 William K. Storck1,2†
William K. Storck1,2† Allison M. May2,3†
Allison M. May2,3† Thomas C. Westbrook1,2†
Thomas C. Westbrook1,2† Zhi Duan1,2
Zhi Duan1,2 Colm Morrissey4
Colm Morrissey4 Joel A. Yates1,2
Joel A. Yates1,2 Joshi J. Alumkal1,2*
Joshi J. Alumkal1,2*- 1Department of Internal Medicine, University of Michigan, Ann Arbor, MI, United States
- 2Rogel Cancer Center, University of Michigan, Ann Arbor, MI, United States
- 3Department of Urology, University of Michigan, Ann Arbor, MI, United States
- 4Department of Urology, University of Washington, Seattle, WA, United States
The androgen receptor (AR) signaling pathway is critical for growth and differentiation of prostate cancer cells. For that reason, androgen deprivation therapy with medical or surgical castration is the principal treatment for metastatic prostate cancer. More recently, new potent AR signaling inhibitors (ARSIs) have been developed. These drugs improve survival for men with metastatic castration-resistant prostate cancer (CRPC), the lethal form of the disease. However, ARSI resistance is nearly universal. One recently appreciated resistance mechanism is lineage plasticity or switch from an AR-driven, luminal differentiation program to an alternate differentiation program. Importantly, lineage plasticity appears to be increasing in incidence in the era of new ARSIs, strongly implicating AR suppression in this process. Lineage plasticity and shift from AR-driven tumors occur on a continuum, ranging from AR-expressing tumors with low AR activity to AR-null tumors that have activation of alternate differentiation programs versus the canonical luminal program found in AR-driven tumors. In many cases, AR loss coincides with the activation of a neuronal program, most commonly exemplified as therapy-induced neuroendocrine prostate cancer (t-NEPC). While genetic events clearly contribute to prostate cancer lineage plasticity, it is also clear that epigenetic events—including chromatin modifications and DNA methylation—play a major role. Many epigenetic factors are now targetable with drugs, establishing the importance of clarifying critical epigenetic factors that promote lineage plasticity. Furthermore, epigenetic marks are readily measurable, demonstrating the importance of clarifying which measurements will help to identify tumors that have undergone or are at risk of undergoing lineage plasticity. In this review, we discuss the role of AR pathway loss and activation of a neuronal differentiation program as key contributors to t-NEPC lineage plasticity. We also discuss new epigenetic therapeutic strategies to reverse lineage plasticity, including those that have recently entered clinical trials.
Introduction
Prostate cancer is the second leading cause of cancer-related death in men in the United States with an estimated 34,500 deaths predicted for 2022 (1). Since the discovery that the majority of prostate cancers respond to androgen depletion (2), androgen deprivation therapy (ADT) has been the principal treatment for metastatic tumors. ADT is also commonly used as adjuvant therapy with surgery or irradiation (3). While most metastatic tumors initially respond to ADT, many tumors will eventually progress to the lethal, castration-resistant form of the disease (4–7). We now know that androgen levels sufficient to activate the androgen receptor (AR) are commonly found in castration-resistant prostate cancer (CRPC) due to intratumoral androgen synthesis or metabolism of adrenally-produced androgen precursors (8). In the past decade, novel AR signaling inhibitors (ARSIs) that inhibit androgen biosynthesis—such as abiraterone acetate (9)—or that competitively bind to the AR and interfere with androgen activation—such as enzalutamide (10), apalutamide (11), and darolutamide (12)—have been tested and approved for the treatment of CRPC.
However, even though ARSIs prolong survival, resistance is nearly universal, and there are limited treatment options once tumors become resistant. Resistance to ARSI treatment may be broken down into two major categories: AR signaling-dependent and AR signaling-independent (13). Maintenance of AR signaling despite continued treatment with ARSIs occurs through multiple mechanisms, including alterations to the AR itself or through compensation by other factors such as the glucocorticoid receptor (14–18).
An AR signaling-independent state is defined by reduced reliance on the AR and activation of other factors, such as MYCN and AURKA that promote cell survival (19). One such AR-independent resistance mechanism is lineage plasticity, wherein tumor cells switch from an AR-driven, luminal differentiation program to an alternate differentiation program (20). Recent work has clarified that lineage plasticity occurs on a continuum and that there are distinct subsets. These subsets include: AR activity-low tumors with decreased AR signaling despite persistent AR expression, amphicrine tumors that have both active AR and neuronal programs in the same cell, double negative tumors that lack AR expression but that do not express a neuronal program, and finally neuroendocrine prostate cancer (NEPC) tumors that lack AR expression, but activate a neuronal program (13).
A key challenge in developing effective treatments for patients with advanced prostate cancer is inter- and even intra-patient tumor heterogeneity (21–25). Therefore, understanding mechanisms by which tumors undergo lineage plasticity may lead to therapeutic approaches to prevent or reverse this virulent form of treatment resistance. In this review, we focus on the NEPC subtype and summarize the epigenetic changes that contribute to NEPC lineage plasticity, discuss methods to identify and classify NEPC, and discuss promising treatment strategies. An overview of selected factors and molecular events discussed in this review that contribute to NEPC lineage plasticity are highlighted in Figure 1.
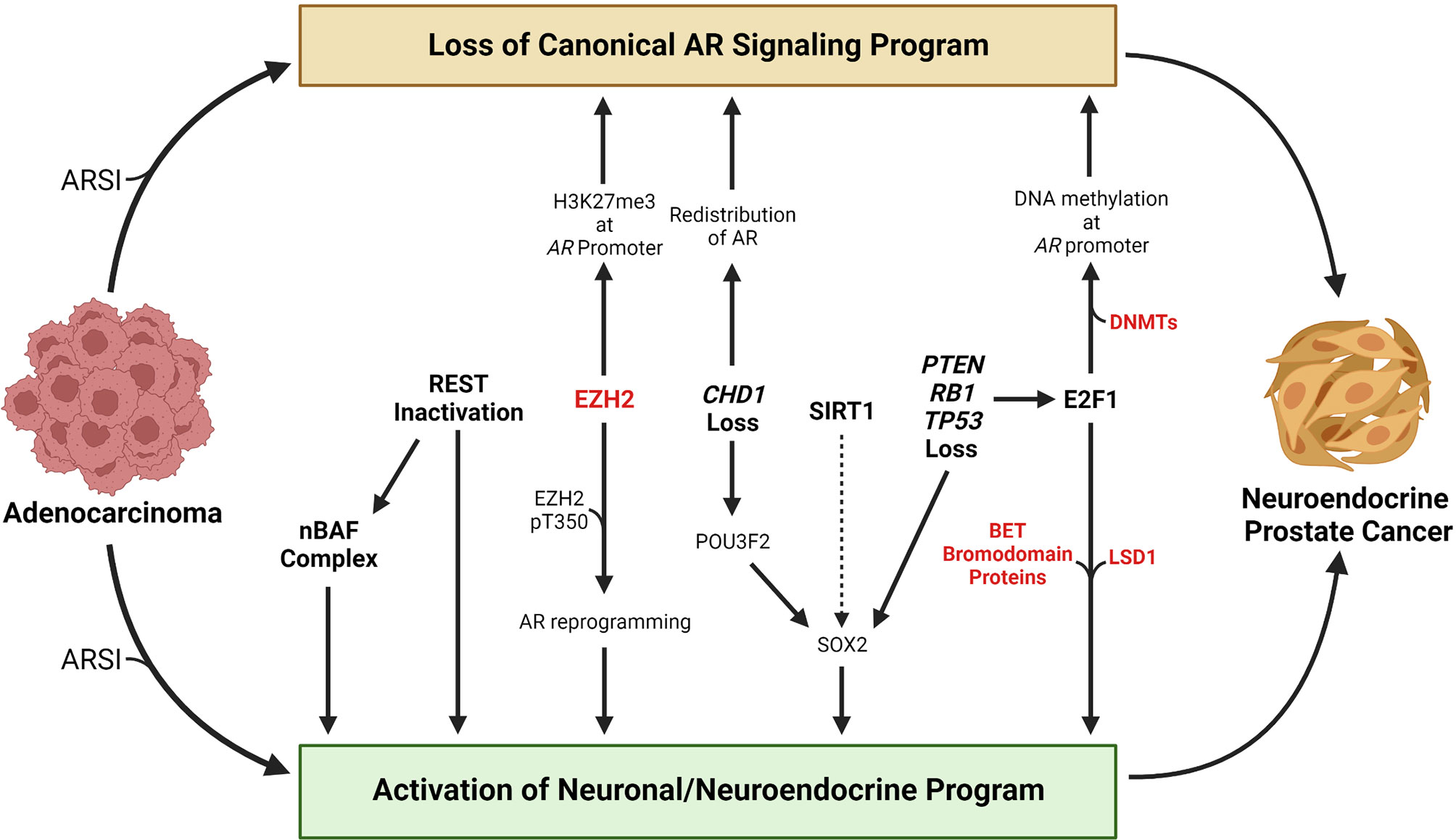
Figure 1 Overview of selected molecular events that contribute to the transition from adenocarcinoma to neuroendocrine prostate cancer. Loss of a canonical AR signaling program and activation of a neuronal or neuroendocrine program are hallmarks of the transition from adenocarcinoma to neuroendocrine prostate cancer. Treatment with AR signaling inhibitors (ARSIs) results in suppression of canonical AR signaling and activation of AR-repressed genes linked with a neuronal or neuroendocrine program in some tumors. Loss of REST’s repressive activity promotes NEPC lineage plasticity, in part through activation of the nBAF chromatin remodeling complex. EZH2 has been shown to silence AR expression by catalyzing H3K27me3 at the AR promoter. Phosphorylation of the T350 residue on EZH2 results in reprograming of the AR cistrome and activation of stemness or neuronal genes. Loss of the chromatin remodeling factor CHD1 results in redistribution of the AR away from canonical AR target genes to tumor-specific AR-bound regions. CHD1 loss also increases chromatin accessibility for the neuronal transcription factor POU3F2 that may contribute to activation of the stemness factor SOX2. The HDAC SIRT1 promotes NEPC lineage plasticity, potentially through activation of SOX2. PTEN, RB1, and TP53 are commonly altered in NEPC, and their loss promotes activation of a neuronal program in part through activation of E2F1 or SOX2. E2F1 has been shown to cooperate with DNA methyltransferases to silence the AR through AR promoter DNA methylation. E2F1 also cooperates with LSD1 or BET bromodomain chromatin readers to activate a cell cycle or neuronal program. Factors that are currently targetable with drugs that are approved by the Food & Drug Administration or that are in clinical trials are shown in red. Created with BioRender.com.
The Role of AR Pathway Loss in NEPC Lineage Plasticity
It is now well-appreciated that the frequency of tumors with AR pathway loss is increasing in the era of novel ARSIs (26, 27). While de novo NEPC comprises less than 1% of newly diagnosed prostate tumors, therapy-induced (t)-NEPC emerging after ARSI treatment represents approximately 20% of CRPC tumors in recent biopsy studies (26, 28). Further, the frequency of NEPC tumors found in a rapid autopsy series appears to be increasing in the era of novel ARSIs (27). This increase in prevalence strongly suggests that ARSIs play a role in the transition of tumors to an NEPC phenotype.
