 Vincent Duval
Vincent Duval Paul Alayrac
Paul Alayrac Jean-Sébastien Silvestre
Jean-Sébastien Silvestre Angélique Levoye
Angélique Levoye- 1Université Paris Cité, Institut National de la Santé Et Recherche Médicale (INSERM), Paris Cardiovascular Research Center PARCC, Paris, France
- 2UFR Santé Médecine Biologie Humaine, Université Sorbonne Paris Nord, Bobigny, France
Chemokines, and their receptors play a crucial role in the pathophysiology of cardiovascular diseases (CVD). Chemokines classically mediate their effects by binding to G-protein-coupled receptors. The discovery that chemokines can also bind to atypical chemokine receptors (ACKRs) and initiate alternative signaling pathways has changed the paradigm regarding chemokine-related functions. Among these ACKRs, several studies have highlighted the exclusive role of ACKR3, previously known as C-X-C chemokine receptor type 7 (CXCR7), in CVD. Indeed, ACKR3 exert atheroprotective, cardioprotective and anti-thrombotic effects through a wide range of cells including endothelial cells, platelets, inflammatory cells, fibroblasts, vascular smooth muscle cells and cardiomyocytes. ACKR3 functions as a scavenger receptor notably for the pleiotropic chemokine CXCL12, but also as a activator of different pathways such as β-arrestin-mediated signaling or modulator of CXCR4 signaling through the formation of ACKR3-CXCR4 heterodimers. Hence, a better understanding of the precise roles of ACKR3 may pave the way towards the development of novel and improved therapeutic strategies for CVD. Here, we summarize the structural determinant characteristic of ACKR3, the molecules targeting this receptor and signaling pathways modulated by ACKR3. Finally, we present and discuss recent findings regarding the role of ACKR3 in CVD.
Introduction
Cardiovascular diseases (CVD) are the leading cause of death worldwide. CVD cover all heart and blood vessel dysfunctions, including hypertension, coronary artery disease, cerebrovascular disease (stroke or transient ischemic attack), myocardial infarction (MI), heart failure and peripheral vascular diseases such as aneurysms. The main pathology behind CVD is atherosclerosis, a chronic inflammatory disease of the arterial wall characterized by the development of lesions caused by increasing endothelial permeability and the accumulation of low density lipids in the intima (1, 2). Subsequent lipid modifications trigger endothelial dysfunction, migration and recruitment of inflammatory cells through the action of several adhesion molecules and chemokines. Monocyte-derived macrophages take up these lipids, resulting in the formation of foam cells (3). Smooth muscle cells migrate from the media to the intima, where they proliferate and extracellular matrix molecules such as collagen and proteoglycans are synthesized, leading to the creation of a fibrous cap. This process promotes plaque development within the arterial wall resulting mainly to vascular occlusion through thrombus formation and subsequent tissue ischemia with severe clinical consequences, such as stroke or MI.
Treatment of risk factors, development of potent anticoagulant and antiplatelet therapies, and the widespread availability of revascularization methods have significantly improved the treatment and prevention of CVD. However, according to the WHO, approximately 17.9 million deaths per year are caused by CVD. In 2030, this number expected to rise to more than 23.6 million deaths (4). This clearly supports the necessity to better understand the cellular and molecular mechanisms regulating CVD development, such as inflammatory response after injury or during atherosclerosis in order to propose novel therapeutic strategies. Chemokines and their receptors play a key role in these processes and thereby represent promising potential therapeutic targets in the treatment of CVD. Several reviews have already highlighted the importance of the Atypical Chemokine Receptors (ACKRs) in CVD (5). The aims of this mini-review are to focus on the specific roles of a ACKR subtype i.e., ACKR3, in CVD, to detail the mechanisms leading to the modulation of the ACKR3-related functions, and to discuss the therapeutic opportunities based on targeting ACKR3.
Atypical Chemokine Receptors
Chemokines are small cytokines (8-12 KDa) that trigger cell chemotaxis along a concentration gradient. To date, chemokines have been classified into 4 families based on the position of the first two cysteine residues (CC, CXC, CX3C, XC). Chemokines can be expressed by different cell types such as endothelial cells, fibroblasts, cardiomyocytes but also inflammatory cells. They can be detected within injury tissues, where they regulate the recruitment of leukocytes to sites of inflammation and control tissue homeostasis through their ability to shape resident cell-related functions (6). Indeed, chemokines can also modulate, through their binding to multiple receptors, a plethora of other cellular functions such as proliferation, survival, differentiation and cytokine release. Conventional (cCKRs) and atypical (ACKRs) comprise the two types of chemokine receptors (7). Most chemokines are recognized by a set of cCKRs belonging to the G-protein-coupled receptors (GPCRs) family associated with G-protein coupling and β arrestin activation leading to functional redundancy within the chemokine system. Currently, they are 23 cCKRs classified according to the type of chemokine they bind. However, the complexity of the chemokine system is increased by the presence of ACKR subtypes (8). ACKRs are seven transmembrane domain receptors structurally homologous to GPCRs. Currently, the ACKR family comprises five major receptors: ACKR1/DARC (Duffy Antigen Receptor for Chemokines), ACKR2/D6, ACKR3/CXCR7, ACKR4/CCRL1 (CC-Chemokine Receptors like 1) and ACKR5/CCRL2 (9). ACKRs can bind to a variety of different ligands to elicit their biological effects. Originally, ACKRs have been characterized as decoy or silent receptors because they fail to induce a classical signalling signature characteristic for GPCRs in response to the binding of the chemokine. The term “Atypical” emerges from the observation that ACKRs have either lacked or modified canonical DRYLAIV motif located at the end of the third transmembrane domain and within the second intracellular loop, essential for binding and activation of G-proteins. Their failure to couple to G-proteins and to induce “classical” GPCR signalling led to their exclusion from the chemokine receptor nomenclature and to their initial designation as “silent” receptor. However, modifications of the DRYLAIV motif present in the CKR XCR1 and CXCR6 both known to signal via PTX-sensitive Gαi proteins have challenged this dogma. This has confirmed that modifications of the DRYLAIV motif were neither a indicator neither the only determinant for the lack of G-protein coupling (10, 11). Hence, ACKRs are atypical related to their ability to activate intracellular pathways through G-protein-independent signalling (12). ACKRs were initially considered as scavenger receptors by their ability to promote the targeting and the degradation of the bound chemokine in lysosomes. The typical D6 receptor is a prototypical example of receptors harbouring scavenging function (13). The transcytosis function allowing the transport of chemokines across biological barriers has also been proposed for some ACKR such as DARC (14). Thus, ACKRs appear as major regulators of chemokine internalization, degradation, and transcytosis (15). These activities may lead to the establishment of chemokine concentration gradient within tissue which may regulate the bioavailability of a defined chemokine and subsequently the cCKR-dependent signalling (16). In addition, ACKRs may also modulate the responses to chemokines through receptor heterodimerization (17) or by sustaining their own signalling pathways.
