 Chenyu Zhu
Chenyu Zhu Shiwei Shen3†
Shiwei Shen3† Mei Huang
Mei Huang Lan Zhang
Lan Zhang Xi Chen
Xi Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 30 June 2022
Sec. Bone Research
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.898634
This article is part of the Research Topic Fat and Skeletal Metastasis View all 11 articles
Bone homeostasis involves bone formation and bone resorption, which are processes that maintain skeletal health. Oxidative stress is an independent risk factor, causing the dysfunction of bone homeostasis including osteoblast-induced osteogenesis and osteoclast-induced osteoclastogenesis, thereby leading to bone-related diseases, especially osteoporosis. Autophagy is the main cellular stress response system for the limination of damaged organelles and proteins, and it plays a critical role in the differentiation, apoptosis, and survival of bone cells, including bone marrow stem cells (BMSCs), osteoblasts, osteoclasts, and osteocytes. High evels of reactive oxygen species (ROS) induced by oxidative stress induce autophagy to protect against cell damage or even apoptosis. Additionally, pathways such as ROS/FOXO3, ROS/AMPK, ROS/Akt/mTOR, and ROS/JNK/c-Jun are involved in the regulation of oxidative stress-induced autophagy in bone cells, including osteoblasts, osteocytes and osteoclasts. This review discusses how autophagy regulates bone formation and bone resorption following oxidative stress and summarizes the potential protective mechanisms exerted by autophagy, thereby providing new insights regarding bone remodeling and potential therapeutic targets for osteoporosis.
Bone is constantly being remodeled to maintain the balance of growth and development of the skeletal system (1). Bone remodeling is essential for the formation and maintenance of bone morphology and the repair of damaged bone (2). Physiological bone remodeling requires a balance between bone formation and bone resorption, while the dynamic balance needs coupling of the activities of different bone cells (e.g., osteoblasts, osteocytes, and osteoclasts) (3). Osteoblasts mainly arise by differentiation of bone marrow mesenchymal stem cells (BMSCs) and play an osteogenic role in the regulation of the synthesis, secretion, and mineralization of the bone matrix (4). At the end stage of bone formation, osteoblasts become encapsulated in the bone matrix and mature into osteocytes, which play a crucial role in bone remodeling (5). Osteoclasts, which are the only bone-resorbing cells in the body, are tissue-specific multinucleated macrophages that arise by the differentiation of monocytes or macrophage precursors on or near the bone surface (6). Bone remodeling consists of four primary stages, including bone resorption, recruitment of osteoblasts and BMSCs, osteoblast differentiation, and completion of bone mineralization (7). Dysfunction of any cell type involved in this process can lead to the failure of bone remodeling followed by the development of bone-related diseases, especially osteoporosis (8).
Oxidative stress plays a pivotal role in the regulation of the balance of bone remodeling processes (9), including effects on bone formation and bone resorption. Reactive oxygen species (ROS) induced by oxidative stress can lead to apoptosis of osteocytes and osteoblasts and inhibit bone mineralization and osteogenesis, which combine with unbalanced osteoclast formation to lead to enhanced bone loss and progression of osteoporosis (10, 11). At physiological levels, ROS can act as signaling molecules involved in cellular processes such as differentiation, proliferation, apoptosis, autophagy, and redox signaling (12). In contrast, excessive ROS levels result in damage to lipids, proteins, and DNA, which can ultimately lead to cell death (13).
Autophagy is an essential metabolic pathway for cell survival in case of nutrient or energy deficiencies, oxidative stress, infections, or hypoxia (14). The cytoplasm or organelles of the cell itself are engulfed into vesicles to form autophagosomes, which are then transported to the lysosome for degradation to remove damaged or aging organelles and to maintain the basal cellular homeostasis (15, 16). In response to oxidative stress, autophagy is regulated by the level of ROS resulting from cellular injury, and it supports cell survival by a cytoprotective mechanism that mitigates the damage resulting from the oxidative stress (17). However, excessive accumulation of ROS can also exacerbate cellular damage by dysregulation of autophagy, leading to mitochondrial dysfunction and increased levels of ROS (18). It appears that the interaction between ROS and autophagy is critical for cellular homeostasis. Therefore, the mode of interaction between autophagy and oxidative stress during bone remodeling warrants further elucidation. Here, we reviewed the mechanism of autophagy in response to oxidative stress during bone remodeling and discussed potential therapeutic targets of the autophagy process for osteoporosis.
Cellular oxidative stress is caused by an imbalance of intracellular redox homeostasis or a relative overload of ROS (19). Mitochondrial are rod-shaped or elongated under normal conditions, whereas under conditions of oxidative stress, the length and density of mitochondria are significantly reduced as they become fragmented, resulting in impaired cellular metabolic function and increased ROS production, and potentially even cell death (20). Oxidative stress is an independent risk factor for postmenopausal, glucocorticoid, and diabetic osteoporosis (20). By impairing bone remodeling as a result of disruption of the coupling of osteoblasts and osteoclasts, oxidative stress-induced ROS may underlie the main cellular mechanism of osteoporosis (21, 22).
At physiological levels, ROS help maintain cellular function, whereas uncontrolled levels of ROS are detrimental (23). As osteoblast differentiation requires energy, BMSCs or preosteoblasts undergo a metabolic transformation whereby mitochondrial respiration and ATP production are increased to ensure an adequate energy supply, which is accompanied by an increase in endogenous ROS (24). Additionally, excessive ROS levels reduce osteogenic differentiation in situations of estrogen deficiency, high glucose, diabetes, inflammation, stress, aging, or other pathophysiological factors, which can decrease metabolic enzyme activity or antioxidant production (25, 26). BMSCs cultured long-term in vitro exhibit decreased antioxidant capacities and elevated ROS levels, leading to reduction or loss of osteogenic differentiation potential (27). Likewise, hydrogen peroxide (H2O2)-induced oxidative stress has been shown to inhibit osteogenic differentiation in rat BMSC as measured by reduction in alkaline phosphatase (ALP) activity and Runx2 and ATF4 expression levels (28) (29). In contrast, reduction in the level of oxidative stress in BMSCs enhanced osteogenic function and restored bone mass and bone microarchitecture in ovariectomized rats (30). In addition, signaling pathways triggered by ROS regulate cell proliferation, growth, differentiation, and even apoptosis, thereby affecting the lifespan of osteoblasts. Mitogen-activated protein kinases (MAPKs) such as c-Jun-N terminal kinase (JNK), extracellular signal-regulated kinase (ERK1/2), and p38 are involved in osteoblasts apoptosis (31–33). High levels of ROS activated the JNK signaling pathway, which increases the transcriptional expression of pro-apoptotic genes such as caspase 3, FASL, and caspase 9 (34). Moreover, ROS induced by H2O2 continuously stimulated the ERK signaling pathway in osteoblasts, which then enhances the expression of Bax and the hyperpolarization of the mitochondrial membrane potential, thereby resulting in cell apoptosis (35).
