 Mengdi Zhang
Mengdi Zhang Yanyan Zhou
Yanyan Zhou Zhiguo Xie
Zhiguo Xie Shuoming Luo
Shuoming Luo Zhiguang Zhou
Zhiguang Zhou Jiaqi Huang
Jiaqi Huang Bin Zhao
Bin Zhao- 1National Clinical Research Center for Metabolic Diseases, Key Laboratory of Diabetes Immunology, Ministry of Education, and Department of Metabolism and Endocrinology, The Second Xiangya Hospital of Central South University, Changsha, China
- 2Department of Critical Care Medicine, The Second Xiangya Hospital of Central South University, Changsha, China
Type 1 diabetes (T1D) is an autoimmune disease mediated by T cells and is becoming a serious public health threat. Despite the increasing incidence rate of T1D worldwide, our understanding of why T1D develops and how T cells lose their self-tolerance in this process remain limited. Recent advances in immunometabolism have shown that cellular metabolism plays a fundamental role in shaping T cell responses. T cell activation and proliferation are supported by metabolic reprogramming to meet the increased energy and biomass demand, and deregulation in immune metabolism can lead to autoimmune disorders. Specific metabolic pathways and factors have been investigated to rectify known deficiencies in several autoimmune diseases, including T1D. Most therapeutic strategies have concentrated on aerobic glycolysis to limit T cell responses, whereas glycolysis is the main metabolic pathway for T cell activation and proliferation. The use of metabolic inhibitors, especially glycolysis inhibitors may largely leave T cell function intact but primarily target those autoreactive T cells with hyperactivated metabolism. In this review, we provide an overview of metabolic reprogramming used by T cells, summarize the recent findings of key metabolic pathways and regulators modulating T cell homeostasis, differentiation, and function in the context of T1D, and discuss the opportunities for metabolic intervention to be employed to suppress autoreactive T cells and limit the progression of β-cell destruction.
Introduction
T1D is a chronic immune-metabolic disease and is becoming a serious public health threat (1). Over the past three decades, the incidence of T1D has escalated worldwide, afflicting as many as 10 million people (2, 3). The pathogenesis of T1D is complicated, and available data suggest that T1D arises due to the combination of genetically determined susceptibility, environmental factors, and impairment of immunity, which eventually leads to the breakdown of immune tolerance to self (4, 5). It was demonstrated that autoreactive CD4+ and CD8+ T cells that infiltrate the islets of T1D patients play a key role in the process of β-cell destruction (6, 7). Thus, those autoreactive T cells are regarded as a potential target for immune-based interventions aiming to combat T1D (8–11).
Recent advances in metabolomics, transgenic mice and immunometabolism have shown that metabolic adaptation plays a crucial role in shaping T cell responses (12–14). T cell activation is linked to metabolic reprogramming to meet the increased energy and biomass demand (15, 16). Binding of antigen to T cell receptor (TCR) initiates the activation of naïve T cells, which leads to a metabolic program shift from oxidative phosphorylation (OXPHOS) to robust aerobic glycolysis for rapid clonal proliferation and effector functions (17–19). In recent years, many exciting findings have uncovered novel metabolic pathways and key molecules that could be applied to improve the governance of autoimmunity and guide the treatment of autoimmune diseases (20). The use of metabolic inhibitors, especially glycolysis inhibitors, may largely leave T cell function intact but primarily target autoreactive T cells with hyperactivated metabolism (9, 20). This review aims to provide an overview of metabolic reprogramming used by T cells, summarize the recent findings of key metabolic pathways and regulators modulating T cell homeostasis, differentiation, and functions in the context of T1D, and discuss the opportunities for metabolic intervention to be employed to suppress autoreactive T cells and limit the progression of β-cell destruction.
Metabolic Reprogramming of T Cells in T1D
The pathogenesis of T1D is mainly mediated by the activation of autoreactive CD4+ and CD8+ T cells, which are fueled by metabolic reprogramming (7, 21). Activated effector T cells are more metabolically active and engage mainly in aerobic glycolysis (22). The transition from naïve into effector T cells is driven by variations in anabolic and catabolic metabolism (23). Notably, researchers have reported that glycolysis is essential for cytotoxic T lymphocytes’ function. Treatment of nonobese diabetic (NOD) mice with glycolysis inhibitors resulted in delayed T1D onset and protected β-cell mass (24). In addition, as part of the OXPHOS program, the movement of electrons generates a substantial amount of reactive oxygen species (ROS) (25). The role of ROS in controlling the differentiation of T cells by modulating metabolism has recently been described (21, 25–30). Investigators have shown that ROS can act as signaling molecules involved in the process of T cell activation, proliferation, and function (26). In T1D, ROS generation leads to the activation of autoreactive T cells and β-cell destruction (30). Regulatory T cells (Tregs) are key mediators of peripheral immune tolerance (31–33). Yet, in some autoimmune diseases, Tregs have been shown to have altered stability or function (32). Several researchers have confirmed that impaired Treg function, decreased Treg numbers, or the transition into Th1 (helper T cell 1)-like Treg, contributed to T1D development (33–38). Tregs have unique metabolic preferences that have not been characterized clearly (31, 39). It is generally recognized that Tregs preferentially use OXPHOS and fatty acid oxidation (FAO) for differentiation and function (35, 40, 41)(Figure 1).
