 Dolores Aguilar-Cazares1†
Dolores Aguilar-Cazares1† Rodolfo Chavez-Dominguez1,2†
Rodolfo Chavez-Dominguez1,2† Mario Marroquin-Muciño1,3†
Mario Marroquin-Muciño1,3† Mario Perez-Medina1,3
Mario Perez-Medina1,3 Jesus J. Benito-Lopez1,2
Jesus J. Benito-Lopez1,2 Angel Camarena4Uriel Rumbo-Nava5
Angel Camarena4Uriel Rumbo-Nava5 Jose S. Lopez-Gonzalez1*
Jose S. Lopez-Gonzalez1*- 1Laboratorio de Investigacion en Cancer Pulmonar, Departamento de Enfermedades Cronico-Degenerativas, Instituto Nacional de Enfermedades Respiratorias “Ismael Cosio Villegas”, Mexico City, Mexico
- 2Posgrado en Ciencias Biologicas, Universidad Nacional Autonoma de Mexico, Mexico City, Mexico
- 3Laboratorio de Quimioterapia Experimental, Departamento de Bioquimica, Escuela Nacional de Ciencias Biologicas, Instituto Politecnico Nacional, Mexico City, Mexico
- 4Laboratorio de Human Leukocyte Antigen (HLA), Instituto Nacional de Enfermedades Respiratorias “Ismael Cosio Villegas”, Mexico City, Mexico
- 5Clinica de Neumo-Oncologia, Instituto Nacional de Enfermedades Respiratorias “Ismael Cosio Villegas”, Mexico City, Mexico
The tumor microenvironment is a dynamic, complex, and redundant network of interactions between tumor, immune, and stromal cells. In this intricate environment, cells communicate through membrane–membrane, ligand–receptor, exosome, soluble factors, and transporter interactions that govern cell fate. These interactions activate the diverse and superfluous signaling pathways involved in tumor promotion and progression and induce subtle changes in the functional activity of infiltrating immune cells.
The immune response participates as a selective pressure in tumor development. In the early stages of tumor development, the immune response exerts anti-tumor activity, whereas during the advanced stages, the tumor establishes mechanisms to evade the immune response, eliciting a chronic inflammation process that shows a pro-tumor effect.
The deregulated inflammatory state, in addition to acting locally, also triggers systemic inflammation that has repercussions in various organs and tissues that are distant from the tumor site, causing the emergence of various symptoms designated as paraneoplastic syndromes, which compromise the response to treatment, quality of life, and survival of cancer patients. Considering the tumor–host relationship as an integral and dynamic biological system, the chronic inflammation generated by the tumor is a communication mechanism among tissues and organs that is primarily orchestrated through different signals, such as cytokines, chemokines, growth factors, and exosomes, to provide the tumor with energetic components that allow it to continue proliferating. In this review, we aim to provide a succinct overview of the involvement of cancer-related inflammation at the local and systemic level throughout tumor development and the emergence of some paraneoplastic syndromes and their main clinical manifestations. In addition, the involvement of these signals throughout tumor development will be discussed based on the physiological/biological activities of innate and adaptive immune cells. These cellular interactions require a metabolic reprogramming program for the full activation of the various cells; thus, these requirements and the by-products released into the microenvironment will be considered. In addition, the systemic impact of cancer-related proinflammatory cytokines on the liver—as a critical organ that produces the leading inflammatory markers described to date—will be summarized. Finally, the contribution of cancer-related inflammation to the development of two paraneoplastic syndromes, myelopoiesis and cachexia, will be discussed.
Introduction
In 2020, GLOBOCAN estimated the global cancer statistics as 19.3 million new cases and 10 million deaths yearly (1). The increasing incidence and mortality rates reflect the growth and aging of the population and the increase in risk factors associated with socioeconomic development. Great efforts have been made to detect cancer early; however, most cases are detected at advanced stages.
Inflammation is a well-conserved process in which a distinct subset of cells from the innate and adaptive immune response is recruited to eliminate harmful agents in the host. This process is essential for the host’s defense against pathogens and is accompanied by tissue repair and wound healing to regulate tissue homeostasis. However, when dysregulated, inflammation contributes to the emergence and development of cancer. Tumor-associated inflammation is a well-recognized tumor-enabling characteristic that promotes or sustains the acquisition of some characteristics termed the hallmarks of cancer (2, 3). During tumor development, tumor-associated inflammation shapes the anti-tumor immune response towards a more permissive and pro-tumoral state (3). In this regard, the relationship between the tumor and the immune response is well known; according to immunoediting theory, at the early stages of tumor development, the immune system exerts anti-tumor activity through immunosurveillance (4). In this setting, as the tumor evolves, so does its microenvironment and the immune response, favoring the establishment of a pro-tumoral immune response. Several reports have indicated that the shift from anti-tumor immunity towards a pro-tumoral response is supported by a myriad of factors released from the tumor, immune, and stromal cells into the tumor microenvironment, which act to establish a persistent tumor-associated inflammatory state (5).
Nonetheless, the tumor-associated inflammatory state not only has repercussions in its immediate local microenvironment, but the release of various components into the bloodstream that promote or sustain inflammatory activity at the systemic level primes a cancer-induced systemic inflammatory response (6). At the plasma level, high concentrations of these proinflammatory factors can affect different organs or systems, such as the endocrine, nervous, dermatological, and hematological systems, among others, resulting in the alteration of the expression of some molecules or set of circulating cells, which are currently used as markers of systemic inflammation associated with cancer (5). In addition to the known cytokines, chemokines, and growth factors, it is now recognized that exosomes are one of the main factors capable of reaching different organs or systems, leading to the development of additional comorbidities called paraneoplastic syndromes (6). Among these paraneoplastic syndromes, neuropathy, hypercalcemia, dermatomyositis, cachexia, and dysregulated hematopoiesis cause detrimental effects on the patient’s quality of life and are sometimes manifested before cancer detection (7). In some instances, the clinical manifestation of paraneoplastic syndromes contributes to the promotion of tumor growth-promoting capabilities, leading to decreased overall survival (8, 9).
The study of cancer initially focused on the tumor’s genetic alterations and biological activity. Recently, the role of the bidirectional interactions between the tumor and its microenvironment as an integral and evolving biological system has been considered. Although human tumors are composed of heterogeneous cell populations, employing tumor cell lines and animal models has allowed us to deepen our understanding of the participation of the microenvironment throughout tumor development.
This review highlights the intricate signaling mediated by the different components released in the tumor microenvironment and their contribution at the systemic level. First, we will describe the interaction between the tumor and the immune cells and its evolution during tumor development. The local production of immune-stimulating factors by the stroma and immune inhibitory mediators induced or produced by the tumor will also be considered. At the systemic level, the effect of the main proinflammatory cytokines reaching their target organs and their impact on the production of inflammation markers will be addressed. Finally, the clinical manifestations associated with the development of inflammatory cytokines-induced paraneoplastic syndromes will be examined.
Tumor microenvironment
According to the multistep carcinogenesis model, a tumor is shaped by a group of heterogeneous cells harboring genomic and epigenomic alterations. Transformed cells carrying driver mutations and epigenetic alterations activate aberrant signaling pathways that hinder the apoptotic process and promote uncontrolled cell proliferation. The growth of these transformed cells leads to changes in tissue architecture, which induces stress in the cells of the surrounding stroma, causing an increase in the production of soluble inflammatory mediators and growth factors and exosome release. These factors maintain a chronic inflammatory microenvironment that enables tumorigenesis (10). As the tumor grows, heterogeneous cell populations are generated due to the high and stochastic proliferation rate. Some of these new populations in the tumor mass acquire immune evasion mechanisms or produce soluble factors that modify immune cell phenotypes to support pro-tumor activity (11).
It has been recognized that the tumor microenvironment (TME) participates in cancer development and promotes the acquisition of some hallmarks of cancer (2). The composition of TME is heterogeneous; it is mainly composed of—but not limited to—cells such as endothelial cells, cancer-associated fibroblasts, pericytes, cancer stem cells, and immune-inflammatory cells, in addition to diverse extracellular matrix components (2).
In this context, the TME is a complex, redundant, and dynamic network that is constantly evolving throughout tumor development and progression. In this network, tumor, immune, and non-immune cells establish membrane–membrane and ligand–receptor interactions as well as communicate through the paracrine, juxtacrine, and internal secretion of various substances, such as proteins, different types of RNA, lipids, and biological mediators, which are delivered through the production of exosomes (12–14). Exosomes are vesicles between 40 and 160 nm in diameter. Exosomes arise from an early endosome in a process mediated by the endosomal sorting complex required for transport (ESCRT) (15). These mature endosomes are also known as multivesicular bodies (MVBs). MVBs can fuse with lysosomes for the degradation of their contents or can fuse with the plasma membrane, releasing their vesicles into the extracellular space (16). Exosomes can contain proteins, RNA, DNA, lipids, and carbohydrates. Initially considered as waste products of cells, exosomes are now known to play an essential role in cell communication (17). Most reports indicate that exosomes play paramount roles in tumor cell invasion, metastasis, and angiogenesis. In addition, exosomes are involved in modulating the TME, altering cellular metabolism, and promoting or inhibiting the immune response (18).
All of these interactions and molecule transfers activate diverse signaling pathways that affect gene expression, support the metabolic demands of different cell types, and induce the synthesis of various proteins that act as critical biomolecules to induce the participation of the immune response against genotoxic insults, incipient tumor formation, and tumor development (19, 20). During the early stage of tumor development, a nascent transformed cell develops in close interaction with the resident immune cells, among which the incipient transformed cell proliferates to form a small group of cells that lead to the distortion of the local tissue morphology. In this regard, and as part of the innate immune response, natural killer (NK) cells and resident macrophages eliminate susceptible tumor cells by releasing cytotoxic molecules that insert themselves into the tumor cell membrane, altering its permeability and causing cell death (21). Throughout this process, the dying cells expose molecules on their membrane or release intracellular molecules that acquire a new function, acting as alarmins or damage-associated molecular patterns (DAMPs) that promote the recruitment of other populations of immune cells, such as those involved in the adaptive immune response.
At this point, some reports have indicated that the exosomes released by tumor cells express class I and II MHC molecules and can prime and activate the immune response. As tumor cells develop and persistent growth occurs, the activation of the immune response continues and chronic inflammation is promoted, which initially stimulates an anti-tumor immune response (see below). However, it is known that chronic inflammation allows for the acquisition of new mutations and increased genome instability. Chronic inflammation causes the cellular composition of the tumor to become heterogeneous, resulting in a progressive change in the activities of the immune and stromal cells to promote a microenvironment that favors progression, invasion, and metastasis (22–24).
Participation of the immune response in cancer
The relationship between chronic inflammation and cancer development is well known and is considered a hallmark of cancer (2). Virchow’s observations led him to propose that chronic inflammation provoked by the presence of an immune infiltrate was associated with the development of cancer (25). Afterward, Dvorak reported similar features between inflammation and cancer, such as proliferation, cell survival, angiogenesis, and migration (26).
The immune system is composed of an intricate network of cells, including NK cells, which are part of the innate lymphoid cells (ILCs) (27) and NKT cells, along with macrophages and dendritic cells (DCs), which are cells of the phagocytic mononuclear system that are involved in antigen presentation. As part of the adaptive immune response includes T lymphocytes, such as CD4+ T and CD8+ T cells, and B lymphocytes (28). The detailed study of tumor-infiltrating immune cells in biopsied material obtained from cancer patients has indicated that immune cells interact with tumor cells through the production of diverse factors, such as cytokines, chemokines, the by-products of cell metabolism, growth factors, and the components of exosomes, which participate during the tumor development stages (29). It has been suggested that immune cells and the soluble factors they secrete induce a particular microenvironment that, in the early stages of tumor development, supports anti-tumor activities; nevertheless, the microenvironment evolves, and in the advanced stages of the tumor, the immune cells are modulated to promote tumor growth (29).
According to emerging knowledge on the biological role and physiological importance of the different cells that compose the immune system, it has been proposed that NK cells patrol the human body to recognize normal self-cells, a process carried out by two types of receptors. Thus, NK cell activation is tightly regulated by an intricate balance between activation and inhibition signals (30, 31). In a normal cell, the peptides derived from self-proteins are loaded onto class I MHC molecules and are recognized by NK cells through the killer cell immunoglobulin-like receptor (KIR). In contrast, the recognition of self-cell ligands, such as the stress-induced proteins MICA, MICB, and ULBP-1, is mediated by the natural cytotoxic receptor (NCR) (32, 33). In tumor cells, tumoral peptides are associated with class I molecules, impeding recognition by KIR receptors and triggering effector activity. For full activation, NK cells depend on glycolysis and oxidative phosphorylation (OXPHOS), which are modulated by mTORC1 (34, 35). NK cells fight tumors by releasing cytolytic molecules, such as perforin, granzymes, and granulysin, causing the death of sensitive tumor cells. Some authors have also shown that NK cells can release exosomes containing these cytolytic molecules that reduce or eliminate malignant cells in both tumor-bearing animal models and human tumor cell lines of distinct origins (36–38).
In addition, activated NK cells release several soluble mediators, such as tumor necrosis factor-alpha (TNF-α); interferon-gamma (IFN-γ); interleukin (IL)-10; chemokines, including CCL3, CCL4, CCL5, XCL1, etc.; and growth factors, such as granulocyte macrophage colony-stimulating factor (GM-CSF), etc. (39). IFN-γ is known to be essential for immune cell activation; in NK cells, it increases cell activity and cytolytic potential (40, 41). From this point of view, NK cell overactivity increases the proportion of dying tumor cells and releases more DAMPs are released, which act as “find me” signals, and tumor antigens. These tumor-released compounds promote the arrival of inflammatory and immune cells, initiating an acute inflammatory process (42, 43). In this setting, tissue-resident macrophages, dendritic cells (DCs), and recruited monocyte-derived DCs comprise the mononuclear phagocytic system (44, 45), playing a critical role in homeostasis, tissue repair, the immune response, and cancer (46). In local tissues, resident and immature DCs (iDCs) exhibit elevated phagocytic activity mediated by the expression of a variety of pattern recognition receptors (PRRs) (47), which recognize the DAMPs and tumor antigens released from dead and dying tumor cells. Then, the iDCs trigger a rigorous metabolic process to meet the cell’s energy demands, including increased aerobic glycolysis, decreased OXPHOS with a concomitant increase in nitric oxide (NO) production, and increased fatty acid (FA) metabolism (48, 49). ROS production regulates the acidification of the lysosomal compartment for the degradation of phagocytosed antigens to peptides, while FA metabolism supplies the components for cell membranes. During these events, the endoplasmic reticulum and Golgi apparatus are expanded for protein synthesis, which assists in the upregulation of class II MHC molecules and antigen cross-presentation by class I MHC molecules; the expression of the costimulatory molecules CD80, CD86, and CD40; the expression of receptors for chemokines; and cytokine secretion, including interleukin (IL)-1, TNF-α, IL-6, IL-8, IL-12, IL-15, IL-18, etc. All of these activities induce the progressive maturation of DCs to become professional antigen-presenting cells (APCs). Then, the APCs travel to the lymph node through the lymphatic vessels, a process in which glucose metabolism plays a critical role (50, 51).
In the lymph node, DCs (mDCs) act as potent APCs that stimulate the proliferation and maturation of naïve antigen-specific CD4+ T cell clones and, by antigen cross-presentation, the activation of naïve antigen-specific CD8+ T cells. In addition to direct cell–cell interactions, some studies have indicated that exosomes released by APCs can also induce T-cell activation. It has been demonstrated that they express peptides associated with class I and II MHC and costimulatory molecules. In addition, they can also activate T and NK cells through the NKG2D–NKG2D ligand interaction (52–58).
Soon after the initial T-lymphocyte priming, T cells upregulate aerobic glycolysis, increasing glucose transporters and enzymes to meet their energetic demands; glutaminolysis and increased amino acid uptake favor OXPHOS for ROS and NO synthesis. In addition to mitochondrial biogenesis, lipogenesis by the endoplasmic reticulum and Golgi apparatus are required. Following this PI3K-AKT-mTORC1-dependent metabolic reprogramming, effector CD4+ T cells secrete several cytokines, such as IL-2, IFN-γ, etc., that induce the activation of specific transcriptional programs for the stimulation of antigen-specific CD8+ T cells and the overactivation of NK cells (59–61). The CD8+ T cells then release various cytokines, such as IFN-γ, IL-2, and TNF-α, and synthesize cytolytic molecules to become effector cytotoxic T lymphocytes (CTLs) (60, 62). After T-cell expansion, effector CD4+ T cells and CTLs migrate through the bloodstream and infiltrate the tumor, becoming critical cells for tumor destruction (63). A recent report from Rezaei R et al. using a CT-26-induced BALB/c mouse model of colorectal cancer indicated that when incorporated into tumor-derived exosomes, miR-124-3p, which acts as a post-transcriptional regulator of gene expression, stimulates a potent antitumor immune response, diminishing T regulatory (Treg) cells, reducing tumor mass, and increasing the overall survival rate (64). This miRNA is downregulated in colon cancer compared to non-malignant tissue, and in vitro studies have indicated that in Treg cells, PD-L1 expression is inhibited by cytokines such as TGF- β and IL-10 (65). All of this information suggests the possible involvement of exosomes released by tumor cells in the induction of a potent anti-tumor immune response. The cytokines released by T cells create a positive feedback loop that perpetuates the inflammatory process, as the array of pro-inflammatory cytokines leads to the overstimulation of innate immune cells. In addition, the overstimulated NK cells upregulate the activity of tissue-resident macrophages and the recruited neutrophils at the tumor site. These phagocytic cells carry out the respiratory burst to further produce pro-inflammatory cytokines and release ROS and NOS, promoting the M1 and N1 cell phenotypes, respectively. The induced anti-tumor activity leads to the additional destruction of tumor cells (66). Exosomes released by these metabolically activated cells mimic the tumoricidal activity of M1 and N1 cells (67–69).
A chronic inflammatory process is induced when this cellular circuit is maintained to eliminate tumor cells which mutations generate immunogenic changes in the synthetized tumor proteins. Reports have indicated that the chronic inflammatory process causes the release of transferrin-bound iron, which accumulates in the extracellular space. It is known that tumor cells take up this element, which promotes the production of DNA-damaging ROS. Increased DNA damage may lead to cell death in some vulnerable tumor cells in a process known as cell death mediated by ferroptosis (70). Conversely, these and other mutation-causing factors could promote genomic instability and epigenetic changes in other cells within the tumor cell population, which could lead to the maintenance of cell viability and enhance tumor proliferation, induce resistance to apoptosis, and, concomitantly, increase tumor heterogeneity (71).
