 Yong Zhou
Yong Zhou Yang Jin
Yang Jin Yuan Wang
Yuan Wang Ruijin Wu
Ruijin Wu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 20 October 2022
Sec. Reproduction
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.945578
This article is part of the Research TopicEndometriosis: Pathogenesis, Diagnosis and Treatment, volume IIView all 10 articles
Endometriosis (EMS) is a chronic gynecological disease that affects women of childbearing age. However, the exact cause remains unclear. The uterus is a highly vascularized organ that continuously exposes endometrial cells to high oxygen concentrations. According to the “planting theory” of EMS pathogenesis, when endometrial cells fall from the uterine cavity and retrograde to the peritoneal cavity, they will face severe hypoxic stress. Hypoxic stress remains a key issue even if successfully implanted into the ovaries or peritoneum. In recent years, increasing evidence has confirmed that hypoxia is closely related to the occurrence and development of EMS. Hypoxia-inducible factor-1α (HIF-1α) can play an essential role in the pathological process of EMS by regulating carbohydrate metabolism, angiogenesis, and energy conversion of ectopic endometrial cells. However, HIF-1α alone is insufficient to achieve the complete program of adaptive changes required for cell survival under hypoxic stress, while the unfolded protein response (UPR) responding to endoplasmic reticulum stress plays an essential supplementary role in promoting cell survival. The formation of a complex signal regulation network by hypoxia-driven UPR may be the cytoprotective adaptation mechanism of ectopic endometrial cells in unfavorable microenvironments.
Endometriosis is a common benign gynecological disorder characterized by the presence and growth of functional endometrial glands and stroma outside the uterine cavity, usually accompanied by reactive fibrosis and muscular metaplasia of affected organs (1, 2). Unfortunately, only a few studies estimated the prevalence and incidence of endometriosis in the general population, reporting that the prevalence of symptomatic endometriosis is about 10%, and approximately 2-7/1000 women are diagnosed with endometriosis every year. However, 11% of cases remain undiagnosed (3, 4).
EMS is closely related to dysmenorrhea, painful intercourse, painful defecation, and infertility in women of reproductive age and may seriously affect their quality of life (5, 6). In addition, patients and physicians face the great challenge of high recurrence rates after conservative EMS surgery (7, 8). Lesions are most commonly found in the pelvic cavity, including the ovaries, uterus, sacral ligaments, vaginal septum, bladder, rectum, and ureters. EMS may also invade organs other than the pelvic cavity, such as abdominal incision (9), diaphragm, and even lungs (10).
The exact pathogenesis of EMS is still unclear. However, the implant theory is still widely accepted as the mainstream theory (11). The hypothesis is based on the concept that EMS lesions’ matrix and glands are derived from ectopic endometrium. Thus, EMS is considered a benign metastasis of ectopic endometrium, which is transferred from the uterine cavity to another position in the body through different pathways. The theory of menstrual blood reflux, based on clinical and anatomical observations, was first proposed by Sampson in 1927 (12). It is believed that most EMS were induced by endometrial fragments entering the pelvic cavity through fallopian tube reflux during menstruation, the transition from the endometrium to ectopic endometrium, and growth on peritoneum as well as ovary.
Although menstrual reflux theory cannot explain all forms of EMS, it remains the most accepted hypothesis of EMS. Some scientists found that 76~90% of women have the phenomenon of menstrual blood regurgitation of fallopian tubes (13), and laparoscopy has revealed that endometrial epithelial cells can be isolated from abdominal fluid at the early stage of endometrial hyperplasia (14). In addition, some researchers have identified viable single endometrial cells and glandular structures from the shedding of menstrual blood (15). The “eutopic endometrium determinism”, proposed by Lang (16), believes that the onset of EMS depends on the characteristics of eutopic endometrial cells in the uterine, so eutopic endometrial cells of patients with EMS may have stronger survival and proliferation ability after falling off. Thus, they are more likely to become ectopic lesions. Among this process, retrograde menstruation is the only bridge from the potential pathogenicity of endometrial cells to the onset of EMS. Recently, based on the physiological phenomenon of periodic stripping and regeneration of endometrium, it is speculated that endometrial stem cells with differentiation potential may exist in menstrual blood. Therefore, some scientists proposed the “stem cell theory” of EMS pathogenesis (17). Subsequently, relevant studies also confirmed that after menstrual blood enters the pelvic cavity from the fallopian tube, endometrial cell fragments with stem cell characteristics can adhere to mesothelial cells and promote the growth of EMS lesions, leading to the occurrence and development of EMS (18, 19). These theories enriched the theoretical basis of planting theory.
The anatomical characteristics of the uterine vessels are that the body branches of the uterine arteries vertically discharge the arch artery along with the muscle layer of the lateral wall of the uterus. The uterine spiral artery then extends vertically into the endometrium, finally forming a blood-rich capillary network in the endometrium (as shown in Figure 1). During menstruation, endometrial vasospasm causes acute hypoxia of the endometrium, leading to necrosis and endometrial dissection. Finally, blood vessels rupture and a mixture of blood and exfoliated endometrium fragments are formed. The main pathogenesis of EMS, including implant theory, menstrual blood reflux theory, endometrial determinism, and stem cell theory, all began with the shedding of endometrial fragments into the pelvic cavity. Under normal circumstances, the shedding endometrial cells immediately convert into a state of severe hypoxia when the blood supply of the endometrial capillaries is lost. Then relevant apoptotic signals are activated to induce apoptosis and endometrial cells are finally cleared by the immune cells in the pelvic. At the same time, this transient physiological hypoxia could promote the timely repair of the exfoliated endometrial surface and prevent excessive menstrual bleeding, which is mostly meditated by hypoxia-induced high expression of vascular endothelial growth factor (VEGF) (20). However, in patients with EMS, various changes of cell biological functions (21) may occur in the shedding endometrial cells under hypoxic stress, such as increased adhesive and invasive capacity, enhanced angiogenesis, and dysregulated immuno-clearance system, thus improving their ability to resist hypoxia or activating certain functions to resist apoptosis, adapt to the hypoxic environment, and survive. Eventually, they will develop into EMS lesions. Therefore, hypoxia can also be the driving factor for EMS, but the relevant pathological mechanism remains unclear.