ACKR3 and its Ligands
ACKR3, initially thought to be an orphan 7 transmembrane domain receptor, has been identified in dog in 1989 (18), human in 1991 (19) and was named RDC1. The murine form was identified in 1998 (20) and showed more than 85% homology with the human and canine RDC1. ACKR3 losts its orphan receptor status when CXCL12 initialy named SDF-1 (Stromal Derived Factor-1) was discovered as one of its ligands (21). CXCL12 mainly acts through its classical CXCR4 receptor (22, 23). Three and six CXCL12 isoforms have been identified in mouse and human, respectively (24). Although, all CXCL12 isoforms are likely able to bind to ACKR3 their specific roles in ACKR3-related functions remain to be defined. However, with a 10-fold higher affinity than CXCR4, RDC1 is able to induce chemotaxis of lymphocytes in response to the CXCL12α isoform (21, 25). As a consequence, RDC1 was then considered to belong to the CXC chemokine receptor family and was renamed CXCR7. Unlike cCKRs, CXCR7 is not always associated with Gαi proteins and does not induce calcium mobilization. Its canonical DRYLAIV motif known as essential for G protein coupling is altered. This structural difference has led to a change in its classification from CXCR7 to ACKR3 (26). However, it should be noted that the inability of ACKR3 to couple heterotrimeric Gα proteins remains debated. Indeed, ACKR3 is able to interact with Gαi1 and Gαi3 proteins in HEK293 cells (27, 28) but not trigger a calcium response. Only, primary rodent astrocytes and human glioma cells respond to CXCL12-dependent activation of ACKR3 with increases in intracellular calcium which can be reversed by pertussis toxin (27, 28) (Table 1). Other chemokines, CXCL11 (or I-TAC, interferon-inducible T-cell alpha chemoattractant), a CXCR3 agonist, has also been identified as a ligand of ACKR3 (21). Recently, vCCL2, also known as vMIPII, a viral chemokine, was found to act as a partial agonist of ACKR3 which can induce β-arrestin recruitment and Mitogen Activated Protein Kinases (MAPK) activation (44). Additional non-chemokine ligands have also been identified including the macrophage migration inhibitory factor (MIF) (29, 45), adrenomedullin, pro-adrenomedullin N-terminal 20 Peptides and its processed form PAMP12 (30, 46) as well as several opioid-derived peptides (30, 31, 47). Furthermore, several synthetic ligands for ACKR3 were synthesized, such as the cyclic peptide agonist TC14012 derived from T140 (32), VUF11207 as well as VUF11403 (33), ACT-1004-1239 (34) and CCX771 (35). AMD3100, a well-known CXCR4 antagonist, has been identified as a allosteric modulator of ACKR3 but only at high concentrations (48) (Table 1).

Table 1 ACKR3: ligands, signalling and functions.
ACKR3 Signalling Pathways
Because of its various ligands, ACKR3 can initiate downstream effects through several modes of action; in particular regarding the CXCL12/CXCR4/ACKR3-related signalling (21, 49, 50) (Figure 1). One of the first functions identified for ACKR3 is its scavenger activity where ACKR3 is internalized upon chemokine binding which leads to the lysosomal degradation of its ligands. Thus, ACKR3 regulates extracellular CXCL12 concentrations leading to modulation of CXCL12/CXCR4-mediated signaling (51–53). ACKR3 also acts as a scavenger receptor to regulate the migration of CXCR4 expressing cells (52).
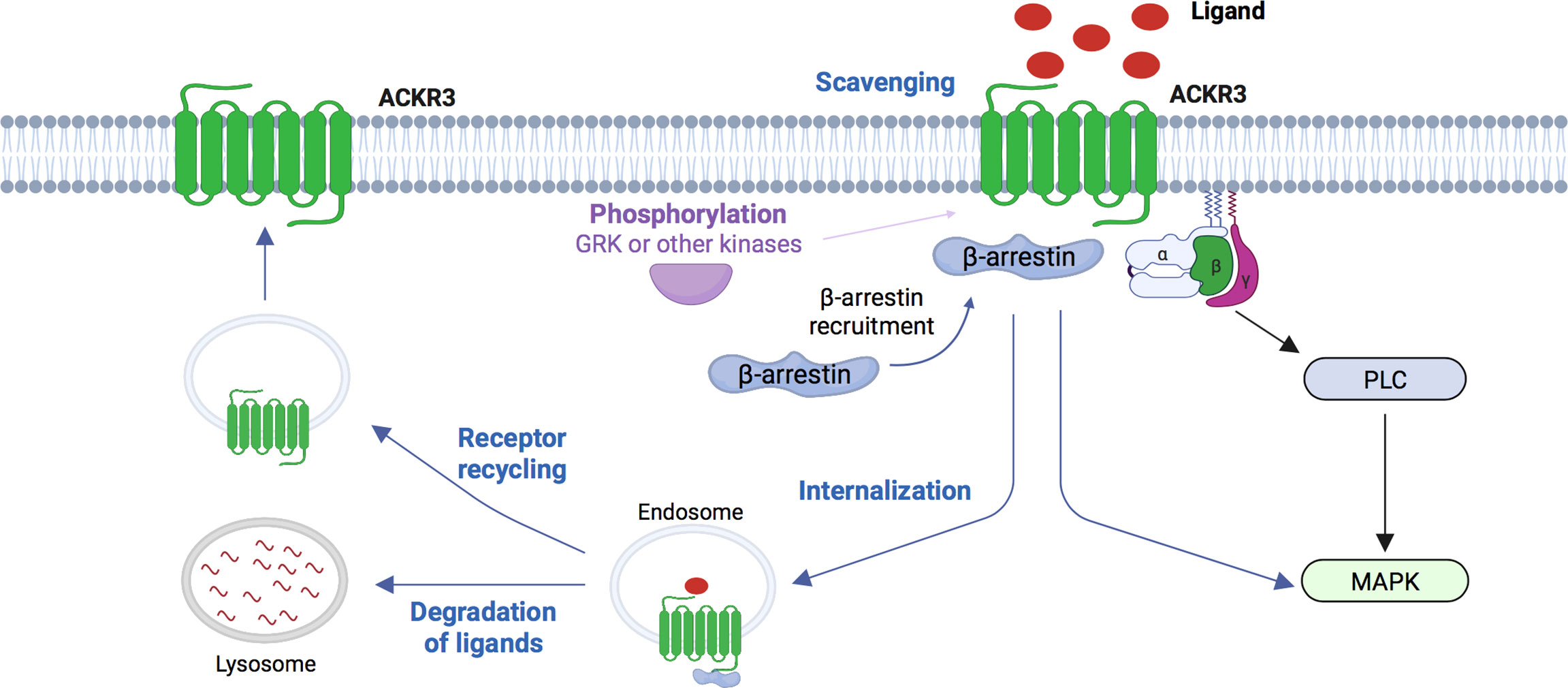
Figure 1 Schematic representation of signaling pathways for ACKR3. Chemokines ligands CXCL12 and CXCL11 can induce phosphorylation of ACKR3 by GRK or others kinases, the recruitment of β-arrestins and trigger intracellular G-protein independent signaling. ACKR3 is also able to scavenge various ligands (CXCL12, CXCL11, ADM, PAMP, PAMP12, opioїd peptides) at the cell surface, internalized these ligands which subsequently result in degradation in lysosome while the receptor recycles to the cell membrame. ACKR3 is also able to interact with G-proteins without to lead to their activation. However, in two specific cellular types, primary rodent astrocytes and human glioma cells, ACKR3 can be coupled to Gα-protein and triggers PLC and MAPK activation. GRK, G-protein receptor kinase; ADM, adrenomedullin; PAMP, proadrenomedullinN-terminal 20 peptide; PLC, phospholipase C; MAPK, mitogen-activated protein kinase. (Figure created with BioRender.com).
Unlike many receptors, ACKR3 is not necessarily degraded after activation and internalization. ACKR3 continuously cycles between the plasma membrane and intracellular compartments (53) and can also regulate the expression of CXCR4 and CXCR4-mediated signalling. Cardiac stem cells (CSCs) express CXCR4 and ACKR3 and their expression and cooperation is necessary for the regulation of cell migration. ACKR3 inhibits CXCR4-dependent migration of CSCs (54). Furthermore, in cardiomyocyte derived from human induced pluripotent stem cells (hiPSCs), it has been shown that Cxcr4 silencing reduced ACKR3 expression at the membrane suggesting a contribution of CXCR4 to the ACKR3 membrane trafficking. Although CXCR4 and ACKR3 downregulation resulted in a delayed cardiac phenotype, only knockdown of CXCR4 impaired the spontaneous beating of hiPSC-derived cardiomyocytes (55), suggesting that CXCR4 and ACKR3 govern differential effects during cardiac lineage differentiation. Interestingly, conditional deletion of Ackr3 in interneurons regulates cortical neuron migration. In this model, ACKR3 controls CXCR4 protein levels that is made available for efficient signaling at the cell surface, thereby adapting chemokine responsiveness to migratory CXCR4-expressing cells (42, 43).