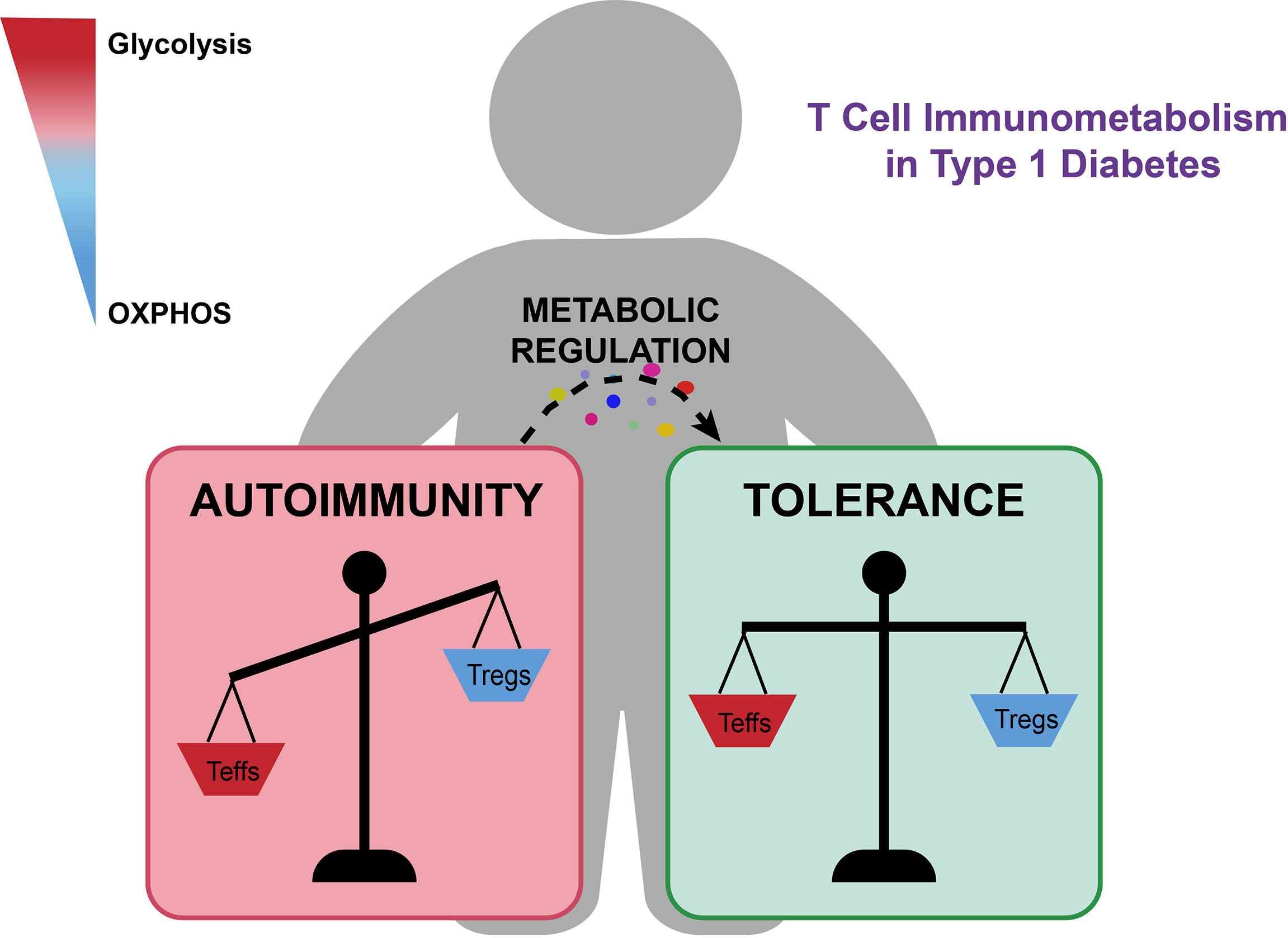
Figure 1 The metabolic imbalance of autoreactive T cells and Tregs contributes to T1D development and progression. Glycolysis supports pro-inflammatory responses, while OXPHOS promotes anti-inflammatory responses and immune tolerance. Immunometabolism regulators of key metabolic pathways, such as metformin and PFK15, could suppress effector T cell responses and facilitate Treg expansion and function, which could provide a promising metabolic intervention strategy for limiting the progression of β-cell destruction and reversing pathologies of T1D.
Metabolic Interventions: A New Opportunity for T1D Treatment
Both mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) are metabolic sensors required for T cell proliferation and function (19, 42–44). Activation of AMPK inhibits anabolic metabolism, such as nucleic acid and lipid synthesis, but favors catabolic metabolism. In contrast, activation of mTOR signaling facilitates glycolysis, fatty acid production, and mitochondrial biogenesis (42, 45, 46). As mentioned above, autoreactive CD4+/CD8+ T cells exhibit a higher level of glycolysis and depend less on OXPHOS, thus suggesting that glycolysis could be used as an attractive therapeutic target (47). Inhibiting mTOR signaling with rapamycin or enhancing the AMPK signaling pathway with metformin are known to reduce glycolysis (19). Given the key role of AMPK in the activation of T cells, multiple studies have investigated the capacity of metformin to suppress autoimmune diseases, notably T1D. Metformin is now the first line of oral antidiabetic medicine and is used to regulate glucose metabolism (48). Mechanistically, metformin inhibits the mitochondrial electron transport chain (ETC) at Complex I and results in a reduction in intracellular ATP production (48–50). Furthermore, laboratory work demonstrated that metformin could reduce the expansion of activated T cells by inhibiting the expression of cellular myelocytomatosis oncogene (c-Myc) and hypoxia-inducible factor 1 alpha (HIF1-α) in an AMPK-independent way (51–53). Metformin exhibits a dose-dependent effect to control T cell proliferation and suppress the differentiation of Th1 and Th17 cells while enhancing Treg development in vitro. NOD mice treated with metformin showed alleviated autoimmune insulitis and reduced amounts of Th1 and Th17 cells in the spleens (50). Furthermore, the anti-inflammatory function of metformin has also been investigated in detail in mouse models of autoimmune arthritis, systemic lupus erythematosus and colitis, all of which portrayed a role of metformin as an anti-inflammatory coordinator and provided the rationale for possible islet protective properties (54–57). Currently, the REMOVAL study and some other smaller trials have proven the clinical advantage of metformin against diverse cardiovascular surrogate endpoints, while the long-term effect of metformin on islet autoimmunity still needs to be further investigated (58–61). As a master regulator of cell metabolism, mTOR has been shown to enhance helper T cell differentiation, especially Th1 and Th17, by modulating glucose metabolism through glucose transporter 1 (Glut1) (62). Therefore, targeting upstream or downstream of mTOR signaling is a potential therapeutic strategy. As a classic mTOR inhibitor, rapamycin decreases the proliferation of Th1 and Th17 cells (63). Furthermore, rapamycin was documented to facilitate Treg expansion and enhance their capability to suppress conventional T cells in a T1D mouse model (34, 64–66). Likewise, augmenting catabolic pathways in CD8+ T cells with metformin or rapamycin decreased the differentiation and proliferation of effector T cells instead of enhancing memory T cell expansion (67). In a phase 2, single-center, randomized, double-blind, placebo-controlled study, rapamycin was shown to decrease insulin requirement in patients with long-term T1D (68). Interestingly, ω-3 polyunsaturated fatty acids (ω-3 PUFAs) have been shown to inhibit CD4+ T cell differentiation via suppressing mTOR complex 1 (mTORC1). The pancreatic enrichment of ω-3 PUFAs could inhibit or avoid T1D progression in streptozotocin (STZ)-induced mice (69, 70).
Upon initial activation of lymphocytes, Glut1, one of the typical glucose transporters, is upregulated by the PI3K-Akt-mTOR signaling pathway to enhance glucose influx as well as concomitant with the increased production of key glycolytic enzymes (71, 72). The upregulation of Glut1 is critical for T cell activation, as deletion of Glut1 greatly suppresses proliferation and function of effector T cells (73, 74). Pharmacological blockade of Glut1 might be an efficient way to inhibit autoreactive T cells. The glycolysis inhibitor 2-deoxy-D-glucose (2-DG) is a glucose analog that selectively targets effector T cells with upregulated glycolytic activity (16, 24, 75, 76). NOD mice treated with 2-DG displayed a reduced frequency of activated T cells, decreased immune infiltration within pancreatic islets and increased β-cell granularity (24, 77, 78). Additionally, 2-DG facilitates the differentiation of naïve T cells into Tregs but represses their polarization to Th17 cells (36). Likewise, studies have demonstrated that the combination of 2-DG and metformin reduces CD4+/CD8+ effector T cell responses while inducing Tregs, probably by increasing FAO (79). However, in the light of translation from preclinical trials to clinical application for T1D patients, one of the most relevant side effects of 2-DG is central nervous system toxicity, which demands a prompt solution (80–82). In addition, various natural or synthetic molecules that function as Glut1 inhibitors have emerged in recent years, such as sodium meta-arsenite, STF-31, WZB117 and BAY876, which give us more therapeutic options (73, 83–89).