Oxidative stress and the consequent production of ROS promotes osteoclast differentiation and osteoclastogenesis (36). Receptor activator of nuclear factor-κB ligand (RANKL) stimulation has been shown to increase ROS production in bone marrow mesenchymal stem cells (BMMs) through a tumor necrosis factor receptor-associated factor 6 (TRAF6)/RAC1/nicotinamide adenine dinucleotide phosphate oxidase 1 (Nox1) signaling cascade, resulting in enhanced differentiation of osteoclasts. Conversely, exposure to the antioxidant N-acetylcysteine (NAC) has been shown to inhibit the response of BMMs to RANKL, involving ROS production, activation of the MAPK pathway, and osteoclastogenesis (37). Likewise, in the glucose-induced diabetic osteoporosis model in rats, increased ROS production in osteoclasts and subsequently enhanced expression of proteins related to MAPKs [phosphorylated (p)-ERK, p-JNK, and p-p38], NF-κB (NF-κB, p-IκB, and IKK), and NACHT-LRR-PYD domains-containing protein 3 (NLRP3)-related protein expression, which promotes osteoclast differentiation and bone resorption, were observed (38).
ROS production not only directly enhances osteoclast differentiation but also interacts with osteoblasts to regulate the formation and differentiation of osteoclasts. OPG/RANK/RANKL form a molecular triad that links osteoblasts and osteoclasts and thus plays a significant role in osteoclastogenesis (39, 40). High levels of H2O2-induced ROS in osteoblasts (including osteoblast-like MG63 cells and primary mice osteoblasts) and BMSCs have been shown to stimulate the expression of RANKL mRNA and protein through ERKs and the PKA-CREB pathway (41). Co-culture of osteoblasts with osteoclast precursor cells has revealed that ethanol (EtOH)-induced RANKL expression depends on intracellular ROS stimulation by NADPH oxidase activity in osteoblast, which promotes osteoclast differentiation (42). These results demonstrate that ROS can promote RANKL secretion by osteoblasts, thereby regulating osteoclast differentiation, thus providing novel insights into the role of ROS production in the regulation of osteoblast-osteoclast communication.
Taken together, these findings suggest that ROS can inhibit osteoblast differentiation and hence also bone formation, in addition to promoting osteoclast differentiation and osteoclastogenesis. The effect of oxidative stress on different cell types and their communication are thought to play an essential role in the development of osteoporosis.
Autophagy plays a significant role in bone formation, including differentiation of BMSCs into osteoblasts to osteocytes, osteogenesis, differentiation, and the formation of bone matrix. BMSC differentiation requires energy, while the products of autophagosomal degradation can be oxidized by mitochondria to provide a suitable energy supply for their differentiation (43). Optimal differentiation of MSCs into osteoblasts involves an early stage of AMP-activated protein kinase (AMPK)/mTOR signaling axis-mediated autophagy as well as a later stage of Akt/mTOR signaling axis activation (44). Conversely, reduction of the level of autophagy directly inhibits the function of endogenous BMSCs and further promotes the development of osteoporosis (45). When MSCs are fully differentiated into osteoblasts, basal autophagy is completely inhibited, but this does not indicate that the differentiated cells are no longer capable of autophagy (46).
Mesenchymal-derived osteoblasts, which are recognized as specialized mineralizing cells in bone formation, are known to play a critical role in the synthesis, secretion, and mineralization of the bone matrix (47, 48). A previous study in vitro found that autophagy defects induced by ablation of FIP200 in osteoblasts led to the dysfunction of osteoblasts differentiation (49). Furthermore, downregulation of the expression of autophagy markers, such as LC3-II and ATG7, has been shown to result in the inhibition of osteoblast differentiation (50, 51). The early stage of osteoblast differentiation requires the activation of AMPK, and the terminal stage is dependent on downregulation of AMPK (52, 53), which is mediated by stimulation of AKT and mTOR (54), and then activates cell autophagy.
Additionally, autophagy is also directly involved in the mineralization process of osteoblasts. Conditional knockdown of ATG7 in osteoblasts led to a reduction of mineralization capacity in vivo (55), and knockout of the autophagy-related genes Fip200 or Atg5 in Osterix-Cre transgenic mice also resulted in impaired mineralization and reduced bone mass in mice (56). These results indicated that autophagy is required in the mineralization process of osteoblasts, which can be attributed to autophagic vesicles acting as carriers for the secretion of apatite crystals to the extracellular matrix (55).
Osteoclasts, which differentiate from hematopoietic mononuclear stem cells in the bone marrow, are critical at the beginning of bone remodeling by bone resorption via following differentiation into multinucleated osteoclasts which then migrate to the surface of the bone (57, 58). HIF1-α, which is produced in response to hypoxic stress, has been reported to upregulated BNIP3, which increases the level of Beclin-1 and then activates autophagic flux accompanied by the autophagy-related genes ATGs, thereby leading to increased osteoclastogenesis by upregulation of CTSK, NFATC1, and MMP9 (59). Another study showed that a microgravity environment (rotary cell culture system) increased autophagy in osteoclasts, which then stimulated osteoclast differentiation and osteoclastogenesis (60). Moreover, the level of autophagy initiation protein Beclin-1 has been reported to increase during osteoclast differentiation. Ctsk-cell expression conditional Beclin-1 deficient mice exhibited an increase in the thickness of cortical bone via attenuated osteoclast function, while overexpression of Beclin-1 in osteoclast precursors has been reported to enhance autophagy-induced osteoclastogenesis in vitro and increase bone resorption (61). Mechanistically, it was concluded that TRAF6-mediated K63-linked ubiquitination at Beclin1-K117 is needed for RANKL-induced osteoclast differentiation (61, 62). These findings support the notion that autophagy in osteoclasts is susceptible to environmental factors such as hypoxic stress and microgravity, which results in further regulation of the differentiation of osteoclasts and osteoclastogenesis.