Several studies have confirmed that t-NEPC tumors—like de novo NEPC tumors—exhibit significantly lower canonical AR signaling. In a series of metastatic biopsies from men whose tumors were progressing on ARSIs, Aggarwal, et al. performed unsupervised clustering analysis on RNA sequencing data. They identified a distinct cluster termed cluster 2 that was highly enriched for t-NEPC tumors (26). Master regulator analysis demonstrated that the AR was predicted to be the most deactivated transcription factor in cluster 2 tumors compared to the other four clusters. In another report by Beltran, et al., AR mutations—often gain of function events—were commonly found in adenocarcinoma CRPC tumors but were notably absent from t-NEPC tumors (29). These data further suggest that canonical AR signaling becomes dispensable in t-NEPC.
Interestingly, in the report by Aggarwal, et al., approximately 75% of t-NEPC tumors continued to express the AR (26). However, canonical AR signaling was absent or decreased. Importantly, patients were still taking ARSIs at the time of biopsy in many cases, which may have contributed to loss of AR function in the t-NEPC tumors with persistent AR expression. The inverse relationship between canonical AR signaling and activation of an NEPC program suggests that loss of AR function—including through pharmacologic agents—may promote lineage plasticity.
A key function of the AR is to regulate proliferation and differentiation of normal and transformed luminal prostate cells (30). The developing prostate is dependent on androgen signaling, and castration induces involution of the prostate (31–34). In vitro, androgen-induced luminal differentiation of prostate epithelial cells is dependent on AR expression (35, 36). Furthermore, tissue recombination experiments using urinary tract epithelia from wild-type and androgen-insensitive mice highlight the importance of the AR for promoting luminal differentiation and the secretory functions of prostatic epithelial cells (37). Previous studies examining the effects of androgen deprivation on prostate cells showed that loss of androgen signaling results in significant apoptosis and dedifferentiation of luminal cells (38–40). Studies using conditional knock out mice deficient for the AR specifically in prostate epithelia revealed that epithelial cell-specific loss of the AR prevents luminal differentiation and leads to loss of glandular in-folding and eventual epithelial sloughing into the prostate lumen (41). These results demonstrate that AR signaling is essential for maintenance of luminal differentiation. Indeed, transcriptional analysis of prostate luminal vs. basal cell populations determined that AR target gene expression is strongly linked with a luminal program, and luminal cells could be distinguished from basal cells based on activation of canonical AR target genes (42, 43). In contrast, the gene expression program of basal cells was activated in aggressive human prostate tumors linked with higher risk of tumor metastasis and shorter survival (43, 44). In our own work, we determined that tumors that exhibit de novo resistance to the ARSI enzalutamide have lower AR activity and activation of a basal stemness program, strongly suggesting that AR-independent tumors may be more aggressive (45). These de novo resistant tumors also had activation of gene signatures linked to NEPC (45, 46). Overall, these results suggest that in some tumors—perhaps due to the cell of origin (47, 48)—loss of AR signaling may contribute to lineage plasticity, at least in part through loss of a luminal differentiation program.
While the correlation between AR pathway loss and lineage plasticity is clear, recent work sheds new light on how loss of AR signaling—including through ARSIs—might contribute to NEPC lineage plasticity. ADT expands stem-like cells while suppressing non-stem-like cells in tumor samples from patients before and after undergoing androgen deprivation (49). Studies on the highly castration-sensitive patient-derived xenograft (PDX) model BM18 revealed that ADT enriches for a population of prostate cancer cells with both stem-like and luminal characteristics and low AR expression (50). Upon androgen stimulation, these cells proliferate, suggesting that these AR-low cells may promote tumor repopulation after ADT (51).
Recent work by Bishop, et al. demonstrated that the AR also represses activation of alternate differentiation programs, thus helping to maintain luminal identity (52). They developed enzalutamide-resistant cell lines called MR42D and MR42F that continue to express the AR but that have low canonical AR activity (52). These cells harbor a stem-like program with activation of the neuronal transcription factor BRN2 (POU3F2) (53)—a direct target gene repressed by the AR (52). BRN2 was found to activate SOX2, which may contribute to BRN2’s effects on promoting NEPC lineage plasticity (52).
Using these same models, our group recently clarified additional mechanisms that contribute to AR pathway loss-induced NEPC lineage plasticity (54). We determined that enzalutamide treatment of MR42D and MR42F—but not AR-driven parental LNCaP or the CRPC LNCaP derivative line V16D—activates an AR-repressed NEPC lineage plasticity program (54). The chromatin state of MR42D cells appeared to be more conducive to activation of this program, and we determined that the master regulator transcription factor E2F1—in cooperation with the chromatin reader BET bromodomain protein BRD4—appeared to be critical for NEPC reprogramming (54). Using both pre-clinical models and pre-treatment biopsies from a recently completed ZEN-3694 BET bromodomain inhibitor clinical trial (55), we determined that a subset of NEPC tumors harboring high expression of the AR-repressed, E2F1-activated NEPC lineage plasticity program we identified in MR42D cells was strongly linked to prolonged tumor control with BET bromodomain inhibitor treatment. This suggests that these tumors may be particularly susceptible to BET bromodomain inhibition (54). Finally, prior work using LNCaP and LNCaP C-33 cell lines grown in androgen-depleted conditions demonstrated that androgen deprivation is sufficient to induce an NEPC phenotype by inducing RPTPα expression, leading to activation of the MEK/ERK pathway to promote neuroendocrine transdifferentiation (56, 57). Thus, loss of AR function may lead to NEPC lineage plasticity through activation of multiple reprogramming factors.
While activation of AR-repressed genes appears to play an important role in lineage plasticity, recent work by Davies, et al. suggests the AR may also be reprogrammed during the transition to NEPC (58). In that report, they used MR42D and MR42F cells and showed that AR cooperates with the polycomb protein EZH2 in a noncanonical polycomb complex to activate a subset of genes linked to a neuronal program (58). They found that MR42D and MR42F cells have phosphorylation of EZH2 at the T350 residue (58). This post-translational modification is associated with EZH2 activation and activation of NEPC and lineage plasticity-related factors (58). AR and pEZH2-T350 colocalized at non-canonical AR target genes—genes associated with stem cell programs—suggesting that phosphorylation of EZH2 may be associated with redistribution of the AR (58). Importantly, the AR- and pEZH2-T350-bound regions they identified by chromatin immunoprecipitation (ChIP) sequencing were not marked by high levels of the histone methylation mark catalyzed by EZH2—H3K27me3—demonstrating histone methylation-independent mechanisms of EZH2 function (58). Interestingly, EZH2 inhibition reversed the NEPC phenotype, restored canonical AR signaling, and re-sensitized cells to enzalutamide (58). Importantly, the authors also deleted the AR with CRISPR-Cas9, which resulted in further NEPC differentiation (58). These results indicate that even though the AR may be reprogrammed to activate stemness and NEPC genes in cooperation with EZH2, AR’s dominant role in the cells examined may be to repress NEPC differentiation.
The Role of PTEN, RB1, and TP53 Loss in NEPC Lineage Plasticity
Several studies have investigated the genomic alterations associated with NEPC tumors (5, 26, 29). Three important tumor suppressor genes, PTEN, RB1, and TP53 are commonly altered in human NEPC vs. AR-dependent prostate cancer (5, 26, 29). Interestingly, these tumor suppressor genes are also known to be important in aggressive neuroendocrine tumors from other organs, including small cell lung cancer (59). Loss of more than one of these tumor suppressor genes is common in NEPC (26, 29).
Pre-clinical studies with cell lines or genetically engineered mouse models have confirmed that loss of PTEN, RB1, and TP53, are synergistic for reprogramming prostate epithelial cells and promoting NEPC lineage plasticity. Indeed, Mu, et al. demonstrated that combined knockdown of RB1 and TP53 in PTEN-null LNCaP cells led to lower AR expression, resistance to enzalutamide treatment, and activation of an NEPC program (60). RB1 and TP53 loss was linked to increased expression of SOX2, which has also been associated with RB1 and TP53 loss in human NEPC (26). SOX2 knockdown abrogated the induction of the lineage plasticity program caused by RB1/TP53 loss and re-sensitized cells to enzalutamide, strongly suggesting that SOX2 was a key downstream effector (60).
Similarly, Ku, et al. developed genetically engineered mouse models to study the impact of PTEN loss alone (SKO); PTEN and RB1 loss (DKO); or PTEN, RB1, and TP53 loss (TKO) (61). Compared to SKO, DKO and TKO tumors exhibited lower AR expression and had activation of an NEPC program (61). DKO and TKO tumors were resistant to castration, and mice bearing these tumors had a shorter overall survival vs. SKO mice (61). The authors studied mechanisms that contribute to tumor survival despite surgical castration of mice bearing DKO tumors (recurrent tumors are termed DKO-Cr). Interestingly, they determined that some DKO-Cr tumors acquired TP53 loss of function mutations (61). This suggests that TP53 genetic loss may be selected for in RB1-deficient tumors after ADT. Like Mu, et al. (60), Ku, et al. determined that DKO and TKO tumors exhibited increased expression of SOX2 (61).
Furthermore, evidence for the importance of RB1 and TP53 loss was provided by Park, et al., who sought to identify factors that cooperate to promote NEPC (62). The authors used normal basal prostate epithelial cells and altered the expression or function of several candidate reprogramming factors to create PARCB tumors which contained a dominant negative TP53, constitutively activated myristoylated AKT1 (myrAKT1), RB1-short hairpin RNA, overexpression of c-Myc, and overexpression of BCL2 (62). These cells were cultured in an organoid system and then transplanted into immunodeficient mice. The authors then measured tumor growth or NEPC differentiation. To define which factors were necessary to induce NEPC, the authors performed leave-one-out analysis and found that no tumors grew in the absence of c-Myc or myrAKT1 (62). RB1 and TP53 loss—together or individually—were indispensable for NEPC (62). This further demonstrates that loss of RB1 and inactivation of TP53 are important factors in the conversion of cells to NEPC.
Despite a very clear association between PTEN, RB1, and TP53 loss and the NEPC phenotype, these genomic changes alone are insufficient to promote lineage plasticity in patient tumors. Indeed, many NEPC tumors do not exhibit these genomic alterations, and some non-NEPC CRPC tumors harbor genomic alterations in these genes (29, 63). For example, Beltran, et al. determined that only 70% of NEPC tumors harbored RB1 loss vs. 32% of CRPC. Sixty-seven percent of NEPC tumors harbored TP53 mutations vs. 31% of CRPC (29). Nyquist, et al. profiled 410 metastatic biopsies from patients with CRPC and found that 40% of those harboring combined RB1/TP53 loss were adenocarcinomas (63). Furthermore, Nyquist, et al. generated combined RB1/TP53 knockouts in PTEN-deficient LNCaP cells using CRISPR-Cas9. While AR transcriptional activity was lower in the knockout cells vs. parental cells, NEPC gene expression was not increased (63). This suggests that other reprogramming factors or the tumor microenvironment that was absent from these in vitro-generated cell lines may be important to induce NEPC. Finally, given the importance of RB1 in restraining proliferation, it is also possible that RB1 loss in a subset of tumor cells that had already undergone NEPC lineage plasticity from an adenocarcinoma phenotype may provide a proliferative advantage, accelerating the shift in the tumor population to an NEPC phenotype.
The Role of Epigenetic Change in NEPC Lineage Plasticity
Gene expression is controlled not only by DNA sequence alterations, but also by epigenetic mechanisms such as DNA methylation, histone modifications, and chromatin accessibility. There is a wealth of data demonstrating the importance of epigenetic change for NEPC lineage plasticity. Indeed, loss of AR signaling in prostate cancer lineage plasticity is strongly associated with extensive epigenetic reprograming (64). Distinct DNA methylation and histone modification profiles have been shown to be correlated with tumor progression (65–68), and various epigenetic factors have been shown to induce stemness or epithelial-mesenchymal transition (EMT) (61, 69, 70).
