 Xuejiao Wei
Xuejiao Wei Yue Hou1
Yue Hou1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 25 July 2022
Sec. Renal Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.927329
This article is part of the Research TopicThe role of Oxidative Stress and Systemic Inflammation in Diabetes and Chronic Kidney DiseaseView all 6 articles
Renal fibrosis is the result of renal tissue damage and repair response disorders. If fibrosis is not effectively blocked, it causes loss of renal function, leading to chronic renal failure. Metabolic reprogramming, which promotes cell proliferation by regulating cellular energy metabolism, is considered a unique tumor cell marker. The transition from oxidative phosphorylation to aerobic glycolysis is a major feature of renal fibrosis. Hypoxia-inducible factor-1 α (HIF-1α), a vital transcription factor, senses oxygen status, induces adaptive changes in cell metabolism, and plays an important role in renal fibrosis and glucose metabolism. This review focuses on the regulation of proteins related to aerobic glycolysis by HIF-1α and attempts to elucidate the possible regulatory mechanism underlying the effects of HIF-1α on glucose metabolism during renal fibrosis, aiming to provide new ideas for targeted metabolic pathway intervention in renal fibrosis.
Chronic kidney disease (CKD) is a global public health concern. With an increase in hypertension and diabetes, the prevalence of CKD is increasing every year (1). Renal fibrosis is a common pathological change in the transition from CKD to end-stage renal disease caused by different pathological changes, mainly manifesting as glomerulosclerosis, renal tubular atrophy, and renal interstitial fibrosis, which are closely related to renal deterioration (2). Currently, there is no effective treatment for renal fibrosis; thus, it is of great clinical significance to find a way to improve or reverse renal fibrosis. Abnormal energy metabolism, often considered a marker of cancer cells, has attracted considerable attention in this research area. In contrast to normal cells, which derive most of the energy from mitochondrial oxidative phosphorylation (OXPHOS), cancer cells rely on aerobic glycolysis as the main energy source (3). This metabolic pattern, similar to that in tumor cells, has also been confirmed in damaged kidneys, and aerobic glycolysis may be a possible mechanism driving renal fibrosis (4). The fibrotic kidney undergoes metabolic reprogramming to adapt, and activating aerobic glycolysis is a way for kidney cells to cope with metabolic stress (5).
Hypoxia-inducible factor-1 α (HIF-1α) is the main regulator of cell responses under hypoxia. The α subunit of HIF-1 binds to the β subunit to form a dimer, which then binds to hypoxia response elements (HRE) in promoters or enhancer regions of downstream target genes to initiate gene transcription (6). Transforming growth factor-β1 (TGF-β1) is the main cytokine that promotes renal fibrosis, and inhibition of TGF-β1 or its downstream signaling pathway can significantly inhibit renal fibrosis (7). The HIF-1α transcription complex induces TGF-β1 activation (8). Subsequently, TGF-β1 stabilizes HIF-1α by selectively inhibiting proline hydroxylase-2 (9). A mutual regulatory relationship between HIF-1α and TGF-β1 promotes renal fibrosis. Furthermore, studies have shown that HIF-1α is an important transcription factor that inhibits mitochondrial OXPHOS and activates the glycolysis pathway, regulating aerobic glycolysis by transcriptional regulation of a series of glycolytic genes (10).
In this review, we summarized the latest knowledge on the mechanisms of action and function of HIF-1α in regulating glucose metabolism reprogramming during renal fibrosis, focusing on the regulation of glycolysis-related enzymes. We hope to provide a new strategy for targeted therapy of renal fibrosis.
Renal fibrosis is a common result of various CKDs, regardless of the underlying cause. Currently, it is believed that renal fibrosis is not merely a static and substantial scar, but a dynamic and evolving process involving multiple complex signaling pathways and intercellular crosstalk. The main manifestations are the release of inflammatory cytokines, accumulation of extracellular matrix (ECM), transformation of renal intrinsic cells to mesenchymal cells, activation of fibrosis-related signaling pathways, and renal microvascular lesions (11, 12). Although a multitude of experimental data are available regarding targeting the above pathogenic processes, the application of anti-fibrotic therapeutic strategies to patient treatment still needs further exploration. Finding more effective and targeted methods to improve renal fibrosis is of great significance.
The change in energy metabolism is increasingly regarded as an important pathogenic mechanism in CKD, and normal energy metabolism is the basis for maintaining the physiology of the kidney (13). Although the kidney is not a metabolic organ, it requires a large amount of energy to filter waste, regulate electrolyte and fluid balance, and perform other processes (14). On the one hand, abnormal energy metabolism leads to renal fibrosis. On the other hand, kidney damage alters energy metabolism. Thus, abnormal energy metabolism and renal fibrosis cause and affect each other, forming a vicious cycle (13).
Most cells rely on mitochondrial OXPHOS for energy production, whereas glycolysis occurs only when oxygen is scarce. Glycolysis is the production of adenosine triphosphate (ATP) through a series of intercellular enzymatic reactions that require ten enzymes, including three rate-limiting enzymes, hexokinases (HKs), phosphofructose kinase 1 (PFK1), and pyruvate kinase M2(PKM2), which catalyze irreversible reactions (15). Under the action of ten enzymes, glucose is gradually degraded to pyruvate, resulting in the formation of two ATP molecules. Pyruvate is reduced to lactate by lactate dehydrogenase-A(LDHA) when the oxygen supply is insufficient. In the presence of oxygen, pyruvate is converted to acetyl-CoA, which then enters the tricarboxylic acid cycle (TCA cycle) to produce nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which enters the electron transport chain (ETC) to promote OXPHOS to produce ATP (15). However, this ubiquitous pathway does not seem to be applicable to tumor cells. Warburg found that even under normoxia, cancer cells could not use mitochondrial OXPHOS to obtain energy, and that pyruvate could not enter the TCA cycle but was instead degraded by LDHA into lactate for energy. This discovery changed our previous understanding of cancer cell metabolism, and the phenomenon was later termed aerobic glycolysis or the “Warburg effect” (16). Subsequently, the Warburg effect was observed and confirmed in various tumor cells. Moreover, continuous studies have shown that the Warburg effect, which may be the mechanism driving renal fibrosis, is also observed in injured kidneys (17). Blocking aerobic glycolysis inhibits renal fibrosis progression. Targeted metabolic analysis of renal tissues of rats with unilateral ureteral obstruction (UUO) showed high levels of lactate in renal tissue (18). This suggests that glycolysis is enhanced in the fibrotic kidney.
Under physiological conditions, proximal renal tubular epithelial cells (TECs) account for 90% of the renal cortex, which is rich in mitochondria, mainly uses fatty acids as fuel, and produces ATP through mitochondrial OXPHOS. When the kidney is damaged, TECs alter their energy metabolism and rely on aerobic glycolysis to adapt to the destruction of mitochondrial OXPHOS. This abnormal change of energy metabolism promotes renal fibrosis (19). Abnormal proliferation of TECs in the UUO model, which is characterized by high levels of glycolytic enzymes, was consistent with the degree of fibrosis (20). Cao et al. found that the activation of mammalian target of rapamycin (mTOR) signaling mediated by tuberous sclerosis complex 1 (a key regulator of glycolysis) promoted TEC proliferation and glycolytic enzyme upregulation, as well as extensive renal interstitial fibrosis, which was significantly improved using a glycolysis inhibitor (21). Zhang et al. observed that in UUO mice and TECs in CKD patients, OXPHOS was interrupted, glycolysis was increased, and mitochondrial proteins were highly acetylated, whereas deacetylation of pyruvate dehydrogenase complex (PDC), a key enzyme linking glycolysis to the TCA cycle, at lysine 385 could be significantly reversed, thereby improving renal fibrosis (22).
Studies have confirmed that the activation of renal interstitial fibroblasts is similar to metabolic reprogramming in cancer cells, and enhanced aerobic glycolysis motivates fibroblast-myofibroblast transformation, leading to renal fibrosis, which is reduced after the use of glycolysis inhibitors (23). In UUO kidneys or TGF-β1-induced renal interstitial fibroblasts, the expression of glycolysis-related enzymes was upregulated and aerobic glycolytic flux (glucose uptake and lactate production) was increased, which was positively correlated with the degree of fibrosis (20). Wei et al. observed that glycolytic genes were upregulated in renal interstitial fibroblasts in UUO mice, and when treated with glycolysis inhibitors, the production of fibronectin and α-smooth muscle actin was reduced, and the degree of renal fibrosis was alleviated (24).
Hypoxia-inducible factors (HIFs) are important heterodimer transcription factors that sense oxygen status. They have an oxygen-sensitive α subunit, which plays the main biological role, and a stably expressed β subunit (25). HIFs perform their biological functions as follows. When oxygen is sufficient, the prolyl residue on the α subunit is hydroxylated by prolyl hydroxylase domain (PHD), combines with the VHL protein, recruits ubiquitin ligases, enters the proteasome for degradation, preventing HIF from exerting its biological function (26). Under hypoxia, hydroxylation of the α subunit is inhibited, and HIF-α instead of being degraded it accumulates. Subsequently, it enters the nucleus, combines with the β subunit to form a dimer, which binds to the HRE of the downstream target gene promoter or enhancer region, and activates the transcription of all target genes (26). Excluding the PHD, the stability and transcriptional activities of HIF-α are regulated by a factor inhibiting hypoxia-inducible factor. The factor inhibiting hypoxia-inducible factor is a JmjC domain 2-oxogluarate and Fe(II)-dependent oxygenase that catalyzes hydroxylation of specific asparagines in the C-terminal transcriptional activation domain of HIF-α isoforms. This modification suppresses the transcriptional activity of HIF by reducing its interaction with the transcriptional coactivators p300/CBP.
There are three α-subunit subtypes: HIF-1α, HIF-2α, and HIF-3α. Presently, there are many studies on HIF-1α and HIF-2α, the biochemical properties of which are very similar. They can identify the same DNA binding region and play a key role in the regulation of the cell hypoxia response (25). However, reports on HIF-3α are limited. Studies have shown that HIF-3α can be a HIF-1α target gene and negatively regulate the activities of HIF-1α and HIF-2α (27). In the kidney, HIF-1α exists in almost all cells, especially TECs, whereas HIF-2α is only expressed in specific cell types, such as vascular endothelial cells and renal interstitial fibroblasts (28).
