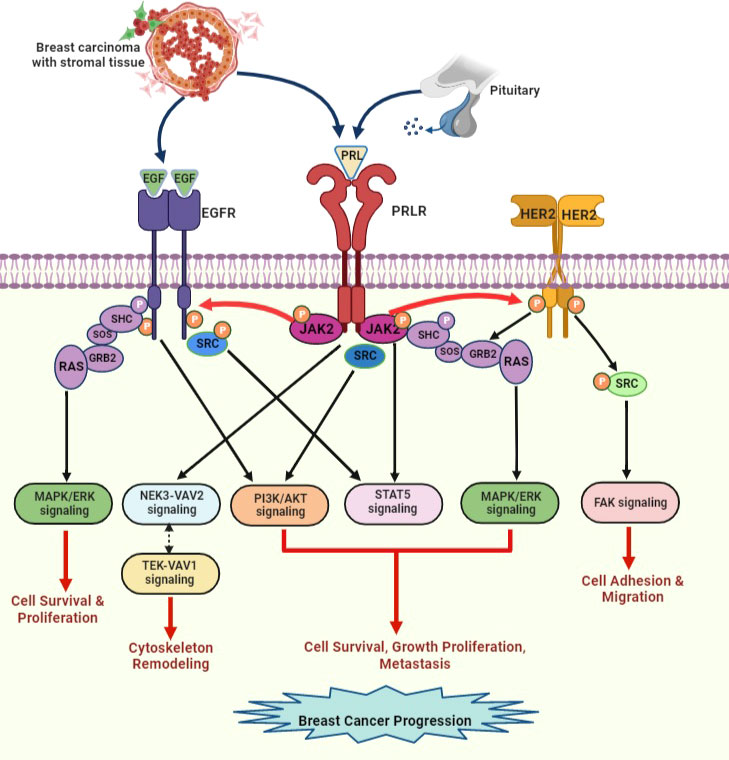
Figure 7 PRLR and EGFR/HER2 signaling crosstalk in breast cancer. EGF released by stromal microenvironment surrounding the breast tumor activates signaling cascades that overlap with PRLR signaling cascades upon activation with PRL secreted by breast tumor cells. PRL stimulates HER2 and EGFR signaling pathways via JAK2. EGF/EGFR also activates STAT5 signaling indirectly via s-SRC. This crosstalk between receptors can increase progression of breast tumor and endocrine resistance [adapted from reference (44)].
Resistance to endocrine therapy in breast cancer and future perspectives
Endocrine therapy is one of most effective forms of targeted adjuvant therapy for hormone receptor-positive breast cancer. Adjuvant therapy has been well established with different types of antiestrogens, including selective ER modulators (tamoxifen, raloxifene), which block the activity of ER, selective ER downregulators, such as fulvestrant, which causes destabilization and degradation of ER, and the third generation of aromatase inhibitors (anastrozole, letrozole and exemestane), which reduce the production of E2 in tumors [ (91), reviewed in (92)]. Although these endocrine therapies for women with ER+/PR+ breast cancer have led to substantial improvements, a significant number of cancer patients develop either intrinsic resistance or acquired resistance, which often results in tumor relapse. There can be multiple reasons for this endocrine resistance which includes mutations of ER, enhanced MAPK and PI3K/mTOR signaling pathways. Other aspects of endocrine resistance could result from overexpression of HER2, and crosstalk of ER with bypass signaling pathways, such as the EGFR/HER2 and PRLR signaling pathways [reviewed in (93–96)]. To overcome HER2 hyperactivation, trastuzumab is still being used as the most effective form of treatment for ER+ and HER+ breast cancer patients. However, some cancer patients develop resistance to trastuzumab and tumor relapse within one year of treatment. Hyperactivation of HER2-induced downstream PI3K/AKT signaling is often observed in trastuzumab-resistant breast cancer patients [reviewed in (97, 98)]. HER2-targeted therapies have been established in recent years, including tyrosine kinase inhibitors, such as lapatinib, neratinib, tucatinib, and pyrotinib (99). Together, these drugs targeting multiple receptors, such as HER2, EGFR and HER4, were studied in the early and advanced stages of breast cancer and revealed some promising outcomes. Furthermore, in clinical trials, the combination of HER family inhibitors with endocrine therapy has been shown to have better results [reviewed in (100)]. PRLR and EGFR/HER2 crosstalk, which greatly increases the activation of the RAS/ERK and PI3K/AKT pathways, are associated with poor prognosis and therapeutic resistance in breast tumor patients. In the case of PRLR, only a few attempts have successfully developed a potential therapeutic small molecule inhibitor or monoclonal antibody (LFA102) to block PRLR signaling induced cell proliferation in breast cancer cell lines (101). Therefore, simultaneous treatments targeting both the HER2 and PRLR signaling cascades may offer better outcomes by efficiently hindering breast tumor progression and ameliorating endocrine resistance. A study using G129R (PRLR antagonist) and trastuzumab (monoclonal antibody targeting HER2) as a combination therapy to inhibit HER2+ breast cancer cells and a nude mouse xenograft model showed inhibition of cell proliferation (102). Additionally, combining PI3K/AKT/mTOR pathway inhibitors with endocrine therapy has been shown to potentially reverse resistance to trastuzumab in HER2+ patients and metastatic breast cancer in early clinical trials. A rational combination of therapeutic agents based on the disease profile would be more beneficial to breast cancer patients [reviewed in (103)].