Thus, the tumor comprises new tumor cell clones, increasing phenotypic heterogeneity, and the tumor mass itself. Oncogenic changes promote the activation of various signaling pathways in the heterogeneous tumor population, increasing the release of exosomes with different molecules or soluble factors that reinforce the inflammatory phase of chronic inflammation. For example, driver mutations in genes, such as MYC, K-RAS, or RET activate signaling pathways that promote the synthesis and release of proinflammatory cytokines, such as IL-8, IL-1, and CXC chemokines (72–74).
Depending on the tumor type, stage of tumor progression, genetic background, clinicopathological characteristics of the patient, etc., the distribution and density of non-malignant cells infiltrating the tumor vary greatly. Tumor and non-malignant cells produce several cytokines, growth and differentiation factors, chemokines, lipids, and nucleic acids that are released or loaded into exosomes, generating a wide array of molecules that promote cancer. Various research groups have published excellent reviews describing the signaling pathways involving cytokines, growth factors, and exosome components that may play a role in cancer (14, 18, 54, 75–77).
In the more advanced stages of cancer development, the surrounding tumor, stromal, and immune cells show a high rate of proliferation that requires high metabolic activity, resulting in the release of various by-products. In addition to the factors released by the various cells that make up the tumor, these metabolic by-products create a complex and changing microenvironment, which gradually promotes cancer cell survival and tumor mass growth. High metabolic activity is indispensable due to the chronic inflammatory process induced by the tumor. The increased energy and biosynthetic requirements mediated by increased glucose uptake and aerobic glycolysis favor the tumor’s proliferation, differentiation, and growth and affect the stromal cells. During this metabolic reprogramming, tumor cells produce—or induce the immune and stromal cells to produce—several cytokines that stimulate tumor growth while inhibiting or blocking the effector activity of immune cells. Some of the cytokines produced include IL-10, IL-6, IL-4, High Mobility Group-Box 1 (HMGB1) protein, etc., while several growth factors are generated as well, such as Epithelial Growth Factor (EGF), vascular endothelial growth factor (VEGF)-A, transforming growth factor-β (TGF-β), platelet-derived growth factor subunit A (PDGF-A), angiopoietin-like 4 (ANGPTL4), etc.; information about these factors is summarized in Table 1. In addition, some chemokines and their receptors play an important role in the TME and are expressed by tumor, immune, and stromal cells. Due to their anti- and pro-tumor effects, α-chemokines, i.e., CXC chemokines that contain a CXC motif at their N-terminus, have attracted attention. Some reports have indicated that CXCR3 and its corresponding ligands, CXCL14 and CXCL16, recruit primary immune cells with immune regulatory functions and pro-tumor activities, such as tumor-associated macrophages (TAMs) and neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), and Treg cells (120–122). Chemerin was initially described as a chemotactic factor for NK cells, macrophages, and myeloid and plasmacytoid DCs (123–125), favoring tumor infiltration by leukocytes and the regulation of cell metabolism. Current reports indicate that different cell types produce chemerin, including fibroblasts, epithelial cells residing in the tumor niche, and cells from distant organs, such as hepatocytes and adipocytes (126–128). Although most tumors downregulate the expression of chemerin, the potential pro- and anti-tumor activities of this molecule have been reported in the TME and have been suggested as a prognosis marker (129). More information on this topic is beyond the scope of this review; however, further information can be obtained from previous studies (130, 131).
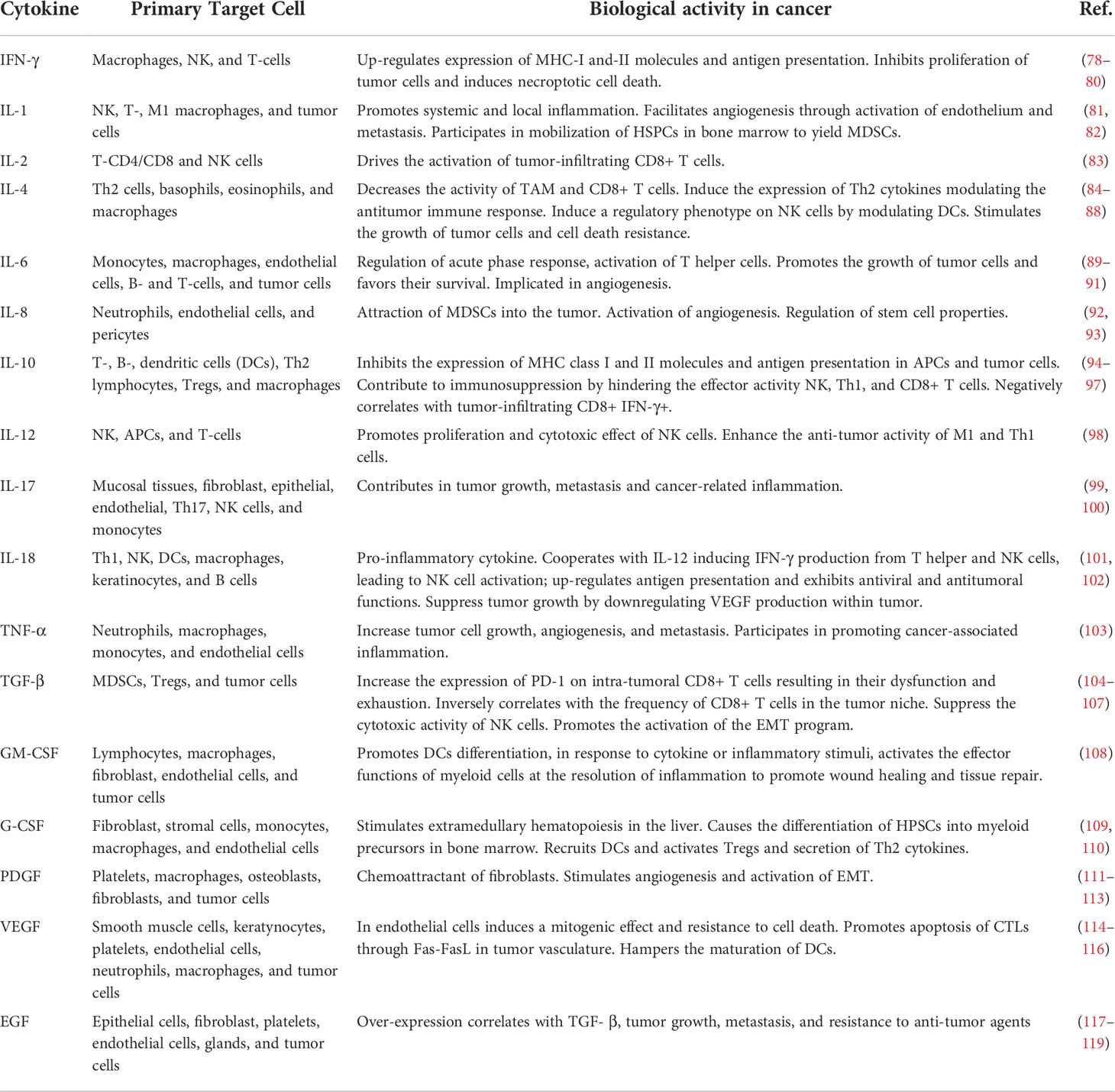
Table 1 Cytokines and Growth Factors associated with cancer-related inflammation.
In the TME, the high proliferation rate of tumor cells induces hypoxia, leading to hypoxia-induced acidosis caused by the release of lactate (132). During this metabolic reprogramming in liver cancer, the lncRNA HULC modulates the activity of crucial glycolytic enzymes through phosphorylation (133). In addition, LINC00261 promotes aerobic glycolysis in pancreatic cancer by activating the miR-222-3p/HIPK2/ERK axis (134). The participation of exosomes during hypoxia is an emerging research area. In one study, the long-intergenic non-coding RNA regulator of reprogramming (LINC-RoR) contained in the exosomes released by hepatocellular cancer cells under hypoxic conditions was shown to lead to increased HIF-α expression and the poorer survival of patients (135). In addition, in colorectal cancer, LINC00152 has been shown to be released in exosomes under hypoxic conditions, participating in the pathogenesis and progression of this cancer (136, 137). The production and secretion of lactate by tumor and infiltrating immune cells induce a gradual reconversion of immune cells from anti-tumor to pro-tumor activity to promote tumor immune evasion and support the migration and invasion of tumor cells (138, 139).
Tumor cells increase their iron uptake to supply their bioenergetic demand. Iron metabolism impacts DNA synthesis, cell cycle progression, and morphogenesis in physiological processes during normal cell life. This metabolic program also participates in invasion, metastasis, and EMT (140). However, as mentioned previously, increased cytoplasmic iron levels could induce a type of cell death known as ferroptosis (141). Cells undergoing ferroptosis release DAMPs associated with cell death, such as HMGB1, ATP, etc. (142). In the TME, extracellular ATP released by dead or dying cancer cells is hydrolyzed by several families of ectonucleotidases (143), mainly CD39 and CD73, which are expressed by immune and endothelial cells (144, 145). These enzymes are responsible for the conversion of extracellular ATP to adenosine. Adenosine accumulation in hypoxic cancer tissue is sensed by A2AR and A2BR receptors on immune cells that hamper the anti-tumor immune response. It is known that A2AR blocks the immune cells secretion of IFN- γ and IL-2. At the same time, A2BR prevents antigen presentation by mDCs and induces the polarization of M1 to M2 macrophages and the stimulation of MDSCs (146, 147).
Additionally, metabolic reprogramming activates signaling pathways that lead to aberrant gene expression due to epigenetic changes that alter tumor and stromal cells. These metabolic changes produce cells that also display the pro-tumor properties, such as cancer-associated fibroblasts (CAFs), induced by the TGF- β and EGF secreted by the tumor (148). In addition, they produce several chemokines; TGF- β ; IL6; some growth factors such as hepatocyte growth factor, insulin-like growth factor, etc.; and release exosomes. Furthermore, this environment favors the recruitment of MDSCs (149), TAMs (150), mesenchymal stem cells (MSCs) (151), and Tregs (152, 153). The role of exosomes in promoting the participation of these subsets of immune cells displaying pro-tumoral activity is discussed in the following reviews (154, 155).
In the TME, the accumulation of cytokines, chemokines, soluble factors, and a mixture of biomolecules in exosomes compromises the immune response during the advanced stages of cancer (156–158). Soluble factors with immunosuppressant activity, such as IL-10, TGF-b, IL-4, IL-6, etc., impair the function of NK cells (159, 160) and CTLs (161, 162) by inhibiting their effector activity and downregulating the expression of transcripts coding for cytolytic molecules. This cytokine environment induces the differentiation of CD4+ T to Treg cells (163–165) and maintains a stage of dedifferentiation in MDSCs by enhancing the expression of TGF-β and IL-10. In addition, tumor-resident MDSCs produce cyclo-oxygenase 2 (COX-2), arginase 1 (ARG1), inducible NO synthase (iNOS), IL-10, and indoleamine 2,3-dioxygenase (IDO), which accentuate the suppressor milieu (166). In hypoxia, pyruvate is reduced to lactate, which decreases the local pH. Moreover, IL-4 or IL-13 stimulates the polarization of the macrophages from the M1 to M2 phenotype to support tumor progression and angiogenesis (167) (Figure 1).
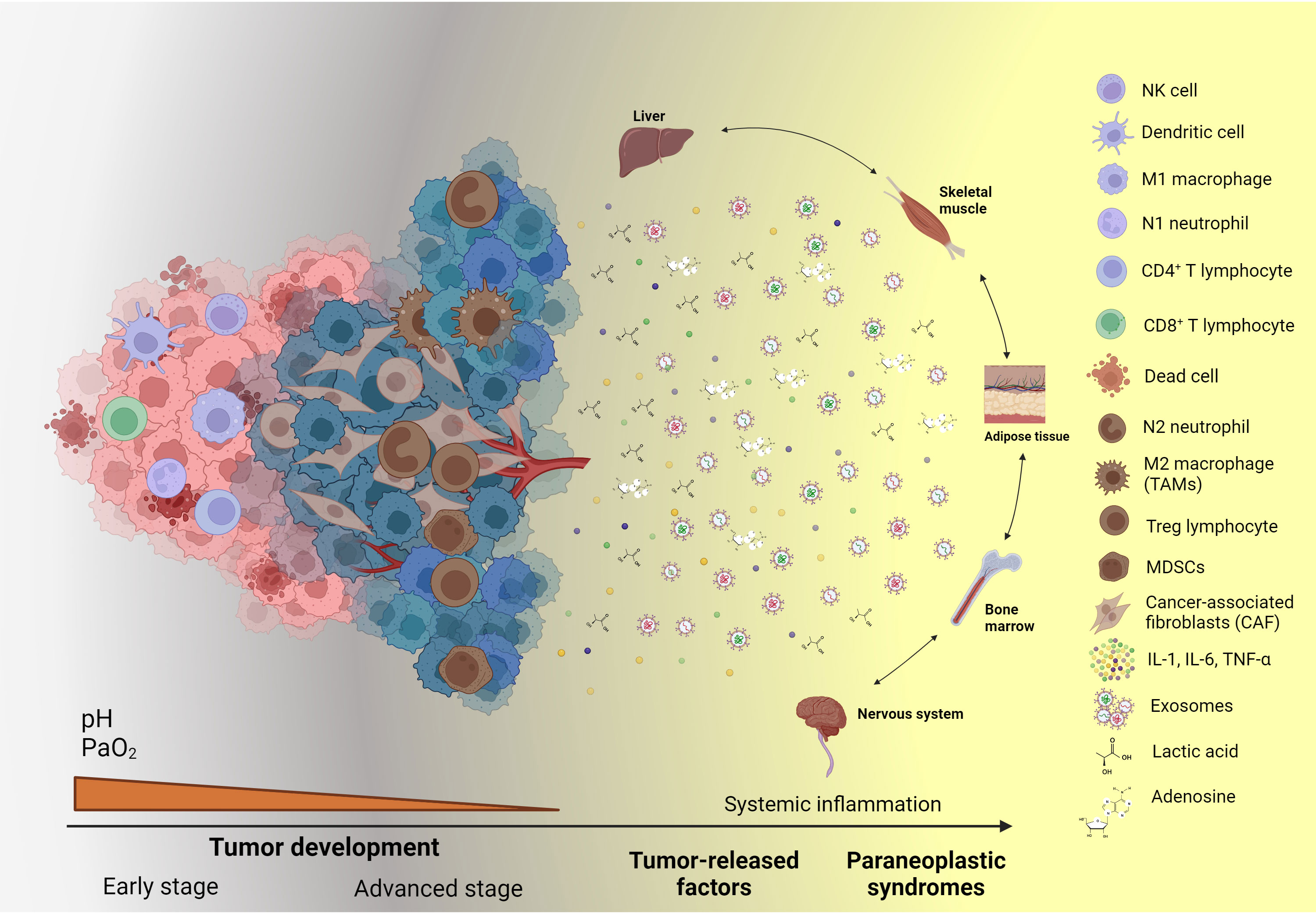
Figure 1 Systemic effects and paraneoplastic syndromes caused by cancer-associated inflammation. During the advanced stages of tumor development, tumor and stromal cells release an array of soluble factors, such as cytokines, chemokines, growth factors, metabolic by-products, exosomes, and ncRNAs, which sustain the local inflammatory state. Moreover, these soluble factors, when released into the bloodstream, reach distinct organs, systems, and tissues, causing alterations in their function and the production of diverse molecules and subsets of cells, which can be employed as biomarkers to assess cancer-related systemic inflammation. Created with BioRender.com.
The systemic effect of tumor microenvironment-derived cytokines
Studies performed with cancer patients and animal models have studied several local and systemic cytokines. In cancer patients, diverse cytokines and soluble factors showing pro- and anti-inflammatory activities may be detected as free circulating or exosome membrane-bound molecules (168). Reports have indicated that the cytokines IL-1, TNF-α, IL-6, G-CSF, and GM-CSF mainly act at the systemic level, affecting the function of some organs (169). Our group quantified eight cytokines at the systemic level in smoking patients with lung adenocarcinoma and found a significant increase in IL-2, IL-4, IL-6, and IL-10 compared to their levels in healthy smoking subjects. Among these cytokines, the concentration of IL-6 was the highest in the peripheral blood of cancer patients, showing an increase of approximately sevenfold. To explain the increased systemic levels of IL-6, several reports from distinct groups, including ours, have pointed out that tumor cells from different types of cancers produce this cytokine (170, 171). Currently, the roles of these cytokines in cancer have gained considerable attention. In particular, those cytokines acting at the systemic level have been associated with the development of some clinical signs or symptoms of paraneoplastic syndromes.
TNF-α
As discussed in the previous section, the tumor and its stroma release cytokines that act on various systems and organs. TNF-α is a pro-inflammatory cytokine with diverse functions that participates in homeostatic and distinct pathological conditions. The biological activity of TNF-α is exerted via binding with its cognate receptors: TNF-α receptor 1 (TNFR1) and 2 (TNFR2). TNFR1 is ubiquitously expressed in cells and is activated by the transmembrane or the soluble form of TNF-α. To the contrary, the expression of TNFR2 is limited to specific cells, such as immune and endothelial cells and cells of the central nervous system, and its activation mainly depends on the transmembrane form of the ligand (172). The role of these receptors in homeostasis has been described. The activation of TNFR1 mainly promotes inflammation, the induction of cell death via apoptosis or necroptosis, and tissue degeneration.
Conversely, the activation of TNFR2 is associated with cell survival and the wound healing process (172). Once TNF-α is produced and released into the bloodstream, this cytokine can reach the adipose tissue, where it modifies the adipocytes glucose and lipid metabolism. In this regard, TNF-α decreases glucose uptake by inhibiting the signaling pathways triggered by insulin and, as a result, downregulates the mRNA and protein expression of GLUT-4 (173, 174). Simultaneously, TNF-α activates lipolysis via the inhibition of the peroxisome proliferator-activated receptor (PPAR)- γ and CCAAT enhancer-binding protein, causing an increase in the expression of neutral lipases (175). These observations might explain the increased levels of serum lipids detected in cancer; however, more studies are required to demonstrate the role of TNF-α during the dyslipidemia observed in cancer (176).
Studies in cancer patients have demonstrated that cancer-associated systemic inflammation is associated with a sense of pain or hyperalgesia from an unknown source. In peripheral nerves, TNF-α is linked with this phenomenon. In this setting, the interaction of TNF-α with TNFR1 causes the activation of the p38/MAPK signaling pathway, which culminates in the activation of Na+ and K+ ionic channels in peripheral nerves, leading to pain generation (177, 178).
In addition, the findings from a recent study indicated that in hepatocellular carcinoma, TNF-α released in exosomes promoted osteoclast differentiation. During this phenomenon, the TNF-α stored in the exosomes produced by hepatocellular Huh-7 cells caused the expression of osteoclast-associated differentiation markers when added to murine macrophage/monocyte cell lines through the activation of the NF-κB/cathepsin K/triiodothyronine receptor auxiliary protein axis. These findings explain the tendency of hepatocellular cancer cells to generate bone metastases (179).