HIF-1α has many important target genes that affect various physiological and pathological processes. Generally, the activation of HIF-1α manifests as two types of cellular adaptive responses. First, HIF-1α can promote angiogenesis by activating vascular endothelial growth factor (VEGF), Angiopoietin 2, notch ligand and ECM remodeling proteins, increasing the supply of oxygen and nutrients, and increasing the utilization rate of oxygen during metabolism (29). Moreover, it is worth noting that the activation of HIF-1α can also cause the upregulation of glucose transporter-1 (GLUT1) and glycolysis-related enzymes, which ultimately leads to the enhancement of aerobic glycolysis ability and inhibition of OXPHOS (10). Pyruvate produced through glycolysis is further metabolized to lactate by LDHA and excessive accumulation of lactate leads to intracellular acidosis. Monocarboxylate transporter 4, whose expression is HIF-1 dependent, plays a role in intracellular pH homeostasis during lactate efflux (30). Further, the expression and functional activation of carbonic anhydrase IX (CAIX) will increase as a coping mechanism (31). CAIX, a transmembrane protein neutralizing intracellular acidosis, is induced by HIF-1 and associated with glycolysis (32). Previous studies have reported that the expression of HIF-1α and its transcription target CAIX increases in tumor cells under hypoxia. This catalyzes the reversible conversion of carbon dioxide to bicarbonate and hydrogen ions and regulates the PH of tumors, leading to acidification of the tumor environment and alkalization of cytoplasm (33).
HIF-1 promotes glucose catabolism and glycogen synthesis. Pouysségur et al. determined that HIF-1 specifically upregulates enzymes involved in glycogen synthesis, such as phosphoglucomutase 1 and glycogen synthase 1, to maintain sufficient energy levels for later use under more severe nutrient-limiting conditions (34). Recently, Ito et al. demonstrated that HIF-1 protects the kidney from ischemia and hypoxia by enhancing glycogen synthesis and inhibiting glycogen decomposition in TECs (35). Fructose-1, 6-bisphosphatase 1 is among the major genes controlling renal gluconeogenesis. Li et al. demonstrated its ability to inhibit cell proliferation, glycolysis, and pentose phosphate pathways through direct interaction with the HIF “inhibitory domain” in pVHL (protein encoded by VHL gene) deficient renal cancer cells, thereby inhibiting nuclear HIF function and the potential Warburg effect.
Recent evidence indicates that many aspects of lipid metabolism are modified in a HIF-dependent manner during hypoxia. Peroxisome proliferator-activated receptor-γ, a member of the nuclear receptor transcription factor family, plays an important role in lipid metabolism by promoting free fatty acid uptake and triglyceride (TG) accumulation. Krishnan et al. demonstrated that HIF-1 can directly activate peroxisome proliferator-activated receptor-γtranscription, leading to the accumulation of TG in cardiomyocytes and promoting cardiomyocyte apoptosis (36). As a lipid metabolism disorder under hypoxia, the excessive accumulation of intracellular free fatty acids could cause lipotoxicity. Fatty acids can be converted to TG that is stored in the form of lipid droplets (LDs) in order to avoid this. TG synthesis is important for metabolic homeostasis and cell growth because LDs act as lipid stores providing both metabolic substrates and components for membrane synthesis. Triantafyllou et al. found that the expression of Acylglycerol-3-phosphate acyltransferase, an enzyme involved in the TG biosynthesis pathway, was directly regulated by HIF-1 and promoted the survival of cancer cells under hypoxia (37). Gimm et al. confirmed that HIF-1 can directly and specifically transactivate hypoxia-inducible protein 2, a novel LDs protein, to increase neutral lipid accumulation and stimulate cytokine expression in cancer cells (38).
A series of studies have confirmed that the expression of HIF-1α increases in renal fibrosis, suggesting that HIF-1α may play a vital role in promoting renal fibrosis (39). How does HIF-1α mediate renal fibrosis? Presently, research has mainly focused on the role of HIF-1α in regulating the release of inflammatory mediators, promoting the epithelial-mesenchymal transition (EMT) process and renal vascular remodeling, activating the transcription of downstream pro-fibrosis-related cytokines, and interacting with various pro-fibrosis signaling pathways (40) (Figure 1). Notably, HIF-2 also plays a key role in the progress of renal fibrosis. Li et al. bred HIF-2α deficient mice and subjected them to renal trauma through UUO surgery, ultimately determining that HIF-2α ablation attenuated renal fibrogenesis induced by UUO injury (41). Studies on HIF-2 inhibiting renal fibrosis are ongoing, and long-term clinical trial data are expected to confirm the renoprotection and anti-fibrosis of HIF-2 in the future.
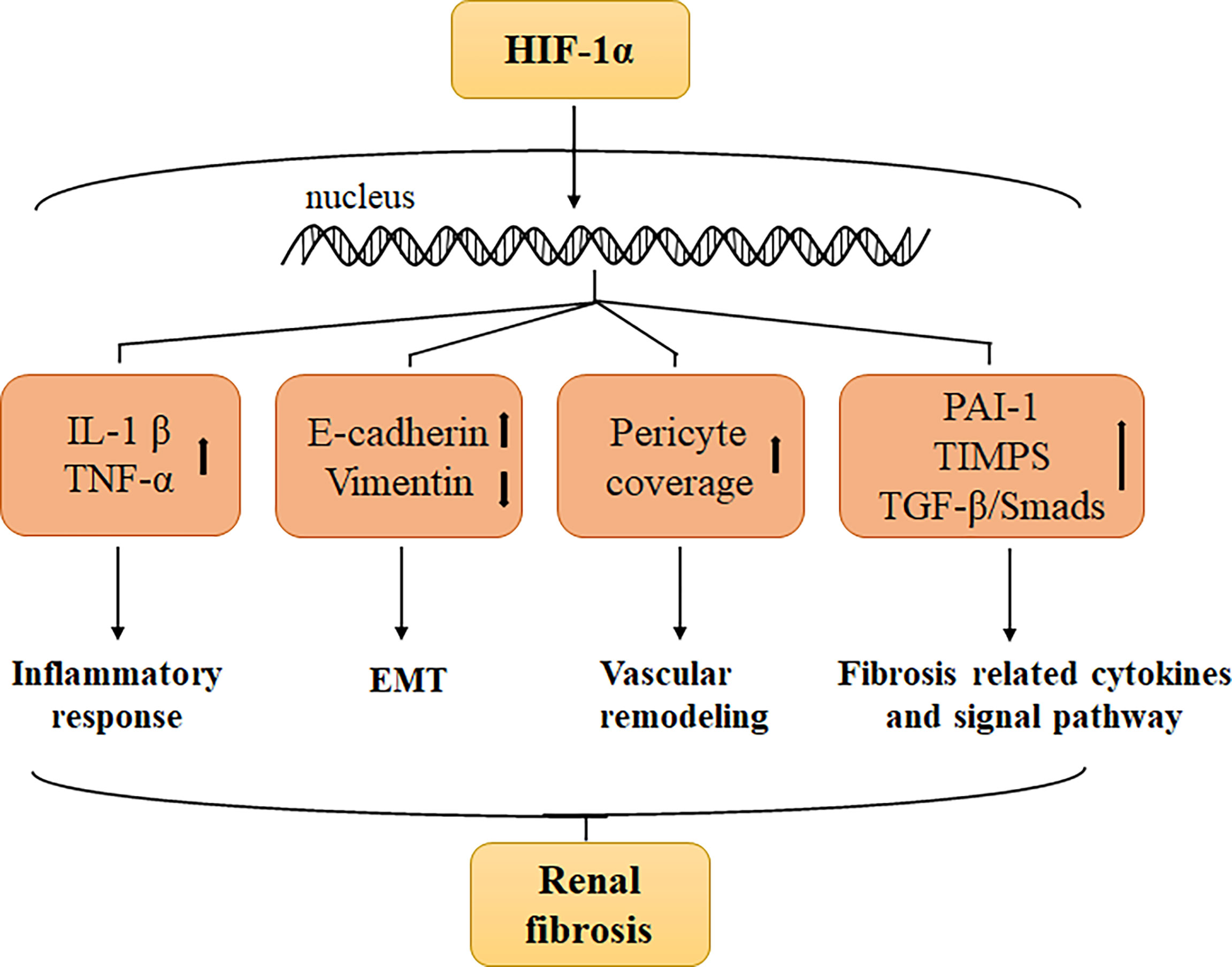
Figure 1 HIF-1α expression increases under hypoxia, which promoted renal fibrosis through the following aspects: Stimulating the secretion of interleukin-1β (IL-1 β) and tumor necrosis factor-α (TNF-α), and increasing the inflammatory reaction, upregulation of the epithelial cell marker E-cadherin and downregulation of the mesenchymal marker vimentin promote EMT, enhance pro-fibrosis cytokines such as plasminogen activator inhibitor 1 (PAI-1) and tissue inhibitor of metalloproteinase (TIMPs), and regulate fibrosis-related signaling pathways such as TGF-β/Smads signaling.
Inflammation exists widely in all stages of CKD and is an important factor in disease progression. Studies have shown that HIF-1α can release inflammatory mediators, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), change the local microenvironment of the kidney, and cause inflammation and fibrosis (42). Studies have shown that TNF-α-induced nuclear factor-κB signaling stimulates muscle glycolytic metabolism through activation of the glycolytic regulator HIF-1α (43). Furthermore, inflammatory factors can upregulate HIF-1α in a nuclear factor-κB-dependent manner, thereby increasing the risk of kidney injury (44). Reactive oxygen species (ROS) are mainly produced by mitochondria, and they can mediate cell differentiation, proliferation, apoptosis and inflammatory responses by activating some signaling pathways (45). Inflammatory mediators such as TNF-α have been shown to induce ROS production (46). Recent studies have demonstrated that HIF-1α stabilization is regulated by ROS (47). Xie et al. investigated the relationship between TNF-α, ROS production and HIF-1α stabilization in hepatocellular carcinoma cells. Their results showed that TNF-α induced HIF-1α activation was ROS dependent, and inhibition of ROS generation significantly reduced HIF-1 α expression (48). Huang et al. found that titanium dioxide nanoparticle exposure increased ROS production and the expression of HIF-1α and TGF-β in TECs, which were reduced by ROS scavenger N-acetylcysteine treatment at the transcription level. These results suggested that titanium dioxide nanoparticle exposure may induce renal fibrosis through an ROS-related HIF-1α up-regulated TGF-β signaling pathway (49). Therefore, HIF-1α is closely related to inflammatory reactions that can promote the expression of inflammatory factors. Inflammatory reactions can also regulate the activity of HIF-1α.
EMT is an important process and a central link in the occurrence and development of renal fibrosis, in which enhancement of cell migration ability is an important feature of epithelial cell dedifferentiation (50). HIF-1α can activate the transcriptional activity of these genes by combining with HRE in the promoters of EMT regulators, such as Twist and Bmi1, downregulating the expression of E-cadherin and tight junction proteins, increasing the levels of fibronectin and vimentin, transforming epithelial cells into fibroblast-like cells, promoting EMT, and aggravating renal interstitial fibrosis (51, 52). Higgins et al. analyzed the regulatory effect of HIF-1α on the dedifferentiation of mouse TECs and proved that HIF-1α could enhance the migration ability of TECs, thus strengthening EMT. By selectively knocking out HIF-1α, collagen deposition, inflammatory infiltration, and fibroblast expression in the renal interstitium decreased (40).