Concluding remarks
PRL is a pleiotropic hormone that plays a crucial role in mammary gland development, lactogenesis, reproduction and immune function. It mediates its actions through PRLR, a member of the lactogen/cytokine receptor family. PRLR activation induces JAK/STAT and mitogen-activated protein kinase signaling pathways implicated in the development of mammary glands and etiology of breast cancer. In this review, we provide an overview of the current understanding of the complex organization of the human PRLR gene and its transcriptional regulation. Preclinical data, epidemiological studies, and patient tumor tissues analyses strongly support the contribution of PRL/PRLR to breast tumorigenesis and cancer progression. This review also stresses the importance of signal transduction pathways (PI3K/AKT, RAF/MEK/ERK, FAK, and SFK) activated by PRL/PRLR in breast cancer. We have summarized how steroid hormones (E2 and PR) and growth factors (EGF/ERBB1 and HER2) can induce the transcription of PRLR, thereby increasing its expression in breast cancer cells and promoting cell proliferation. Therefore, in this era of precision medicine, we conclude that combination therapy involving pathway-selective kinase inhibitors and PRLR inhibitors depending on the status of the breast cancer can provide better outcomes in clinical studies.
Authors contributions
RK and MD were responsible for manuscript writing and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health Intramural Research Program through the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Acknowledgments
The authors thank all laboratory members who participated in the studies covered by this review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bern HA, Nicoll CS. The comparative endocrinology of prolactin. Recent Prog Horm Res (1968) 24:681–720. doi: 10.1016/B978-1-4831-9827-9.50019-8
2. Freeman ME, Kanyicska B, Lerant A, Nagy G. Prolactin: Structure, function, and regulation of secretion. Physiol Rev (2000) 80(4):1523–631. doi: 10.1152/physrev.2000.80.4.1523
3. Ben-Jonathan N, Mershon JL, Allen DL, Steinmetz RW. Extrapituitary prolactin: distribution, regulation, functions, and clinical aspects. Endocr Rev (1996) 17(6):639–69. doi: 10.1210/edrv-17-6-639
4. Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev (1998) 19(3):225–68. doi: 10.1210/edrv.19.3.0334
5. Boutin JM, Jolicoeur C, Okamura H, Gagnon J, Edery M, Shirota M, et al. Cloning and expression of the rat prolactin receptor, a member of the growth hormone/prolactin receptor gene family. Cell (1988) 53(1):69–77. doi: 10.1016/0092-8674(88)90488-6
6. Boutin JM, Edery M, Shirota M, Jolicoeur C, Lesueur L, Ali S, et al. Identification of a cDNA encoding a long form of prolactin receptor in human hepatoma and breast cancer cells. Mol Endocrinol (1989) 3(9):1455–61. doi: 10.1210/mend-3-9-1455
7. Zhang R, Buczko E, Tsai-Morris CH, Hu ZZ, Dufau ML. Isolation and characterization of two novel rat ovarian lactogen receptor cDNA species. Biochem Biophys Res Commun (1990) 168(2):415–22. doi: 10.1016/0006-291X(90)92337-Y
8. Bugge K, Papaleo E, Haxholm GW, Hopper JT, Robinson CV, Olsen JG, et al. A combined computational and structural model of the full-length human prolactin receptor. Nat Commun (2016) 7:11578. doi: 10.1038/ncomms11578
9. Hu ZZ, Zhuang L, Meng J, Leondires M, Dufau ML. The human prolactin receptor gene structure and alternative promoter utilization: the generic promoter hPIII and a novel human promoter hP(N). J Clin Endocrinol Metab (1999) 84(3):1153–6. doi: 10.1210/jcem.84.3.5659
