 Graeme Eisenhofer
Graeme Eisenhofer Mirko Peitzsch
Mirko Peitzsch Nicole Bechmann
Nicole Bechmann Angela Huebner
Angela Huebner- 1Institute of Clinical Chemistry and Laboratory Medicine, Universitätsklinikum Carl Gustav Carus Dresden, Technische Universität Dresden, Dresden, Germany
- 2Department of Internal Medicine III, Universitätsklinikum Carl Gustav Carus Dresden, Technische Universität Dresden, Dresden, Germany
- 3Department of Pediatrics, Universitätsklinikum Carl Gustav Carus Dresden, Technische Universität Dresden, Dresden, Germany
Catecholamine-producing tumors of childhood include most notably neuroblastoma, but also pheochromocytoma and paraganglioma (PPGL). Diagnosis of the former depends largely on biopsy-dependent histopathology, but this is contraindicated in PPGL where diagnosis depends crucially on biochemical tests of catecholamine excess. Such tests retain some importance in neuroblastoma though continue to largely rely on measurements of homovanillic acid (HVA) and vanillylmandelic acid (VMA), which are no longer recommended for PPGL. For PPGL, urinary or plasma metanephrines are the recommended most accurate tests. Addition of methoxytyramine to the plasma panel is particularly useful to identify dopamine-producing tumors and combined with normetanephrine also shows superior diagnostic performance over HVA and VMA for neuroblastoma. While use of metanephrines and methoxytyramine for diagnosis of PPGL in adults is established, there are numerous pitfalls for use of these tests in children. The establishment of pediatric reference intervals is particularly difficult and complicated by dynamic changes in metabolites during childhood, especially in infants for both plasma and urinary measurements, and extending to adolescence for urinary measurements. Interpretation of test results is further complicated in children by difficulties in following recommended preanalytical precautions. Due to this, the slow growing nature of PPGL and neglected consideration of the tumors in childhood the true pediatric prevalence of PPGL is likely underappreciated. Earlier identification of disease, as facilitated by surveillance programs, may uncover the true prevalence and improve therapeutic outcomes of childhood PPGL. For neuroblastoma there remain considerable obstacles in moving from entrenched to more accurate tests of catecholamine excess.
Introduction
Catecholamine-producing tumors include neuroblastoma, as well as pheochromocytoma and paraganglioma (PPGL), all derived from cells of the neural crest. Neuroblastoma occurs almost exclusively in childhood and originate from immature embryonic neuroblast cells that undergo transformation to form tumors at intra-adrenal and extra-adrenal locations. PPGL similarly occur at respective intra-adrenal and extra-adrenal locations, but originate from chromaffin cells or their chromoblast precursors, and are usually detected in adulthood though may be overlooked in childhood.
It is now apparent that neuroblasts, sympathoblasts, chromoblasts and mature chromaffin cells originate from neural crest derived Schwann cell precursors by way of different transitions and manifest by variable stages of differentiation (1, 2). These emerging concepts about development from different neural crest derivatives are fundamental to a complete understanding of the utility of catecholamine-related biomarkers for diagnosis of neuroblastoma and PPGL. In particular, differences in transition of neural-crest derived tumor precursors appear to be recapitulated in the considerable heterogeneity of presentations of both neuroblastoma and PPGL, including the nature of catecholamine production.
While neuroblastomas are characterized by poorly developed catecholamine biosynthetic and secretory pathways, there is nevertheless variation in this that relates to differences in catecholamine-associated features important for biochemical testing and disease aggressiveness (3–6). PPGL on the other hand show more mature catecholamine biosynthetic and secretory pathways compared to neuroblastoma, though even within those two intra- and extra-adrenal groups of tumors there is considerable heterogeneity in differentiation that impacts the nature of catecholamine secretory and metabolic products employed for biochemical diagnosis (7).
Of relevance to this article, PPGL detected in childhood and young adulthood tend to be poorly differentiated and often occur secondary to a particular group of mutated genes (i.e., so called cluster 1 group mutations) that predispose to more often extra-adrenal paraganglioma and multifocal adrenal and extra-adrenal tumors than the tumors that originate from cluster 2 group mutations (7–9). As detailed later, such differences have relevance to biochemical testing strategies and interpretation of biochemical test results in children with suspected chromaffin cell tumors.
It is also important to appreciate that PPGL are typically slow growing with a volume doubling time of 5-7 years (10, 11). From this it can be expected that it would take at least 15 years for a tumor to enlarge from 1.5 to 3 cm in diameter (Figure 1). Furthermore, in the early stages when tumors are less than 1.5 cm, they are unlikely to produce enough catecholamines for detection by standard biochemical tests let alone to evoke the signs and symptoms that may alert clinicians to the possibility of the tumor. Even when tumors do attain a size and level of catecholamine secretion sufficient to evoke signs and symptoms, there is usually considerable delay in recognizing the significance of this (12). Given that the median diameter of PPGL detected on the basis of signs and symptoms is 4 cm (13), it can be appreciated that for any PPGL first diagnosed in adults under an age of 30 years, the tumor is likely to have originated in childhood. The prevalence of childhood PPGL commonly cited in the literature is 10-20% (14–16); however, with the considerations outlined above, this likely represents an underestimate, particularly for patients with cluster 1 type gene mutations who have a median age of tumor diagnosis of 29 to 32 years (17).
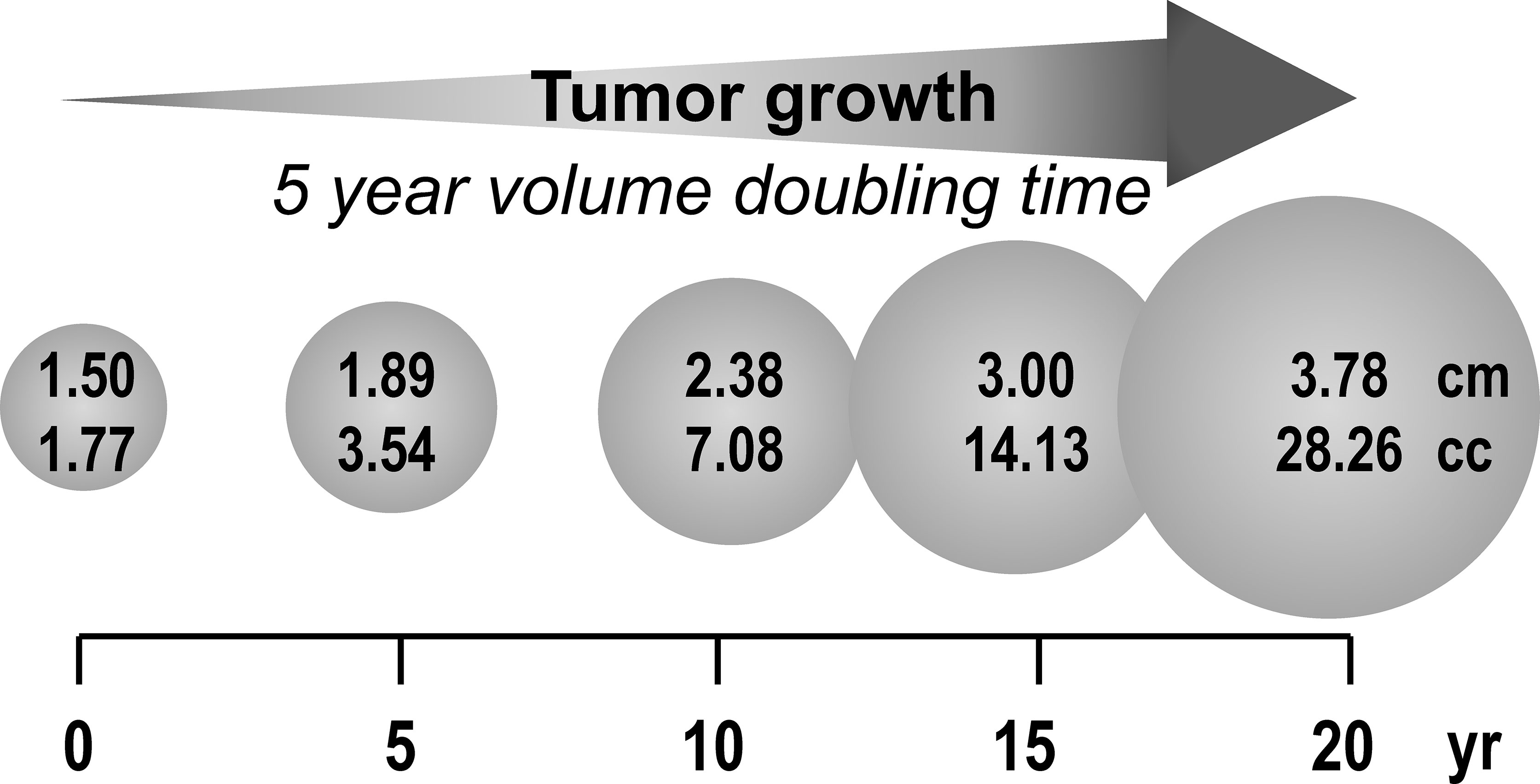
Figure 1 Estimated duration (years) for a 1.5 cm PPGL to reach 3 to 4 cm based on reported minimum tumor volume doubling time of 5 years.
As in adulthood the most important factor for an early diagnosis of PPGL in children and adolescents is attentiveness to the clinical clues of these tumors by pediatric caregivers. For familial cases involving offspring or siblings identified with germline mutations of tumor susceptibility genes an earlier diagnosis can be facilitated by enrolment into routine surveillance programs beginning as early as 5 years of age depending on the mutated gene (18–20).
Modes of Clinical Suspicion
The initial mode of clinical suspicion of a PPGL is usually based on signs and symptoms of catecholamine excess. Signs and symptoms in children as in adults include hypertension, particularly paroxysmal hypertension, as well as palpitations, excessive sweatiness, headache, visual disturbances, pallor, anxiety, tremor, panic/anxiety, constipation, and nausea and/or vomiting (14, 21, 22). Among these signs and symptoms, any of a few may be present and mostly in episodes. Weight loss associated with a lowered body mass index and high heart rate are also useful to consider, in fact, more so than hypertension, which is relatively common in adults and of limited discriminatory value (23). High blood pressure in children and adolescents is, however, uncommon and in combination with any other signs and symptoms should always be considered as a potential indicator of a catecholamine-producing tumor. Nighttime sweatiness, polyuria and disturbances of vision or mental status have been reported in pediatric cases of PPGL (15), and might warrant particular attention.
As in adults, some children with PPGLs may be normotensive and asymptomatic (24), particularly when tumors are found as part of surveillance programs involving family members with a known mutation of a tumor susceptibility gene (25). Children with incidentally discovered adrenal or extra-adrenal masses based on imaging studies for reasons other than a suspected PPGL can also be normotensive and asymptomatic and have a catecholamine-producing tumor (26, 27).
For neuroblastoma, which have a more limited hereditary background compared to PPGL and do not usually secrete catecholamines in amounts sufficient to cause signs and symptoms, the mode of initial clinical suspicion is different from that for chromaffin cell tumors. Since routine screening for the tumors is now out of favor, neuroblastoma are usually suspected based on findings of a palpable abdominal mass or as a mass found incidentally during ultrasonography (28), including on occasion during prenatal ultrasound (29). Masses in the neck or thoracic regions may be discovered on the basis of Horner’s syndrome. Children with neuroblastoma may also present with fever, weight loss, bone or joint pain, or other symptoms that may evoke discovery during imaging studies. Hematological abnormalities from bone marrow involvement can also lead to discovery of these tumors and eventual diagnosis achieved by histopathology often after percutaneous needle biopsy.
Catecholamine Synthesis, Metabolism and Secretion
Both PPGL and neuroblastoma are characterized by synthesis and metabolism of catecholamines within tumor cells. For any appreciation of the use of catecholamine-related biomarkers for diagnosis of these tumors it is useful to understand the pathways of catecholamine biosynthesis, storage, metabolism and secretion (30).