In addition to its role in osteoclast differentiation, autophagy has also been demonstrated to be essential in osteoclast function. Terminally differentiated osteoclasts are tightly attached to the bone surface by pedicles. F-actin, talin, vinculin, and α-actinin are the key anchor targets for osteoclast attachment, and lysosomes then migrate to the bone surface and resorbing bone (57). The autophagy-related proteins ATG4B, ATG5, ATG7, and LC3 have all been shown to play crucial roles in promoting bone resorption activity. For example, knockdown of ATG5 and ATG7 in osteoclasts has been shown to significantly reduce the depth and volume of bone traps and reduce the ability to deliver lysosomes to the fold membrane boundary, although this does not appear to affect osteoclast formation. The lysosomal secretory function requires ATG5-ATG12 coupling to facilitate LC3 binding to phosphatidylethanolamine. ATG5 deficiency inhibits LC3II production as well as CTSK localization (63, 64).
Oxidative stress is involved in the development of osteoporosis and aging, as evidenced by both ovariectomy and age-increased oxidative stress and reduction of the antioxidant system in rat femurs, which promotes the development of osteoporosis (65). As mentioned above, oxidative stress disrupts the balance of bone formation and resorption by inhibiting osteoblast function and promoting osteoclast activity. In response to oxidative stress-induced autophagy, ROS may act as an antioxidant during dysregulation of bone remodeling to protect from bone loss and osteoporosis. In the following, we describe the regulation of autophagy in response to oxidative stress in osteoblasts, osteocytes, and osteoclasts (Figure1).
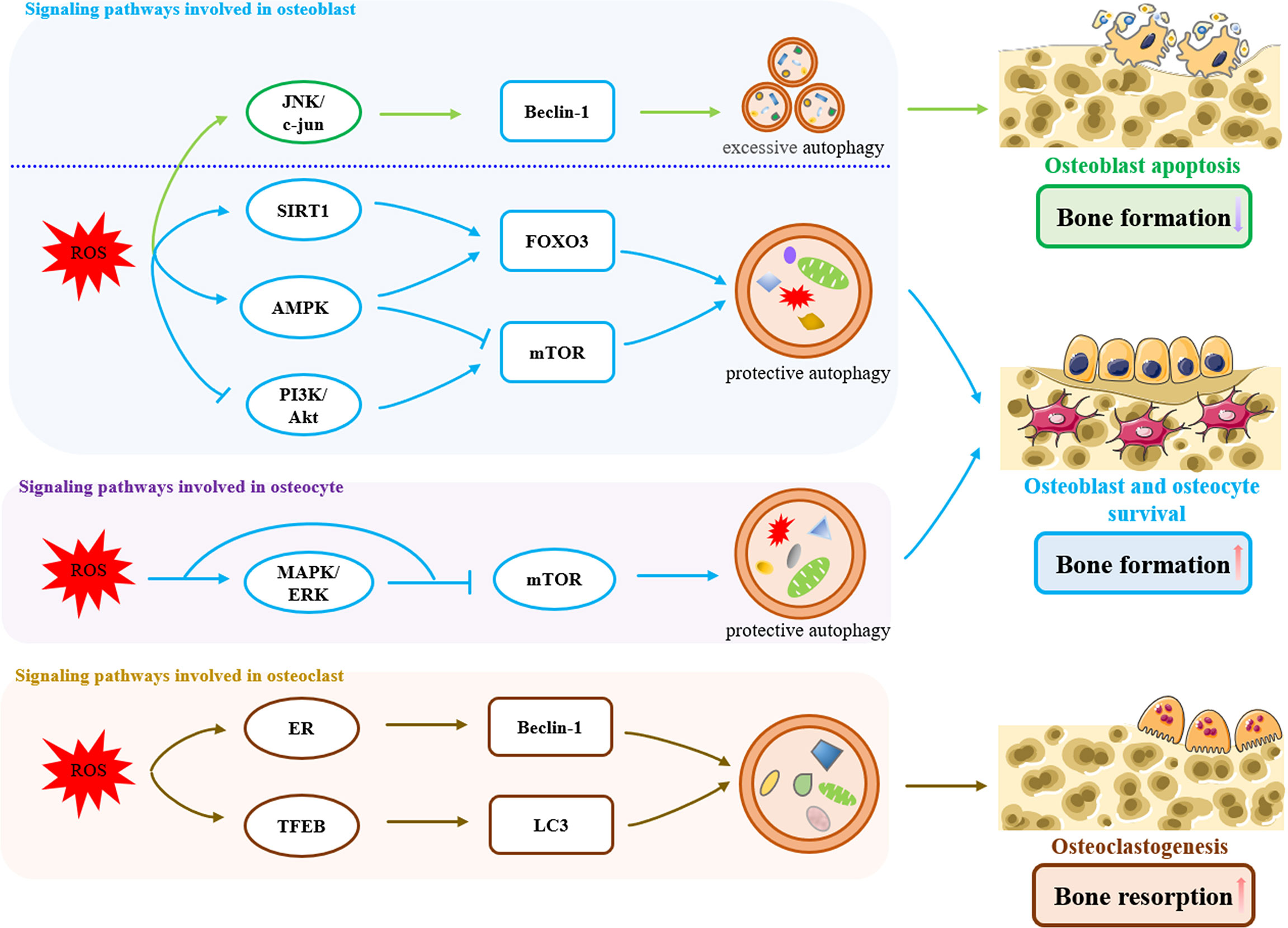
Figure 1 Signaling pathways involved in oxidative stress-induced autophagy in bone remodeling. In osteoblasts, ROS induced an excessive increase in Beclin-1 levels by activation of the JNK/c-jun pathway, which triggered excessive autophagy, exacerbated osteoblasts apoptosis, and reduced bone formation. On the other hand, oxidative stress activates protective autophagy through ROS/SIRT1/FOXO3, ROS/AMPK/FOXO3, ROS/AMPK/mTOR and ROS/PI3K/Akt/mTOR pathways to remove excessive ROS within a certain range, promoting the survival of osteoblasts and increasing bone formation. Likewise in osteoblasts, oxidative stress-induced protective autophagy is also present in osteocytes, which is achieved by ROS/MAPK/ERK/mTOR pathway. In osteoclasts, Oxidative stress-induced autophagy promotes osteoclastogenesis and bone resorption through the ROS/ER and ROS/TFEB pathways.