10. Hu ZZ, Meng J, Dufau ML. Isolation and characterization of two novel forms of the human prolactin receptor generated by alternative splicing of a newly identified exon 11. J Biol Chem (2001) 276(44):41086–94. doi: 10.1074/jbc.M102109200
11. Brooks CL. Molecular mechanisms of prolactin and its receptor. Endocr Rev (2012) 33(4):504–25. doi: 10.1210/er.2011-1040
12. Kang JH, Hassan SA, Zhao P, Tsai-Morris CH, Dufau ML. Impact of subdomain D1 of the short form S1b of the human prolactin receptor on its inhibitory action on the function of the long form of the receptor induced by prolactin. Biochim Biophys Acta (2014) 1840(7):2272–80. doi: 10.1016/j.bbagen.2014.04.006
13. Clevenger CV, Furth PA, Hankinson SE, Schuler LA. The role of prolactin in mammary carcinoma. Endocr Rev (2003) 24:1–27. doi: 10.1210/er.2001-0036
15. Ramírez-de-Arellano A, Villegas-Pineda JC, Hernández-Silva CD, Pereira-Suárez AL. The relevant participation of prolactin in the genesis and progression of gynecological cancers. Front Endocrinol (2021) 12:747810. doi: 10.3389/fendo.2021.747810
16. Buckley AR, Putnam CW, Russell DH. Prolactin as a mammalian mitogen and tumor promoter. Adv Enzyme Regul (1988) 27:371–91. doi: 10.1016/0065-2571(88)90027-1
17. Tworoger SS, Hankinson SE. Prolactin and breast cancer etiology: An epidemiologic perspective. J Mammary Gland Biol Neoplasia (2008) 13:41–3. doi: 10.1007/s10911-008-9063-y
18. Bernichtein S, Touraine P, Goffin V. New concepts in prolactin biology. J Endocrinol (2010) 206(1):1–11. doi: 10.1677/JOE-10-0069
19. Goffin V. Prolactin receptor targeting in breast and prostate cancers: New insights into an old challenge. Pharmacol Ther (2017) 179:111–26. doi: 10.1016/j.pharmthera.2017.05.009
20. Tworoger SS, Eliassen AH, Zhang X, Qian J, Sluss PM, Rosner BA, et al. A 20-year prospective study of plasma prolactin as a risk marker of breast cancer development. Cancer Res (2013) 73:4810–9. doi: 10.1158/0008-5472.CAN-13-0665
21. O'Sullivan CC, Bates SE. Targeting prolactin receptor (PRLR) signaling in PRLR-positive breast and prostate cancer. Oncol (2016) 21,5:523–6. doi: 10.1634/theoncologist.2016-0108
22. Bogorad RL, Courtillot C, Mestayer C, Bernichtein S, Harutyunyan L, Jomain JB, et al. Identification of a gain-of-function mutation of the prolactin receptor in women with benign breast tumors. Proc Natl Acad Sci U.S.A. (2008) 105(38):14533–8. doi: 10.1073/pnas.0800685105
23. Ouhtit A, Morel G, Kelly PA. Visualization of gene expression of short and long forms of prolactin receptor in the rat. Endocrinology (1993) 133(1):135–44. doi: 10.1210/endo.133.1.8319561
24. Royster M, Driscoll P, Kelly PA, Freemark M. The prolactin receptor in the fetal rat: cellular localization of messenger ribonucleic acid, immunoreactive protein, and ligand-binding activity and induction of expression in late gestation. Endocrinology (1995) 136(9):3892–900. doi: 10.1210/endo.136.9.7649097
25. Dagil R, Knudsen MJ, Olsen JG, O'Shea C, Franzmann M, Goffin V, et al. The WSXWS motif in cytokine receptors is a molecular switch involved in receptor activation: insight from structures of the prolactin receptor. Structure (2012) 20(2):270–82. doi: 10.1016/j.str.2011.12.010