PFK15, a competitive inhibitor of the rate-limiting glycolysis enzyme, has been found to suppress glycolysis utilization of CD4+ T cells and decrease the response of CD4+ T cells to β-cell antigens. Additionally, treatment of PFK15 in NOD mouse models delayed T1D onset due to metabolic and functional exhaustion of T cells (47). In addition, peroxisome proliferator-activated receptors (PPARs) are transcription factors that control genes involved in glucose and lipid metabolism and FAO (90–92). PPARs are expressed in multiple immune cells including T cells, and modulation of FAO through PPARs provides the possibility to promote immunological intervention therapy (93, 94). Activation of PPARβ/δ inhibits Th1 and Th17 cell differentiation due to the transition from glycolysis to FAO and suppresses the proliferative burst of T cells upon activation (95, 96). Researchers have shown that the PPARα activator fenofibrate and the PPARγ activators troglitazone and rosiglitazone have the capability to decrease the incidence of T1D (95, 97, 98). With the treatment of troglitazone, STZ-induced T1D mice exhibited reduced hyperglycemia and insulitis (99, 100).
Another potential approach to improving T1D is to regulate ROS production. T1D is known to be highly actuated by oxidative stress, as CD4+ T cells require high levels of ROS for optimal activation (26, 101). Utilizing manganese metalloporphyrin (MnP), a ROS scavenger and potent antioxidant, delayed T1D progression through modulating aerobic glycolysis and the mTOR/AMPK axis (102–104). Given the critical role of ROS in autoimmune diseases, researchers have applied superoxide dismutase (SOD) mimetics in a T1D mouse model to promote the longevity and stability of antioxidants to delay β-cell damage (25, 105). T1D was significantly delayed or prevented in NOD mice treated with SOD mimic, partly owing to the decrease in proliferation of CD4+/CD8+ T cells as well as reduced production of pro-inflammatory factors (26, 53, 106, 107). Additionally, lymphocyte activation gene 3 (LAG-3) is an inhibitory receptor expressed on the CD4+ T cell surface, whose deficiency would result in their homeostatic expansion. Studies have reported that the expression of LAG-3 in naïve CD4+ T cells contributes to the restriction of mitochondrial biogenesis and cellular metabolism to keep T cells quiescent. Loss of LAG-3 in NOD mice leads to accelerated T1D progression, potentially by enhancing OXPHOS and glycolytic metabolism and promoting mitochondrial biogenesis of CD4+ T cells (102, 108, 109).
Bacillus Calmette-Guérin (BCG) has been reported as a conducive environmental qualifier of the immune system that could reduce the incidence of autoimmune diseases such as T1D (110). Recent studies indicate that BCG vaccination in patients with long-term T1D showed promising antidiabetic effects, including death of autoimmune T cells as well as expansion of beneficial Tregs (111–113). In an 8-year human study with T1D, BCG vaccination was demonstrated to promote the transition from OXPHOS to aerobic glycolysis of immune cells, improving Treg generation and function, and conferring an immunotolerance effect (114–116). High-mobility group box 1 (HMGB1), an evolutionarily conserved chromosomal protein, was demonstrated to impair the stability and function of Tregs by enhancing PI3K-AKT-mTOR signaling. NOD mice with HMGB1 blockage could protect islet isografts from autoimmune attacks and delay or even reverse T1D development (117).
Therapeutic Applications of Immunometabolism in Combination Therapy
The complex etiology of T1D is the consequence of failures in controlling autoimmunity as well as perturbations of β cells (118). In addition to controlling autoimmune responses, ideal therapies would also aim to preserve β-cell function and promote β-cell regeneration (119, 120). To date, several immunometabolism-related interventions combined with other therapy regimens have been proven to be successful in NOD mouse models (63, 121–124). For example, the combination treatment regimen of rapamycin and a CD28 antagonist was reported to inhibit T cell activation and migration into pancreatic islets, hence suppressing the progression of T1D (122). Treatment of NOD mice with rapamycin and IL-2 limits T cell expansion and effectively protects islet β-cells from autoimmune attacks (125). Furthermore, combination therapy with rapamycin, islet autoantigen peptides, and IL-2/IL-2 monoclonal antibody complexes increases Treg numbers and protects against autoimmune diabetes in NOD mice (121). However, a phase 1 clinical trial of a rapamycin/IL-2 combination in 10 T1D patients led to transient dysfunction of β cells despite an enrichment of Treg cells (63). Laboratory evidence has demonstrated that IL-21 signaling plays a critical role in promoting lymphocyte infiltration into the pancreas and rewiring T cell metabolism to form long-lived memory CD8+ T cells, which are the predominantly presented T cell subsets in the pancreatic islets of T1D mouse model (126–128). Matthias von Herrath et al. evaluated the combination of immunotherapy (IL-21) and β-cell-directed treatment (liraglutide) in a randomized, double-blind and phase 2 trial in 308 adults with new-onset T1D (129). After fifty-four weeks of treatment and follow-up, C-peptide secretion was prominently improved in the combination therapy group compared with the placebo, but the effect disappeared after therapy cessation in the follow-up period. In conclusion, the effectiveness of combination therapies in animal models and the first large clinical trial provides a promising approach for the development of novel combination therapies (130).
Conclusion
Our understanding of immunometabolism has considerably advanced over the past few years. Multiple studies have demonstrated that key metabolic enzymes and regulators are involved in different processes of T cell responses by alternating the metabolic pathways and networks to match their specific functional requirements (18, 131, 132). Modulating T cell metabolism has the capability of selectively enhancing or inhibiting particular T cell subsets with distinct functions (133). Of note, although gene knockout mice have presented valuable information, an inescapable limitation is that there are differences between mouse and human immune systems as well as metabolic programs. Moreover, cellular metabolism in vivo is distinct from that in vitro, while a large number of studies have assessed the metabolism of immune cells during their differentiation, proliferation, and responses in vitro. Collectively, targeting T cell metabolism could be a promising strategy for the next wave of immunotherapies treating human diseases, including T1D.