A high glucose (HG) environment, glucocorticoids or estrogen deficiency cause a pathological increase in ROS levels, thereby impairing the osteoblast function (66–68). In response to ROS, autophagy is activated and promotes osteoblast function as a negative feedback loop. Alberto et al. found that HG increased protein oxidation and the ROS levels, thereby activating autophagy in MC3T3-E1 cells, which reduced damage from HG and protected the cells, whereas inhibition of autophagy increased cell apoptosis (66). In addition, increased ROS levels caused the LC3II/LC3I ratios to increase and p62/SQSTMI to decrease, as observed in advanced glycation end products (AGE)-treated osteoblasts. Furthermore, the autophagy agonist rapamycin (RA) attenuated AGE-induced apoptosis, while the autophagy inhibitor 3-methyladenine (3-MA) increased AGE-induced apoptosis, indicating that autophagy plays a critical role in protecting osteoblasts from AGE-induced apoptosis (69). Likewise, other studies have also demonstrated that osteoblast activity is regulated by glucocorticoids in a dose-dependent manner. Low doses of dexamethasone promoted osteoblast autophagy, protected from damage by ROS, and attenuated apoptosis in osteoblasts. However, as the dose and the duration of the dexamethasone treatment increased, the antioxidant effects of autophagy were overwhelmed, which then lead to apoptosis (67). These results reveal that the protective effect of ROS-induced autophagy is limited and dependent on the dose of ROS level and the duration of stimulation.
Osteoblasts experiencing stress from aging or ovariectomy in mice have been reported to have increased levels of ROS and swollen mitochondria, followed by a 95% decrease in LC3-II levels. Further research has indicated that osteoblast conditional autophagy deficiency in mice results in enhanced aging and estrogen deficiency-related bone loss (68). Conversely, estradiol administration has been shown to increase ULK1, Beclin1, and LC3II protein levels in osteoblasts, decrease oxidative stress levels, and significantly reduced the expression of apoptotic biomarkers through the ER-ERK-mTOR pathway (70). Thus, autophagy can be an important potential target for protection against damage from oxidative stress or ROS, but how autophagy responds to ROS signaling needs to be further explored. Following is a review of ROS/FOXO3, ROS/AMPK, ROS/Akt/mTOR, and ROS/JNK/c-Jun pathways that are involved in the autophagic response to oxidative stress in osteoblasts.
Forkhead box O3(FOXO3) protein is a member of the FOXO family, which can be activated by catalase, SOD2, and glutathione peroxide in antioxidant reactions (71). In response to oxidative stress, MAPK8, MAPK14/p38α, and serine/threonine-protein kinase 4 (STK4)/MST1 phosphorylate FOXOs, causing their nuclear translocation as well as transcriptional activation of target genes such as manganese superoxide dismutase (MnSOD) and catalase (72, 73). During BMSC differentiation into osteoblasts, and then osteoblasts differentiation into osteocytes, the increasing level of ROS activates FOXO3 serine 294 phosphorylation, and FOXO3-induced autophagy then downregulates the increased ROS levels as a negative feedback loop to ensure proper differentiation (74). In addition, inhibition of MAPK11/12/14 kinase can reduce the nuclear translocation of FOXO3 by MSC exposure to oxidative stress, while LC3B and GABARAPL1 are significantly upregulated upon FOXO activation, suggesting that MAPK11/12/14 participate in the activation of FOXO3 by ROS and then activate autophagy. PARK2, a ubiquitin ligase that is indispensable for inducing mitochondrial autophagy, was also significantly increased when FOXO3 was induced by ROS, which is an important process for the clearance of ROS, while the process was impaired when FOXO3 was knocked down (74).
SIRT1 is another key factor involved in ROS-mediated FOXO3 activation. Gu et al. found that ROS/SIRT1/FOXO3 may be involved in the survival of the damage from fluoride in MC3T3-E1 osteoblasts. ROS-mediated activation of SIRT1 has been shown to increase the level of FOXO3 deacetylation and to promote the expression of its substrate Bnip3, which promotes the upregulation of autophagy levels and reduces fluoride-induced osteoblast apoptosis. Conversely, inhibition of SIRT1 expression has been shown to impair FOXO3-induced autophagy (75).
AMPK is a heterotrimeric complex comprising a catalytic subunit (α-subunit) and two regulatory subunits (β- and γ-subunits) (76). In addition to its role in energy metabolism, AMPK also acts as an oxidative stress sensor to regulate cell survival under stressful conditions (77). ROS activates AMPK by phosphorylating the AMPK alpha1 threonine 172 (78), and activated AMPK directly phosphorylates the mTORC1 subunit Raptor, which can then suppress the inhibitory effect of mTORC1 on ULK1 to promote autophagy. Moreover, AMPK also directly phosphorylates Ser 317 and Ser 777 of the UKL1 complex to activate autophagy (79). However, inhibition of autophagy enhances ROS-induced cell apoptosis. H2O2 can induce phosphorylation of ULK1 and upregulation of LC3B-II via activation of AMPK, while treatment with the autophagy inhibitors 3-MA and bafilomycin A1 increases H2O2-induced cell death. Furthermore, AMPKα knockdown has been reported to further inhibit ULK1 phosphorylation and LC3B-II upregulation, indicating that ROS/autophagy activation in osteoblasts requires AMPK, which can act as a negative feedback loop in the regulation of ROS levels when exposed to oxidative stress (80). Consistent with these results, the AMPK activators GSK621 or A-769662 enhance the protective autophagic response as evidenced by phosphorylation of ULK1 on Ser-317, upregulation of ATG5 and Beclin-1, and downregulation of p62 (81, 82) in case of H2O2-induced oxidative stress in osteoblasts.
The PI3K/Akt/mTOR pathway plays an essential role in stress responses, autophagy, cell survival, and apoptosis (83). The PI3K/Akt signaling axis activates mTOR by phosphorylation of p70S6K and 4EBP1, thereby inhibiting autophagy (84, 85). ROS initially regulate PI3K/Akt, and the PI3K/Akt pathway in turn regulates ROS homeostasis to promote cell survival (86). It has been reported that ROS levels are significantly elevated under high glucose conditions, and p-Akt and p-mTOR protein expression was significantly downregulated in MC3T3-E1 cells, while the antioxidant NAC reversed their expression and reduced osteoblasts apoptosis, suggesting that high levels of ROS promoted the protective autophagy by inhibition of the Akt/mTOR axis (87). Further study has revealed that the inactivation of phosphatase and tensin homologs (PTEN) when ROS activates PI3K may be the main reason for ROS inhibition of the Akt/mTOR signaling pathway, as PTEN inhibits the synthesis of PIP3 and thus activation of Akt signaling (88). The Chinese traditional medicine monotropein has been reported to protect against the damage from H2O2-induced oxidative stress in osteoblasts. Monotropein was found to decrease phosphorylation of Akt, mTOR, p70S6K, and 4EBP1, as well as upregulate Beclin-1 expression and LC3-II/LC3-I ratios, which then activated autophagy to increase osteoblastic bone formation (89). Monotropein, hence, appears to have potential for treatment or prevention of aging or estrogen-deficiency osteoporosis.
