 Kelly A. Mason
Kelly A. Mason Alan D. Rogol
Alan D. Rogol
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 12 July 2022
Sec. Pediatric Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.935354
This article is part of the Research Topic Insights in Pediatric Endocrinology: 2022 View all 13 articles
Since cystic fibrosis (CF) was first described in 1938, there have been many discoveries and innovations in the field, each having a profound impact on survival, growth and quality of life. For example, the introduction of enteric-coated pancreatic enzyme microspheres increased fat absorption and improved nutritional status. Early detection of CF through newborn screening facilitated prompt nutritional intervention for infants at high risk of malnutrition. Use of anti-pseudomonal therapy, such as inhaled tobramycin, increased weight gain and pulmonary function in addition to reducing pulmonary exacerbations. Similarly, DNAse and hypertonic saline improved pulmonary function and reduced exacerbations. The identification of the CFTR gene and its protein product were fundamental in understanding the pathophysiology of CF and paved the way for advances in both diagnosis and management. In fact, CFTR modulator therapies have revolutionized the care for individuals with CF. Here, we examine the impact of these interventions on the nutritional status, growth and pubertal maturation of children and adolescents with CF.
Cystic Fibrosis (CF) affects nearly 70,000 individuals worldwide. It is caused by an autosomal recessive mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which results in a dysfunctional CFTR protein. This, in turn, impairs chloride ion transport to the cell surface, resulting in viscous secretions in the lungs, pancreas, intestine and hepatobiliary ducts, leading to obstruction and fibrosis (1). Since its initial description by Dorothy Andersen in 1938 (2), there have been many landmark discoveries that have led to remarkable improvements in life expectancy and quality of life.
For decades, chronic infection, malabsorption of essential nutrients, inflammation and the frequent use of corticosteroids set the child with CF on a course of diminished weight gain and linear growth restriction. It should be noted that increased energy requirements (expenditure) due to inflammation and chronic pulmonary disease also contribute to the energy deficit. Growth restriction may even begin in utero since the birthweight of children with CF is approximately 250 g less than that of healthy newborns (3). It may be that the absent CFTR affects placental function.
Over the decades the treatment goals have been to optimize pulmonary function and growth through proper nutrition and reduced inflammation. We aim to characterize the trends in growth and maturation over time. Notably, many studies report anthropometric data differently, using weight or height-for-age or weight-for-height as a % or z-score or % of reference median. In their study of 13,116 children with CF, Lai et al. discovered inconsistencies in classifications when using various criteria, underscoring the importance of standardized definitions (4).
Nevertheless, it is evident that interventions such as multi-disciplinary care and the introduction of newborn screening have contributed to the increased growth of children with CF, demonstrated by an increase in the average height z-score at age 6 years from -0.69 to 0.39 SD (5).
Cystic Fibrosis was first described in 1938 by Dorothy Andersen who carefully reviewed the clinical histories and post-mortem examinations from 49 individuals with pancreatic fibrosis, many of whom died during infancy or early childhood (2). Infants who died within the first week of life were felt to have died from intestinal obstruction. The remaining children were characterized by failure to gain weight beginning in the neonatal period, hunger, distended abdomens, intolerance of dietary fat with large fatty stools in the absence of vomiting and diarrhea, and chronic respiratory tract infections. Andersen termed the condition cystic fibrosis of the pancreas based on the observation that the pancreatic acinar tissue “was replaced by epithelium-lined cysts containing concretions and surrounded by fibrous tissue”; there was also evidence of vitamin A deficiency. The lungs demonstrated “bronchitis, bronchiectasis, pulmonary abscesses arising in the bronchi”, and/or lobular pneumonia with S. aureus as a common “bacteriologic agent”. The oldest child in the cohort was 14.5 years (2). In 1935 Parmelee described this girl’s stature as closely approximating normal until age 11, after which time her growth was described as “retarded”, but the “development of secondary sex characteristics was not.” Her weight was reported to be considerably below average throughout much of her childhood and adolescence (6). Based on the findings in this report, this child likely had more mild disease.
Anthropometric data in children with CF throughout the 1940’s are largely unavailable.
Given the short life span in those diagnosed with CF in the 1930’s and 1940’s, there are few data on growth and puberty for these children. However, the life expectancy in the 1950’s (1951-1956), increased to 59 months (7) allowing for more detailed descriptions of growth and nutritional status.
Rustin McIntosh, for example, recorded observations on a cohort of 23 patients with CF who survived to at least 10 years of age. In this group of children, malnutrition occurred more commonly during infancy compared to childhood, but height and weight were “retarded” (average height z-score 2 SD below mean and average weight 1.8 SD below mean). Height and weight were noted to increase in response to appropriate therapy for staphylococcal infections, suggesting that chronic infection and inflammation play a role in the poor growth observed in children with CF (8).
Several years later, Shwachman and Kulczycki, described a larger cohort of 105 children who were followed longitudinally for at least 5 years after diagnosis. The majority (87/105) received antibiotic therapy. Their patient population consumed a liberal diet high in protein with limited fat intake. Those who had pancreatic insufficiency took pancreatic enzyme replacement with each meal and double the typical daily dose of multivitamins. The patients were divided into groups based on the age of diagnosis and each was assigned a score based on their level of tolerated activity, physical and radiographic findings and nutritional status (Shwachman-Kulczycki or the SK score). The nutritional score was based on height and weight percentiles for age, stool characteristics, muscle mass and tone, and degree of abdominal distension. The nutritional score was not reported for all subgroups but was documented for the group diagnosed between the age of 7 and 16 years both at baseline and at follow-up. At baseline, none of the 9 children with CF were categorized as marked malnutrition, 4/9 (45%) had a weight and height for age under the 3rd centile and 2 (22%) had height and weight for age over the 25th centile. At follow-up 5 to 10 years later, 2 (22%) were categorized as having marked malnutrition, 4/9 (44%) had a weight and height for age less than the 3rd centile and none had a height and weight for age above the 25th centile, suggesting a decline in nutritional status and linear growth over time (7) (Table 1).