DNA Methylation
DNA methylation is an important epigenetic mark wherein a methyl group is covalently attached to the 5’ carbon of the pyrimidine rings on cytosines, and these methylation patterns are retained after cell division (71). Maintenance DNA methylation is catalyzed by the DNA methyltransferase DNMT1 that prefers hemimethylated DNA as its substrate. DNMT3A and DNMT3B are capable of methylating both hemimethylated and non-methylated DNA and are referred to as de novo methyltransferases (71).
Expression of DNMT1 is increased in prostate epithelial cells with loss of RB1 (72). Mechanistically, RB1 negatively regulates the transcription factor E2F1, and E2F1 can activate DNMT1 expression through interactions with the DNMT1 promoter (72). Accordingly, overexpression of E2F1 increases DNMT1 expression (72). E2F1 overexpression was sufficient to repress expression of the AR and AR promoter-driven reporter constructs while DNMT1 knockdown reactivated AR expression in AR-negative human primary prostate epithelial cells (73). These results suggest that DNMT1 is an important mediator of E2F1-induced AR silencing.
In vivo studies utilizing the murine prostate cancer model TRAMP (74) demonstrated the potential of DNA methyltransferase inhibition for the treatment of prostate cancer and AR reactivation (75, 76). Upon puberty, the TRAMP model expresses SV40 large T antigen specifically in prostate epithelial cells. SV40 large T antigen blocks RB1 and TP53 function and leads to the development of poorly differentiated tumors (74). Recent work suggests that the poorly-differentiated tumor cells that emerge may arise from pre-existing malignantly transformed progenitor cells, rather than adenocarcinoma cells that undergo transdifferentiation (77).
TRAMP mice exhibit elevated E2F1 and DNMT1 levels in premalignant and malignant prostate cancer lesions (75). Treatment with the DNA demethylating agent 5-aza-2’-deoxycytidine (5-aza) reactivated AR expression, prevented the development of poorly differentiated prostate cancer and lymph node metastases, and significantly extended survival vs. control-treated mice (75). 5-aza treatment also caused tumor regression in TRAMP mice with established tumors (76). Finally, treatment with 5-aza with or without castration in TRAMP mice demonstrated that combined 5-aza + castration treatment had the lowest number of poorly differentiated prostate cancer tumors and metastases, suggesting combination treatment is a promising strategy to reverse lineage plasticity and castration-resistance (76).
DNA methylation has been also implicated in regulating AR expression in AR-null cell line models and patient tumor samples (78–80). Treatment with 5-aza led to re-expression of the AR with concomitant DNA demethylation of the AR promoter in the AR-negative DuPro, TSU-PR1 and DU145 cell lines (78, 80). Thus, DNMT inhibitors may be a promising class of drugs to restore AR expression in subsets of AR-null prostate cancer.
Genome-wide patterns of DNA methylation can also be used to distinguish between benign prostate tissue, primary prostate cancer, and metastatic CRPC (68). Using differential DNA methylation profiling, Zhao, et al. was able to classify t-NEPC patient tumor samples (68). In an analysis of AR+, amphicrine, double negative, and NEPC PDXs, Brennen, et al. determined that loss of AR expression was correlated with AR promoter hypermethylation specifically in NEPC models (81). Beltran, et al. examined DNA methylation in NEPC vs. adenocarcinoma tumors and found differences in pathways linked to cell-cell adhesion, EMT, and stem cell programs (29). Another study by Beltran, et al. examined DNA methylation in cell-free DNA (cfDNA) samples from patients with t-NEPC or adenocarcinoma to determine unique features found in NEPC (82). Hypermethylation of the ASXL3 and SPDEF genes was observed in the NEPC samples (82). Additionally, NEPC tumors exhibited hypomethylation of both the NEPC marker INSM1 and the plasticity gene CDH2 (82). Altogether, these studies suggest that DNA methylation may play an important role in regulating expression of the AR and genes linked to lineage plasticity.
Histone Methyltransferases, Demethylases, and Deacetylases
Histones possess unstructured N-terminal tails that are subject to post-translational modifications that influence gene expression. Among the most well-studied are lysine methylation and acetylation that alter chromatin organization and accessibility (83). Two classes of histone writers—histone methyltransferases and histone acetyltransferases that methylate or acetylate specific lysines, respectively—are responsible for catalyzing the addition of these marks. Histone methylation and acetylation are tightly regulated processes and are reversible through the activity of histone erasers—histone demethylases and histone deacetylases (HDACs). Finally, chromatin readers are a third major class of epigenetic factors that are thought to “read,” or interpret, histone modifications by binding these marks and recruiting transcription factors and other regulatory proteins to chromatin.
Histone methylation can either activate or repress gene transcription depending on the specific lysine residue that is modified, and repressive histone methylation has been linked to AR expression loss (84). Using AR- and AR+ NEPC PDX models, Kleb, et al. demonstrated that the AR promoter was enriched with the repressive histone modifications H3K27me3 and H3K9me2 in AR-null tumors (84). EZH2, the histone methyltransferase responsible for the H3K27me3 mark, has been shown to be upregulated in NEPC (29, 69, 85, 86). Ku, et al. showed that EZH2 catalytic inhibition in DKO and TKO tumors reactivated AR signaling and re-sensitized tumors to enzalutamide while simultaneously decreasing expression of NEPC target genes (61). These data support the importance of histone methylation mediated by EZH2 for AR repression in subsets of NEPC.
In addition to histone methyltransferases, histone demethylases may also play a role in CRPC. Lysine specific demethylase 1 (LSD1/KDM1A) is known to demethylate lysine 4 on histone H3 and lysine residues on several non-histone proteins, including TP53, E2F1, DNMT1, and HIF-1α (87). LSD1 may also indirectly demethylate lysine 9 on histone H3, including at canonical AR target genes (87) or cell cycle genes (88). Han, et al. demonstrated the importance of LSD1 for E2F1 chromatin binding in RB1-deficient CRPC C4-2 and VCaP cell lines (89). Collectively, these data suggest that RB1 inactivation may confer vulnerability to LSD1 inhibition.
Recent work from our group demonstrates that LSD1 promotes AR-independent survival of CRPC cells independently of its catalytic function (90). We also recently identified a neuronal-specific isoform of LSD1 called LSD1+8a that was specifically expressed in NEPC vs. adenocarcinoma in PDX models and metastatic biopsies (91). The splicing factor SRRM4 that is overexpressed in NEPC was shown to mediate the alternative splicing of LSD1+8a (91). Through gain of function studies, LSD1+8a and SRRM4 were shown to co-regulate a distinct set of genes from canonical LSD1 (91). SRRM3 has also been shown to mediate alternative splicing of LSD1+8a (92). Collectively, these data suggest LSD1 splice variants such as LSD1+8a may be biomarkers for NEPC or contribute to NEPC lineage plasticity.
HDACs lead to changes in chromatin accessibility and are important in AR signaling and prostate cancer (93–96). Ruan, et al. demonstrated that the HDAC SIRT1 is upregulated in NEPC and showed that overexpressing SIRT1 promotes NEPC lineage plasticity (94). SIRT1 has been shown to promote upregulation of SOX2 in breast cancer (97), liver cancer stem cells (98), and bone marrow-derived mesenchymal stem cells (99). Thus, it is possible that SOX2 upregulation by SIRT1 may also contribute to NEPC lineage plasticity. In summary, although the precise mechanisms by which HDACs promote NEPC remain unclear, these data suggest that HDACs may be important therapeutic targets in this disease.
Histone Acetylation, Chromatin Accessibility, and Chromatin Readers
The accessibility, or openness, of chromatin is an essential determinant of gene expression. Histone acetylation reduces the positive charge on histones and is thought to de-compact chromatin by weakening histone binding to negatively charged DNA (83). Histone acetylation is also recognized by bromodomain proteins that recruit transcriptional machinery (100). Chromatin accessibility is regulated by chromatin remodelers, which regulate transcription by controlling the positioning of nucleosomes, the basic repeating unit of eukaryotic chromatin (101). Recent studies show that ARSI treatment may induce widespread changes in chromatin accessibility that may contribute to lineage plasticity (54, 102–104).
Pomerantz, et al. evaluated the epigenomes of human prostate samples and PDXs (103). They identified reprogramming of the AR cistrome between benign prostate tissues, hormone sensitive prostate cancer tissues, and CRPC tissues, identifying over 17,000 AR binding sites and over 16,000 H3K27ac sites enriched in CRPC (103). Assay for Transposase Accessible Chromatin (ATAC)-sequencing in both normal and primary tumor specimens demonstrated chromatin accessibility at the AR binding sites, and the DNA was hypomethylated in these regions (103). In metastatic CRPC, AR was reprogrammed to sites associated with developmental prostate programs (103). These data demonstrate that chromatin accessibility and the AR cistrome change with prostate cancer progression, contributing to reactivation of prostate developmental pathways in CRPC cells (103). While this study did not specifically examine NEPC, it is quite possible that reactivation of a more primitive developmental program may facilitate eventual commitment to non-luminal lineages.
A well-studied chromatin remodeler in prostate cancer is CHD1. Deletion of CHD1 in normal prostate cells altered the chromatin landscape and led to AR redistribution from lineage commitment regions to tumor-specific AR-bound regions (105). Similarly, CHD1 loss resulted in enzalutamide resistance by promoting neuronal differentiation (104). CHD1 loss increased chromatin accessibility for four factors associated with activation of non-luminal lineage programs: NR3C1, POU3F2, TBX2, and NR2F1 (104). Importantly, deletion of each factor by CRISPR-Cas9 re-sensitized CHD1 knockout cells to enzalutamide (104).
The restrictive element-1 silencing transcription factor (REST) is a repressor of neuronal differentiation (106). REST has been shown to cooperate with the AR to repress neuronal gene expression (107). Downregulation of REST resulting from hypoxic conditions induces neuroendocrine differentiation in CRPC (108), and REST knockdown increases stemness and EMT gene expression in NEPC models (109). Loss of REST’s repressor activity through a splicing-in event by SRRM3/4 promotes BAF53B (ACTL6B) expression in both amphicrine and NEPC tumors (13). BAF53B is a component of the neuron-specific nBAF chromatin remodeling complex that regulates gene expression and differentiation (110). The exchange of BAF53A and BAF45A subunits within the BAF complex for homologous BAF53B and BAF45B subunits within neuron-specific BAF (nBAF) complexes promotes a chromatin switch to a differentiated neuronal phenotype in post-mitotic neurons (111–113). Cytra, et al. demonstrated that BAF53B and BAF45B are highly expressed in NEPC but absent from benign prostate, localized prostate cancer, or CRPC adenocarcinoma samples, demonstrating high specificity for the neuroendocrine phenotype (102). Interestingly, neither BAF53B nor BAF45B knockdown had an effect on NEPC cell proliferation. Therefore, the authors suggest that BAF53B and BAF45B expression may be specific for the NEPC phenotype, but not a critical mediator of NEPC aggressiveness (102). Finally, BET bromodomain proteins—such as BRD4 that recognizes the H3K27ac mark—have been shown to play an important role in NEPC. Our prior work demonstrated that BRD4 cooperates with E2F1 to drive lineage plasticity (54). We found strong colocalization of H3K27ac and BRD4 signals in t-NEPC cell lines (54). Furthermore, BET bromodomain inhibition abrogated E2F1 induction of NEPC lineage plasticity genes and suppressed growth of E2F1-high t-NEPC cell lines, strongly suggesting these factors cooperate (54). Collectively, these studies demonstrate that epigenetic factors play a key role in promoting NEPC lineage plasticity. Because of the diversity of epigenetic regulators that are altered in NEPC, it may be necessary to target multiple epigenetic regulators simultaneously to reverse or prevent lineage plasticity.