Fluoride-mediated ROS triggers oxidative cell damage and apoptosis through N-terminal kinase (JNK)/c-Jun signaling. In contrast, the ROS-induced JNK/c-Jun pathway activates SIRT1 and triggers autophagy as an adaptive reaction to protect cells from fluoride damage (90). However, it has also been shown that the ROS-autophagy process mediated by the JNK pathway enhanced osteoblast apoptosis. Glucocorticoids upregulated JNK and c-Jun phosphorylation in osteoblasts, thereby activating JNK/c-Jun signaling pathway-induced autophagy, which then leads to increased apoptosis (91). ROS inhibitors have been reported to downregulate the JNK/c-Jun signaling pathway, but JNK inhibitors did not reduce ROS, indicating that ROS is an upstream signal for JNK, while autophagy and apoptosis occur in response to ROS/JNK/c-Jun signaling (91). Further studies have shown that JNK causes the degradation of the Beclin-1/Bcl-2 complex by phosphorylating Bcl2, and Beclin-1 excessively stimulates the onset of autophagy (92, 93), and a low level of Beclin-1 promotes autophagy for cell survival, while a high level of Beclin-1 induces autophagic cell death (94, 95). These findings indicated that JNK may be a potential target involved in the balance between oxidative stress-induced autophagy and apoptosis.
In the above studies, autophagy induced by oxidative stress may be a double-edged sword for osteoblasts. On the one hand, in response to aberrant ROS signaling, the MAPK/FOXO3, SIRT1/FOXO3, and AMPK pathways are activated, and the Akt/mTOR pathway is inhibited, leading to activation of autophagy and the scavenging of excessive ROS within a certain range, thereby promoting osteoblast survival and increasing bone formation. On the other hand, when ROS levels are so high as to exceed the clearance effect of protective autophagy, they can activate the JNK pathway and subsequently induce excessive autophagy, thereby enhancing apoptosis of osteoblasts and thus reducing bone formation. A large cascade of interdependent responses between autophagy and JNK-mediated apoptosis has been documented, but how the JNK pathway regulates the balance of autophagy and apoptosis in osteoblasts in response to ROS signaling remains to be fully elucidated.
As in osteoblasts, oxidative stress-induced autophagy in osteocytes is also a protective response. Decreased estrogen levels are a prominent cause of postmenopausal osteoporosis. Yang et al. established an ovariectomized rat model that mimics the decrease in estrogen levels in vivo. They found a significant decrease in bone mineral density and bone mass in ovariectomized rats, accompanied by a decrease in antioxidant parameters such as the total antioxidant capacity, superoxide dismutase activity, catalase activity, and an increase in the expression level of osteocyte autophagy-related factors such as ATG5, LC3, and Beclin-1. In contrast, estrogen treatment prevented the decrease in bone mass and the abnormal increase in oxidative stress levels, and it restored autophagy to normal levels (96). These data suggest that estrogen deficiency can lead to an increase in oxidative stress levels in vivo, which in turn triggers its downstream protective autophagic response, but ultimately leads to the development of osteoporosis due to its limited protective effect. Further exploration of the negative feedback protection mechanism of autophagy in osteocytes has revealed that ROS/MAPK/ERK and ROS/mTOR/ULK1 signaling axes appear to play important roles (97, 98).
ERK is one of the classical signal transduction components of the MAPK family, and it can directly induce autophagy by upregulation of the expression of autophagy-related proteins such as LC3 and p62 (99). Rekha et al. found that treatment with low doses of glucocorticoids increased oxidative stress levels and basal autophagy levels in osteocytes without increasing osteocyte apoptosis, whereas high doses of glucocorticoids enhanced osteocyte apoptosis. Further studies have revealed that glucocorticoid treatment significantly increases MAPK and ERK phosphorylation in osteocytes, while the ERK-specific inhibitor U0126 completely abolished glucocorticoid-induced elevated LC3 expression. These data suggest that low-dose glucocorticoid-induced oxidative stress activates the MAPK/ERK signaling pathway, which in turn enhances autophagy levels and protects osteocytes from oxidative stress damage, whereas the protective effect of autophagy induced by high levels of glucocorticoids has a range and does not respond to abnormally elevated ROS levels, thus manifesting as excessive apoptosis of osteocytes (97).
ULK1 is a key initiator protein in the induction of autophagy, and inhibition of mTOR activity can enhance autophagy levels by binding to and phosphorylating the serine site of ULK1 (100). Bisphenol A (BPA) is an environmental endocrine disruptor that can perturb bone metabolism and bone homeostasis (101). BPA has been reported to increase malondialdehyde and ROS levels in osteocytes and decrease the expression of the antioxidant enzymes nuclear factor E2-related factor 2 (Nrf2) and heme oxygenase-1 (HO-1), leading to oxidative stress. BPA has also been shown to significantly inhibited mTOR phosphorylation and promoted ULK1 phosphorylation, there inducing activation of autophagy. In contrast, treatment with the mTOR activator MHY1485 (MHY) or the ULK1 inhibitor SBI-0206965 (SBI) inhibited BPA-induced autophagy and enhanced apoptosis in osteocytes, but did not reduce ROS levels. Furthermore, NAC treatment attenuated the level of ROS-mediated autophagy. This suggests that the high level of ROS caused by BPA acts upstream of the mTOR/ULK1 signaling axis and that the autophagic response that it triggers is protective against the cytotoxic effects of BPA (98).
ROS acts as intracellular signaling mediators in osteoclast differentiation. RANKL stimulation of osteoclast precursor cells increases intracellular ROS production, and reduction of RANKL-induced ROS by NAC treatment down-regulates of MAPK, ERK, and other signaling pathways, thereby leading to attenuated osteoclast precursor differentiation (102, 103). Unlike autophagy acting as the cytoprotective role in osteoblasts, ROS-induced autophagy even promotes osteoclast differentiation and formation. High levels of ROS induced by glucocorticoids or inflammatory conditions act as a catalyst for osteoclastogenesis. Sul et al. found that lipopolysaccharide promoted autophagy and led to osteoclastogenesis by stimulating ROS production, while reduction of ROS by siNOX1 and siNOX2 dramatically diminished LC3II levels accumulation as well as the expression of osteoclast-specific genes expression (104). Interestingly, osteoclastogenesis was upregulated by glucocorticoids at high doses, but low doses had no effect (105). The accumulation of intracellular ROS in the presence of high glucocorticoid levels was synchronized with the upregulation of autophagic activity, which was prevented by the ROS scavenger NAC. While 3-MA administration blocked the promotion of osteoclast formation by glucocorticoids, it failed to reduce intracellular ROS accumulation. We further explored how ROS mediates autophagy to enhance osteoclastogenesis and we found that the ROS/ER and ROS/TFEB pathways may be involved in this process.