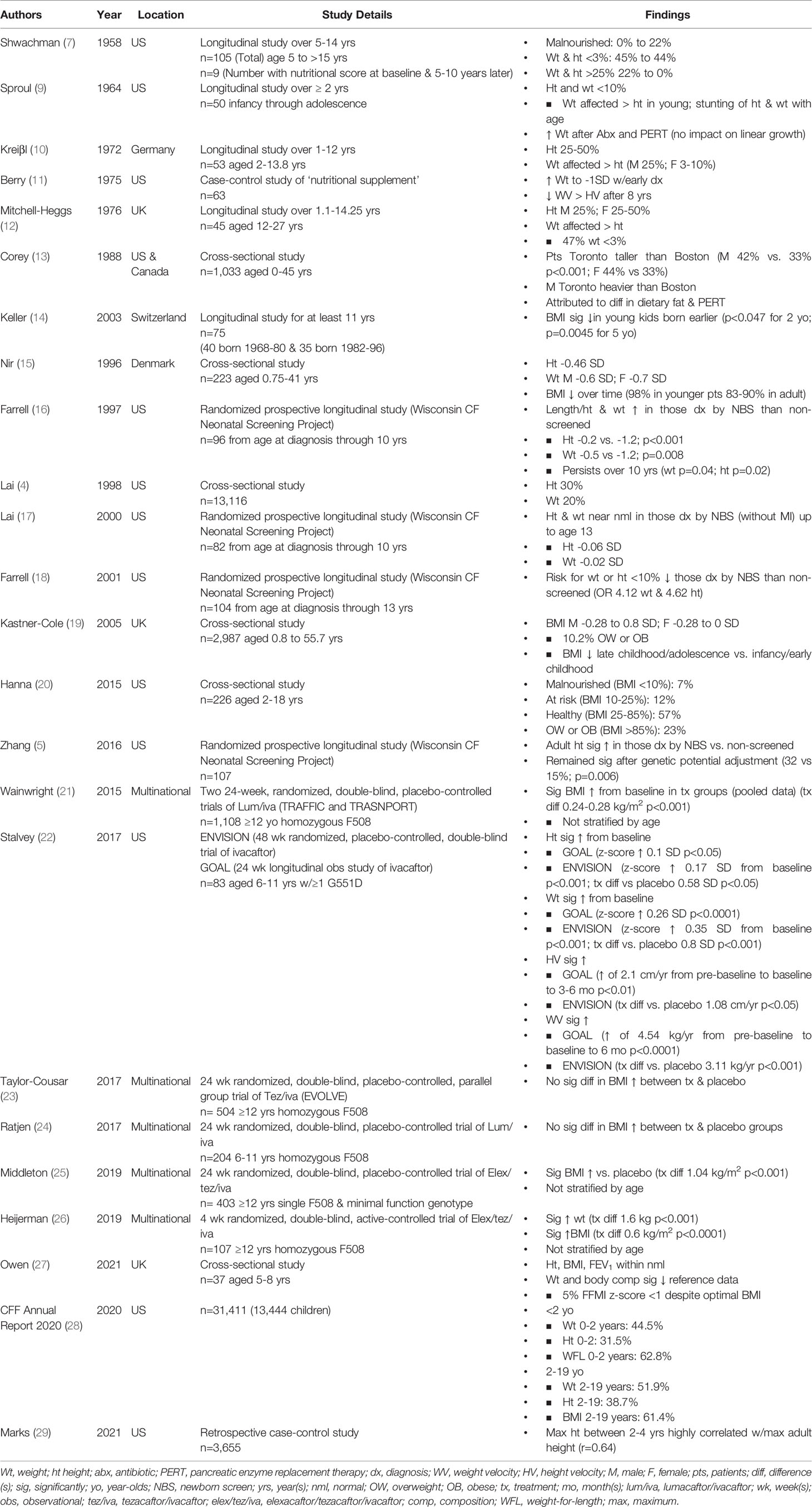
Table 1 Growth Data over Time.
The birthweights of infants with CF were significantly below those of both the general population (30) and unaffected siblings (31), suggesting that the slow growth was not solely due to infections, pancreatic insufficiency or nutritional status, but began in utero.
The 1960’s marked the beginning of a specific focus on growth in children with CF. Sproul and Huang studied the growth patterns in 50 children and noted a period of accelerated weight gain after initiation of therapy that included antibiotics and pancreatic enzyme replacement therapy (PERT), with the longest duration of effect occurring when therapy was initiated during infancy. They did not find an effect on linear growth despite a minimum observation period of 2 years. The authors observed that the median height and weight for all age groups were under the 10th centile with weight more affected than height in the pre-school and school-age children and growth “retardation” more prominent in the preadolescent and adolescent age groups, a finding that was attributed to lack of the pubertal growth spurt (Table 1). Skeletal maturation was evaluated in 40 of the 50 children and was “retarded” in 25%. Notably, the authors also observed an inverse relationship between severity of respiratory disease and growth (p=0.005 for height; p<0.001 for weight), though the methods used for categorizing respiratory status were not reported (9) (Table 2).
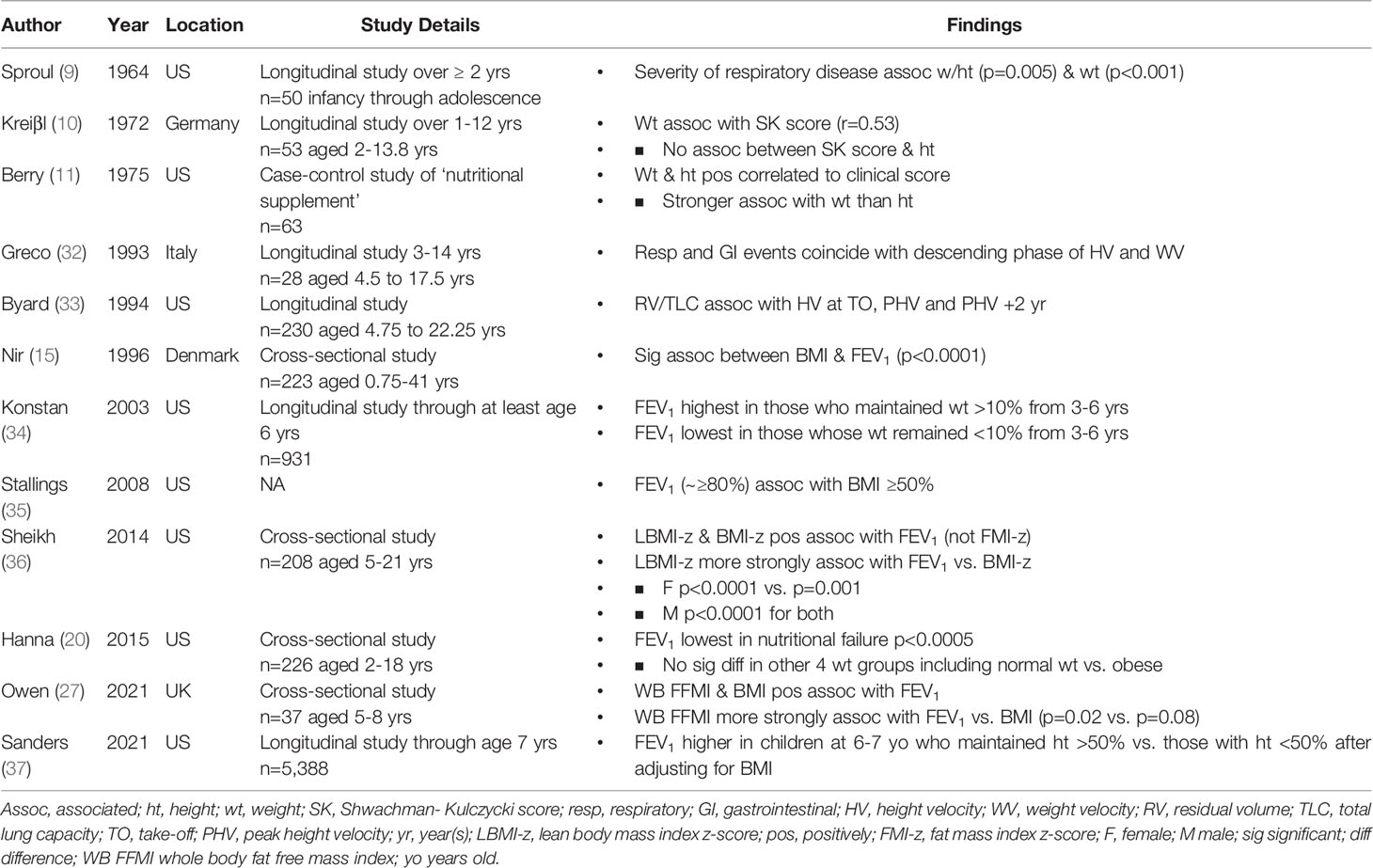
Table 2 Relationship between Growth and Pulmonary Function.