A stable vascular network is important for maintaining tissue and organ homeostasis. Vascular remodeling is the adaptive response to various physiological and pathophysiological alterations that are closely related to aging and vascular diseases. The remodeling of the vascular network may initially be adaptive, however, eventually it becomes maladaptive and compromises organ function (53). Emerging evidence is suggesting that HIF-1α plays a pivotal role in vascular remodeling. Liu et al. found that interleukin-33 initiates vascular remodeling of hypoxic pulmonary hypertension by up-regulating HIF-1α and VEGF expression in vascular endothelial cells (54). Luo et al. showed that CD146 was up-regulated in pulmonary artery smooth muscle cells in proportion to disease severity, and CD146 expression and HIF-1α transcriptional program reinforced each other to drive vascular remodeling and pulmonary arterial hypertension (55). Accumulating evidence is demonstrating that the HIF-α hydroxylase system plays a critical role in vascular remodeling. Three PHD isoforms have been identified in mammals, PHD1, PHD2 and PHD3. Based on the expression pattern and dominant effects under normoxic conditions, PHD2 was considered as the critical oxygen sensor to hydroxylate HIF-α (56). Wang et al. confirmed that the deletion of endothelial PHD2 promoted renal vascular remodeling and fibrosis by up-regulating Notch3/TGF-β1 signaling and increasing the coverage of pericytes (57).Studies have shown that HIF-1α can combine with HRE in the promoters of plasminogen activator inhibitor 1 (PAI-1) and tissue inhibitor of metalloproteinase (TIMPs), trans-activate the transcription of target genes, and inhibit ECM degradation and collagen decomposition, thus promoting renal fibrosis (58, 59). TGF-β1 has long been regarded as a key molecule in the promotion of renal fibrosis, and the TGF-β/Smads signaling pathway is a classic profibrogenic signaling pathway. TGF-β1 mainly induces renal scar formation by activating downstream Smad signaling (7). Kushida et al. found that the increase in Smad3 binding was mainly caused by HIF-1α under hypoxia. Specifically, HIF-1α could bind to Smad3, upregulate the level of fibrosis-related proteins, and promote renal fibrosis (60). Arginase-II(Arg-II) is a mitochondrial enzyme which is highly expressed in renal proximal TECs and up-regulated by hypoxia. Recent studies have indicated that inhibition of Arg-II can reduce renal injury in diabetic nephropathy mouse models (61). Liang et al. (62) demonstrated that HIFs can mediate the up-regulation of Arg-II induced by hypoxia in TECs, and showed hypoxia promoting the expression of TGF-β1 dependent on Arg-II as the silence of Arg-II can prevent the expression of TGF-β1. These results indicate that hypoxia activates renal epithelial HIFs-Arg-II-TGF-β1 signaling, exacerbating renal injury and fibrosis.HIF-1α Promotes Glycolysis
One of the adaptive responses of cell survival mediated by HIF-1α is HIF-1α dependent glucose metabolism reprogramming. This metabolic reprogramming promotes an increase in glycolysis flux and inhibits OXPHOS by upregulating the expression of glucose transporters and glycolytic enzymes (10) (Figure 2). Studies have shown that genes encoding glycolytic enzymes, such as HKs, PFK1, aldolase, phosphoglyceride kinase-1, and LDHA, contain HIF-1α-binding sites in their promoter or enhancer regions, and are directly upregulated by HIF-1α in response to hypoxia (10). Interestingly, Semenza et al. observed that the consensus sequence recognized by HIF-1α in human aldolase and phosphoglyceride kinase-1 was evolutionarily conserved between humans and mice, which undoubtedly suggests that the HIF-1α-induced upregulation of these two genes would be of great significance in the regulation of glycolysis in mammals, and it warrants further study in the future (63).
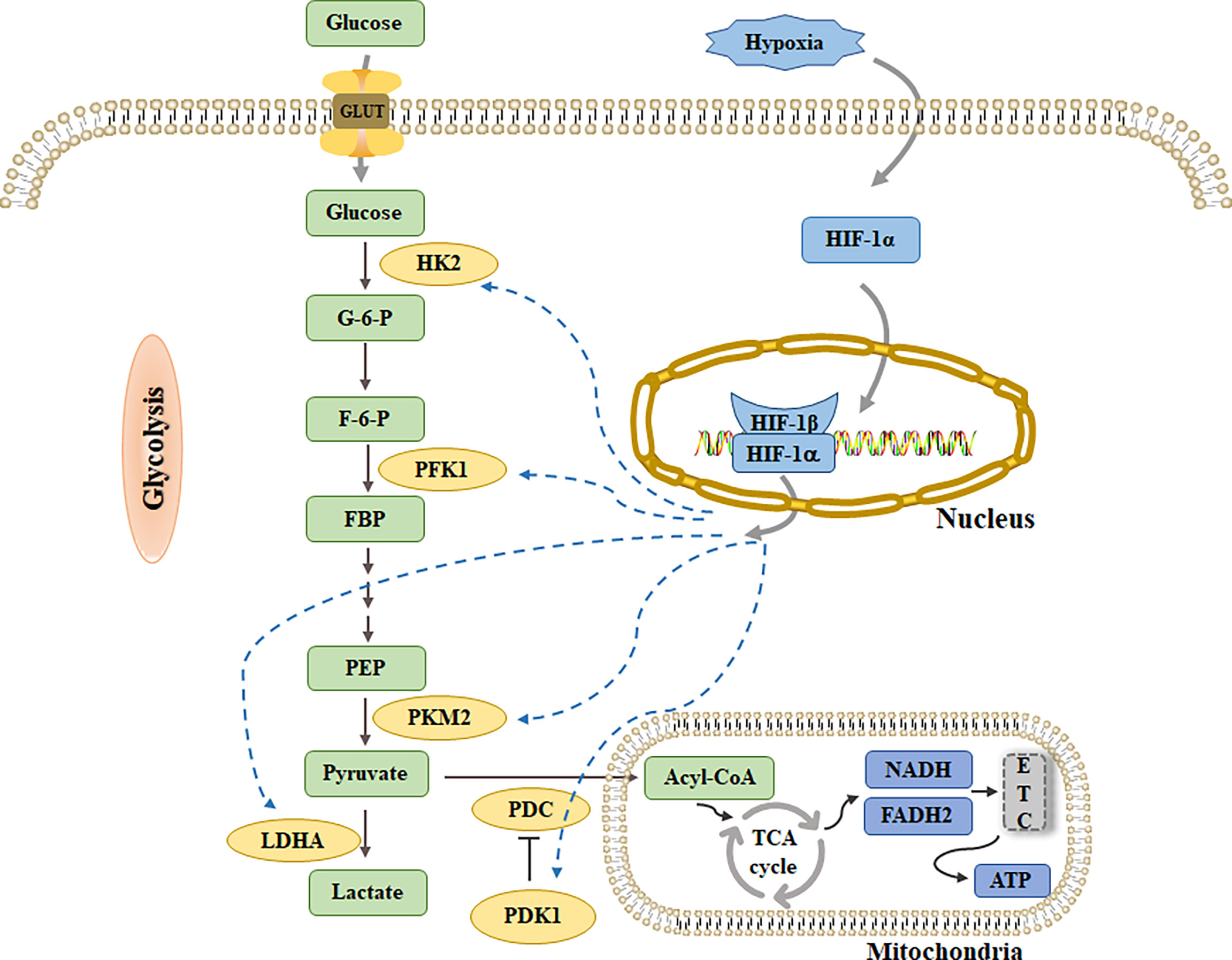
Figure 2 Glucose transport into cells through GLUT and decompose into pyruvate by a series of glycolytic enzymes such as HK2, PFK1 and PKM2. PDC catalyzes the conversion of pyruvate into acetyl-CoA, enters TCA cycle, generates NADH and FADH2, and then enters ETC to produce ATP. Under hypoxia, pyruvate is activated by LDHA to generate lactate. HIF-1α can increase glycolysis flux by up-regulating HK2, PFK1 and PKM2. At the same time, increasing the levels of PDK1 and LDHA inhibited pyruvate from entering TCA cycle.
HIF-1α precisely regulates the reabsorption and release of glucose into the blood by the kidneys. As glucose is soluble in water and cannot pass through the phospholipid bilayer of the cell membrane, carriers are required for its transport. Glucose transporters are cell membrane (transmembrane) glycoproteins that mediate extracellular glucose cell uptake. Three families of glucose transporters have been identified in humans: diffusion-prone glucose transporters (GLUTs) encoded by SLC2A, sodium-dependent glucose transporter encoded by SLC5A, and glucose efflux transporters encoded by SLC50A (64). In the kidney, only GLUTs and sodium-dependent glucose transporter are involved in the transport of renal glucose (64). To date, 14 subtypes of GLUTs (GLUT1–GLUT14) have been identified. Their distribution, function, and affinity with glucose differ, and they have different glucose transport efficiencies. GLUT1 and GLUT3 have the highest affinities for glucose, GLUT2 has the lowest affinity, and the glucose affinities of the rest are between those of GLUT1, 2, and 3. GLUT1 plays an important role in kidney and tumor cell metabolism and is closely related to glycolysis activity (65). In laryngeal carcinoma cells, the expression of GLUT1 is positively correlated with that of E-cadherin and negatively correlated with that of vimentin, indicating that GLUT1 is involved in EMT (66). Moreover, some studies have shown a correlation between GLUT1 expression and pathological fibrosis. Transgenic mice overexpressing GLUT1 showed an increase in the expression of TGF-β1 and nuclear factor-κB in the glomeruli and pathological manifestations similar to those in diabetic glomerulosclerosis (67). This finding suggests that GLUT1 may cooperate with cytokines or growth factors to induce renal fibrosis. The specific pathogenic mechanism involved requires further clarification.
GLUTs are one of the target genes activated by HIF-1α, but glucose transport into cells is undoubtedly detrimental to glycolysis (68). Studies have shown that an important way by which HIF-1α affects glycolysis is the upregulation of GLUTs. HIF-1α can induce the transcription and translation of SLC2A1 and SLC2A3, leading to the upregulation of GLUT1 and GLUT3, and finally to an increase in glycolysis flux. Wound healing and tumorigenesis are both involved in cell proliferation and tissue remodeling. Elson et al. found that HIF-1α and GLUT1 increased to their highest levels in re-epithelialized basal keratinocytes during the healing period of skin wounds. They believed that the increased GLUT1 level was related to the increased glucose uptake of cells, which was necessary to provide the energy needed for cell migration and proliferation during hypoxia (69).