Catecholamine biosynthesis starts with conversion of tyrosine to 3,4-dihydroxyphenylalanine (DOPA) by the rate limiting enzyme, tyrosine hydroxylase. DOPA is then converted to dopamine by aromatic-L-amino acid decarboxylase, an enzyme with a wide tissue distribution and broad substrate specificity. Dopamine is then transported by vesicular monoamine transporters into vesicular storage granules, where it is further converted to norepinephrine by dopamine β-hydroxylase, an enzyme with a unique presence in vesicular storage granules. Presence of phenylethanolamine N- methyltransferase (PNMT) in adrenal chromaffin cells leads to further conversion of norepinephrine to epinephrine (Figure 2); however, since PNMT is a cytosolic enzyme, this step depends on leakage of norepinephrine from vesicular storage granules into the cell cytoplasm. Epinephrine is then translocated back into storage granules from where it can be actively secreted as a circulating hormone.
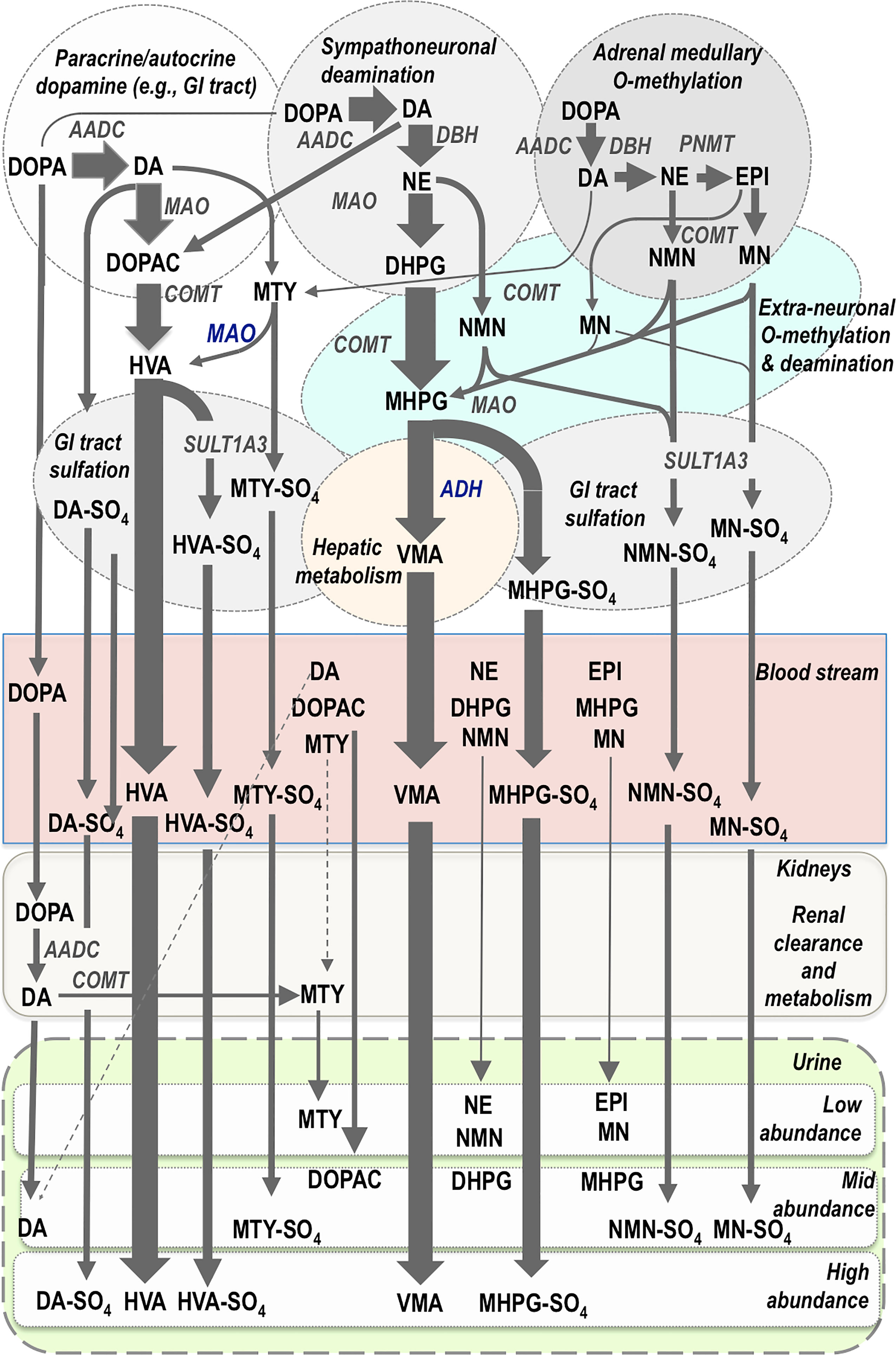
Figure 2 Pathways of catecholamine metabolism according to initial biosynthesis of dopamine (DA) in paracrine and autocrine systems, of DA and norepinephrine (NE) in sympathoneuronal systems and of DA, NE and epinephrine (EPI) in chromaffin cells of the adrenal medulla. Thereafter the main metabolic pathways are shown according different compartments of metabolism and according to entry into the bloodstream and thereafter clearance and metabolism by the kidneys for elimination in the urine. Flux through metabolic pathways is reflected by arrow size and final abundance of metabolites in the blood stream and urine. The normetanephrine (NMN), metanephrine (MN) and methoxytyramine (MTY) in the bloodstream are produced in low abundance, primarily from adrenal chromaffin cells, and thereby provide better diagnostic signals for PPGLs than metabolites produced in higher abundance and largely from sources other than adrenal chromaffin cells. Almost all catecholamines and their metabolites undergo some degree of sulfate conjugation in humans, but only the main sulfate-conjugated metabolites are displayed. Note also the high abundance of dopamine metabolites, despite their minor source from sympatho-adrenal medullary systems. AADC, aromatic amino acid decarboxylase; DBH, dopamine-beta-hydroxylase; PNMT, phenylethanolamine-N-methyltransferase; MAO, monoamine oxidase; COMT, catechol-O-methyltransferase; SULT1A3; sulfotransferase isoenzyme 1A3; ADH, alcohol dehydrogenase; DOPA, 3,4-dihydroxyphenylalanine; DOPAC, 3,4-dihydroxyphenylacetic acid; DHPG, 3,4-dihydroxyphenylglycol; MHPG, 3-methoxy-4-hydroxyphenylglycol; HVA, homovanillic acid; VMA, vanillylmandelic acid; MTY-SO4, methoxytyramine-sulfate; NMN-SO4, normetanephrine-sulfate; MN-SO4, metanephrine-sulfate; HVA-SO4, homovanillic acid-sulfate; MHPG-SO4, 3-methoxy-4-hydroxyphenylglycol-sulfate; DA-SO4, dopamine-sulfate.
Importantly and contrary to usual textbook depictions, vesicular stores of catecholamines do not exist in a static state until exocytotic secretion (31). Rather, vesicular stores of catecholamines exist in a highly dynamic equilibrium with the surrounding cytoplasm, with passive outward leakage into the cytoplasm counterbalanced by inward active transport under the control of vesicular monoamine transporters. The processes of active exocytotic secretion versus passive leakage of catecholamines from vesicular stores are entirely different and contribute differently to the metabolism of catecholamines produced at different sites including within tumor cells (32). These differences, however, are rarely appreciated despite the importance of this understanding for clinical applications of catecholamine-related biomarkers for diagnosis of catecholamine-producing tumors (30).
Exocytotic secretion of catecholamines involves the emptying of contents of vesicular catecholamine stores into the surrounding extracellular space. The process for hormonal secretion from the adrenal medulla involves closer proximity to the bloodstream than for norepinephrine secreted by sympathetic neurons, which acts locally rather than systemically. Close to 90% of norepinephrine secreted by sympathetic nerves is removed back into nerves by neuronal uptake so that only a small proportion escapes into the bloodstream or is metabolized at extra-neuronal locations before entry into the bloodstream (31). Some of the norepinephrine recaptured by sympathetic nerves is deaminated intraneuronally to 3,4-dihydroxyphenylglycol (DHPG) by monoamine oxidase (MAO), but most is returned to vesicular stores via vesicular monoamine transporters. By far the most DHPG is produced not after neuronal reuptake but rather after vesicular leakage of norepinephrine into the cytoplasm. DHPG is thereby the main initial metabolite produced from norepinephrine (Figure 2). DHPG is further metabolized at extraneuronal sites by catechol-O-methyltransferase (COMT) to 3-methoxy-4-hydroxyphenylglycol (MHPG). Thereafter most MHPG is metabolized in the liver by alcohol dehydrogenase to vanillylmandelic acid (VMA), the main urinary metabolic end-product of norepinephrine metabolism (33).
The processes of metabolism for catecholamines synthesized and secreted by adrenal chromaffin cells are somewhat similar to those for catecholamine-producing tumors, but entirely different from those for the norepinephrine produced in sympathetic nerves (31). First and foremost, while sympathetic nerves express only MAO, chromaffin cells also express COMT and thereby produce O-methylated metabolites (31). These include metanephrine from epinephrine, normetanephrine from norepinephrine and methoxytyramine from dopamine (Figure 2). The same former two metabolites can also be produced at extraneuronal locations from the catecholamines secreted into the bloodstream from the adrenals or from the norepinephrine secreted by sympathetic nerves. For circulating metanephrine, over 90% is derived from epinephrine metabolized within adrenal chromaffin cells rather than from epinephrine secreted from the same chromaffin cells (34). For circulating normetanephrine, about 25% is produced within adrenal chromaffin cells and 75% from extraneuronal metabolism of norepinephrine secreted by sympathetic nerves.
The processes for metabolism of dopamine are somewhat different in that substantial amounts of this catecholamine are not produced in the sympatho-adrenal system, but rather in diffuse paracrine systems of the gastrointestinal tract, kidneys and other tissues (35, 36) (Figure 2). In the kidneys, dopamine is produced after renal uptake and local metabolism of circulating DOPA (37). Thus, more than 90% of urinary dopamine is formed from circulating DOPA rather than circulating dopamine (38). Urinary methoxytyramine appears to be derived by similar processes that may also include renal O-methylation of the dopamine produced from circulating DOPA (39). Finally, the end-product of dopamine metabolism, homovanillic acid (HVA), is derived from the combined actions of COMT and MAO; unlike VMA, the production of HVA does not require any additional actions of hepatic alcohol dehydrogenase so that HVA has distinctly different sources from VMA (35).
Apart from final metabolism of catecholamines and catecholamine metabolites to HVA and VMA, all compounds also undergo varying degrees of sulfate conjugation (Figure 2). This process occurs mainly in gastrointestinal tissues, the site of expression of the required sulfotransferase isoenzyme, SULT1A3 (40). The sulfated metabolic end-products are primarily removed by the kidneys and excreted in urine. This is particularly important for dopamine and its metabolites, but also provides an important metabolic pathway for O-methylated catecholamine metabolites. Thus, the normetanephrine, metanephrine and methoxytyramine commonly measured in urine after acid hydrolysis mainly reflect sulfate conjugates that have partly different sources from the more rapidly cleared and thus much lower concentrations of circulating free metabolites (41).
Tumoral Catecholamine Metabolism
With the considerations outlined above concerning the subcellular, cellular and organ wide compartmentalized disposition of catecholamines it can be better appreciated why the O-methylated metabolites of catecholamines offer the best biomarkers of catecholamine-producing tumors, as also displayed according to the simplifications of Figure 3. For cluster 1 norepinephrine-producing noradrenergic PPGL typical of childhood (Figure 3, panel A), the tumor-derived signal for free normetanephrine commonly shows a stronger and larger proportional increase above normal plasma concentrations than for circulating norepinephrine, 90% of which is derived from sympathetic nerves (34). That large proportion serves to dilute the diagnostic signal from tumors considerably more than for circulating normetanephrine. Moreover, the tumoral production of normetanephrine is continuous, whereas the exocytotic secretion of norepinephrine by tumors can be intermittent or minimal unless provoked. The signal produced by VMA is also largely diluted by the considerable amounts of this metabolite originally derived from the DHPG produced in sympathetic nerves, which lack COMT. Similarly, the normetanephrine sulfate produced in gastrointestinal tissues from locally secreted normetanephrine dilutes the signal for this metabolite, thereby explaining lower diagnostic signal for urinary deconjugated normetanephrine than for free normetanephrine (42).