Author Contributions
JH and BZ: conceptualization and guidance. MZ and YZ wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (grant numbers 82170795 and 82100949), and the Outstanding Young Investigator of Hunan Province (2022JJ10094).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Norris JM, Johnson RK, Stene LC. Type 1 Diabetes-Early Life Origins and Changing Epidemiology. Lancet Diabetes Endocrinol (2020) 8:226–38. doi: 10.1016/S2213-8587(19)30412-7
2. Sharp SA, Rich SS, Wood AR, Jones SE, Beaumont RN, Harrison JW, et al. Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident Diagnosis. Diabetes Care (2019) 42:200–7. doi: 10.2337/dc18-1785
3. Weng J, Zhou Z, Guo L, Zhu D, Ji L, Luo X, et al. Incidence of Type 1 Diabetes in China, 2010-13: Population Based Study. BMJ (2018) 360:j5295. doi: 10.1136/bmj.j5295
4. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 Diabetes. Lancet Lond Engl (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
5. Chen W, Xie A, Chan L. Mechanistic Basis of Immunotherapies for Type 1 Diabetes Mellitus. Transl Res J Lab Clin Med (2013) 161:217–29. doi: 10.1016/j.trsl.2012.12.017
6. Stechova K, Sklenarova-Labikova J, Kratzerova T, Pithova P, Filipp D. Not Only Glycaemic But Also Other Metabolic Factors Affect T Regulatory Cell Counts and Proinflammatory Cytokine Levels in Women With Type 1 Diabetes. J Diabetes Res (2017) 2017:1–12. doi: 10.1155/2017/5463273
7. Burrack AL, Martinov T, Fife BT. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front Endocrinol (2017) 8:343. doi: 10.3389/fendo.2017.00343
8. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 Diabetes Immunotherapy Using Polyclonal Regulatory T Cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
9. Jacobsen LM, Newby BN, Perry DJ, Posgai AL, Haller MJ, Brusko TM. Immune Mechanisms and Pathways Targeted in Type 1 Diabetes. Curr Diabetes Rep (2018) 18:90. doi: 10.1007/s11892-018-1066-5
10. Smith EL, Peakman M. Peptide Immunotherapy for Type 1 Diabetes-Clinical Advances. Front Immunol (2018) 9:392. doi: 10.3389/fimmu.2018.00392
11. Bone RN, Evans-Molina C. Combination Immunotherapy for Type 1 Diabetes. Curr Diabetes Rep (2017) 17:50. doi: 10.1007/s11892-017-0878-z
12. Huang H, Zhou P, Wei J, Long L, Shi H, Dhungana Y, et al. In Vivo CRISPR Screening Reveals Nutrient Signaling Processes Underpinning CD8+ T Cell Fate Decisions. Cell (2021) 184:1245–1261.e21. doi: 10.1016/j.cell.2021.02.021
13. Ceballos B, Alexander M, Lakey JRT. Advanced Approaches in Immunotherapy for the Treatment of Type 1 Diabetes Mellitus. EMJ Diabetes (2020). doi: 10.33590/emjdiabet/20-00062
14. Lee M-S, Bensinger SJ. Reprogramming Cholesterol Metabolism in Macrophages and Its Role in Host Defense Against Cholesterol-Dependent Cytolysins. Cell Mol Immunol (2022) 19:327–36. doi: 10.1038/s41423-021-00827-0
15. Chi H. Immunometabolism at the Intersection of Metabolic Signaling, Cell Fate, and Systems Immunology. Cell Mol Immunol (2022) 19:299–302. doi: 10.1038/s41423-022-00840-x
16. Lercher A, Baazim H, Bergthaler A. Systemic Immunometabolism: Challenges and Opportunities. Immunity (2020) 53:496–509. doi: 10.1016/j.immuni.2020.08.012
17. Martins CP, Piganelli J. Targeting T Cell Metabolism to Combat Autoimmunity: Implications for the Future of Type 1 Diabetes Therapeutics. (2020). doi: 10.20900/immunometab20200010
18. Nm C HC. Metabolic Adaptation of Lymphocytes in Immunity and Disease. Immunity (2022) 55(1):14–30. doi: 10.1016/j.immuni.2021.12.012
19. Galgani M, De Rosa V, Matarese G. T Cell Metabolism and Susceptibility to Autoimmune Diseases. Mol Immunol (2015) 68:558–63. doi: 10.1016/j.molimm.2015.07.035
20. Pålsson-McDermott EM, O’Neill LAJ. Targeting Immunometabolism as an Anti-Inflammatory Strategy. Cell Res (2020) 30:300–14. doi: 10.1038/s41422-020-0291-z
21. Previte DM, Piganelli JD. Reactive Oxygen Species and Their Implications on CD4+ T Cells in Type 1 Diabetes. Antioxid Redox Signal (2018) 29:1399–414. doi: 10.1089/ars.2017.7357
22. Dimeloe S, Burgener A, Grählert J, Hess C. T-Cell Metabolism Governing Activation, Proliferation and Differentiation; a Modular View. Immunology (2017) 150:35–44. doi: 10.1111/imm.12655
23. Chen H, Yang T, Zhu L, Zhao Y. Cellular Metabolism on T-Cell Development and Function. Int Rev Immunol (2015) 34:19–33. doi: 10.3109/08830185.2014.902452
24. Garyu JW, Uduman M, Stewart A, Rui J, Deng S, Shenson J, et al. Characterization Of Diabetogenic Cd8+ T Cells: Immune Therapy With Metabolic Blockade. J Biol Chem (2016) 291:11230–40. doi: 10.1074/jbc.M115.713362
25. Neyestani TR, Ghandchi Z, Eshraghian M-R, Kalayi A, Shariatzadeh N, Houshiarrad A. Evidence for Augmented Oxidative Stress in the Subjects With Type 1 Diabetes and Their Siblings: A Possible Preventive Role for Antioxidants. Eur J Clin Nutr (2012) 66:1054–8. doi: 10.1038/ejcn.2012.81
26. Chávez MD, Tse HM. Targeting Mitochondrial-Derived Reactive Oxygen Species in T Cell-Mediated Autoimmune Diseases. Front Immunol (2021) 12:703972. doi: 10.3389/fimmu.2021.703972
27. Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang C-H, Sanin DE, et al. Mitochondrial Dynamics Controls T Cell Fate Through Metabolic Programming. Cell (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
28. Klein Geltink RI, O’Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, et al. Mitochondrial Priming by CD28. Cell (2017) 171:385–397.e11. doi: 10.1016/j.cell.2017.08.018
29. Chen J, Stimpson SE, Fernandez-Bueno GA, Mathews CE. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid Redox Signal (2018) 29:1361–72. doi: 10.1089/ars.2017.7346
30. Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The Role of Reactive Oxygen Species and Proinflammatory Cytokines in Type 1 Diabetes Pathogenesis. Ann N Y Acad Sci (2013) 1281:16–35. doi: 10.1111/j.1749-6632.2012.06826.x