ACKR3 can also recruit and activate the β-arrestin signalling constitutively or after binding of its ligand (16, 32, 35) (56) (Table 1). CXCL12, but also CXCL11 and TC14012, a synthetic CXCR4 inverse agonist peptide and ACKR3 agonist, induce ß-arrestin 2 recruitment (16, 32, 57) and activate MAPK-associated signalling pathways (16, 35). β-arrestin 2 pathways are known to play an important role in cell survival and proliferation via ERK1/2 and Akt activation (16, 58). In addition, ACKR3 can modulate the signalling and functions of other GPCRs receptors or membrane proteins (Table 1). ACKR3 can modulate CXCL12 signalling either by regulating CXCR4 activity through ACKR3-CXCR4 heterodimers formation or by regulating the CXCL12 gradient through its scavenging activity (17, 36). Furthermore, it has also been shown that this heterodimer potentiates β-arrestin activation with increased stimulation of the ERK1/2 cascade and activation of p38, MAPK and SAPK (36, 38). Beyond interactions with cCKRs, ACKR3 is also able to interrelate with other membrane proteins. Indeed, ACKR3 can interact with Connexin-43 (Cx-43) in a CXCL12 dependent-manner. ACKR3 activation by CXCL12 promotes β-arrestin-2 and Cx-43 internalization which inhibits gap junctional intercellular communication in primary astrocytes (28). Cx-43 is a major protein of cardiac ventricular gap junctions and is crucial to cell-cell communication and cardiac function. Thus, this study suggests that ACKR3 and Cx-43 could interact in the heart, modulates cardiomyocyte phenotypes and thus cardiac function. However, the lack of tools such as molecules or synthetic peptides that prevent heterodimerisation does not allow confirmation of the existence of these interactions in vivo.
Adrenomedullin (ADM) is a peptide hormone critical for cardiac vascular and lymphatic development (59). ADM binds the GPCR Calcitonin receptor-Like Receptor (CLR) which can only be exported to the cell surface through its heterodimerization with one of the three Receptor Activity-Modify Proteins (RAMPs). A complex with either RAMP2 or RAMP3 confers a receptor selective for ADM and those with RAMP1 creates a receptor for calcitonin gene-related peptide (CGRP) (60). Phenotypic analysis of Ackr3-/- and Adm-/- mice revealed that these mice exhibited similar cardiac and lymphatic vascular defects. Overexpressing of Adm in mice caused cardiac hypertrophy during embryogenesis similar to that observed in Ackr3-/- animals (61). Interestingly, Ackr3 deficiency onto genetic background with Adm haploinsufficiency (Adm+/- mice) partially rescued the lethal defects in Ackr3-/- mice by preventing cardiac hyperplasia suggesting that ACKR3 scavenging activity is required for controlling ADM concentrations during development (46). Recently, RAMP3 was identified as an interacting partner of ACKR3 in vitro able to control its signalling and trafficking properties without affecting β-arrestin coupling in response to either AM or CXCL12 ligands. Thus, ACKR3-RAMP3 interactions scavenge ADM thereby reducing bioavailability of ADM and signaling mediated by CLR-RAMP3 receptor (62). Recent studies confirmed the interaction between ACKR3 and RAMP3 and the absence of impact of RAMP on ACKR3 activity (β-arrestin recruitment) in response to ADM. However, the ability of ACKR3-RAMP3 to scavenge ADM is lower than those of CLR/RAMP2 or CLR/RAMP3 receptors (30, 63). These data question the role of ACKR3 as ADM receptor. Further and more precise binding studies are needed before adding ADM to the ACKR3 ligand. Collectively, these studies suggest that the regulatory role of ACKR3 in the ADM signaling axis is either indirect, requires an accessory protein or occurs in a particular microenvironment.
Expression of ACKR3 in a wide variety of neuronal cells, vascular cells and immune cells as well as its up-regulation under hypoxic and inflammatory conditions, which are features of ischemic CVD, have made ACKR3 a central player in the regulation of homeostatic processes but also in inflammatory and ischemic diseases (Table 2).

Table 2 ACKR3 in cardiovascular diseases.
ACKR3 in Cardiovascular Diseases
Cardiovascular Development
Ackr3 deficient mice (Ackr3-/-) develop normally during early embryonic stages. At embryonic gestation days 18.5, animals die in utero or shortly after birth due to significant defects in the development of the cardiovascular system (38–40). Analysis of these animals showed cardiac hyperplasia associated with cardiomyocyte hypertrophy, aortic and pulmonary valve stenosis without mitral and tricuspid valve involvement, as well as atrial and ventricular septal defects, suggesting a major role of ACKR3 in cardiovascular development. Transcriptomic analyses also revealed a decrease in the expression of certain factors essential to vascular growth in semilunar valves (38). Of those that do survive to adulthood (30%), most animals have impaired heart function, cardiac hyperplasia, severe aortic valve calcification and thickening of aortic leaflets causing sudden death (40). These abnormalities were mainly associated to endothelial cell dysfunction because these defects were similar in endothelial cell specific Ackr3 knockout mice (38). Ackr3 has also been deleted in cardiomyocytes and fibroblasts but no changes were observed in cardiac phenotypes compared to control wild-type mice (68). More recently, conditional smooth muscle specific deficiency of Cxcl12 (SM22CRE-Cxcl12flox/flox) revealed that CXCL12 was involved in cardiac and vascular homeostasis by regulating ACKR3 expression and its signalling. Indeed, in these mice, a high embryonic lethality characterized by developmental defects of the cardiovascular system was observed concomitant with alterations of coronary arteries structure (67). Adult mice developed cardiac hypertrophy and severe vascular defects associated with increased fibrosis, apoptosis and dilated arteries with thinner walls leading to impaired cardiac function. Transcriptomic data analysis of these hearts revealed the activation of genes involved in signalling pathways associated with hypertrophic cardiomyopathy, collagen synthesis and extracellular matrix reorganization. CXCL12-mediated signalling pathways such as Akt and ERK1/2 were also increased. Interestingly, this phenotype was associated with downregulation of ACKR3 expression in coronary artery endothelial cells, indicating an important role of ACKR3 in vessel maturation and cardiac remodelling. Furthermore, treatment of these mice with ACKR3 agonist TC14012 attenuated these defects and restored cardiac function with a marked activation of pERK signalling, suggesting that activation of pERK signalling pathways induced by the binding of TC14012 to ACKR3 prevented the progression of cardiac hypertrophy (67) (Figure 1).
Atherosclerosis
ACKR3 expression is significantly upregulated in macrophages and endothelial cells in aortic atheroma of hypercholesterolemic mice and human (64, 72, 73). Using atherosclerotic model of ApoE-/- mice, Li et al. demonstrated that ubiquitous and conditional knockout of Ackr3 increased neointima formation and macrophage accumulation at lesion site after vascular injury. Administration of a pharmacological agonist of ACKR3 (CCX771) to ApoE-/- mice, inhibited lesion formation by reducing blood cholesterol through activation of its uptake within white adipose tissue. Thus, the athero-protective effects of ACKR3 are mainly mediated by the impaired cholesterol uptake within the adipose tissue (65). However, these findings also highlighted a cell specific functional response in particular in macrophages. Activation of ACKR3 by TC14012 in ApoE-/- mice fed a high-fat diet was also reported to limit atherosclerosis initiation by promoting endothelial repair and reducing atherosclerotic lesions through inhibition of pyroptosis signaling pathways. ACKR3 expression is significantly upregulated in both lesional and neointimal endothelial cells in murine and human arteries (64). Moreover, endothelial Ackr3 deletion in mice also exacerbated neointimal hyperplasia following endothelial denudation injury, impaired re-endothelialization and promoted the development of atherosclerosis (64) (Figure 2).