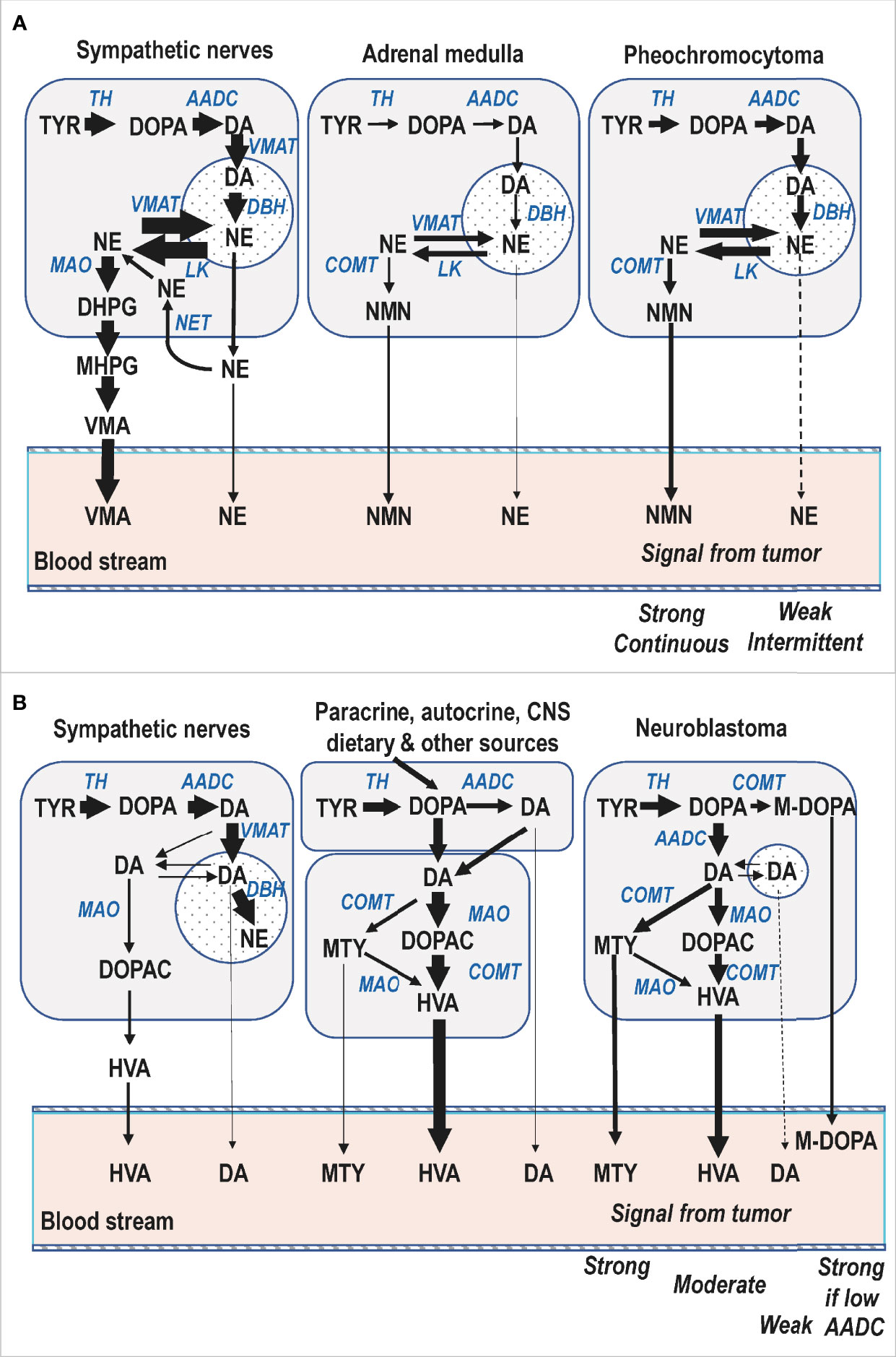
Figure 3 Simplified diagrammatic representations of catecholamine biosynthetic and metabolic pathways in a pediatric patient with a cluster 1 norepinephrine-producing PPGL (A) and another with a dopamine-producing neuroblastoma (B). For both panels the storage vesicles where dopamine is converted to norepinephrine and from where catecholamines are secreted by exocytosis are depicted by the circular compartments. For neuroblastoma the smaller size of that compartment serves to illustrate lower numbers of vesicular storage granules and associated minimal catecholamine secretory activity of these tumors. AADC, aromatic amino acid decarboxylase; DBH, dopamine-beta-hydroxylase; MAO, monoamine oxidase; COMT, catechol-O-methyltransferase; VMAT, vesicular monoamine transporter; LK, leakage of catecholamines from vesicles; NET, cell membrane norepinephrine transporter; DOPA, 3,4-dihydroxyphenylalanine; M-DOPA, 3-O-methyl-dopa; DA, dopamine; NE, norepinephrine; DOPAC, 3,4-dihydroxyphenylacetic acid; DHPG, 3,4-dihydroxyphenylglycol; MHPG, 3-methoxy-4-hydroxyphenylglycol; HVA, homovanillic acid; VMA, vanillylmandelic acid; MTY, methoxytyramine; NMN, normetanephrine.
Similar considerations to those outlined above also clarify why measurements of plasma free normetanephrine provide a stronger diagnostic signal than urinary VMA for children with neuroblastoma (4). However, unlike PPGL, in neuroblastoma it is the dopamine metabolites and not the norepinephrine metabolites that provide the more consistently increased biomarkers of excess catecholamine production. The considerations for catecholamine metabolism in neuroblastoma are also somewhat different than in PPGL where COMT is expressed in larger abundance than MAO, the expression of which is also lower than in adrenal medullary chromaffin cells (43, 44). Neuroblastomas express not only COMT (45), but also MAO in high abundance (46), and thus produce significant amounts of both deaminated and O-methylated catecholamine metabolites (Figure 3, panel B). Nevertheless, since HVA is normally produced in substantial amounts by an array of different pathways, this production acts to dilute the diagnostic signal for this metabolite when produced by neuroblastoma. In contrast, free methoxytyramine circulates at very low plasma concentrations so the diagnostic signal for this metabolite is stronger than for urinary HVA (4). For some tumors plasma concentrations of DOPA and 3-O-methyldopa provide the only signal (4, 47), presumably due to their poorly differentiated nature, including minimal expression of aromatic-L-amino acid decarboxylase, the enzyme that converts DOPA to dopamine.
Biochemical Tests of Catecholamine Excess
For neuroblastoma the value of measuring catecholamine metabolites as biomarkers, rather than the catecholamines themselves, was established in the 1970s from the observations of LaBrosse and colleagues (48). As shown by these investigators, neuroblastomas display a relative lack of the catecholamine storage vesicles characteristic of mature chromaffin cells and their PPGL derivatives. Thus, these tumors usually do not present with hypertension or increased plasma or urinary catecholamines, so that biochemical tests have traditionally depended on measurements of catecholamine metabolites, in particular HVA and VMA.
Intra-tumoral metabolism of catecholamines in PPGLs was also described in the 1960s by Crout and Sjoerdsma (49). Nevertheless, because PPGLs are characterized by hypertension and symptoms of catecholamine excess, those early findings were largely ignored and diagnosis continued to focus on measurements of catecholamines. It was not until after the turn of the 21st century that emphasis moved from catecholamines to their O-methylated metabolites, the metanephrines (50). Shift in emphasis from catecholamines to metanephrines for diagnosis of PPGLs followed the understanding outlined earlier about how catecholamines are synthesized, stored and metabolized independently of secretion and by different pathways. Consequently, recommendations today mandate measurements of plasma free or urinary fractionated metanephrines as initial screening tests for PPGLs (51). Among these tests, plasma measurements offer superior diagnostic accuracy than urinary measurements (42). For the latter it should be appreciated that these may involve either the free metabolites or the more commonly used and much higher concentrations of sulfate-conjugated metabolites measured together with the free metabolites after an acid hydrolysis deconjugation step. The measurements of urinary free metabolites offer some diagnostic advantages over the combined free and deconjugated metabolites, though the value of urinary free methoxytyramine for identification of dopamine producing tumors is limited (42). Most likely this reflects origins of urinary free methoxytyramine from renal uptake, clearance and metabolism of circulating DOPA (Figure 2).
The vast majority of studies that have examined the diagnostic performance of biochemical tests for patients with PPGL have been in adults, with only a few isolated reports in children some of which employed outdated measurements of urinary VMA or spectrophotometric measurements of total metanephrines rather than fractionated normetanephrine and metanephrine (Table 1). Nevertheless, there have been two reports that documented high diagnostic accuracy of plasma free metanephrines for childhood cases of PPGL (25, 52).
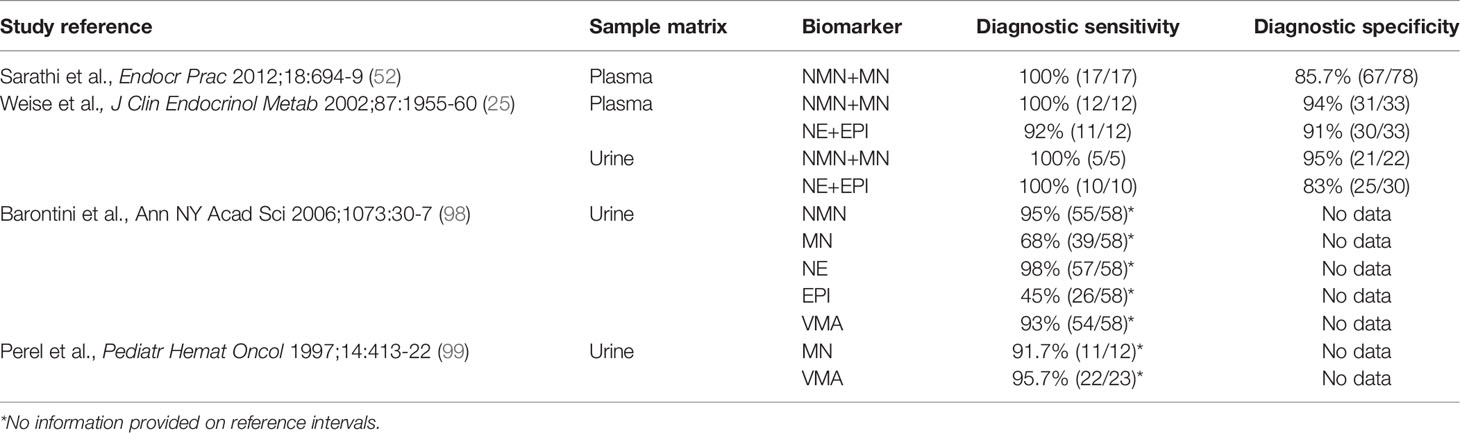
Table 1 Diagnostic performance of biochemical biomarkers for pediatric PPGL.
The two catecholamine metabolites that continue to provide the mainstay for biochemical testing of neuroblastoma are HVA and VMA, usually measured in urine. Since the HVA and VMA derived from neuroblastoma or PPGL are diluted by considerable amounts of the same metabolites produced from other sources, these metabolites are relatively poor diagnostic markers for catecholamine-producing tumors. For adult pheochromocytoma, diagnostic sensitivity of VMA reaches only to 46-77% compared to 97-99% for plasma free metanephrines at similar specificities (53). In a prospective trial involving 1.5 million neonates only 73% of all infants detected at follow-up with neuroblastoma had elevated urinary excretion of HVA or VMA at screening (54). Moreover, many that were detected by screening were those that spontaneously regressed while those that were missed were usually aggressive. Consequently, screening programs involving urinary HVA and VMA have been abandoned. Today, diagnosis of neuroblastoma depends primarily on biopsy-dependent histopathology, which combined with genomic biomarkers (e.g., MYCN amplification) can provide information for staging and therapeutic intervention (55).
Reflecting historical precedence, almost all reports that have examined the utility of tests of catecholamine excess to identify children with neuroblastoma have included measurements of urinary HVA and VMA (Table 2). Nevertheless, there have been recent reports that have examined utility of plasma free or urinary measurements of normetanephrine and methoxytyramine. Thus, similar to PPGL, there is now evidence that measurements of plasma normetanephrine and methoxytyramine provide excellent biomarkers for identification of patients with neuroblastoma (4, 56, 57), including one study that showed the expected superiority over urinary HVA and VMA (4). Introduction of new biochemical tests for neuroblastoma is, nevertheless, largely made futile by reliance on biopsy-dependent histopathological diagnosis and measurements of urinary HVA and VMA for assessing tumoral catecholamine production.