Identifying NEPC Tumors
Currently, several approaches are used to identify tumors that have undergone NEPC lineage plasticity. Histology and examination of morphologic features are the gold standard to distinguish de novo NEPC from adenocarcinoma (114). However, use of morphology alone is challenging in tumor specimens from patients who have been treated with agents such as ADT or ARSIs that may alter cellular morphology and give a false impression of the phenotype of a tumor. Another approach is to use immunohistochemistry for canonical NEPC markers, including CHGA, SYP, NSE, CD56 (NCAM1), or INSM1 that are highly expressed in de novo NEPC (114–116). However, many t-NEPC tumors do not have high expression of these genes, presumably because they are not as far along the continuum of NEPC differentiation as a de novo NEPC tumor (26).
Another approach to determine the program of a cell more accurately is through simultaneous measurement of multiple markers through RNA-sequencing. Indeed, we now know that lineage plasticity exists on a continuum (13). As previously discussed, Labrecque, et al. used rapid autopsy samples and identified five tumor subtypes based on IHC and gene expression that may represent a more accurate classification system than current standard pathologic assessment (13). Other studies have created gene expression signatures associated with NEPC or t-NEPC using clinical cohorts (26, 29). These studies had high levels of cross validation and accuracy when applied to external cohorts and further suggest molecular classification of tumors through gene expression may be preferable. However, the development of assays that are easy to use and interpret will be critical before transcriptional profiling is routinely used for molecular subtyping in clinical practice.
Though only a minority of patients will eventually develop t-NEPC after ARSI treatment, patients with t-NEPC are often not identified because metastatic biopsies are not routinely done in practice and assessment for an NEPC phenotype remains challenging. NEPC detection is a key area that must be improved upon—both to identify men for whom conventional therapies such as ARSIs will not be effective and to identify patients who might be eligible for NEPC-focused clinical trials. To date, clinical trials in NEPC have generally used histologic assessments, clinical phenotype (e.g., liver metastasis in absence of PSA progression), and blood markers (e.g., serum or tissue CGA) as enrollment criteria. Several other therapeutic trials have focused on specific clinical markers to enrich for patients with “aggressive variant prostate cancer” (AVPC), though this clinical classification likely includes a mixture of tumor subtypes that may or may not be AR-driven.
Some phase II studies (NCT04592237, NCT03263650) enrolling AVPC used mutations or loss of function events in the tumor suppressors PTEN, RB1, and TP53. Given the key role these genes play in both suppressing lineage plasticity and proliferation (60, 61), enriching clinical trials for patients whose tumors harbor loss of these genes is a pragmatic approach to test treatments predicted to be active in highly proliferative tumors, including NEPC. Detection of genomic loss in these genes is relatively straightforward vs. RNA-based assays and may enrich for NEPC tumors. However, loss of these genes is clearly not specific for NEPC (60–63, 117).
There remains an unmet need to identify tumors that have undergone lineage plasticity via non-invasive methods. Molecular imaging may be one promising avenue. Radionuclide scans using tracers conjugated to prostate specific membrane antigen (PSMA) and dihydrotestosterone (DHT) have been widely studied in prostate cancer (118). PSMA or DHT low/negative tumors that are metabolically active — FDG or choline positive — are enriched for aggressive tumors (119, 120) or those that are AR-independent, such as NEPC (121). DLL3 has recently gained recognition as an NEPC marker and drug target (122). The authors of a recent study developed a PET imaging method using a (89Zr)-labeled DLL3 targeting antibody that specifically detected neuroendocrine PDXs in vivo (123).
Promising strategies for identification of NEPC non-invasively via circulating blood markers have also been identified. Beltran, et al. published a proof of concept study in which they identified NEPC circulating tumor cells (CTCs) via morphology and immunofluorescence staining (124). A later study found that detection of an NEPC phenotype in CTCs was associated with significantly worse overall survival after starting an ARSI (125). Another blood-based method is cfDNA assays. One recent study detected NEPC features via whole exome sequencing and whole genome bisulfite sequencing (82). An NEPC score was created using a targeted panel of key DNA genomic alterations and 20 hyper- or hypomethylated sites (82). Another study created a NEPC signature from tissue samples using methylated DNA immunoprecipitation and sequencing (MeDIP-seq) that was subsequently used to predict the presence of NEPC via cfDNA (126). The optimal cutoff produced results with 100% sensitivity and 95% specificity in a validation cohort. The clear implication from these studies is that non-invasive identification of NEPC may soon become a reality in patients with tumor burden significant enough to lead to detectable CTCs.
Clinical Trials in NEPC
Aggarwal, et al. determined that patients with small cell histologic features or the cluster 2 transcriptional program in their CRPC biopsies were associated with poor overall survival (26). Thus, t-NEPC tumors appear to be more aggressive than adenocarcinoma CRPC tumors. The standard of care for both de novo and t-NEPC is chemotherapy based on the treatment regimens used in small cell lung cancer (127–132). Most commonly, platinum doublets (e.g., cisplatin or carboplatin) with etoposide or a taxane (e.g., docetaxel or cabazitaxel) are utilized. High initial rates of response were also found to docetaxel and carboplatin in the clinically-defined AVPC subset that included some NEPC tumors (130). While most patients respond, relapse is universal with a median survival of 1-2 years from the time of diagnosis (128, 130). Though a trial of the single agent anti-PD1 inhibitor avelumab showed limited efficacy in men with NEPC (NCT03179410) (133) we await the results of trials incorporating checkpoint inhibitors with standard platinum doublets — as is now standard of care in small cell lung cancer.
There are few published reports of clinical trials using targeted agents focused on NEPC patients, specifically. One of the earliest phase II trials tested the aurora kinase inhibitor alisertib (NCT01799278) (134). MYCN is a known regulator of lineage plasticity that is upregulated in subsets of NEPC (135), and AURKA appears to be important for stabilizing MYCN (136). Therefore, these investigators tested alisertib in patients with NEPC. Of note, MYCN or AURKA upregulation was not required for enrollment. This trial did not meet its primary endpoint of 6-month progression free survival. However, the subpopulation of patients with increased AURKA expression — 16% of study population — did appear to have longer overall survival (134).
Another targeted agent, rovalpituzumab tesirine (an antibody drug conjugate against DLL3), was tested in a phase I/II trial (NCT02709889) that included 18 patients with NEPC and found an objective response rate of 10% (137). There are several ongoing targeted trials specifically enrolling patients with NEPC or AVPC with targeted agents summarized in Table 1.
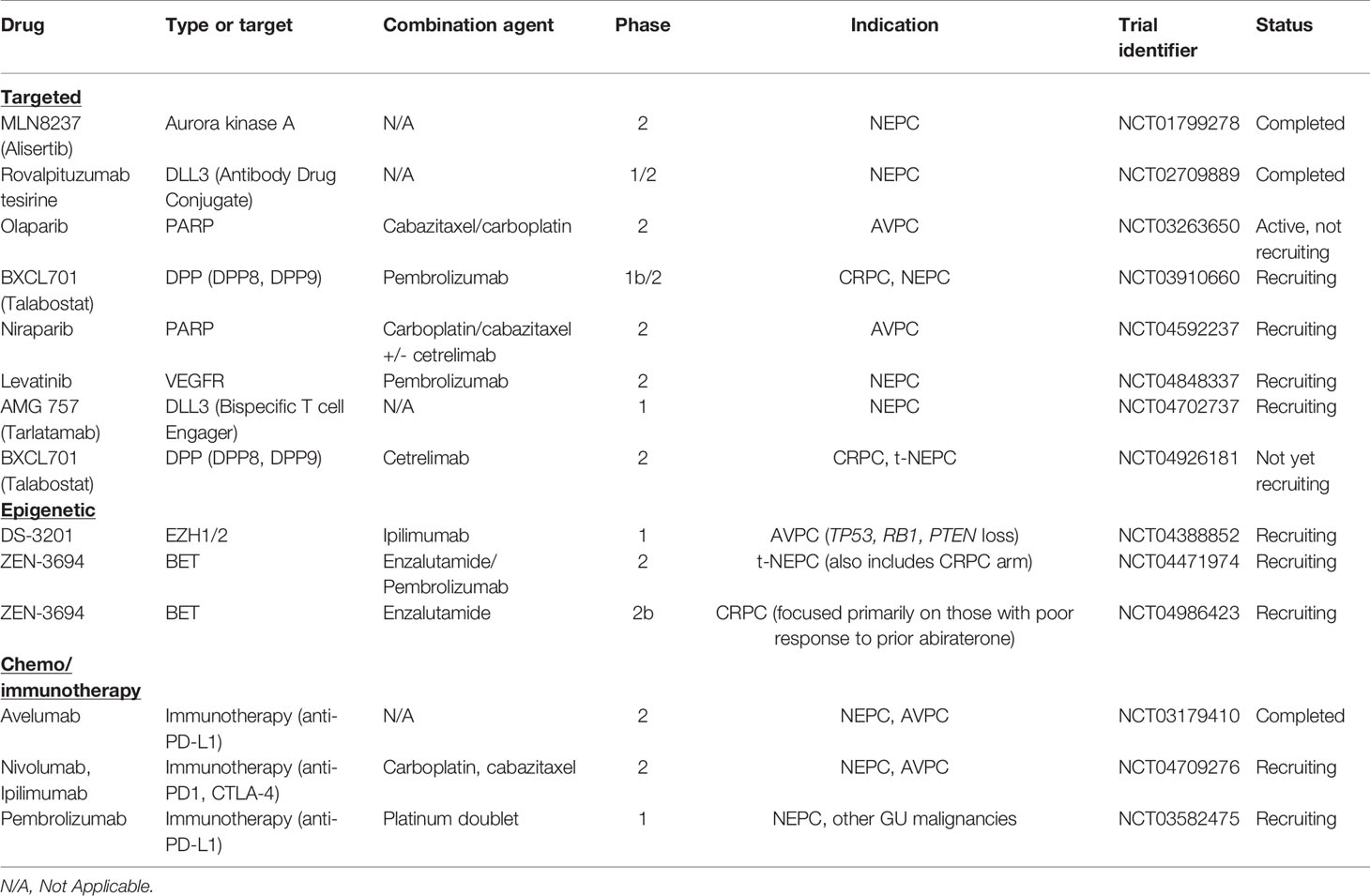
Table 1 Recent clinical trials in neuroendocrine prostate cancer.
Multiple phase I-II studies have tested epigenetic targeted agents in CRPC. The majority of completed and ongoing trials of epigenetic targeted agents recruited from the general CRPC population and were not specifically focused on NEPC, thus making it difficult to determine the effectiveness of these agents in this molecular subset.
As stated previously, there are marked DNA methylation differences between adenocarcinoma and NEPC tumors, and DNMTs have been implicated as key lineage plasticity factors (29). While DNMT inhibition showed promise in preclinical models of NEPC (75, 76), clinical trials of DNMT inhibitors in general CRPC populations (NCT00384839) have reported a low proportion of responders (138).
HDAC regulate AR activity (139) and have been found to be upregulated in NEPC cell lines (94). HDAC inhibitors are among the best studied epigenetic therapies in prostate cancer in both preclinical (95) and clinical trials (93, 140–142). In the CRPC population, single agent activity has been low, but combination therapy with the ARSI casodex appeared promising (140).