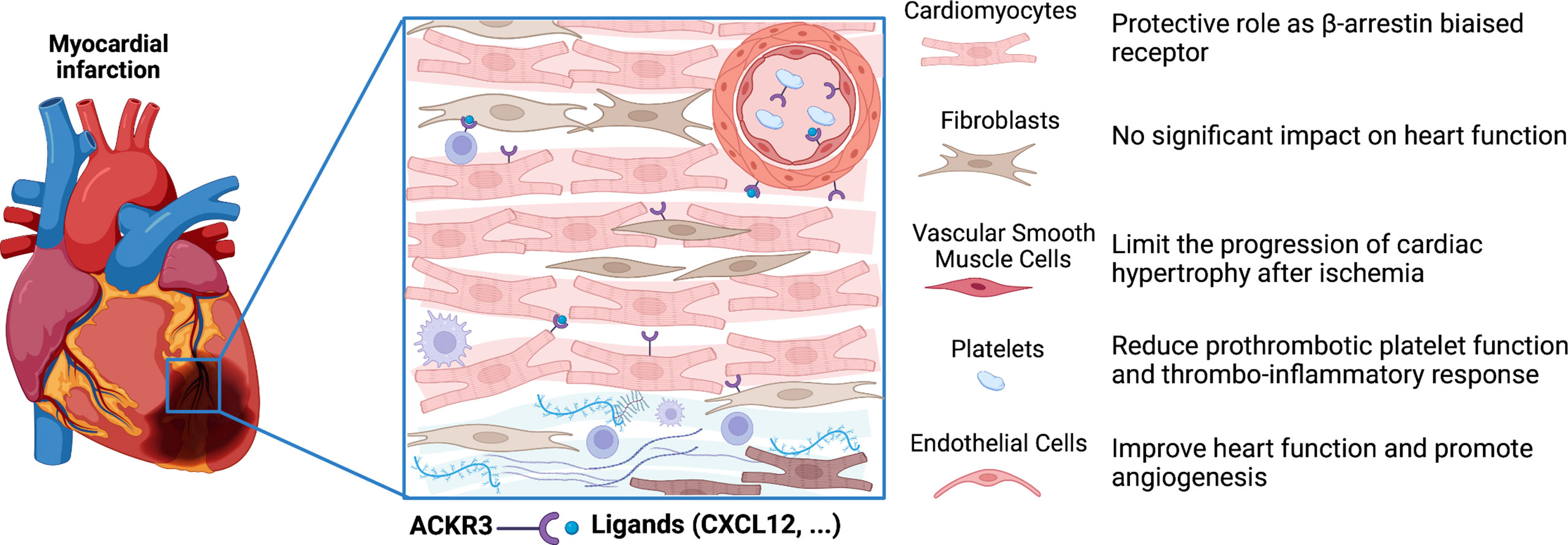
Figure 2 Role of ACKR3 in the infarcted heart. ACKR3 expression is increased in several cells of the heart following myocardial infarction, and can play multiple roles after activation by endogenous and exogenous ligands. ACKR3 exerts cardioprotective effects by acting either as a rheostat for certain ligands through scavenging activity or as a co-partner through dimerization with other receptors. In addition, ACKR3 may trigger activation of β-arrestin-dependent pathways. (Figure created with BioRender.com).
ACKR3 is also constitutively expressed in human and murine platelets ant its expression is upregulated in patients with acute coronary syndrome (74, 75). Recently, ACKR3 activation, by the VUF11207 agonist, has been shown to exert anti-thrombotic actions through the modulation of the platelet lipidome (66). Indeed, its activation limited the generation of phospholipase C-derived lysophosphatidylcholine, a marker of plaque instability with distinct atherogenic properties. Thus, ACKR3 may regulate the generation of pro-thrombotic and pro-atherogenic lipids carried by platelets to the site of vascular injury (Figure 2).
Although, these studies demonstrated an athero-protective role of ACKR3, some studies suggested potential detrimental effects. Indeed, ACKR3 expression was detected in macrophage in aortic atheroma of ApoE-/- mice but not in healthy aorta, suggesting that ACKR3 plays a role in inflammation at the lesion. In line with this reasoning, ACKR3 expression is also correlated with monocyte to macrophage differentiation as well as with the degree of maturation of B cells or that of dendritic cells (73, 76, 77). The differentiation of monocyte to macrophage is a key event in the formation of foam cells in atherosclerotic lesions. During this process, ACKR3 plays a crucial role in the regulation monocyte-macrophage function and induces pro-inflammatory signaling pathways in macrophages (73, 78). In addition, B cells regulate atherosclerotic plaque development through production of antibodies and cytokines. Thus, secretion of pro-inflammatory cytokines and antibodies influenced by ACKR3 may foster pro-atherosclerotic events. Future investigations are needed to understand whether the regulation of ACKR3 expression during this differentiation processes could contribute to the progression of atherosclerosis. Taken together, these findings demonstrate that ACKR3 has cell specific and process-specific functional outcomes.
Stroke
CXCL12, CXCR4 and ACKR3 are widely expressed in the adult mouse and human brain (20, 79, 80) and plays a critical role in brain repair after ischemic stroke (81–84). Over-expression of CXCL12 by gene therapy shows protective effects and improves brain repair after ischemic injury (85). After focal ischemic stroke in mice, CXCL12, CXCR4 and ACKR3 expression is rapidly upregulated. In mice, CXCL12 and CXCR4 are strongly increased in the ischemic region and ACKR3 expression was largely expressed in the blood vessels in the peri-infarcted area, suggesting that ACKR3-dependent pathways may influence vascular and neuronal functions (86–88). In human, expression levels of both CXCL12 and ACKR3, but not that of CXCR4, are increased in the peri-infarction area of ischemic cerebral cortex, suggesting that CXCL12 modulates the repair of the brain after a stroke through ACKR3 activation (89). Stumm et al. showed a differential distribution of CXCL12 isoforms in the brain after focal cerebral ischemia suggesting an isoform-specific regulation of CXCL12 after injury (90). Indeed, CXCL12β isoform was selectively expressed by vascular endothelial cells and its upregulation was associated with a concomitant infiltration of CXCR4-expressing inflammatory cells whereas CXCL12α is expressed in neurons and modulates neuronal plasticity. Thus, one can speculate that isoform-specific regulation of CXCL12 may be due to the differential expression of ACKR3 and/or to the capacity of ACKR3 to scavenge CXCL12 and thereby modulate the CXCL12-dependent response. Recently, neutralization of ACKR3 with a specific blocking antibody following cerebral ischemia resulted in significantly enhanced neurogenesis in the hippocampal associated to cognitive functional recovery (71). These effects could be explained by the elevated levels of CXCL12 resulting from ACKR3 neutralization or by the modulation of CXCR4 signaling through CXCR4-ACKR3 heterodimers. However, further studies are needed to investigate the direct effect of ACKR3 on neurogenesis using knockout Ackr3 mice models or by distinct RNA silencing of ACKR3.
Myocardial Infarction
ACKR3 is the most highly expressed 7-transmembrane domain receptor in mouse heart (68). Single-cell RNA sequencing in mouse heart showed its expression in cardiomyocytes, fibroblasts, endothelial cells and smooth muscle cells (68). Expression of ACKR3 is significantly increased in the infarct border zone of mouse heart and in heart failure patients (68). Conditional endothelial Ackr3 invalidation model (Cxcr7flox/flox Cdh5-Cre model) showed excess mortality, impaired cardiac function associated with increased MI size, reduced angiogenesis and pro-fibrotic remodelling after experimental MI. The absence of ACKR3 could promote CXCR4-dependent signalling known to induce fibrosis. Conversely, cardiac overexpression of ACKR3 or ACKR3 agonist TC14012 treatment preserve cardiac function, enhance angiogenesis and reduce infarct size after MI in C57BL/6J mice (64, 69). These effects underlined the crucial role of endothelial ACKR3 in cardiac remodelling after MI. Activation of ACKR3 in vitro by the agonist TC14012 promotes proliferation and vascular tube formation of murine endothelial cells through ERK and PI3K/Akt pathways (64, 69, 91). In addition, ACKR3 is a critical regulator of mesenchymal stem cells-mediated postnatal vasculogenesis and arterial specification in vitro and in vivo via Notch signalling. These effects have been associated with the activation of ACKR3 expression on mesenchymal stem cells by VEGF and PDGF (41). Alternatively, several studies showed that ACKR3 governs the survival and proliferation of endothelial progenitor cells (EPCs) and their adhesion onto the endothelium contributing to angiogenesis (92, 93). The absence of ACKR3 in endothelial cells also decreases post-ischemic revascularization (64). In addition, EPC from diabetic mice showed decreased expression of ACKR3 and reduced capacity for vascular tube formation. CXCL12/ACKR3 interactions allow EPCs to better resist oxidative stress and improve their angiogenic potential in the context of diabetes-related lower limb ischemia (92).