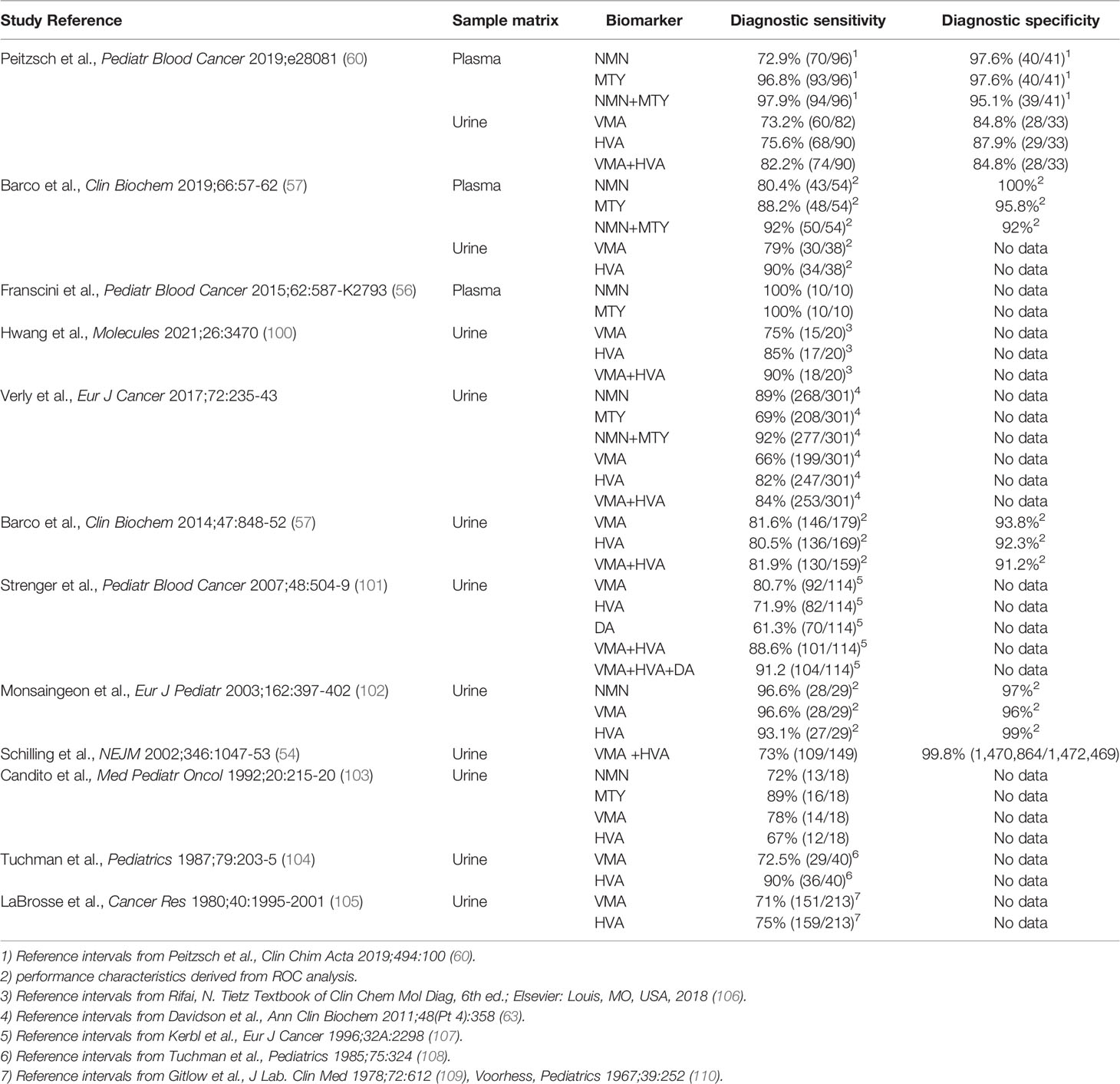
Table 2 Diagnostic performance of biochemical biomarkers for neuroblastoma.
Urinary rather than plasma methoxytyramine has also recently been advanced as an alternative to urinary HVA for identification of neuroblastoma (58). This metabolite, similar to urinary dopamine, appears to be largely derived from DOPA and is thus not a particularly good biomarker of tumoral dopamine metabolism (42). Nevertheless, these measurements appear to have prognostic utility (5). Since plasma DOPA also shows prognostic utility in neuroblastoma (47, 59), it is possible that this may underly the prognostic utility of urinary methoxytyramine. With prognostic rather than diagnostic utility in mind, there may be a rationale to advance from current antiquated reliance on urinary HVA and VMA to new and improved methods for biochemical testing of neuroblastoma.
Reference Intervals
The need to establish reference intervals for biochemical tests of catecholamine excess represents another impediment to moving from outdated methods of diagnosis to new and improved biochemical tests to detect catecholamine-producing tumors. For adults it is relatively simple to obtain blood or urine specimens to establish reference intervals and further test utility of those reference intervals in patient populations. The practical and ethical barriers to procure blood or urine specimens for establishing reference intervals are more complex to negotiate for pediatric than adult populations (60). Consequently, advances in place for adults can be considerably delayed to implement for children.
As with many biomarkers, those involving tests of catecholamine excess can show variable differences between adults and children and within the pediatric population differences according to developmental age and sex. For plasma free normetanephrine and methoxytyramine there are highly dynamic changes in early childhood with markedly higher plasma concentrations in neonates that drop rapidly within the first year (Figure 4). Thereafter, concentrations level off, though for normetanephrine slowly climb later in adolescence and continue to increase throughout adulthood. In contrast, plasma concentrations of metanephrine increase during early infancy, are higher in young children than adults and remain higher in males than females (25). The reciprocal changes of methoxytyramine and normetanephrine compared to metanephrine in early childhood are suggested to reflect apoptosis of neural crest-derived cells of the paraganglia from which the former metabolites derive, changes that contrast with development of the adrenal chromaffin cells responsible for almost all circulating metanephrine (60). For use of O-methylated catecholamine metabolites as biomarkers of neuroblastoma it is essential to employ age-specific reference intervals, which can be achieved from polynomial curve fitting (56, 60). For children over 5 years of age, plasma concentrations of methoxytyramine and normetanephrine remain relatively constant throughout childhood and all that is required are reference intervals for that broader age range. For metanephrine, concentrations are higher in boys than girls and particularly in younger children, when slightly higher cut-offs may be preferable compared to adolescents and adults.
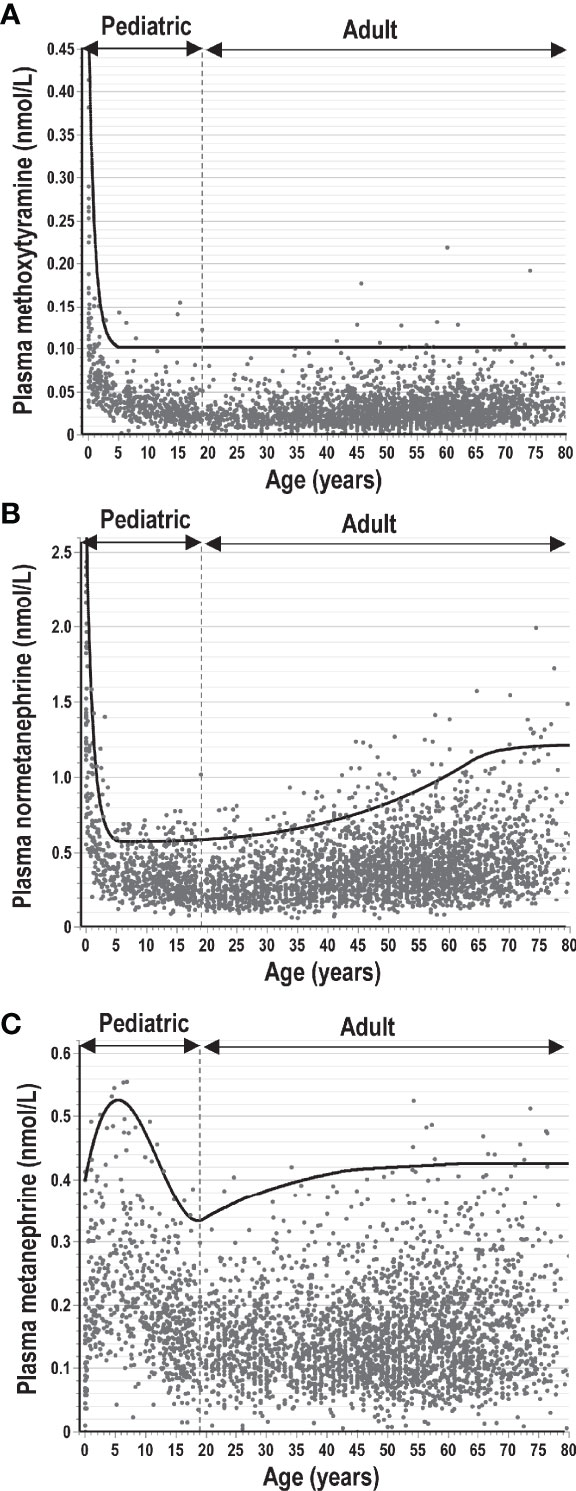
Figure 4 Plasma concentrations of methoxytyramine (A), normetanephrine (B) and metanephrine (C) from infancy and early childhood through adolescence to later ages in adults. Concentrations are derived from measurements in 3706 individuals (including 707 children from 0-18 years of age) without catecholamine producing tumors according to previously published data (4, 25, 42, 60). The solid lines indicate upper limits of reference intervals, which for adults and children over 5 years of age were optimized to provide optimum diagnostic sensitivity for normetanephrine at optimal specificity and lower proportions of false-positive results (<1%) for metanephrine and methoxytyramine.
Since diagnosis of catecholamine-producing tumors of childhood have historically involved measurements in urine, reference intervals for this sample matrix are those that have received the most attention (61–65). However, such measurements are not without associated problems for establishing reference intervals. Since 24-hr collections of urine from both infants and older children are unreliable and difficult, spot urines are the method commonly employed with dilutional differences corrected using creatinine (61). This, however, has the problem of introducing another variable as a denominator (Figure 5). Urinary outputs of creatinine vary according to diet, exercise and most importantly muscle mass (66, 67). Consequently, urinary outputs of creatinine are higher in males than females and are positively correlated to body mass or body surface area and increase substantially from early infancy throughout childhood (67–69). There are also body size related increases in urinary outputs of catecholamines and their metabolites, but these are not as substantial as those for creatinine. Thus, although 24-hr urinary outputs of catecholamines and metabolites increase throughout childhood, when expressed as a ratio to urinary outputs of creatinine there are marked decreases throughout childhood and particularly in infancy (61–65).