By 1974, life expectancy reached 16 years (38) and several investigators began to describe the growth patterns and pubertal maturation of children and adolescents with CF (10–12, 38). Antibiotic agents and treatment intensity improved between the 1960’s and 1970’s (10), such that standard therapy in the 1970’s included use of antimicrobials based on susceptibility and clinical status, aerosol inhalations or nightly mist therapy, postural drainage and chest physiotherapy, vitamin supplementation and PERT (11, 38). In 1979, encapsulated enteric-coated pancreatic enzyme microspheres were introduced. This formulation was designed to reduce gastric acid and pepsin-mediated inactivation, thereby delivering more active enzyme to the duodenum (39) and was shown to be effective in the treatment of pancreatic insufficiency (40).
In 1972, Kreiβl and colleagues evaluated growth parameters in a cohort of 53 patients. Similar to Sproul and Huang, their group identified an association between weight percentiles and disease severity as determined by Shwachman-Kulczycki scores. However, they found no association between disease severity and height (Table 2). The median heights in this particular group ranged from the 25th to the 50th centile, with a normal height velocity in all but 7 of the 53 children. As observed in previous studies, weight tended to be more affected than height (Table 1). Weight velocity, however, was considered normal in all but 11 of the 53 children (10).
As in the study by Kreiβl, Mitchell-Heggs and colleagues reported a median height at the 25th centile in boys and between the 25-50th centiles in girls. The authors noted that the mean weight percentiles tended to be lower than the mean height percentiles for most (Table 1). They also reported that puberty tended to be delayed, but there was no significant relationship between skeletal maturity and disease severity (12).
Results from Berry et al. were similar to those that Sproul and Huang reported in 1964 (9). They too observed an increase in weight following early diagnosis that stabilized 1 SD below the mean from 2 to 8 years of age. This was followed by a gradual decline in height and weight velocity with weight more significantly affected than height (11) (Table 1).
In 1976, Stern et al. reported on a cohort of 95 patients and noted that weight-for-height was generally ‘deficient’ with weight often under the 3rd centile and height below average. Menarche was noticeably delayed with a mean age of >14 years (38).
Throughout the 1970’s, many studies revealed that weight was more significantly affected than height and that puberty and/or skeletal maturity tended to be delayed.
By the early 1980’s the mean survival for those with CF increased to 19 years in the United States and 21 years in Canada (13). One notable change to the care of individuals with CF was early detection through newborn screening. In 1985, a comprehensive evaluation of a newborn screening program in Wisconsin began (16). There were also significant changes to nutrition. In the early 1980’s, for example, enteric-coated pancreatic enzyme microspheres significantly increased fat absorption over conventional pancreatic enzyme formulations (41). A seminal paper by Corey and colleagues in 1982 demonstrated a significant growth discrepancy between children treated at the CF center in Toronto and those treated in Boston and speculated that these discrepancies were due to differences in PERT dosing and dietary fat intake. Both males and females in Toronto were significantly taller than those in Boston; males in Toronto were also significantly heavier than those treated in Boston, where dietary fat intake was limited (13) (Table 1). These findings led to the adoption of the high-calorie, high-fat diets that became fundamental in the care of children with CF.
Soutter et al. reported that most of the children in their cohort had a weight under the 50th centile with a decline in late childhood and pre-adolescence. However, weight and height velocity nearly doubled in those receiving overnight gastrostomy tube feeds, highlighting the importance of caloric (energy) intake. The mean height in their cohort was under the 50th centile (often ranging between the 25 and 50th centiles) (42). These data are similar to those published by Kreiβl and Mitchell-Heggs in the 1970’s but reflect a significant improvement from those reported by Sproul in the 1960’s (9, 10, 12).
Similar to Soutter et al., Keller and colleagues noted that the mean height in their population was significantly lower than the general population, as were weight and BMI z-scores. However, BMI z-scores in young children were significantly higher in the cohort born in the 1980’s and 1990’s compared to the cohort born in the 1960’s and 1970’s, supporting an overall improvement in nutritional status over time (14) (Table 1).
Puberty in individuals with CF tended to be delayed in the 1980’s with reports of delayed peak height velocity (PHV) (15 years) in boys (43) and delayed menarche (14.5 years) in girls (44). Some authors attributed the delayed puberty to disease severity and nutritional status (44), while others found no such association (45).
Throughout the 1980’s, height, weight and BMI in children with CF remained below the general population, though they appeared to be greater than earlier cohorts. The median survival had also increased such that it approximated 30 years by the late 1980’s (15).
In the 1990’s there was a focus on airway clearance through use of inhaled DNAse to thin the DNA-rich viscous secretions as well as anti-pseudomonal therapy. In a landmark study in 1994, inhaled DNAse (Pulmozyme) improved pulmonary function more than placebo and resulted in fewer pulmonary exacerbations (46). Use of inhaled tobramycin, an anti-pseudomonal therapy, every other month improved pulmonary function, reduced the need for IV antibiotics, and increased weight compared to placebo (47).
The 1990’s appear to be a turning point with a general improvement in growth patterns. In fact, the mean height of Swedish children approximated that of the general population (z-score of -0.3 SD) by age 5 y (48). Early identification of CF through newborn screening likely contributed to the improvement in growth observed throughout the decade. In fact, infants diagnosed by newborn screening (mean age 12 weeks) had significantly greater length or height z-scores (-0.2 SD vs. -1.2 SD), weight z-scores (-0.5 SD vs. -1.2 SD) and head circumferences than those identified by symptoms (mean age 72 weeks) (16). Furthermore, those diagnosed by newborn screening were taller and heavier up to 10 years later compared to those diagnosed by symptoms alone (16) (Table 1).
Authors from the UK reported mean weight z-scores ranging from -0.25 and -0.5 SD in their population under age 23 (49). Although these were lower than the general population, this reflects a general improvement from data published in the 1970’s (11, 38). However, as seen in previous studies (11, 42), these authors also reported a decline in weight z-scores after the first decade of life with BMI z-scores declining from 0 to -0.5 SD to -0.5 to -1 SD (49).
Nir et al. studied a cohort of 223 patients in Denmark after high fat diets and routine anti-pseudomonal therapy became standard. Their group also observed a decline in BMI over time, such that BMI values were lower than the general population by late childhood and early adolescence (Table 1). In addition, they identified a strong positive correlation between nutritional status (as indicated by BMI) and FEV1 (p<0.0001) (15) (Table 2). Greco et al. reported a regular intermittent (pulsatile) pattern in height and weight velocity with most respiratory or GI events coinciding with a descending phase of height and weight velocity (32) (Table 2). Similarly, Byard demonstrated a significant correlation between height velocity and pulmonary function (residual volume/total lung capacity) at the onset of the pubertal growth spurt, PHV and 2 years later (33) (Table 2).