31. Kitz A, Singer E, Hafler D. Regulatory T Cells: From Discovery to Autoimmunity. Cold Spring Harb Perspect Med (2018) 8:a029041. doi: 10.1101/cshperspect.a029041
32. Hua J, Inomata T, Chen Y, Foulsham W, Stevenson W, Shiang T, et al. Pathological Conversion of Regulatory T Cells is Associated With Loss of Allotolerance. Sci Rep (2018) 8:7059. doi: 10.1038/s41598-018-25384-x
33. Visperas A, Vignali DAA. Are Regulatory T Cells Defective in Type 1 Diabetes and Can We Fix Them? J Immunol Baltim Md 1950 (2016) 197:3762–70. doi: 10.4049/jimmunol.1601118
34. Galgani M, De Rosa V, La Cava A, Matarese G. Role of Metabolism in the Immunobiology of Regulatory T Cells. J Immunol Baltim Md 1950 (2016) 197:2567–75. doi: 10.4049/jimmunol.1600242
35. Newton R, Priyadharshini B, Turka LA. Immunometabolism of Regulatory T Cells. Nat Immunol (2016) 17:618–25. doi: 10.1038/ni.3466
36. Tanimine N, Germana SK, Fan M, Hippen K, Blazar BR, Markmann JF, et al. Differential Effects of 2-Deoxy-D-Glucose on In Vitro Expanded Human Regulatory T Cell Subsets. PloS One (2019) 14:e0217761. doi: 10.1371/journal.pone.0217761
37. Chen Y, Chen S, Gu Y, Feng Y, Shi Y, Fu Q, et al. CTLA-4 +49 G/A, a Functional T1D Risk SNP, Affects CTLA-4 Level in Treg Subsets and IA-2A Positivity, But Not Beta-Cell Function. Sci Rep (2018) 8:10074. doi: 10.1038/s41598-018-28423-9
38. Lawson JM, Tremble J, Dayan C, Beyan H, Leslie RDG, Peakman M, et al. Increased Resistance to CD4+CD25hi Regulatory T Cell-Mediated Suppression in Patients With Type 1 Diabetes. Clin Exp Immunol (2008) 154:353–9. doi: 10.1111/j.1365-2249.2008.03810.x
39. Tsutsumi Y, Jie X, Ihara K, Nomura A, Kanemitsu S, Takada H, et al. Phenotypic and Genetic Analyses of T-Cell-Mediated Immunoregulation in Patients With Type 1 Diabetes. Diabetes Med J Br Diabetes Assoc (2006) 23:1145–50. doi: 10.1111/j.1464-5491.2006.01951.x
40. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol Baltim Md 1950 (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
41. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25:1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018
42. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. Mtorc1 Couples Immune Signals and Metabolic Programming to Establish T(reg)-Cell Function. Nature (2013) 499:485–90. doi: 10.1038/nature12297
43. Sun I-H, Oh M-H, Zhao L, Patel CH, Arwood ML, Xu W, et al. mTOR Complex 1 Signaling Regulates the Generation and Function of Central and Effector Foxp3+ Regulatory T Cells. J Immunol Baltim Md 1950 (2018) 201:481–92. doi: 10.4049/jimmunol.1701477
44. Chou W-C, Rampanelli E, Li X, Ting JP-Y. Impact of Intracellular Innate Immune Receptors on Immunometabolism. Cell Mol Immunol (2022) 19:337–51. doi: 10.1038/s41423-021-00780-y
45. Pollizzi KN, Powell JD. Regulation of T Cells by mTOR: The Known Knowns and the Known Unknowns. Trends Immunol (2015) 36:13–20. doi: 10.1016/j.it.2014.11.005
46. Yang K, Shrestha S, Zeng H, Karmaus PWF, Neale G, Vogel P, et al. T Cell Exit From Quiescence and Differentiation Into Th2 Cells Depend on Raptor-Mtorc1-Mediated Metabolic Reprogramming. Immunity (2013) 39:1043–56. doi: 10.1016/j.immuni.2013.09.015
47. Martins CP, New LA, O’Connor EC, Previte DM, Cargill KR, Tse IL, et al. Glycolysis Inhibition Induces Functional and Metabolic Exhaustion of CD4+ T Cells in Type 1 Diabetes. Front Immunol (2021) 12:669456. doi: 10.3389/fimmu.2021.669456
48. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J Clin Invest (2001) 108:1167–74. doi: 10.1172/JCI13505
49. Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, et al. Metformin Activates AMP-Activated Protein Kinase in Primary Human Hepatocytes by Decreasing Cellular Energy Status. Diabetologia (2011) 54:3101–10. doi: 10.1007/s00125-011-2311-5
50. Duan W, Ding Y, Yu X, Ma D, Yang B, Li Y, et al. Metformin Mitigates Autoimmune Insulitis by Inhibiting Th1 and Th17 Responses While Promoting Treg Production. Am J Transl Res (2019) 11(4):2393–402.
51. Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin Inhibits Hepatic Gluconeogenesis in Mice Independently of the LKB1/AMPK Pathway via a Decrease in Hepatic Energy State. J Clin Invest (2010) 120:2355–69. doi: 10.1172/JCI40671
52. Kim YD, Park K-G, Lee Y-S, Park Y-Y, Kim D-K, Nedumaran B, et al. Metformin Inhibits Hepatic Gluconeogenesis Through AMP-Activated Protein Kinase-Dependent Regulation of the Orphan Nuclear Receptor SHP. Diabetes (2008) 57:306–14. doi: 10.2337/db07-0381
53. Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. Adenosine-Mono-Phosphate-Activated Protein Kinase-Independent Effects of Metformin in T Cells. PloS One (2014) 9:e106710. doi: 10.1371/journal.pone.0106710
54. de Bari L, Atlante A. Including the Mitochondrial Metabolism of L-Lactate in Cancer Metabolic Reprogramming. Cell Mol Life Sci CMLS (2018) 75:2763–76. doi: 10.1007/s00018-018-2831-y
55. Lee S-Y, Lee SH, Yang E-J, Kim E-K, Kim J-K, Shin D-Y, et al. Metformin Ameliorates Inflammatory Bowel Disease by Suppression of the STAT3 Signaling Pathway and Regulation of the Between Th17/Treg Balance. PloS One (2015) 10:e0135858. doi: 10.1371/journal.pone.0135858
56. Kim EK, Lee SH, Jhun JY, Byun JK, Jeong JH, Lee S-Y, et al. Metformin Prevents Fatty Liver and Improves Balance of White/Brown Adipose in an Obesity Mouse Model by Inducing Fgf21. Mediators Inflammation (2016) 2016:5813030. doi: 10.1155/2016/5813030
57. Yin Y, Choi S-C, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T Cell Metabolism Reverses Lupus. Sci Transl Med (2015) 7:274ra18. doi: 10.1126/scitranslmed.aaa0835