26. Haxholm GW, Nikolajsen LF, Olsen JG, Fredsted J, Larsen FH, Goffin V, et al. Intrinsically disordered cytoplasmic domains of two cytokine receptors mediate conserved interactions with membranes. Biochem J (2015) 468(3):495–506. doi: 10.1042/BJ20141243
27. Tsai-Morris CH, Dufau ML. PRLR (prolactin receptor). In: Atlas Genet cytogenet oncol haematol (2011). Available at: http://atlasgeneticsoncology.org/gene/42891/
28. Ali S, Pellegrini I, Kelly PA. A prolactin-dependent immune cell line (Nb2) expresses a mutant form of prolactin receptor. J Biol Chem (1991) 266(30):20110–7. doi: 10.1016/S0021-9258(18)54897-3
29. Grible JM, Zot P, Olex AL, Hedrick SE, Harrell JC, Woock AE, et al. The human intermediate prolactin receptor is a mammary proto-oncogene. NPJ Breast Cancer (2021) 7(1):37. doi: 10.1038/s41523-021-00243-7
30. Pujianto DA, Curry BJ, Aitken RJ. Prolactin exerts a prosurvival effect on human spermatozoa via mechanisms that involve the stimulation of akt phosphorylation and suppression of caspase activation and capacitation. Endocrinology (2010) 151(3):1269–79. doi: 10.1210/en.2009-0964
31. Qazi AM, Tsai-Morris CH, Dufau ML. Ligand-independent homo- and heterodimerization of human prolactin receptor variants: inhibitory action of the short forms by heterodimerization. Mol Endocrinol (2006) 20(8):1912–23. doi: 10.1210/me.2005-0291
32. Tan D, Walker AM. Short form 1b human prolactin receptor down-regulates expression of the long form. J Mol Endocrinol (2010) 44(3):187–94. doi: 10.1677/JME-09-0101
33. Tan D, Huang KT, Ueda E, Walker AM. S2 deletion variants of human PRL receptors demonstrate that extracellular domain conformation can alter conformation of the intracellular signaling domain. Biochemistry (2008) 47(1):479–89. doi: 10.1021/bi7013882
34. Binart N, Bachelot A, Bouilly J. Impact of prolactin receptor isoforms on reproduction. Trends Endocrinol Metab (2010) 21(6):362–8. doi: 10.1016/j.tem.2010.01.008
35. Meng J, Tsai-Morris CH, Dufau ML. Human prolactin receptor variants in breast cancer: low ratio of short forms to the long-form human prolactin receptor associated with mammary carcinoma. Cancer Res (2004) 64(16):5677–82. doi: 10.1158/0008-5472.CAN-04-1019
36. Xie YL, Hassan SA, Qazi AM, Tsai-Morris CH, Dufau ML. Intramolecular disulfide bonds of the prolactin receptor short form are required for its inhibitory action on the function of the long form of the receptor. Mol Cell Biol (2009) 29(10):2546–55. doi: 10.1128/MCB.01716-08
37. Devi YS, Shehu A, Halperin J, Stocco C, Le J, Seibold AM, et al. Prolactin signaling through the short isoform of the mouse prolactin receptor regulates DNA binding of specific transcription factors, often with opposite effects in different reproductive issues. Reprod Biol Endocrinol (2009) 7:87. doi: 10.1186/1477-7827-7-87
38. Hu ZZ, Zhuang L. Dufau prolactin receptor gene diversity: Structure and regulation. Trends Endocrinol Metab (1998) 9:94–102. doi: 10.1016/S1043-2760(98)00027-7
39. Hu ZZ, Zhuang L, Meng J, Tsai-Morris CH, Dufau ML. Complex 5' genomic structure of the human prolactin receptor: multiple alternative exons 1 and promoter utilization. Endocrinology (2002) 143(6):2139–42. doi: 10.1210/endo.143.6.8949
40. Dong J, Tsai-Morris CH, Dufau ML. A novel estradiol/estrogen receptor alpha-dependent transcriptional mechanism controls expression of the human prolactin receptor. J Biol Chem (2006) 281(27):18825–36. doi: 10.1074/jbc.M512826200