Lai et al. analyzed data from 13,116 children participating in the National Cystic Fibrosis Patient Registry in 1993 and reported that the median height fell along the 30th centile with weight more significantly affected (20th centile), as observed in previous studies (4, 10–12, 38) (Table 1). Malnutrition, as defined by height or weight under the 5th centile was common during infancy. Similar to Nir et al., Lai and colleagues found that malnutrition was also common in adolescence (15). More girls (29%) than boys (19%) were short (height less than the 5th centile) between the ages of 11-14 with the opposite trend noted between the ages of 15-18 (4), potentially related to differences in pubertal timing.
Indeed, Byard reported on 230 children in the US and noted that PHV was lower in magnitude and occurred later than the general population (33). Similarly, in a small study of 17 girls with CF, both age at PHV and age of menarche were significantly later than the general population (50).
Previous studies from the 1960’s and 1970’s demonstrated an increase in weight upon initiation of appropriate therapy for CF (9, 11), likely due, in part, to the association between growth and disease severity identified by several groups in the 1990’s (15, 32, 33).
Despite the overall increase in growth parameters in the 1990’s, additional studies continued to report a general decline in weight z-scores after the first decade of life (4, 15, 49), comparable to findings in earlier decades (11, 42).
Airway clearance and anti-pseudomonal therapy remained important therapeutic factors in the 2000’s. Similar to inhaled DNAse therapy, inhaled hypertonic saline, used to rehydrate the airway surface and improve mucociliary clearance, reduced pulmonary exacerbations and improved pulmonary function over placebo (51). In addition, oral azithromycin was associated with a reduced risk of pulmonary exacerbations and improved pulmonary function over placebo for those with chronic pseudomonas aeruginosa infections (52).
Studies on newborn screening continued to show a benefit of early detection. The mean height (-0.6 SD) and weight z-scores (-0.23 SD) of those without MI (less likely to be identified early by symptoms alone) approximated those of the general population (17) (Table 1). Furthermore, when studied for up to 13 years, the odds ratios for the risk of height or weight under the 10th centile in the non-screened group compared to the screened group were 4.62 and 4.12 respectively (18, 53) (Table 1). Data continued to support an association between growth and pulmonary function (34) and, in 2008, evidence-based practice recommendations advocated for individuals aged 2-20 years to maintain a BMI ≥50%, given the association with improved pulmonary function (35) (Table 2).
Pulmonary function was also associated with PHV. Specifically, those classified as having severe disease based on a diminished FEV1 had a PHV that was delayed and of lower magnitude than those with milder disease (54).
In a study of 84 Polish children, mean height SD was -0.57 with only 11% of the population having a height z-score more than 2 SD below the mean (55), a stark contrast from the nearly 45% in a notably smaller cohort reported by Shwachman and Kulczycki almost 5 decades earlier (7). The mean BMI z-score in this cohort was -0.77 SD with 33% being classified as malnourished (BMI <10%) and 1 child categorized as obese (55). In the UK, 10.2% of children were categorized as overweight or obese with mean BMI z-score ranging from -0.28 to 0.8 SD in boys and -0.28 to 0.00 SD in girls (19). As observed in earlier studies (15, 49), mean BMI tended to be lower in later childhood and adolescence compared to infancy and early childhood (19) (Table 1).
Data published in 2004 demonstrated that height and weight z-scores were not significantly different in a cohort of 15 non-oxygen dependent children with CF compared with their age and sex-matched controls. Furthermore, there were no significant differences in body fat percentage, fat mass, fat-free mass or resting energy expenditure between the two groups (56).
Studies from the 2000’s continue to demonstrate an association between growth and pulmonary function. In general, height and weight appear greater than those recorded in previous decades (Table 2) with some children meeting criteria for overweight or obesity (19, 55).
Nationwide newborn screening in the United States occurred by 2010 (57). This allowed for early detection and early nutritional intervention for infants with CF. It also permitted an opportunity for aggressive PERT dosing, which was associated with a favorable change in weight-for-age and weight-for-length at age 2 years (58).
In a multi-center longitudinal observational cohort study of 231 infants diagnosed with CF by newborn screening (BONUS study), infants achieved a weight z-score of -0.04 SD by 12 months of age, a significant increase over a cohort who did not undergo screening 20 years earlier (59). Although length z-scores increased over the earlier cohort, they remained low through 12 months of age (Table 1). In an additional study, adult height was significantly greater in those diagnosed by newborn screening (50 vs. 29th centile) compared to those diagnosed by symptoms, a finding that remained significant after adjusting for genetic height potential (32 vs. 15th centile) (5) (Table 1).
Similarly, VanDevanter reported an increase in mean height z-scores and pulmonary function that occurred in parallel to an increase in the proportion of infants diagnosed by newborn screening between 1994 and 2012 (60).
As with studies in the 2000’s (19, 55), Marília da Silva Garrote and colleagues in Brazil categorized nearly 1/3 of their population as malnourished (BMI <10%) and approximately 15% as overweight or obese (defined as BMI >75%). The risk of colonization with pseudomonas aeruginosa was 2.3 times higher in those who were malnourished compared to those who were not (61), a finding that may have contributed to the overall improvement in nutritional metrics and possibly inflammatory status with the initiation of anti-pseudomonal therapy.
These data are in contrast to those presented by Hanna and Weiner, in which only 7% of children in a single United States center were categorized as malnourished (BMI <10%), over half as healthy (BMI 25-85%) and nearly a quarter as overweight or obese (BMI >85%) (20) (Table 1).
By the early 2010’s, the median childhood BMI reported in the Cystic Fibrosis Foundation Annual Report approached the 50% (28). This decade marked the beginning of more targeted therapies, namely CFTR-modulatory agents (Table 3). The first CFTR potentiator, ivacaftor, was FDA approved in 2012 for use in individuals ≥6 years of age with relatively uncommon (gating) mutations, comprising approximately 5% of the CF population (62, 63).
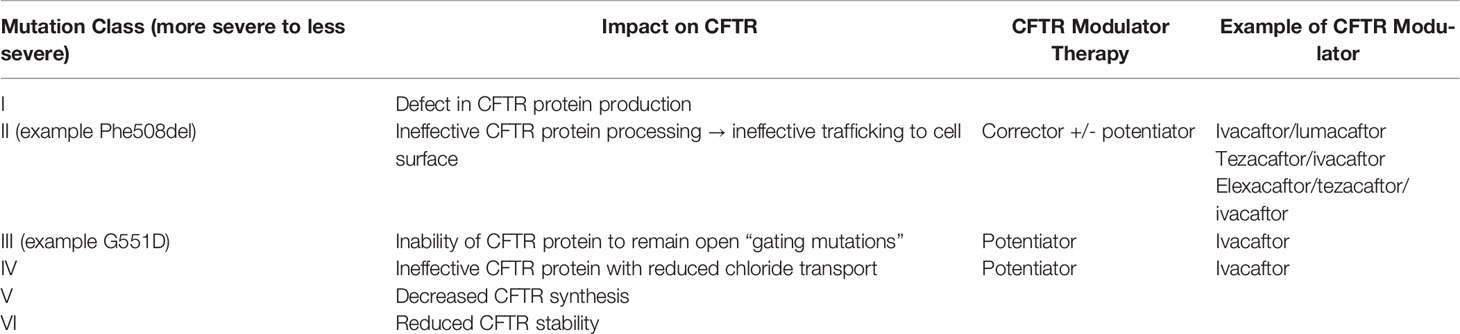
Table 3 CFTR Mutations and Modulator Therapies.