Cardioprotective effects of ACKR3 could also be mediated by its capacity to impact platelet-related activities. Expression of ACKR3 in platelets is increased post-MI in mice and patients with acute coronary syndrome (94) and is associated with improved cardiac function and prognosis (78, 94). Pro-survival and anti-thrombotic effects mediated through the binding of MIF to ACKR3 have been demonstrated in vivo in murine model of atherothrombosis (95). Administration of specific and selective ACKR3 agonist, VUF11207, reduced thrombo-inflammatory response, infarct size and improved cardiac function in a ischemia-reperfusion mice model (66). In these mice, reduced platelet-leukocyte aggregation in peripheral circulation and lower plasma levels of several inflammatory mediators were observed supporting the cardioprotective role of platelet ACKR3. Interestingly, treatment with ACKR3 agonist does not affect basal hemostatic or coagulation response. Thus, the anti-thrombotic actions of ACKR3 through the modulation of the platelet lipidome to limit metabolism and release of thrombotic and atherogenics mediators strengthened the therapeutic potential of ACKR3 in CVD (96). However, the mechanisms underlying this anti-thrombotic action of ACKR3 in platelets remain ascertained and further studies are needed.
ACKR3 expression is increased in cardiomyocytes of patients with heart failure (68). Genomic analysis of single-cardiomyocyte RNA-seq data from human heart biopsies of pre- and post-implanted patients with left ventricular assist device revealed that implantation normalized this Ackr3 upregulation in both ischemic and non-ischemic cardiomyopathies (97) indicating a crucial role of ACKR3 in human cardiomyocytes. Interestingly, conditional specific deletion of Ackr3 in cardiomyocytes (αMHC-Cre+/− CXCR7flox/flox model) in mice leads to excessive left ventricular dilatation and major systolic dysfunction after MI demonstrating that cardiomyocyte ACKR3 protects the heart after ischemia (68). This cardioprotective effect of ACKR3 involves ERK activation induced by β-arrestin signalling in cardiomyocytes. In agreement with this result, the expression of ACKR3 is significantly upregulated concomitantly with ERK activation at the border zone of infarcted heart. These results indicate that ACKR3 may play a cardioprotective role by acting as a β-arrestin-biased receptor. Therapeutic targeting of β-arrestin-biased signalling through activation of β-adrenergic (carvediol) (98) and angiotensin II receptors (TRV120067) (99) showed cardioprotective effect in pre-clinical models. Collectively, these data support the fact that β-arrestin-biased signalling mediated by ACKR3 may be a new target in the treatment of CVD. Although ACKR3 is also expressed in cardiac fibroblasts, which are involved in cardiac repair after MI, no impact on cardiac function was observed in infarcted mice with conditional ACKR3 deletion in fibroblasts (Col1a2-CreERT2 x Cxcr7flox/flox model) (68) (Figure 1).
Cells of the immune system have been shown to mediate both protective and damaging effects in heart remodeling (100–102). ACKR3 is expressed in inflammatory cells (103) and its expression is especially correlated with monocyte to macrophage differentiation (73), the degree of maturation of B cells (76) or that of dendritic cells (77). ACKR3 expression in macrophages is upregulated during polarization in vitro and following MI with positive correlation with M1 but not M2 (70, 73) like macrophage markers. In these macrophages, ACKR3 controls CXCL12 and CXCL11-mediated chemotaxis as well as the production of pro-inflammatory cytokines such as IL-1 and Il-6 after MI indicating a deleterious role of ACKR3 in macrophages in this setting. Invalidation of Ackr3 by shRNA before induction of MI inhibited macrophage polarization, chemotaxis and inflammation and reduced infarct size leading to an improvement of cardiac function post-MI (70). Unfortunately, the role of ACKR3 in different immune cell sub-populations in post-MI cardiac remodeling are incomplete. Hence, studies investigating the specific role of ACKR3 in each immune cell type would allow to better understand its role in CVD.
Conclusions and Future Directions
Various pharmacological tools targeting ACKR3 have been tested in murine model of atherosclerosis, myocardial infarction, stroke and different knockout murine models for ACKR3 have been developed to highlight its role crucial in CVD. However, although these studies clearly demonstrated that ACKR3 may elicit a cell- and tissue-specific functional responses, the molecular mechanisms controlling ACKR3-related functions in CVD are a topic still debated. Tissue-dependent functioning can be related to the capacity of ACKR3 to interact with numerous ligands and can depend on surrounding cells producing and processing these ligands. Up-regulation of ACKR3 expression and those of its ligands in response to hypoxia, inflammation or infection suggests that ACKR3 function may vary under these different pathologic conditions. The fact that ACKR3 heterodimerizes with other proteins or that ACKR3 signaling can interfere with numerous intracellular pathways add another level of complexity. One main issue will be to analyze the functional consequences of these interactions ligand or protein/ACKR3 to identify new potential targets. As mentioned in this review, an unique function cannot be assigned to ACKR3. Therefore, the molecular mechanisms underlying distinct signaling pathways and functional responses downstream of ACKR3 need to be further investigated to better understand ACKR3 functions, and develop tailored therapeutics for CVD.
Author Contributions
VD and AL wrote the initial manuscript draft. PA conceived the Table and Figure. AL and J-SS edited the draft and with all other authors contributed to editing manuscript and approved the submitted version.
Funding
This work was supported by Inserm, Université Paris Cité. PA and VD were supported by the "Fondation pour la Recherche Médicale".
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Figures were created using BioRender.com.
Abbreviations
ACKR3, atypical chemokine receptor 3; ADM, adrenomedullin; cCKR, conventional chemokine receptor; CGRP, calcitonin gene-related peptide; CVD, cardiovascular diseases; CXCL11, C-X-C motif chemokine ligand 11; CXCL12, C-X-C motif chemokine ligand 12; CXCR7, C-X-C motif chemokine receptor 7; GPCR, G-protein-coupled receptor; MIF, macrophage migration inhibitory factor; PAMP, proadrenomedullin N-terminal 20 peptide; SDF-1, stromal derived factor-1; RAMP, receptor activity-modify proteins.
References
1. Back M, Weber C, Lutgens E. Regulation of Atherosclerotic Plaque Inflammation. J Intern Med (2015) 278:462–82.
2. Michel JB, Martin-Ventura JL, Nicoletti A, Ho-Tin-Noe B. Pathology of Human Plaque Vulnerability: Mechanisms and Consequences of Intraplaque Haemorrhages. Atherosclerosis (2014) 234:311–9.
3. Hansson GK, Libby P, Tabas I. Inflammation and Plaque Vulnerability. J Intern Med (2015) 278:483–93.
4. Aifah A, Iwelunmor J, Akwanalo C, Allison J, Amberbir A, Asante KP, et al. The Kathmandu Declaration on Global CVD/Hypertension Research and Implementation Science: A Framework to Advance Implementation Research for Cardiovascular and Other Noncommunicable Diseases in Low- and Middle-Income Countries. Glob Heart (2019) 14:103–7.
5. Gencer S, van der Vorst EPC, Aslani M, Weber C, Doring Y, Duchene J. Atypical Chemokine Receptors in Cardiovascular Disease. Thromb Haemost (2019) 119:534–41.
6. Raman D, Sobolik-Delmaire T, Richmond A. Chemokines in Health and Disease. Exp Cell Res (2011) 317:575–89.
7. Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, et al. International Union of Pharmacology. XXII Nomenclature Chemokine Receptors Pharmacol Rev (2000) 52:145–76.
9. Bachelerie F, Graham GJ, Locati M, Mantovani A, Murphy PM, Nibbs R, et al. New Nomenclature for Atypical Chemokine Receptors. Nat Immunol (2014) 15:207–8.
10. Yoshida T, Imai T, Kakizaki M, Nishimura M, Takagi S, Yoshie O. Identification of Single C Motif-1/Lymphotactin Receptor XCR1. J Biol Chem (1998) 273:16551–4.
11. Chandrasekar B, Bysani S, Mummidi S. CXCL16 Signals via Gi, Phosphatidylinositol 3-Kinase, Akt, I Kappa B Kinase, and Nuclear Factor-Kappa B and Induces Cell-Cell Adhesion and Aortic Smooth Muscle Cell Proliferation. J Biol Chem (2004) 279:3188–96.