Interleukin-1
IL-1 is a pleiotropic cytokine that is involved in various inflammatory processes. IL-1 belongs to the Ig-like receptor superfamily, which is characterized by the presence of the Toll/interleukin-1 receptor (TIR) domain. The TIR domain is essential for the biological activity of IL-1 (180, 181). The ligands of the IL-1 family are IL-1, IL-18, IL-33, and IL-36. These agonists bind to three receptors: IL-1α and IL-1β perform cell activation when bound to IL-1R1; IL-18 is a ligand of IL-18Ra; IL-33 binds to ST2 (IL-1R4); and IL-36α, β, and γ are agonists of IL-1Rp2 (IL-1R6). The primary function of this family of ligands and receptors is to participate in inflammatory processes (182, 183).
IL-1α and IL-1β are encoded by different genes and have minimal homology. IL-1α is synthesized as pro-IL-1α, which can be proteolytically cleaved by calpain, granzyme B, elastase, and chymase. When the mature form of IL-1α is released, it acts as an “alarmin,” activating the innate immune response against noxious stimuli (184). IL-1β is a critical player in the inflammasome. The activation of the inflammasome in innate immune response cells, such as monocytes and neutrophils, occurs when pathogen-associated molecular patterns (PAMPs) and DAMPs bind to Toll-like (TLRs) and NOD-like (NLRs) receptors. The assembly of the inflammasome causes the cleavage of pro-caspase 1, and caspase 1 is released, which then cleaves pro-IL-1β and pro-IL-18. IL-1α and IL-1β establish chronic inflammation in the process of carcinogenesis (185). In IL-1/IL-1R1 knockout murine models, it was observed that IL-1 was a critical factor in the inflammatory process associated with 3-methylcholanthrene carcinogenesis (186). Both IL-1α and IL-1β participate in the systemic inflammation associated with cancer. Tumor and stromal cells in the tumor microenvironment produce and release IL-1.
Recent reports have indicated that cancer-derived exosomes from prostate and lung cancer as well as glioblastoma cell lines stimulate the production and release of IL-1β in immune and non-immune cells (187–189). In this setting, cancer-derived exosomes may trigger distinct intracellular signaling pathways in receptor cells, culminating in NF-κB activation and the subsequent expression of the IL-1β gene or the activation of inflammasomes through NLRP3 for the cleavage of pro-IL-1β into its active form (190, 191). In support of this, Linton et al. reported that exosomes derived from pancreatic ductal adenocarcinoma (PDAC) cell lines caused the polarization of M0 into immunosuppressive pro-tumoral M2 macrophages, which increased the production and release of IL-1β (327). In this regard, PDAC-derived exosomes were shown to contain increased levels of arachidonic acid, leading to its subsequent metabolism into free fatty acids, which activate inflammasomes through NLRP3 (193). Several reports have indicated that IL-1 in circulation affects several organs and may contribute to establishing paraneoplastic syndromes, such as cachexia (see below) (194–197).
IL-6
Low blood levels of IL-6 ranging between 1 and 5 pg/mL have been reported under normal conditions (198). In addition, soluble forms of the receptors IL-6R and gp130 have been detected. After local production by tumor cells in the inflammatory niche, IL-6 is released into the bloodstream and eventually reaches the liver, where it has several biological effects on hepatocytes. In one study, an increase in the production of acute phase proteins (APPs), such as C-reactive protein (CRP), serum amyloid A (SAA), fibrinogen, haptoglobin, and α1-antichymotrypsin, and an opposing decrease in albumin, fibronectin, and transferrin were detected. In addition, IL-6 regulates the transporters of iron and zinc associated with anemia that are detected in chronic inflammation (199). IL-6 also promotes megakaryocyte maturation in the bone marrow, increasing the serum platelet count (200).
The varied effects induced by IL-6 are detected mainly in chronic inflammatory diseases. In addition, this cytokine exerts various biological actions on the distinct types of immune cells, maintaining a deregulated and persistent positive feedback loop. IL-6 has evident pleiotropic effects in hematopoiesis, inflammation, the immune response, and cancer.
Parameters to assess systemic immune-inflammatory markers
The liver is a primordial organ that eliminates waste and toxic compounds, mainly found in dietary products, or harmful particles from pathogens. In addition, it provides nutrients and produces mediators that alert immune cells to induce an inflammatory response that eliminates harmful agents and induces the restoration of tissue homeostasis. The products of pathogens (PAMPs) or those derived from the host’s damaged cells (DAMPs) induce the release of acute phase proteins from the liver (201, 202). Kupffer cells and macrophages produce proinflammatory cytokines, such as IL-6, IL-1β, and TNF-α, to generate a series of products associated with the inflammatory response (203, 204). These cytokines activate resident cells, such as hepatocytes, endothelial cells, hepatic stellate cells, and diverse immune cells, from the hepatic arteries and portal vein in the liver. In addition, the liver releases diverse enzymes that inactivate harmful drugs and produces serum proteins, such as albumin and coagulation factors (205) (Figure 1).
Local inflammation appears to be reflected at the systemic level, and this is supported by several studies (6, 206, 207). Routine hematological parameters have been used in recent years as indicators of systemic inflammation. Among these simple parameters, the proportions of circulating inflammatory cells, including the white blood cell (WBC) count, lymphocyte count, neutrophil count, platelet count (PLTs), mean platelet volume (MPV), and levels of hemoglobin (Hb) and serum CRP, have been screened (208, 209). Analyte-based scores or ratios of some of these parameters have been reported to assess systemic inflammation, and their correlation with the prognosis of numerous pathologies, including cancer, has been described.
The Glasgow prognostic score/modified Glasgow prognostic score (GPS/mGPS) is an inflammatory indicator (210, 211). The GPS/mGPS reflects the systematic inflammatory response and nutritional status. Recent studies have shown that the GPS/mGPS is a novel inflammatory index that can predict outcomes in various cancers (212–214). However, the molecular mechanisms underlying the relationship between the GPS/mGPS and poor prognostic outcomes are still unclear. A plausible explanation is that an elevated GPS/mGPS may reflect an individual’s immune and nutritional status. The GPS/mGPS comprises albumin and CRP; both are acute-phase proteins synthesized in the liver. The CRP level is regulated by several pro-inflammatory cytokines, such as IL-1, TNF-α, TGF-β, IFN-γ, and IL-6 (215). Studies have shown that IL-6 correlates with OS and SOC in CRC, and its effect could explain the promotion of tumorigenesis and metastasis (216, 217).
Additionally, CRP is associated with the activity of infiltrated immune cells, including DCs, T cells, and NK cells (218, 219). Many studies have shown that CRP is an independent biomarker for predicting prognostic outcomes in various cancers (220, 221). The serum albumin level is used to evaluate liver function and nutritional status. Hypoalbuminemia is a common feature of the systemic inflammatory response, cancer recurrence, and metastasis. In addition, it has been shown to be positively correlated with the OS and CSS of patients with various cancers, including CRC and ovarian cancer (222, 223).
Albumin
Albumin is a low-molecular-weight protein of 66 kDa consisting of a single polypeptide chain with 585 amino acid residues that is fully synthesized in the liver, which produces approximately 15 g daily. Albumin maintains a plasma concentration of 35–45 g/L and is the most abundant protein in plasma, contributing to the maintenance of oncotic pressure and the permeability of the microvasculature. In addition, it has been implicated in important metabolic functions as it transports several endogenous ligands, such as free FAs, bilirubin, and ion metals, as well as some exogenous ligands (224, 225). Albumin expression is mainly regulated at the transcriptional level. Decreased albumin synthesis leads to hypoalbuminemia, which contributes to the development of edema by the transudation of fluids into extravascular spaces. TNF-α is a key cytokine involved in the inhibition of albumin synthesis, although other cytokines, such as IL-1β and IL-6, may contribute as well.
The serum albumin level is a marker of nutritional status, and a level less than 35 g/L is considered to indicate hypoalbuminemia. Albumin acts as an anti-inflammatory molecule; its increase is associated with blocking the migration of neutrophils through the endothelium by decreasing the expression of VCAM-1 in a TNF-α-dependent manner (226). Regardless of the disease, decreased serum albumin has been proposed as a risk factor and predictor for morbidity and mortality.
Platelets
Platelets, or thrombocytes, are nonnucleated and discoidal fragments derived from precursor megakaryocytes during megakaryopoiesis. Platelets maintain normal hemostasis and participate in several biological processes, such as the control of blood vessels and their interactions with endothelial cells. They also form a platelet plug with various extracellular matrix components, which inhibits vascular leakage. In addition, they are vital in acute and chronic inflammation due to their release of cytokines and chemokines that attract leukocytes and favor immune cells to reach the damaged tissue for wound healing. Furthermore, they are critical participants in the pathophysiology of several diseases, including cancer (227–229). Platelet characteristics have been found to be significantly associated with the clinical outcomes of several pathologies. Two of these features are the number of circulating platelets, designated as the platelet count, and the mean platelet volume (MPV).
The complete blood count includes the platelet count and the MPV. The platelet count quantifies the number of platelets in the blood; there are usually between 150,000 and 450,000 platelets in each microliter. Platelet parameters are used in the diagnosis of a patient’s general condition and have a prognostic value in some pathologies. A platelet count is related to pathologies associated with a chronic condition (229). MPV is a measure of platelet size that results from a higher production by megakaryocytes; hence, it indicates if there are more young platelets circulating in the bloodstream. The MPV ranges between 7.5 and 12.0 fl (229). The value is inversely related to the platelet count, hemostasis maintenance, and the preservation of a constant platelet mass. MPV is a marker of platelet activity, and it has been related to prothrombotic and proinflammatory diseases (230).
Under inflammatory conditions, the increase in IL-6 causes an increase in the ploidy of megakaryocytic nuclei and an increase in the cytoplasmic volume, leading to the generation of a large number of platelets. These platelets migrate to the site of inflammation, where they undergo activation and are depleted, triggering a decrease in the MPV of the patient’s blood during the development of the inflammatory process (229).
Fibrinogen
Fibrinogen is produced in the liver during general acute-phase inflammatory provocation. Inflammatory cytokines induce fibrinogen synthesis by hepatocytes and Kupffer cells. Several reports have indicated that IL-6 is a key cytokine that promotes fibrinogen production due to the presence of several IL-6 response elements in the fibrinogen genes. In addition, IL-6 regulates fibrinogen transcripts via the MEK-ERK signaling pathway (231, 232).
In association with other molecules, fibrinogen participates in coagulation, fibrinolysis, and cellular and matrix interactions that support cell migration, inflammation, and wound healing. An increased risk of cardiovascular disease has been associated with elevated fibrinogen levels (233). Cancer patients present with malfunctions in the coagulation process, and this molecule has been associated with cancer development. Fibrinogen binds to growth factors, such as fibroblast growth factor-2 (FGF-2, bFGF) and (VEGF, to enhance tumor growth and increase the migration, invasion, and metastasis of tumor cells, as well as angiogenesis—processes which are considered to be hallmarks of cancer. The fibrinogen/albumin (F/A) ratio is considered a promising inflammation-based marker. A high FAR was shown to be associated with clinical–pathological features and survival in some cancers (234). However, multicentral clinical trials are needed to analyze the clinical impact of this relationship on medical oncology.
The evidence presented thus far provides an explanation for the usage of these hematological parameters and their diverse relationships to analyze the systemic inflammatory state. These ratios are associated with prognosis and survival in inflammatory diseases, including cancer (235). Owing to the extensive information indicating the prognostic value of these ratios, we only refer to those works in which these indices were assessed in the most frequent types of cancer. The reported ratios are indicated in Table 2.

Table 2 Available systemic inflammation indices predicting prognosis and outcome in cancer patients.
Paraneoplastic syndromes
Understanding the local inflammation associated with cancer allows for a better understanding of the interaction between tumor cells, immune cells, stromal cells, and the soluble factors released by this system. The release of proinflammatory factors from tumor or stromal cells—in the form of soluble factors or those contained in exosomes—into the bloodstream alters the production of APPs, metabolites, and cells, which impacts organs and systems, causing the appearance of symptoms unrelated to the tumor itself, called paraneoplastic syndromes (6). Approximately 8% of cancer patients present one or more paraneoplastic syndromes. Paraneoplastic syndromes are classified as neurologic, rheumatologic, dermatologic, or hematologic, depending on the type of tumor (264, 265). The manifestation of paraneoplastic syndromes has detrimental effects on patient’s quality of life and outcome. In addition, some paraneoplastic syndromes may exhibit tumor-promoting capabilities (7). In this section, we will discuss the development of myelopoiesis and cachexia.
Stimulation of myelopoiesis in cancer-related inflammation
One of the main targets of pro-inflammatory mediators in the bone marrow is a process known as hematopoiesis, whereby hematopoietic stem and progenitor cells (HSPCs) differentiate into mature blood cells. Under physiological conditions, denominated as steady-state hematopoiesis, local and external signals promote the retention of HSPCs in the bone marrow. For this purpose, the bone marrow stroma cells, including endothelial, vascular, and osteolineage cells and macrophages, participate in a well-orchestrated network to control the proliferation and differentiation of HSPCs (266). However, during acute systemic infection or inflammation, factors released from pathogens and damaged cells, as well as proinflammatory cytokines (IL-6, IL-1β, and TNF-α), impact HSPCs and stromal cells, causing the rapid mobilization and differentiation of HSPCs into myeloid cells, a process known as emergency myelopoiesis (267, 268).
In cancer, it has been demonstrated that tumors take advantage of emergency myelopoiesis to rapidly expand pools of myeloid-derived cells showing immune suppressive and tumor-promoting activities (266). These cells are known as MDSCs and can be classified as granulocytic (CD11b+CD14-CD15+) or monocytic (CD11b+CD14+CD15-) depending on the myeloid progenitor from which they were derived (269).
As mentioned above, the plasma concentration of proinflammatory cytokines, such as IL-1β, TNF-α, and IL-6, increases in several types of cancer due to the sustained inflammatory environment caused by the tumor and its stroma. These cytokines, along with other bioactive soluble factors, such as GM-CSF and G-CSF, travel freely in circulation or are stored in exosomes reaching the bone marrow, causing the proliferation, mobilization, and skewed differentiation of HSPCs towards myeloid cells (270). Reports on G-CSF have indicated that as the G-CSF level in the bone marrow decreases, the expression of maintenance molecules for HPSCs, such as CXCL12, osteopontin, Kit-1, angiopoietin, and vascular cell adhesion molecule 1 (271) increases, thus promoting their proliferation. In the bone marrow, IL-6 binds to IL-6R—which is mainly expressed on hematopoietic multipotent progenitors but not in short- or long-term repopulating HPSCs—suppressing the differentiation of lymphoid cells and stimulating myeloid differentiation (272). Marigo et al. demonstrated in mice that GM-CSF, G-CSF, and IL-6 in the bone marrow caused the activation of the master regulator of emergency granulopoiesis, the C/EBPβ transcription factor, which is responsible for the differentiation of HSPCs into MDSCs and their immunosuppressive activity (273). Initial studies in humans by Wu et al. showed that increased frequencies of HSPCs and granulocytic progenitors in different cancer types were found in cancer patient’s blood compared to those of healthy donors (274). The increase in these cell populations was negatively correlated with several clinical parameters, including the time to progression and the stage of the disease, suggesting that activated bone marrow myelopoiesis is a phenomenon that promotes tumor progression.
However, these proinflammatory cytokines are not only responsible for the expansion and differentiation of HSPCs in the bone marrow. In a mouse model of breast and lung cancer, Sayed et al. found that hematopoiesis was biased towards myelopoiesis due to the action of TNF-α released from infiltrating CD4+ T cells (275). During this process, TNF-α caused a skewed differentiation of HSPCs towards MDSCs, with a concomitant decrease in erythroid and lymphoid precursors (275). In this scenario, TNF-α acted on HSPCs and MDSCs through TNFR-2, upregulating the expression of the caspase-8 inhibitor c-FLIP and promoting their survival (276). In these works, it has been demonstrated that tumor-promoting inflammation has profound systemic effects during the advanced stages of cancer, causing biased hematopoiesis toward the production of immunosuppressive myeloid cells and sustaining tumor progression. In addition, the results from these studies have helped to explain the altered percentages of hematological inflammatory parameters detected in cancer patients by the effect of these pro-inflammatory cytokines and growth factors in the bone marrow.
In recent years, owing to the use of NGS approaches and elegant in vivo models, it has been shown that hematopoiesis is altered and biased towards myelopoiesis in the early stages of tumor development (277). Surprisingly, pro-inflammatory cytokines play a significant role during this process by activating hematopoiesis in the bone marrow to direct the production of myeloid progenitors and micro-RNAs (miRNAs) delivered from the tumor. The TME plays a critical role in controlling this process. Some of these tumor-associated miRNAs, such as miR-23b-3p, miR-27a-3p, and miR-671-5p, in bone marrow downregulate the expression of genes involved in B-cell receptor signaling and antigen processing. With this work, it is tempting to speculate that other subsets of non-coding RNA, in addition to miRNAs, are released from the tumor or its stroma to activate myelopoiesis in the bone marrow. However, studies to demonstrate this proposal are required.
In addition to proinflammatory cytokines and non-coding regulatory RNA, myelopoiesis is controlled directly or indirectly by the metabolites released from the tumor as part of the metabolic reprogramming of cancer cells (2, 278). For example, the direct control of myelopoiesis by metabolites involves the participation of oxysterols and desmosterol in the differentiation and expansion of MDSCs. These cholesterol metabolites bind to retinoic acid-related orphan receptors expressed in the cell precursors of MDSCs (278). They have been detected in distinct pathologies, such as obesity, metabolic disorder, diabetes, and cancer (279). Recent evidence from Vladimirov et al. demonstrated that colorectal cancer patients had significantly increased values of cholesterol serum precursors compared to healthy donors, suggesting that cholesterol metabolism is altered in cancer (280). This evidence allows us to speculate that such patients might present altered myelopoiesis and thus increased numbers of MDSCs and other immunoregulatory cell populations; however, intensive research using in vitro and in vivo models in distinct types of cancer is needed to confirm this hypothesis. The activation of aerobic glycolysis indirectly affects cancer metabolic reprogramming in myelopoiesis. In a triple-negative breast cancer mouse model, Li et al. demonstrated that aerobic glycolysis stimulated the release of G-CSF and GM-CSF via C/EBP-β and liver-enriched activator protein. This glycolytic-dependent production of G-CSF and GM-CSF was correlated with increased MDSCs and low T lymphocyte counts in breast cancer patients (281). Although these results are pioneering, more studies are necessary to uncover the complete impact of altered metabolism on myelopoiesis in cancer.