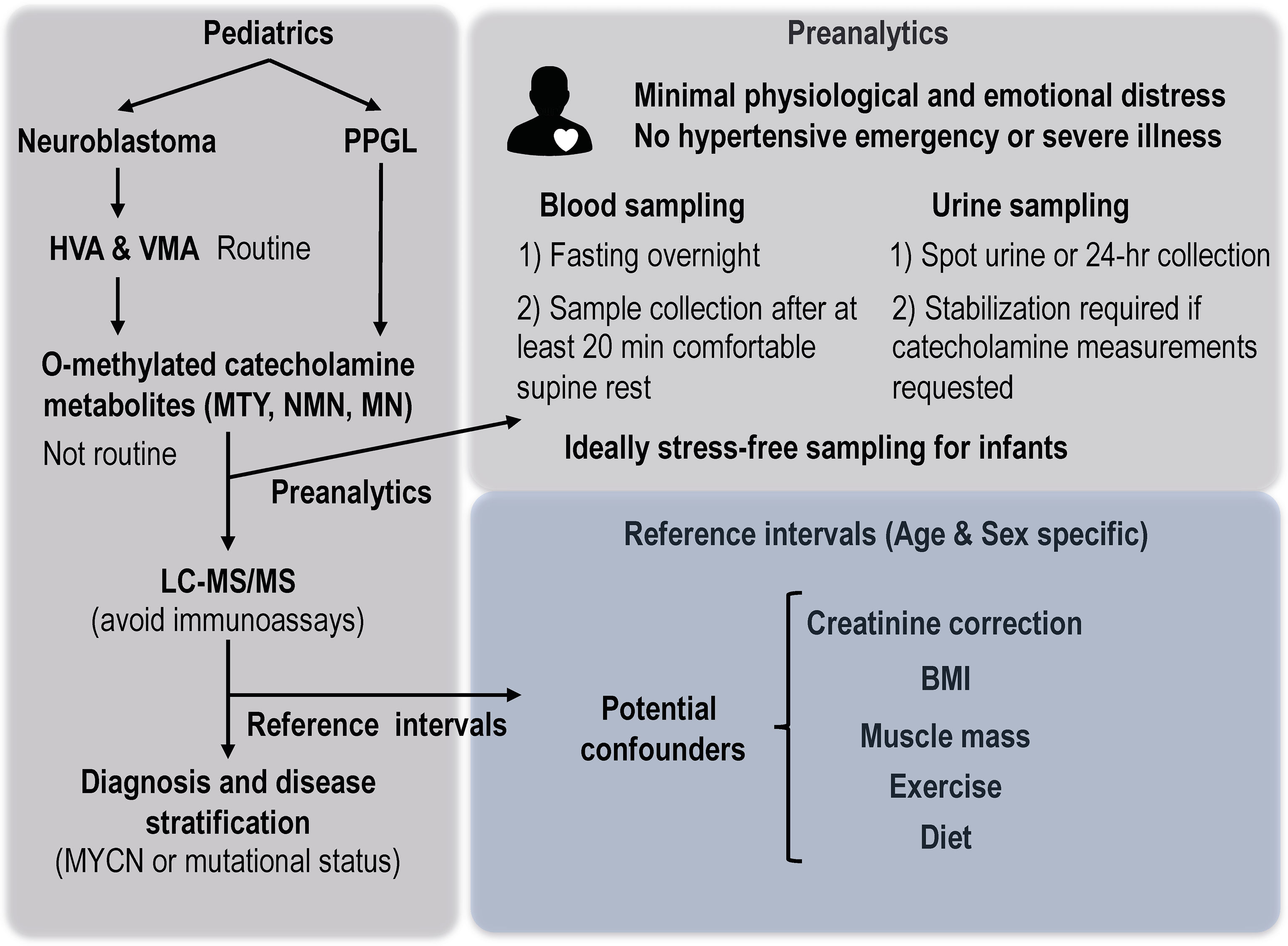
Figure 5 Considerations of preanalytics and reference intervals for interpretation of biochemical tests of catecholamine excess in children. MTY, methoxytyramine; NMN, normetanephrine; MN, metanephrine; BMI, body mass index.
Due to the dynamic changes in urinary excretion of both creatinine and catecholamine metabolites, reference intervals must be established for different age groups (61–65). Thus, for example, as reported by Davidson et al. (63), upper limits for urinary excretion of normetanephrine as a ratio to creatinine vary considerably from 0.529 nmol/mol for infants below one year to 0.123 nmol/mol for children 8 to 10 years of age and 0.086 nmol/mol for those between 14 to 19 years of age. As reported by Cole et al. (61), use of creatinine to correct for dilutional influences on measured concentrations of VMA and HVA in spot urine samples suffers from several limitations that impact reliability of measurements as a laboratory test for neuroblastoma. These investigators proposed further adjustments for sex and body weight.
For all reference intervals due consideration must also be given to the methods of measurement. Early spectrophotometric methods as well as more recent immunoassay methods can be inaccurate and suffer from interferences so that reference intervals developed by these methods can be unreliable (70). Even for techniques employing gas or liquid chromatographic separation there can be differences in measurements between laboratories so that reference intervals should be validated by each laboratory and not simply involve those reported in the literature. Mass spectrometric methods offer opportunities for both improved analytical specificity and accuracy. By participation in interlaboratory proficiency programs or comparison studies there is now the possibility for both harmonized measurements and reference intervals (71).
Preanalytical Considerations
Apart from appropriately established reference intervals and accurate measurement methods, consideration of preanalytics and other patient specific variables is crucial to interpretation of analytical test results (72), particularly those that fall close to either side of upper limits of reference intervals. For both plasma and urinary measurements, it is important that samples are procured with patients under conditions of minimal physiological or emotional distress (Figure 5). In adults it is well established that emergency situations and acute illness can result in plasma concentrations or urinary outputs of metanephrines that can be indistinguishable from those in patients with catecholamine-producing tumors (73). The same can also be expected for children, so that ideally testing for a catecholamine-producing tumor should be undertaken after recovery from a hypertensive emergency or severe illness.
Exercise or other forms of physical activity should be avoided or minimized before blood collections or preceding and during urine collections. First morning urine collections are associated with lower urinary outputs of catecholamines and metabolites due to the preceding nighttime rest and have been proposed as a means to minimize false-positive results (74). However, as yet this has not been verified for diagnosis of catecholamine-producing tumors in childhood.
For measurements of plasma metanephrines it is important that blood samples are taken after at least 20 minutes of comfortable supine rest, but for young children this can be difficult and requires close supervision (25). Similarly, although it is standard practice to acquire blood samples by direct venipuncture, this is established in adults to result in higher plasma concentrations and rates of false-positive results than sampling using a previously inserted intravenous cannula (75). For children, the emotional distress of venipuncture can be expected to be more troublesome than for adults. With preanalytical precautions in place high diagnostic sensitivity is preserved and false-positive results are minimal according to upper limits of reference intervals appropriately determined for children (25).
For younger children, stress-free blood sampling can be highly problematic, which represents a limitation of plasma compared to the urinary measurements typically used when testing children for suspected neuroblastoma. The volume of blood required can also be a limiting factor for neonates. However, new advances in sample preparation procedures before mass spectrometry is now allowing accurate and precise measurements of plasma methoxytyramine and other O-methylated metabolites in as little as 50 µl of plasma (76). With such advances it might even be possible to obtain blood by heel prick, which might provide an advantage of a blood versus a urine test.
Dietary influences and medications can also be important to consider. Certain foods can increase plasma concentrations of methoxytyramine as well as urinary outputs of catecholamine metabolites (77). For blood sampling such influences can be avoided by an overnight fast; however, for infants and urine collections, dietary restrictions may be preferable or more practical (60). Drugs that significantly impact monoamine systems, such as norepinephrine uptake blockers, can be troublesome in adults but are rarely administered to children. Although some antihypertensives and other medications can result in analytical interferences for some methods of analysis, such drugs are not usually troublesome with modern mass spectrometric methods (78).
Biochemical Test Interpretation
Apart from assessment of whether test results are positive or negative, the pattern and nature of increases of the three O-methylated catecholamine metabolites can provide other information about a catecholamine producing tumor that cannot be so easily gleaned from measurements of the catecholamines themselves or other metabolites such as HVA and VMA. Extents and patterns of increases above reference intervals not only allow assessments of relative probabilities of a tumor, but also other information such as location, size, disease aggressiveness and type of disease-causing mutations (79, 80). For neuroblastoma the information is a little more limited than for PPGL, but from increases in methoxytyramine can be used to assess likelihood of MYCN amplification (4, 5), which has prognostic significance.
For PPGL, increases in plasma methoxytyramine and metanephrine can be used to assess likelihood of extra- versus intra-adrenal tumor locations (80), the former more common in pediatric than adult patients. Increases in methoxytyramine relative to normetanephrine can also be used to assess risk of metastatic disease (79), which as in neuroblastoma may also relate to the developmental aspects of the tumors and associated degree of differentiation. Related to this, relative increases of the three metabolites can be used to assess probabilities of underlying mutations in groups of genes (80), these differing in prevalence among children and young adults compared to older adults with the tumors (7, 8).
Magnitudes of increases above normal of all combined O-methylated metabolites correlate with disease burden and can thereby be used to predict tumor size (80). The generally slow growing nature of PPGLs is also reflected by time-dependent increases in plasma concentrations of the O-methylated metabolites, which can be useful in follow-up to better confirm a PPGL. This can be particularly useful in surveillance programs, such as in children with identified mutations of the von-Hippel Lindau (VHL) gene whereby relative changes over time can signal development of a PPGL better than a single point measurement (25).
Differential Diagnosis
Neuroblastoma is more common in children than PPGL and since clinical presentations can overlap, there can be difficulties in the differential diagnosis of the two tumor entities (22). This difficulty has been highlighted in this issue by a case series of five children with PPGL who were initially diagnosed mistakenly with neuroblastoma, including some who underwent biopsy and all of whom were inadequately prepared before surgical intervention (81). Though commonly employed for neuroblastoma, percutaneous biopsy of a PPGL is potentially dangerous and is therefore contraindicated for those tumors (82, 83). Furthermore, because all patients, including children, with PPGL should be prepared for surgery using adrenergic blocking drugs (22, 84), it is imperative that there is no mistaking a PPGL for a neuroblastoma before scheduling fine needle biopsy or surgical intervention. Adding to the difficulties in differential diagnosis, cases have also been reported of composite neuroblastoma and pheochromocytoma (85–87). Furthermore, some gene mutations, such as those impacting the myc-associated factor X (MAX) gene, can predispose to both childhood neuroblastoma and pheochromocytoma (88, 89).
For PPGL at least, identification of disease depends crucially on biochemical tests of catecholamine excess; thus, if there is any doubt about a presumed neuroblastoma, the same tests should be considered in those cases. In the case series presented in this issue, involving five patients with PPGL and a mistaken initial diagnosis of neuroblastoma, only two underwent any form of testing for catecholamine excess (81). The problem remains, however, whether such tests can be used to distinguish neuroblastoma from PPGL.
Since both PPGL and neuroblastoma show overlapping patterns of increases in plasma dopamine and norepinephrine metabolites, as well as in some cases the precursor catecholamines themselves, these measurements alone are unlikely to offer a solution for differential diagnosis in all pediatric cases of these tumors. Plasma concentrations of 3-O-methydopa are negligibly impacted in patients with PPGL (personal observations), but show increases above cut-offs in about 75% of patients with neuroblastoma (4). Such increases may therefore be useful for distinguishing neuroblastoma from PPGL, but cannot solve the problem for more fully differentiated cases of neuroblastoma. As outlined earlier, neuroblastoma differ from PPGL in their relative expression of the primary catecholamine-metabolizing enzymes, MAO and COMT, with the former deaminating enzyme more highly expressed in neuroblastoma than PPGL. Thus, it is possible that, in addition to measurements of 3-O-methyldopa, different patterns of deaminated and O-methylated metabolites might be useful for differential diagnosis. This, however, remains to be determined.
Technologies such as those involving circulating tumor cells or DNA and protein biomarkers offer potential molecular diagnostic strategies for therapeutic stratification and monitoring in neuroblastoma (90, 91). Such approaches might also be also useful for differential diagnosis. Nevertheless, it will take considerable time to develop and establish liquid biopsies for these purposes, let alone prospectively determine any clinical utility, particularly in pediatric solid tumors (92).
Differences in imaging features between neuroblastoma and PPGL, such as presence of calcifications in the former, could also offer clues to help distinguish the two types of tumors (93). However, calcifications can occasionally occur in PPGL (94). Imaging characteristics of encasement, invasion and infiltration are relatively common in neuroblastoma (95), but far less so in PPGL and might thereby offer other clues to differentiate the two tumor types. Nevertheless, as indicated in the case series covered in this issue, there can be exceptions. As yet any use of functional imaging for differential diagnosis appears undocumented. Therefore, without histopathological confirmation, differential diagnosis of neuroblastoma and PPGL must continue to rely on a combination of considerations, including presentation and clinical features of the patient, imaging characteristics as well as whatever laboratory tests may be available.
Biochemical Testing in Disease Surveillance Programs
While surveillance programs for neuroblastoma have all but disappeared, those for PPGLs are becoming increasingly important due to recognition of the high hereditability of the tumors. For families with identified germline mutations in tumor susceptibility genes, screening is recommended to start in childhood as early as 5 years for some gene mutations, such as for succinate dehydrogenase subunit B (SDHB) and VHL, that lead to activation of pseudohypoxia pathways (18, 19). A slightly later start at about 10 years is currently recommended for mutations of other pseudohypoxia genes and later still for mutations of genes impacting kinase signaling pathways, such as those encoding the rearranged during transfection proto-oncogene (RET) and the neurofibromatosis type 1 (NF1) gene (96, 97).
Since mutations of genes such as VHL and SDHB that result in activation of pseudohypoxia pathways lead to PPGLs that do not produce epinephrine, the focus of biochemical testing in children as in adults with these mutations should be on normetanephrine. However, for mutations of succinate dehydrogenase subunit genes, the resulting tumors often also produce methoxytyramine and sometimes only methoxytyramine (80). Therefore, for these children biochemical testing should include measurements of methoxytyramine, which for assessing tumoral dopamine production should involve measurements in plasma rather than urine (42). On the other hand, for children with mutations of RET, NF1 or other genes that primarily impact kinase signaling pathways, interpretation of biochemical tests must include measurements of metanephrine since the associated tumors express PNMT and invariably produce epinephrine (80).