Endoplasmic reticulum stress (ER) is induced by the accumulation of misfolded proteins leading to an unfolded protein response. ROS can cause aggregation and misfolding of proteins (106). Activation of ER regulates autophagy, which in turn regulates cell survival and death (107). MCP-1 is an important protein in the differentiation of monocytes into osteoclast precursors, and p47PHOX expression and its membrane translocation expressions induced by MCP-1 have been reported to promote ROS production, which induced ER and subsequently promoted upregulation of the autophagy markers Beclin-1 and LC3II as well as expression of osteoclast-associated markers such as TRAP and Ctsk. 3-MA treatment or knockdown of Beclin-1 significantly suppressed TRAP and Ctsk expression without affecting ER or its upstream ROS levels (108). These results indicate that osteoclast precursor cell differentiation is mediated by ROS production, which leads to ER stress, thereby inducing autophagy and ultimately promoting osteoclastogenesis.
TFEB is a key transcription factor that controls the autophagy-lysosome system. Stress conditions such as lysosomal dysfunction or starvation cause nuclear translocation of TFEB and promote transcription of its target genes (109). ROS can directly oxidize TFEB, reduce its association with RRAG GTPase on lysosomes, and rapidly induce nuclear localization (110). Sul et al. found that high levels of ROS induced by 7-ketocholesterol (7-KC) significantly increased the nuclear translocation of TFEB and upregulated the lipidated form of LC3II in osteoclasts as well as the number and the activity of osteoclasts. In contrast, TFEB knockdown significantly downregulated autophagy levels and osteoclastogenesis. This suggests that 7-KC-mediated ROS induced oxidation of TFEB and promoted its nuclear translocation to enhance autophagy, leading to increased osteoclast numbers and activity (111).
We have provided an overview of the function of oxidative stress-mediated autophagy in bone remodeling. Oxidative stress-induced ROS impair bone formation by osteoblasts and osteocytes and promote bone resorption by osteoclasts, thereby disrupting the homeostasis of bone and enhancing the progression of osteoporosis. In addition, ROS also activates autophagy and then regulates osteoblasts and osteocytes in a negative feedback loop. However, ROS-mediated autophagy enhances osteoclast differentiation, which can overwhelm the protective effect in osteoblasts and osteocytes, as bone tissue exposed to oxidative stress leads to the development of osteoporosis. Therefore, further studies of the regulatory mechanisms of autophagy in redox signaling during pathological bone remodeling are needed. Furthermore, it may be possible to exploit the potential targets of autophagy for protective or therapeutic strategies against osteoporosis.
XC and LZ designed this review and supervised the whole program; CZ, SS and SZ searched the articles and offered advice; MH prepared the figure; CZ, SS and XC wrote the paper. All the authors reviewed and approved the manuscript.
The work is supported by funding from Wenzhou basic scientific research project (Grant No. 2019Y0848) and the national undergraduate innovation and entrepreneurship training program (Grant No.202110343040).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Feng X, McDonald JM. Disorders of Bone Remodeling. Annu Rev Pathol (2011) 6:121–45. doi: 10.1146/annurev-pathol-011110-130203
2. Martin T, Gooi JH, Sims NA. Molecular Mechanisms in Coupling of Bone Formation to Resorption. Crit Rev Eukaryot Gene Expr (2009) 19(1):73–88. doi: 10.1615/critreveukargeneexpr.v19.i1.40
3. Wang L, You X, Lotinun S, Zhang L, Wu N, Zou W. Mechanical Sensing Protein Piezo1 Regulates Bone Homeostasis Via Osteoblast-Osteoclast Crosstalk. Nat Commun (2020) 11(1):282. doi: 10.1038/s41467-019-14146-6
4. Dirckx N, Moorer MC, Clemens TL, Riddle RC. The Role of Osteoblasts in Energy Homeostasis. Nat Rev Endocrinol (2019) 15(11):651–65. doi: 10.1038/s41574-019-0246-y
5. Robling AG, Bonewald LF. The Osteocyte: New Insights. Annu Rev Physiol (2020) 82:485–506. doi: 10.1146/annurev-physiol-021119-034332
6. Feng X, Teitelbaum SL. Osteoclasts: New Insights. Bone Res (2013) 1(1):11–26. doi: 10.4248/BR201301003
7. Martin TJ, Seeman E. New Mechanisms and Targets in the Treatment of Bone Fragility. Clin Sci (Lond) (2007) 112(2):77–91. doi: 10.1042/CS20060046
8. Sućur A, Katavić V, Kelava T, Jajić Z, Kovačić N, Grčević D. Induction of Osteoclast Progenitors in Inflammatory Conditions: Key to Bone Destruction in Arthritis. Int Orthop (2014) 38(9):1893–903. doi: 10.1007/s00264-014-2386-y
9. Wauquier F, Leotoing L, Coxam V, Guicheux J, Wittrant Y. Oxidative Stress in Bone Remodelling and Disease. Trends Mol Med (2009) 15(10):468–77. doi: 10.1016/j.molmed.2009.08.004
10. Manolagas SC. From Estrogen-Centric to Aging and Oxidative Stress: A Revised Perspective of the Pathogenesis of Osteoporosis. Endocr Rev (2010) 31(3):266–300. doi: 10.1210/er.2009-0024
11. Baek KH, Oh KW, Lee WY, Lee SS, Kim MK, Kwon HS, et al. Association of Oxidative Stress With Postmenopausal Osteoporosis and the Effects of Hydrogen Peroxide On Osteoclast Formation in Human Bone Marrow Cell Cultures. Calcif Tissue Int (2010) 87(3):226–35. doi: 10.1007/s00223-010-9393-9