GLUTs can effectively transport glucose into cells, provide substrates for glycolysis, and lay a foundation for glycolysis. Therefore, GLUTs will become a potential therapeutic target to limit the glycolysis rate and block energy sources. Presently, many GLUT1 inhibitors are in the research stage and potentially available, such as phloretin (the first discovered flavonoid GLUT1 inhibitor) (70) and resveratrol (71), which show good biological activities. Unfortunately, there is a lack of in-depth research on how HIF-1α regulates GLUT1 to affect energy metabolism in renal fibrosis. However, targeting GLUT1 also provides a new technical means for exploring the changes in energy metabolism in renal fibrosis.
The first rate-limiting reaction in glycolysis is the phosphorylation of glucose to form glucose-6-phosphate (G-6-P), which is catalyzed by HKs. Four HKs (HK1, HK2, HK3, and HK4) have been identified in mammals, but their tissue distribution differs. HK1 is constitutively expressed in most normal tissues, HK2 is widely expressed in insulin-sensitive tissues, such as muscle and fat, and is also expressed in the kidney, blood vessels, and retina (72). Most immortalized and malignant cells show increased expression of HK2, which may contribute to an increase in glycolysis and provide energy and metabolites for tumor DNA synthesis (73). Existing research shows that the increase in HK2 activity is the initiator of glycolysis overload and downstream metabolic dysfunction, and the increase in HK2 and G-6-P causes metabolic dysfunction and accelerates cell aging and tissue damage (74).
Studies have shown that HIF-1α activation can upregulate the levels of HK1 and HK2 and that it plays an important role in tumor cell proliferation by enhancing glycolysis activity (75). Research shows that the increase in HK2 not only enhances glycolysis but also promotes EMT under hypoxia (76). Similar to GLUTs, HK2, a rate-limiting enzyme in glycolysis, is a potential target of glucose metabolism. Studies have proved that 3-Bromopyruvic acid and 2-deoxy-d-glucose can significantly inhibit HK activity and glycolysis (77).
PFK1 converts fructose-6-phosphate (F-6-P) into fructose-1,6-diphosphate (FBP), which is the rate-limiting enzyme in glycolysis and the key control point for regulating glycolytic flux. PFK1 is a tetrameric protein with three subtypes: PFK-M (muscle), PFK-L (liver), and PFK-P (platelets). The dimerization of the PFK1 monomer is beneficial for maintaining the tertiary structure of the enzyme (78). The PFK1 dimer showed the lowest catalytic activity, while complete catalytic activity was achieved when it combined to form a tetramer (78). PFK1 is subject to complex allosteric regulation and is inhibited by high ATP concentrations and activated by adenosine monophosphate (AMP), which prevents excessive glucose degradation when ATP is sufficient. Moreover, it is inhibited by citrate and long-chain fatty acids produced in the TCA cycle (79). It must be said that the most effective allosteric activator of PFK1 is fructose- 2,6-diphosphate, and that the level of fructose- 2,6-diphosphate is closely related to that of fructose-6-phosphate-2-kinase/fructose-2,6-diphosphatase (PFK2, PFKFB) (79). PFKFB contains two catalytic domains with kinase and phosphatase activities and has dual functions. It increases the affinity of PFK1 to F-6-P, releases enzymes from ATP-mediated inhibition, and synergistically increases the affinity of PFK1 to AMP. Furthermore, PFKFB stabilizes PFK1 and promotes its association with tetramers and activates higher oligomers (80). Therefore, changes in the PFKFB activity are important for glucose metabolism.
PFKFB, an important regulator of glycolysis, has four isozymes (PFKFB1, PFKFB2, PFKFB3, and PFKFB4), among which PFKFB3 has approximately 700 times higher kinase activity than bisphosphatase activity, which is beneficial for the generation of intracellular F-2, 6-BP, and glycolysis (81). Studies have shown that HRE in the PFKFB3 promoter can be activated by HIF-1α under hypoxia, leading to an increase in glucose uptake and glycolysis flux. Glycolysis and pro-inflammatory activation of macrophages mainly depend on the accumulation of HIF-1α, which can increase glucose uptake and upregulate HK2 and PFKFB3 under hypoxia (82). In renal cell carcinoma, PFKFB3 knockout inhibits glycolysis and cell proliferation, suggesting that PFKFB3 could be used as a potential therapeutic target (83). Emerging evidence has shown that PFKFB3 also plays an important role in regulating physiological and pathological angiogenesis (84). Endothelial proliferation depends on glycolysis: 85% of ATP is produced by converting glucose into lactate, and pharmacological inhibition of PFKFB3 damages the development of the mouse retinal vascular system (85). In diabetic retinopathy, HIF-1α/PFKFB3 signaling not only promotes pathological angiogenesis, but also drives glycolysis, which damages the antioxidant capacity of neurons, leading to neuronal degeneration and an increase in the number of reactive glia (86). The kidney is a highly vascularized organ with a rich microvascular structure. Exploring the relationship between glucose metabolism in the renal microcirculation and renal fibrosis may provide new insights into understanding the mechanism underlying CKD development.
PK is the rate-limiting enzyme in glycolysis, which converts phosphoenolpyruvate (PEP) to pyruvate. PK has four subtypes (PKL, PKR, PKM1, and PKM2), which can be specifically expressed according to the different metabolic needs of tissues. PKR is only expressed in red blood cells, PKL is mainly expressed in the liver, PKM1 is mainly expressed in the skeletal muscle, heart, and brain, and PKM2, which exists in various differentiated tissues, is the main subtype in the kidney (87). According to reports, the expression level of PKM2 is increased in highly proliferating cells (such as stem cells and tumor cells), which is beneficial to the accumulation of glycolytic metabolites and stimulates tumor proliferation. PKM2 exists in four different enzyme states: inactive monomer, near-inactive dimer, inactive T-tetramer (low enzyme activity), and active R-tetramer (high enzyme activity). There is a conformational switch between the inactive T-state and active R-state during glycolysis (88).
PKM2 plays a key role in reprogramming tumor metabolism and is considered an important regulator of the Warburg effect. This downregulation reduces the glucose utilization and glycolysis rates and inhibits tumor growth (89). Research has shown that the change in the metabolic state of PKM2 is mainly mediated by the interaction between HIF-1α and c-Myc (an oncogenic transcription factor), which can upregulate the transcription of PKM2 and increase its protein synthesis (90). Wang et al. reported that HIF‐1α not only directly activates PKM2 but also upregulates the expression of PKM2 in an m5C-dependent manner by activating ALYREF (an m5C RNA binding protein), increasing glycolysis, and promoting bladder cancer cell proliferation (91). PKM2 is also involved in EMT, and the expression and nuclear accumulation of PKM2 increase when EMT is induced (92). Luo et al. demonstrated that PKM2 can also act as a co-activator, which can stimulate the transactivation of HIF-1α encoding GLUT1, LDHA, and PDK1 target genes in tumor cells by combining with the transactivation domain of HIF-1α, which accelerates glucose metabolic reprogramming and cancer progression mediated by HIF-1α (93).
Furthermore, increasing evidence indicates that PKM2 can be used as a biomarker of nephrotoxicity. In the conditioned medium of human TECs pretreated with cisplatin or other nephrotoxic substances (such as cyclosporine A and HgCl2), the secretion of PKM2 increased, and this effect was not observed in liver and breast cancer cells, suggesting that PKM2 is specific to renal cells and can be used as an early marker of nephrotoxicity (94). It is well established that hyperglycemia promotes CKD. Hyperglycemia reduces the formation and activity of the PKM2 tetramer in the glomerulus, TECs, and podocytes through sulfinylation, resulting in the increase of toxic glucose metabolites and mitochondrial dysfunction (95, 96). The activation of PKM2 can restore mitochondrial function by inhibiting the accumulation of toxic glucose metabolites, increasing glycolysis and TCA cycle flux, and inducing mitochondrial biogenesis (96). However, the protective role of glycolysis flux may be relative, and extreme or long-term hyperglycemia may alter this protective mechanism. In diabetic nephropathy, knocking out sirtuin 3 can induce TGF-β/smad3 signaling, which increases the accumulation of HIF-1α and formation of PKM2 dimers, and enhances the transcription of glycolytic enzymes, leading to the accumulation of glycolytic intermediates before pyruvate, induces abnormal glycolysis, and helps to drive EMT, thus accelerating renal fibrosis (97). Hence, targeting PKM2 may be a new strategy to prevent or treat kidney diseases.
HIF-1α upregulates glycolytic enzymes and reduces the amount of metabolites entering the TCA cycle by upregulating pyruvate dehydrogenase kinases (PDKs), thus inhibiting OXPHOS (98). PDKs can inhibit the activity of PDC, which is an important nodal enzyme that catalyzes the conversion of pyruvate into acetyl-CoA and is connected to the TCA cycle (98). There are four subtypes of PDKs (PDK1, PDK2, PDK3, and PDK4) with different distributions and binding affinities. PDK2 and PDK4 are the most widely distributed in the heart, liver, and kidney, while the levels of PDK1 and PDK3 are limited in tissues (99). Regarding binding affinity, PDK4 has the lowest affinity, PDK3 has the highest affinity, PDK1 and PDK2 have medium affinity, and PDK1-3 can interact with various signal transduction factors, which is helpful in cancer progression (99). However, PDK4 can act as both an oncogene and tumor suppressor (99).
Research shows that HIF-1α can effectively inhibit PDC activity by activating the transcription of PDK1, preventing pyruvate from being converted into acetyl-CoA, thus hindering the start of the TCA cycle (100). Notably, the blocking effect of HIF-1α on the TCA cycle not only activates PDK1 but also promotes LDHA expression (101). LDHA is the key enzyme responsible for converting pyruvate into lactate in the last step of glycolysis, and it also produces NADH2, which is a cofactor necessary for maintaining glycolytic activity (102). HIF-1α can increase lactate by activating LDHA, reducing the conversion of pyruvate into acetyl-CoA, and further keeping pyruvate away from the TCA cycle (101). He et al. selected six active molecules from a database of 500,000 molecules using high-throughput virtual screening, optimized their structures, and developed a new dual-target molecule with nanomolar activity against PDK1 and LDHA. The results showed that this molecule inhibited the activity of PDK1 and LDHA, significantly reduced the production of lactate in tumor cells, increased oxygen consumption, and regulated glucose metabolism (103).
Moreover, HIF-1α interacts with other transcription factors to regulate glucose metabolism. c-Myc regulates cell metabolism and cell proliferation, either alone (under normal oxygen) or in combination with HIF-1α, and can activate PDK1 and LDHA, increase the conversion of pyruvate to lactate, and weaken mitochondrial respiration (104). Estrogen-related receptors (ERRs) are orphan nuclear receptors that regulate cell energy metabolism by coordinating mitochondrial biogenesis and OXPHOS (105). Cai et al. showed that ERRs can be combined with the promoter of glycolytic genes alone to activate gene transcription, and can also be combined with c-Myc to activate the transcription of glycolytic genes. At the same time, ERRs can also be used as a cofactor of HIF-1α to enhance HIF-1α-dependent transcription of glycolytic genes under hypoxia, thus contributing to the Warburg effect (106).