Recent studies have demonstrated that hematopoiesis can occur outside the bone marrow under distinct inflammatory conditions, a phenomenon known as extramedullary hematopoiesis (269). In cancer, extramedullary hematopoiesis is mainly found in the liver and spleen, where HPSCs or myeloid progenitors arrive (282). In this setting, the accumulation of immunosuppressive cells, such as MDSCs, Tregs, and erythroid progenitor cells, in the spleen was found in a mouse model of breast cancer. Several reports have demonstrated that splenectomy in animals bearing tumors is associated with a decrease in the number of peripheral MDSCs cells and cytokines related to HPSCs mobilization (9, 283). In this scenario, a survival benefit was achieved due to decreased tumor growth following the splenectomy, suggesting that extramedullary hematopoiesis has a critical role in sustaining tumor growth and progression. However, in humans, a concomitant splenectomy following colon, liver, gastric, and pancreatic cancer has not shown any beneficial effects on patient’s overall survival (284). Nevertheless, owing to technical and experimental barriers, little is known about this process in humans; thus, most of the studies in this field are performed using animal models. For this reason, further research exploring the implications of extramedullary hematopoiesis in solid human tumors is required.
In solid tumors, the shift in anti-tumoral immunity towards a pro-tumoral response not only depends on the reshaping of the local tumor microenvironment by cytokines, chemokines, metabolic by-products, exosomes, or other released factors. The development of populations showing immune regulatory functions occurs in distinct and distant organs away from the primary lesion due to the systemic release of the factors mentioned above. In this setting, a considerable increase in the production of MDSCs in the bone marrow and secondary lymphoid organs and their subsequent migration to the tumor support the development of some cancer hallmarks by providing growth and angiogenic factors, cytokines, chemokines, and extracellular matrix remodeling enzymes (285) (Figure 1). In addition, these MDSCs strengthen the immunosuppressive tumor microenvironment by releasing immunomodulatory cytokines, metabolites, and other soluble factors and by expressing immune checkpoints on their surface, thus causing the recruitment of M2 macrophages, N2 neutrophils, and Tregs (286).
Cachexia in cancer-related inflammation
Advanced tumors release a spectrum of factors that induce systemic inflammation, which is reflected in paraneoplastic syndromes, such as cachexia. Cancer cachexia is a multifactorial syndrome characterized by progressive sarcopenia that may be accompanied by a loss in adipose tissue. This pathology involves a harmful protein and energy balance and an abnormal metabolism due to anorexia. Cachexia cannot be reversed entirely with nutritional support, resulting in a detriment to the quality of life of cancer patients (287). In addition, cachexia is responsible for 20–30% of deaths in patients with advanced cancer. Tumors in which cachexia may develop include pancreatic, gastric, colon, lung, Hodgkin’s and non-Hodgkin’s lymphoma, breast, sarcoma, and leukemia (288).
The cellular mechanisms that induce cachexia are not yet fully understood; thus far, it is known to be a syndrome that involves several organs and is orchestrated at the systemic level by inflammatory factors released by the tumor and its microenvironment. Although systemic inflammation is not considered in the current classification as a parameter for diagnosing cancer cachexia, several authors have considered the measurement of inflammatory markers, such as CRP and IL-6, at the blood level (289). Biswas and Acharyya (290) mentioned that factors secreted directly by the tumor and non-tumor cells of the neoplastic microenvironment can mediate cachexia and other syndromes. These factors can directly interact with various tissues, such as muscle, liver, brain, and adipose tissue, and can induce metabolic reprogramming, leading to a negative metabolic energy state at the systemic level.
As mentioned above, as tumors continue to proliferate, the lack of oxygen and nutrients induces hypoxia. Hypoxia generates critical changes, for example, the metabolic turnover of tumor cells for energy. A hypoxic microenvironment releases several factors, including VEGF (291), which induces angiogenesis and recruits inflammatory cells, such as macrophages. In addition to the above components of the tumor microenvironment, the release of DAMPs by dead cells increases inflammation and generates extra-tumoral cytokines. Another important metabolite in cancer that contributes to tumor development and results from metabolic turnover is lactate (292, 293). The hypermetabolism of tumor cells leads them to consume large amounts of glucose and excrete lactic acid, which has pleiotropic activity, is involved in energy metabolism, has an immunosuppressive function, and promotes angiogenesis (294).
Lactate in the liver is converted to glucose in a process called gluconeogenesis. The glucose generated can be utilized by both the tumor cells and the host organism, increasing blood glucose. This hyperglycemia leads to insulin resistance, which is related to muscle wasting. The relationship between the tumor and liver via lactate is called the Cori cycle and is considered as a futile cycle in cancer cachexia (292). Insulin resistance in patients with cachexia, as well as in murine models, has been associated with muscle wasting and is induced by TNF-α (295). Noguchi et al. found a high correlation between TNF-α expression in muscle tissue obtained from the intestine of cancer patients and insulin resistance and muscle wasting. After tumor resection, the patients showed complete improvement in insulin resistance (296). More recent studies using a murine model of C-26 colon adenocarcinoma cell cachexia showed that insulin resistance occurred before weight loss. In the quadriceps muscle of these mice, the expression of four muscle atrophy-promoting genes, Atrogin-1, and MuRF-1, ubiquitin ligases E3 and Bnip3, was increased (297). Systemic inflammation is also associated with skeletal muscle wasting during cancer cachexia. The ubiquitin-proteasome pathway is activated during cachexia and is responsible for the degradation of most skeletal muscle proteins. Type IIB muscle fibers in an ApcMin/+ mouse model, which produced colon polyps, were shown to be highly susceptible to IL-6-mediated muscle wasting, as it induced the overexpression of the Atrogin-1 gene (298). In mouse bladder and colorectal cancer models, increased TNF-α, IL-6, and IL-1 in circulation and decreased muscle mass were associated with high Atrogin-1 and MurRF1 expression (299).
Among the factors that tumors can release are exosomes, which may contain molecules that promote muscle wasting. In a mouse model of cachexia, a group of researchers found that cachexigenic tumors released Hsp70/90 heat shock proteins into extracellular vesicles; furthermore, they participated in muscle wasting through TLR4 activation (300). Similarly, miR-181a-3p found in exosomes from a conditioned medium of oral squamous cell carcinoma induced endoplasmic reticulum stress and skeletal muscle wasting (301). Another study showed that HMGB1 protein, a DAMP contained in the exosomes of CT26 mouse colon cancer cells, induced the expression of the Atrogin1 and MuRF1 genes, leading to muscle wasting through the activation of TLR4/NF-κB axis (302). Another factor in exosomes that promotes muscle wasting is growth differentiation factor 15 (GDF-15), identified in the exosomes of CT26 colon cancer cells. GDF-15 can interact with C2C12 myotube cells, regulate Bcl-2/caspase, and induce cell apoptosis, favoring muscle atrophy in cancer cachexia (303).
The insulin resistance and systemic inflammatory cytokines in cancer cachexia patients also impact adipose tissue (304). In adults, adipose tissue is composed primarily of white adipocytes (WAT), although brown adipocyte fat deposits (BAT) exist in specific anatomic locations, such as the perivascular viscera and periviscus. BAT can also be found in supraclavicular, axillary, and inguinal subcutaneous fat and the intestinal walls (305). The difference between WAT and BAT is that the latter contain many mitochondria expressing uncoupling protein-1 (UCP-1). This molecule “uncouples” an electron in the process of ATP synthesis via OXPHOS across the inner membrane of the mitochondrion, which generates heat (306). Virtually all fat ingested in food is stored as WAT. In states of excessive exercise or prolonged fasting, triglycerides in WAT are degraded by three lipases: adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL). The regulation of lipases is highly influenced by several hormones, such as insulin, catecholamines, and growth hormone (307, 308).
In cancer cachexia, various cytokines, such as TNF-α and IL-6, promote the lipolysis of triglycerides in WAT (309). Shaw and Wolfe found increased lipolysis associated with increased blood fatty acids in patients with cachexia (310), indicating that lipid metabolism is dysregulated in cachexia states. In a muscle stem cell model, as well as in models of cachexia induced by human kidney neoplasia in mice, researchers observed that the process of muscle atrophy was preceded by an increase in FA β-oxidation as well as inflammatory factors, such as IL-1 β, IL-6, IL-8, and TNF-α. In this work, blocking FA β -oxidation using a carnitine-palmitoyltransferase-1 inhibitor also blocked muscle wasting (311). Another mechanism of adipose tissue degradation is the browning of WAT. For a long time, it was believed that the function of WAT was lipid storage, while BAT was involved in heat dissipation and the regulation of temperature (312). It is now known that WAT can be converted to BAT via specific mechanisms, such as hypothermia. The browning of WAT has been associated with cachexia.
Petruzzelli et al., using several genetically engineered mouse models, observed that the browning of WAT was an event that preceded muscle wasting. They also showed that IL-6 and catecholamines increased UCP-1 expression in the WAT of these cachexic mice (312). Recently, a study in cancer patients with and without cachexia showed an association between tumor-derived factors and inflammatory changes in the adipose tissue of the cachectic patients (313), suggesting that factors released by the tumor and its microenvironment modify adipose tissue metabolism. Exosomes have been found to play an important role in adipose tissue wasting.
Exosomes derived from cell lines and the plasma of gastric cancer patients contain ciRS-133, which induces the browning of WAT (314). Exosomes derived from lung adenocarcinoma cell lines containing TGF- β were shown to inhibit adipogenesis in primary adipocyte cultures from healthy subjects (315). The miR155 in gastric cancer exosomes was shown to promote the browning of adipose tissue through the transcription factor CCAAT/enhancer-binding protein β, which upregulates UCP1 (316).
Finally, circulating inflammatory cytokines produced by the tumor microenvironment affect the central nervous system, amplifying and orchestrating the symptoms of cancer-associated cachexia and causing anorexia, fatigue, and the wasting of muscle and fat tissue. In particular, a loss of appetite has been associated with hypothalamus inflammation. The nucleus of the hypothalamus regulates energy homeostasis (290, 317, 318). Increased cytokine expression in the brain alters the neurochemistry of the hypothalamus nucleus, where cytokines activate pro-opiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART) neurons, which mediate satiety and reduce food intake. The activation of these neurons induces serotonin release, suppressing appetite. In addition, cytokines are likely to inhibit neuropeptide Y (NPY) and agouti-related peptide (AgRP) neurons, which mediate appetite and energy intake. These changes in the neurochemistry of the hypothalamus result in a “resistance” to signals that inform the brain of energy deficits in the periphery. As mentioned above, adipose tissue wasting leads to the circulation of free FA, which generates a satiety signal in the hypothalamus, contributing to anorexia (290, 317, 318). On the other hand, some reports have indicated that the stimulation of the hypothalamic–pituitary–adrenal axis with IL-1 induces the release of glucocorticoids that act on skeletal muscle and accelerate protein degradation (317, 319, 320).
Cancer patients present two types of damage: that produced locally by the tumor, which can be direct damage to the organ where it is located, and immunopathological damage that occurs when the tumor and the tumor microenvironment release compounds that cause metabolic derangement and systemic inflammation, such as in cachexia. IL-1, IL-6, and TNF-α play an essential role at the systemic level as inducers of cancer cachexia (321). In the 1970s, although there was not yet a methodology with sufficient sensitivity to measure these cytokines in cancer patients and obtain consistent results, treatment with anti-TNF- and IL-6 antibodies was proven to be effective in reducing cachexia in mouse tumor models (322).
Jafri et al. proposed a cachexia index to estimate the degree of cachexia in patients with advanced non-small-cell lung carcinoma (NSCLC) and to identify which patients might respond to cancer cachexia treatment (323). This index considers albumin; the skeletal muscle index, which results from comparing abdominal and paraspinal muscle scans between the time of diagnosis and one month later; and the NLR (323). In another paper, a sarcopenia index was defined as the muscle area at the third vertebra/height2, and values of ≤ 55 cm2/m2 for men and ≤ 39 cm2/m2 for women indicated sarcopenia. This index was correlated with CRP and the neutrophil/lymphocyte ratio in patients with small-cell lung carcinoma. Sarcopenia was found to have a linear relationship with CRP (324). Finally, Barrer et al. found that the neutrophil/lymphocyte ratio was associated with weight loss in patients with colon, lung, and prostate cancer cachexia (325).
The cachexia syndrome is characterized by the metabolic dysregulation of carbohydrates, lipids, and proteins in various organs and the sympathetic activation of the nervous system. All this leads to a poor quality of life for patients, and their diminished physical condition may not be suitable for treatment. The incidence of the anorexia–cachexia syndrome is high in cancer patients, affecting the evolution of the underlying disease at the clinical level. Unfortunately, its clinical management is complex in cancer patients. Consequently, cancer patients severely compromised nutritional status and weight loss remain standard features. It is essential to recognize and treat this syndrome early, together with antitumor therapy, to prolong survival and positively influence the quality of life of cancer patients.
Concluding remarks and perspectives
During the uncontrolled growth of tumors, several inflammatory factors, including—but not limited to—cytokines, chemokines, growth factors, metabolites, and ncRNAs, are produced and released by both tumor and stromal cells. The continuous release of these factors causes impacts at the local level; their delivery into the bloodstream reaches other systems or organs, such as the liver, nervous system, bone marrow, adipose tissue, skeletal muscle, etc. In this setting, the continuous presence of these factors, in particular IL-6, IL-1, TNF-α, G-CSF, and GM-CSF, promotes cancer-associated systemic inflammation. Due to their pleiotropic activity, these molecules impact distinct subsets of cells, such as endothelial, epithelial, mesenchymal, neurologic, and hematologic cells, amplifying the inflammatory state and the clinical manifestations of the aberrant function of organs and systems, known as paraneoplastic syndromes. Paraneoplastic syndromes have detrimental effects on the patient’s quality of life and can sometimes cause their demise.
In addition, paraneoplastic syndromes can be exacerbated during the administration of cytotoxic antitumor therapies focused on eliminating tumor cells. Because advanced-stage tumors deregulate communication between the immune, endocrine, and neurological systems, a deeper we believe that, in addition to using antitumor agents, knowledge of the regulation of neuro–endocrine–immune intercommunication during cancer-associated inflammation will to could favor the development of upcoming therapies that will impact patient survival and quality of life. In support of this proposal, recent reports have demonstrated that nonsteroidal anti-inflammatory drugs may be used in cancer to reduce systemic inflammation (326). However, in clinically advanced tumors, cancer-associated systemic inflammation is dysregulated at another level; thus, these anti-inflammatory drugs would have no effect. Furthermore, the use of steroid drugs, such as glucocorticoids, could suppress the antitumor immune response, as they block the function of CD8+ effector T and NK cells (327). To address this issue, models that capture the complexity of tumor–host organ interactions will help clarify the picture and offer new therapeutic alternatives to improve patient outcomes.
Author contributions
Conceptualization and design of the entire manuscript and draft: DA-C, RC-D, MM-M, and JL-G. JL-G, DA-C, and MM-M wrote the microenvironment section. JL-G, RC-D, and AC wrote the participation of the immune response in cancer section. DA-C, RC-D, JB-L, and MP-M wrote the systemic tumor microenvironment-derived cytokines section. JL-G, UR-N, and MM-M wrote the parameters to assess systemic immune-inflammatory markers section. DA-C, RC-D, and AC wrote the paraneoplastic syndromes section. Figure 1 was designed by RC-D, DA-C, and JB-L. Tables 1, 2 was designed by JL-G, RC-D and MM-M. All authors contributed to the article and approved the submitted version.
Funding
This manuscript was partially funded by Consejo Nacional de Ciencia y Tecnologia (CONACYT) (grant number: 284775).