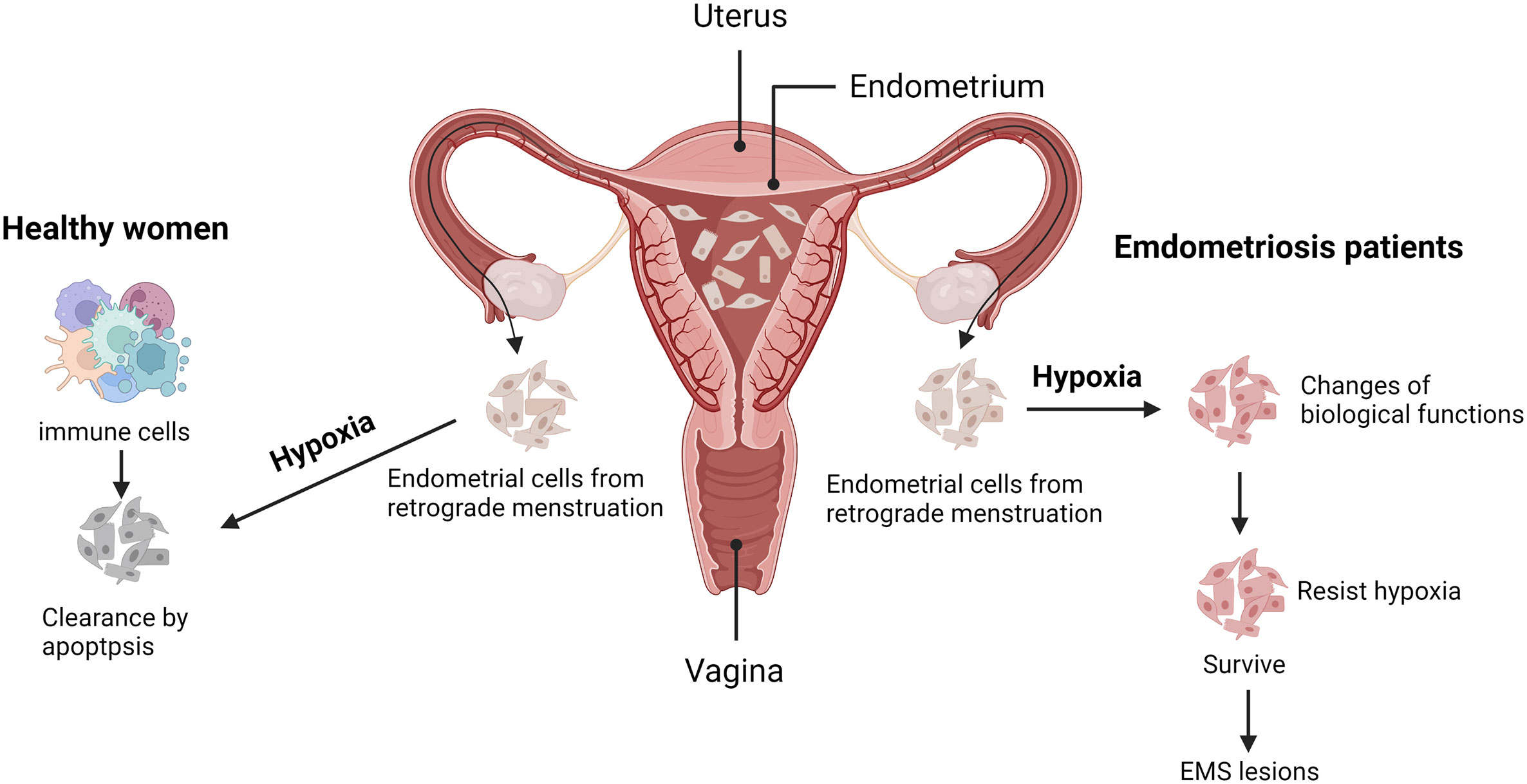
Figure 1 Possible adaptive mechanism for endometriosis under hypoxia. During menstruation, the exfoliated endometrium fragments flow into the pelvic cavity with retrograde menstruation. Under normal circumstances (as shown in the left part), the shedding endometrial cells will face acute hypoxic stress, activating apoptotic signals and being cleared by the immune cells. Conversely, those endometrial cells of endometriosis (EMS) patients (as shown in the right part), possess with altered biological functions, may resist hypoxia or apoptosis and finally develop into EMS lesions. Created with BioRender.com.
Hypoxia (22) generally refers to the pathological process in which oxygen is not adequately supplied or consumed excessively, resulting in insufficient oxygen concentration, reduced availability, and the inability to maintain normal cellular functions. If hypoxia persists, it will lead to a metabolic crisis and ultimately threaten the cell’s survival. The concentration of oxygen in the atmosphere is approximately 20%, but the content of oxygen in human tissues is much lower. Generally, the normal oxygen content of human tissues is approximately 3%~5% (as shown in Figure 2) (23), which is essential for the maintenance of cells’ normal life. When the intracellular oxygen content ranges in 1%~3%, it is called mild hypoxia. In this condition, molecular O2 will play a role as a critical signal to regulate cell fate, activating various adaptive mechanisms to promote cell survival and proliferation.
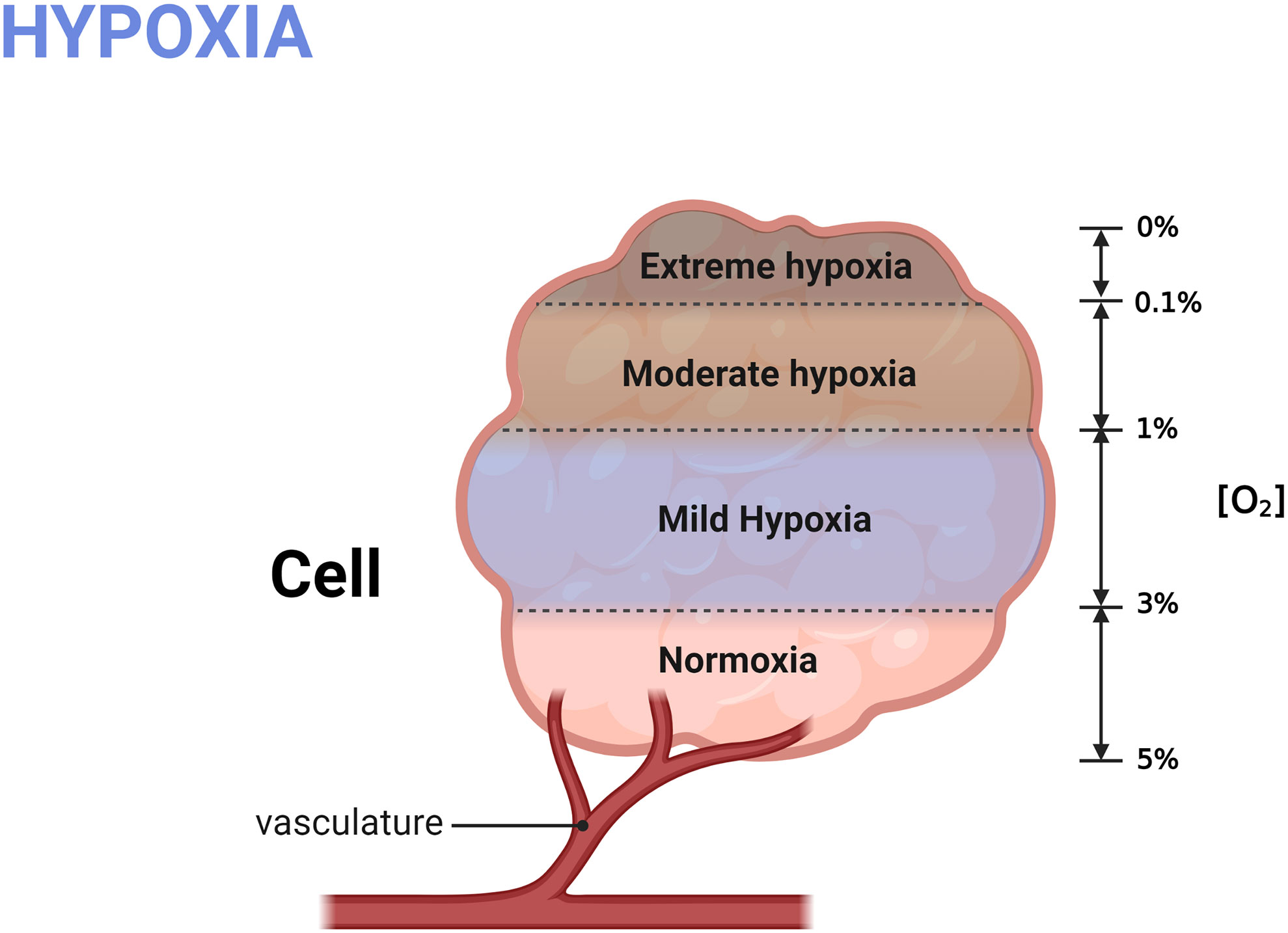
Figure 2 Grading of hypoxia. The normal oxygen content in human cell is 3%~5%; mild hypoxia refers to the oxygen concentration raging in 1%-3%; Oxygen content less than 1% is defined as severe hypoxia. Created with BioRender.com.