Generally, HIF-1α-dependent transcription reduces PDH activity by upregulating PDK1 and increases the LDH-mediated conversion of pyruvate to lactate, thus reducing pyruvate to the TCA cycle effectively, leading to a decrease in TCA cycle flux and OXPHOS. In view of this, PDK1 can be used as an effective target for precise therapy, in which the pyruvate analog dichloroacetate, a naturally occurring inhibitor, has been shown to inhibit PDK activity in vitro and in vivo (107). Unfortunately, there are no reports on the effect of HIF-1α on OXPHOS in renal fibrosis. In the future, more in-depth research in this area should be conducted.
PI3K is an intracellular heterodimer lipid kinase that responds to extracellular stimuli, such as growth factors, cytokines, and hormones, and participates in regulating cell survival, proliferation, and metabolism (108). Akt, a serine/threonine kinase, is the central molecule of the PI3K pathway, which can be combined with lipid products of PI3K and thus localize on the cell membrane to activate many cell processes, such as glucose metabolism, apoptosis, and cell proliferation (109). Many studies have shown that PI3K/Akt directly or indirectly upregulates glycolytic enzymes through post-translational modifications (such as phosphorylation). For example, Akt phosphorylation can regulate GLUT1 expression and increase glucose uptake (110). Akt also enhances the phosphorylation level of glycolytic enzymes, such as HK2 and PFK1, and increases the glycolysis rate (110). Moreover, Akt indirectly coordinates glycolysis and the TCA cycle by inducing the expression of HIF-1α (111). The transcription factor forkhead box O3a directly inhibits c-Myc transcription. Akt indirectly activates c-Myc by phosphorylating forkhead box O3a, upregulates glycolytic enzymes induced by c-Myc, and catalyzes glycolysis (112). Therefore, the activation of PI3K/Akt is closely related to the glycolytic activity of cells.
mTOR is a conserved serine/threonine kinase that regulates anabolism and cell growth, and its phosphorylation level can be regulated by Akt (113). Studies have shown that mTOR mediated by the PI3K/Akt pathway is of great significance in regulating cell survival, metabolism, and angiogenesis. Abnormalities in this pathway are common in human tumor diseases, and targeting cytokines in this pathway is helpful in increasing the therapeutic effect on tumors (114). Notably, this pathway plays a key role in promoting glycolysis and lactate production. Activating this signaling pathway can increase the transcription of glycolytic genes by promoting HIF-1α-dependent mechanisms, upregulating the activities of PDK1 and LDHA, and inhibit OXPHOS (115) (Figure 3). Weng et al. first confirmed that fasting upregulated the level of the cholesterol steroid gene farnesyl-diphosphate farnesyl transferase 1 (FDFT1) and then decreased glycolysis by negatively regulating AKT/mTOR/HIF1α signaling, thus prolonging the survival time of colorectal cancer patients (116). Phosphatase and tensin homolog deleted on chromosome ten (PTEN) is a tumor suppressor, and its biological effect is mainly the negative regulation of PI3K signaling (117). Other studies have shown that deletion of PTEN leads to many metabolic changes (118). Xiao et al. confirmed that overexpression of miR-21 can mediate the PI3K/Akt/mTOR axis by increasing PTEN and then reducing the expression of HIF-1α, resulting in a decrease in aerobic glycolysis in bladder cancer cells (119). Interestingly, some studies have shown that the metabolic pathway mediated by PI3K signaling also plays an important role in tissue fibrosis. Hu et al. reported that lipopolysaccharide upregulated the expression of PFKFB3 by activating the PI3K/Akt/mTOR pathway, increasing aerobic glycolysis in lung fibroblasts, promoting collagen synthesis, and accelerating pulmonary fibrosis (120). Studies have shown that activating the PI3K/Akt/mTOR and HIF-1α signaling pathways can effectively inhibit inflammatory reactions, reduce ECM deposition, and reverse EMT, and thus play a protective role in renal fibrosis (121). The possible pathogenic mechanism underlying the glycolysis process of renal fibrosis needs to be elucidated.
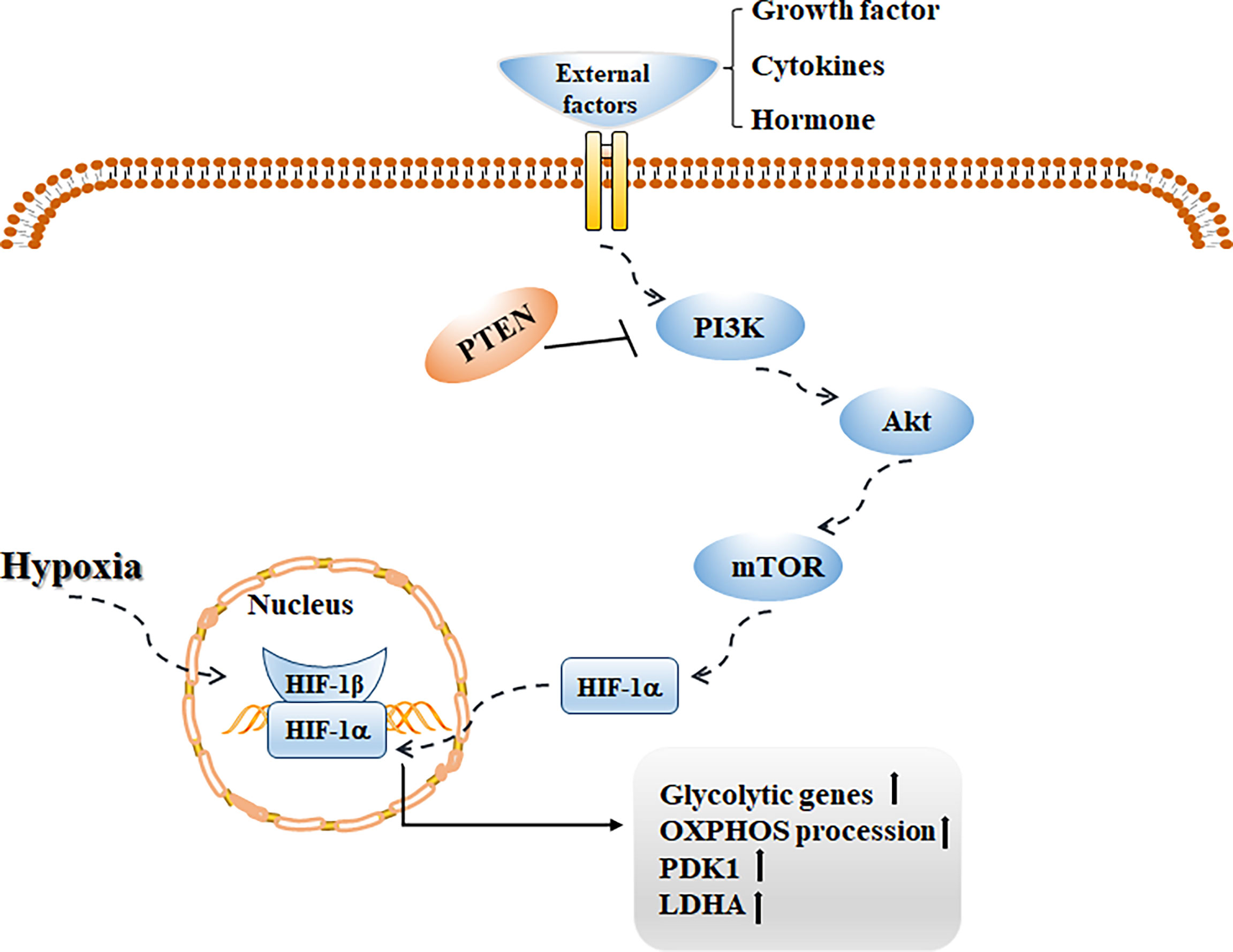
Figure 3 With the stimulation of external factors such as growth factor, cytokines and hormone, PI3K/Akt/mTOR signaling pathway is activated, which can promote HIF-1α transcription activity, increase the transcription of glycolytic genes, up-regulate the levels of PDK1 and LDHA, and then increase glycolysis while inhibit OXPHOS.
HIFs, a key factor in the metabolic regulation of eukaryotic cells under hypoxia, controls the expression of downstream target genes, involving multiple links such as oxygen transport, angiogenesis and cell metabolism, and plays an important regulatory role in diseases. At present, the clinical application of HIFs mainly focuses on two aspects: the stabilizers of HIF to treat renal anemia, and HIF inhibitors for the treatment of tumors.
CKD often progresses to end-stage renal disease, and most patients suffer renal anemia. The pathogenesis of anemia is multifactorial, including chronic inflammation, iron deficiency, and shortened half-life of erythrocytes, and the deficiency of erythropoietin is the primary cause (122). Erythropoiesis-stimulating agents combined with oral or intravenous iron supplementation is the standard treatment for renal anemia, however, some safety reports have demonstrated that high-dose erythropoiesis-stimulating agent therapy is associated with increased risk of cardiovascular events (123). Roxadustat (FG-4592) is an oral HIF prolyl hydroxylase inhibitor (HIF-PHI) that stimulates erythropoiesis and regulates iron metabolism. It has been approved in China for the treatment of dialysis-dependent (124) and non-dialysis-dependent CKD (125). Although clinical data have demonstrated that Roxadustat is effective and well-tolerated, information is still required on the range of non-erythropoiesis effects of HIF stabilizers, such as a potential proangiogenic effect related to the VEGF and VEGF receptors, the progression of pulmonary arterial hypertension (55), and the potential development of renal fibrosis related to the HIF activation over long periods (126). Considering that HIFs regulate a broad spectrum of genes, HIF-PHI are expected to protect against metabolic disorders in addition to erythropoiesis. Sugahara et al. determined the potential renoprotective effects of a HIF-PHI, enarodustat, which can improve glucose and lipid metabolism in obese and diabetic mice and reduce the infiltration of macrophages in kidney and adipose tissue (127). Studies have shown that the metabolites of glycolysis and the TCA cycle were accumulated and amino acids were reduced in kidney tissues of diabetic animals, and these metabolic disturbances were alleviated by enarodustat. Meanwhile, enarodustat can also reduce oxidative stress by increasing the ratio of glutathione to glutathione disulfide (128). Thus, stabilizing HIF can counteract the changes of renal energy metabolism in early diabetic nephropathy.