41. Kang J-H, Tsai-Morris C-H, Dufau ML. Complex formation and interactions between transcription factors essential for human prolactin receptor gene transcription. Mol Cell Biol (2011) 31(16):3208–22. doi: 10.1128/MCB.05337-11
42. Gutzman JH, Miller KK, Schuler LA. Endogenous human prolactin and not exogenous human prolactin induces estrogen receptor alpha and prolactin receptor expression and increases estrogen responsiveness in breast cancer cells. J Steroid Biochem Mol Biol (2004) 88(1):69–77. doi: 10.1016/j.jsbmb.2003.10.008
43. Goldhar AS, Duan R, Ginsburg E, Vonderhaar BK. Progesterone induces expression of the prolactin receptor gene through cooperative action of Sp1 and C/EBP. Mol Cell Endocrinol (2011) 335(2):148–57. doi: 10.1016/j.mce.2011.01.004
44. Kavarthapu R, Anbazhagan R, Dufau ML. Crosstalk between PRLR and EGFR/HER2 signaling pathways in breast cancer. Cancers (Basel) (2021) 13(18):4685. doi: 10.3390/cancers13184685
45. Lebrun JJ, Ali S, Goffin V, Ullrich A, Kelly PA. A single phosphotyrosine residue of the prolactin receptor is responsible for activation of gene transcription. Proc Natl Acad Sci U.S.A. (1995) 92(9):4031–5. doi: 10.1073/pnas.92.9.4031
46. Finidori J, Kelly PA. Cytokine receptor signalling through two novel families of transducer molecules: Janus kinases, and signal transducers and activators of transcription. J Endocrinol (1995) 147(1):11–23. doi: 10.1677/joe.0.1470011
47. Horseman ND, Yu-Lee LY. Transcriptional regulation by the helix bundle peptide hormones: Growth hormone, prolactin, and hematopoietic cytokines. Endocr Rev (1994) 15(5):627–49. doi: 10.1210/edrv-15-5-627
48. Ihle JN. STATs: signal transducers and activators of transcription. Cell (1996) 84(3):331–4. doi: 10.1016/S0092-8674(00)81277-5
49. Das R, Vonderhaar BK. Involvement of SHC, GRB2, SOS and RAS in prolactin signal transduction in mammary epithelial cells. Oncogene (1996) 13(6):1139–45.
50. Das R, Vonderhaar BK. Activation of raf-1, MEK, and MAP kinase in prolactin responsive mammary cells. Breast Cancer Res Treat (1996) 40(2):141–9. doi: 10.1007/BF01806209
51. Ihle JN. STATs and MAPKs: Obligate or opportunistic partners in signaling. Bioessays (1996) 18(2):95–8. doi: 10.1002/bies.950180204
52. al-Sakkaf KA, Dobson PR, Brown BL. Prolactin induced tyrosine phosphorylation of p59fyn may mediate phosphatidylinositol 3-kinase activation in Nb2 cells. J Mol Endocrinol (1997) 19(3):347–50. doi: 10.1677/jme.0.0190347
53. Miller SL, DeMaria JE, Freier DO, Riegel AM, Clevenger CV. Novel association of Vav2 and Nek3 modulates signaling through the human prolactin receptor. Mol Endocrinol (2005) 19(4):939–49. doi: 10.1210/me.2004-0443
54. Miller SL, Antico G, Raghunath PN, Tomaszewski JE, Clevenger CV. Nek3 kinase regulates prolactin-mediated cytoskeletal reorganization and motility of breast cancer cells. Oncogene (2007) 26(32):4668–78. doi: 10.1038/sj.onc.1210264
55. Reynolds C, Montone KT, Powell CM, Tomaszewski JE, Clevenger CV. Expression of prolactin and its receptor in human breast carcinoma. Endocrinology (1997) 138:5555–60. doi: 10.1210/endo.138.12.5605
56. Gorvin CM. The prolactin receptor: Diverse and emerging roles in pathophysiology. J Clin Transl Endocrinol (2015) 2(3):85–91. doi: 10.1016/j.jcte.2015.05.001
57. Tran-Thanh D, Arneson NC, Pintilie M, Deliallisi A, Warren KS, Bane A, et al. Amplification of the prolactin receptor gene in mammary lobular neoplasia. Breast Cancer Res Treat (2011) 128:31–40. doi: 10.1007/s10549-010-1025-6