12. Finkel T. Signal Transduction by Reactive Oxygen Species. J Cell Biol (2011) 194(1):7–15. doi: 10.1083/jcb.201102095
13. Finkel T, Holbrook NJ. Oxidants, Oxidative Stress and the Biology of Ageing. Nature (2000) 408(6809):239–47. doi: 10.1038/35041687
14. Dikic I, Elazar Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat Rev Mol Cell Bio (2018) 19(6):349–64. doi: 10.1038/s41580-018-0003-4
15. Kim KH, Lee MS. Autophagy–A Key Player in Cellular and Body Metabolism. Nat Rev Endocrinol (2014) 10(6):322–37. doi: 10.1038/nrendo.2014.35
16. Klionsky DJ, Emr SD. Autophagy as a Regulated Pathway of Cellular Degradation. Science (2000) 290(5497):1717–21. doi: 10.1126/science.290.5497.1717
17. Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and Disease: Always Two Sides to a Problem. J Pathol (2012) 226(2):255–73. doi: 10.1002/path.3025
18. Yun HR, Jo YH, Kim J, Shin Y, Kim SS, Choi TG. Roles of Autophagy in Oxidative Stress. Int J Mol Sci (2020) 21(9):3289. doi: 10.3390/ijms21093289
19. Yoo BH, Wu X, Derouet M, Haniff M, Eskelinen EL, Rosen K. Hypoxia-Induced Downregulation of Autophagy Mediator Beclin 1 Reduces the Susceptibility of Malignant Intestinal Epithelial Cells to Hypoxia-Dependent Apoptosis. Autophagy (2009) 5(8):1166–79. doi: 10.4161/auto.5.8.10167
20. Li Q, Gao Z, Chen Y, Guan MX. The Role of Mitochondria in Osteogenic, Adipogenic and Chondrogenic Differentiation of Mesenchymal Stem Cells. Protein Cell (2017) 8(6):439–45. doi: 10.1007/s13238-017-0385-7
21. Cervellati C, Bonaccorsi G, Cremonini E, Bergamini CM, Patella A, Castaldini C, et al. Bone Mass Density Selectively Correlates With Serum Markers of Oxidative Damage in Post-Menopausal Women. Clin Chem Lab Med (2013) 51(2):333–8. doi: 10.1515/cclm-2012-0095
22. Fatokun AA, Stone TW, Smith RA. Hydrogen Peroxide-Induced Oxidative Stress in Mc3T3-E1 Cells: The Effects of Glutamate and Protection by Purines. Bone (2006) 39(3):542–51. doi: 10.1016/j.bone.2006.02.062
23. Sart S, Song L, Li Y. Controlling Redox Status for Stem Cell Survival, Expansion, and Differentiation. Oxid Med Cell Longev (2015) 2015:105135. doi: 10.1155/2015/105135
24. Atashi F, Modarressi A, Pepper MS. The Role of Reactive Oxygen Species in Mesenchymal Stem Cell Adipogenic and Osteogenic Differentiation: A Review. Stem Cells Dev (2015) 24(10):1150–63. doi: 10.1089/scd.2014.0484
25. Wang L, Zhao X, Wei BY, Liu Y, Ma XY, Wang J, et al. Insulin Improves Osteogenesis of Titanium Implants Under Diabetic Conditions by Inhibiting Reactive Oxygen Species Overproduction Via the PI3K-Akt Pathway. Biochimie (2015) 108:85–93. doi: 10.1016/j.biochi.2014.10.004
26. Shi C, Wu J, Yan Q, Wang R, Miao D. Bone Marrow Ablation Demonstrates That Estrogen Plays an Important Role in Osteogenesis and Bone Turnover Via an Antioxidative Mechanism. Bone (2015) 79:94–104. doi: 10.1016/j.bone.2015.05.034
27. Geissler S, Textor M, Kühnisch J, Könnig D, Klein O, Ode A, et al. Functional Comparison of Chronological and in Vitro Aging: Differential Role of the Cytoskeleton and Mitochondria in Mesenchymal Stromal Cells. PLoS One (2012) 7(12):e52700. doi: 10.1371/journal.pone.0052700
28. Chen T, Wang H, Jiang C, Lu Y. Pkd1 Alleviates Oxidative Stress-Inhibited Osteogenesis of Rat Bone Marrow-Derived Mesenchymal Stem Cells Through Taz Activation. J Cell Biochem (2021) 122(11):1715–25. doi: 10.1002/jcb.30124
29. Tan J, Xu X, Tong Z, Lin J, Yu Q, Lin Y, et al. Decreased Osteogenesis of Adult Mesenchymal Stem Cells by Reactive Oxygen Species Under Cyclic Stretch: A Possible Mechanism of Age Related Osteoporosis. Bone Res (2015) 3:15003. doi: 10.1038/boneres.2015.3
30. Chen L, Shi X, Xie J, Weng SJ, Xie ZJ, Tang JH, et al. Apelin-13 Induces Mitophagy in Bone Marrow Mesenchymal Stem Cells to Suppress Intracellular Oxidative Stress and Ameliorate Osteoporosis by Activation of Ampk Signaling Pathway. Free Radic Biol Med (2021) 163:356–68. doi: 10.1016/j.freeradbiomed.2020.12.235
31. Fontani F, Marcucci G, Iantomasi T, Brandi ML, Vincenzini MT. Glutathione, N-Acetylcysteine and Lipoic Acid Down-Regulate Starvation-Induced Apoptosis, Rankl/Opg Ratio and Sclerostin in Osteocytes: Involvement of Jnk and Erk1/2 Signalling. Calcif Tissue Int (2015) 96(4):335–46. doi: 10.1007/s00223-015-9961-0
32. Plotkin LI, Aguirre JI, Kousteni S, Manolagas SC, Bellido T. Bisphosphonates and Estrogens Inhibit Osteocyte Apoptosis Via Distinct Molecular Mechanisms Downstream of Extracellular Signal-Regulated Kinase Activation. J Biol Chem (2005) 280(8):7317–25. doi: 10.1074/jbc.M412817200
33. Marathe N, Rangaswami H, Zhuang S, Boss GR, Pilz RB. Pro-Survival Effects of 17β-Estradiol On Osteocytes Are Mediated by Nitric Oxide/Cgmp Via Differential Actions of Cgmp-Dependent Protein Kinases I and Ii. J Biol Chem (2012) 287(2):978–88. doi: 10.1074/jbc.M111.294959
34. Li X, Han Y, Guan Y, Zhang L, Bai C, Li Y. Aluminum Induces Osteoblast Apoptosis Through the Oxidative Stress-Mediated Jnk Signaling Pathway. Biol Trace Elem Res (2012) 150(1-3):502–8. doi: 10.1007/s12011-012-9523-5
35. Park BG, Yoo CI, Kim HT, Kwon CH, Kim YK. Role of Mitogen-Activated Protein Kinases in Hydrogen Peroxide-Induced Cell Death in Osteoblastic Cells. Toxicology (2005) 215(1-2):115–25. doi: 10.1016/j.tox.2005.07.003