58. Snaith JR, Holmes-Walker DJ, Greenfield JR. Reducing Type 1 Diabetes Mortality: Role for Adjunctive Therapies? Trends Endocrinol Metab (2020) 31:150–64. doi: 10.1016/j.tem.2019.11.007
59. Vella S, Buetow L, Royle P, Livingstone S, Colhoun HM, Petrie JR. The Use of Metformin in Type 1 Diabetes: A Systematic Review of Efficacy. Diabetologia (2010) 53:809–20. doi: 10.1007/s00125-009-1636-9
60. Standl E. Metformin in Type 1 Diabetes. Lancet Diabetes Endocrinol (2017) 5:567–9. doi: 10.1016/S2213-8587(17)30216-4
61. Sciannimanico S, Grimaldi F, Vescini F, De Pergola G, Iacoviello M, Licchelli B, et al. Metformin: Up to Date. Endocr Metab Immune Disord - Drug Targets (2020) 20:172–81. doi: 10.2174/1871530319666190507125847
62. Waickman AT, Powell JD. mTOR, Metabolism, and the Regulation of T-Cell Differentiation and Function. Immunol Rev (2012) 249:43–58. doi: 10.1111/j.1600-065X.2012.01152.x
63. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 Combination Therapy in Patients With Type 1 Diabetes Augments Tregs Yet Transiently Impairs β-Cell Function. Diabetes (2012) 61:2340–8. doi: 10.2337/db12-0049
64. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo M-G. Rapamycin Promotes Expansion of Functional CD4+CD25+FOXP3+ Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients. J Immunol Baltim Md 1950 (2006) 177:8338–47. doi: 10.4049/jimmunol.177.12.8338
65. Monti P, Scirpoli M, Maffi P, Piemonti L, Secchi A, Bonifacio E, et al. Rapamycin Monotherapy in Patients With Type 1 Diabetes Modifies CD4+CD25+FOXP3+ Regulatory T-Cells. Diabetes (2008) 57:2341–7. doi: 10.2337/db08-0138
66. Battaglia M, Stabilini A, Draghici E, Migliavacca B, Gregori S, Bonifacio E, et al. Induction of Tolerance in Type 1 Diabetes via Both CD4+CD25+ T Regulatory Cells and T Regulatory Type 1 Cells. Diabetes (2006) 55:1571–80. doi: 10.2337/db05-1576
67. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, et al. Enhancing CD8 T-Cell Memory by Modulating Fatty Acid Metabolism. Nature (2009) 460:103–7. doi: 10.1038/nature08097
68. Bolla AM, Gandolfi A, Borgonovo E, Laurenzi A, Caretto A, Molinari C, et al. Rapamycin Plus Vildagliptin to Recover β-Cell Function in Long-Standing Type 1 Diabetes: A Double-Blind, Randomized Trial. J Clin Endocrinol Metab (2021) 106:e507–19. doi: 10.1210/clinem/dgaa791
69. Li X, Bi X, Wang S, Zhang Z, Li F, Zhao AZ. Therapeutic Potential of ω-3 Polyunsaturated Fatty Acids in Human Autoimmune Diseases. Front Immunol (2019) 10:2241. doi: 10.3389/fimmu.2019.02241
70. Bellenger J, Bellenger S, Bataille A, Massey KA, Nicolaou A, Rialland M, et al. High Pancreatic N-3 Fatty Acids Prevent STZ-Induced Diabetes in Fat-1 Mice: Inflammatory Pathway Inhibition. Diabetes (2011) 60:1090–9. doi: 10.2337/db10-0901
71. Swainson L, Kinet S, Manel N, Battini J-L, Sitbon M, Taylor N. Glucose Transporter 1 Expression Identifies a Population of Cycling CD4 + CD8 + Human Thymocytes With High CXCR4-Induced Chemotaxis. Proc Natl Acad Sci (2005) 102:12867–72. doi: 10.1073/pnas.0503603102
72. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The Transcription Factor Myc Controls Metabolic Reprogramming Upon T Lymphocyte Activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
73. Di Dedda C, Vignali D, Piemonti L, Monti P. Pharmacological Targeting of GLUT1 to Control Autoreactive T Cell Responses. Int J Mol Sci (2019) 20:4962. doi: 10.3390/ijms20194962
74. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The Glucose Transporter Glut1 is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
75. Apaya MK, Kuo T-F, Yang M-T, Yang G, Hsiao C-L, Chang S-B, et al. Phytochemicals as Modulators of β-Cells and Immunity for the Therapy of Type 1 Diabetes: Recent Discoveries in Pharmacological Mechanisms and Clinical Potential. Pharmacol Res (2020) 156:104754. doi: 10.1016/j.phrs.2020.104754
76. Zhang D, Li J, Wang F, Hu J, Wang S, Sun Y. 2-Deoxy-D-Glucose Targeting of Glucose Metabolism in Cancer Cells as a Potential Therapy. Cancer Lett (2014) 355:176–83. doi: 10.1016/j.canlet.2014.09.003
77. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting Glycolytic Metabolism Enhances CD8+ T Cell Memory and Antitumor Function. J Clin Invest (2013) 123:4479–88. doi: 10.1172/JCI69589
78. Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose Deprivation Inhibits Multiple Key Gene Expression Events and Effector Functions in CD8+ T Cells. Eur J Immunol (2008) 38:2438–50. doi: 10.1002/eji.200838289
79. Bordignon C, Canu A, Dyczko A, Leone S, Monti P. T-Cell Metabolism as a Target to Control Autoreactive T Cells in β-Cell Autoimmunity. Curr Diabetes Rep (2017) 17:24. doi: 10.1007/s11892-017-0848-5
80. Leen WG, Taher M, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA. GLUT1 Deficiency Syndrome Into Adulthood: A Follow-Up Study. J Neurol (2014) 261:589–99. doi: 10.1007/s00415-014-7240-z
81. De Giorgis V, Veggiotti P. GLUT1 Deficiency Syndrome 2013: Current State of the Art. Seizure (2013) 22:803–11. doi: 10.1016/j.seizure.2013.07.003
82. Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Phenotypic Spectrum of Glucose Transporter Type 1 Deficiency Syndrome (Glut1 Ds). Curr Neurol Neurosci Rep (2013) 13:342. doi: 10.1007/s11910-013-0342-7
83. Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, et al. A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell Growth In Vitro and In Vivo. Mol Cancer Ther (2012) 11:1672–82. doi: 10.1158/1535-7163.MCT-12-0131
84. Vignali D, Cantarelli E, Bordignon C, Canu A, Citro A, Annoni A, et al. Detection and Characterization of CD8+ Autoreactive Memory Stem T Cells in Patients With Type 1 Diabetes. Diabetes (2018) 67:936–45. doi: 10.2337/db17-1390
85. Siebeneicher H, Cleve A, Rehwinkel H, Neuhaus R, Heisler I, Müller T, et al. Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY-876. ChemMedChem (2016) 11:2261–71. doi: 10.1002/cmdc.201600276