As previously discussed, BET bromodomain proteins are chromatin readers that cooperate with several transcription factors, including the AR and E2F1 (54, 143). Several clinical trials with BETi have been conducted in prostate cancer, though none was specifically focused on NEPC (55, 144, 145). In a recent study with the BETi ZEN-3694, pre-treatment biopsies and clinical factors were examined to identify markers of response (NCT02711956) (55). In an exploratory analysis, shorter time on ARSI treatment prior to study entry was linked to better chance of response to ZEN-3694 (55). Further, examination of RNA-sequencing from baseline, pre-treatment biopsies demonstrated that lower canonical AR transcriptional activity was associated with longer time to progression while on treatment (19 vs. 45 weeks) (55). Four patients enrolled on this trial exhibited an NEPC program at baseline (54). Of those, two were long-term responders and had high expression of BRD4 and E2F1 and activation of an AR-repressed, E2F1-activated NEPC lineage plasticity program vs. the other two patients whose tumors progressed more rapidly. This suggests that E2F1/BRD4-activity-high NEPC tumors may be particularly susceptible to BET bromodomain inhibition. ZEN-3694 is currently being tested in combination with enzalutamide + pembrolizumab in a phase II trial that includes a t-NEPC cohort (NCT04471974). A separate study of ZEN-3694 in combination with enzalutamide vs. enzalutamide alone is recruiting a cohort that had poor response to abiraterone (NCT04986423)—based on results from the prior phase I ZEN-3694 trial (55)—with the hope that doing so will enrich for tumors that have undergone lineage plasticity.
Other epigenetic targets are also being assessed in ongoing trials. The histone demethylase LSD1 is being studied in a phase I/II study that includes neuroendocrine tumors (NCT02712905). Inhibitors of EZH2 are also being tested in ongoing trials alone or in combination with ARSIs (NCT04986423). Notably, a phase I trial of DS3201 (EZH1/2 inhibitor) and ipilimumab is specifically recruiting AVPC in addition to other genitourinary cancers (NCT04388852).
Identifying Patients at Risk for Therapy-Induced NEPC Lineage Plasticity
The current focus of t-NEPC research is how to identify these tumors more accurately and treat them more effectively once they develop after ARSI treatment. However, identification of tumors at greatest risk of ARSI-induced AR pathway loss and t-NEPC lineage plasticity before these changes occur may lead to better outcomes. Indeed, recent reports indicate that significant heterogeneity exists in tumors that have undergone lineage plasticity (13), which may contribute to resistance to NEPC-directed therapies. Thus, it may be preferable to identify tumors before they have undergone ARSI-induced NEPC lineage plasticity.
Understanding lineage plasticity risk requires a longitudinal assessment of patients with matched assays (before treatment and at the time of progression). We do not yet know the best measures of risk of NEPC lineage plasticity, and there are no markers that exist to risk-stratify patients. Therefore, we believe it will be important to measure several factors that may contribute to NEPC lineage plasticity risk: genomic, epigenomic, and microenvironmental features. Whole genome or whole exome DNA sequencing can identify genomic loss of known tumor suppressor genes (e.g., PTEN, RB1, and TP53). Single cell assays such as ATAC sequencing paired with RNA sequencing may provide information about the baseline gene expression program of distinct cell populations and their capacity to turn on other gene expression programs, including those linked with NEPC lineage plasticity. Spatial profiling may help to unravel immune and microenvironmental contributions to lineage plasticity and will be crucial for understanding the communication and cooperativity of different cell populations present in a tumor. Inclusion of non-invasive assays using CTCs or cfDNA may help facilitate serial collection of samples that may give clues about risk of NEPC lineage plasticity that do not require invasive, repeated sampling of patients.
Provided that the correct samples and analytes are collected at baseline, such studies to determine NEPC lineage plasticity risk could be prospective or retrospective. Our group has recently initiated a prospective study called the MIchigan ONCOlogy Multi-omic Assessment of Tumor Change and Heterogeneity (Mi-ONCOMATCH) that incorporates many of the aforementioned assays in an effort to improve our ability to identify those at greatest risk of developing t-NEPC after ARSI treatment. Once studies establishing markers of lineage plasticity risk are completed, prospective validation will be necessary. It is our hope that improvements in non-invasive detection through blood-based assays or molecular imaging will hasten completion of such studies and improve our ability to identify those at greatest risk of developing t-NEPC. The next critical step will be to develop clinical trials testing drugs that suppress induction of an NEPC program or that maintain AR expression and signaling. We believe that epigenetic therapies may be a particularly promising approach for these patients.
Conclusion
Both genetic and epigenetic changes play a key role in therapy-induced AR pathway loss and t-NEPC lineage plasticity. Understanding mechanisms by which t-NEPC lineage plasticity occurs and markers that indicate this transition has taken place are critical for making progress for this group of patients. We predict that combination clinical trials with ARSIs + lineage plasticity-modifying epigenetic therapies alone or with other targeted agents will be critical for making progress. Advances in detection of lineage plasticity risk will be important to prevent this newly appreciated, aggressive form of ARSI resistance from happening in the first place.
Author Contributions
WKS, AMM, TCW, and JJA conceived the article. WKS, AMM, TCW, CM, and JJA reviewed the literature and wrote the first draft of the manuscript. WKS made the figure. TCW made the table. WKS, ZD, CM, JAY, and JJA edited the manuscript.
Funding
This work was supported by National Cancer Institute (NCI) R01 (CA251245); The Pacific Northwest Prostate Cancer Specialized Programs of Research Excellence (SPORE) NCI P50 CA097186; the Michigan Prostate SPORE NCI P50 CA186786; the NCI Drug Resistance and Sensitivity Network NCI P50 CA186786-07S1; NCI Training Grant T32 CA009676; NCI Training Grant 5-T32 CA180984-07; NCI Training Grant T32 CA009357; Department of Defense Idea Award (W81XWH-20-1-0405); National Comprehensive Cancer Network (NCCN)/Astellas Pharma Global Development Award; a Prostate Cancer Foundation Challenge Award; and a Sheppard Family Foundation Sheppard Scholar Award.
Conflict of Interest
JJA reports consulting and speaker’s fees from Astellas Pharma, consulting fees from Dendreon, consulting fees from Merck, consulting fees from Bristol Myers Squibb, and research support to his institution from Astellas Pharma, Zenith Epigenetics, and Beactica.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
2. Huggins C, Hodges CV. Studies on Prostatic Cancer. I. The Effect of Castration, of Estrogen and Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. CA Cancer J Clin (1972) 22(4):232–40. doi: 10.3322/canjclin.22.4.232
3. Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen Deprivation Therapy: Progress in Understanding Mechanisms of Resistance and Optimizing Androgen Depletion. Nat Clin Pract Urol (2009) 6(2):76–85. doi: 10.1038/ncpuro1296
4. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular Determinants of Resistance to Antiandrogen Therapy. Nat Med (2004) 10(1):33–9. doi: 10.1038/nm972
5. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature (2012) 487(7406):239–43. doi: 10.1038/nature11125
6. Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, et al. A Mechanism for Androgen Receptor-Mediated Prostate Cancer Recurrence After Androgen Deprivation Therapy. Cancer Res (2001) 61(11):4315–9.
7. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell (2015) 161(5):1215–28. doi: 10.1016/j.cell.2015.05.001
8. Cai C, Balk SP. Intratumoral Androgen Biosynthesis in Prostate Cancer Pathogenesis and Response to Therapy. Endocr Relat Cancer (2011) 18(5):R175–82. doi: 10.1530/ERC-10-0339
9. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. N Engl J Med (2011) 364(21):1995–2005. doi: 10.1056/NEJMoa1014618
10. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased Survival With Enzalutamide in Prostate Cancer After Chemotherapy. N Engl J Med (2012) 367(13):1187–97. doi: 10.1056/NEJMoa1207506
11. Chi KN, Agarwal N, Bjartell A, Chung BH, Pereira de Santana Gomes AJ, Given R, et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N Engl J Med (2019) 381(1):13–24. doi: 10.1056/NEJMoa1903307
12. Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Nonmetastatic, Castration-Resistant Prostate Cancer and Survival With Darolutamide. N Engl J Med (2020) 383(11):1040–9. doi: 10.1056/NEJMoa2001342
13. Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. Molecular Profiling Stratifies Diverse Phenotypes of Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer. J Clin Invest (2019) 129(10):4492–505. doi: 10.1172/JCI128212
14. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell (2013) 155(6):1309–22. doi: 10.1016/j.cell.2013.11.012
15. Asangani I, Blair IA, Van Duyne G, Hilser VJ, Moiseenkova-Bell V, Plymate S, et al. Using Biochemistry and Biophysics to Extinguish Androgen Receptor Signaling in Prostate Cancer. J Biol Chem (2021) 296:100240. doi: 10.1074/jbc.REV120.012411
16. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a Novel Androgen Receptor Exon Generates a Constitutively Active Androgen Receptor That Mediates Prostate Cancer Therapy Resistance. Cancer Res (2008) 68(13):5469–77. doi: 10.1158/0008-5472.CAN-08-0594
17. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-Independent Androgen Receptor Variants Derived From Splicing of Cryptic Exons Signify Hormone-Refractory Prostate Cancer. Cancer Res (2009) 69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764
18. Shen MM, Abate-Shen C. Molecular Genetics of Prostate Cancer: New Prospects for Old Challenges. Genes Dev (2010) 24(18):1967–2000. doi: 10.1101/gad.1965810
19. Davies AH, Beltran H, Zoubeidi A. Cellular Plasticity and the Neuroendocrine Phenotype in Prostate Cancer. Nat Rev Urol (2018) 15(5):271–86. doi: 10.1038/nrurol.2018.22
20. Quintanal-Villalonga A, Chan JM, Yu HA, Pe'er D, Sawyers CL, Sen T, et al. Lineage Plasticity in Cancer: A Shared Pathway of Therapeutic Resistance. Nat Rev Clin Oncol (2020) 17(6):360–71. doi: 10.1038/s41571-020-0340-z
21. Beltran H, Demichelis F. Prostate Cancer: Intrapatient Heterogeneity in Prostate Cancer. Nat Rev Urol (2015) 12(8):430–1. doi: 10.1038/nrurol.2015.182
22. Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, Lalonde E, et al. Spatial Genomic Heterogeneity Within Localized, Multifocal Prostate Cancer. Nat Genet (2015) 47(7):736–45. doi: 10.1038/ng.3315
23. Carm KT, Hoff AM, Bakken AC, Axcrona U, Axcrona K, Lothe RA, et al. Interfocal Heterogeneity Challenges the Clinical Usefulness of Molecular Classification of Primary Prostate Cancer. Sci Rep (2019) 9(1):13579. doi: 10.1038/s41598-019-49964-7
24. Tiwari R, Manzar N, Ateeq B. Dynamics of Cellular Plasticity in Prostate Cancer Progression. Front Mol Biosci (2020) 7:130. doi: 10.3389/fmolb.2020.00130
25. Brady NJ, Bagadion AM, Singh R, Conteduca V, Van Emmenis L, Arceci E, et al. Temporal Evolution of Cellular Heterogeneity During the Progression to Advanced AR-Negative Prostate Cancer. Nat Commun (2021) 12(1):3372. doi: 10.1038/s41467-021-23780-y
26. Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-Institutional Prospective Study. J Clin Oncol (2018) 36(24):2492–503. doi: 10.1200/JCO.2017.77.6880
27. Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained Through FGF Signaling. Cancer Cell (2017) 32(4):474–89.e6. doi: 10.1016/j.ccell.2017.09.003
28. Zaffuto E, Pompe R, Zanaty M, Bondarenko HD, Leyh-Bannurah SR, Moschini M, et al. Contemporary Incidence and Cancer Control Outcomes of Primary Neuroendocrine Prostate Cancer: A SEER Database Analysis. Clin Genitourin Cancer (2017) 15(5):e793–800. doi: 10.1016/j.clgc.2017.04.006
29. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent Clonal Evolution of Castration-Resistant Neuroendocrine Prostate Cancer. Nat Med (2016) 22(3):298–305. doi: 10.1038/nm.4045
30. Zhou Y, Bolton EC, Jones JO. Androgens and Androgen Receptor Signaling in Prostate Tumorigenesis. J Mol Endocrinol (2015) 54(1):R15–29. doi: 10.1530/JME-14-0203
31. Cunha GR. The Dual Origin of Vaginal Epithelium. Am J Anat (1975) 143(3):387–92. doi: 10.1002/aja.1001430309
32. Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, et al. Generation and Characterization of Androgen Receptor Knockout (ARKO) Mice: An In Vivo Model for the Study of Androgen Functions in Selective Tissues. Proc Natl Acad Sci U S A (2002) 99(21):13498–503. doi: 10.1073/pnas.212474399
33. Isaacs JT, Lundmo PI, Berges R, Martikainen P, Kyprianou N, English HF. Androgen Regulation of Programmed Death of Normal and Malignant Prostatic Cells. J Androl (1992) 13(6):457–64. doi: 10.1002/j.1939-4640.1992.tb00337.x
34. Kyprianou N, Isaacs JT. Activation of Programmed Cell Death in the Rat Ventral Prostate After Castration. Endocrinology (1988) 122(2):552–62. doi: 10.1210/endo-122-2-552
35. Ling MT, Chan KW, Choo CK. Androgen Induces Differentiation of a Human Papillomavirus 16 E6/E7 Immortalized Prostate Epithelial Cell Line. J Endocrinol (2001) 170(1):287–96. doi: 10.1677/joe.0.1700287
36. Whitacre DC, Chauhan S, Davis T, Gordon D, Cress AE, Miesfeld RL. Androgen Induction of In Vitro Prostate Cell Differentiation. Cell Growth Differ (2002) 13(1):1–11.