For all surveillance involving children, reference intervals established for pediatric populations are essential. With plasma measurements, use of reference intervals established for adults is likely to lead to false-negative results when applied to children over 5 years of age. This is particularly important for normetanephrine, which shows higher plasma concentrations in adults than in children (25).
Future Perspective
While biochemical tests of catecholamine excess in neuroblastoma are of limited importance for diagnosis compared to biopsy based histopathological diagnosis and molecular analyses, there are situations where biopsy is either not possible or may be contra-indicated. For such situations biochemical tests of catecholamine excess remain useful, but for the most part continue to rely on measurements of VMA and HVA, tests that have long been abandoned for PPGL. There are now better tests of catecholamine excess than urinary HVA and VMA, but it will likely take considerable time, if ever, before these will enter the clinical mainstream for neuroblastoma. For PPGL such tests have been widely available for the past two decades, but still need to be better integrated into pediatric care. The need for appropriate transition of pediatric to adult care for childhood cases of PPGL is another challenge, and particularly important in light of the high rate of disease recurrence and/or metastasis for tumors that originate in childhood. PPGL, however, remain rare tumors and patient management requires a depth of knowledge not within reach for most pediatricians who might first encounter affected children. Appropriate management of all patients with PPGL, but especially those in childhood, requires a multidisciplinary approach. This need is now recognized by patient-led organizations that are establishing procedures for review and accreditation of centers with appropriate resources and expertise. Through these and other efforts it is possible that earlier diagnosis of PPGL might be achieved and the true prevalence of childhood PPGL clarified. On the other hand, biochemical tests of catecholamine excess in children with neuroblastoma are likely to follow the trend of the past six decades and continue to remain sub-optimally based on historical precedence rather than more ideally on contemporary understanding.
Author Contributions
GE drafted the first version of manuscript, while MP, NB and AH contributed to the editing and revision of the manuscript. All authors provided conceptual input and MP additionally assisted with compilation of data from the literature.
Funding
The authors gratefully acknowledge support from the Deutsche Forschungsgemeinschaft (DGE, 314061271-TRR/CRC 205 to GE, MP, NB and AH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
PPGL, pheochromocytoma and paraganglioma; LC-MS/MS, liquid chromatography with tandem mass spectrometry; VMA, vanillylmandelic acid; HVA, homovanillic acid; PNMT, phenylethanolamine N-methyltransferase; COMT, catechol-O-methyltransferase; MAO, monoamine oxidase; DHPG, 3,4-dihydroxyphenylglycol; MHPG, 3-methoxy-4-hydroxyphenylglycol; MAX, myc-associated factor X; VHL, von-Hippel Lindau; SDHB, succinate dehydrogenase subunit B; RET, rearranged during transfection proto-oncogene; NF1, neurofibromatosis type 1.
References
1. Kameneva P, Artemov AV, Kastriti ME, Faure L, Olsen TK, Otte J, et al. Single-Cell Transcriptomics of Human Embryos Identifies Multiple Sympathoblast Lineages With Potential Implications for Neuroblastoma Origin. Nat Genet (2021) 53(5):694–706. doi: 10.1038/s41588-021-00818-x
2. Jansky S, Sharma AK, Korber V, Quintero A, Toprak UH, Wecht EM, et al. Single-Cell Transcriptomic Analyses Provide Insights Into the Developmental Origins of Neuroblastoma. Nat Genet (2021) 53(5):683–93. doi: 10.1038/s41588-021-00806-1
3. Berthold F, Hunneman DH, Harms D, Kaser H, Zieschang J. Serum Vanillylmandelic Acid/Homovanillic Acid Contributes to Prognosis Estimation in Patients With Localised But Not With Metastatic Neuroblastoma. Eur J Cancer (1992) 28A(12):1950–4. doi: 10.1016/0959-8049(92)90234-S
4. Peitzsch M, Butch ER, Lovorn E, Mangelis A, Furman WL, Santana VM, et al. Biochemical Testing for Neuroblastoma Using Plasma Free 3-O-Methyldopa, 3-Methoxytyramine, and Normetanephrine. Pediatr Blood Cancer (2020) 67(2):e28081. doi: 10.1002/pbc.28081
5. Verly IRN, Matser YAH, Leen R, Meinsma R, Fiocco M, Koster J, et al. Urinary 3-Methoxytyramine Is a Biomarker for MYC Activity in Patients With Neuroblastoma. JCO Precis Oncol (2022) 6:e2000447. doi: 10.1200/PO.20.00447
6. Verly IRN, Leen R, Meinsma JR, Hooijer GKJ, Savci-Heijink CD, van Nes J, et al. Catecholamine Excretion Profiles Identify Clinical Subgroups of Neuroblastoma Patients. Eur J Cancer (2019) 111:21–9. doi: 10.1016/j.ejca.2019.01.014
7. Pamporaki C, Hamplova B, Peitzsch M, Prejbisz A, Beuschlein F, Timmers HJ, et al. Characteristics of Pediatric vs Adult Pheochromocytomas and Paragangliomas. J Clin Endocrinol Metab (2017) 102(4):1122–1132. doi: 10.1210/jc.2016-3829
8. Redlich A, Pamporaki C, Lessel L, Fruhwald MC, Vorwerk P, Kuhlen M. Pseudohypoxic Pheochromocytomas and Paragangliomas Dominate in Children. Pediatr Blood Cancer (2021):e28981. doi: 10.1002/pbc.28981
9. Petenuci J, Guimaraes AG, Fagundes GFC, Benedetti AFF, Afonso ACF, Pereira MAA, et al. Genetic and Clinical Aspects of Paediatric Pheochromocytomas and Paragangliomas. Clin Endocrinol (Oxf) (2021) 95(1):117–24. doi: 10.1111/cen.14467
10. Michalowska I, Cwikla JB, Michalski W, Wyrwicz LS, Prejbisz A, Szperl M, et al. Growth Rate of Paragangliomas Related to Germline Mutations of the Sdhx Genes. Endocr Pract (2017) 23(3):342–52. doi: 10.4158/EP161377.OR
11. Powers JF, Pacak K, Tischler AS. Pathology of Human Pheochromocytoma and Paraganglioma Xenografts in NSG Mice. Endocr Pathol (2017) 28(1):2–6. doi: 10.1007/s12022-016-9452-5
12. Olson SW, Yoon S, Baker T, Prince LK, Oliver D, Abbott KC. Longitudinal Plasma Metanephrines Preceding Pheochromocytoma Diagnosis: A Retrospective Case-Control Serum Repository Study. Eur J Endocrinol (2016) 174(3):289–95. doi: 10.1530/EJE-15-0651
13. Rogowski-Lehmann N, Geroula A, Prejbisz A, Timmers H, Megerle F, Robledo M, et al. Missed Clinical Clues in Patients With Pheochromocytoma/Paraganglioma Discovered by Imaging. Endocr Connect (2018). doi: 10.1530/EC-18-0318
14. Ciftci AO, Tanyel FC, Senocak ME, Buyukpamukcu N. Pheochromocytoma in Children. J Pediatr Surg (2001) 36(3):447–52. doi: 10.1053/jpsu.2001.21612
15. Sullivan J, Groshong T, Tobias JD. Presenting Signs and Symptoms of Pheochromocytoma in Pediatric-Aged Patients. Clin Pediatr (Phila) (2005) 44(8):715–9. doi: 10.1177/000992280504400811
16. Waguespack SG, Rich T, Grubbs E, Ying AK, Perrier ND, Ayala-Ramirez M, et al. A Current Review of the Etiology, Diagnosis, and Treatment of Pediatric Pheochromocytoma and Paraganglioma. J Clin Endocrinol Metab (2010) 95(5):2023–37. doi: 10.1210/jc.2009-2830
17. Eisenhofer G, Timmers HJ, Lenders JW, Bornstein SR, Tiebel O, Mannelli M, et al. Age at Diagnosis of Pheochromocytoma Differs According to Catecholamine Phenotype and Tumor Location. J Clin Endocrinol Metab (2011) 96(2):375–84. doi: 10.1210/jc.2010-1588
18. Rednam SP, Erez A, Druker H, Janeway KA, Kamihara J, Kohlmann WK, et al. Von Hippel-Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res (2017) 23(12):e68–75. doi: 10.1158/1078-0432.CCR-17-0547
19. Wong MY, Andrews KA, Challis BG, Park SM, Acerini CL, Maher ER, et al. Clinical Practice Guidance: Surveillance for Phaeochromocytoma and Paraganglioma in Paediatric Succinate Dehydrogenase Gene Mutation Carriers. Clin Endocrinol (Oxf) (2019) 90(4):499–505. doi: 10.1111/cen.13926
20. Tufton N, Shapiro L, Sahdev A, Kumar AV, Martin L, Drake WM, et al. An Analysis of Surveillance Screening for SDHB-Related Disease in Childhood and Adolescence. Endocr Connect (2019) 8(3):162–72. doi: 10.1530/EC-18-0522
21. Revillon Y, Daher P, Jan D, Buisson C, Bonnerot V, Martelli H, et al. Pheochromocytoma in Children: 15 Cases. J Pediatr Surg (1992) 27(7):910–1. doi: 10.1016/0022-3468(92)90396-o
22. Virgone C, Andreetta M, Avanzini S, Chiaravalli S, De Pasquale D, Crocoli A, et al. Pheochromocytomas and Paragangliomas in Children: Data From the Italian Cooperative Study (TREP). Pediatr Blood Cancer (2020) 67(8):e28332. doi: 10.1002/pbc.28332
23. Geroula A, Deutschbein T, Langton K, Masjkur J, Pamporaki C, Peitzsch M, et al. Pheochromocytoma and Paraganglioma: Clinical Feature-Based Disease Probability in Relation to Catecholamine Biochemistry and Reason for Disease Suspicion. Eur J Endocrinol (2019) 181(4):409–20. doi: 10.1530/EJE-19-0159
24. Igaki J, Nishi A, Sato T, Hasegawa T. A Pediatric Case of Pheochromocytoma Without Apparent Hypertension Associated With Von Hippel-Lindau Disease. Clin Pediatr Endocrinol (2018) 27(2):87–93. doi: 10.1297/cpe.27.87
25. Weise M, Merke DP, Pacak K, Walther MM, Eisenhofer G. Utility of Plasma Free Metanephrines for Detecting Childhood Pheochromocytoma. J Clin Endocrinol Metab (2002) 87(5):1955–60. doi: 10.1210/jcem.87.5.8446
26. Bonacruz Kazzi G. Asymptomatic Bladder Phaeochromocytoma in a 7-Year-Old Boy. J Paediatr Child Health (2001) 37(6):600–2. doi: 10.1046/j.1440-1754.2001.00716.x
27. Pozo J, Munoz MT, Martos G, Argente J. Sporadic Phaeochromocytoma in Childhood: Clinical and Molecular Variability. J Pediatr Endocrinol Metab (2005) 18(6):527–32. doi: 10.1515/jpem.2005.18.6.527
28. Powell JE, Esteve J, Mann JR, Parker L, Frappaz D, Michaelis J, et al. Neuroblastoma in Europe: Differences in the Pattern of Disease in the UK. SENSE. Study Group for the Evaluation of Neuroblastoma Screening in Europe. Lancet (1998) 352(9129):682–7. doi: 10.1016/S0140-6736(97)11239-9
29. Ho PT, Estroff JA, Kozakewich H, Shamberger RC, Lillehei CW, Grier HE, et al. Prenatal Detection of Neuroblastoma: A Ten-Year Experience From the Dana-Farber Cancer Institute and Children's Hospital. Pediatrics (1993) 92(3):358–64. doi: 10.1542/peds.92.3.358
30. Eisenhofer G, Huynh TT, Hiroi M, Pacak K. Understanding Catecholamine Metabolism as a Guide to the Biochemical Diagnosis of Pheochromocytoma. Rev Endocr Metab Disord (2001) 2(3):297–311. doi: 10.1023/A:1011572617314
31. Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine Metabolism: A Contemporary View With Implications for Physiology and Medicine. Pharmacol Rev (2004) 56(3):331–49. doi: 10.1124/pr.56.3.1
32. Eisenhofer G, Keiser H, Friberg P, Mezey E, Huynh TT, Hiremagalur B, et al. Plasma Metanephrines are Markers of Pheochromocytoma Produced by Catechol-O-Methyltransferase Within Tumors. J Clin Endocrinol Metab (1998) 83(6):2175–85. doi: 10.1210/jcem.83.6.4870
33. Eisenhofer G, Aneman A, Hooper D, Rundqvist B, Friberg P. Mesenteric Organ Production, Hepatic Metabolism, and Renal Elimination of Norepinephrine and its Metabolites in Humans. J Neurochem (1996) 66(4):1565–73. doi: 10.1046/j.1471-4159.1996.66041565.x
34. Eisenhofer G, Rundquist B, Aneman A, Friberg P, Dakak N, Kopin IJ, et al. Regional Release and Removal of Catecholamines and Extraneuronal Metabolism to Metanephrines. J Clin Endocrinol Metab (1995) 80(10):3009–17. doi: 10.1210/jcem.80.10.7559889
35. Eisenhofer G, Aneman A, Friberg P, Hooper D, Fandriks L, Lonroth H, et al. Substantial Production of Dopamine in the Human Gastrointestinal Tract. J Clin Endocrinol Metab (1997) 82(11):3864–71. doi: 10.1210/jcem.82.11.4339
36. Goldstein DS, Mezey E, Yamamoto T, Aneman A, Friberg P, Eisenhofer G. Is There a Third Peripheral Catecholaminergic System? Endogenous Dopamine as an Autocrine/Paracrine Substance Derived From Plasma DOPA and Inactivated by Conjugation. Hypertens Res (1995) 18 Suppl 1:S93–9. doi: 10.1291/hypres.18.SupplementI_S93
37. Brown MJ, Allison DJ. Renal Conversion of Plasma DOPA to Urine Dopamine. Br J Clin Pharmacol (1981) 12(2):251–3. doi: 10.1111/j.1365-2125.1981.tb01210.x
38. Wolfovitz E, Grossman E, Folio CJ, Keiser HR, Kopin IJ, Goldstein DS. Derivation of Urinary Dopamine From Plasma Dihydroxyphenylalanine in Humans. Clin Sci (Lond) (1993) 84(5):549–57. doi: 10.1042/cs0840549
39. Baines AD, Craan A, Chan W, Morgunov N. Tubular Secretion and Metabolism of Dopamine, Norepinephrine, Methoxytyramine and Normetanephrine by the Rat Kidney. J Pharmacol Exp Ther (1979) 208(1):144–7.