However, in a state of moderate hypoxia, which means that the oxygen content is less than 1%, many cells can still survive. To survive in a moderate hypoxic environment, the expression of various genes is needed to allow cells to adapt to the stress response induced by hypoxia. This process is heavily controlled by the hypoxia-inducible factors (HIFs) family. HIFs, first discovered by Semenza et al. (24) in 1992, are activated through a transcription mechanism under hypoxic conditions and then promote the expression of many hypoxia-regulated gene products to form an adaptive mechanism for cell protection. Acting as the master switch in the body in response to hypoxia, the downstream mechanisms of HIFs are multifaceted (25). For example, HIFs promotes increased secretion of erythropoietin to accelerate red blood cell production, enhancing tissue’s ability to transport oxygen, ensuring that cells have an adequate supply of oxygen (26). In addition, switching of metabolic patterns during hypoxia is essential to reduce oxygen consumption. HIFs can activate glycolytic genes and inhibit tricarboxylic acid cycle metabolism, thereby reducing oxygen consumption and maintaining cell survival in hypoxia condition (27). HIFs can also induce increased secretion of vascular endothelial growth factor (VEGF) in cells and increase the blood supply of tissues by promoting angiogenesis, thus protecting cells from ischemic damage (28). Hypoxia can also promote the synthesis of some glucose transporters and maintain the production of high-energy molecular ATP in cells (29). Overall, HIFs are the most sensitive and important nuclear transcription regulators response to hypoxia. They are prevalent in various mammalian cells even in the simplest animal (30). HIFs regulate the expression of various genes, and they are widely involved in regulating oxygen homeostasis in response to hypoxia and other changes in the cell’s internal environment of the cell. Thus, HIF-1 is a key oxygen sensor and a hypoxic adaptive response regulator.
Regarding EMS, when endometrial cells enter the pelvic cavity with retrograde menstrual blood and have not established effective blood circulation, continuous hypoxia can induce the overexpression of HIF-1 in endometrial cells, thus inducing the secretion of various factors within the endometrial environment and prompting the development of endometriosis through multiple mechanisms (31). Angiogenesis is the primary challenge for the survival of endometrial cells flowed into the pelvis, and it is also a major prerequisite for the initiation and progression of endometriosis (32). In this process, HIFs can induce the expression of numerous downstream factors to promote the angiogenesis of endometrial cells. Among them, VEGF family are the most classical factors which were found up-regulated both in vivo and in vitro (33, 34). Other cytokines and chemokines, such as IL-8, leptin, CYR61 and osteopontin also participant in the HIF-induced angiogenesis process (35–37). In addition, HIFs could also enhance cell adhesive ability through the regulation of Transforming growth factor β1 (TGF-β1), Enhancer of zeste homolog 2 (EZH2), and anthrax toxin receptor 2 (ANTXR2) (34, 38). Hypoxia also participants in the regulation of inflammation and immune system via mediating the expression of IL-6, DUPS2, and COX-2 in endometrial cells (39). Furthermore, although EMS is a benign disorder, it displays some pathogenic characteristics of malignant diseases (40), such as the tendency to invade and relapse, and adaptation to hypoxia (41). A latest research showed that the expression of early growth response 1 (EGR1) and its downstream target carbonic anhydrase 9 (CA9), which were up-regulated in hypoxic cancers, are significantly increased in ectopic lesions (42). In addition, malignant cells possess a unique energy metabolism feature which is called the “Warburg effect” (43), also known as aerobic glycolysis. Specifically, even in anoxic conditions, malignant tumor cells could remain powered by glycolysis, increasing glucose consumption and lactic acid production. This progress also provides abundant energy for the growth of malignant cells, ensuring their ability to proliferate rapidly, and HIF-1α plays a vital role in this energy metabolic pathway and may protect cells from hypoxia (44). Interestingly, Young et al. (45) found that the “Warburg effect” also existed in the EMS lesion, and HIF-1α also showed same effect on the aerobic glycolysis of EMS.
Recent evidence suggests that HIFs alone cannot achieve the whole program of adaptive changes required for cell survival under hypoxic stress and the unfolded protein response (UPR) under endoplasmic reticulum stress (ERS) plays an essential complementary role in this process (46, 47). UPR and HIF pathway interacts with HIF-independent pathways, forming a highly correlated regulatory network under hypoxia stress.
The endoplasmic reticulum (ER) is an extensive intracellular membranous network extending to the entire cytoplasm. The key site for lipid and glucose metabolism, calcium homeostasis, detoxifying drugs, and metabolisms is the central processing unit responsible for protein translation, folding, and modification (48). In protein synthesis, folding the protein spatial structure depends on the oxygen content, which is also called oxidizing protein folding (49). When intracellular oxygen availability is reduced (hypoxia), protein folding will be disturbed, leading to accumulation of misfolded or unfolded proteins. These changes break the protein dynamics in the endoplasmic reticulum and activate the UPR signaling network, thus inducing the production of self-protection mechanisms in cells. This process is called endoplasmic reticulum stress (ERS), which aims to restore homeostasis and function of the intracellular environment (50–52).