58. Tikk K, Sookthai D, Johnson T, Rinaldi S, Romieu I, Tjønneland A, et al. Circulating prolactin and breast cancer risk among pre- and postmenopausal women in the EPIC cohort. Ann Oncol (2014) 25(7):1422–8. doi: 10.1093/annonc/mdu150 Rinaldi S, Romieu I, Tjønneland A
59. Hankinson SE, Willett WC, Michaud DS, Manson JE, Colditz GA, Longcope C. Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst (1999) 91:629–34. doi: 10.1093/jnci/91.7.629
60. Tworoger SS, Hankinson SE. Prolactin and breast cancer risk. Cancer Lett (2006) 243:160–9. doi: 10.1016/j.canlet.2006.01.032
61. Tworoger SS, Sluss P, Hankinson SE. Association between plasma prolactin concentrations and risk of breast cancer among predominately premenopausal women. Cancer Res (2006) 66:2476–82. doi: 10.1158/0008-5472.CAN-05-3369
62. Tworoger SS, Eliassen AH, Sluss P, Hankinson SE. A prospective study of plasma prolactin concentrations and risk of premenopausal and postmenopausal breast cancer. J Clin Oncol (2007) 25:1482–8. doi: 10.1200/JCO.2006.07.6356
63. Ferreira M, Mesquita M, Quaresma M, André S. Prolactin receptor expression in gynaecomastia and male breast carcinoma. Histopathology (2008) 53(1):56–61. doi: 10.1111/j.1365-2559.2008.03059.x
64. O’Leary KA, Shea MP, Schuler LA. Modeling prolactin actions in breast cancer in vivo: Insights from the NRL-PRL mouse. Adv Exp Med Biol (2015) 846:201–20. doi: 10.1007/978-3-319-12114-7_9
65. Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA. Prolactin induces ERalpha-positive and ERalpha-negative mammary cancer in transgenic mice. Oncogene (2003) 22(30):4664–74. doi: 10.1038/sj.onc.1206619
66. Courtillot C, Chakhtoura Z, Bogorad R, Genestie C, Bernichtein S, Badachi Y. Characterization of two constitutively active prolactin receptor variants in a cohort of 95 women with multiple breast fibroadenomas. J Clin Endocrinol Metab (2010) 95:271–9. doi: 10.1210/jc.2009-1494
67. Chakhtoura Z, Laki F, Bernadet M, Cherifi I, Chiche A, Pigat N, et al. Gain-of-function prolactin receptor variants are not associated with breast cancer and multiple fibroadenoma risk. J Clin Endocrinol Metab (2016) 101(11):4449–60. doi: 10.1210/jc.2016-2372
68. Mong FY, Kuo YL, Liu CW, Liu WS, Chang LC. Association of gene polymorphisms in prolactin and its receptor with breast cancer risk in Taiwanese women. Mol Biol Rep (2011) 38:4629–36. doi: 10.1007/s11033-010-0596-y
69. Acosta JJ, Munoz RM, Gonzalez L, Subtil-Rodriguez A, Dominguez-Caceres MA, Garcia-Martinez JM, et al. Src mediates prolactin-dependent proliferation of T47D and MCF7 cells via the activation of focal adhesion kinase/Erk1/2 and phosphatidylinositol 3-kinase pathways. Mol Endocrinol (2003) 17:2268–82. doi: 10.1210/me.2002-0422
70. Kavarthapu R, Tsai Morris CH, Dufau ML. Prolactin induces up-regulation of its cognate receptor in breast cancer cells via transcriptional activation of its generic promoter by cross-talk between ERα and STAT5. Oncotarget (2014) 5:90 79–9091. doi: 10.18632/oncotarget.2376
71. Aksamitiene E, Achanta S, Kolch W, Kholodenko BN, Hoek JB, Kiyatkin A. Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell Signal (2011) 23:1794–805. doi: 10.1016/j.cellsig.2011.06.014
72. Tune BXJ, Sim MS, Poh CL, Guad RM, Woon CK, Hazarika I, et al. Matrix metalloproteinases in chemoresistance: Regulatory roles, molecular interactions, and potential inhibitors. J Oncol (2022) 2022:3249766. doi: 10.1155/2022/3249766
73. Barcus CE, et al. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. J Biol Chem (2013) 288(18):12722–32. doi: 10.1074/jbc.M112.447631