37. Donjacour AA, Cunha GR. Assessment of Prostatic Protein Secretion in Tissue Recombinants Made of Urogenital Sinus Mesenchyme and Urothelium From Normal or Androgen-Insensitive Mice. Endocrinology (1993) 132(6):2342–50. doi: 10.1210/endo.132.6.7684975
38. Evans GS, Chandler JA. Cell Proliferation Studies in the Rat Prostate: II. The Effects of Castration and Androgen-Induced Regeneration Upon Basal and Secretory Cell Proliferation. Prostate (1987) 11(4):339–51. doi: 10.1002/pros.2990110406
39. Lee SO, Tian J, Huang CK, Ma Z, Lai KP, Hsiao H, et al. Suppressor Role of Androgen Receptor in Proliferation of Prostate Basal Epithelial and Progenitor Cells. J Endocrinol (2012) 213(2):173–82. doi: 10.1530/JOE-11-0474
40. Mirosevich J, Bentel JM, Zeps N, Redmond SL, D'Antuono MF, Dawkins HJ. Androgen Receptor Expression of Proliferating Basal and Luminal Cells in Adult Murine Ventral Prostate. J Endocrinol (1999) 162(3):341–50. doi: 10.1677/joe.0.1620341
41. Wu CT, Altuwaijri S, Ricke WA, Huang SP, Yeh S, Zhang C, et al. Increased Prostate Cell Proliferation and Loss of Cell Differentiation in Mice Lacking Prostate Epithelial Androgen Receptor. Proc Natl Acad Sci U S A (2007) 104(31):12679–84. doi: 10.1073/pnas.0704940104
42. Wang F, Koul HK. Androgen Receptor (AR) Cistrome in Prostate Differentiation and Cancer Progression. Am J Clin Exp Urol (2017) 5(3):18–24.
43. Zhang D, Park D, Zhong Y, Lu Y, Rycaj K, Gong S, et al. Stem Cell and Neurogenic Gene-Expression Profiles Link Prostate Basal Cells to Aggressive Prostate Cancer. Nat Commun (2016) 7:10798. doi: 10.1038/ncomms10798
44. Smith BA, Sokolov A, Uzunangelov V, Baertsch R, Newton Y, Graim K, et al. A Basal Stem Cell Signature Identifies Aggressive Prostate Cancer Phenotypes. Proc Natl Acad Sci U S A (2015) 112(47):E6544–52. doi: 10.1073/pnas.1518007112
45. Alumkal JJ, Sun D, Lu E, Beer TM, Thomas GV, Latour E, et al. Transcriptional Profiling Identifies an Androgen Receptor Activity-Low, Stemness Program Associated With Enzalutamide Resistance. Proc Natl Acad Sci U S A (2020) 117(22):12315–23. doi: 10.1073/pnas.1922207117
46. Taavitsainen S, Engedal N, Cao S, Handle F, Erickson A, Prekovic S, et al. Single-Cell ATAC and RNA Sequencing Reveal Pre-Existing and Persistent Cells Associated With Prostate Cancer Relapse. Nat Commun (2021) 12(1):5307. doi: 10.1038/s41467-021-25624-1
47. Kwon OJ, Zhang L, Jia D, Xin L. Sox2 Is Necessary for Androgen Ablation-Induced Neuroendocrine Differentiation From Pten Null Sca-1(+) Prostate Luminal Cells. Oncogene (2021) 40(1):203–14. doi: 10.1038/s41388-020-01526-2
48. Ben-Salem S, Hu Q, Liu Y, Alshalalfa M, Zhao X, Wang I, et al. Diversity in Androgen Receptor Action Among Treatment-Naive Prostate Cancers Is Reflected in Treatment Response Predictions and Molecular Subtypes. Eur Urol Open Sci (2020) 22:34–44. doi: 10.1016/j.euros.2020.10.002
49. Lee SO, Ma Z, Yeh CR, Luo J, Lin TH, Lai KP, et al. New Therapy Targeting Differential Androgen Receptor Signaling in Prostate Cancer Stem/Progenitor vs. Non-Stem/Progenitor Cells. J Mol Cell Biol (2013) 5(1):14–26. doi: 10.1093/jmcb/mjs042
50. Germann M, Wetterwald A, Guzman-Ramirez N, van der Pluijm G, Culig Z, Cecchini MG, et al. Stem-Like Cells With Luminal Progenitor Phenotype Survive Castration in Human Prostate Cancer. Stem Cells (2012) 30(6):1076–86. doi: 10.1002/stem.1087
51. Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, et al. Androgen Deprivation Causes Epithelial-Mesenchymal Transition in the Prostate: Implications for Androgen-Deprivation Therapy. Cancer Res (2012) 72(2):527–36. doi: 10.1158/0008-5472.CAN-11-3004
52. Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov (2017) 7(1):54–71. doi: 10.1158/2159-8290.CD-15-1263
53. Ishii J, Sato H, Sakaeda M, Shishido-Hara Y, Hiramatsu C, Kamma H, et al. POU Domain Transcription Factor BRN2 Is Crucial for Expression of ASCL1, ND1 and Neuroendocrine Marker Molecules and Cell Growth in Small Cell Lung Cancer. Pathol Int (2013) 63(3):158–68. doi: 10.1111/pin.12042
54. Kim DH, Sun D, Storck WK, Welker Leng K, Jenkins C, Coleman DJ, et al. BET Bromodomain Inhibition Blocks an AR-Repressed, E2F1-Activated Treatment-Emergent Neuroendocrine Prostate Cancer Lineage Plasticity Program. Clin Cancer Res (2021) 27(17):4923–36. doi: 10.1158/1078-0432.CCR-20-4968
55. Aggarwal RR, Schweizer MT, Nanus DM, Pantuck AJ, Heath EI, Campeau E, et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in Combination With Enzalutamide in Patients With Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res (2020) 26(20):5338–47. doi: 10.1158/1078-0432.CCR-20-1707
56. Yuan TC, Veeramani S, Lin FF, Kondrikou D, Zelivianski S, Igawa T, et al. Androgen Deprivation Induces Human Prostate Epithelial Neuroendocrine Differentiation of Androgen-Sensitive LNCaP Cells. Endocr Relat Cancer (2006) 13(1):151–67. doi: 10.1677/erc.1.01043
57. Zhang XQ, Kondrikov D, Yuan TC, Lin FF, Hansen J, Lin MF. Receptor Protein Tyrosine Phosphatase Alpha Signaling Is Involved in Androgen Depletion-Induced Neuroendocrine Differentiation of Androgen-Sensitive LNCaP Human Prostate Cancer Cells. Oncogene (2003) 22(43):6704–16. doi: 10.1038/sj.onc.1206764
58. Davies A, Nouruzi S, Ganguli D, Namekawa T, Thaper D, Linder S, et al. An Androgen Receptor Switch Underlies Lineage Infidelity in Treatment-Resistant Prostate Cancer. Nat Cell Biol (2021) 23(9):1023–34. doi: 10.1038/s41556-021-00743-5
59. Gazdar AF, Bunn PA, Minna JD. Small-Cell Lung Cancer: What We Know, What We Need to Know and the Path Forward. Nat Rev Cancer (2017) 17(12):725–37. doi: 10.1038/nrc.2017.87
60. Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 Promotes Lineage Plasticity and Antiandrogen Resistance in TP53- and RB1-Deficient Prostate Cancer. Science (2017) 355(6320):84–8. doi: 10.1126/science.aah4307
61. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 Cooperate to Suppress Prostate Cancer Lineage Plasticity, Metastasis, and Antiandrogen Resistance. Science (2017) 355(6320):78–83. doi: 10.1126/science.aah4199
62. Park JW, Lee JK, Sheu KM, Wang L, Balanis NG, Nguyen K, et al. Reprogramming Normal Human Epithelial Tissues to a Common, Lethal Neuroendocrine Cancer Lineage. Science (2018) 362(6410):91–5. doi: 10.1126/science.aat5749
63. Nyquist MD, Corella A, Coleman I, De Sarkar N, Kaipainen A, Ha G, et al. Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cell Rep (2020) 31(8):107669. doi: 10.1016/j.celrep.2020.107669
64. Formaggio N, Rubin MA, Theurillat JP. Loss and Revival of Androgen Receptor Signaling in Advanced Prostate Cancer. Oncogene (2021) 40(7):1205–16. doi: 10.1038/s41388-020-01598-0
65. Bianco-Miotto T, Chiam K, Buchanan G, Jindal S, Day TK, Thomas M, et al. Global Levels of Specific Histone Modifications and an Epigenetic Gene Signature Predict Prostate Cancer Progression and Development. Cancer Epidemiol Biomarkers Prev (2010) 19(10):2611–22. doi: 10.1158/1055-9965.EPI-10-0555
66. Brocks D, Assenov Y, Minner S, Bogatyrova O, Simon R, Koop C, et al. Intratumor DNA Methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Rep (2014) 8(3):798–806. doi: 10.1016/j.celrep.2014.06.053
67. Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, et al. Global Histone Modification Patterns Predict Risk of Prostate Cancer Recurrence. Nature (2005) 435(7046):1262–6. doi: 10.1038/nature03672
68. Zhao SG, Chen WS, Li H, Foye A, Zhang M, Sjostrom M, et al. The DNA Methylation Landscape of Advanced Prostate Cancer. Nat Genet (2020) 52(8):778–89. doi: 10.1038/s41588-020-0648-8
69. Clermont P, Lin D, Crea F, Wu R, Xue H, Wang Y, et al. Polycomb-Mediated Silencing in Neuroendocrine Prostate Cancer. Clin Epigenet (2015) 40. doi: 10.1186/s13148-015-0074-4
70. Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov (2017) 7(7):736–49. doi: 10.1158/2159-8290.CD-16-1174
71. Breiling A, Lyko F. Epigenetic Regulatory Functions of DNA Modifications: 5-Methylcytosine and Beyond. Epigenet Chromatin (2015) 8:24. doi: 10.1186/s13072-015-0016-6
72. McCabe MT, Davis JN, Day ML. Regulation of DNA Methyltransferase 1 by the pRb/E2F1 Pathway. Cancer Res (2005) 65(9):3624–32. doi: 10.1158/0008-5472.CAN-04-2158
73. Valdez CD, Davis JN, Odeh HM, Layfield TL, Cousineau CS, Berton TR, et al. Repression of Androgen Receptor Transcription Through the E2F1/DNMT1 Axis. PloS One (2011) 6(9):e25187. doi: 10.1371/journal.pone.0025187
74. Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, et al. Prostate Cancer in a Transgenic Mouse. Proc Natl Acad Sci U S A (1995) 92(8):3439–43. doi: 10.1073/pnas.92.8.3439
75. McCabe MT, Low JA, Daignault S, Imperiale MJ, Wojno KJ, Day ML. Inhibition of DNA Methyltransferase Activity Prevents Tumorigenesis in a Mouse Model of Prostate Cancer. Cancer Res (2006) 66(1):385–92. doi: 10.1158/0008-5472.CAN-05-2020
76. Zorn CS, Wojno KJ, McCabe MT, Kuefer R, Gschwend JE, Day ML. 5-Aza-2'-Deoxycytidine Delays Androgen-Independent Disease and Improves Survival in the Transgenic Adenocarcinoma of the Mouse Prostate Mouse Model of Prostate Cancer. Clin Cancer Res (2007) 13(7):2136–43. doi: 10.1158/1078-0432.CCR-06-2381
77. Huss WJ, Gray DR, Tavakoli K, Marmillion ME, Durham LE, Johnson MA, et al. Origin of Androgen-Insensitive Poorly Differentiated Tumors in the Transgenic Adenocarcinoma of Mouse Prostate Model. Neoplasia (2007) 9(11):938–50. doi: 10.1593/neo.07562
78. Jarrard DF, Kinoshita H, Shi Y, Sandefur C, Hoff D, Meisner LF, et al. Methylation of the Androgen Receptor Promoter CpG Island is Associated With Loss of Androgen Receptor Expression in Prostate Cancer Cells. Cancer Res (1998) 58(23):5310–4.