Several studies demonstrated a significant increase in growth and nutritional status in children with at least one G551D mutation treated with ivacaftor. Specifically, ivacaftor treatment for 48 weeks resulted in more weight gain (1.9 kg, p<0.001) and a greater increase in BMI z-score (0.45 SD, p<0.001) in children aged 6-11 years when compared to placebo (ENVISION) (64).
Stalvey and colleagues performed a post-hoc analysis of 83 children ages 6-11 years enrolled in the ENVISION and the GOAL study, a 6-month longitudinal observational study of ivacaftor therapy in those with at least one G551D mutation. Height and weight z-scores increased significantly from baseline in both studies (height z-score increase of 0.1 SD in GOAL and 0.17 SD in ENVISION; weight z-score increase of 0.26 SD in GOAL and 0.35 SD in ENVISION). Height velocity increased significantly in both studies (increase of 2.1 cm/yr between 3-6 months of treatment in GOAL and 1.08 cm/yr greater than placebo in ENVISION). Furthermore, weight velocity was significantly greater in the ivacaftor group compared to the placebo group in ENVISION (treatment difference of 3.06 kg/yr in boys and 2.81 kg/yr in girls) (22) (Table 1).
In a 3-month observational study of 23 patients with at least one gating mutation ranging in age from 5 to 61 years, those treated with ivacaftor had a significant increase in weight, which was positively correlated to the change in FEV1 (p=0.028). Furthermore, there was an increase in fat free mass with a reduction in resting energy expenditure in response to ivacaftor therapy (65).
Ivacaftor therapy was also associated with a significantly greater increase in BMI and BMI z-score in children ≥ 6 years of age with non-G551D gating mutations when compared to placebo (treatment effect 0.7 kg/m2 and 0.28 SD respectively) (66). However, when used to treat individuals ≥ 12 years of age who were homozygous for Phe508del, there were no significant differences in change in weight or BMI between those treated with ivacaftor or placebo (67).
A few years after the FDA approval of ivacaftor, lumacafator/ivacaftor (corrector/potentiator) received FDA approval for individuals ≥12 years of age who were homozygous for the most common mutation in CFTR, the Phe508del (Table 1) (63). Data pooled from two large 24-week randomized double-blind placebo-controlled trials of lumacaftor/ivacaftor in individuals ≥12 years of age homozygous for Phe508del (TRAFFIC and TRANSPORT) demonstrated a significant increase in BMI in the treatment groups (treatment difference of 0.24-0.28 kg/m2). Unfortunately, these data were not stratified by age, and the specific impact in children was not reported (21) (Table 1). When these data were stratified by severity of pulmonary disease, the increase in BMI in the treatment group remained significant across all levels of pulmonary function (68).
In a separate 24-week randomized double-blind, placebo-controlled trial of lumacaftor/ivacaftor in children aged 6-11 years who were homozygous for Phe508del, there was an increase in BMI in both the treatment and placebo groups without a significant treatment difference (24).
Tezacaftor/ivacaftor (corrector/potentiator) was approved in 2018 for individuals homozygous for the Phe508del mutation as well as those with a single Phe508del mutation and one of 26 other mutations (62). In a study of individuals ≥12 years of age homozygous for Phe508del, BMI increased from baseline to 24 weeks in both the tezacaftor/ivacaftor and placebo groups with no significant treatment effect reported (23).
In 2019, elexacaftor/tezacaftor/ivacaftor (next generation corrector/corrector/potentiator) received FDA approval to treat the nearly 90% of individuals harboring at least 1 copy of the Phe508del mutation (62).
In a large multi-center, randomized, double-blind, active controlled trial of elexacaftor/tezacaftor/ivacaftor for individuals aged ≥12 years homozygous for Phe508del, there was a significant increase in mean weight (1.6 kg) and BMI (0.6 kg/m2) compared to those treated with tezacaftor/ivacaftor alone. Unfortunately, these data were not stratified by age and the impact on the pediatric population was not reported (26). The treatment difference in BMI (mean treatment difference of 1.04 kg/m2) was also significant when elexacaftor/tezacaftor/ivacaftor was compared to placebo in individuals aged ≥12 years with a single Phe508del mutation and a minimal function genotype. Again, these data were not stratified by age and the specific impact on the pediatric population could not be assessed (25) (Table 1). Additional study is needed to evaluate the impact of modulator therapies, including triple combination therapy, on long-term growth outcomes in children.
The nutritional status of children with CF in the 2020’s remains comparable to the general population. In fact, median weight and BMI for children and adolescents aged 2-19 are 51.9% and 61.4% respectively. Likewise, the median weight and weight-for-length during infancy and early childhood are 44.5% and 62.8% respectively (28) (Table 1).
Despite overall improvements in nutritional metrics, however, length and height remain below those of the general population (median length 31.5% for ages <24 months; median height 38.7% for ages 2-19 years) (28) (Table 1). This continues to be an important area of investigation, as early childhood height is associated with adult height. In fact, Marks et al., found that the maximum height measurements between ages 2 and 4 years are highly correlated with maximum adult height at age 18 or 19 (r=0.64) (29) (Table 1). Furthermore, children who maintain a height-for-age over the 50th centile have a higher FEV1 at age 6 or 7 than those who have a height-for-age less than the 50th centile for at least 1 year. The relationship between height and pulmonary function in this study was not affected by BMI adjustment (37), suggesting that linear growth alone plays an important role in pulmonary function (Table 2).
Recent evidence suggests that BMI may not be a reliable indicator of nutritional status in children with CF. Owen et al. reported on body composition and its association with pulmonary function in 37 pre-pubertal children (27). They found that 5% of their population had a fat-free mass index (FFMI) z-score of < -1 [near the value of hidden depletion previously published in adult CF studies (69)] despite optimal BMI z-scores. FEV1 was more strongly associated with whole body FFMI than with BMI (27), supporting the importance of body composition on pulmonary function previously observed in both children and adults (36, 70) (Table 2).
Since cystic fibrosis was first described in 1938, there have been many breakthroughs in diagnosis and management. Such developments include the use of antibiotics to treat pulmonary infections, dietary modifications to increase fat and caloric intake, use of pancreatic enzymes, anti-pseudomonal therapies, early identification and intervention through newborn screening, use of mucolytics, and treatment with CFTR modulators, all of which have played a critical role in improving the life expectancy and quality of life for individuals with CF.
Growth in children with CF has historically been poor with weights and heights far below the general population, attributed to malabsorption, reduced caloric intake, increased resting energy expenditure, glucocorticoid exposure and systemic inflammation (71). In addition, historical data support an indirect impact of CFTR genotype on nutritional status through the association with pancreatic exocrine function (72). Whether there is a direct effect of CFTR on growth remains unknown.
Previous studies reported a greater impact on weight than height, resulting in BMI z-scores less than 0 SD. Over the last several decades, however, the weight and height z-scores seem to be increasing with some children now meeting criteria for overweight or obesity. While this could reflect an improvement in both weight and height with a more significant improvement in weight relative to height, it appears to be the result of improved weight with minimal increase in height (28, 73). One plausible explanation is a direct effect of CFTR on the GH-IGF-1 axis. However, the exact mechanism remains unclear and additional study is warranted.