UPR is mediated by a partner molecule specific for the endoplasmic reticulum, namely glucose-regulated protein78(GRP78), and three transmembrane protein stress sensors (53), namely protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring protein 1α (IRE1α)(as shown in Figure 3). UPR can avoid the ERS-caused damage by reducing the accumulation of unfolded proteins and improving the correct folding of proteins, and restoring normal physiological functions of cells. However, if ERS becomes persistent or damage is too severe, the signal will change from pro-survival to pro-apoptosis (54). Under normal physiological conditions, these three UPR-related transmembrane proteins all bind to GRP78 and remain in an inactive state. When ERS damage occurs, GRP78 will dissociate from these transmembrane proteins (55). Activated PERK phosphorylates the α subunit of eukaryotic initiation factor 2 (eIF2), initiating selective translation of activating transcription factor 4 (ATF4) under stress conditions. ATF4 is mainly involved in regulating peptide chain biosynthesis, antioxidant action, protein folding, and gene expression related to the maintenance of redox homeostasis (56). If the stress persists, ATF4 can also promote the transcription of C/EBP homologous protein (CHOP), a pro-apoptotic protein, to induce cell apoptosis (57). Activated ATF6 can also initiate related transcriptional procedures to restore endoplasmic reticulum homeostasis, including inducing GRP78 expression, promoting protein chaperone and lipid synthesis, stimulating endoplasmic reticulum degradation, and improving N-glycosylation (58, 59), and ATF6 also induces CHOP expression, leading to UPR-related apoptosis (60, 61).
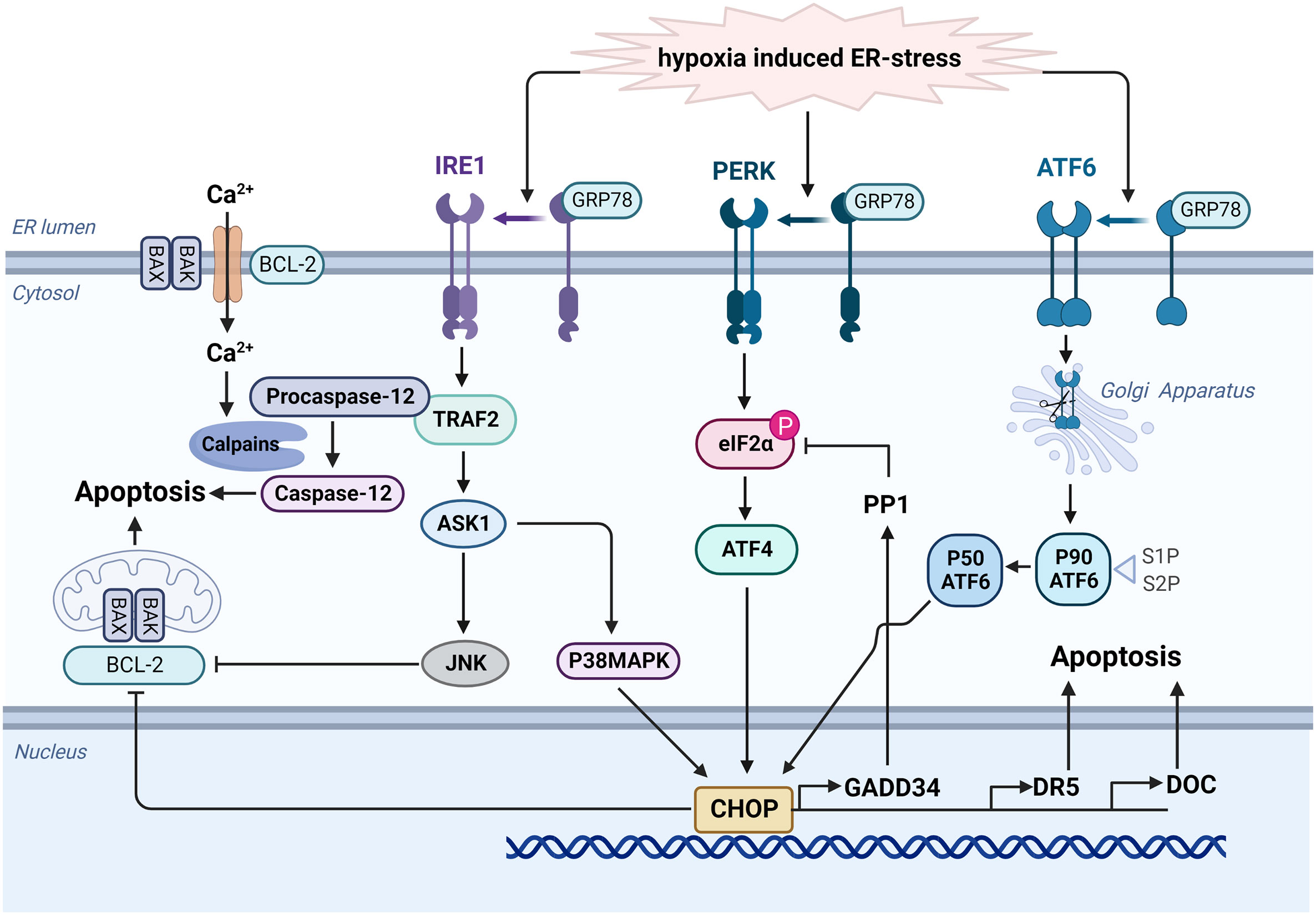
Figure 3 Hypoxia activates the UPR signaling network to mediate apoptosis. When ERS damage occurs, GRP78 dissociates from PERK, ATF6 and IRE1α. Activated ATF6 can be transported to Golgi apparatus. After modification, it could induce CHOP expression and lead to UPR-related apoptosis via the inhibition of BCL-2 protein family and the activation of multiple genes including GADD34, DR5, and DOC. Similarly, activated PERK phosphorylates the α subunit of eIF2 and initiates translation of ATF4, then promoting the transcription of CHOP. Additionally, activated IRE1α stimulates the expression of TRAF2, then activate ASK1 and JNK to promote apoptosis. TRAF2 also promotes clustering of procaspase-12, and it will be activated into caspase 12 by calpains under ER stress. Created with BioRender.com.
Activated IRE1α regulates gene expression by increasing ER protein folding capacity via TRAF2. It also promotes the expression of proteins related to disulfide bond formation and molecular chaperones and proteins involved in ER degradation and vesicle transport (62). TRAF2 also promotes clustering of procaspase-12. Under ER stress, sustained Ca2+ release from the ER and activate calpains, which may induce the activation of caspase 12 to mediate apoptosis (63). In addition, IRE1α kinase can up-regulated the expression of apoptosis signal regulated kinase 1(ASK1) and then activate c-Jun N-terminal kinase (JNK) to promote apoptosis through the inhibition of BCL-2 protein family (64, 65). CHOP, a member of C/EBP transcription factor family, is an ERS-specific effector molecule representing an important cell transition signal to apoptosis (66). Under normal physiological conditions, CHOP expression remains at a very low level, while in the ERS state, activation of PERK, ATF6, and IRE1α can induce CHOP transcription, significantly increasing its expression and migration into the nucleus, promoting cell apoptosis via the inhibition of BCL-2 protein family (67, 68). CHOP also prompt the expression of GADD34 (69), which complexes with protein phosphatase 1 (PP1) to induce the dephosphorylation of eIF2α, thus forming a negative feedback loop. Other downstream target genes, including death receptor 5 (DR5) and downstream of CHOP (DOC) have also been proposed to lead to apoptosis (70).