Acknowledgments
The authors acknowledge Instituto Nacional de Enfermedades Respiratorias Ismael Cosio Villegas. RC-D, JB-L, MM-M, and MP-M are students from the Posgrado en Ciencias Biologicas Universidad Nacional Autonoma de Mexico, Mexico and Posgrado en Ciencias en Biomedicina y Biotecnologia Molecular, Instituto Politecnico Nacional, Mexico, Mexico, respectively. All are recipients of a fellowship from CONACYT (RC-D 631047) (JJB-L 1085486) (MM-M 718959) (MP-M 740805).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
3. Greten FR, Grivennikov SI. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
4. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
5. Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14:1014–22. doi: 10.1038/ni.2703
6. Dolan RD, Lim J, McSorley ST, Horgan PG, McMillan DC. The role of the systemic inflammatory response in predicting outcomes in patients with operable cancer: Systematic review and meta-analysis. Sci Rep (2017) 7:16717. doi: 10.1038/s41598-017-16955-5
7. Pelosof LC, Gerber DE. Paraneoplastic syndromes: An approach to diagnosis and treatment. Mayo Clin Proc (2010) 85:838–54. doi: 10.4065/mcp.2010.0099
8. Oh SY, Jun HJ, Park SJ, Park IK, Lim GJ, Yu Y, et al. A randomized phase II study to assess the effectiveness of fluid therapy or intensive nutritional support on survival in patients with advanced cancer who cannot be nourished via enteral route. J Palliat Med (2014) 17:1266–70. doi: 10.1089/jpm.2014.0082
9. Wu C, Ning H, Liu M, Lin J, Luo S, Zhu W, et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J Clin Invest (2018) 128:3425–38. doi: 10.1172/JCI97973
10. Almagro J, Messal HA, Elosegui-Artola A, van Rheenen J, Behrens A. Tissue architecture in tumor initiation and progression. Trends Cancer (2022) 8:494–505. doi: 10.1016/j.trecan.2022.02.007
11. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res (2015) 21:687–92. doi: 10.1158/1078-0432.CCR-14-1860
12. Song W, Li D, Tao L, Luo Q, Chen L. Solute carrier transporters: the metabolic gatekeepers of immune cells. Acta Pharm Sin B (2020) 10:61–78. doi: 10.1016/j.apsb.2019.12.006
13. Wang S, Wang J, Wei W, Ma G. Exosomes: The indispensable messenger in tumor pathogenesis and the rising star in antitumor applications. Adv Biosyst (2019) 3:1900008. doi: 10.1002/adbi.201900008
14. Kalluri R, LeBleu VS. The biology, function , and biomedical applications of exosomes. Science (2020) 367:eaau6977. doi: 10.1126/science.aau6977
15. Ruivo CF, Adem B, Silva M, Melo SA. The biology of cancer exosomes: Insights and new perspectives. Cancer Res (2017) 77:6480–8. doi: 10.1158/0008-5472.CAN-17-0994
16. Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem (2019) 88:487–514. doi: 10.1146/annurev-biochem-013118-111902
17. Beach A, Zhang H-G, Ratajczak MZ, Kakar SS. Exosomes: An overview of biogenesis, composition and role in ovarian cancer. J Ovarian Res (2014) 7:14. doi: 10.1186/1757-2215-7-14
18. Boussadia Z, Zanetti C, Parolini I. Role of microenvironmental acidity and tumor exosomes in cancer immunomodulation. Transl Cancer Res (2020) 9:5775–86. doi: 10.21037/tcr.2020.03.69
19. Qu X, Tang Y, Hua S. Immunological approaches towards cancer and inflammation: A cross talk. Front Immunol (2018) 9:563. doi: 10.3389/fimmu.2018.00563
20. Bebelman MP, Smit MJ, Pegtel DM, Baglio SR. Biogenesis and function of extracellular vesicles in cancer. Pharmacol Ther (2018) 188:1–11. doi: 10.1016/j.pharmthera.2018.02.013
21. Paul S, Kulkarni N, Shilpi, Lal G. Intratumoral natural killer cells show reduced effector and cytolytic properties and control the differentiation of effector Th1 cells. OncoImmunology (2016) 5:e1235106. doi: 10.1080/2162402X.2016.1235106
22. Ronca R, Van Ginderachter JA, Turtoi A. Paracrine interactions of cancer-associated fibroblasts, macrophages and endothelial cells: tumor allies and foes. Curr Opin Oncol (2018) 30:45–53. doi: 10.1097/CCO.0000000000000420
23. Obacz J, Pastorekova S, Vojtesek B, Hrstka R. Cross-talk between HIF and p53 as mediators of molecular responses to physiological and genotoxic stresses. Mol Cancer (2013) 12:93. doi: 10.1186/1476-4598-12-93
24. Abhange K, Makler A, Wen Y, Ramnauth N, Mao W, Asghar W, et al. Small extracellular vesicles in cancer. Bioact Mat (2021) 6:3705–43. doi: 10.1016/j.bioactmat.2021.03.015
25. Virchow R. As based upon physiological and pathological histology. Nutr Rev (2009) 47:23–5. doi: 10.1111/j.1753-4887.1989.tb02747.x
26. Dvorak HF. Tumors: wounds that do not heal. similarities between tumor stroma generation and wound healing. N Engl J Med (1986) 315:1650–9. doi: 10.1056/nejm198612253152606
27. Daniotti JL, Lardone RD, Vilcaes AA. Dysregulated expression of glycolipids in tumor cells: From negative modulator of anti-tumor immunity to promising targets for developing therapeutic agents. Front Oncol (2016) 5:300. doi: 10.3389/fonc.2015.00300
28. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6:24–37. doi: 10.1038/nrc1782
29. Chimal-Ramírez GK, Espinoza-Sánchez NA, Fuentes-Pananá EM. Protumor activities of the immune response: Insights in the mechanisms of immunological shift, oncotraining, and oncopromotion. J Oncol (2013) 2013:1–16. doi: 10.1155/2013/835956
30. Diefenbach A, Raulet DH. Innate immune recognition by stimulatory immunoreceptors. Curr Opin Immunol (2003) 15:37–44. doi: 10.1016/S0952-7915(02)00007-9
31. Sivori S, Della Chiesa M, Carlomagno S, Quatrini L, Munari E, Vacca P, et al. Inhibitory receptors and checkpoints in human NK cells, implications for the immunotherapy of cancer. Front Immunol (2020) 11:2156. doi: 10.3389/fimmu.2020.02156
32. Malmberg KJ, Carlsten M, Björklund A, Sohlberg E, Bryceson YT, Ljunggren HG. Natural killer cell-mediated immunosurveillance of human cancer. Semin Immunol (2017) 31:20–9. doi: 10.1016/j.smim.2017.08.002
33. Dulberger CL, McMurtrey CP, Hölzemer A, Neu KE, Liu V, Steinbach AM, et al. Human leukocyte antigen F presents peptides and regulates immunity through interactions with NK cell receptors. Immunity (2017) 46:1018–29.e7. doi: 10.1016/j.immuni.2017.06.002
34. Isaacson B, Mandelboim O. Sweet killers: NK cells need glycolysis to kill tumors. Cell Metab (2018) 28:183–4. doi: 10.1016/j.cmet.2018.07.008
35. Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol (2014) 193:4477–84. doi: 10.4049/jimmunol.1401558
36. Zhu L, Gangadaran P, Kalimuthu S, Oh JM, Baek SH, Jeong SY, et al. Novel alternatives to extracellular vesicle-based immunotherapy – exosome mimetics derived from natural killer cells. Artif Cells Nanomed Biotechnol (2018) 46:S166–79. doi: 10.1080/21691401.2018.1489824
37. Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, et al. Immune surveillance properties of human NK cell-derived exosomes. J Immunol (2012) 189:2833–42. doi: 10.4049/jimmunol.1101988
38. Cochran AM, Kornbluth J. Extracellular vesicles from the human natural killer cell line NK3.3 have broad and potent anti-tumor activity. Front Cell Dev Biol (2021) 9:698639. doi: 10.3389/fcell.2021.698639
39. Fauriat C, Long EO, Ljunggren H-G, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood (2010) 115:2167–76. doi: 10.1182/blood-2009-08-238469
40. Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-γ in tumor progression and regression: A review. biomark Res (2020) 8:49. doi: 10.1186/s40364-020-00228-x
41. Aquino-López A, Senyukov VV, Vlasic Z, Kleinerman ES, Lee DA. Interferon gamma induces changes in natural killer (NK) cell ligand expression and alters NK cell-mediated lysis of pediatric cancer cell lines. Front Immunol (2017) 8:391. doi: 10.3389/fimmu.2017.00391
42. Chavez-Dominguez R, Perez-Medina M, Aguilar-Cazares D, Galicia-Velasco M, Meneses-Flores M, Islas-Vazquez L, et al. Old and new players of inflammation and their relationship with cancer development. Front Oncol (2021) 11:722999. doi: 10.3389/fonc.2021.722999
43. Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene (2016) 35:5931–41. doi: 10.1038/onc.2016.104
44. Haniffa M, Bigley V, Collin M. Human mononuclear phagocyte system reunited. Semin Cell Dev Biol (2015) 41:59–69. doi: 10.1016/j.semcdb.2015.05.004
45. Gardner A, de Mingo Pulido Á, Ruffell B. Dendritic cells and their role in immunotherapy. Front Immunol (2020) 11:924. doi: 10.3389/fimmu.2020.00924
46. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
47. André F, Schartz NEC, Chaput N, Flament C, Raposo G, Amigorena S, et al. Tumor-derived exosomes: A new source of tumor rejection antigens. Vaccine (2002) 20:A28–31. doi: 10.1016/S0264-410X(02)00384-5
48. Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15:323–32. doi: 10.1038/ni.2833
49. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
50. Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, et al. Hypoxia and hypoxia-inducible factor-1α modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol (2008) 180:4697–705. doi: 10.4049/jimmunol.180.7.4697
51. Sinclair C, Bommakanti G, Gardinassi L, Loebbermann J, Johnson MJ, Hakimpour P, et al. mTOR regulates metabolic adaptation of APCs in the lung and controls the outcome of allergic inflammation. Science (2017) 357:1014–21. doi: 10.1126/science.aaj2155
52. Hao S, Bai O, Li F, Yuan J, Laferte S, Xiang J. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology (2007) 90–120. doi: 10.1111/j.1365-2567.2006.02483.x
53. Wang Y, Xiang Y, Xin VW, Wang X-W, Peng X-C, Liu X-Q, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol OncolJ Hematol Oncol (2020) 13:107. doi: 10.1186/s13045-020-00939-6
54. Record M, Subra C, Silvente-Poirot S, Poirot M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem Pharmacol (2011) 81:1171–82. doi: 10.1016/j.bcp.2011.02.011
55. Lindenbergh MFS, Stoorvogel W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu Rev Immunol (2018) 36:435–59. doi: 10.1146/annurev-immunol-041015-055700
56. Théry C, Duban L, Segura E, Véron P, Lantz O, Amigorena S. Indirect activation of naïve CD4+ T cells by dendritic cell–derived exosomes. Nat Immunol (2002) 3:1156–62. doi: 10.1038/ni854
57. Du Z, Huang Z, Chen X, Jiang G, Peng Y, Feng W, et al. Modified dendritic cell-derived exosomes activate both NK cells and T cells through the NKG2D/NKG2D-l pathway to kill CML cells with or without T315I mutation. Exp Hematol Oncol (2022) 11:36. doi: 10.1186/s40164-022-00289-8
58. Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol (2009) 9:581–93. doi: 10.1038/nri2567
59. Greyer M, Whitney PG, Stock AT, Davey GM, Tebartz C, Bachem A, et al. T Cell help amplifies innate signals in CD8 + DCs for optimal CD8 + T cell priming. Cell Rep (2016) 14:586–97. doi: 10.1016/j.celrep.2015.12.058
60. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity (2010) 32:79–90. doi: 10.1016/j.immuni.2009.11.012
61. Zhang H, Zhu Z, Modrak S, Little A. Tissue-resident memory CD4 + T cells play a dominant role in the initiation of antitumor immunity. J Immunol (2022) 208:2837–46. doi: 10.4049/jimmunol.2100852
62. Nicolet BP, Guislain A, van Alphen FPJ, Gomez-Eerland R, Schumacher TNM, van den Biggelaar M, et al. CD29 identifies IFN-γ–producing human CD8 + T cells with an increased cytotoxic potential. Proc Natl Acad Sci (2020) 117:6686–96. doi: 10.1073/pnas.1913940117
63. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol (2018) 18:635–47. doi: 10.1038/s41577-018-0044-0
64. Rezaei R, Baghaei K, Hashemi SM, Zali MR, Ghanbarian H, Amani D. Tumor-derived exosomes enriched by miRNA-124 promote anti-tumor immune response in CT-26 tumor-bearing mice. Front Med (2021) 8:619939. doi: 10.3389/fmed.2021.619939
65. Roshani Asl E, Rasmi Y, Baradaran B. MicroRNA-124-3p suppresses PD-L1 expression and inhibits tumorigenesis of colorectal cancer cells via modulating STAT3 signaling. J Cell Physiol (2021) 236:7071–87. doi: 10.1002/jcp.30378
66. Rogers T, DeBerardinis RJ. Metabolic plasticity of neutrophils: Relevance to pathogen responses and cancer. Trends Cancer (2021) 7:700–13. doi: 10.1016/j.trecan.2021.04.007
67. Yang D, Yang L, Cai J, Li H, Xing Z, Hou Y. Phosphoinositide 3-kinase/Akt and its related signaling pathways in the regulation of tumor-associated macrophages polarization. Mol Cell Biochem (2022). doi: 10.1007/s11010-022-04461-w
68. Pu Y, Ji Q. Tumor-associated macrophages regulate PD-1/PD-L1 immunosuppression. Front Immunol (2022) 13:874589. doi: 10.3389/fimmu.2022.874589
69. Li Z, Suo B, Long G, Gao Y, Song J, Zhang M, et al. Exosomal miRNA-16-5p derived from M1 macrophages enhances T cell-dependent immune response by regulating PD-L1 in gastric cancer. Front Cell Dev Biol (2020) 8:572689. doi: 10.3389/fcell.2020.572689
70. Bebber CM, Müller F, Prieto Clemente L, Weber J, von Karstedt S. Ferroptosis in cancer cell biology. Cancers (2020) 12:164. doi: 10.3390/cancers12010164
71. Kundu J, Surh Y. Inflammation: Gearing the journey to cancer. Mutat Res Mutat Res (2008) 659:15–30. doi: 10.1016/j.mrrev.2008.03.002
72. Borrello MG, Alberti L, Fischer A, Degl’innocenti D, Ferrario C, Gariboldi M, et al. Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc Natl Acad Sci U A (2005) 102:14825–30. doi: 10.1073/pnas.0503039102
73. Shchors K, Shchors E, Rostker F, Lawlor ER, Brown-Swigart L, Evan GI. The myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev (2006) 20:2527–38. doi: 10.1101/gad.1455706
74. Hamarsheh S, Osswald L, Saller BS, Unger S, De Feo D, Vinnakota JM, et al. Oncogenic KrasG12D causes myeloproliferation via NLRP3 inflammasome activation. Nat Commun (2020) 11:1659. doi: 10.1038/s41467-020-15497-1
75. Corrado C, Raimondo S, Chiesi A, Ciccia F, De Leo G, Alessandro R. Exosomes as intercellular signaling organelles involved in health and disease: Basic science and clinical applications. Int J Mol Sci (2013) 14:5338–66. doi: 10.3390/ijms14035338
76. Othman N, Jamal R, Abu N. Cancer-derived exosomes as effectors of key inflammation-related players. Front Immunol (2019) 10:2103. doi: 10.3389/fimmu.2019.02103
77. Shen M, Ren X. New insights into the biological impacts of immune cell-derived exosomes within the tumor environment. Cancer Lett (2018) 431:115–22. doi: 10.1016/j.canlet.2018.05.040
78. Martini M, Testi MG, Pasetto M, Picchio MC, Innamorati G, Mazzocco M, et al. IFN-γ-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine (2010) 28:3548–57. doi: 10.1016/j.vaccine.2010.03.007
79. Thapa RJ, Basagoudanavar SH, Nogusa S, Irrinki K, Mallilankaraman K, Slifker MJ, et al. NF-κB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol Cell Biol (2011) 31:2934–46. doi: 10.1128/MCB.05445-11
80. Song M, Ping Y, Zhang K, Yang L, Li F, Zhang C, et al. Low-dose IFNγ induces tumor cell stemness in tumor microenvironment of non–small cell lung cancer. Cancer Res (2019) 79:3737–48. doi: 10.1158/0008-5472.CAN-19-0596
81. Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M, et al. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J Immunol (2009) 183:4705–14. doi: 10.4049/jimmunol.0901511
82. Carmi Y, Rinott G, Dotan S, Elkabets M, Rider P, Voronov E, et al. Microenvironment-derived IL-1 and IL-17 interact in the control of lung metastasis. J Immunol (2011) 186:3462–71. doi: 10.4049/jimmunol.1002901
83. Sun Z, Ren Z, Yang K, Liu Z, Cao S, Deng S, et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nat Commun (2019) 10:3874. doi: 10.1038/s41467-019-11782-w
84. Nappo G, Handle F, Santer FR, McNeill RV, Seed RI, Collins AT, et al. The immunosuppressive cytokine interleukin-4 increases the clonogenic potential of prostate stem-like cells by activation of STAT6 signalling. Oncogenesis (2017) 6:e342–2. doi: 10.1038/oncsis.2017.23
85. Li Z, Jiang J, Wang Z, Zhang J, Xiao M, Wang C, et al. Endogenous interleukin-4 promotes tumor development by increasing tumor cell resistance to apoptosis. Cancer Res (2008) 68:8687–94. doi: 10.1158/0008-5472.CAN-08-0449
86. Gocheva V, Wang H-W, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev (2010) 24:241–55. doi: 10.1101/gad.1874010
87. Apte SH, Groves P, Olver S, Baz A, Doolan DL, Kelso A, et al. IFN-γ inhibits IL-4–induced type 2 cytokine expression by CD8 T cells In vivo and modulates the anti-tumor response. J Immunol (2010) 185:998–1004. doi: 10.4049/jimmunol.0903372
88. Agaugué S, Marcenaro E, Ferranti B, Moretta L, Moretta A. Human natural killer cells exposed to IL-2, IL-12, IL-18, or IL-4 differently modulate priming of naive T cells by monocyte-derived dendritic cells. Blood (2008) 112:1776–83. doi: 10.1182/blood-2008-02-135871
89. De Simone V, Franzè E, Ronchetti G, Colantoni A, Fantini MC, Di Fusco D, et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene (2015) 34:3493–503. doi: 10.1038/onc.2014.286
90. Lin M-T, Juan C-Y, Chang K-J, Chen W-J, Kuo M-L. IL-6 inhibits apoptosis and retains oxidative DNA lesions in human gastric cancer AGS cells through up-regulation of anti-apoptotic gene mcl-1. Carcinogenesis (2001) 22:1947–53. doi: 10.1093/carcin/22.12.1947
91. Huang Y-H, Yang H-Y, Huang S-W, Ou G, Hsu Y-F, Hsu M-J. Interleukin-6 induces vascular endothelial growth factor-c expression via src-FAK-STAT3 signaling in lymphatic endothelial cells. PloS One (2016) 11:e0158839. doi: 10.1371/journal.pone.0158839
92. Alfaro C, Sanmamed MF, Rodríguez-Ruiz ME, Teijeira Á, Oñate C, González Á, et al. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat Rev (2017) 60:24–31. doi: 10.1016/j.ctrv.2017.08.004
93. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest (2010) 120:485–97. doi: 10.1172/JCI39397
94. Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: A target for anticancer immunotherapy. Immunol Res (2011) 51:170–82. doi: 10.1007/s12026-011-8262-6
95. Wang X, Wong K, Ouyang W, Rutz S. Targeting IL-10 family cytokines for the treatment of human diseases. Cold Spring Harb Perspect Biol (2019) 11:a028548. doi: 10.1101/cshperspect.a028548
96. Hamidullah, Changkija B, Konwar R. Role of interleukin-10 in breast cancer. Breast Cancer Res Treat (2012) 133:11–21. doi: 10.1007/s10549-011-1855-x
97. Li L, Ma Y, Xu Y. Follicular regulatory T cells infiltrated the ovarian carcinoma and resulted in CD8 T cell dysfunction dependent on IL-10 pathway. Int Immunopharmacol (2019) 68:81–7. doi: 10.1016/j.intimp.2018.12.051