74. Barcus CE, Keely PJ, Eliceiri KW, Schuler LA. Prolactin signaling through focal adhesion complexes is amplified by stiff extracellular matrices in breast cancer cells. Oncotarget (2016) 7:48093–106. doi: 10.18632/oncotarget.10137
75. Barcus CE, Holt EC, Keely PJ, Eliceiri KW, Schuler LA. Dense collagen-I matrices enhance pro-tumorigenic estrogen-prolactin crosstalk in MCF-7 and T47D breast cancer cells. PloS One (2015) 10(1):e0116891. doi: 10.1371/journal.pone.0116891
76. Harrington KM, Clevenger CV. Identification of NEK3 kinase threonine 165 as a novel regulatory phosphorylation site that modulates focal adhesion remodeling necessary for breast cancer cell migration. J Biol Chem (2016) 291(41):21388–406. doi: 10.1074/jbc.M116.726190
77. González L, Zambrano A, Lazaro-Trueba I, Lopéz E, González JJ, Martín-Pérez J, et al. Activation of the unliganded estrogen receptor by prolactin in breast cancer cells. Oncogene (2009) 28(10):1298–308. doi: 10.1038/onc.2008.473
78. Oladimeji P, Skerl R, Rusch C, Diakonova M. Synergistic activation of ERα by estrogen and prolactin in breast cancer cells requires tyrosyl phosphorylation of PAK1. Cancer Res (2016) 76(9):2600–11. doi: 10.1158/0008-5472.CAN-15-1758
79. Halim CE, Deng S, Ong MS, Yap CT. Involvement of STAT5 in oncogenesis. Biomedicines (2020) 8:316. doi: 10.3390/biomedicines8090316
80. Walker SR, Xiang M, Frank DA. Distinct roles of STAT3 and STAT5 in the pathogenesis and targeted therapy of breast cancer. Mol Cell Endocrinol (2014) 382(1):616–21. doi: 10.1016/j.mce.2013.03.010
81. Nevalainen MT, Xie J, Torhorst J, Bubendorf L, Haas P, Kononen J, et al. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. J Clin Oncol (2004) 22:2053–60. doi: 10.1200/JCO.2004.11.046
82. Woock AE, Grible JM, Olex AL, Harrell JC, Zot P, Idowu M, et al. Serine residues 726 and 780 have nonredundant roles regulating STAT5a activity in luminal breast cancer. Sci Rep (2021) 11(1):13506. doi: 10.1038/s41598-021-92830-8
83. Johnson KJ, Peck AR, Liu C, Tran TH, Utama FE, Sjolund AB, et al. PTP1B suppresses prolactin activation of Stat5 in breast cancer cells. Am J Pathol (2010) 177(6):2971–83. doi: 10.2353/ajpath.2010.090399
84. Sultan AS, Xie J, LeBaron MJ, Ealley EL, Nevalainen MT, Rui H. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene (2005) 24(5):746–60. doi: 10.1038/sj.onc.1208203
85. Kavarthapu R, Dufau ML. Essential role of endogenous prolactin and CDK7 in estrogen-induced upregulation of the prolactin receptor in breast cancer cells. Oncotarget (2017) 8(16):27353–63. doi: 10.18632/oncotarget.16040
86. Galbaugh T, Feeney YB, Clevenger CV. Prolactin receptor-integrin cross-talk mediated by SIRPα in breast cancer cells. Mol Cancer Res (2010) 8(10):1413–24. doi: 10.1158/1541-7786.MCR-10-0130
87. Rasmussen LM, Frederiksen KS, Din N, Galsgaard E, Christensen L, Berchtold MW, et al. Prolactin and oestrogen synergistically regulate gene expression and proliferation of breast cancer cells. Endocrine-Related Cancer (2010) 17:809–22. doi: 10.1677/ERC-09-0326
88. Gutzman JH, Nikolai SE, Rugowski DE, Watters JJ, Schuler LA. Prolactin and estrogen enhance the activity of activating protein 1 in breast cancer cells: role of extracellularly regulated kinase 1/2-mediated signals to c-fos. Mol Endocrinol (2005) 19(7):1765–78. doi: 10.1210/me.2004-0339