40. Goldstein DS, Swoboda KJ, Miles JM, Coppack SW, Aneman A, Holmes C, et al. Sources and Physiological Significance of Plasma Dopamine Sulfate. J Clin Endocrinol Metab (1999) 84(7):2523–31. doi: 10.1210/jcem.84.7.5864
41. Eisenhofer G. Free or Total Metanephrines for Diagnosis of Pheochromocytoma: What is the Difference? Clin Chem (2001) 47(6):988–9.
42. Eisenhofer G, Prejbisz A, Peitzsch M, Pamporaki C, Masjkur J, Rogowski-Lehmann N, et al. Biochemical Diagnosis of Chromaffin Cell Tumors in Patients at High and Low Risk of Disease: Plasma Versus Urinary Free or Deconjugated O-Methylated Catecholamine Metabolites. Clin Chem (2018) 64(11):1646–56. doi: 10.1373/clinchem.2018.291369
43. Feldman JM, Blalock JA, Zern RT, Wells SA Jr. The Relationship Between Enzyme Activity and the Catecholamine Content and Secretion of Pheochromocytomas. J Clin Endocrinol Metab (1979) 49(3):445–51.
44. Grouzmann E, Matter M, Bilz S, Herren A, Triponez F, Henzen C, et al. Monoamine Oxidase A Down-Regulation Contributes to High Metanephrine Concentration in Pheochromocytoma. J Clin Endocrinol Metab (2012) 97(8):2773–81. doi: 10.1210/jc.2012-1557
45. Labrosse EH, Karon M. Catechol-O-Methyltransferase Activity in Neuroblastoma Tumour. Nature (1962) 196:1222–1223. doi: 10.1038/1961222a0
46. Goldstein M, Anagoste B, Goldstein MN. Tyramine-H3: Deaminated Metabolites in Neuroblastoma Tumors and in Continuous Cell Line of a Neuroblastoma. Science (1968) 160(3829):767–8. doi: 10.1126/science.160.3829.767
47. Ikeda H, Suzuki N, Takahashi A, Kuroiwa M, Matsuyama S. 3,4-Dihydroxyphenylalanine (DOPA) Metabolism in Screening-Detected and non-Screening-Detected Neuroblastoma. Pediatr Hematol Oncol (1996) 13(1):21–32. doi: 10.3109/08880019609033369
48. LaBrosse EH, Comoy E, Bohuon C, Zucker JM, Schweisguth O. Catecholamine Metabolism in Neuroblastoma. J Natl Cancer Inst (1976) 57(3):633–8. doi: 10.1093/jnci/57.3.633
49. Crout JR, Sjoerdsma A. Turnover and Metabolism of Catecholamines in Patients With Pheochromocytoma. J Clin Invest (1964) 43(1):94–102. doi: 10.1172/JCI104898
50. Eisenhofer G, Goldstein DS, Kopin IJ, Crout JR. Pheochromocytoma: Rediscovery as a Catecholamine-Metabolizing Tumor. Endocr Pathol (2003) 14(4):193–212. doi: 10.1007/s12022-003-0012-4
51. Lenders JWM, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2014) 99(6):1915–1915. doi: 10.1210/jc.2014-1498
52. Sarathi V, Pandit R, Patil VK, Lia AR, Bandgar TR, Shah NS. Performance of Plasma Fractionated Free Metanephrines by Enzyme Immunoassay in the Diagnosis of Pheochromocytoma and Paraganglioma in Children. Endocr Pract (2012) 18(5):694–9. doi: 10.4158/EP12050.OR
53. Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, et al. Biochemical Diagnosis of Pheochromocytoma: Which Test is Best? JAMA (2002) 287(11):1427–34. doi: 10.1001/jama.287.11.1427
54. Schilling FH, Spix C, Berthold F, Erttmann R, Fehse N, Hero B, et al. Neuroblastoma Screening at One Year of Age. N Engl J Med (2002) 346(14):1047–53. doi: 10.1056/NEJMoa012277
55. Overman RE, Kartal TT, Cunningham AJ, Fialkowski EA, Naik-Mathuria BJ, Vasudevan SA, et al. Optimization of Percutaneous Biopsy for Diagnosis and Pretreatment Risk Assessment of Neuroblastoma. Pediatr Blood Cancer (2020) 67(5):e28153. doi: 10.1002/pbc.28153
56. Franscini LC, Vazquez-Montes M, Buclin T, Perera R, Dunand M, Grouzmann E, et al. Pediatric Reference Intervals for Plasma Free and Total Metanephrines Established With a Parametric Approach: Relevance to the Diagnosis of Neuroblastoma. Pediatr Blood Cancer (2015) 62(4):587–93. doi: 10.1002/pbc.25385
57. Barco S, Verly I, Corrias MV, Sorrentino S, Conte M, Tripodi G, et al. Plasma Free Metanephrines for Diagnosis of Neuroblastoma Patients. Clin Biochem (2019) 66:57–62. doi: 10.1016/j.clinbiochem.2019.02.012
58. Lam L, Woollard GA, Teague L, Davidson JS. Clinical Validation of Urine 3-Methoxytyramine as a Biomarker of Neuroblastoma and Comparison With Other Catecholamine-Related Biomarkers. Ann Clin Biochem (2017) 54(2):264–72. doi: 10.1177/0004563216654723
59. Boomsma F, Ausema L, Hakvoort-Cammel FG, Oosterom R, Man in't Veld AJ, Krenning EP, et al. Combined Measurements of Plasma Aromatic L-Amino Acid Decarboxylase and DOPA as Tumour Markers in Diagnosis and Follow-Up of Neuroblastoma. Eur J Cancer Clin Oncol (1989) 25(7):1045–52. doi: 10.1016/0277-5379(89)90386-6
60. Peitzsch M, Mangelis A, Eisenhofer G, Huebner A. Age-Specific Pediatric Reference Intervals for Plasma Free Normetanephrine, Metanephrine, 3-Methoxytyramine and 3-O-Methyldopa: Particular Importance for Early Infancy. Clin Chim Acta (2019) 494:100–5. doi: 10.1016/j.cca.2019.03.1620
61. Cole M, Craft AW, Parker L, Bell S, Seviour JA, McGill AC, et al. Urinary Creatinine Adjusted Reference Ranges for Homovanillic and Vanillylmandelic Acid in Children and Adults. Clin Chim Acta (1995) 236(1):19–32. doi: 10.1016/0009-8981(95)06031-1
62. Pussard E, Neveux M, Guigueno N. Reference Intervals for Urinary Catecholamines and Metabolites From Birth to Adulthood. Clin Biochem (2009) 42(6):536–9. doi: 10.1016/j.clinbiochem.2008.10.022
63. Davidson DF, Hammond PJ, Murphy DL, Carachi R. Age-Related Medical Decision Limits for Urinary Free (Unconjugated) Metadrenalines, Catecholamines and Metabolites in Random Urine Specimens From Children. Ann Clin Biochem (2011) 48:358–6. doi: 10.1258/acb.2011.011023
64. Griffin A, O'Shea P, FitzGerald R, O'Connor G, Tormey W. Establishment of a Paediatric Age-Related Reference Interval for the Measurement of Urinary Total Fractionated Metanephrines. Ann Clin Biochem (2011) 48(Pt 1):41–4. doi: 10.1258/acb.2010.010062
65. Barco S, Gennai I, Reggiardo G, Galleni B, Barbagallo L, Maffia A, et al. Urinary Homovanillic and Vanillylmandelic Acid in the Diagnosis of Neuroblastoma: Report From the Italian Cooperative Group for Neuroblastoma. Clin Biochem (2014) 47(9):848–52. doi: 10.1016/j.clinbiochem.2014.04.015
66. Wang ZM, Gallagher D, Nelson ME, Matthews DE, Heymsfield SB. Total-Body Skeletal Muscle Mass: Evaluation of 24-H Urinary Creatinine Excretion by Computerized Axial Tomography. Am J Clin Nutr (1996) 63(6):863–9. doi: 10.1093/ajcn/63.6.863
67. Modi N, Hutton JL. Urinary Creatinine Excretion and Estimation of Muscle Mass in Infants of 25-34 Weeks Gestation. Acta Paediatr Scand (1990) 79(12):1156–62. doi: 10.1111/j.1651-2227.1990.tb11404.x
68. Al-Dahhan J, Stimmler L, Chantler C, Haycock GB. Urinary Creatinine Excretion in the Newborn. Arch Dis Child (1988) 63(4):398–402. doi: 10.1136/adc.63.4.398
69. Skinner AM, Addison GM, Price DA. Changes in the Urinary Excretion of Creatinine, Albumin and N-Acetyl-Beta-D-Glucosaminidase With Increasing Age and Maturity in Healthy Schoolchildren. Eur J Pediatr (1996) 155(7):596–602. doi: 10.1007/BF01957912
70. Weismann D, Peitzsch M, Raida A, Prejbisz A, Gosk M, Riester A, et al. Measurements of Plasma Metanephrines by Immunoassay vs Liquid Chromatography With Tandem Mass Spectrometry for Diagnosis of Pheochromocytoma. Eur J Endocrinol (2015) 172(3):251–60. doi: 10.1530/EJE-14-0730
71. Peitzsch M, Novos T, Kaden D, Kurlbaum M, van Herwaarden AE, Muller D, et al. Harmonization of LC-MS/MS Measurements of Plasma Free Normetanephrine, Metanephrine, and 3-Methoxytyramine. Clin Chem (2021) 67(8):1098–112. doi: 10.1093/clinchem/hvab060
72. Davies SL, Davison AS. Liquid Chromatography Tandem Mass Spectrometry for Plasma Metadrenalines. Clin Chim Acta (2019) 495:512–21. doi: 10.1016/j.cca.2019.05.024
73. Kline GA, Boyd J, Sadrzadeh HSM, Leung AA. Inpatient Measurements of Urine Metanephrines are Indistinguishable From Pheochromocytoma: Retrospective Cohort Study. Am J Med (2021) 134(8):1039–46.e3. doi: 10.1016/j.amjmed.2021.03.015
74. Peitzsch M, Kaden D, Pamporaki C, Langton K, Constantinescu G, Conrad C, et al. Overnight/first-Morning Urine Free Metanephrines and Methoxytyramine for Diagnosis of Pheochromocytoma and Paraganglioma: Is This an Option? Eur J Endocrinol (2020) 182(5):499–509. doi: 10.1530/EJE-19-1016
75. Eijkelenkamp K, van Geel EH, Kerstens MN, van Faassen M, Kema IP, Links TP, et al. Blood Sampling for Metanephrines Comparing Venipuncture vs. Indwelling Intravenous Cannula in Healthy Subjects. Clin Chem Lab Med (2020) 58(10):1681–6. doi: 10.1515/cclm-2020-0022
76. van Faassen M, Bischoff R, Eijkelenkamp K, de Jong WHA, van der Ley CP, Kema IP. In Matrix Derivatization Combined With LC-MS/MS Results in Ultrasensitive Quantification of Plasma Free Metanephrines and Catecholamines. Anal Chem (2020) 92(13):9072–8. doi: 10.1021/acs.analchem.0c01263