12. Sun Y, Huang J, Xiang Y, Bastepe M, Juppner H, Kobilka BK, et al. Dosage-Dependent Switch From G Protein-Coupled to G Protein-Independent Signaling by a GPCR. EMBO J (2007) 26:53–64.
13. Fra AM, Locati M, Otero K, Sironi M, Signorelli P, Massardi ML, et al. Cutting Edge: Scavenging of Inflammatory CC Chemokines by the Promiscuous Putatively Silent Chemokine Receptor D6. J Immunol (2003) 170:2279–82.
14. Pruenster M, Mudde L, Bombosi P, Dimitrova S, Zsak M, Middleton J, et al. The Duffy Antigen Receptor for Chemokines Transports Chemokines and Supports Their Promigratory Activity. Nat Immunol (2009) 10:101–8.
15. Mantovani A, Bonecchi R, Locati M. Tuning Inflammation and Immunity by Chemokine Sequestration: Decoys and More. Nat Rev Immunol (2006) 6:907–18.
16. Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, et al. Beta-Arrestin- But Not G Protein-Mediated Signaling by the "Decoy" Receptor CXCR7. Proc Natl Acad Sci U S A (2010) 107:628–32.
17. Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 Heterodimerizes With CXCR4 and Regulates CXCL12-Mediated G Protein Signalling. Blood (2009). 24:6085–93
18. Libert F, Parmentier M, Lefort A, Dumont JE, Vassart G. Complete Nucleotide Sequence of a Putative G Protein Coupled Receptor: RDC1. Nucleic Acids Res (1990) 18:1917.
19. Sreedharan SP, Robichon A, Peterson KE, Goetzl EJ. Cloning and Expression of the Human Vasoactive Intestinal Peptide Receptor. Proc Natl Acad Sci U S A (1991) 88:4986–90.
20. Heesen M, Berman MA, Charest A, Housman D, Gerard C, Dorf ME. Cloning and Chromosomal Mapping of an Orphan Chemokine Receptor: Mouse RDC1. Immunogenetics (1998) 47:364–70.
21. Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, et al. A Novel Chemokine Receptor for SDF-1 and I-TAC Involved in Cell Survival, Cell Adhesion, and Tumor Development. J Exp Med (2006) 203:2201–13.
22. Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The Lymphocyte Chemoattractant SDF-1 is a Ligand for LESTR/fusin and Blocks HIV-1 Entry. Nature (1996) 382:829–33.
23. Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, et al. The CXC Chemokine SDF-1 is the Ligand for LESTR/fusin and Prevents Infection by T-Cell-Line-Adapted HIV-1. Nature (1996) 382:833–5.
25. Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, et al. The Chemokine SDF-1/CXCL12 Binds to and Signals Through the Orphan Receptor RDC1 in T Lymphocytes. J Biol Chem (2005) 280:35760–6.
26. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International Union of Basic and Clinical Pharmacology. [Corrected]. LXXXIX. Update on the Extended Family of Chemokine Receptors and Introducing a New Nomenclature for Atypical Chemokine Receptors. Pharmacol Rev (2014) 66:1–79.
27. Odemis V, Lipfert J, Kraft R, Hajek P, Abraham G, Hattermann K, et al. The Presumed Atypical Chemokine Receptor CXCR7 Signals Through G(i/o) Proteins in Primary Rodent Astrocytes and Human Glioma Cells. Glia (2012) 60:372–81.
28. Fumagalli A, Heuninck J, Pizzoccaro A, Moutin E, Koenen J, Seveno M, et al. The Atypical Chemokine Receptor 3 Interacts With Connexin 43 Inhibiting Astrocytic Gap Junctional Intercellular Communication. Nat Commun (2020) 11:4855.
29. Alampour-Rajabi S, El Bounkari O, Rot A, Muller-Newen G, Bachelerie F, Gawaz M, et al. MIF Interacts With CXCR7 to Promote Receptor Internalization, ERK1/2 and ZAP-70 Signaling, and Lymphocyte Chemotaxis. FASEB J (2015) 29:4497–511.
30. Meyrath M, Palmer CB, Reynders N, Vanderplasschen A, Ollert M, Bouvier M, et al. Proadrenomedullin N-Terminal 20 Peptides (PAMPs) Are Agonists of the Chemokine Scavenger Receptor Ackr3/Cxcr7. ACS Pharmacol Transl Sci (2021) 4:813–23.
31. Meyrath M, Szpakowska M, Zeiner J, Massotte L, Merz MP, Benkel T, et al. The Atypical Chemokine Receptor ACKR3/CXCR7 is a Broad-Spectrum Scavenger for Opioid Peptides. Nat Commun (2020) 11:3033.
32. Gravel S, Malouf C, Boulais PE, Berchiche YA, Oishi S, Fujii N, et al. The Peptidomimetic CXCR4 Antagonist TC14012 Recruits Beta-Arrestin to CXCR7: Roles of Receptor Domains. J Biol Chem (2010) 285:37939–43.
33. Wijtmans M, Maussang D, Sirci F, Scholten DJ, Canals M, Mujic-Delic A, et al. Synthesis, Modeling and Functional Activity of Substituted Styrene-Amides as Small-Molecule CXCR7 Agonists. Eur J Med Chem (2012) 51:184–92.
34. Richard-Bildstein S, Aissaoui H, Pothier J, Schafer G, Gnerre C, Lindenberg E, et al. Discovery of the Potent, Selective, Orally Available CXCR7 Antagonist ACT-1004-1239. J Med Chem (2020) 63:15864–82.
35. Zabel BA, Wang Y, Lewen S, Berahovich RD, Penfold ME, Zhang P, et al. Elucidation of CXCR7-Mediated Signaling Events and Inhibition of CXCR4-Mediated Tumor Cell Transendothelial Migration by CXCR7 Ligands. J Immunol (2009) 183:3204–11.
36. Decaillot FM, Kazmi MA, Lin Y, Ray-Saha S, Sakmar TP, Sachdev P. CXCR7/CXCR4 Heterodimer Constitutively Recruits Beta-Arrestin to Enhance Cell Migration. J Biol Chem (2011) 286:32188–97.
37. Odemis V, Boosmann K, Heinen A, Kury P, Engele J. CXCR7 is an Active Component of SDF-1 Signalling in Astrocytes and Schwann Cells. J Cell Sci (2010) 123:1081–8.
38. Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, et al. Disrupted Cardiac Development But Normal Hematopoiesis in Mice Deficient in the Second CXCL12/SDF-1 Receptor, CXCR7. Proc Natl Acad Sci U S A (2007) 104:14759–64.
39. Yu S, Crawford D, Tsuchihashi T, Behrens TW, Srivastava D. The Chemokine Receptor CXCR7 Functions to Regulate Cardiac Valve Remodeling. Dev Dynamics an Off Publ Am Assoc Anatomists (2011) 240:384–93.
40. Gerrits H, van Ingen Schenau DS, Bakker NE, van Disseldorp AJ, Strik A, Hermens LS, et al. Early Postnatal Lethality and Cardiovascular Defects in CXCR7-Deficient Mice. Genesis (2008) 46:235–45.
41. Wei ST, Huang YC, Hsieh ML, Lin YJ, Shyu WC, Chen HC, et al. Atypical Chemokine Receptor ACKR3/CXCR7 Controls Postnatal Vasculogenesis and Arterial Specification by Mesenchymal Stem Cells via Notch Signaling. Cell Death Dis (2020) 11:307.
42. Wang Y, Li G, Stanco A, Long JE, Crawford D, Potter GB, et al. CXCR4 and CXCR7 Have Distinct Functions in Regulating Interneuron Migration. Neuron (2011) 69:61–76.
43. Sanchez-Alcaniz JA, Haege S, Mueller W, Pla R, Mackay F, Schulz S, et al. Cxcr7 Controls Neuronal Migration by Regulating Chemokine Responsiveness. Neuron (2011) 69:77–90.
44. Szpakowska M, Dupuis N, Baragli A, Counson M, Hanson J, Piette J, et al. Human Herpesvirus 8-Encoded Chemokine Vccl2/vMIP-II is an Agonist of the Atypical Chemokine Receptor ACKR3/CXCR7. Biochem Pharmacol (2016) 114:14–21.
45. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a Noncognate Ligand of CXC Chemokine Receptors in Inflammatory and Atherogenic Cell Recruitment. Nat Med (2007) 13:587–96.
46. Klein KR, Karpinich NO, Espenschied ST, Willcockson HH, Dunworth WP, Hoopes SL, et al. Decoy Receptor CXCR7 Modulates Adrenomedullin-Mediated Cardiac and Lymphatic Vascular Development. Dev Cell (2014) 30:528–40.
47. Ikeda Y, Kumagai H, Skach A, Sato M, Yanagisawa M. Modulation of Circadian Glucocorticoid Oscillation via Adrenal Opioid-CXCR7 Signaling Alters Emotional Behavior. Cell (2013) 155:1323–36.
48. Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 Ligand With Allosteric Agonist Properties. Mol Pharmacol (2009) 75:1240–7.
49. Puchert M, Engele J. The Peculiarities of the SDF-1/CXCL12 System: In Some Cells, CXCR4 and CXCR7 Sing Solos, in Others, They Sing Duets. Cell Tissue Res (2014) 355:239–53.
50. Pawig L, Klasen C, Weber C, Bernhagen J, Noels H. Diversity and Inter-Connections in the CXCR4 Chemokine Receptor/Ligand Family: Molecular Perspectives. Front Immunol (2015) 6:429.
51. Valentin G, Haas P, Gilmour D. The Chemokine SDF1a Coordinates Tissue Migration Through the Spatially Restricted Activation of Cxcr7 and Cxcr4b. Curr Biol (2007) 17:1026–31.
52. Boldajipour B, Mahabaleshwar H, Kardash E, Reichman-Fried M, Blaser H, Minina S, et al. Control of Chemokine-Guided Cell Migration by Ligand Sequestration. Cell (2008) 132:463–73.
53. Naumann U, Cameroni E, Pruenster M, Mahabaleshwar H, Raz E, Zerwes HG, et al. CXCR7 Functions as a Scavenger for CXCL12 and CXCL11. PLoS One (2010) 5:e9175.
54. Chen D, Xia Y, Zuo K, Wang Y, Zhang S, Kuang D, et al. Crosstalk Between SDF-1/CXCR4 and SDF-1/CXCR7 in Cardiac Stem Cell Migration. Sci Rep (2015) 5:16813.
55. Ceholski DK, Turnbull IC, Pothula V, Lecce L, Jarrah AA, Kho C, et al. CXCR4 and CXCR7 Play Distinct Roles in Cardiac Lineage Specification and Pharmacologic Beta-Adrenergic Response. Stem Cell Res (2017) 23:77–86.
56. Tripathi V, Kumar R, Dinda AK, Kaur J, Luthra K. CXCL12-CXCR7 Signaling Activates ERK and Akt Pathways in Human Choriocarcinoma Cells. Cell Commun Adhes (2014) 21:221–8.
57. Luker KE, Gupta M, Steele JM, Foerster BR, Luker GD. Imaging Ligand-Dependent Activation of CXCR7. Neoplasia (2009) 11:1022–35.
58. Torossian F, Anginot A, Chabanon A, Clay D, Guerton B, Desterke C, et al. CXCR7 Participates in CXCL12-Induced CD34+ Cell Cycling Through Beta-Arrestin-Dependent Akt Activation. Blood (2014) 123:191–202.
59. Caron KM, Smithies O. Extreme Hydrops Fetalis and Cardiovascular Abnormalities in Mice Lacking a Functional Adrenomedullin Gene. Proc Natl Acad Sci U S A (2001) 98:615–9.
60. Hay DL, Garelja ML, Poyner DR, Walker CS. Update on the Pharmacology of Calcitonin/CGRP Family of Peptides: IUPHAR Review 25. Br J Pharmacol (2018) 175:3–17.
61. Wetzel-Strong SE, Li M, Klein KR, Nishikimi T, Caron KM. Epicardial-Derived Adrenomedullin Drives Cardiac Hyperplasia During Embryogenesis. Dev Dynamics an Off Publ Am Assoc Anatomists (2014) 243:243–56.
62. Mackie DI, Nielsen NR, Harris M, Singh S, Davis RB, Dy D, et al. RAMP3 Determines Rapid Recycling of Atypical Chemokine Receptor-3 for Guided Angiogenesis. Proc Natl Acad Sci U S A (2019) 116:24093–9.
63. Szpakowska M, Meyrath M, Reynders N, Counson M, Hanson J, Steyaert J, et al. Mutational Analysis of the Extracellular Disulphide Bridges of the Atypical Chemokine Receptor ACKR3/CXCR7 Uncovers Multiple Binding and Activation Modes for its Chemokine and Endogenous non-Chemokine Agonists. Biochem Pharmacol (2018) 153:299–309.
64. Hao H, Hu S, Chen H, Bu D, Zhu L, Xu C, et al. Loss of Endothelial CXCR7 Impairs Vascular Homeostasis and Cardiac Remodeling After Myocardial Infarction: Implications for Cardiovascular Drug Discovery. Circulation (2017) 135:1253–64.
65. Li X, Zhu M, Penfold ME, Koenen RR, Thiemann A, Heyll K, et al. Activation of CXCR7 Limits Atherosclerosis and Improves Hyperlipidemia by Increasing Cholesterol Uptake in Adipose Tissue. Circulation (2014) 129:1244–53.
66. Cebo M, Dittrich K, Fu X, Manke MC, Emschermann F, Rheinlaender J, et al. Platelet ACKR3/CXCR7 Favors Anti-Platelet Lipids Over an Atherothrombotic Lipidome and Regulates Thrombo-Inflammation. Blood (2021) 11:1722–1742.
67. Ghadge SK, Messner M, Seiringer H, Maurer T, Staggl S, Zeller T, et al. Smooth Muscle Specific Ablation of CXCL12 in Mice Downregulates CXCR7 Associated With Defective Coronary Arteries and Cardiac Hypertrophy. Int J Mol Sci 22 (2021) 22:5908.
68. Ishizuka M, Harada M, Nomura S, Ko T, Ikeda Y, Guo J, et al. CXCR7 Ameliorates Myocardial Infarction as a Beta-Arrestin-Biased Receptor. Sci Rep (2021) 11:3426.
69. Zhang S, Yue J, Ge Z, Xie Y, Zhang M, Jiang L. Activation of CXCR7 Alleviates Cardiac Insufficiency After Myocardial Infarction by Promoting Angiogenesis and Reducing Apoptosis. BioMed Pharmacother (2020) 127:110168.
70. Zhang J, Zhang Y, Xin S, Wu M, Zhang Y, Sun L. CXCR7 Suppression Modulates Macrophage Phenotype and Function to Ameliorate Post-Myocardial Infarction Injury. Inflammation Res (2020) 69:523–32.
71. Dong BC, Li MX, Wang XY, Cheng X, Wang Y, Xiao T, et al. Effects of CXCR7-Neutralizing Antibody on Neurogenesis in the Hippocampal Dentate Gyrus and Cognitive Function in the Chronic Phase of Cerebral Ischemia. Neural Regener Res (2020) 15:1079–85.
72. Merckelbach S, van der Vorst EPC, Kallmayer M, Rischpler C, Burgkart R, Doring Y, et al. Expression and Cellular Localization of CXCR4 and CXCL12 in Human Carotid Atherosclerotic Plaques. Thromb Haemost (2018) 118:195–206.
73. Ma W, Liu Y, Ellison N, Shen J. Induction of C-X-C Chemokine Receptor Type 7 (CXCR7) Switches Stromal Cell-Derived Factor-1 (SDF-1) Signaling and Phagocytic Activity in Macrophages Linked to Atherosclerosis. J Biol Chem (2013) 288:15481–94.
74. Rath D, Chatterjee M, Borst O, Muller K, Stellos K, Mack AF, et al. Expression of Stromal Cell-Derived Factor-1 Receptors CXCR4 and CXCR7 on Circulating Platelets of Patients With Acute Coronary Syndrome and Association With Left Ventricular Functional Recovery. Eur Heart J (2014) 35:386–94.