98. Mirlekar B, Pylayeva-Gupta Y. IL-12 family cytokines in cancer and immunotherapy. Cancers (2021) 13:167. doi: 10.3390/cancers13020167
99. Schmidt-Weber CB, Akdis M, Akdis CA. TH17 cells in the big picture of immunology. J Allergy Clin Immunol (2007) 120:247–54. doi: 10.1016/j.jaci.2007.06.039
100. Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev (2003) 14:155–74. doi: 10.1016/S1359-6101(03)00002-9
101. Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev (2008) 223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x
102. Xing Y, Tian Y, Kurosawa T, Matsui S, Touma M, Wu Q, et al. Inhibition of blood vessel formation in tumors by IL-18-polarized M1 macrophages. Genes Cells (2016) 21:287–95. doi: 10.1111/gtc.12329
103. Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin (2008) 29:1275–88. doi: 10.1111/j.1745-7254.2008.00889.x
104. Chen X, Wang L, Li P, Song M, Qin G, Gao Q, et al. Dual TGF-β and PD-1 blockade synergistically enhances MAGE-A3-specific CD8 + T cell response in esophageal squamous cell carcinoma. Int J Cancer (2018) 143:2561–74. doi: 10.1002/ijc.31730
105. Shen Y, Teng Y, Lv Y, Zhao Y, Qiu Y, Chen W, et al. PD-1 does not mark tumor-infiltrating CD8+ T cell dysfunction in human gastric cancer. J Immunother Cancer (2020) 8:e000422. doi: 10.1136/jitc-2019-000422
106. Yu H, Yang J, Jiao S, Wang J, Li Y. TGF-β1 precursor and CD8 are potential prognostic and predictive markers in operated breast cancer. J Huazhong Univ Sci Technolog Med Sci (2014) 34:51–8. doi: 10.1007/s11596-014-1231-2
107. Fujii R, Jochems C, Tritsch SR, Wong HC, Schlom J, Hodge JW. An IL-15 superagonist/IL-15Rα fusion complex protects and rescues NK cell-cytotoxic function from TGF-β1-mediated immunosuppression. Cancer Immunol Immunother (2018) 67:675–89. doi: 10.1007/s00262-018-2121-4
108. Kumar A, Taghi Khani A, Sanchez Ortiz A, Swaminathan S. GM-CSF: A double-edged sword in cancer immunotherapy. Front Immunol (2022) 13:901277. doi: 10.3389/fimmu.2022.901277
109. Rahmati M, Petitbarat M, Dubanchet S, Bensussan A, Chaouat G, Ledee N. Granulocyte-colony stimulating factor related pathways tested on an endometrial ex-vivo model. PloS One (2014) 9:e102286. doi: 10.1371/journal.pone.0102286
110. He K, Liu X, Hoffman RD, Shi R, Lv G, Gao J. G-CSF / GM-CSF -induced hematopoietic dysregulation in the progression of solid tumors. FEBS Open Bio (2022) 12:1268–85. doi: 10.1002/2211-5463.13445
111. Dong J, Grunstein J, Tejada M, Peale F, Frantz G, Liang W-C, et al. VEGF-null cells require PDGFR α signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J (2004) 23:2800–10. doi: 10.1038/sj.emboj.7600289
112. Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PloS One (2010) 5:e12445. doi: 10.1371/journal.pone.0012445
113. Grismaldo A, Sobrevia L, Morales L. Role of platelet-derived growth factor c on endothelial dysfunction in cardiovascular diseases. Biochim Biophys Acta BBA - Gen Subj (2022) 1866:130188. doi: 10.1016/j.bbagen.2022.130188
114. Geindreau M, Bruchard M, Vegran F. Role of cytokines and chemokines in angiogenesis in a tumor context. Cancers (2022) 14:2446. doi: 10.3390/cancers14102446
115. Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med (2014) 20:607–15. doi: 10.1038/nm.3541
116. Bai W, Zhang W, Hu B. Vascular endothelial growth factor suppresses dendritic cells function of human prostate cancer. OncoTargets Ther (2018) 11:1267–74. doi: 10.2147/OTT.S161302
117. Singh B, Carpenter G, Coffey RJ. EGF receptor ligands: recent advances. F1000Research (2016) 5:2270. doi: 10.12688/f1000research.9025.1
118. Sasaki T, Hiroki K, Yamashita Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. BioMed Res Int (2013) 2013:1–8. doi: 10.1155/2013/546318
119. Ferreira AS, Lopacinski A, Batista M, Hiraiwa PM, Guimarães BG, Zanchin NIT. A toolkit for recombinant production of seven human EGF family growth factors in active conformation. Sci Rep (2022) 12:5034. doi: 10.1038/s41598-022-09060-9
120. Do HTT, Lee CH, Cho J. Chemokines and their receptors: Multifaceted roles in cancer progression and potential value as cancer prognostic markers. Cancers (2020) 12:287. doi: 10.3390/cancers12020287
121. Krawczyk KM, Nilsson H, Allaoui R, Lindgren D, Arvidsson M, Leandersson K, et al. Papillary renal cell carcinoma-derived chemerin, IL-8, and CXCL16 promote monocyte recruitment and differentiation into foam-cell macrophages. Lab Invest (2017) 97:1296–305. doi: 10.1038/labinvest.2017.78
122. Redjimi N, Raffin C, Raimbaud I, Pignon P, Matsuzaki J, Odunsi K, et al. CXCR3 + T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res (2012) 72:4351–60. doi: 10.1158/0008-5472.CAN-12-0579
123. Wittamer V, Franssen J-D, Vulcano M, Mirjolet J-F, Le Poul E, Migeotte I, et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med (2003) 198:977–85. doi: 10.1084/jem.20030382
124. Samson M, Edinger AL, Stordeur P, Rucker J, Verhasselt V, Sharron M, et al. ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. Eur J Immunol (1998) 28:1689–700. doi: 10.1002/(SICI)1521-4141(199805)28:05<1689::AID-IMMU1689>3.0.CO;2-I
125. Parolini S, Santoro A, Marcenaro E, Luini W, Massardi L, Facchetti F, et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood (2007) 109:3625–32. doi: 10.1182/blood-2006-08-038844
126. Eisinger K, Bauer S, Schäffler A, Walter R, Neumann E, Buechler C, et al. Chemerin induces CCL2 and TLR4 in synovial fibroblasts of patients with rheumatoid arthritis and osteoarthritis. Exp Mol Pathol (2012) 92:90–6. doi: 10.1016/j.yexmp.2011.10.006
127. Goralski KB, Jackson AE, McKeown BT, Sinal CJ. More than an adipokine: The complex roles of chemerin signaling in cancer. Int J Mol Sci (2019) 20:4778. doi: 10.3390/ijms20194778
128. Banas M, Zabieglo K, Kasetty G, Kapinska-Mrowiecka M, Borowczyk J, Drukala J, et al. Correction: Chemerin is an antimicrobial agent in human epidermis. PloS One (2013) 1–8. doi: 10.1371/annotation/4dfd522c-f0fd-40db-aadc-44cbef367a40
129. Dubois-Vedrenne I, Al Delbany D, De Henau O, Robert V, Vernimmen M, Langa F, et al. The antitumoral effects of chemerin are independent from leukocyte recruitment and mediated by inhibition of neoangiogenesis. Oncotarget (2021) 12:1903–19. doi: 10.18632/oncotarget.28056
130. Bhat AA, Nisar S, Singh M, Ashraf B, Masoodi T, Prasad CP, et al. Cytokine- and chemokine-induced inflammatory colorectal tumor microenvironment: Emerging avenue for targeted therapy. Cancer Commun (2022) 1–27. doi: 10.1002/cac2.12295
131. Gowhari Shabgah A, Jadidi-Niaragh F, Ebrahimzadeh F, Mohammadi H, Askari E, Pahlavani N, et al. A comprehensive review of chemokine CXC17 (VCC1) in cancer, infection, and inflammation. Cell Biol Int (2022) 1–14. doi: 10.1002/cbin.11846
132. Helmlinger G, Sckell A, Dellian M, Forbes NS, Jain RK. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin Cancer Res Off J Am Assoc Cancer Res (2002) 8:1284–91.
133. Wang C, Li Y, Yan S, Wang H, Shao X, Xiao M, et al. Interactome analysis reveals that lncRNA HULC promotes aerobic glycolysis through LDHA and PKM2. Nat Commun (2020) 11:3162. doi: 10.1038/s41467-020-16966-3
134. Zhai S, Xu Z, Xie J, Zhang J, Wang X, Peng C, et al. Epigenetic silencing of LncRNA LINC00261 promotes c-myc-mediated aerobic glycolysis by regulating miR-222-3p/HIPK2/ERK axis and sequestering IGF2BP1. Oncogene (2021) 40:277–91. doi: 10.1038/s41388-020-01525-3
135. Takahashi K, Yan IK, Haga H, Patel T. Modulation of hypoxia-signaling pathways by extracellular long non-coding RNA regulator of reprogramming. J Cell Sci (2014) (7):1585–94. doi: 10.1242/jcs.141069
136. Nishizawa Y, Konno M, Asai A, Koseki J, Kawamoto K, Miyoshi N, et al. Hypoxia stimulates the cytoplasmic localization of oncogenic long noncoding RNA LINC00152 in colorectal cancer. Int J Oncol (2017) (52):453–60. doi: 10.3892/ijo.2017.4218
137. Ng CT, Azwar S, Yip WK, Zahari Sham SY, Faisal Jabar M, Sahak NH, et al. Isolation and identification of long non-coding RNAs in exosomes derived from the serum of colorectal carcinoma patients. Biology (2021) 10:918. doi: 10.3390/biology10090918
138. Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-jun activation. Int J Cancer (2012) 131:633–40. doi: 10.1002/ijc.26410
139. Raychaudhuri D, Bhattacharya R, Sinha BP, Liu CSC, Ghosh AR, Rahaman O, et al. Lactate induces pro-tumor reprogramming in intratumoral plasmacytoid dendritic cells. Front Immunol (2019) 10:1878. doi: 10.3389/fimmu.2019.01878
140. Brown RAM, Richardson KL, Kabir TD, Trinder D, Ganss R, Leedman PJ. Altered iron metabolism and impact in cancer biology, metastasis, and immunology. Front Oncol (2020) 10:476. doi: 10.3389/fonc.2020.00476
141. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Diff (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
142. Efimova I, Catanzaro E, van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer (2020) 8:e001369. doi: 10.1136/jitc-2020-001369
143. Yegutkin GG. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: Functional implications and measurement of activities. Crit Rev Biochem Mol Biol (2014) 49:473–97. doi: 10.3109/10409238.2014.953627
144. Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD 7 3. Immunol Rev (1998) 161:95–109. doi: 10.1111/j.1600-065X.1998.tb01574.x
145. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev (2017) 276:121–44. doi: 10.1111/imr.12528
146. Raskovalova T, Lokshin A, Huang X, Jackson EK, Gorelik E. Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-Activated NK cells: Involvement of protein kinase a isozyme I (PKA I). Immunol Res (2006) 36:91–100. doi: 10.1385/IR:36:1:91
147. Csóka B, Selmeczy Z, Koscsó B, Németh ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J (2012) 26:376–86. doi: 10.1096/fj.11-190934
148. Augsten M, Hägglöf C, Olsson E, Stolz C, Tsagozis P, Levchenko T, et al. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi-modal stimulator of prostate tumor growth. Proc Natl Acad Sci (2009) 106:3414–9. doi: 10.1073/pnas.0813144106
149. Shi H, Han X, Sun Y, Shang C, Wei M, Ba X, et al. Chemokine (C-X-C motif) ligand 1 and CXCL 2 produced by tumor promote the generation of monocytic myeloid-derived suppressor cells. Cancer Sci (2018) 109:3826–39. doi: 10.1111/cas.13809
150. Unver N, Esendagli G, Yilmaz G, Guc D. CXCL7-induced macrophage infiltration in lung tumor is independent of CXCR2 expression. Cytokine (2015) 75:330–7. doi: 10.1016/j.cyto.2015.07.018
151. Kasashima H, Yashiro M, Nakamae H, Kitayama K, Masuda G, Kinoshita H, et al. CXCL1–chemokine (C-X-C motif) receptor 2 signaling stimulates the recruitment of bone marrow–derived mesenchymal cells into diffuse-type gastric cancer stroma. Am J Pathol (2016) 186:3028–39. doi: 10.1016/j.ajpath.2016.07.024
152. Deng S, Deng Q, Zhang Y, Ye H, Yu X, Zhang Y, et al. Non-platelet-derived CXCL4 differentially regulates cytotoxic and regulatory T cells through CXCR3 to suppress the immune response to colon cancer. Cancer Lett (2019) 443:1–12. doi: 10.1016/j.canlet.2018.11.017
153. Yan M, Jene N, Byrne D, Millar EK, O’Toole SA, McNeil CM, et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res (2011) 13:R47. doi: 10.1186/bcr2869
154. Shao C, Yang F, Miao S, Liu W, Wang C, Shu Y, et al. Role of hypoxia-induced exosomes in tumor biology. Mol Cancer (2018) 17:120. doi: 10.1186/s12943-018-0869-y
155. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin J-P, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest (2010) (2):457–71. doi: 10.1172/JCI40483
156. Jafari R, Rahbarghazi R, Ahmadi M, Hassanpour M, Rezaie J. Hypoxic exosomes orchestrate tumorigenesis: molecular mechanisms and therapeutic implications. J Transl Med (2020) 18:474. doi: 10.1186/s12967-020-02662-9
157. Zhang Q, Zhong C, Shen J, Chen S, Jia Y, Duan S. Emerging role of LINC00461 in cancer. BioMed Pharmacother (2022) 152:113239. doi: 10.1016/j.biopha.2022.113239
158. Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs bind to toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci (2012) 109:E2110–6. doi: 10.1073/pnas.1209414109
159. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC: Innate immunity. Eur J Immunol (2013) 43:2756–64. doi: 10.1002/eji.201343448
160. Xu Y, Zeng H, Jin K, Liu Z, Zhu Y, Xu L, et al. Immunosuppressive tumor-associated macrophages expressing interlukin-10 conferred poor prognosis and therapeutic vulnerability in patients with muscle-invasive bladder cancer. J Immunother Cancer (2022) 10:e003416. doi: 10.1136/jitc-2021-003416
161. Chang L-Y, Lin Y-C, Mahalingam J, Huang C-T, Chen T-W, Kang C-W, et al. Tumor-derived chemokine CCL5 enhances TGF-β–mediated killing of CD8 + T cells in colon cancer by T-regulatory cells. Cancer Res (2012) 72:1092–102. doi: 10.1158/0008-5472.CAN-11-2493
162. Chen M-L, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proc Natl Acad Sci (2005) 102:419–24. doi: 10.1073/pnas.0408197102
163. Donkor MK, Sarkar A, Li MO. Tgf-β1 produced by activated CD4 + T cells antagonizes T cell surveillance of tumor development. OncoImmunology (2012) 1:162–71. doi: 10.4161/onci.1.2.18481
164. Barilla RM, Diskin B, Caso RC, Lee KB, Mohan N, Buttar C, et al. Specialized dendritic cells induce tumor-promoting IL-10+IL-17+ FoxP3neg regulatory CD4+ T cells in pancreatic carcinoma. Nat Commun (2019) 10:1424. doi: 10.1038/s41467-019-09416-2
165. Ye S-B, Zhang H, Cai T-T, Liu Y-N, Ni J-J, He J, et al. Exosomal miR-24-3p impedes T-cell function by targeting FGF11 and serves as a potential prognostic biomarker for nasopharyngeal carcinoma: Role of exosomal miR-24 in immune regulation of nasopharyngeal carcinoma. J Pathol (2016) 240:329–40. doi: 10.1002/path.4781
166. Li Y, Patel SP, Roszik J, Qin Y. Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment: New approaches for combinational immunotherapy. Front Immunol (2018) 9:1591. doi: 10.3389/fimmu.2018.01591
167. Sawa-Wejksza K, Dudek A, Lemieszek M, Kaławaj K, Kandefer-Szerszeń M. Colon cancer–derived conditioned medium induces differentiation of THP-1 monocytes into a mixed population of M1/M2 cells. Tumor Biol (2018) 40:101042831879788. doi: 10.1177/1010428318797880
168. Walsh SA, Davis TA. Key early proinflammatory signaling molecules encapsulated within circulating exosomes following traumatic injury. J Inflammation (2022) 19:6. doi: 10.1186/s12950-022-00303-0
169. Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci (2011) 7:651–8. doi: 10.7150/ijbs.7.651
170. Islas-Vazquez L, Aguilar-Cazares D, Galicia-Velasco M, Rumbo-Nava U, Meneses-Flores M, Luna-Rivero C, et al. IL-6, NLR, and SII markers and their relation with alterations in CD8+ T-lymphocyte subpopulations in patients treated for lung adenocarcinoma. Biology (2020) 9:376. doi: 10.3390/biology9110376
171. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci (2011) 108:1397–402. doi: 10.1073/pnas.1018898108
172. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol (2016) 12:49–62. doi: 10.1038/nrrheum.2015.169
173. Stephens JM, Pekala PH. Transcriptional repression of the GLUT4 and C/EBP genes in 3T3-L1 adipocytes by tumor necrosis factor-alpha. J Biol Chem (1991) 266:21839–45. doi: 10.1016/S0021-9258(18)54714-1
174. Palacios-Ortega S, Varela-Guruceaga M, Algarabel M, Milagro FI, Martínez JA, de Miguel C. Effect of TNF-alpha on caveolin-1 expression and insulin signaling during adipocyte differentiation and in mature adipocytes. Cell Physiol Biochem (2015) 36:1499–516. doi: 10.1159/000430314
175. Xing H, Northrop JP, Grove JR, Kilpatrick KE, Su J-L, Ringold GM. TNFα-mediated inhibition and reversal of adipocyte differentiation is accompanied by suppressed expression of PPARγ without effects on pref-1 expression*. Endocrinology (1997) 138:2776–83. doi: 10.1210/endo.138.7.5242
176. Zhang L, Han L, He J, Lv J, Pan R, Lv T. A high serum-free fatty acid level is associated with cancer. J Cancer Res Clin Oncol (2020) 146:705–10. doi: 10.1007/s00432-019-03095-8
177. Watkins LR, Goehler LE, Relton J, Brewer MT, Maier SF. Mechanisms of tumor necrosis factor-α (TNF-α) hyperalgesia. Brain Res (1995) 692:244–50. doi: 10.1016/0006-8993(95)00715-3
178. Czeschik JC, Hagenacker T, Schäfers M, Büsselberg D. TNF-α differentially modulates ion channels of nociceptive neurons. Neurosci Lett (2008) 434:293–8. doi: 10.1016/j.neulet.2008.01.070
179. Li C, Palanisamy K, Li X, Yu S, Wang I, Li C, et al. Exosomal tumor necrosis factor-α from hepatocellular cancer cells (Huh-7) promote osteoclast differentiation. J Cell Biochem (2021) 122:1749–60. doi: 10.1002/jcb.30127
180. Fields JK, Günther S, Sundberg EJ. Structural basis of IL-1 family cytokine signaling. Front Immunol (2019) 10:1412. doi: 10.3389/fimmu.2019.01412
181. Gelfo V, Romaniello D, Mazzeschi M, Sgarzi M, Grilli G, Morselli A, et al. Roles of IL-1 in cancer: From tumor progression to resistance to targeted therapies. Int J Mol Sci (2020) 21:6009. doi: 10.3390/ijms21176009
182. Zhang W, Borcherding N, Kolb R. “IL-1 signaling in tumor microenvironment.,”. In: Birbrair A, editor. Tumor microenvironment. advances in experimental medicine and biology. Cham: Springer International Publishing (2020). p. 1–23. doi: 10.1007/978-3-030-38315-2_1
183. Malik A, Kanneganti T-D. Function and regulation of IL-1α in inflammatory diseases and cancer. Immunol Rev (2018) 281:124–37. doi: 10.1111/imr.12615
184. Afonina IS, Müller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: Variations on a common theme. Immunity (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
185. Santoni G, Cardinali C, Morelli M, Santoni M, Nabissi M, Amantini C. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J Neuroinflamm (2015) 12:21. doi: 10.1186/s12974-015-0239-2
186. Huang F-Y, Chan AO-O, Rashid A, Wong DK-H, Seto W-K, Cho C-H, et al. Interleukin-1β increases the risk of gastric cancer through induction of aberrant DNA methylation in a mouse model. Oncol Lett (2016) 11:2919–24. doi: 10.3892/ol.2016.4296
187. Mezzasoma L, Costanzi E, Scarpelli P, Talesa VN, Bellezza I. Extracellular vesicles from human advanced-stage prostate cancer cells modify the inflammatory response of microenvironment-residing cells. Cancers (2019) 11:1276. doi: 10.3390/cancers11091276