Since protein synthesis and oxygen-dependent protein folding are energy-intensive processes, and hypoxia significantly reduces the level of intracellular ATP, regulating mRNA translation is an important cellular response to hypoxia (27). Hypoxia activates PERK, leading to the inhibition of eIF2α phosphorylation and overall translation, while ATF4 translation increases after PERK/eIF2α is activated (71, 72). This is a rapid response independent of HIF-1α and usually occurs within minutes of exposure to hypoxic conditions in cells. The phosphorylation of eIF2α is transient, attributed to the negative feedback caused by GADD34 dependent on ATF4 upregulation. Dephosphorylation of eIF2α will increase the production of various intracellular reactive oxygen species (ROS), thus stimulating various biological reactions, while mitochondria are the main source of oxygen-deficient ROS (73). Mitochondrial hypoxic ROS activates and integrates the stress response to maintain energy and REDOX homeostasis and then constitutes an early adaptive response to hypoxia. Some enzyme antioxidants, such as catalase and glutathione peroxidase, can reduce eIF2α phosphorylation due to hypoxia. In contrast, ATF4 can promote cell survival by enhancing the upregulation of HIF-1α-mediated downstream targets (74). Brief exposure to ERS can modulate cells and enable them to survive in more severe stress. Survival-promoting genes may induce this pre-adaptation, and integration of stress response is also a sufficient survival-promoting mechanism under hypoxia. Cells with impaired PERK-eIF2a-ATF4 signal transduction are more sensitive to hypoxic stress in vitro, indicating that the PERK-eIF2a-ATF4 pathway provides a survival advantage for cells under hypoxic conditions (75, 76), which is crucial for resistance to intracellular hypoxia, metabolic stress, and starvation.
Scientists have analyzed variations of gene expression under hypoxia conditions, but UPR-related genes can be excessively induced in the state of “extreme” hypoxia or “moderate” hypoxia. For example, X-box binding protein 1 (XBP1), a transcription factor containing zinc finger structure, is critical for UPR signaling network and acts on the folding of multiple proteins regulated by downstream target molecules. Under normal conditions, XBP1 exists in XBP1 unspliced (XBP1u). In the hypoxia state, XBP1u is activated into XBP1 spliced (XBP1s) by IRE1, enabling effective transcription of multiple target genes in the nucleus. Meanwhile, hypoxia can also induce XBP1 expression and activate its mRNA splicing in a HIF-1α dependent pathway, leading to increased XBP1s (77). When XBP1s were co-localized in tumors with hypoxia markers, the loss of XBP1 increased the sensitivity of transformed cells to hypoxia-induced apoptosis and inhibited tumor growth (78). Studies have demonstrated that XBP1 is critical for carcinogenicity and progression of triple-negative breast cancer (TNBC). TNBC is a type of breast cancer that lacks estrogen receptors, progesterone, and HER2, in which HIF-1α is overactivated. However, XBP1 splicing is not directly regulated by HIF-1α. XBP1 mainly enhances the transcriptional activity of HIF-1α and regulates its transcriptional program by binding to HIF-1α and forming the transcription complex to drive the carcinogenicity of TNBC. In contrast, XBP1 knockout can reduce the formation of breast cancer lesions under hypoxia conditions. The characteristics of XBP1 gene expression in TNBC patients are closely related to those in the state of hypoxia, indicating a poor prognosis.
UPR is a protection mechanism to maintain the stability of the intracellular environment under hypoxia circumstances, and its activation will promote cell survival. To determine the role of UPR in the pathogenesis of EMS, Guzel (79) et al. detected GRP78 expression in normal and ectopic endometrial cells, finding that the level of GRP78 in ectopic endometrial cells is significantly higher than that of normal endometrial cells. This indicated that the UPR cascade reaction was activated in EMS, and the upregulated expression of GRP78 significantly reduced the sensitivity of ectopic endometrial cells to apoptosis and increased their anti-apoptosis ability, which was beneficial for the survival of ectopic endometrial cells. Other UPR proteins, p-IRE1 and p-PERK, were also found increased in endometrial cells when they were treated with peritoneal fluid obtained from women with endometriosis (80). Taylor (81) et al. also reported that UPR induced the increased expression of IL-8, indicating that UPR may be involved in the pathogenesis of EMS by promoting neovascularization and cell survival via IL-8 related pathways. The protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway plays an essential role in enhancing the invasiveness of cells, and inhibition of this pathway can effectively reduce the invasiveness of various cancer cells (82). Several studies (83, 84) have displayed that UPR can inhibit AKT/mTOR pathway through CCAAT/CHOP/(Tribbles Homolog 3, TRIB3) signal transduction and regulate the invasiveness of ectopic endometrial cells. EMS is an estrogen-dependent disease and is related to progesterone resistance (85). UPR-induced apoptosis can be inhibited by estrogen, thus promoting endometrial cell survival (86). In contrast, in the secretory phase, due to the antagonistic effect of progesterone, the inhibition effect of estrogen on the UPR-mediated apoptotic pathway can be reduced. Then, UPR upregulation may help reduce the invasiveness of endometrial cells (87). However, progesterone resistance exists in ectopic and endometrial stromal cells of women with EMS, and in the secretory phase, progesterone resistance in ectopic endometrium stromal cells may alter the effect induced by UPR as described above (88, 89). In vitro studies on UPR regulating endometrial cell invasiveness also suggest that (90), in endometrial cells with estrogen added alone in the proliferative phase, the expressions of GRP78, CHOP, and TRIB3 increased significantly with increased progesterone during the secretory phase. In contrast, endometrial cell invasiveness was significantly suppressed when AKT and mTOR activity was inhibited. Thus, progesterone can upregulate the expression of UPR-related CHOP and TRIB3 by antagonizing estrogen and inhibiting the AKT/mTOR pathway, reducing the invasiveness of endometrial cells. These progesterone-induced signal regulations can occur in normal endometrial cells. In ectopic cells, progesterone has no significant effect on CHOP/TRIB3 or AKT/mTOR signaling, so it does not play a role in the invasiveness of endometrial cells.
EMS severely affects the physical and mental health and quality of life of women of reproductive age. The implantation theory on the pathogenesis of EMS indicates the key role of hypoxia in the occurrence and development of EMS, which involves cell biological processes such as apoptosis, adhesion, proliferation, invasion, and metastasis of ectopic endometrial cells. The mechanism of EMS is complex, and its etiology and pathology cannot be clearly explained with a onefold theory. Hypoxia-induced UPR may be one of the potential mechanisms by which ectopic endometrium cells could resist apoptosis and develop into endometriotic lesions, and some drugs (91, 92) targeting this mechanism may become potential effective therapeutic agents for the treatment of the disease. But the exact mechanism of hypoxia-stimulated UPR in EMS remains to be explored, and we look forward to new break-throughs in this field.