77. de Jong WH, Eisenhofer G, Post WJ, Muskiet FA, de Vries EG, Kema IP. Dietary Influences on Plasma and Urinary Metanephrines: Implications for Diagnosis of Catecholamine-Producing Tumors. J Clin Endocrinol Metab (2009) 94(8):2841–9. doi: 10.1210/jc.2009-0303
78. Joyce CM, Melvin A, O'Shea PM, Costelloe SJ, O'Halloran DJ. Case Report of a Phantom Pheochromocytoma. Biochem Med (Zagreb) (2020) 30(2):21003. doi: 10.11613/BM.2020.021003
79. Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma Methoxytyramine: A Novel Biomarker of Metastatic Pheochromocytoma and Paraganglioma in Relation to Established Risk Factors of Tumour Size, Location and SDHB Mutation Status. Eur J Cancer (2012) 48:1739–49. doi: 10.1016/j.ejca.2011.07.016
80. Eisenhofer G, Deutschbein T, Constantinescu G, Langton K, Pamporaki C, Calsina B, et al. Plasma Metanephrines and Prospective Prediction of Tumor Location, Size and Mutation Type in Patients With Pheochromocytoma and Paraganglioma. Clin Chem Lab Med (2020) 59(2):353–63. doi: 10.1515/cclm-2020-0904
81. Kuhlen M, Pamporaki C, Kunstreich M, Wudy SA, Hartmann A, Peitzsch M, et al. Adrenocortical Tumors and Pheochromocytoma/Paraganglioma Initially Mistaken as Neuroblastoma – Experiences From the GPOH-MET Registry. Front Endocrinol (Lausanne) (2022). doi: 10.3389/fendo.2022.918435
82. McCorkell SJ, Niles NL. Fine-Needle Aspiration of Catecholamine-Producing Adrenal Masses: A Possibly Fatal Mistake. AJR Am J Roentgenol (1985) 145(1):113–4. doi: 10.2214/ajr.145.1.113
83. Quayle FJ, Spitler JA, Pierce RA, Lairmore TC, Moley JF, Brunt LM. Needle Biopsy of Incidentally Discovered Adrenal Masses is Rarely Informative and Potentially Hazardous. Surgery (2007) 142(4):497–502; discussion 502-4. doi: 10.1016/j.surg.2007.07.013
84. Hack HA. The Perioperative Management of Children With Phaeochromocytoma. Paediatr Anaesth (2000) 10(5):463–76. doi: 10.1046/j.1460-9592.2000.00504.x
85. Tatekawa Y, Muraji T, Nishijima E, Yoshida M, Tsugawa C. Composite Pheochromocytoma Associated With Adrenal Neuroblastoma in an Infant: A Case Report. J Pediatr Surg (2006) 41(2):443–5. doi: 10.1016/j.jpedsurg.2005.11.024
86. Comstock JM, Willmore-Payne C, Holden JA, Coffin CM. Composite Pheochromocytoma: A Clinicopathologic and Molecular Comparison With Ordinary Pheochromocytoma and Neuroblastoma. Am J Clin Pathol (2009) 132(1):69–73. doi: 10.1309/AJCPN76VTIGWPOAG
87. Tran L, Fitzpatrick C, Cohn SL, Pytel P. Composite Tumor With Pheochromocytoma and Immature Neuroblastoma: Report of Two Cases With Cytogenetic Analysis and Discussion of Current Terminology. Virchows Arch (2017) 471(4):553–7. doi: 10.1007/s00428-017-2225-9
88. Seabrook AJ, Harris JE, Velosa SB, Kim E, McInerney-Leo AM, Dwight T, et al. Multiple Endocrine Tumors Associated With Germline MAX Mutations: Multiple Endocrine Neoplasia Type 5? J Clin Endocrinol Metab (2021) 106(4):1163–82. doi: 10.1210/clinem/dgaa957
89. Duarte DB, Ferreira L, Santos AP, Costa C, Lima J, Santos C, et al. Case Report: Pheochromocytoma and Synchronous Neuroblastoma in a Family With Hereditary Pheochromocytoma Associated With a MAX Deleterious Variant. Front Endocrinol (Lausanne) (2021) 12:609263. doi: 10.3389/fendo.2021.609263
90. Egler RA, Li Y, Dang TA, Peters TL, Leung E, Huang S, et al. An Integrated Proteomic Approach to Identifying Circulating Biomarkers in High-Risk Neuroblastoma and Their Potential in Relapse Monitoring. Proteomics Clin Appl (2011) 5(9-10):532–41. doi: 10.1002/prca.201000089
91. Van Paemel R, Vandeputte C, Raman L, Van Thorre J, Willems L, Van Dorpe J, et al. The Feasibility of Using Liquid Biopsies as a Complementary Assay for Copy Number Aberration Profiling in Routinely Collected Paediatric Cancer Patient Samples. Eur J Cancer (2022) 160:12–23. doi: 10.1016/j.ejca.2021.09.022
92. Van Paemel R, Vlug R, De Preter K, Van Roy N, Speleman F, Willems L, et al. The Pitfalls and Promise of Liquid Biopsies for Diagnosing and Treating Solid Tumors in Children: A Review. Eur J Pediatr (2020) 179(2):191–202. doi: 10.1007/s00431-019-03545-y
93. Hanafy AK, Mujtaba B, Roman-Colon AM, Elsayes KM, Harrison D, Ramani NS, et al. Imaging Features of Adrenal Gland Masses in the Pediatric Population. Abdom Radiol (NY) (2020) 45(4):964–81. doi: 10.1007/s00261-019-02213-x
94. Kenney PJ, Stanley RJ. Calcified Adrenal Masses. Urol Radiol (1987) 9(1):9–15. doi: 10.1007/BF02932620
95. Chen AM, Trout AT, Towbin AJ. A Review of Neuroblastoma Image-Defined Risk Factors on Magnetic Resonance Imaging. Pediatr Radiol (2018) 48(9):1337–47. doi: 10.1007/s00247-018-4117-9
96. Machens A, Lorenz K, Dralle H. Peak Incidence of Pheochromocytoma and Primary Hyperparathyroidism in Multiple Endocrine Neoplasia 2: Need for Age-Adjusted Biochemical Screening. J Clin Endocrinol Metab (2013) 98(2):E336–45. doi: 10.1210/jc.2012-3192
97. Gruber LM, Erickson D, Babovic-Vuksanovic D, Thompson GB, Young WF Jr., Bancos I. Pheochromocytoma and Paraganglioma in Patients With Neurofibromatosis Type 1. Clin Endocrinol (Oxf) (2017) 86(1):141–9. doi: 10.1111/cen.13163
98. Barontini M, Levin G, Sanso G. Characteristics of Pheochromocytoma in a 4- to 20-Year-Old Population. Ann N Y Acad Sci (2006) 1073:30–7. doi: 10.1196/annals.1353.003
99. Perel Y, Schlumberger M, Marguerite G, Alos N, Revillon Y, Sommelet D, et al. Pheochromocytoma and Paraganglioma in Children: A Report of 24 Cases of the French Society of Pediatric Oncology. Pediatr Hematol Oncol (1997) 14(5):413–22. doi: 10.3109/08880019709028771
100. Hwang N, Chong E, Oh H, Cho HW, Lee JW, Sung KW, et al. Application of an LC-MS/MS Method for the Simultaneous Quantification of Homovanillic Acid and Vanillylmandelic Acid for the Diagnosis and Follow-Up of Neuroblastoma in 357 Patients. Molecules (2021) 26(11):3470. doi: 10.3390/molecules26113470
101. Strenger V, Kerbl R, Dornbusch HJ, Ladenstein R, Ambros PF, Ambros IM, et al. Diagnostic and Prognostic Impact of Urinary Catecholamines in Neuroblastoma Patients. Pediatr Blood Cancer (2007) 48(5):504–9. doi: 10.1002/pbc.20888
102. Monsaingeon M, Perel Y, Simonnet G, Corcuff JB. Comparative Values of Catecholamines and Metabolites for the Diagnosis of Neuroblastoma. Eur J Pediatr (2003) 162(6):397–402. doi: 10.1007/s00431-003-1175-1
103. Candito M, Thyss A, Albertini M, Deville A, Politano S, Mariani R, et al. Methylated Catecholamine Metabolites for Diagnosis of Neuroblastoma. Med Pediatr Oncol (1992) 20(3):215–20. doi: 10.1002/mpo.2950200306
104. Tuchman M, Ramnaraine ML, Woods WG, Krivit W. Three Years of Experience With Random Urinary Homovanillic and Vanillylmandelic Acid Levels in the Diagnosis of Neuroblastoma. Pediatrics (1987) 79(2):203–5.
105. LaBrosse EH, Com-Nougué C, Zucker JM, Comoy E, Bohuon C, Lemerle J, et al. Urinary Excretion of 3-Methoxy-4-Hydroxymandelic Acid and 3-Methoxy-4-Hydroxyphenylacetic Acid by 288 Patients With Neuroblastoma and Related Neural Crest Tumors. Cancer Res (1980) 40(6):1995–2001.
106. Rifai N, Horvath R, Wittwer C, Comoy E, Bohuon C, Lemerle J, et al. Tietz Textbook of Clinical Chemistry and Molecular Diagnostics. (Philadelphia:Elsevier Science Inc,) (2018) 1421.
107. Kerbl R, Urban CE, Ambros PF, Lackner H, Ladenstein R, Spuller E, et al. Screening for neuroblastoma in late infancy by use of EIA (enzyme-linked immunoassay) method: 115000 screened infants in Austria. Eur J Cancer (1996) 32A(13):2298–305. doi: 10.1016/s0959-8049(96)00361-9
108. Tuchman M, Morris CL, Ramnaraine ML, Bowers LD, Krivit W. Value of random urinary homovanillic acid and vanillylmandelic acid levels in the diagnosis and management of patients with neuroblastoma: comparison with 24-hour urine collections. Pediatrics (1985) 75(2):324–8.
109. Gitlow SE, Mendlowitz M, Wilk EK, Wilk S, Wolf RL, Bertani LM. Excretion of catecholamine catabolites by normal children. J Lab Clin Med (1968) 72(4):612–20.
Keywords: pediatric, pheochromocytoma, paraganglioma, neuroblastoma, catecholamines, metanephrines, methoxytyramine, homovanillic acid
Citation: Eisenhofer G, Peitzsch M, Bechmann N and Huebner A (2022) Biochemical Diagnosis of Catecholamine-Producing Tumors of Childhood: Neuroblastoma, Pheochromocytoma and Paraganglioma. Front. Endocrinol. 13:901760. doi: 10.3389/fendo.2022.901760
Received: 22 March 2022; Accepted: 09 June 2022;
Published: 26 July 2022.
Edited by:
Karel Pacak, National Institutes of Health (NIH), United StatesReviewed by:
Madson Almeida, University of São Paulo, BrazilMassimo Mannelli, University of Florence, Italy
Copyright © 2022 Eisenhofer, Peitzsch, Bechmann and Huebner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Graeme Eisenhofer, R3JhZW1lLkVpc2VuaG9mZXJAdW5pa2xpbmlrdW0tZHJlc2Rlbi5kZQ==