188. Chen J, Sun W, Zhang H, Ma J, Xu P, Yu Y, et al. Macrophages reprogrammed by lung cancer microparticles promote tumor development via release of IL-1β. Cell Mol Immunol (2020) 17:1233–44. doi: 10.1038/s41423-019-0313-2
189. Arienti C, Pignatta S, Zanoni M, Zamagni A, Cortesi M, Sarnelli A, et al. High-pressure oxygen rewires glucose metabolism of patient-derived glioblastoma cells and fuels inflammasome response. Cancer Lett (2021) 506:152–66. doi: 10.1016/j.canlet.2021.02.019
190. Wu L, Zhang X, Zhang B, Shi H, Yuan X, Sun Y, et al. Exosomes derived from gastric cancer cells activate NF-κB pathway in macrophages to promote cancer progression. Tumor Biol (2016) 37:12169–80. doi: 10.1007/s13277-016-5071-5
191. Liang M, Chen X, Wang L, Qin L, Wang H, Sun Z, et al. Cancer-derived exosomal TRIM59 regulates macrophage NLRP3 inflammasome activation to promote lung cancer progression. J Exp Clin Cancer Res (2020) 39:176. doi: 10.1186/s13046-020-01688-7
192. Linton SS, Abraham T, Liao J, Clawson GA, Butler PJ, Fox T, et al. Tumor-promoting effects of pancreatic cancer cell exosomes on THP-1-derived macrophages. PloS One (2018) 13:e0206759. doi: 10.1371/journal.pone.0206759
193. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT-H, et al. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol (2011) 12:408–15. doi: 10.1038/ni.2022
194. Laird BJ, McMillan D, Skipworth RJE, Fallon MT, Paval DR, McNeish I, et al. The emerging role of interleukin 1β (IL-1β) in cancer cachexia. Inflammation (2021) 44:1223–8. doi: 10.1007/s10753-021-01429-8
195. Song X, Voronov E, Dvorkin T, Fima E, Cagnano E, Benharroch D, et al. Differential effects of IL-1α and IL-1β on tumorigenicity patterns and invasiveness. J Immunol (2003) 171:6448–56. doi: 10.4049/jimmunol.171.12.6448
196. Fonsatti E, Altomonte M, Coral S, Cattarossi I, Nicotra M, Gasparollo A, et al. Tumour-derived interleukin 1α (IL-1α) up-regulates the release of soluble intercellular adhesion molecule-1 (sICAM-1) by endothelial cells. Br J Cancer (1997) 76:1255–61. doi: 10.1038/bjc.1997.545
197. Mishra D, Banerjee D. “Metabolic interactions between tumor and stromal cells in the tumor microenvironment. In: Banerjee D, Tiwari RK, editors. Tumor microenvironment: Cellular, metabolic and immunologic interactions. advances in experimental medicine and biology. Cham: Springer International Publishing (2021). p. 101–21. doi: 10.1007/978-3-030-83282-7_5
198. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int J Biol Sci (2012) 8:1237–47. doi: 10.7150/ijbs.4989
199. Bode JG, Albrecht U, Häussinger D, Heinrich PC, Schaper F. Hepatic acute phase proteins – regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur J Cell Biol (2012) 91:496–505. doi: 10.1016/j.ejcb.2011.09.008
200. Ishibashi T, Kimura H, Uchida T, Kariyone S, Friese P, Burstein SA. Human interleukin 6 is a direct promoter of maturation of megakaryocytes in vitro. Proc Natl Acad Sci (1989) 86:5953–7. doi: 10.1073/pnas.86.15.5953
201. Gauldie J, Richards C, Harnish D, Lansdorp P, Baumann H. Interferon beta 2/B-cell stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc Natl Acad Sci (1987) 84:7251–5. doi: 10.1073/pnas.84.20.7251
202. Liu C, Chu D, Kalantar-Zadeh K, George J, Young HA, Liu G. Cytokines: From clinical significance to quantification. Adv Sci (2021) 8:2004433. doi: 10.1002/advs.202004433
203. Geiger T, Andus T, Klapproth J, Hirano T, Kishimoto T, Heinrich PC. Induction of rat acute-phase proteins by interleukin 6 in vivo. Eur J Immunol (1988) 18:717–21. doi: 10.1002/eji.1830180510
204. Andus T, Geiger T, Hirano T, Kishimoto T, Heinrich PC. Action of recombinant human interleukin 6, interleukin 1β and tumor necrosis factor α on the mRNA induction of acute-phase proteins. Eur J Immunol (1988) 18:739–46. doi: 10.1002/eji.1830180513
205. Duvigneau JC, Luís A, Gorman AM, Samali A, Kaltenecker D, Moriggl R, et al. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine (2019) 124:154577. doi: 10.1016/j.cyto.2018.10.018
206. Germolec DR, Shipkowski KA, Frawley RP, Evans E. Markers of inflammation. Methods Mol Biol (2018) 1803:57–79. doi: 10.1007/978-1-4939-8549-4_5
207. Rumba R, Cipkina S, Cukure F, Vanags A. Systemic and local inflammation in colorectal cancer. Acta Med Litu (2019) 25:185–96. doi: 10.6001/actamedica.v25i4.3929
208. Proctor MJ, McMillan DC, Morrison DS, Fletcher CD, Horgan PG, Clarke SJ. A derived neutrophil to lymphocyte ratio predicts survival in patients with cancer. Br J Cancer (2012) 107:695–9. doi: 10.1038/bjc.2012.292
209. Schiefer S, Wirsik NM, Kalkum E, Seide SE, Nienhüser H, Müller B, et al. Systematic review of prognostic role of blood cell ratios in patients with gastric cancer undergoing surgery. Diagnostics (2022) 12:593. doi: 10.3390/diagnostics12030593
210. Laird BJA, Fallon M, Hjermstad MJ, Tuck S, Kaasa S, Klepstad P, et al. Quality of life in patients with advanced cancer: Differential association with performance status and systemic inflammatory response. J Clin Oncol (2016) 34:2769–75. doi: 10.1200/JCO.2015.65.7742
211. McMillan DC. The systemic inflammation-based Glasgow prognostic score: A decade of experience in patients with cancer. Cancer Treat Rev (2013) 39:534–40. doi: 10.1016/j.ctrv.2012.08.003
212. Yamamura K, Sugimoto H, Kanda M, Yamada S, Nomoto S, Nakayama G, et al. Comparison of inflammation-based prognostic scores as predictors of tumor recurrence in patients with hepatocellular carcinoma after curative resection. J Hepato-Biliary-Pancreat Sci (2014) 21:682–8. doi: 10.1002/jhbp.114
213. Kurahara H, Maemura K, Mataki Y, Sakoda M, Iino S, Hiwatashi K, et al. Prognostication by inflammation-based score in patients with locally advanced pancreatic cancer treated with chemoradiotherapy. Pancreatology (2015) 15:688–93. doi: 10.1016/j.pan.2015.09.015
214. Crumley ABC, McMillan DC, McKernan M, McDonald AC, Stuart RC. Evaluation of an inflammation-based prognostic score in patients with inoperable gastro-oesophageal cancer. Br J Cancer (2006) 94:637–41. doi: 10.1038/sj.bjc.6602998
215. Morris-Stiff G, Gomez D, Prasad KR. C-reactive protein in liver cancer surgery. Eur J Surg Oncol EJSO (2008) 34:727–9. doi: 10.1016/j.ejso.2008.01.016
216. Mauer J, Denson JL, Brüning JC. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol (2015) 36:92–101. doi: 10.1016/j.it.2014.12.008
217. Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol (2016) 17:230–40. doi: 10.1038/ni.3384
218. Inatsu A, Kinoshita M, Nakashima H, Shimizu J, Saitoh D, Tamai S, et al. Novel mechanism of c-reactive protein for enhancing mouse liver innate immunity. Hepatology (2009) 49:2044–54. doi: 10.1002/hep.22888
219. Van Vré EA, Bult H, Hoymans VY, Van Tendeloo VFI, Vrints CJ, Bosmans JM. Human c-reactive protein activates monocyte-derived dendritic cells and induces dendritic cell-mediated T-cell activation. Arterioscler Thromb Vasc Biol (2008) 28:511–8. doi: 10.1161/ATVBAHA.107.157016
220. Rasmussen LJH, Schultz M, Gaardsting A, Ladelund S, Garred P, Iversen K, et al. Inflammatory biomarkers and cancer: CRP and suPAR as markers of incident cancer in patients with serious nonspecific symptoms and signs of cancer: Biomarkers in the DOC. Int J Cancer (2017) 141:191–9. doi: 10.1002/ijc.30732
221. Pletnikoff PP, Laukkanen JA, Tuomainen T-P, Kauhanen J, Rauramaa R, Ronkainen K, et al. Cardiorespiratory fitness, C-reactive protein and lung cancer risk: A prospective population-based cohort study. Eur J Cancer (2015) 51:1365–70. doi: 10.1016/j.ejca.2015.04.020
222. Komrokji RS, Corrales-Yepez M, Kharfan-Dabaja MA, Al Ali NH, Padron E, Rollison DE, et al. Hypoalbuminemia is an independent prognostic factor for overall survival in myelodysplastic syndromes. Am J Hematol (2012) 87:1006–9. doi: 10.1002/ajh.23303
223. Ataseven B, du Bois A, Reinthaller A, Traut A, Heitz F, Aust S, et al. Pre-operative serum albumin is associated with post-operative complication rate and overall survival in patients with epithelial ovarian cancer undergoing cytoreductive surgery. Gynecol Oncol (2015) 138:560–5. doi: 10.1016/j.ygyno.2015.07.005
224. Tabata F, Wada Y, Kawakami S, Miyaji K. Serum albumin redox states: More than oxidative stress biomarker. Antioxidants (2021) 10:503. doi: 10.3390/antiox10040503
225. Chojkier M. Inhibition of albumin synthesis in chronic diseases: Molecular mechanisms. J Clin Gastroenterol (2005) 39:S143–6. doi: 10.1097/01.mcg.0000155514.17715.39
226. Zhang W. Albumin selectively inhibits TNFα-induced expression of vascular cell adhesion molecule-1 in human aortic endothelial cells. Cardiovasc Res (2002) 55:820–9. doi: 10.1016/S0008-6363(02)00492-3
227. Tijssen MR, Ghevaert C. Transcription factors in late megakaryopoiesis and related platelet disorders. J Thromb Haemost (2013) 11:593–604. doi: 10.1111/jth.12131
228. Shameer K, Denny JC, Ding K, Jouni H, Crosslin DR, de Andrade M, et al. A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum Genet (2014) 133:95–109. doi: 10.1007/s00439-013-1355-7
229. Korniluk A, Koper-Lenkiewicz OM, Kamińska J, Kemona H, Dymicka-Piekarska V. Mean platelet volume (MPV): New perspectives for an old marker in the course and prognosis of inflammatory conditions. Mediators Inflammation (2019) 2019:1–14. doi: 10.1155/2019/9213074
230. Shen X-B, Wang Y, Shan B-J, Lin L, Hao L, Liu Y, et al. Prognostic significance of platelet-To-Lymphocyte ratio (PLR) and mean platelet volume (MPV) during etoposide-based first-line treatment in small cell lung cancer patients. Cancer Manag Res (2019) 11:8965–75. doi: 10.2147/CMAR.S215361
231. Rein-Smith CM, Anderson NW, Farrell DH. Differential regulation of fibrinogen γ chain splice isoforms by interleukin-6. Thromb Res (2013) 131:89–93. doi: 10.1016/j.thromres.2012.09.017
232. Duan HO, Simpson-Haidaris PJ. Functional analysis of interleukin 6 response elements (IL-6REs) on the human γ-fibrinogen promoter. J Biol Chem (2003) 278:41270–81. doi: 10.1074/jbc.M304210200
233. Fuller GM, Zhang Z. Transcriptional control mechanism of fibrinogen gene expression. Ann N Y Acad Sci (2006) 936:469–79. doi: 10.1111/j.1749-6632.2001.tb03534.x
234. Zhang Y, Xiao G. Prognostic significance of the ratio of fibrinogen and albumin in human malignancies: A meta-analysis. Cancer Manag Res (2019) 11:3381–93. doi: 10.2147/CMAR.S198419
235. Kubota K, Ito R, Narita N, Tanaka Y, Furudate K, Akiyama N, et al. Utility of prognostic nutritional index and systemic immune-inflammation index in oral cancer treatment. BMC Cancer (2022) 22:368. doi: 10.1186/s12885-022-09439-x
236. Nozoe T, Matono R, Ijichi H, Ohga T, Ezaki T. Glasgow Prognostic score (GPS) can be a useful indicator to determine prognosis of patients with colorectal carcinoma. Int Surg (2014) 99:512–7. doi: 10.9738/INTSURG-D-13-00118.1
237. Aurello P, Tierno SM, Berardi G, Tomassini F, Magistri P, D’Angelo F, et al. Value of preoperative inflammation-based prognostic scores in predicting overall survival and disease-free survival in patients with gastric cancer. Ann Surg Oncol (2014) 21:1998–2004. doi: 10.1245/s10434-014-3533-9
238. Hoshimoto S, Hishinuma S, Shirakawa H, Tomikawa M, Ozawa I, Ogata Y. Validation and clinical usefulness of pre- and postoperative systemic inflammatory parameters as prognostic markers in patients with potentially resectable pancreatic cancer. Pancreatology (2020) 20:239–46. doi: 10.1016/j.pan.2019.12.004
239. Hu X, Wang Y, Yang W-X, Dou W-C, Shao Y-X, Li X. Modified Glasgow prognostic score as a prognostic factor for renal cell carcinomas: A systematic review and meta-analysis. Cancer Manag Res (2019) 11:6163–73. doi: 10.2147/CMAR.S208839
240. Proctor MJ, Morrison DS, Talwar D, Balmer SM, O’Reilly DSJ, Foulis AK, et al. An inflammation-based prognostic score (mGPS) predicts cancer survival independent of tumour site: A Glasgow inflammation outcome study. Br J Cancer (2011) 104:726–34. doi: 10.1038/sj.bjc.6606087
241. Fan H, Shao Z-Y, Xiao Y-Y, Xie Z-H, Chen W, Xie H, et al. Comparison of the Glasgow prognostic score (GPS) and the modified Glasgow prognostic score (mGPS) in evaluating the prognosis of patients with operable and inoperable non-small cell lung cancer. J Cancer Res Clin Oncol (2016) 142:1285–97. doi: 10.1007/s00432-015-2113-0
242. Sun F, Peng H-X, Gao Q-F, Li S-Q, Zhang J, Chen Q-G, et al. Preoperative circulating FPR and CCF score are promising biomarkers for predicting clinical outcome of stage II–III colorectal cancer patients. Cancer Manag Res (2018) 10:2151–61. doi: 10.2147/CMAR.S167398
243. Martin D, Rödel F, Balermpas P, Winkelmann R, Fokas E, Rödel C. C-reactive protein-to-Albumin ratio as prognostic marker for anal squamous cell carcinoma treated with chemoradiotherapy. Front Oncol (2019) 9:1200. doi: 10.3389/fonc.2019.01200
244. Zhou Q, Li X. C-reactive protein to albumin ratio in colorectal cancer: A meta-analysis of prognostic value. Dose-Response (2019) 17:155932581988981. doi: 10.1177/1559325819889814
245. Xiao Y, Xie Z, Shao Z, Chen W, Xie H, Qin G, et al. Neutrophil and lymphocyte counts at diagnosis are associated with overall survival of pancreatic cancer: A retrospective cohort study. Med (Baltimore) (2016) 95:e5024. doi: 10.1097/MD.0000000000005024
246. Chen L, Wang X, Shu J, Xu S, Wu Q, Yu Y. Diagnostic value of serum D-dimer, CA 125, and neutrophil-to-lymphocyte ratio in differentiating ovarian cancer and endometriosis. Int J Gynecol Obstet (2019) 147:212–8. doi: 10.1002/ijgo.12949
247. Wang J, Zhang F, Jiang F, Hu L, Chen J, Wang Y. Distribution and reference interval establishment of neutral-to-lymphocyte ratio (NLR), lymphocyte-to-monocyte ratio (LMR), and platelet-to-lymphocyte ratio (PLR) in Chinese healthy adults. J Clin Lab Anal (2021) 35:e23935. doi: 10.1002/jcla.23935
248. Cho U, Park HS, Im SY, Yoo CY, Jung JH, Suh YJ, et al. Prognostic value of systemic inflammatory markers and development of a nomogram in breast cancer. PloS One (2018) 13:e0200936. doi: 10.1371/journal.pone.0200936
249. Song S, Li C, Li S, Gao H, Lan X, Xue Y. Derived neutrophil to lymphocyte ratio and monocyte to lymphocyte ratio may be better biomarkers for predicting overall survival of patients with advanced gastric cancer. OncoTargets Ther (2017) 10:3145–54. doi: 10.2147/OTT.S138039
250. Troppan K, Deutsch A, Gerger A, Stojakovic T, Beham-Schmid C, Wenzl K, et al. The derived neutrophil to lymphocyte ratio is an independent prognostic factor in patients with diffuse large B-cell lymphoma. Br J Cancer (2014) 110:369–74. doi: 10.1038/bjc.2013.763
251. Yang T, Hao L, Yang X, Luo C, Wang G, Lin Cai C, et al. Prognostic value of derived neutrophil-to-lymphocyte ratio (dNLR) in patients with non-small cell lung cancer receiving immune checkpoint inhibitors: a meta-analysis. BMJ Open (2021) 11:e049123. doi: 10.1136/bmjopen-2021-049123
252. Grenader T, Nash S, Adams R, Kaplan R, Fisher D, Maughan T, et al. Derived neutrophil lymphocyte ratio is predictive of survival from intermittent therapy in advanced colorectal cancer: a post hoc analysis of the MRC COIN study. Br J Cancer (2016) 114:612–5. doi: 10.1038/bjc.2016.23
253. Xu Z, Zhang J, Zhong Y, Mai Y, Huang D, Wei W, et al. Predictive value of the monocyte-to-lymphocyte ratio in the diagnosis of prostate cancer. Med (Baltimore) (2021) 100:e27244. doi: 10.1097/MD.0000000000027244
254. Jakubowska K, Koda M, Grudzińska M, Kańczuga-Koda L, Famulski W. Monocyte-to-lymphocyte ratio as a prognostic factor in peripheral whole blood samples of colorectal cancer patients. World J Gastroenterol (2020) 26:4639–55. doi: 10.3748/wjg.v26.i31.4639
255. Huszno J, Kołosza Z, Mrochem-Kwarciak J, Zajusz A. Prognostic value of the neutrophil-lymphocyte, platelet-lymphocyte, and monocyte-lymphocyte ratios in Male breast cancer patients. Oncology (2020) 98:487–92. doi: 10.1159/000505627
256. Luo H, He L, Zhang G, Yu J, Chen Y, Yin H, et al. Normal reference intervals of neutrophil-To-Lymphocyte ratio, platelet-To-Lymphocyte ratio, lymphocyte-To-Monocyte ratio, and systemic immune inflammation index in healthy adults: a Large multi-center study from Western China. Clin Lab (2019) 65. doi: 10.7754/Clin.Lab.2018.180715
257. Chen J-H, Zhai E-T, Yuan Y-J, Wu K-M, Xu J-B, Peng J-J, et al. Systemic immune-inflammation index for predicting prognosis of colorectal cancer. World J Gastroenterol (2017) 23:6261. doi: 10.3748/wjg.v23.i34.6261
258. Deng M, Ma X, Liang X, Zhu C, Wang M. Are pretreatment neutrophil-lymphocyte ratio and platelet-lymphocyte ratio useful in predicting the outcomes of patients with small-cell lung cancer? Oncotarget (2017) 8:37200–7. doi: 10.18632/oncotarget.16553
259. Eraslan E, Adas YG, Yildiz F, Gulesen AI, Karacin C, Arslan UY. Systemic immune-inflammation index (SII) predicts pathological complete response to neoadjuvant chemoradiotherapy in locally advanced rectal cancer. J Coll Phys Surg Pak (2021) 31:399–404. doi: 10.29271/jcpsp.2021.04.399
260. Guo W, Cai S, Zhang F, Shao F, Zhang G, Zhou Y, et al. Systemic immune-inflammation index (SII) is useful to predict survival outcomes in patients with surgically resected non-small cell lung cancer. Thorac Cancer (2019) 10:761–8. doi: 10.1111/1759-7714.12995
261. Atasever Akkas E, Yucel B. Prognostic value of systemic ımmune ınflammation ındex in patients with laryngeal cancer. Eur Arch Otorhinolaryngol (2021) 278:1945–55. doi: 10.1007/s00405-021-06798-2
262. Rice SJ, Belani CP. Diversity and heterogeneity of immune states in non-small cell lung cancer and small cell lung cancer. PloS One (2021) 16:e0260988. doi: 10.1371/journal.pone.0260988
263. Zinellu A, Collu C, Nasser M, Paliogiannis P, Mellino S, Zinellu E, et al. The aggregate index of systemic inflammation (AISI): A novel prognostic biomarker in idiopathic pulmonary fibrosis. J Clin Med (2021) 10:4134. doi: 10.3390/jcm10184134
264. Zuffa M, Kubancok J, Rusnák I, Mensatoris K, Horváth A. Early paraneoplastic syndrome in medical oncology: clinicopathological analysis of 1694 patients treated over 20 years. Neoplasma (1984) 31:231–6.