HIF has recently been recognized as an important drug target for cancer treatment. Many studies have determined that the increase of HIF level promotes the formation of tumor microvessels, tumor cell proliferation, cell survival and progression, and metastatic spread (129). There are two major categories of HIF inhibitors: small-molecule inhibitors that directly affect the expression or function of HIF, and indirect HIF inhibitors that block other molecules in related signaling pathways. Clear cell renal cell carcinoma (ccRCC) is the most common subtype of renal cell carcinoma, and the inactivation of VHL gene is the main molecular feature. The loss of VHL leads to the accumulation of HIF-α and then the uncontrolled activation of HIF target genes, many of which promote angiogenesis, cell proliferation and metastasis of ccRCC (130). A large number of clinical trials have indicated that drugs that inhibit HIF/VEGF signaling demonstrated overall survival advantages for patients with advanced or metastatic renal cell carcinoma (131).The latest research focuses on Belzutifan, an inhibitor of HIF-2α, which was first approved in the USA to treat solid tumors including ccRCC and von Hippel-Lindau disease-associated renal cell carcinoma (132).
In recent years, the annual incidence of CKD has been on the rise worldwide. Renal fibrosis is a dynamic process driven by many complicated factors and a key pathogenic factor that leads to end-stage renal disease. However, there is no clinical treatment or drug available to cure or reverse renal fibrosis. Exploring the pathogenesis of renal fibrosis and blocking or reversing the renal fibrosis process is of great clinical significance. Changes in energy metabolism in various diseases have become a hot research topic. Recent studies have shown that there is a metabolic pattern similar to that of tumor cells in the process of renal fibrosis, that is, a switch from OXPHOS to aerobic glycolysis. After kidney injury, enhancing aerobic glycolysis is a compensatory energy supply mechanism. Therefore, it is of great significance to study the related proteins that change during glucose metabolism. HIF-1α is a key transcription regulatory factor that mediates the adaptive response of cells to hypoxic microenvironments. It promotes renal fibrosis by regulating inflammatory reactions, promoting EMT, activating the transcription of downstream cytokines related to fibrosis, and interacting with fibrogenic signaling pathways. It is worth noting that HIF-1α-dependent glucose metabolism reprogramming plays an important role in the adaptive response of cells, which is mainly manifested by upregulating the expression of glucose transporters and glycolysis-related enzymes, promoting glycolysis while inhibiting OXPHOS. In summary, targeting metabolic pathways, especially HIF-1α-mediated aerobic glycolysis, may be beneficial in renal fibrosis. Second, an in-depth study of glycolysis inhibitors will be a reliable means of improving renal fibrosis. However, it should be noted that the specific influence of anti-fibrotic treatments on renal function and their final curative effects, the best action time and dose, and other issues need to be clarified, for patients to obtain the greatest benefits.
YD and XW conceived and designed the review; XW wrote the original draft; YH updated the references; ML and LJ wrote the conclusions and sections of the manuscript; All authors have read and agreed to the published version of the manuscript.
This research was funded by Finance Department of Jilin Province, grant number JLSWSRCZX2021-006.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
All aspects of manuscript writing were carried out by the authors.
1. Obrador GT, Levin A. CKD hotspots: Challenges and areas of opportunity. Semin Nephrol (2019) 39:308–14. doi: 10.1016/j.semnephrol.2019.02.009
2. Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet (2017) 389:1238–52. doi: 10.1016/S0140-6736(16)32064-5
3. Moreno-Sánchez R, Rodríguez-Enríquez S, Saavedra E, Marín-Hernández A, Gallardo-Pérez JC. The bioenergetics of cancer: is glycolysis the main ATP supplier in all tumor cells? BioFactors (2009) 35:209–25. doi: 10.1002/biof.31
4. Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov (2020) 19:57–75. doi: 10.1038/s41573-019-0040-5
5. Ung CY, Onoufriadis A, Parsons M, McGrath JA, Shaw TJ. Metabolic perturbations in fibrosis disease. Int J Biochem Cell Biol (2021) 139:106073. doi: 10.1016/j.biocel.2021.106073
6. Semenza GL. HIF-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol (2001) 13:167–71. doi: 10.1016/S0955-0674(00)00194-0
7. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol (2016) 12:325–38. doi: 10.1038/nrneph.2016.48
8. Selnø ATH, Schlichtner S, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, et al. Transforming growth factor beta type 1 (TGF-β) and hypoxia-inducible factor 1 (HIF-1) transcription complex as master regulators of the immunosuppressive protein galectin-9 expression in human cancer and embryonic cells. Aging (2020) 12:23478–96. doi: 10.18632/aging.202343
9. McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem (2006) 281:24171–81. doi: 10.1074/jbc.M604507200
10. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol (2021) 599:23–37. doi: 10.1113/JP280572
11. Nastase MV, Zeng-Brouwers J, Wygrecka M, Schaefer L. Targeting renal fibrosis: Mechanisms and drug delivery systems. Advanced Drug Deliv Rev (2018) 129:295–307. doi: 10.1016/j.addr.2017.12.019
12. Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol (2011) 7:684–96. doi: 10.1038/nrneph.2011.149
13. Inagi R. Organelle stress and metabolic derangement in kidney disease. Int J Mol Sci (2022) 23(3):1723. doi: 10.3390/ijms23031723
14. Alsahli M, Gerich JE. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res Clin Pract (2017) 133:1–9. doi: 10.1016/j.diabres.2017.07.033
15. Fernie AR, Carrari F, Sweetlove LJ. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol (2004) 7:254–61. doi: 10.1016/j.pbi.2004.03.007
16. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11:325–37. doi: 10.1038/nrc3038
17. Hewitson TD, Smith ER. A metabolic reprogramming of glycolysis and glutamine metabolism is a requisite for renal fibrogenesis-why and how? Front Physiol (2021) 12:645857. doi: 10.3389/fphys.2021.645857
18. Li Z, Li A, Gao J, Li H, Qin X. Kidney tissue targeted metabolic profiling of unilateral ureteral obstruction rats by NMR. Front Pharmacol (2016) 7:307. doi: 10.3389/fphar.2016.00307
19. Li Z, Lu S, Li X. The role of metabolic reprogramming in tubular epithelial cells during the progression of acute kidney injury. Cell Mol Life Sci (2021) 78:5731–41. doi: 10.1007/s00018-021-03892-w
20. Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q, et al. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol (2017) 313:F561–f575. doi: 10.1152/ajprenal.00036.2017
21. Cao H, Luo J, Zhang Y, Mao X, Wen P, Ding H, et al. Tuberous sclerosis 1 (Tsc1) mediated mTORC1 activation promotes glycolysis in tubular epithelial cells in kidney fibrosis. Kidney Int (2020) 98:686–98. doi: 10.1016/j.kint.2020.03.035
22. Zhang Y, Wen P, Luo J, Ding H, Cao H, He W, et al. Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis (2021) 12:847. doi: 10.1038/s41419-021-04134-4
23. Yu H, Zhu J, Chang L, Liang C, Li X, Wang W. 3-bromopyruvate decreased kidney fibrosis and fibroblast activation by suppressing aerobic glycolysis in unilateral ureteral obstruction mice model. Life Sci (2021) 272:119206. doi: 10.1016/j.lfs.2021.119206
24. Wei Q, Su J, Dong G, Zhang M, Huo Y, Dong Z. Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am J Physiol Renal Physiol (2019) 316:F1162–f1172. doi: 10.1152/ajprenal.00422.2018
25. Loboda A, Jozkowicz A, Dulak J. HIF-1 and HIF-2 transcription factors–similar but not identical. Molecules Cells (2010) 29:435–42. doi: 10.1007/s10059-010-0067-2
26. Corrado C, Fontana S. Hypoxia and HIF signaling: One axis with divergent effects. Int J Mol Sci (2020) 21(16):5611. doi: 10.3390/ijms21165611
27. Tanaka T, Wiesener M, Bernhardt W, Eckardt KU, Warnecke C, The human HIF. (hypoxia-inducible factor)-3alpha gene is a HIF-1 target gene and may modulate hypoxic gene induction. Biochem J (2009) 424:143–51. doi: 10.1042/BJ20090120
28. Shu S, Wang Y, Zheng M, Liu Z, Cai J, Tang C, et al. Hypoxia and hypoxia-inducible factors in kidney injury and repair. Cells (2019) 8(3):207. doi: 10.3390/cells8030207
29. Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW. Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp Mol Med (2004) 36:1–12. doi: 10.1038/emm.2004.1
30. Rosafio K, Pellerin L. Oxygen tension controls the expression of the monocarboxylate transporter MCT4 in cultured mouse cortical astrocytes via a hypoxia-inducible factor-1α-mediated transcriptional regulation. Glia (2014) 62:477–90. doi: 10.1002/glia.22618
31. Silagi ES, Schoepflin ZR, Seifert EL, Merceron C, Schipani E, Shapiro IM, et al. Bicarbonate recycling by HIF-1-Dependent carbonic anhydrase isoforms 9 and 12 is critical in maintaining intracellular pH and viability of nucleus pulposus cells. J Bone mineral Res (2018) 33:338–55. doi: 10.1002/jbmr.3293
32. Sowa T, Menju T, Chen-Yoshikawa TF, Takahashi K, Nishikawa S, Nakanishi T, et al. Hypoxia-inducible factor 1 promotes chemoresistance of lung cancer by inducing carbonic anhydrase IX expression. Cancer Med (2017) 6:288–97. doi: 10.1002/cam4.991
33. McDonald PC, Chafe SC, Brown WS, Saberi S, Swayampakula M, Venkateswaran G, et al. Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology (2019) 157:823–37. doi: 10.1053/j.gastro.2019.05.004