79. Kinoshita H, Shi Y, Sandefur C, Meisner LF, Chang C, Choon A, et al. Methylation of the Androgen Receptor Minimal Promoter Silences Transcription in Human Prostate Cancer. Cancer Res (2000) 60(13):3623–30.
80. Nakayama T, Watanabe M, Suzuki H, Toyota M, Sekita N, Hirokawa Y, et al. Epigenetic Regulation of Androgen Receptor Gene Expression in Human Prostate Cancers. Lab Invest (2000) 80(12):1789–96. doi: 10.1038/labinvest.3780190
81. Brennen WN, Zhu Y, Coleman IM, Dalrymple SL, Antony L, Patel RA, et al. Resistance to Androgen Receptor Signaling Inhibition Does Not Necessitate Development of Neuroendocrine Prostate Cancer. JCI Insight (2021) 6(8). doi: 10.1172/jci.insight.146827
82. Beltran H, Romanel A, Conteduca V, Casiraghi N, Sigouros M, Franceschini GM, et al. Circulating Tumor DNA Profile Recognizes Transformation to Castration-Resistant Neuroendocrine Prostate Cancer. J Clin Invest (2020) 130(4):1653–68. doi: 10.1172/JCI131041
83. Bannister AJ, Kouzarides T. Regulation of Chromatin by Histone Modifications. Cell Res (2011) 21(3):381–95. doi: 10.1038/cr.2011.22
84. Kleb B, Estecio MR, Zhang J, Tzelepi V, Chung W, Jelinek J, et al. Differentially Methylated Genes and Androgen Receptor Re-Expression in Small Cell Prostate Carcinomas. Epigenetics (2016) 11(3):184–93. doi: 10.1080/15592294.2016.1146851
85. Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell (2016) 30(4):563–77. doi: 10.1016/j.ccell.2016.09.005
86. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov (2011) 1(6):487–95. doi: 10.1158/2159-8290.CD-11-0130
87. Perillo B, Tramontano A, Pezone A, Migliaccio A. LSD1: More Than Demethylation of Histone Lysine Residues. Exp Mol Med (2020) 52(12):1936–47. doi: 10.1038/s12276-020-00542-2
88. He Y, Zhao Y, Wang L, Bohrer LR, Pan Y, Wang L, et al. LSD1 Promotes S-Phase Entry and Tumorigenesis via Chromatin Co-Occupation With E2F1 and Selective H3K9 Demethylation. Oncogene (2018) 37(4):534–43. doi: 10.1038/onc.2017.353
89. Han W, Liu M, Han D, Li M, Toure AA, Wang Z, et al. RB1 Loss in Castration-Resistant Prostate Cancer Confers Vulnerability to LSD1 Inhibition. Oncogene (2022) 41(6):852–64. doi: 10.1038/s41388-021-02135-3
90. Sehrawat A, Gao L, Wang Y, Bankhead A III, McWeeney SK, King CJ, et al. LSD1 Activates a Lethal Prostate Cancer Gene Network Independently of Its Demethylase Function. Proc Natl Acad Sci U S A (2018) 115(18):E4179–E88. doi: 10.1158/1538-7445.AM2018-LB-240
91. Coleman DJ, Sampson DA, Sehrawat A, Kumaraswamy A, Sun D, Wang Y, et al. Alternative Splicing of LSD1+8a in Neuroendocrine Prostate Cancer is Mediated by SRRM4. Neoplasia (2020) 22(6):253–62. doi: 10.1016/j.neo.2020.04.002
92. Labrecque MP, Brown LG, Coleman IM, Lakely B, Brady NJ, Lee JK, et al. RNA Splicing Factors SRRM3 and SRRM4 Distinguish Molecular Phenotypes of Castration-Resistant Neuroendocrine Prostate Cancer. Cancer Res (2021) 81(18):4736–50. doi: 10.1158/0008-5472.CAN-21-0307
93. Eigl BJ, North S, Winquist E, Finch D, Wood L, Sridhar SS, et al. A Phase II Study of the HDAC Inhibitor SB939 in Patients With Castration Resistant Prostate Cancer: NCIC Clinical Trials Group Study IND195. Invest N Drugs (2015) 33(4):969–76. doi: 10.1007/s10637-015-0252-4
94. Ruan L, Wang L, Wang X, He M, Yao X. SIRT1 Contributes to Neuroendocrine Differentiation of Prostate Cancer. Oncotarget (2018), 9(2):2002–16. doi: 10.18632/oncotarget.23111
95. Ruscetti M, Dadashian EL, Guo W, Quach B, Mulholland DJ, Park JW, et al. HDAC Inhibition Impedes Epithelial-Mesenchymal Plasticity and Suppresses Metastatic, Castration-Resistant Prostate Cancer. Oncogene (2016) 35(29):3781–95. doi: 10.1038/onc.2015.444
96. Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, et al. Histone Deacetylases are Required for Androgen Receptor Function in Hormone-Sensitive and Castrate-Resistant Prostate Cancer. Cancer Res (2009) 69(3):958–66. doi: 10.1158/0008-5472.CAN-08-2216
97. Jia Y, Zhao J, Yang J, Shao J, Cai Z. miR-301 Regulates the SIRT1/SOX2 Pathway via CPEB1 in the Breast Cancer Progression. Mol Ther Oncolytics (2021) 22:13–26. doi: 10.1016/j.omto.2021.03.007
98. Liu L, Liu C, Zhang Q, Shen J, Zhang H, Shan J, et al. SIRT1-Mediated Transcriptional Regulation of SOX2 Is Important for Self-Renewal of Liver Cancer Stem Cells. Hepatology (2016) 64(3):814–27. doi: 10.1002/hep.28690
99. Yoon DS, Choi Y, Jang Y, Lee M, Choi WJ, Kim SH, et al. SIRT1 Directly Regulates SOX2 to Maintain Self-Renewal and Multipotency in Bone Marrow-Derived Mesenchymal Stem Cells. Stem Cells (2014) 32(12):3219–31. doi: 10.1002/stem.1811
100. Marmorstein R, Zhou MM. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harb Perspect Biol (2014) 6(7):a018762. doi: 10.1101/cshperspect.a018762
101. Reyes AA, Marcum RD, He Y. Structure and Function of Chromatin Remodelers. J Mol Biol (2021) 433(14):166929. doi: 10.1016/j.jmb.2021.166929
102. Cyrta J, Augspach A, De Filippo MR, Prandi D, Thienger P, Benelli M, et al. Role of Specialized Composition of SWI/SNF Complexes in Prostate Cancer Lineage Plasticity. Nat Commun (2020) 11(1):5549. doi: 10.1038/s41467-020-19328-1
103. Pomerantz MM, Qiu X, Zhu Y, Takeda DY, Pan W, Baca SC, et al. Prostate Cancer Reactivates Developmental Epigenomic Programs During Metastatic Progression. Nat Genet (2020) 52(8):790–9. doi: 10.1038/s41588-020-0664-8
104. Zhang Z, Zhou C, Li X, Barnes SD, Deng S, Hoover E, et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell (2020) 37(4):584–98.e11. doi: 10.1016/j.ccell.2020.03.001
105. Augello MA, Liu D, Deonarine LD, Robinson BD, Huang D, Stelloo S, et al. CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell (2019) 35(4):603–17.e8. doi: 10.1016/j.ccell.2019.03.001
106. Hwang JY, Zukin RS. REST, a Master Transcriptional Regulator in Neurodegenerative Disease. Curr Opin Neurobiol (2018) 48:193–200. doi: 10.1016/j.conb.2017.12.008
107. Svensson C, Ceder J, Iglesias-Gato D, Chuan YC, Pang ST, Bjartell A, et al. REST Mediates Androgen Receptor Actions on Gene Repression and Predicts Early Recurrence of Prostate Cancer. Nucleic Acids Res (2014) 42(2):999–1015. doi: 10.1093/nar/gkt921
108. Lin TP, Chang YT, Lee SY, Campbell M, Wang TC, Shen SH, et al. REST Reduction is Essential for Hypoxia-Induced Neuroendocrine Differentiation of Prostate Cancer Cells by Activating Autophagy Signaling. Oncotarget (2016) 7(18):26137–51. doi: 10.18632/oncotarget.8433
109. Chang YT, Lin TP, Campbell M, Pan CC, Lee SH, Lee HC, et al. REST Is a Crucial Regulator for Acquiring EMT-Like and Stemness Phenotypes in Hormone-Refractory Prostate Cancer. Sci Rep (2017) 7:42795. doi: 10.1038/srep42795
110. Alfert A, Moreno N, Kerl K. The BAF Complex in Development and Disease. Epigenet Chromatin (2019) 12(1):19. doi: 10.1186/s13072-019-0264-y
111. Tang J, Yoo AS, Crabtree GR. Reprogramming Human Fibroblasts to Neurons by Recapitulating an Essential microRNA-Chromatin Switch. Curr Opin Genet Dev (2013) 23(5):591–8. doi: 10.1016/j.gde.2013.07.001
112. Olave I, Wang W, Xue Y, Kuo A, Crabtree GR. Identification of a Polymorphic, Neuron-Specific Chromatin Remodeling Complex. Genes Dev (2002) 16(19):2509–17. doi: 10.1101/gad.992102
113. Yoo AS, Staahl BT, Chen L, Crabtree GR. MicroRNA-Mediated Switching of Chromatin-Remodelling Complexes in Neural Development. Nature (2009) 460(7255):642–6. doi: 10.1038/nature08139
114. Parimi V, Goyal R, Poropatich K, Yang XJ. Neuroendocrine Differentiation of Prostate Cancer: A Review. Am J Clin Exp Urol (2014) 2(4):273–85.