265. Sardiña González C, Martínez Vivero C, López Castro J. Paraneoplastic syndromes review: The great forgotten ones. Crit Rev Oncol Hematol (2022) 174:103676. doi: 10.1016/j.critrevonc.2022.103676
266. Mitroulis I, Kalafati L, Bornhäuser M, Hajishengallis G, Chavakis T. Regulation of the bone marrow niche by inflammation. Front Immunol (2020) 11:1540. doi: 10.3389/fimmu.2020.01540
267. Chiba Y, Mizoguchi I, Hasegawa H, Ohashi M, Orii N, Nagai T, et al. Regulation of myelopoiesis by proinflammatory cytokines in infectious diseases. Cell Mol Life Sci (2018) 75:1363–76. doi: 10.1007/s00018-017-2724-5
268. Helbling PM, Piñeiro-Yáñez E, Gerosa R, Boettcher S, Al-Shahrour F, Manz MG, et al. Global transcriptomic profiling of the bone marrow stromal microenvironment during postnatal development, aging, and inflammation. Cell Rep (2019) 29:3313–3330.e4. doi: 10.1016/j.celrep.2019.11.004
269. Wildes TJ, DiVita Dean B, Flores CT. Myelopoiesis during solid cancers and strategies for immunotherapy. Cells (2021) 10:968. doi: 10.3390/cells10050968
270. Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol (2007) 178:6867–75. doi: 10.4049/jimmunol.178.11.6867
271. Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, MacArthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature (2010) 466:829–34. doi: 10.1038/nature09262
272. Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell (2011) 20:661–73. doi: 10.1016/j.ccr.2011.10.012
273. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity (2010) 32:790–802. doi: 10.1016/j.immuni.2010.05.010
274. Wu W-C, Sun H-W, Chen H-T, Liang J, Yu X-J, Wu C, et al. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc Natl Acad Sci (2014) 111:4221–6. doi: 10.1073/pnas.1320753111
275. Al Sayed MF, Amrein MA, Bührer ED, Huguenin A-L, Radpour R, Riether C, et al. T-cell–secreted TNFα induces emergency myelopoiesis and myeloid-derived suppressor cell differentiation in cancer. Cancer Res (2019) 79:346–59. doi: 10.1158/0008-5472.CAN-17-3026
276. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest (2012) 122:4094–104. doi: 10.1172/JCI64115
277. Chiodoni C, Cancila V, Renzi TA, Perrone M, Tomirotti AM, Sangaletti S, et al. Transcriptional profiles and stromal changes reveal bone marrow adaptation to early breast cancer in association with deregulated circulating microRNAs. Cancer Res (2019). doi: 10.1158/0008-5472.CAN-19-1425
278. Strauss L, Guarneri V, Gennari A, Sica A. Implications of metabolism-driven myeloid dysfunctions in cancer therapy. Cell Mol Immunol (2021) 18:829–41. doi: 10.1038/s41423-020-00556-w
279. Yamauchi Y, Rogers MA. Sterol metabolism and transport in atherosclerosis and cancer. Front Endocrinol (2018) 9:509. doi: 10.3389/fendo.2018.00509
280. Vladimirov S, Gojkovic T, Zeljkovic A, Jelic-Ivanovic Z, Zeljkovic D, Antonic T, et al. Can non-cholesterol sterols indicate the presence of specific dysregulation of cholesterol metabolism in patients with colorectal cancer? Biochem Pharmacol (2022) 196:114595. doi: 10.1016/j.bcp.2021.114595
281. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab (2018) 28:87–103.e6. doi: 10.1016/j.cmet.2018.04.022
282. Bao Y, Liu Z, Guo M, Li B, Sun X, Wang L. Extramedullary hematopoiesis secondary to malignant solid tumors: a case report and literature review. Cancer Manag Res (2018) 10:1461–70. doi: 10.2147/CMAR.S161746
283. Levy L, Mishalian I, Bayuch R, Zolotarov L, Michaeli J, Fridlender ZG. Splenectomy inhibits non-small cell lung cancer growth by modulating anti-tumor adaptive and innate immune response. OncoImmunology (2015) 4:e998469. doi: 10.1080/2162402X.2014.998469
284. Cadili A, de Gara C. Complications of splenectomy. Am J Med (2008) 121:371–5. doi: 10.1016/j.amjmed.2008.02.014
285. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
286. Yang Y, Li C, Liu T, Dai X, Bazhin AV. Myeloid-derived suppressor cells in tumors: From mechanisms to antigen specificity and microenvironmental regulation. Front Immunol (2020) 11:1371. doi: 10.3389/fimmu.2020.01371
287. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol (2011) 12:489–95. doi: 10.1016/S1470-2045(10)70218-7
288. Ni J, Zhang L. Cancer cachexia: Definition, staging, and emerging treatments. Cancer Manag Res (2020) 12:5597–605. doi: 10.2147/CMAR.S261585
289. Evans WJ, Morley JE, Argilés J, Bales C, Baracos V, Guttridge D, et al. Cachexia: A new definition. Clin Nutr (2008) 27:793–9. doi: 10.1016/j.clnu.2008.06.013
290. Biswas AK, Acharyya S. Cancer-associated cachexia: A systemic consequence of cancer progression. Annu Rev Cancer Biol (2020) 4:391–411. doi: 10.1146/annurev-cancerbio-030419-033642
291. Aguilar-Cazares D, Chavez-Dominguez R, Carlos-Reyes A, Lopez-Camarillo C, Hernadez de la Cruz ON, Lopez-Gonzalez JS. Contribution of angiogenesis to inflammation and cancer. Front Oncol (2019) 9:1399. doi: 10.3389/fonc.2019.01399
292. Ippolito L, Morandi A, Giannoni E, Chiarugi P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem Sci (2019) 44:153–66. doi: 10.1016/j.tibs.2018.10.011
293. Ferguson BS, Rogatzki MJ, Goodwin ML, Kane DA, Rightmire Z, Gladden LB. Lactate metabolism: historical context, prior misinterpretations, and current understanding. Eur J Appl Physiol (2018) 118:691–728. doi: 10.1007/s00421-017-3795-6
294. Choi SYC, Collins CC, Gout PW, Wang Y. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol (2013) 230:350–5. doi: 10.1002/path.4218
295. Rohm M, Zeigerer A, Machado J, Herzig S. Energy metabolism in cachexia. EMBO Rep (2019) 20:e47258. doi: 10.15252/embr.201847258
296. Noguchi Y, Yoshikawa T, Marat D, Doi C, Makino T, Fukuzawa K, et al. Insulin resistance in cancer patients is associated with enhanced tumor necrosis factor-α expression in skeletal muscle. Biochem Biophys Res Commun (1998) 253:887–92. doi: 10.1006/bbrc.1998.9794
297. Asp ML, Tian M, Wendel AA, Belury MA. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int J Cancer (2010) 126:756–63. doi: 10.1002/ijc.24784
298. Baltgalvis KA, Berger FG, Peña MMO, Davis JM, White JP, Carson JA. Muscle wasting and interleukin-6-induced atrogin-I expression in the cachectic apc min/+ mouse. Pflüg Arch - Eur J Physiol (2009) 457:989–1001. doi: 10.1007/s00424-008-0574-6
299. Chen M-C, Hsu W-L, Hwang P-A, Chen Y-L, Chou T-C. Combined administration of fucoidan ameliorates tumor and chemotherapy-induced skeletal muscle atrophy in bladder cancer-bearing mice. Oncotarget (2016) 7:51608–18. doi: 10.18632/oncotarget.9958
300. Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, Sin KWT, et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat Commun (2017) 8:589. doi: 10.1038/s41467-017-00726-x
301. Qiu L, Chen W, Wu C, Yuan Y, Li Y. Exosomes of oral squamous cell carcinoma cells containing miR-181a-3p induce muscle cell atrophy and apoptosis by transmissible endoplasmic reticulum stress signaling. Biochem Biophys Res Commun (2020) 533:831–7. doi: 10.1016/j.bbrc.2020.09.066
302. Li L, Liu H, Tao W, Wen S, Fu X, Yu S. Pharmacological inhibition of HMGB1 prevents muscle wasting. Front Pharmacol (2021) 12:731386. doi: 10.3389/fphar.2021.731386
303. Zhang W, Sun W, Gu X, Miao C, Feng L, Shen Q, et al. GDF-15 in tumor-derived exosomes promotes muscle atrophy via bcl-2/caspase-3 pathway. Cell Death Discovery (2022) 8:162. doi: 10.1038/s41420-022-00972-z
304. Masi T, Patel BM. Altered glucose metabolism and insulin resistance in cancer-induced cachexia: a sweet poison. Pharmacol Rep (2021) 73:17–30. doi: 10.1007/s43440-020-00179-y
305. Sacks H, Symonds ME. Anatomical locations of human brown adipose tissue. Diabetes (2013) 62:1783–90. doi: 10.2337/db12-1430
306. Weber BZC, Arabaci DH, Kir S. Metabolic reprogramming in adipose tissue during cancer cachexia. Front Oncol (2022) 12:848394. doi: 10.3389/fonc.2022.848394
307. Bolsoni-Lopes A, Alonso-Vale MIC. Lipolysis and lipases in white adipose tissue – an update. Arch Endocrinol Metab (2015) 59:335–42. doi: 10.1590/2359-3997000000067
308. Dalal S. Lipid metabolism in cancer cachexia. Ann Palliat Med (2019) 8:13–23. doi: 10.21037/apm.2018.10.01
309. Das SK, Hoefler G. The role of triglyceride lipases in cancer associated cachexia. Trends Mol Med (2013) 19:292–301. doi: 10.1016/j.molmed.2013.02.006
310. Shaw JHF, Wolfe RR. Fatty acid and glycerol kinetics in septic patients and in patients with gastrointestinal cancer: The response to glucose infusion and parenteral feeding. Ann Surg (1987) 205:368–76. doi: 10.1097/00000658-198704000-00005
311. Fukawa T, Yan-Jiang BC, Min-Wen JC, Jun-Hao ET, Huang D, Qian C-N, et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med (2016) 22:666–71. doi: 10.1038/nm.4093
312. Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab (2014) 20:433–47. doi: 10.1016/j.cmet.2014.06.011
313. de Matos-Neto EM, Lima JDCC, de Pereira WO, Figuerêdo RG, Riccardi DM dos R, Radloff K, et al. Systemic inflammation in cachexia – is tumor cytokine expression profile the culprit? Front Immunol (2015) 6:629. doi: 10.3389/fimmu.2015.00629
314. Zhang H, Zhu L, Bai M, Liu Y, Zhan Y, Deng T, et al. Exosomal circRNA derived from gastric tumor promotes white adipose browning by targeting the miR-133/PRDM16 pathway. Int J Cancer (2019) 144:2501–15. doi: 10.1002/ijc.31977
315. Wang S, Li X, Xu M, Wang J, Zhao RC. Reduced adipogenesis after lung tumor exosomes priming in human mesenchymal stem cells via TGFβ signaling pathway. Mol Cell Biochem (2017) 435:59–66. doi: 10.1007/s11010-017-3056-3
316. Liu Y, Wang M, Deng T, Liu R, Ning T, Bai M, et al. Exosomal miR-155 from gastric cancer induces cancer-associated cachexia by suppressing adipogenesis and promoting brown adipose differentiation via C/EPBβ. Cancer Biol Med (2022) 19. doi: 10.20892/j.issn.2095-3941.2021.0220
317. Argilés JM, Stemmler B, López-Soriano FJ, Busquets S. Inter-tissue communication in cancer cachexia. Nat Rev Endocrinol (2019) 15:9–20. doi: 10.1038/s41574-018-0123-0
318. Olson B, Diba P, Korzun T, Marks DL. Neural mechanisms of cancer cachexia. Cancers (2021) 13:3990. doi: 10.3390/cancers13163990
319. Gwosdow AR, Kumar MS, Bode HH. Interleukin 1 stimulation of the hypothalamic-pituitary-adrenal axis. Am J Physiol-Endocrinol Metab (1990) 258:E65–70. doi: 10.1152/ajpendo.1990.258.1.E65
320. Gądek-Michalska A, Bugajski J. Interleukin-1 (IL-1) in stress-induced activation of limbic-hypothalamic-pituitary adrenal axis. Pharmacol Rep (2010) 62:969–82. doi: 10.1016/S1734-1140(10)70359-5
321. Maccio A, Sanna E, Neri M, Oppi S, Madeddu C. Cachexia as evidence of the mechanisms of resistance and tolerance during the evolution of cancer disease. Int J Mol Sci (2021) 22:2890. doi: 10.3390/ijms22062890
322. Noguchi Y, Yoshikawa T, Matsumoto A, Svaninger G, Gelin J. Are cytokines possible mediators of cancer cachexia? Surg Today (1996) 26:467–75. doi: 10.1007/BF00311551
323. Jafri SHR, Previgliano C, Khandelwal K, Shi R. Cachexia index in advanced non-Small-Cell lung cancer patients. Clin Med Insights Oncol (2015) 9. doi: 10.4137/CMO.S30891
324. Kim EY, Kim YS, Seo J-Y, Park I, Ahn HK, Jeong YM, et al. The relationship between sarcopenia and systemic inflammatory response for cancer cachexia in small cell lung cancer. PloS One (2016) 11:e0161125. doi: 10.1371/journal.pone.0161125
325. Barker T, Fulde G, Moulton B, Nadauld LD, Rhodes T. An elevated neutrophil-to-lymphocyte ratio associates with weight loss and cachexia in cancer. Sci Rep (2020) 10:7535. doi: 10.1038/s41598-020-64282-z
326. Zappavigna S, Cossu AM, Grimaldi A, Bocchetti M, Ferraro GA, Nicoletti GF, et al. Anti-inflammatory drugs as anticancer agents. Int J Mol Sci (2020) 21:2605. doi: 10.3390/ijms21072605
327. Chiossone L, Vitale C, Cottalasso F, Moretti S, Azzarone B, Moretta L, et al. Molecular analysis of the methylprednisolone-mediated inhibition of NK-cell function: evidence for different susceptibility of IL-2– versus IL-15–activated NK cells. Blood (2007) 109:3767–75. doi: 10.1182/blood-2006-07-037846
Keywords: cancer, tumor microenvironment, inflammatory mediators, cytokines, systemic inflammation, paraneoplastic syndromes, systemic immune-inflammatory markers
Citation: Aguilar-Cazares D, Chavez-Dominguez R, Marroquin-Muciño M, Perez-Medina M, Benito-Lopez JJ, Camarena A, Rumbo-Nava U and Lopez-Gonzalez JS (2022) The systemic-level repercussions of cancer-associated inflammation mediators produced in the tumor microenvironment. Front. Endocrinol. 13:929572. doi: 10.3389/fendo.2022.929572
Received: 27 April 2022; Accepted: 01 August 2022;
Published: 22 August 2022.
Edited by:
Yang Mi, Henan Key Laboratory for Helicobacter pylori & Microbiota and Gastrointestinal Cancer, ChinaReviewed by:
Miguel Luiz Batista Júnior, Boston Medical Center, United StatesLianjun Zhang, Suzhou Institute of Systems Medicine (ISM), China
Colt Egelston, City of Hope National Medical Center, United States
Copyright © 2022 Aguilar-Cazares, Chavez-Dominguez, Marroquin-Muciño, Perez-Medina, Benito-Lopez, Camarena, Rumbo-Nava and Lopez-Gonzalez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jose S. Lopez-Gonzalez, c2xvcGV6Z29uemFsZXpAeWFob28uY29t
†These authors share first authorship