89. Leehy KA, Truong TH, Mauro LJ, Lange CA. Progesterone receptors (PR) mediate STAT actions: PR and prolactin receptor signaling crosstalk in breast cancer models. J Steroid Biochem Mol Biol (2018) 176:88–93. doi: 10.1016/j.jsbmb.2017.04.011
90. Hagan CR, Daniel AR, Dressing GE, Lange CA. Role of phosphorylation in progesterone receptor signaling and specificity. Mol Cell Endocrinol (2012) 357:43–9. doi: 10.1016/j.mce.2011.09.017
91. Rugo HS, Rumble RB, Macrae E, Barton DL, Connolly HK, Dickler MN, et al. Endocrine therapy for hormone receptor-positive metastatic breast cancer: American society of clinical oncology guideline. J Clin Oncol (2016) 34:3069–103. doi: 10.1200/JCO.2016.67.1487
92. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers (2019) 5(1):66. doi: 10.1038/s41572-019-0111-2
93. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med (2011) 62(1):233–47. doi: 10.1146/annurev-med-070909-182917
94. Murphy CG, Dickler MN. Endocrine resistance in hormone-responsive breast cancer: mechanisms and therapeutic strategies. Endocr Relat Cancer (2016) 23(8):R337–52. doi: 10.1530/ERC-16-0121
95. Rani A, Stebbing J, Giamas G, Murphy J. Endocrine resistance in hormone receptor positive breast cancer-from mechanism to therapy. Front Endocrinol (2019) 10:245. doi: 10.3389/fendo.2019.00245
96. Kavarthapu R, Dufau ML. Role of EGF/ERBB1 in the transcriptional regulation of the prolactin receptor independent of estrogen and prolactin in breast cancer cells. Oncotarget (2016) 7(40):65602–13. doi: 10.18632/oncotarget.11579
97. Nahta R. Molecular mechanisms of trastuzumab-based treatment in HER2-overexpressing breast cancer. ISRN Oncol (2012) 2012:428062. doi: 10.5402/2012/428062
98. Orphanos G, Kountourakis P. Targeting the HER2 receptor in metastatic breast cancer. Hematol Oncol Stem Cell Ther (2012) 5:127–37. doi: 10.5144/1658-3876.2012.127
99. Schlam I, Swain SM. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. NPJ Breast Cancer (2021) 7:56. doi: 10.1038/s41523-021-00265-1
100. Zanardi E, Bregni G, de Braud F, Di Cosimo S. Better together: Targeted combination therapies in breast cancer. Semin Oncol (2015) 42(6):887–95. doi: 10.1053/j.seminoncol.2015.09.029
101. Minami H, Ando Y, Tamura K, Tajima T, Isaacs R. Phase I study of LFA102 in patients with advanced breast cancer or castration-resistant prostate cancer. Anticancer Res (2020) 40(9):5229–35. doi: 10.21873/anticanres.14526
102. Scotti ML, Langenheim JF, Tomblyn S, Springs AE, Chen WY. Additive effects of a prolactin receptor antagonist, G129R, and herceptin on inhibition of HER2-overexpressing breast cancer cells. Breast Cancer Res Treat (2008) 111(2):241–50. doi: 10.1007/s10549-007-9789-z
Keywords: prolactin receptor (PRLR), transcriptional regulation, gene structure, signal transduction, breast cancer, prolactin (PRL)
Citation: Kavarthapu R and Dufau ML (2022) Prolactin receptor gene transcriptional control, regulatory modalities relevant to breast cancer resistance and invasiveness. Front. Endocrinol. 13:949396. doi: 10.3389/fendo.2022.949396
Received: 20 May 2022; Accepted: 19 August 2022;
Published: 15 September 2022.
Edited by:
Vincent GOFFIN, INSERM U1151 Institut Necker Enfants Malades, FranceReviewed by:
Linda Schuler, University of Wisconsin-Madison, United StatesAmeae M Walker, University of California, Riverside, United States
Copyright © 2022 Kavarthapu and Dufau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria L. Dufau, ZHVmYXVtQG1haWwubmloLmdvdg==