While historical data demonstrate a clear association between nutritional status and lung function (34, 74, 75), more recent data support an association between height in early childhood and subsequent pulmonary function regardless of nutritional status (BMI) (37). Therefore, it will be important to identify factors that contribute to early life growth impairment and monitor the impact of highly effective CFTR modulator therapy on linear growth.
Given the minimal increase in height z-scores relative to weight, updated nutritional guidelines will be of great importance in the era of early detection and early treatment with highly effective modulator therapy to optimize growth and pulmonary function while avoiding obesity and its comorbidities. Body composition may provide better insight into nutritional status and its impact on clinical outcomes than BMI alone.
KM and AR conceived the idea for the topic, both contributed to the outline; KM wrote the first draft and KM and AR contributed to the multiple revisions. All authors contributedto the article and approved the submitted version.
This work was supported by the Cystic Fibrosis Foundation Envision CF: Emerging Leaders in CF Endocrinology II Program [grant number MASON19GE0].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Dr. Michael Stalvey for his expertise and contribution to the paper and would also like to thank Ms. Kate Joshua and the Claude Moore Medical Library. KM would also like to thank the Cystic Fibrosis Foundation and the mentors from the EnVision: Emerging Leaders in CF Endocrinology II program, including Drs. Trang Le and Scott Blackman, for their support and mentorship.
1. Cystic Fibrosis Foundation CF Genetics: The Basics. Cystic Fibrosis Foundation (2022) Bethesda, MD. Available at: https://www.cff.org/intro-cf/cf-genetics-basics.
2. Andersen DH. Cystic Fibrosis of the Pancreas and Its Relation to Celiac Disease: A Clinical and Pathologic Study. Am J Dis Child (1938) 56(2):344–99. doi: 10.1001/archpedi.1938.01980140114013
3. Scaparrotta A, Di Pillo S, Attanasi M, Consilvio NP, Cingolani A, Rapino D, et al. Growth Failure in Children With Cystic Fibrosis. J Pediatr Endocrinol Metab (2012) 25(5-6):393–405. doi: 10.1515/jpem-2012-0012
4. Lai HC, Kosorok MR, Sondel SA, Chen ST, FitzSimmons SC, Green CG, et al. Growth Status in Children With Cystic Fibrosis Based on the National Cystic Fibrosis Patient Registry Data: Evaluation of Various Criteria Used to Identify Malnutrition. J Pediatr (1998) 132(3):478–85. doi: 10.1016/S0022-3476(98)70024-1
5. Zhang Z, Lindstrom MJ, Farrell PM, Lai HJ. Pubertal Height Growth and Adult Height in Cystic Fibrosis After Newborn Screening. Pediatrics (2016) 137(5): e20152907. doi: 10.1542/peds.2015-2907
6. Parmelee AH. The Pathology of Steatorrhea. Am J Dis Child (1935) 50(6):1418–28. doi: 10.1001/archpedi.1935.01970120070006
7. Shwachman H, Kulczycki LL. Long-Term Study of One Hundred Five Patients With Cystic Fibrosis: Studies Made Over a Five-to Fourteen-Year Period. AMA J Dis Child (1958) 96(1):6–15. doi: 10.1001/archpedi.1958.02060060008002
8. McIntosh R. Cystic Fibrosis of the Pancreas in Patients Over Ten Years of Age. Acta Pediatr (1954) 43:469–80. doi: 10.1111/j.1651-2227.1954.tb15492.x
9. Sproul A, Huang N. Growth Patterns in Children With Cystic Fibrosis. J Pediatr (1964) 65(5):664–76. doi: 10.1016/S0022-3476(64)80151-7
10. Kreißl T, Bender SW, Mörchen R, Hövels O. The Physical Development of Children With Cystic Fibrosis. Z Fuer Kinderheilkd (1972) 113(2):93–110. doi: 10.1007/BF00473404
11. Berry HK, Kellogg FW, Hunt MM, Ingberg RL, Richter L, Gutjahr C. Dietary Supplement and Nutrition in Children With Cystic Fibrosis. Am J Dis Child (1975) 129(2):165–71. doi: 10.1001/archpedi.1975.02120390009003
12. Mitchell-Heggs P, Mearns M, Batten JC. Cystic Fibrosis in Adolescents and Adults. QJM: Int J Med (1976) 45(3):479–504. doi: 10.1093/oxfordjournals.qjmed.a067476
13. Corey MF, McLaughlin FJ, Williams M, Levison H. A Comparison of Survival, Growth, and Pulmonary Function in Patients With Cystic Fibrosis in Boston and Toronto. J Clin Epidemiol (1988) 41(6):583–91. doi: 10.1016/0895-4356(88)90063-7
14. Keller BM, Aebischer CC, Kraemer R, Schöni MH. Growth in Prepubertal Children With Cystic Fibrosis, Homozygous for the ΔF508 Mutation. J Cyst Fibros (2003) 2(2):76–83. doi: 10.1016/S1569-1993(03)00023-7
15. Nir M, Lanng S, Johansen HK, Koch C. Long-Term Survival and Nutritional Data in Patients With Cystic Fibrosis Treated in a Danish Centre. Thorax (1996) 51(10):1023–7. doi: 10.1136/thx.51.10.1023
16. Farrell PM, Kosorok MR, Laxova A, Shen G, Koscik RE, Bruns WT, et al. Nutritional Benefits of Neonatal Screening for Cystic Fibrosis. New Engl J Med (1997) 337(14):963–9. doi: 10.1056/NEJM199710023371403
17. Lai HC, Kosorok MR, Laxova A, Davis LA, FitzSimmon SC, Farrell PM. Nutritional Status of Patients With Cystic Fibrosis With Meconium Ileus: A Comparison With Patients Without Meconium Ileus and Diagnosed Early Through Neonatal Screening. Pediatrics (2000) 105(1):53–61. doi: 10.1542/peds.105.1.53
18. Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, et al. Early Diagnosis of Cystic Fibrosis Through Neonatal Screening Prevents Severe Malnutrition and Improves Long-Term Growth. Pediatrics (2001) 107(1):1–3. doi: 10.1542/peds.107.1.1
19. Kastner-Cole D, Palmer CN, Ogston SA, Mehta A, Mukhopadhyay S. Overweight and Obesity in ΔF508 Homozygous Cystic Fibrosis. J Pediatr (2005) 147(3):402–4. doi: 10.1016/j.jpeds.2005.06.003
20. Hanna RM, Weiner DJ. Overweight and Obesity in Patients With Cystic Fibrosis: A Center-Based Analysis. Pediatr Pulmonol (2015) 50(1):35–41. doi: 10.1002/ppul.23033
21. Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor–ivacaftor in Patients With Cystic Fibrosis Homozygous for Phe508del CFTR. New Engl J Med (2015) 373(3):220–31. doi: 10.1056/NEJMoa1409547
22. Stalvey MS, Pace J, Niknian M, Higgins MN, Tarn V, Davis J, et al. Growth in Prepubertal Children With Cystic Fibrosis Treated With Ivacaftor. Pediatrics (2017) 139(2): e20162522. doi: 10.1542/peds.2016-2522
23. Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, et al. Tezacaftor–ivacaftor in Patients With Cystic Fibrosis Homozygous for Phe508del. New Engl J Med (2017) 377(21):2013–23. doi: 10.1056/NEJMoa1709846
24. Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, et al. Efficacy and Safety of Lumacaftor and Ivacaftor in Patients Aged 6–11 Years With Cystic Fibrosis Homozygous for F508del-CFTR: A Randomised, Placebo-Controlled Phase 3 Trial. Lancet Respir Med (2017) 5(7):557–67. doi: 10.1016/S1569-1993(17)30234-5
25. Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor–tezacaftor–ivacaftor for Cystic Fibrosis With a Single Phe508del Allele. New Engl J Med (2019) 381(19):1809–19. doi: 10.1056/NEJMoa1908639
26. Heijerman HG, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. Efficacy and Safety of the Elexacaftor Plus Tezacaftor Plus Ivacaftor Combination Regimen in People With Cystic Fibrosis Homozygous for the F508del Mutation: A Double-Blind, Randomised, Phase 3 Trial. Lancet (2019) 394(10212):1940–8. doi: 10.1016/S0140-6736(19)32597-8
27. Owen E, Williams JE, Davies G, Wallis C, Grant RL, Fewtrell MS. Growth, Body Composition, and Lung Function in Prepubertal Children With Cystic Fibrosis Diagnosed by Newborn Screening. Nutr Clin Pract (2021) 36(6):1240–6. doi: 10.1002/ncp.10604
28. Foundation, C.F. Cystic Fibrosis Foundation Patient Registry 2020 Annual Data Report. Bethesda, MD, USA: Cystic Fibrosis Foundation (2022).