86. Adams DJ, Ito D, Rees MG, Seashore-Ludlow B, Puyang X, Ramos AH, et al. NAMPT Is the Cellular Target of STF-31-Like Small-Molecule Probes. ACS Chem Biol (2014) 9:2247–54. doi: 10.1021/cb500347p
87. Ma Y, Wang W, Idowu MO, Oh U, Wang X-Y, Temkin SM, et al. Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti-Tumor Activity of BAY-876. Cancers (2018) 11:E33. doi: 10.3390/cancers11010033
88. Ojelabi OA, Lloyd KP, Simon AH, De Zutter JK, Carruthers A. WZB117 (2-Fluoro-6-(M-Hydroxybenzoyloxy) Phenyl M-Hydroxybenzoate) Inhibits GLUT1-Mediated Sugar Transport by Binding Reversibly at the Exofacial Sugar Binding Site. J Biol Chem (2016) 291:26762–72. doi: 10.1074/jbc.M116.759175
89. Lee YS, Kim D, Lee EK, Kim S, Choi CS, Jun HS. Sodium Meta-Arsenite Prevents the Development of Autoimmune Diabetes in NOD Mice. Toxicol Appl Pharmacol (2015) 284:254–61. doi: 10.1016/j.taap.2014.12.016
90. Wahli W, Michalik L. PPARs at the Crossroads of Lipid Signaling and Inflammation. Trends Endocrinol Metab TEM (2012) 23:351–63. doi: 10.1016/j.tem.2012.05.001
91. Gross B, Pawlak M, Lefebvre P, Staels B. PPARs in Obesity-Induced T2DM, Dyslipidaemia and NAFLD. Nat Rev Endocrinol (2017) 13:36–49. doi: 10.1038/nrendo.2016.135
92. Zhang S, Yang X, Luo J, Ge X, Sun W, Zhu H, et al. Pparα Activation Sensitizes Cancer Cells to Epigallocatechin-3-Gallate (EGCG) Treatment via Suppressing Heme Oxygenase-1. Nutr Cancer (2014) 66:315–24. doi: 10.1080/01635581.2014.868909
93. Holm LJ, Mønsted MØ, Haupt-Jorgensen M, Buschard K. PPARs and the Development of Type 1 Diabetes. PPAR Res (2020) 2020:1–11. doi: 10.1155/2020/6198628
94. Cariello M, Piccinin E, Moschetta A. Transcriptional Regulation of Metabolic Pathways via Lipid-Sensing Nuclear Receptors PPARs, FXR, and LXR in NASH. Cell Mol Gastroenterol Hepatol (2021) 11:1519–39. doi: 10.1016/j.jcmgh.2021.01.012
95. Fu Z, Zhen W, Yuskavage J, Liu D. Epigallocatechin Gallate Delays the Onset of Type 1 Diabetes in Spontaneous non-Obese Diabetic Mice. Br J Nutr (2011) 105:1218–25. doi: 10.1017/S0007114510004824
96. Holm LJ, Krogvold L, Hasselby JP, Kaur S, Claessens LA, Russell MA, et al. Abnormal Islet Sphingolipid Metabolism in Type 1 Diabetes. Diabetologia (2018) 61:1650–61. doi: 10.1007/s00125-018-4614-2
97. Beales PE, Pozzilli P. Thiazolidinediones for the Prevention of Diabetes in the non-Obese Diabetic (NOD) Mouse: Implications for Human Type 1 Diabetes. Diabetes Metab Res Rev (2002) 18:114–7. doi: 10.1002/dmrr.262
98. Ogawa J, Takahashi S, Fujiwara T, Fukushige J, Hosokawa T, Izumi T, et al. Troglitazone can Prevent Development of Type 1 Diabetes Induced by Multiple Low-Dose Streptozotocin in Mice. Life Sci (1999) 65:1287–96. doi: 10.1016/s0024-3205(99)00364-1
99. Weiss L, Zeira M, Reich S, Har-Noy M, Mechoulam R, Slavin S, et al. Cannabidiol Lowers Incidence of Diabetes in non-Obese Diabetic Mice. Autoimmunity (2006) 39:143–51. doi: 10.1080/08916930500356674
100. Castro CN, Barcala Tabarrozzi AE, Winnewisser J, Gimeno ML, Antunica Noguerol M, Liberman AC, et al. Curcumin Ameliorates Autoimmune Diabetes. Evidence in Accelerated Murine Models of Type 1 Diabetes. Clin Exp Immunol (2014) 177:149–60. doi: 10.1111/cei.12322
101. Chen J, Chernatynskaya AV, Li J-W, Kimbrell MR, Cassidy RJ, Perry DJ, et al. T Cells Display Mitochondria Hyperpolarization in Human Type 1 Diabetes. Sci Rep (2017) 7:10835. doi: 10.1038/s41598-017-11056-9
102. Previte DM, O’Connor EC, Novak EA, Martins CP, Mollen KP, Piganelli JD. Reactive Oxygen Species are Required for Driving Efficient and Sustained Aerobic Glycolysis During CD4+ T Cell Activation. PloS One (2017) 12:e0175549. doi: 10.1371/journal.pone.0175549
103. Delmastro-Greenwood MM, Tse HM, Piganelli JD. Effects of Metalloporphyrins on Reducing Inflammation and Autoimmunity. Antioxid Redox Signal (2014) 20:2465–77. doi: 10.1089/ars.2013.5257
104. Delmastro-Greenwood MM, Votyakova T, Goetzman E, Marre ML, Previte DM, Tovmasyan A, et al. Mn Porphyrin Regulation of Aerobic Glycolysis: Implications on the Activation of Diabetogenic Immune Cells. Antioxid Redox Signal (2013) 19:1902–15. doi: 10.1089/ars.2012.5167
105. Coudriet GM, Delmastro-Greenwood MM, Previte DM, Marré ML, O’Connor EC, Novak EA, et al. Treatment With a Catalytic Superoxide Dismutase (SOD) Mimetic Improves Liver Steatosis, Insulin Sensitivity, and Inflammation in Obesity-Induced Type 2 Diabetes. Antioxid Basel Switz (2017) 6:E85. doi: 10.3390/antiox6040085
106. Sklavos MM, Tse HM, Piganelli JD. Redox Modulation Inhibits CD8 T Cell Effector Function. Free Radic Biol Med (2008) 45:1477–86. doi: 10.1016/j.freeradbiomed.2008.08.023
107. Piganelli JD, Flores SC, Cruz C, Koepp J, Batinic-Haberle I, Crapo J, et al. A Metalloporphyrin-Based Superoxide Dismutase Mimic Inhibits Adoptive Transfer of Autoimmune Diabetes by a Diabetogenic T-Cell Clone. Diabetes (2002) 51:347–55. doi: 10.2337/diabetes.51.2.347
108. Previte DM, Martins CP, O’Connor EC, Marre ML, Coudriet GM, Beck NW, et al. Lymphocyte Activation Gene-3 Maintains Mitochondrial and Metabolic Quiescence in Naive CD4+ T Cells. Cell Rep (2019) 27:129–141.e4. doi: 10.1016/j.celrep.2019.03.004
109. Mouat IC, Morse ZJ, Jean-Baptiste VSE, Allanach JR, Horwitz MS. Fresh Ideas, Foundational Experiments (FIFE): Immunology and Diabetes 2016 FIFE Symposium. Front Endocrinol (2017) 8:238. doi: 10.3389/fendo.2017.00238
110. Singh AK, Netea MG, Bishai WR. BCG Turns 100: Its Nontraditional Uses Against Viruses, Cancer, and Immunologic Diseases. J Clin Invest (2021) 131:e148291. doi: 10.1172/JCI148291