YZ and YJ contributed equally to the manuscript. YZ contributed to the conception and design of the review. YZ and YJ drafted the manuscript. YJ performed the descriptive figures. YW and RW revised the manuscript and provided critical advice on the content of the manuscript. All authors read and approved the final manuscript.
This work was supported by National Natural Science foundation of China (82101723), Department of Education of Zhejiang Province (Y202045691), and Zhejiang Province Key R&D Program (2021C03095).
We gratefully acknowledge the assistance of Home for Researchers for language modification.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Giudice LC. Clinical practice. endometriosis. N Engl J Med (2010) 362(25):2389–98. doi: 10.1056/NEJMcp1000274
2. Vercellini P, Viganò P, Somigliana E, Fedele L. Endometriosis: pathogenesis and treatment. Nat Rev Endocrinol (2014) 10(5):261–75. doi: 10.1038/nrendo.2013.255
3. Viganò P, Parazzini F, Somigliana E, Vercellini P. Endometriosis: epidemiology and aetiological factors. Best Pract Res Clin Obstet Gynaecol (2004) 18(2):177–200. doi: 10.1016/j.bpobgyn.2004.01.007
4. Buck Louis GM, Hediger ML, Peterson CM, Croughan M, Sundaram R, Stanford J, et al. Incidence of endometriosis by study population and diagnostic method: the ENDO study. Fertil Steril (2011) 96(2):360–5. doi: 10.1016/j.fertnstert.2011.05.087
5. Laganà AS, La Rosa V, Petrosino B, Vitale SG. Comment on “Risk of developing major depression and anxiety disorders among women with endometriosis: A longitudinal follow-up study”. J Affect Disord (2017) 208:672–3. doi: 10.1016/j.jad.2016.07.016
6. Matalliotakis M, Zervou MI, Matalliotaki C, Rahmioglu N, Koumantakis G, Kalogiannidis I, et al. The role of gene polymorphisms in endometriosis. Mol Med Rep (2017) 16(5):5881–6. doi: 10.3892/mmr.2017.7398
7. Raffaelli R, Garzon S, Baggio S, Genna M, Pomini P, Laganà AS, et al. Mesenteric vascular and nerve sparing surgery in laparoscopic segmental intestinal resection for deep infiltrating endometriosis. Eur J Obstet Gynecol Reprod Biol (2018) 231:214–9. doi: 10.1016/j.ejogrb.2018.10.057
8. Thomin A, Belghiti J, David C, Marty O, Bornes M, Ballester M, et al. Maternal and neonatal outcomes in women with colorectal endometriosis. Bjog (2018) 125(6):711–8. doi: 10.1111/1471-0528.14221
9. Khan Z, Zanfagnin V, El-Nashar SA, Famuyide AO, Daftary GS, Hopkins MR. Risk factors, clinical presentation, and outcomes for abdominal wall endometriosis. J Minim Invasive Gynecol (2017) 24(3):478–84. doi: 10.1016/j.jmig.2017.01.005
10. Chamié LP, Ribeiro D, Tiferes DA, Macedo Neto AC, Serafini PC. Atypical sites of deeply infiltrative endometriosis: Clinical characteristics and imaging findings. Radiographics (2018) 38(1):309–28. doi: 10.1148/rg.2018170093
11. Sampson JA. The development of the implantation theory for the origin of peritoneal endometriosis. Am J Obstet Gynecol (1940) 40:549–57. doi: 10.1016/s0002-9378(40)91238-8
12. Sampson JA. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol (1927) 14:422–69. doi: 10.1016/s0002-9378(15)30003-x
13. Halme J, Hammond MG, Hulka JF, Raj SG, Talbert LM. Retrograde menstruation in healthy women and in patients with endometriosis. Obstet Gynecol (1984) 64(2):151–4. Available at: https://pubmed.ncbi.nlm.nih.gov/6234483/.
14. Kruitwagen RF, Poels LG, Willemsen WN, Jap PH, Thomas CM, Rolland R. Retrograde seeding of endometrial epithelial cells by uterine-tubal flushing. Fertil Steril (1991) 56(3):414–20. doi: 10.1016/s0015-0282(16)54533-6
15. Koks CA, Dunselman GA, De Goeij AF, Arends JW, Evers JL. Evaluation of a menstrual cup to collect shed endometrium for in vitro studies. Fertil Steril (1997) 68(3):560–4. doi: 10.1016/s0015-0282(97)00250-1
16. Lang J. A new milestone in endometriosis research. Chin J Obstet Gynecol (2005) 40:3–4. doi: 10.3760/j.issn:0529-567X.2005.01.002
17. Figueira PG, Abrão MS, Krikun G, Taylor HS. Stem cells in endometrium and their role in the pathogenesis of endometriosis. Ann N Y Acad Sci (2011) 1221(1):10–7. doi: 10.1111/j.1749-6632.2011.05969.x
18. Hapangama DK, Kamal A, Saretzki G. Implications of telomeres and telomerase in endometrial pathology. Hum Reprod Update (2017) 23(2):166–87. doi: 10.1093/humupd/dmw044
19. Liu Y, Zhang Z, Yang F, Wang H, Liang S, Wang H, et al. The role of endometrial stem cells in the pathogenesis of endometriosis and their application to its early diagnosis†. Biol Reprod (2020) 102(6):1153–9. doi: 10.1093/biolre/ioaa011
20. Maybin JA, Murray AA, Saunders PTK, Hirani N, Carmeliet P, Critchley HOD. Hypoxia and hypoxia inducible factor-1α are required for normal endometrial repair during menstruation. Nat Commun (2018) 9(1):295. doi: 10.1038/s41467-017-02375-6
21. Li WN, Wu MH, Tsai SJ. HYPOXIA AND REPRODUCTIVE HEALTH: The role of hypoxia in the development and progression of endometriosis. Reproduction (2021) 161(1):F19–f31. doi: 10.1530/rep-20-0267
22. Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol (2008) 9(4):285–96. doi: 10.1038/nrm2354
23. Koumenis C, Wouters BG. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res (2006) 4(7):423–36. doi: 10.1158/1541-7786.Mcr-06-0150
24. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol (1992) 12(12):5447–54. doi: 10.1128/mcb.12.12.5447-5454.1992
25. West JB. Physiological effects of chronic hypoxia. N Engl J Med (2017) 376(20):1965–71. doi: 10.1056/NEJMra1612008
26. Kourembanas S, Marsden PA, Mcquillan LP, Faller DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest (1991) 88(3):1054–7. doi: 10.1172/jci115367
27. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3(3):177–85. doi: 10.1016/j.cmet.2006.02.002
28. Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature (1992) 359(6398):843–5. doi: 10.1038/359843a0
29. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem (1994) 269(38):23757–63. doi: 10.1016/S0021-9258(17)31580-6