115. Aggarwal R, Zhang T, Small EJ, Armstrong AJ. Neuroendocrine Prostate Cancer: Subtypes, Biology, and Clinical Outcomes. J Natl Compr Cancer Netw (2014) 12(5):719–26. doi: 10.6004/jnccn.2014.0073
116. Maleki Z, Nadella A, Nadella M, Patel G, Patel S, Kholová I. INSM1, a Novel Biomarker for Detection of Neuroendocrine Neoplasms: Cytopathologists’ View. Diagnostics (2021) 11(12):2172. doi: 10.3390/diagnostics11122172
117. Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc Natl Acad Sci U S A (2019) 116(23):11428–36. doi: 10.1073/pnas.1902651116
118. Boustani AM, Pucar D, Saperstein L. Molecular Imaging of Prostate Cancer. Br J Radiol (2018) 91(1084):20170736. doi: 10.1259/bjr.20170736
119. Fox JJ, Gavane SC, Blanc-Autran E, Nehmeh S, Gonen M, Beattie B, et al. Positron Emission Tomography/Computed Tomography-Based Assessments of Androgen Receptor Expression and Glycolytic Activity as a Prognostic Biomarker for Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol (2018) 4(2):217–24. doi: 10.1001/jamaoncol.2017.3588
120. Thang SP, Violet J, Sandhu S, Iravani A, Akhurst T, Kong G, et al. Poor Outcomes for Patients With Metastatic Castration-Resistant Prostate Cancer With Low Prostate-Specific Membrane Antigen (PSMA) Expression Deemed Ineligible for (177)Lu-Labelled PSMA Radioligand Therapy. Eur Urol Oncol (2019) 2(6):670–6. doi: 10.1016/j.euo.2018.11.007
121. Bakht MK, Derecichei I, Li Y, Ferraiuolo RM, Dunning M, Oh SW, et al. Neuroendocrine Differentiation of Prostate Cancer Leads to PSMA Suppression. Endocr Relat Cancer (2018) 26(2):131–46. doi: 10.1530/ERC-18-0226
122. Puca L, Gavyert K, Sailer V, Conteduca V, Dardenne E, Sigouros M, et al. Delta-Like Protein 3 Expression and Therapeutic Targeting in Neuroendocrine Prostate Cancer. Sci Transl Med (2019) 11(484). doi: 10.1126/scitranslmed.aav0891
123. Korsen JA, Kalidindi TM, Khitrov S, Samuels ZV, Chakraborty G, Gutierrez JA, et al. Molecular Imaging of Neuroendocrine Prostate Cancer by Targeting Delta-Like Ligand 3. J Nucl Med (2022). doi: 10.2967/jnumed.121.263221
124. Beltran H, Jendrisak A, Landers M, Mosquera JM, Kossai M, Louw J, et al. The Initial Detection and Partial Characterization of Circulating Tumor Cells in Neuroendocrine Prostate Cancer. Clin Cancer Res (2016) 22(6):1510–9. doi: 10.1158/1078-0432.CCR-15-0137
125. Brown LC, Halabi S, Schonhoft JD, Yang Q, Luo J, Nanus DM, et al. Circulating Tumor Cell Chromosomal Instability and Neuroendocrine Phenotype by Immunomorphology and Poor Outcomes in Men With mCRPC Treated With Abiraterone or Enzalutamide. Clin Cancer Res (2021) 27(14):4077–88. doi: 10.1158/1078-0432.CCR-20-3471
126. Berchuck JE, Baca SC, McClure HM, Korthauer K, Tsai HK, Nuzzo PV, et al. Detecting Neuroendocrine Prostate Cancer Through Tissue-Informed Cell-Free DNA Methylation Analysis. Clin Cancer Res (2021). 28(5):928–938. doi: 10.1158/1078-0432.CCR-21-3762
127. Amato RJ, Logothetis CJ, Hallinan R, Ro JY, Sella A, Dexeus FH. Chemotherapy for Small Cell Carcinoma of Prostatic Origin. J Urol (1992) 147(3):935–7. doi: 10.1016/S0022-5347(17)37427-X
128. Papandreou CN, Daliani DD, Thall PF, Tu S-M, Wang X, Reyes A, et al. Results of a Phase II Study With Doxorubicin, Etoposide, and Cisplatin in Patients With Fully Characterized Small-Cell Carcinoma of the Prostate. J Clin Oncol (2002) 20(14):3072–80. doi: 10.1200/JCO.2002.12.065
129. Culine S, El Demery M, Lamy PJ, Iborra F, Avances C, Pinguet F. Docetaxel and Cisplatin in Patients With Metastatic Androgen Independent Prostate Cancer and Circulating Neuroendocrine Markers. J Urol (2007) 178(3 Pt 1):844–8. doi: 10.1016/j.juro.2007.05.044
130. Aparicio AM, Harzstark AL, Corn PG, Wen S, Araujo JC, Tu SM, et al. Platinum-Based Chemotherapy for Variant Castrate-Resistant Prostate Cancer. Clin Cancer Res (2013) 19(13):3621–30. doi: 10.1158/1078-0432.CCR-12-3791
131. Conteduca V, Oromendia C, Eng KW, Bareja R, Sigouros M, Molina A, et al. Clinical Features of Neuroendocrine Prostate Cancer. Eur J Cancer (2019) 121:7–18. doi: 10.1016/j.ejca.2019.08.011
132. Mitrou PS, Graubner M, Berdel WE, Mende S, Gropp C, Diehl V, et al. Cis-Platinum (DDP) and VP 16-213 (Etoposide) Combination Chemotherapy for Advanced non-Small Cell Lung Cancer. A Phase II Clin Trial. Eur J Cancer Clin Oncol (1984) 20(3):347–51. doi: 10.1016/0277-5379(84)90080-4
133. Brown LC, Halabi S, Somarelli JA, Humeniuk M, Wu Y, Oyekunle T, et al. A Phase 2 Trial of Avelumab in Men With Aggressive-Variant or Neuroendocrine Prostate Cancer. Prostate Cancer Prostatic Dis (2022). doi: 10.1038/s41391-022-00524-7
134. Beltran H, Oromendia C, Danila DC, Montgomery B, Hoimes C, Szmulewitz RZ, et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients With Castration-Resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin Cancer Res (2019) 25(1):43–51. doi: 10.1158/1078-0432.CCR-18-1912
135. Berger A, Brady NJ, Bareja R, Robinson B, Conteduca V, Augello MA, et al. N-Myc-Mediated Epigenetic Reprogramming Drives Lineage Plasticity in Advanced Prostate Cancer. J Clin Invest (2019) 129(9):3924–40. doi: 10.1172/JCI127961
136. Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell (2009) 15(1):67–78. doi: 10.1016/j.ccr.2008.12.005
137. Mansfield AS, Hong DS, Hann CL, Farago AF, Beltran H, Waqar SN, et al. A Phase I/II Study of Rovalpituzumab Tesirine in Delta-Like 3-Expressing Advanced Solid Tumors. NPJ Precis Oncol (2021) 5(1):74. doi: 10.1038/s41698-021-00214-y
138. Singal R, Ramachandran K, Gordian E, Quintero C, Zhao W, Reis IM. Phase I/II Study of Azacitidine, Docetaxel, and Prednisone in Patients With Metastatic Castration-Resistant Prostate Cancer Previously Treated With Docetaxel-Based Therapy. Clin Genitourin Cancer (2015) 13(1):22–31. doi: 10.1016/j.clgc.2014.07.008
139. Fu M, Liu M, Sauve A, Jiao X, Zhang X, Wu X, et al. Hormonal Control of Androgen Receptor Function Through SIRT1. Mol Cell Biol (2006), 26(21):8122–35. doi: 10.1128/MCB.00289-06
140. Ferrari AC, Alumkal JJ, Stein MN, Taplin ME, Babb J, Barnett ES, et al. Epigenetic Therapy With Panobinostat Combined With Bicalutamide Rechallenge in Castration-Resistant Prostate Cancer. Clin Cancer Res (2019) 25(1):52–63. doi: 10.1158/1078-0432.CCR-18-1589
141. Rathkopf DE, Picus J, Hussain A, Ellard S, Chi KN, Nydam T, et al. A Phase 2 Study of Intravenous Panobinostat in Patients With Castration-Resistant Prostate Cancer. Cancer Chemother Pharmacol (2013) 72(3):537–44. doi: 10.1007/s00280-013-2224-8
142. Wheler JJ, Janku F, Falchook GS, Jackson TL, Fu S, Naing A, et al. Phase I Study of Anti-VEGF Monoclonal Antibody Bevacizumab and Histone Deacetylase Inhibitor Valproic Acid in Patients With Advanced Cancers. Cancer Chemother Pharmacol (2014) 73(3):495–501. doi: 10.1007/s00280-014-2384-1
143. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic Targeting of BET Bromodomain Proteins in Castration-Resistant Prostate Cancer. Nature (2014) 510(7504):278–82. doi: 10.1038/nature13229
144. Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J Clin Oncol (2018) 36(30):3007–14. doi: 10.1200/JCO.2018.78.2292
145. Piha-Paul SA, Sachdev JC, Barve M, LoRusso P, Szmulewitz R, Patel SP, et al. First-In-Human Study of Mivebresib (ABBV-075), an Oral Pan-Inhibitor of Bromodomain and Extra Terminal Proteins, in Patients With Relapsed/Refractory Solid Tumors. Clin Cancer Res (2019) 25(21):6309–19. doi: 10.1158/1078-0432.CCR-19-0578
Keywords: Neuroendocrine prostate cancer (NEPC), Lineage plasticity, transdifferentiation, Androgen Receptor (AR), epigenetics
Citation: Storck WK, May AM, Westbrook TC, Duan Z, Morrissey C, Yates JA and Alumkal JJ (2022) The Role of Epigenetic Change in Therapy-Induced Neuroendocrine Prostate Cancer Lineage Plasticity. Front. Endocrinol. 13:926585. doi: 10.3389/fendo.2022.926585
Received: 22 April 2022; Accepted: 19 May 2022;
Published: 14 July 2022.
Edited by:
Amina Zoubeidi, University of British Columbia, CanadaReviewed by:
Kerry L Burnstein, University of Miami Miller School of Medicine, United StatesNahuel Peinetti, University of Miami Miller School of Medicine, United States, contributed to the review of KB
Joshua Russo, Beth Israel Deaconess Medical Center and Harvard Medical School, United States
Copyright © 2022 Storck, May, Westbrook, Duan, Morrissey, Yates and Alumkal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshi J. Alumkal, amFsdW1rYWxAbWVkLnVtaWNoLmVkdQ==
†These authors share first authorship