34. Brahimi-Horn MC, Bellot G, Pouysségur J. Hypoxia and energetic tumour metabolism. Curr Opin Genet Dev (2011) 21:67–72. doi: 10.1016/j.gde.2010.10.006
35. Ito M, Tanaka T, Ishii T, Wakashima T, Fukui K, Nangaku M. Prolyl hydroxylase inhibition protects the kidneys from ischemia via upregulation of glycogen storage. Kidney Int (2020) 97:687–701. doi: 10.1016/j.kint.2019.10.020
36. Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab (2009) 9:512–24. doi: 10.1016/j.cmet.2009.05.005
37. Triantafyllou EA, Georgatsou E, Mylonis I, Simos G, Paraskeva E. Expression of AGPAT2, an enzyme involved in the glycerophospholipid/triacylglycerol biosynthesis pathway, is directly regulated by HIF-1 and promotes survival and etoposide resistance of cancer cells under hypoxia. Biochim Biophys Acta Mol Cell Biol Lipids (2018) 1863:1142–52. doi: 10.1016/j.bbalip.2018.06.015
38. Gimm T, Wiese M, Teschemacher B, Deggerich A, Schödel J, Knaup KX, et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J (2010) 24:4443–58. doi: 10.1096/fj.10-159806
39. Zhao H, Han Y, Jiang N, Li C, Yang M, Xiao Y, et al. Effects of HIF-1α on renal fibrosis in cisplatin-induced chronic kidney disease. Clin Sci (London Engl (2021) 1979) 135:1273–88. doi: 10.1042/CS20210061
40. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest (2007) 117:3810–20. doi: 10.1172/JCI30487
41. Li P, Liu Y, Qin X, Chen K, Wang R, Yuan L, et al. SIRT1 attenuates renal fibrosis by repressing HIF-2α. Cell Death Discov (2021) 7:59. doi: 10.1038/s41420-021-00443-x
42. Scholz CC, Cavadas MA, Tambuwala MM, Hams E, Rodríguez J, von Kriegsheim A, et al. Regulation of IL-1β-induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc Natl Acad Sci USA (2013) 110:18490–5. doi: 10.1073/pnas.1309718110
43. Remels AH, Gosker HR, Verhees KJ, Langen RC, Schols AM. TNF-α-induced NF-κB activation stimulates skeletal muscle glycolytic metabolism through activation of HIF-1α. Endocrinology (2015) 156:1770–81. doi: 10.1210/en.2014-1591
44. Yamaguchi J, Tanaka T, Eto N, Nangaku M. Inflammation and hypoxia linked to renal injury by CCAAT/enhancer-binding protein δ. Kidney Int (2015) 88:262–75. doi: 10.1038/ki.2015.21
45. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol (2018) 80:50–64. doi: 10.1016/j.semcdb.2017.05.023
46. Dada LA, Sznajder JI. Mitochondrial Ca²+ and ROS take center stage to orchestrate TNF-α-mediated inflammatory responses. J Clin Invest (2011) 121:1683–5. doi: 10.1172/JCI57748
47. Horak P, Crawford AR, Vadysirisack DD, Nash ZM, DeYoung MP, Sgroi D, et al. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci USA (2010) 107:4675–80. doi: 10.1073/pnas.0907705107
48. Xia L, Mo P, Huang W, Zhang L, Wang Y, Zhu H, et al. The TNF-α/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis (2012) 33:2250–9. doi: 10.1093/carcin/bgs249
49. Huang KT, Wu CT, Huang KH, Lin WC, Chen CM, Guan SS, et al. Titanium nanoparticle inhalation induces renal fibrosis in mice via an oxidative stress upregulated transforming growth factor-β pathway. Chem Res Toxicol (2015) 28:354–64. doi: 10.1021/tx500287f
50. Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol (2005) 17:548–58. doi: 10.1016/j.ceb.2005.08.001
51. Ning X, Zhang K, Wu Q, Liu M, Sun S. Emerging role of Twist1 in fibrotic diseases. J Cell Mol Med (2018) 22:1383–91. doi: 10.1111/jcmm.13465
52. Du R, Xia L, Ning X, Liu L, Sun W, Huang C, et al. Hypoxia-induced Bmi1 promotes renal tubular epithelial cell-mesenchymal transition and renal fibrosis via PI3K/Akt signal. Mol Biol Cell (2014) 25:2650–9. doi: 10.1091/mbc.e14-01-0044
53. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res (2016) 365:563–81. doi: 10.1007/s00441-016-2431-9
54. Liu J, Wang W, Wang L, Chen S, Tian B, Huang K, et al. IL-33 initiates vascular remodelling in hypoxic pulmonary hypertension by up-regulating HIF-1α and VEGF expression in vascular endothelial cells. EBioMedicine (2018) 33:196–210. doi: 10.1016/j.ebiom.2018.06.003
55. Luo Y, Teng X, Zhang L, Chen J, Liu Z, Chen X, et al. CD146-HIF-1α hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension. Nat Commun (2019) 10:3551. doi: 10.1038/s41467-019-11500-6
56. Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J (2003) 22:4082–90. doi: 10.1093/emboj/cdg392
57. Wang S, Zeng H, Chen ST, Zhou L, Xie XJ, He X, et al. Ablation of endothelial prolyl hydroxylase domain protein-2 promotes renal vascular remodelling and fibrosis in mice. J Cell Mol Med (2017) 21:1967–78. doi: 10.1111/jcmm.13117
58. Li X, Kimura H, Hirota K, Kasuno K, Torii K, Okada T, et al. Synergistic effect of hypoxia and TNF-alpha on production of PAI-1 in human proximal renal tubular cells. Kidney Int (2005) 68:569–83. doi: 10.1111/j.1523-1755.2005.00435.x
59. Norman JT, Clark IM, Garcia PL. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int (2000) 58:2351–66. doi: 10.1046/j.1523-1755.2000.00419.x
60. Kushida N, Nomura S, Mimura I, Fujita T, Yamamoto S, Nangaku M, et al. Hypoxia-inducible factor-1α activates the transforming growth factor-β/SMAD3 pathway in kidney tubular epithelial cells. Am J Nephrol (2016) 44:276–85. doi: 10.1159/000449323
61. You H, Gao T, Cooper TK, Morris SM Jr., Awad AS. Diabetic nephropathy is resistant to oral l-arginine or l-citrulline supplementation. Am J Physiol Renal Physiol (2014) 307:F1292–301. doi: 10.1152/ajprenal.00176.2014
62. Liang X, Potenza DM, Brenna A, Ma Y, Ren Z, Cheng X, et al. Hypoxia induces renal epithelial injury and activates fibrotic signaling through up-regulation of arginase-II. Front Physiol (2021) 12:773719. doi: 10.3389/fphys.2021.773719
63. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem (1994) 269:23757–63. doi: 10.1016/S0021-9258(17)31580-6
64. Szablewski L. Distribution of glucose transporters in renal diseases. J Biomed Sci (2017) 24:64. doi: 10.1186/s12929-017-0371-7
65. Sędzikowska A, Szablewski L. Human glucose transporters in renal glucose homeostasis. Int J Mol Sci (2021) 22(24):13522. doi: 10.3390/ijms222413522
66. Zuo J, Wen J, Lei M, Wen M, Li S, Lv X, et al. Hypoxia promotes the invasion and metastasis of laryngeal cancer cells via EMT. Med Oncol (2016) 33:15. doi: 10.1007/s12032-015-0716-6
67. Wang Y, Heilig K, Saunders T, Minto A, Deb DK, Chang A, et al. Transgenic overexpression of GLUT1 in mouse glomeruli produces renal disease resembling diabetic glomerulosclerosis. Am J Physiol Renal Physiol (2010) 299:F99–f111. doi: 10.1152/ajprenal.00466.2009
68. Young RJ, Möller A. Immunohistochemical detection of tumour hypoxia. Methods Mol Biol (2010) 611:151–9. doi: 10.1007/978-1-60327-345-9_12
69. Elson DA, Ryan HE, Snow JW, Johnson R, Arbeit JM. Coordinate up-regulation of hypoxia inducible factor (HIF)-1alpha and HIF-1 target genes during multi-stage epidermal carcinogenesis and wound healing. Cancer Res (2000) 60:6189–95.
70. Samec M, Liskova A, Koklesova L, Samuel SM, Zhai K, Buhrmann C, et al. Flavonoids against the warburg phenotype-concepts of predictive, preventive and personalised medicine to cut the Gordian knot of cancer cell metabolism. EPMA J (2020) 11:377–98. doi: 10.1007/s13167-020-00217-y
71. Zambrano A, Molt M, Uribe E, Salas M. Glut 1 in cancer cells and the inhibitory action of resveratrol as a potential therapeutic strategy. Int J Mol Sci (2019) 20(13):3374. doi: 10.3390/ijms20133374
72. Rabbani N, Xue M, Thornalley PJ. Hexokinase-2-Linked glycolytic overload and unscheduled glycolysis-driver of insulin resistance and development of vascular complications of diabetes. Int J Mol Sci (2022) 23(4):2165. doi: 10.3390/ijms23042165
73. Zuo J, Tang J, Lu M, Zhou Z, Li Y, Tian H, et al. Glycolysis rate-limiting enzymes: Novel potential regulators of rheumatoid arthritis pathogenesis. Front Immunol (2021) 12:779787. doi: 10.3389/fimmu.2021.779787
74. Rabbani N, Thornalley PJ. Hexokinase-2 glycolytic overload in diabetes and ischemia-reperfusion injury. Trends Endocrinol metabolism: TEM (2019) 30:419–31. doi: 10.1016/j.tem.2019.04.011
75. Wu Z, Wu J, Zhao Q, Fu S, Jin J. Emerging roles of aerobic glycolysis in breast cancer. Clin Trans Oncol (2020) 22:631–46. doi: 10.1007/s12094-019-02187-8
76. Chen G, Zhang Y, Liang J, Li W, Zhu Y, Zhang M, et al. Deregulation of hexokinase II is associated with glycolysis, autophagy, and the epithelial-mesenchymal transition in tongue squamous cell carcinoma under hypoxia. BioMed Res Int (2018) 2018:8480762. doi: 10.1155/2018/8480762
77. Du D, Liu C, Qin M, Zhang X, Xi T, Yuan S, et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sinica B (2022) 12:558–80. doi: 10.1016/j.apsb.2021.09.019
78. Sola-Penna M, Da Silva D, Coelho WS, Marinho-Carvalho MM, Zancan P. Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life (2010) 62:791–6. doi: 10.1002/iub.393
79. Mor I, Cheung EC, Vousden KH. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harbor Symp Quantitative Biol (2011) 76:211–6. doi: 10.1101/sqb.2011.76.010868
80. Bartrons R, Simon-Molas H, Rodríguez-García A, Castaño E, Navarro-Sabaté À., Manzano A, et al. Fructose 2,6-bisphosphate in cancer cell metabolism. Front Oncol (2018) 8:331. doi: 10.3389/fonc.2018.00331
81. Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem (2009) 284:24223–32. doi: 10.1074/jbc.M109.016816
82. Tawakol A, Singh P, Mojena M, Pimentel-Santillana M, Emami H, MacNabb M, et al. HIF-1α and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arteriosclerosis thrombosis Vasc Biol (2015) 35:1463–71. doi: 10.1161/ATVBAHA.115.305551
83. Li J, Zhang S, Liao D, Zhang Q, Chen C, Yang X, et al. Overexpression of PFKFB3 promotes cell glycolysis and proliferation in renal cell carcinoma. BMC Cancer (2022) 22:83. doi: 10.1186/s12885-022-09183-2