30. Loenarz C, Coleman ML, Boleininger A, Schierwater B, Holland PW, Ratcliffe PJ, et al. The hypoxia-inducible transcription factor pathway regulates oxygen sensing in the simplest animal, trichoplax adhaerens. EMBO Rep (2011) 12(1):63–70. doi: 10.1038/embor.2010.170
31. Wu MH, Hsiao KY, Tsai SJ. Hypoxia: The force of endometriosis. J Obstet Gynaecol Res (2019) 45(3):532–41. doi: 10.1111/jog.13900
32. Laschke MW, Menger MD. In vitro and in vivo approaches to study angiogenesis in the pathophysiology and therapy of endometriosis. Hum Reprod Update (2007) 13(4):331–42. doi: 10.1093/humupd/dmm006
33. Lu Z, Zhang W, Jiang S, Zou J, Li Y. Effect of oxygen tensions on the proliferation and angiogenesis of endometriosis heterograft in severe combined immunodeficiency mice. Fertil Steril (2014) 101(2):568–76. doi: 10.1016/j.fertnstert.2013.10.039
34. Yu YX, Xiu YL, Chen X, Li YL. Transforming growth factor-beta 1 involved in the pathogenesis of endometriosis through regulating expression of vascular endothelial growth factor under hypoxia. Chin Med J (Engl) (2017) 130(8):950–6. doi: 10.4103/0366-6999.204112
35. Wu MH, Chen KF, Lin SC, Lgu CW, Tsai SJ. Aberrant expression of leptin in human endometriotic stromal cells is induced by elevated levels of hypoxia inducible factor-1alpha. Am J Pathol (2007) 170(2):590–8. doi: 10.2353/ajpath.2007.060477
36. Lin SC, Wang CC, Wu MH, Yang SH, Li YH, Tsai SJ. Hypoxia-induced microRNA-20a expression increases ERK phosphorylation and angiogenic gene expression in endometriotic stromal cells. J Clin Endocrinol Metab (2012) 97(8):E1515–1523. doi: 10.1210/jc.2012-1450
37. Hsiao KY, Chang N, Lin SC, Li YH, Wu MH. Inhibition of dual specificity phosphatase-2 by hypoxia promotes interleukin-8-mediated angiogenesis in endometriosis. Hum Reprod (2014) 29(12):2747–55. doi: 10.1093/humrep/deu255
38. Lin SC, Lee HC, Hsu CT, Huang YH, Li WN, Hsu PL, et al. Targeting anthrax toxin receptor 2 ameliorates endometriosis progression. Theranostics (2019) 9(3):620–32. doi: 10.7150/thno.30655
39. Hsiao KY, Chang N, Tsai JL, Lin SC, Tsai SJ, Wu MH. Hypoxia-inhibited DUSP2 expression promotes IL-6/STAT3 signaling in endometriosis. Am J Reprod Immunol (2017) 78(4):e12690. doi: 10.1111/aji.12690
40. Terzic M, Aimagambetova G, Kunz J, Bapayeva G, Aitbayeva B, Terzic S, et al. Molecular basis of endometriosis and endometrial cancer: Current knowledge and future perspectives. Int J Mol Sci (2021) 22(17):9274. doi: 10.3390/ijms22179274
41. Liang Y, Wu J, Wang W, Xie H, Yao S. Pro-endometriotic niche in endometriosis. Reprod BioMed Online (2019) 38(4):549–59. doi: 10.1016/j.rbmo.2018.12.025
42. Maurya VK, Szwarc MM, Fernandez-Valdivia R, Lonard DM, Yong S, Joshi N, et al. Early growth response 1 transcription factor is essential for the pathogenic properties of human endometriotic epithelial cells. Reproduction (2022) 164(2):41–54. doi: 10.1530/rep-22-0123
43. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
44. Li R, Li H, Zhu L, Zhang X, Liu D, Li Q, et al. Reciprocal regulation of LOXL2 and HIF1α drives the warburg effect to support pancreatic cancer aggressiveness. Cell Death Dis (2021) 12(12):1106. doi: 10.1038/s41419-021-04391-3
45. Young VJ, Brown JK, Maybin J, Saunders PT, Duncan WC, Horne AW. Transforming growth factor-β induced warburg-like metabolic reprogramming may underpin the development of peritoneal endometriosis. J Clin Endocrinol Metab (2014) 99(9):3450–9. doi: 10.1210/jc.2014-1026
46. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest (2005) 115(10):2656–64. doi: 10.1172/jci26373
47. Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol (2006) 18(4):444–52. doi: 10.1016/j.ceb.2006.06.005
48. Gidalevitz T, Stevens F, Argon Y. Orchestration of secretory protein folding by ER chaperones. Biochim Biophys Acta (2013) 1833(11):2410–24. doi: 10.1016/j.bbamcr.2013.03.007
49. Eletto D, Chevet E, Argon Y, Appenzeller-Herzog C. Redox controls UPR to control redox. J Cell Sci (2014) 127(Pt 17):3649–58. doi: 10.1242/jcs.153643
50. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science (2011) 334(6059):1081–6. doi: 10.1126/science.1209038
51. Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell (2017) 168(4):692–706. doi: 10.1016/j.cell.2016.12.004
52. Dufey E, Bravo-San Pedro JM, Eggers C, González-Quiroz M, Urra H, Sagredo AI, et al. Genotoxic stress triggers the activation of IRE1α-dependent RNA decay to modulate the DNA damage response. Nat Commun (2020) 11(1):2401. doi: 10.1038/s41467-020-15694-y
53. Marciniak SJ. Endoplasmic reticulum stress: A key player in human disease. FEBS J (2019) 286(2):228–31. doi: 10.1111/febs.14740
54. Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol (2018) 69(4):927–47. doi: 10.1016/j.jhep.2018.06.008
55. Pan WW, Myers MG Jr. Leptin and the maintenance of elevated body weight. Nat Rev Neurosci (2018) 19(2):95–105. doi: 10.1038/nrn.2017.168
56. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol (2013) 15(5):481–90. doi: 10.1038/ncb2738
57. Yang Z, Tsuchiya H, Zhang Y, Lee S, Liu C, Huang Y, et al. REV-ERBα activates C/EBP homologous protein to control small heterodimer partner-mediated oscillation of alcoholic fatty liver. Am J Pathol (2016) 186(11):2909–20. doi: 10.1016/j.ajpath.2016.07.014
58. Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-y and YY1. Mol Cell Biol (2000) 20(14):5096–106. doi: 10.1128/mcb.20.14.5096-5106.2000
59. Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem (2004) 279(25):25935–8. doi: 10.1074/jbc.R400008200
60. Iurlaro R, Muñoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J (2016) 283(14):2640–52. doi: 10.1111/febs.13598
61. Bartoszewski R, Gebert M, Janaszak-Jasiecka A, Cabaj A, Króliczewski J, Bartoszewska S, et al. Genome-wide mRNA profiling identifies RCAN1 and GADD45A as regulators of the transitional switch from survival to apoptosis during ER stress. FEBS J (2020) 287(14):2923–47. doi: 10.1111/febs.15195
62. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell (2001) 107(7):881–91. doi: 10.1016/s0092-8674(01)00611-0
63. Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem (2001) 276(17):13935–40. doi: 10.1074/jbc.M010677200
64. Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell (2009) 138(3):562–75. doi: 10.1016/j.cell.2009.07.017
65. Wu Q, Wu W, Jacevic V, Franca TCC, Wang X, Kuca K. Selective inhibitors for JNK signalling: A potential targeted therapy in cancer. J Enzyme Inhib Med Chem (2020) 35(1):574–83. doi: 10.1080/14756366.2020.1720013
66. Liu R, Li X, Huang Z, Zhao D, Ganesh BS, Lai G, et al. C/EBP homologous protein-induced loss of intestinal epithelial stemness contributes to bile duct ligation-induced cholestatic liver injury in mice. Hepatology (2018) 67(4):1441–57. doi: 10.1002/hep.29540
67. Rutkowski DT, Kaufman RJ. All roads lead to ATF4. Dev Cell (2003) 4(4):442–4. doi: 10.1016/s1534-5807(03)00100-x
68. Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol (2011) 31(12):2792–7. doi: 10.1161/atvbaha.111.224881
69. Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol (2001) 153(5):1011–22. doi: 10.1083/jcb.153.5.1011
70. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol (2011) 54(4):795–809. doi: 10.1016/j.jhep.2010.11.005
71. Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J (2005) 24(19):3470–81. doi: 10.1038/sj.emboj.7600777
72. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer (2008) 8(11):851–64. doi: 10.1038/nrc2501
73. Liu L, Wise DR, Diehl JA, Simon MC. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J Biol Chem (2008) 283(45):31153–62. doi: 10.1074/jbc.M805056200
74. Pereira ER, Frudd K, Awad W, Hendershot LM. Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF). J Biol Chem (2014) 289(6):3352–64. doi: 10.1074/jbc.M113.507194
75. Fels DR, Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther (2006) 5(7):723–8. doi: 10.4161/cbt.5.7.2967
76. Dekervel J, Bulle A, Windmolders P, Lambrechts D, Van Cutsem E, Verslype C, et al. Acriflavine inhibits acquired drug resistance by blocking the epithelial-to-Mesenchymal transition and the unfolded protein response. Transl Oncol (2017) 10(1):59–69. doi: 10.1016/j.tranon.2016.11.008
77. Spiotto MT, Banh A, Papandreou I, Cao H, Galvez MG, Gurtner GC, et al. Imaging the unfolded protein response in primary tumors reveals microenvironments with metabolic variations that predict tumor growth. Cancer Res (2010) 70(1):78–88. doi: 10.1158/0008-5472.Can-09-2747
78. Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature (2014) 508(7494):103–7. doi: 10.1038/nature13119
79. Guzel E, Arlier S, Guzeloglu-Kayisli O, Tabak MS, Ekiz T, Semerci N, et al. Endoplasmic reticulum stress and homeostasis in reproductive physiology and pathology. Int J Mol Sci (2017) 18(4):792. doi: 10.3390/ijms18040792
80. Ekiz-Yilmaz T, Isildar B, Gezer A, Kankaya D, Cansiz-Ersoz C, Kayisli UA, et al. The role of unfolded protein response in the pathogenesis of endometriosis: contribution of peritoneal fluid. Reprod BioMed Online (2021) 42(1):1–15. doi: 10.1016/j.rbmo.2020.09.012
81. Taylor RN, Yu J, Torres PB, Schickedanz AC, Park JK, Mueller MD, et al. Mechanistic and therapeutic implications of angiogenesis in endometriosis. Reprod Sci (2009) 16(2):140–6. doi: 10.1177/1933719108324893
82. Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin Cancer Biol (2019) 59:125–32. doi: 10.1016/j.semcancer.2019.07.009
83. Lin S, Yang L, Shi H, Du W, Qi Y, Qiu C, et al. Endoplasmic reticulum-targeting photosensitizer hypericin confers chemo-sensitization towards oxaliplatin through inducing pro-death autophagy. Int J Biochem Cell Biol (2017) 87:54–68. doi: 10.1016/j.biocel.2017.04.001
84. Xu X, Huang E, Tai Y, Zhao X, Chen X, Chen C, et al. Nupr1 modulates methamphetamine-induced dopaminergic neuronal apoptosis and autophagy through CHOP-Trib3-Mediated endoplasmic reticulum stress signaling pathway. Front Mol Neurosci (2017) 10:203. doi: 10.3389/fnmol.2017.00203
85. Kapoor R, Stratopoulou CA, Dolmans MM. Pathogenesis of endometriosis: New insights into prospective therapies. Int J Mol Sci (2021) 22(21):11700. doi: 10.3390/ijms222111700
86. Reis FM, Petraglia F, Taylor RN. Endometriosis: hormone regulation and clinical consequences of chemotaxis and apoptosis. Hum Reprod Update (2013) 19(4):406–18. doi: 10.1093/humupd/dmt010
87. Choi JY, Jo MW, Lee EY, Lee DY, Choi DS. Ovarian steroid dependence of endoplasmic reticulum stress involvement in endometrial cell apoptosis during the human endometrial cycle. Reproduction (2018) 155(6):493–503. doi: 10.1530/rep-17-0713
88. Attia GR, Zeitoun K, Edwards D, Johns A, Carr BR, Bulun SE. Progesterone receptor isoform a but not b is expressed in endometriosis. J Clin Endocrinol Metab (2000) 85(8):2897–902. doi: 10.1210/jcem.85.8.6739
89. Bulun SE, Cheng YH, Yin P, Imir G, Utsunomiya H, Attar E, et al. Progesterone resistance in endometriosis: link to failure to metabolize estradiol. Mol Cell Endocrinol (2006) 248(1-2):94–103. doi: 10.1016/j.mce.2005.11.041
90. Choi J, Jo M, Lee E, Lee DY, Choi D. Dienogest regulates apoptosis, proliferation, and invasiveness of endometriotic cyst stromal cells via endoplasmic reticulum stress induction. Mol Hum Reprod (2020) 26(1):30–9. doi: 10.1093/molehr/gaz064
91. Dioufa N, Kassi E, Papavassiliou AG, Kiaris H. Atypical induction of the unfolded protein response by mifepristone. Endocrine (2010) 38(2):167–73. doi: 10.1007/s12020-010-9362-0
Keywords: hypoxia, unfolded protein response, endometriosis, endoplasmic reticulum stress, hypoxia-inducible factor
Citation: Zhou Y, Jin Y, Wang Y and Wu R (2022) Hypoxia activates the unfolded protein response signaling network: An adaptive mechanism for endometriosis. Front. Endocrinol. 13:945578. doi: 10.3389/fendo.2022.945578
Received: 16 May 2022; Accepted: 05 October 2022;
Published: 20 October 2022.
Edited by:
Tiziano Baroni, University of Perugia, ItalyReviewed by:
Sahar Houshdaran, University of California, San Francisco, United StatesCopyright © 2022 Zhou, Jin, Wang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruijin Wu, d3VyakB6anUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.