29. Marks MP, Heltshe SL, Baines A, Ramsey BW, Hoffman LR, Stalvey MS. Most Short Children With Cystic Fibrosis Do Not Catch Up by Adulthood. Nutrients (2021) 13(12):4414. doi: 10.3390/nu13124414
30. Boyer PH. Low Birth Weight in Fibrocystic Disease of the Pancreas. Pediatrics (1955) 16(6):778–84. doi: 10.1542/peds.16.6.778
31. Hsia DY. Birth Weight in Cystic Fibrosis of the Pancreas. Ann Hum Genet (1959) 23(3):289–99. doi: 10.1111/j.1469-1809.1959.tb01472.x
32. Greco L, Santamaria F, Salvatore D, Ritis GD. Growth Dynamics in Cystic Fibrosis. Acta Pediatr (1993) 82(3):254–60. doi: 10.1111/j.1651-2227.1993.tb12654.x
33. Byard PJ. The Adolescent Growth Spurt in Children With Cystic Fibrosis. Ann Hum Biol (1994) 21(3):229–40. doi: 10.1080/03014469400003242
34. Konstan MW, Butler SM, Wohl ME, Stoddard M, Matousek R, Wagener JS, et al. Growth and Nutritional Indexes in Early Life Predict Pulmonary Function in Cystic Fibrosis. J Pediatr (2003) 142(6):624–30. doi: 10.1067/mpd.2003.152
35. Stallings VA, Stark LJ, Robinson KA, Feranchak AP, Quinton H, Growth CP, et al. Evidence-Based Practice Recommendations for Nutrition-Related Management of Children and Adults With Cystic Fibrosis and Pancreatic Insufficiency: Results of a Systematic Review. J Am Diet Assoc (2008) 108(5):832–9. doi: 10.1016/j.jada.2008.02.020
36. Sheikh S, Zemel BS, Stallings VA, Rubenstein RC, Kelly A. Body Composition and Pulmonary Function in Cystic Fibrosis. Front Pediatr (2014) 2:33. doi: 10.3389/fped.2014.00033
37. Sanders DB, Slaven JE, Maguiness K, Chmiel JF, Ren CL. Early-Life Height Attainment in Cystic Fibrosis Is Associated With Pulmonary Function at Age 6 Years. Ann Am Thorac Soc (2021) 18(8):1335–42. doi: 10.1513/AnnalsATS.202008-933OC
38. Stern RC, Boat TF, Doershuk CF, Tucker AS, Primiano FP Jr., Matthews LW. Course of Cystic Fibrosis in 95 Patients. J Pediatr (1976) 89(3):406–11. doi: 10.1016/S0022-3476(76)80537-9
39. Altman K, McDonald CM, Michel SH, Maguiness K. Nutrition in Cystic Fibrosis: From the Past to the Present and Into the Future. Pediatr Pulmonol (2019) 54:S56–73. doi: 10.1002/ppul.24521
40. Graham DY. An Enteric-Coated Pancreatic Enzyme Preparation That Works. Dig Dis Sci (1979) 24(12):906–9. doi: 10.1007/BF01311943
41. Mischler EH, Parrell S, Farrell PM, Odell GB. Comparison of Effectiveness of Pancreatic Enzyme Preparations in Cystic Fibrosis. Am J Dis Child (1982) 136(12):1060–3. doi: 10.1001/archpedi.1982.03970480026006
42. Soutter VL, Kristidis P, Gruca MA, Gaskin KJ. Chronic Undernutrition/Growth Retardation in Cystic Fibrosis. Clinics Gastroenterol (1986) 15(1):137–55. doi: 10.1016/S0300-5089(21)00676-3
43. Landon C, Rosenfeld RG. Short Stature and Pubertal Delay in Male Adolescents With Cystic Fibrosis: Androgen Treatment. Am J Dis Child (1984) 138(4):388–91. doi: 10.1001/archpedi.1984.02140420054017
44. Moshang T, Holsclaw DS. Menarchal Determinants in Cystic Fibrosis. Am J Dis Child (1980) 134(12):1139–42. doi: 10.1001/archpedi.1980.02130240023008
45. Mahaney MC, Mahaney MS, McCoy KS. Developmental Delays and Pulmonary Disease Severity in Cystic Fibrosis. Hum Biol (1986) 1:445–60.
46. Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients With Cystic Fibrosis. New Engl J Med (1994) 331(10):637–42. doi: 10.1056/NEJM199409083311003
47. Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, et al. Intermittent Administration of Inhaled Tobramycin in Patients With Cystic Fibrosis. New Engl J Med (1999) 340(1):23–30. doi: 10.1056/NEJM199901073400104
48. Karlberg J, Kjellmer I, Kristiansson B. Linear Growth in Children With Cystic Fibrosis: I. Birth 8 Years Age Acta Paediatr (1991) 80(5):508–14. doi: 10.1111/j.1651-2227.1991.tb11894.x
49. Morison S, Dodge JA, Cole TJ, Lewis PA, Coles EC, Geddes D, et al. Height and Weight in Cystic Fibrosis: A Cross Sectional Study. Arch Dis Child (1997) 77(6):497–500. doi: 10.1136/adc.77.6.497
50. Johannesson M, Gottlieb C, Hjelte L. Delayed Puberty in Girls With Cystic Fibrosis Despite Good Clinical Status. Pediatrics (1997) 99(1):29–34. doi: 10.1542/peds.99.1.29
51. Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, et al. A Controlled Trial of Long-Term Inhaled Hypertonic Saline in Patients With Cystic Fibrosis. New Engl J Med (2006) 354(3):229–40. doi: 10.1056/NEJMoa043900
52. Saiman L, Marshall BC, Mayer-Hamblett N, Burns JL, Quittner AL, Cibene DA, et al. Azithromycin in Patients With Cystic Fibrosis Chronically Infected With Pseudomonas Aeruginosa: A Randomized Controlled Trial. Jama (2003) 290(13):1749–56. doi: 10.1001/jama.290.13.1749
53. Southern KW, Mérelle MM, Dankert-Roelse JE, Nagelkerke AD. Newborn Screening for Cystic Fibrosis. Cochrane Database Syst Rev (2009) 1: CD001402. doi: 10.1002/14651858.CD001402.pub2
54. Assael BM, Casazza G, Iansa P, Volpi S, Milani S. Growth and Long-Term Lung Function in Cystic Fibrosis: A Longitudinal Study of Patients Diagnosed by Neonatal Screening. Pediatr Pulmonol (2009) 44(3):209–15. doi: 10.1002/ppul.21001
55. Umławska W, Krzyżanowska M, Zielińska A, Sands D. Effect of Selected Factors Associated With the Clinical Course of the Disease on Nutritional Status in Children With Cystic Fibrosis. Adv Clin Exp Med (2014) 23(5):775–83. doi: 10.17219/acem/37251
56. Marín VB, Velandia S, Hunter B, Gattas V, Fielbaum O, Herrera O, et al. Energy Expenditure, Nutrition Status, and Body Composition in Children With Cystic Fibrosis. Nutrition (2004) 20(2):181–6. doi: 10.1016/j.nut.2003.10.010
57. Cystic Fibrosis Foundation. All Fifty States to Screen Newborns for Cystic Fibrosis by 2010. Cystic Fibrosis Foundation (2022) Bethesda, MD. Available at: https://www.cff.org/node/766.
58. Schechter MS, Michel S, Liu S, Seo BW, Kapoor M, Khurmi R, et al. Relationship of Initial Pancreatic Enzyme Replacement Therapy Dose With Weight Gain in Infants With Cystic Fibrosis. J Pediatr Gastroenterol Nutr (2018) 67(4):520. doi: 10.1097/MPG.0000000000002108
59. Leung DH, Heltshe SL, Borowitz D, Gelfond D, Kloster M, Heubi JE, et al. Effects of Diagnosis by Newborn Screening for Cystic Fibrosis on Weight and Length in the First Year of Life. JAMA Pediatr (2017) 171(6):546–54. doi: 10.1001/jamapediatrics.2017.0206
60. VanDevanter DR, Pasta DJ, Konstan MW. Improvements in Lung Function and Height Among Cohorts of 6-Year-Olds With Cystic Fibrosis From 1994 to 2012. J Pediatr (2014) 165(6):1091–7. doi: 10.1016/j.jpeds.2014.06.061
61. Garrote MD, Camargo Costa LD, Garrote Filho MD, Francisco Duarte WD, Baragatti RF, Fernandes RV. Respiratory Colonization and Nutritional Status of Children and Adolescents in the Cystic Fibrosis Clinic From Clinical Hospital-UFG. Pediatr Pulmonol (2016) 51:S42.
62. Collins FS. Realizing the Dream of Molecularly Targeted Therapies for Cystic Fibrosis. New Engl J Med (2019) 381(19):1863–5. doi: 10.1056/NEJMe1911602
63. Frink L. Our History. Cystic Fibrosis Foundation (2020) Bethesda, MD. Available at: https://www.cff.org/about-us/our-history?msclkid=5af6e566c03411ec91fbed7d065f5867.
64. Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Efficacy and Safety of Ivacaftor in Patients Aged 6 to 11 Years With Cystic Fibrosis With a G551D Mutation. Am J Respir Crit Care Med (2013) 187(11):1219–25. doi: 10.1164/rccm.201301-0153OC
65. Stallings VA, Sainath N, Oberle M, Bertolaso C, Schall JI. Energy Balance and Mechanisms of Weight Gain With Ivacaftor Treatment of Cystic Fibrosis Gating Mutations. J Pediatr (2018) 201:229–37. doi: 10.1016/j.jpeds.2018.05.018
66. De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, et al. Efficacy and Safety of Ivacaftor in Patients With Cystic Fibrosis and a Non-G551D Gating Mutation. J Cyst Fibros (2014) 13(6):674–80. doi: 10.1016/j.jcf.2014.09.005
67. Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordoñez CL, et al. Ivacaftor in Subjects With Cystic Fibrosis Who are Homozygous for the F508del-CFTR Mutation. Chest (2012) 142(3):718–24. doi: 10.1378/chest.11-2672
68. Elborn JS, Ramsey BW, Boyle MP, Konstan MW, Huang X, Marigowda G, et al. Efficacy and Safety of Lumacaftor/Ivacaftor Combination Therapy in Patients With Cystic Fibrosis Homozygous for Phe508del CFTR by Pulmonary Function Subgroup: A Pooled Analysis. Lancet Respir Med (2016) 4(8):617–26. doi: 10.1016/S2213-2600(16)30121-7
69. King SJ, Nyulasi IB, Strauss BJ, Kotsimbos T, Bailey M, Wilson JW. Fat-Free Mass Depletion in Cystic Fibrosis: Associated With Lung Disease Severity But Poorly Detected by Body Mass Index. Nutrition (2010) 26(7-8):753–9. doi: 10.1016/j.nut.2009.06.026
70. Alvarez JA, Ziegler TR, Millson EC, Stecenko AA. Body Composition and Lung Function in Cystic Fibrosis and Their Association With Adiposity and Normal-Weight Obesity. Nutrition (2016) 32(4):447–52. doi: 10.1016/j.nut.2015.10.012
71. Le TN, Anabtawi A, Putman MS, Tangpricha V, Stalvey MS. Growth Failure and Treatment in Cystic Fibrosis. J Cyst Fibros (2019) 18:S82–7. doi: 10.1016/j.jcf.2019.08.010
72. Kerem E, Corey M, Kerem B, Rommens J, Markiewicz D. Association Between the Deltaf508 Mutation and Phenotypes in Cystic Fibrosis. N Engl J Med (1990) 323:1517–22. doi: 10.1056/NEJM199011293232203
73. Konstan MW, Pasta DJ, Wagener JS, VanDevanter DR, Morgan WJ. BMI Fails to Identify Poor Nutritional Status in Stunted Children With CF. J Cyst Fibros (2017) 16(1):158–60. doi: 10.1016/j.jcf.2016.11.005
74. Yen EH, Quinton H, Borowitz D. Better Nutritional Status in Early Childhood is Associated With Improved Clinical Outcomes and Survival in Patients With Cystic Fibrosis. J Pediatr (2013) 162(3):530–5. doi: 10.1016/j.jpeds.2012.08.040
Keywords: cystic fibrosis, growth, height, weight, puberty, body composition
Citation: Mason KA and Rogol AD (2022) Trends in Growth and Maturation in Children with Cystic Fibrosis Throughout Nine Decades. Front. Endocrinol. 13:935354. doi: 10.3389/fendo.2022.935354
Received: 03 May 2022; Accepted: 13 June 2022;
Published: 12 July 2022.
Edited by:
Madhusmita Misra, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Jiunn-Tyng Yeh, National Yang Ming Chiao Tung University, TaiwanCopyright © 2022 Mason and Rogol. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelly A. Mason, a2VsbHkubWFzb25AdmlyZ2luaWEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.