111. Huppmann M, Baumgarten A, Ziegler A-G, Bonifacio E. Neonatal Bacille Calmette-Guerin Vaccination and Type 1 Diabetes. Diabetes Care (2005) 28:1204–6. doi: 10.2337/diacare.28.5.1204
112. Faustman DL. Benefits of BCG-Induced Metabolic Switch From Oxidative Phosphorylation to Aerobic Glycolysis in Autoimmune and Nervous System Diseases. J Intern Med (2020) 288:641–50. doi: 10.1111/joim.13050
113. Faustman DL, Wang L, Okubo Y, Burger D, Ban L, Man G, et al. Proof-Of-Concept, Randomized, Controlled Clinical Trial of Bacillus-Calmette-Guerin for Treatment of Long-Term Type 1 Diabetes. PloS One (2012) 7:e41756. doi: 10.1371/journal.pone.0041756
114. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis Controls the Induction of Human Regulatory T Cells by Modulating the Expression of FOXP3 Exon 2 Splicing Variants. Nat Immunol (2015) 16:1174–84. doi: 10.1038/ni.3269
115. Kühtreiber WM, Tran L, Kim T, Dybala M, Nguyen B, Plager S, et al. Long-Term Reduction in Hyperglycemia in Advanced Type 1 Diabetes: The Value of Induced Aerobic Glycolysis With BCG Vaccinations. NPJ Vaccines (2018) 3:23. doi: 10.1038/s41541-018-0062-8
116. Faustman DL. TNF, TNF Inducers, and TNFR2 Agonists: A New Path to Type 1 Diabetes Treatment. Diabetes Metab Res Rev (2018) 34(1). doi: 10.1002/dmrr.2941
117. Zhang J, Chen L, Wang F, Zou Y, Li J, Luo J, et al. Extracellular HMGB1 Exacerbates Autoimmune Progression and Recurrence of Type 1 Diabetes by Impairing Regulatory T Cell Stability. Diabetologia (2020) 63:987–1001. doi: 10.1007/s00125-020-05105-8
118. Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and β-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes (2016) 65:719–31. doi: 10.2337/db15-0779
119. Matthews JB, Staeva TP, Bernstein PL, Peakman M, Von Herrath M. Developing Combination Immunotherapies for Type 1 Diabetes: Recommendations From the ITN–JDRF Type 1 Diabetes Combination Therapy Assessment Group. Clin Exp Immunol (2010) 160:176–84. doi: 10.1111/j.1365-2249.2010.04153.x
120. von Scholten BJ, Kreiner FF, Gough SCL, von Herrath M. Current and Future Therapies for Type 1 Diabetes. Diabetologia (2021) 64:1037–48. doi: 10.1007/s00125-021-05398-3
121. Manirarora JN, Wei C-H. Combination Therapy Using IL-2/IL-2 Monoclonal Antibody Complexes, Rapamycin, and Islet Autoantigen Peptides Increases Regulatory T Cell Frequency and Protects Against Spontaneous and Induced Type 1 Diabetes in Nonobese Diabetic Mice. J Immunol Baltim Md 1950 (2015) 195:5203–14. doi: 10.4049/jimmunol.1402540
122. Besançon A, Goncalves T, Valette F, Mary C, Vanhove B, Chatenoud L, et al. A Selective CD28 Antagonist and Rapamycin Synergise to Protect Against Spontaneous Autoimmune Diabetes in NOD Mice. Diabetologia (2018) 61:1811–6. doi: 10.1007/s00125-018-4638-7
123. Chen N, Kroger CJ, Tisch RM, Bachelder EM, Ainslie KM. Prevention of Type 1 Diabetes With Acetalated Dextran Microparticles Containing Rapamycin and Pancreatic Peptide P31. Adv Healthc Mater (2018) 7:e1800341. doi: 10.1002/adhm.201800341
124. Guclu M, Oz Gul O, Cander S, Unal O, Ozkaya G, Sarandol E, et al. Effect of Rosiglitazone and Insulin Combination Therapy on Inflammation Parameters and Adipocytokine Levels in Patients With Type 1 Dm. J Diabetes Res (2015) 2015:807891. doi: 10.1155/2015/807891
125. Rabinovitch A, Suarez-Pinzon WL, Shapiro AMJ, Rajotte RV, Power R. Combination Therapy With Sirolimus and Interleukin-2 Prevents Spontaneous and Recurrent Autoimmune Diabetes in NOD Mice. Diabetes (2002) 51:638–45. doi: 10.2337/diabetes.51.3.638
126. Loschinski R, Böttcher M, Stoll A, Bruns H, Mackensen A, Mougiakakos D. IL-21 Modulates Memory and Exhaustion Phenotype of T-Cells in a Fatty Acid Oxidation-Dependent Manner. Oncotarget (2018) 9:13125–38. doi: 10.18632/oncotarget.24442
127. Monteleone G, Pallone F, Macdonald TT. Interleukin-21 as a New Therapeutic Target for Immune-Mediated Diseases. Trends Pharmacol Sci (2009) 30:441–7. doi: 10.1016/j.tips.2009.05.006
128. Ren HM, Lukacher AE, Rahman ZSM, Olsen NJ. New Developments Implicating IL-21 in Autoimmune Disease. J Autoimmun (2021) 122:102689. doi: 10.1016/j.jaut.2021.102689
129. von Herrath M, Bain SC, Bode B, Clausen JO, Coppieters K, Gaysina L, et al. Anti-Interleukin-21 Antibody and Liraglutide for the Preservation of β-Cell Function in Adults With Recent-Onset Type 1 Diabetes: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Diabetes Endocrinol (2021) 9:212–24. doi: 10.1016/S2213-8587(21)00019-X
130. Felton JL. Timing of Immunotherapy in Type 1 Diabetes: The Earlier, the Better? ImmunoHorizons (2021) 5:535–42. doi: 10.4049/immunohorizons.2000105
131. Balyan R, Gautam N, Gascoigne NRJ. The Ups and Downs of Metabolism During the Lifespan of a T Cell. Int J Mol Sci (2020) 21:7972. doi: 10.3390/ijms21217972
132. Leone RD, Powell JD. Metabolism of Immune Cells in Cancer. Nat Rev Cancer (2020) 20:516–31. doi: 10.1038/s41568-020-0273-y
Keywords: type 1 diabetes, T cell, T cell differentiation and function, T cell metabolism, autoimmunity
Citation: Zhang M, Zhou Y, Xie Z, Luo S, Zhou Z, Huang J and Zhao B (2022) New Developments in T Cell Immunometabolism and Therapeutic Implications for Type 1 Diabetes. Front. Endocrinol. 13:914136. doi: 10.3389/fendo.2022.914136
Received: 06 April 2022; Accepted: 09 May 2022;
Published: 10 June 2022.
Edited by:
Mei Zhang, Nanjing Medical University, ChinaReviewed by:
Jon D. Piganelli, University of Pittsburgh, United StatesCopyright © 2022 Zhang, Zhou, Xie, Luo, Zhou, Huang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Zhao, Ymluemhhb0Bjc3UuZWR1LmNu; YmluLnpoYW9AbGl2ZS5jb20=; Jiaqi Huang, amlhcWkuaHVhbmdAY3N1LmVkdS5jbg==; amlhcWkuaHVhbmdAbGl2ZS5jb20=
†These authors have contributed equally to this work and share first authorship
‡These authors jointly supervised this work