84. Zhou ZY, Wang L, Wang YS, Dou GR. PFKFB3: A potential key to ocular angiogenesis. Front Cell Dev Biol (2021) 9:628317. doi: 10.3389/fcell.2021.628317
85. Li X, Kumar A, Carmeliet P. Metabolic pathways fueling the endothelial cell drive. Annu Rev Physiol (2019) 81:483–503. doi: 10.1146/annurev-physiol-020518-114731
86. Min J, Zeng T, Roux M, Lazar D, Chen L, Tudzarova S. The role of HIF1α-PFKFB3 pathway in diabetic retinopathy. J Clin Endocrinol Metab (2021) 106:2505–19. doi: 10.1210/clinem/dgab362
87. Alquraishi M, Puckett DL, Alani DS, Humidat AS, Frankel VD, Donohoe DR, et al. Pyruvate kinase M2: A simple molecule with complex functions. Free Radical Biol Med (2019) 143:176–92. doi: 10.1016/j.freeradbiomed.2019.08.007
88. Wang P, Sun C, Zhu T, Xu Y. Structural insight into mechanisms for dynamic regulation of PKM2. Protein Cell (2015) 6:275–87. doi: 10.1007/s13238-015-0132-x
89. Lebelo MT, Joubert AM, Visagie MH. Warburg effect and its role in tumourigenesis. Arch Pharmacal Res (2019) 42:833–47. doi: 10.1007/s12272-019-01185-2
90. Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol (2007) 27:7381–93. doi: 10.1128/MCB.00440-07
91. Wang JZ, Zhu W, Han J, Yang X, Zhou R, Lu HC, et al. The role of the HIF-1α/ALYREF/PKM2 axis in glycolysis and tumorigenesis of bladder cancer. Cancer Commun (2021) 41:560–75. doi: 10.1002/cac2.12158
92. Hamabe A, Konno M, Tanuma N, Shima H, Tsunekuni K, Kawamoto K, et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc Natl Acad Sci USA (2014) 111:15526–31. doi: 10.1073/pnas.1407717111
93. Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell (2011) 145:732–44. doi: 10.1016/j.cell.2011.03.054
94. Kim SY, Sohn SJ, Won AJ, Kim HS, Moon A. Identification of noninvasive biomarkers for nephrotoxicity using HK-2 human kidney epithelial cells. Toxicol Sci (2014) 140:247–58. doi: 10.1093/toxsci/kfu096
95. Li J, Liu H, Takagi S, Nitta K, Kitada M, Srivastava SP, et al. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight (2020) 5(6):e129034. doi: 10.1172/jci.insight.129034
96. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med (2017) 23:753–62. doi: 10.1038/nm.4328
97. Srivastava SP, Li J, Kitada M, Fujita H, Yamada Y, Goodwin JE, et al. SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis (2018) 9:997. doi: 10.1038/s41419-018-1057-0
98. McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem (2008) 283:22700–8. doi: 10.1074/jbc.M801765200
99. Anwar S, Shamsi A, Mohammad T, Islam A, Hassan MI. Targeting pyruvate dehydrogenase kinase signaling in the development of effective cancer therapy. Biochim Biophys Acta Rev Cancer (2021) 1876:188568. doi: 10.1016/j.bbcan.2021.188568
100. Woolbright BL, Rajendran G, Harris RA, Taylor JA. 3rd, metabolic flexibility in cancer: Targeting the pyruvate dehydrogenase Kinase:Pyruvate dehydrogenase axis. Mol Cancer Ther (2019) 18:1673–81. doi: 10.1158/1535-7163.MCT-19-0079
101. Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J (2007) 405:1–9. doi: 10.1042/BJ20070389
102. Rosenbaum FP, Poehlein A, Egelkamp R, Daniel R, Harder S, Schlüter H, et al. Lactate metabolism in strictly anaerobic microorganisms with a soluble NAD(+) -dependent l-lactate dehydrogenase. Environ Microbiol (2021) 23:4661–72. doi: 10.1111/1462-2920.15657
103. Xiang S, Huang D, He Q, Li J, Tam KY, Zhang SL, et al. Development of dual inhibitors targeting pyruvate dehydrogenase kinases and human lactate dehydrogenase a: High-throughput virtual screening, synthesis and biological validation. Eur J Med Chem (2020) 203:112579. doi: 10.1016/j.ejmech.2020.112579
104. Podar K, Anderson KC. A therapeutic role for targeting c-Myc/Hif-1-dependent signaling pathways. Cell Cycle (2010) 9:1722–8. doi: 10.4161/cc.9.9.11358
105. Deblois G, Giguère V. Oestrogen-related receptors in breast cancer: control of cellular metabolism and beyond. Nat Rev Cancer (2013) 13:27–36. doi: 10.1038/nrc3396
106. Cai Q, Lin T, Kamarajugadda S, Lu J. Regulation of glycolysis and the warburg effect by estrogen-related receptors. Oncogene (2013) 32:2079–86. doi: 10.1038/onc.2012.221
107. Stacpoole PW. Therapeutic targeting of the pyruvate dehydrogenase Complex/Pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst (2017) 109(11):djx071. doi: 10.1093/jnci/djx071
108. Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol (2017) 45:62–71. doi: 10.1016/j.ceb.2017.02.007
109. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell (2017) 170:605–35. doi: 10.1016/j.cell.2017.07.029
110. Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z, et al. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis (2020) 11:797. doi: 10.1038/s41419-020-02998-6
111. Gao T, Zhang X, Zhao J, Zhou F, Wang Y, Zhao Z, et al. SIK2 promotes reprogramming of glucose metabolism through PI3K/AKT/HIF-1α pathway and Drp1-mediated mitochondrial fission in ovarian cancer. Cancer Lett (2020) 469:89–101. doi: 10.1016/j.canlet.2019.10.029
112. Park JH, Pyun WY, Park HW. Cancer metabolism: Phenotype, signaling and therapeutic targets. Cells (2020) 9(10):2308. doi: 10.1053/j.ajkd.2006.03.010
113. Karar J, Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci (2011) 4:51. doi: 10.3389/fnmol.2011.00051
114. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin Cancer Biol (2019) 59:147–60. doi: 10.1016/j.semcancer.2019.05.012
115. Gong T, Cui L, Wang H, Wang H, Han N. Knockdown of KLF5 suppresses hypoxia-induced resistance to cisplatin in NSCLC cells by regulating HIF-1α-dependent glycolysis through inactivation of the PI3K/Akt/mTOR pathway. J Trans Med (2018) 16:164. doi: 10.1186/s12967-018-1543-2
116. Weng ML, Chen WK, Chen XY, Lu H, Sun ZR, Yu Q, et al. Fasting inhibits aerobic glycolysis and proliferation in colorectal cancer via the Fdft1-mediated AKT/mTOR/HIF1α pathway suppression. Nat Commun (2020) 11:1869. doi: 10.1038/s41467-020-15795-8
117. Chen CY, Chen J, He L, Stiles BL. PTEN: Tumor suppressor and metabolic regulator. Front Endocrinol (2018) 9:338. doi: 10.3389/fendo.2018.00338
118. Crespo-Masip M, Pérez-Gómez A, Guzmán C, Rayego S, Doladé N, García-Carrasco A, et al. PTEN deletion in adult mice induces hypoinsulinemia with concomitant low glucose levels. Front Endocrinol (2022) 13:850214. doi: 10.3389/fendo.2022.850214
119. Yang X, Cheng Y, Li P, Tao J, Deng X, Zhang X, et al. A lentiviral sponge for miRNA-21 diminishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumour Biol (2015) 36:383–91. doi: 10.1007/s13277-014-2617-2
120. Hu X, Xu Q, Wan H, Hu Y, Xing S, Yang H, et al. PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab Invest (2020) 100:801–11. doi: 10.1038/s41374-020-0404-9
121. Zhou S, Ai Z, Li W, You P, Wu C, Li L, et al. Deciphering the pharmacological mechanisms of taohe-chengqi decoction extract against renal fibrosis through integrating network pharmacology and experimental validation In vitro and in vivo. Front Pharmacol (2020) 11:425. doi: 10.3389/fphar.2020.00425
122. Levin A, Rocco M. KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis (2006) 47:S11–145.
123. Thavarajah S, Choi MJ. The use of erythropoiesis-stimulating agents in patients with CKD and cancer: A clinical approach. Am J Kidney Dis (2019) 74:667–74. doi: 10.1053/j.ajkd.2019.04.022
124. Chen N, Hao C, Liu BC, Lin H, Wang C, Xing C, et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. New Engl J Med (2019) 381:1011–22. doi: 10.1056/NEJMoa1901713
125. Provenzano R, Szczech L, Leong R, Saikali KG, Zhong M, Lee TT, et al. Efficacy and cardiovascular safety of roxadustat for treatment of anemia in patients with non-Dialysis-Dependent CKD: Pooled results of three randomized clinical trials. Clin J Am Soc Nephrol (2021) 16:1190–200. doi: 10.2215/CJN.16191020
126. Li ZL, Lv LL, Wang B, Tang TT, Feng Y, Cao JY, et al. The profibrotic effects of MK-8617 on tubulointerstitial fibrosis mediated by the KLF5 regulating pathway. FASEB J (2019) 33:12630–43. doi: 10.1096/fj.201901087RR
127. Sugahara M, Tanaka S, Tanaka T, Saito H, Ishimoto Y, Wakashima T, et al. Prolyl hydroxylase domain inhibitor protects against metabolic disorders and associated kidney disease in obese type 2 diabetic mice. J Am Soc Nephrol (2020) 31:560–77. doi: 10.1681/ASN.2019060582
128. Hasegawa S, Tanaka T, Saito T, Fukui K, Wakashima T, Susaki EA, et al. The oral hypoxia-inducible factor prolyl hydroxylase inhibitor enarodustat counteracts alterations in renal energy metabolism in the early stages of diabetic kidney disease. Kidney Int (2020) 97:934–50. doi: 10.1016/j.kint.2019.12.007
129. Masoud GN, Li W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sinica B (2015) 5:378–89. doi: 10.1016/j.apsb.2015.05.007
130. Lai Y, Tang F, Huang Y, He C, Chen C, Zhao J, et al. The tumour microenvironment and metabolism in renal cell carcinoma targeted or immune therapy. J Cell Physiol (2021) 236:1616–27. doi: 10.1002/jcp.29969
131. Considine B, Hurwitz ME. Current status and future directions of immunotherapy in renal cell carcinoma. Curr Oncol Rep (2019) 21:34. doi: 10.1007/s11912-019-0779-1
Keywords: hypoxia-inducible factor-1α, aerobic glycolysis, oxidative phosphorylation, metabolic reprogramming, renal fibrosis
Citation: Wei X, Hou Y, Long M, Jiang L and Du Y (2022) Molecular mechanisms underlying the role of hypoxia-inducible factor-1 α in metabolic reprogramming in renal fibrosis. Front. Endocrinol. 13:927329. doi: 10.3389/fendo.2022.927329
Received: 24 April 2022; Accepted: 30 June 2022;
Published: 25 July 2022.
Edited by:
Nehal Mohsen Elsherbiny, Mansoura University, EgyptReviewed by:
Ilias Mylonis, University of Thessaly, GreeceCopyright © 2022 Wei, Hou, Long, Jiang and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yujun Du, ZHV5akBqbHUuZWRjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.