75. Rath D, Chatterjee M, Meyer L, Tekath N, Olma C, Krumm P, et al. Relative Survival Potential of Platelets Is Associated With Platelet CXCR4/CXCR7 Surface Exposure and Functional Recovery Following STEMI. Atherosclerosis (2018) 278:269–77.
76. Infantino S, Moepps B, Thelen M. Expression and Regulation of the Orphan Receptor RDC1 and its Putative Ligand in Human Dendritic and B Cells. J Immunol (2006) 176:2197–207.
77. Radice E, Ameti R, Melgrati S, Foglierini M, Antonello P, Stahl RAK, et al. Marginal Zone Formation Requires ACKR3 Expression on B Cells. Cell Rep (2020) 32:107951.
78. Chatterjee M, von Ungern-Sternberg SN, Seizer P, Schlegel F, Buttcher M, Sindhu NA, et al. Platelet-Derived CXCL12 Regulates Monocyte Function, Survival, Differentiation Into Macrophages and Foam Cells Through Differential Involvement of CXCR4-Cxcr7. Cell Death Dis (2015) 6:e1989.
79. Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the Chemokine Receptor CXCR4 in Haematopoiesis and in Cerebellar Development. Nature (1998) 393:595–9.
80. Li M, Ransohoff RM. Multiple Roles of Chemokine CXCL12 in the Central Nervous System: A Migration From Immunology to Neurobiology. Prog Neurobiol (2008) 84:116–31.
81. Shimizu S, Brown M, Sengupta R, Penfold ME, Meucci O. CXCR7 Protein Expression in Human Adult Brain and Differentiated Neurons. PLoS One (2011) 6:e20680.
82. Puchert M, Pelkner F, Stein G, Angelov DN, Boltze J, Wagner DC, et al. Astrocytic Expression of the CXCL12 Receptor, CXCR7/ACKR3 Is a Hallmark of the Diseased, But Not Developing CNS. Mol Cell Neurosci (2017) 85:105–18.
83. Banisadr G, Podojil JR, Miller SD, Miller RJ. Pattern of CXCR7 Gene Expression in Mouse Brain Under Normal and Inflammatory Conditions. J Neuroimmune Pharmacol (2016) 11:26–35.
84. Schonemeier B, Kolodziej A, Schulz S, Jacobs S, Hoellt V, Stumm R. Regional and Cellular Localization of the CXCl12/SDF-1 Chemokine Receptor CXCR7 in the Developing and Adult Rat Brain. J Comp Neurol (2008) 510:207–20.
85. Li Y, Tang G, Liu Y, He X, Huang J, Lin X, et al. CXCL12 Gene Therapy Ameliorates Ischemia-Induced White Matter Injury in Mouse Brain. Stem Cells Transl Med (2015) 4:1122–30.
86. Hill WD, Hess DC, Martin-Studdard A, Carothers JJ, Zheng J, Hale D, et al. SDF-1 (CXCL12) is Upregulated in the Ischemic Penumbra Following Stroke: Association With Bone Marrow Cell Homing to Injury. J Neuropathol Exp Neurol (2004) 63:84–96.
87. Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, et al. Directed Migration of Neural Stem Cells to Sites of CNS Injury by the Stromal Cell-Derived Factor 1alpha/CXC Chemokine Receptor 4 Pathway. Proc Natl Acad Sci U S A (2004) 101:18117–22.
88. Bogoslovsky T, Spatz M, Chaudhry A, Maric D, Luby M, Frank J, et al. Stromal-Derived Factor-1[Alpha] Correlates With Circulating Endothelial Progenitor Cells and With Acute Lesion Volume in Stroke Patients. Stroke (2011) 42:618–25.
89. Zhang Y, Zhang H, Lin S, Chen X, Yao Y, Mao X, et al. SDF-1/CXCR7 Chemokine Signaling is Induced in the Peri-Infarct Regions in Patients With Ischemic Stroke. Aging Dis (2018) 9:287–95.
90. Stumm RK, Rummel J, Junker V, Culmsee C, Pfeiffer M, Krieglstein J, et al. A Dual Role for the SDF-1/CXCR4 Chemokine Receptor System in Adult Brain: Isoform-Selective Regulation of SDF-1 Expression Modulates CXCR4-Dependent Neuronal Plasticity and Cerebral Leukocyte Recruitment After Focal Ischemia. J Neurosci (2002) 22:5865–78.
91. Zhang M, Qiu L, Zhang Y, Xu D, Zheng JC, Jiang L. CXCL12 Enhances Angiogenesis Through CXCR7 Activation in Human Umbilical Vein Endothelial Cells. Sci Rep (2017) 7:8289.
92. Dai X, Tan Y, Cai S, Xiong X, Wang L, Ye Q, et al. The Role of CXCR7 on the Adhesion, Proliferation and Angiogenesis of Endothelial Progenitor Cells. J Cell Mol Med (2011) 15:1299–309.
93. Yan X, Cai S, Xiong X, Sun W, Dai X, Chen S, et al. Chemokine Receptor CXCR7 Mediates Human Endothelial Progenitor Cells Survival, Angiogenesis, But Not Proliferation. J Cell Biochem (2012) 113:1437–46.
94. Stellos K, Bigalke B, Borst O, Pfaff F, Elskamp A, Sachsenmaier S, et al. Circulating Platelet-Progenitor Cell Coaggregate Formation is Increased in Patients With Acute Coronary Syndromes and Augments Recruitment of CD34+ Cells in the Ischaemic Microcirculation. Eur Heart J (2013) 34:2548–56.
95. Chatterjee M, Borst O, Walker B, Fotinos A, Vogel S, Seizer P, et al. Macrophage Migration Inhibitory Factor Limits Activation-Induced Apoptosis of Platelets via CXCR7-Dependent Akt Signaling. Circ Res (2014) 115:939–49.
96. Chatterjee M. Atypical Roles of the Chemokine Receptor ACKR3/CXCR7 in Platelet Pathophysiology. Cells (2022) 11, 213.
97. Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, et al. Deep RNA Sequencing Reveals Dynamic Regulation of Myocardial Noncoding RNAs in Failing Human Heart and Remodeling With Mechanical Circulatory Support. Circulation (2014) 129:1009–21.
98. Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A Unique Mechanism of Beta-Blocker Action: Carvedilol Stimulates Beta-Arrestin Signaling. Proc Natl Acad Sci U S A (2007) 104:16657–62.
99. Ryba DM, Li J, Cowan CL, Russell B, Wolska BM, Solaro RJ. Long-Term Biased Beta-Arrestin Signaling Improves Cardiac Structure and Function in Dilated Cardiomyopathy. Circulation (2017) 135:1056–70.
100. Kologrivova I, Shtatolkina M, Suslova T, Ryabov V. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair After Myocardial Infarction. Front Immunol (2021) 12:664457.
101. Gomez I, Duval V, Silvestre JS. Cardiomyocytes and Macrophages Discourse on the Method to Govern Cardiac Repair. Front Cardiovasc Med (2018) 5:134.
102. Sun Y, Pinto C, Camus S, Duval V, Alayrac P, Zlatanova I, et al. Splenic Marginal Zone B Lymphocytes Regulate Cardiac Remodeling After Acute Myocardial Infarction in Mice. J Am Coll Cardiol (2022) 79:632–47.
Keywords: atypical chemokine receptors, ACKR3, chemokine, cardiovascular diseases, signalling
Citation: Duval V, Alayrac P, Silvestre J-S and Levoye A (2022) Emerging Roles of the Atypical Chemokine Receptor 3 (ACKR3) in Cardiovascular Diseases. Front. Endocrinol. 13:906586. doi: 10.3389/fendo.2022.906586
Received: 28 March 2022; Accepted: 27 May 2022;
Published: 29 June 2022.
Edited by:
Julien Hanson, University of Liège, BelgiumReviewed by:
Tony Ngo, Sanford Burnham Prebys Medical Discovery Institute, United StatesStephen John Hill, University of Nottingham, United Kingdom
Copyright © 2022 Duval, Alayrac, Silvestre and Levoye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angélique Levoye, YW5nZWxpcXVlLmxldm95ZUBpbnNlcm0uZnI=