36. Agidigbi TS, Kim C. Reactive Oxygen Species in Osteoclast Differentiation and Possible Pharmaceutical Targets of Ros-Mediated Osteoclast Diseases. Int J Mol Sci (2019) 20(14):3576. doi: 10.3390/ijms20143576
37. Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, et al. A Crucial Role for Reactive Oxygen Species in Rankl-Induced Osteoclast Differentiation. Blood (2005) 106(3):852–9. doi: 10.1182/blood-2004-09-3662
38. An Y, Zhang H, Wang C, Jiao F, Xu H, Wang X, et al. Activation of Ros/Mapks/Nf-κb/Nlrp3 and Inhibition of Efferocytosis in Osteoclast-Mediated Diabetic Osteoporosis. FASEB J (2019) 33(11):12515–27. doi: 10.1096/fj.201802805RR
39. Yang B, Li S, Chen Z, Feng F, He L, Liu B, et al. Amyloid β Peptide Promotes Bone Formation by Regulating Wnt/β-Catenin Signaling and the Opg/Rankl/Rank System. FASEB J (2020) 34(3):3583–93. doi: 10.1096/fj.201901550R
40. Udagawa N, Koide M, Nakamura M, Nakamichi Y, Yamashita T, Uehara S, et al. Osteoclast Differentiation by Rankl and Opg Signaling Pathways. J Bone Miner Metab (2021) 39(1):19–26. doi: 10.1007/s00774-020-01162-6
41. Bai XC, Lu D, Liu AL, Zhang ZM, Li XM, Zou ZP, et al. Reactive Oxygen Species Stimulates Receptor Activator of NF-KappaB Ligand Expression in Osteoblast. J Biol Chem (2005) 280(17):17497–506. doi: 10.1074/jbc.M409332200
42. Chen JR, Shankar K, Nagarajan S, Badger TM, Ronis MJ. Protective Effects of Estradiol On Ethanol-Induced Bone Loss Involve Inhibition of Reactive Oxygen Species Generation in Osteoblasts and Downstream Activation of the Extracellular Signal-Regulated Kinase/Signal Transducer and Activator of Transcription 3/Receptor Activator of Nuclear Factor-Kappab Ligand Signaling Cascade. J Pharmacol Exp Ther (2008) 324(1):50–9. doi: 10.1124/jpet.107.130351
43. Oliver L, Hue E, Priault M, Vallette FM. Basal Autophagy Decreased During the Differentiation of Human Adult Mesenchymal Stem Cells. Stem Cells Dev (2012) 21(15):2779–88. doi: 10.1089/scd.2012.0124
44. Pantovic A, Krstic A, Janjetovic K, Kocic J, Harhaji-Trajkovic L, Bugarski D, et al. Coordinated Time-Dependent Modulation of Ampk/Akt/mTOR Signaling and Autophagy Controls Osteogenic Differentiation of Human Mesenchymal Stem Cells. Bone (2013) 52(1):524–31. doi: 10.1016/j.bone.2012.10.024
45. Qi M, Zhang L, Ma Y, Shuai Y, Li L, Luo K, et al. Autophagy Maintains the Function of Bone Marrow Mesenchymal Stem Cells to Prevent Estrogen Deficiency-Induced Osteoporosis. Theranostics (2017) 7(18):4498–516. doi: 10.7150/thno.17949
46. Priault M, Hue E, Marhuenda F, Pilet P, Oliver L, Vallette FM. Differential Dependence On Beclin1 for the Regulation of Pro-Survival Autophagy by Bcl-2 and Bcl-Xl in Hct116 Colorectal Cancer Cells. PLoS One (2010) 5(1):e8755. doi: 10.1371/journal.pone.0008755
47. Wu M, Chen G, Li YP. TGF-β and Bmp Signaling in Osteoblast, Skeletal Development, and Bone Formation, Homeostasis and Disease. Bone Res (2016) 4:16009. doi: 10.1038/boneres.2016.9
48. Huitema LF, Vaandrager AB. What Triggers Cell-Mediated Mineralization? Front Biosci (2007) 12. doi: 10.2741/2260
49. Liu F, Fang F, Yuan H, Yang D, Chen Y, Williams L, et al. Suppression of Autophagy by Fip200 Deletion Leads to Osteopenia in Mice Through the Inhibition of Osteoblast Terminal Differentiation. J Bone Miner Res (2013) 28(11):2414–30. doi: 10.1002/jbmr.1971
50. Chen L, Shi X, Weng SJ, Xie J, Tang JH, Yan DY, et al. Vitamin K2 Can Rescue the Dexamethasone-Induced Downregulation of Osteoblast Autophagy and Mitophagy Thereby Restoring Osteoblast Function in Vitro and in Vivo. Front Pharmacol (2020) 11:1209. doi: 10.3389/fphar.2020.01209
51. Li H, Li D, Ma Z, Qian Z, Kang X, Jin X, et al. Defective Autophagy in Osteoblasts Induces Endoplasmic Reticulum Stress and Causes Remarkable Bone Loss. Autophagy (2018) 14(10):1726–41. doi: 10.1080/15548627.2018.1483807
52. Shah M, Kola B, Bataveljic A, Arnett TR, Viollet B, Saxon L, et al. Amp-Activated Protein Kinase (Ampk) Activation Regulates in Vitro Bone Formation and Bone Mass. Bone (2010) 47(2):309–19. doi: 10.1016/j.bone.2010.04.596
53. Kasai T, Bandow K, Suzuki H, Chiba N, Kakimoto K, Ohnishi T, et al. Osteoblast Differentiation Is Functionally Associated With Decreased Amp Kinase Activity. J Cell Physiol (2009) 221(3):740–9. doi: 10.1002/jcp.21917
54. Xi G, Rosen CJ, Clemmons DR. GF-I and IGFBP-2 Stimulate AMPK Activation and Autophagy, Which are Required for Osteoblast Differentiation. Endocrinology (2016) 157(1):268–81. doi: 10.1210/en.2015-1690
55. Nollet M, Santucci-Darmanin S, Breuil V, Al-Sahlanee R, Cros C, Topi M, et al. Autophagy in Osteoblasts Is Involved in Mineralization and Bone Homeostasis. Autophagy (2014) 10(11):1965–77. doi: 10.4161/auto.36182
56. Thomas N, Choi HK, Wei X, Wang L, Mishina Y, Guan JL, et al. Autophagy Regulates Craniofacial Bone Acquisition. Calcif Tissue Int (2019) 105(5):518–30. doi: 10.1007/s00223-019-00593-2
57. Teitelbaum SL. Bone Resorption by Osteoclasts. Science (2000) 289(5484):1504–8. doi: 10.1126/science.289.5484.1504
58. Shapiro IM, Layfield R, Lotz M, Settembre C, Whitehouse C. Boning Up On Autophagy: The Role of Autophagy in Skeletal Biology. Autophagy (2014) 10(1):7–19. doi: 10.4161/auto.26679
59. Zhao Y, Chen G, Zhang W, Xu N, Zhu JY, Jia J, et al. Autophagy Regulates Hypoxia-Induced Osteoclastogenesis Through the Hif-1α/Bnip3 Signaling Pathway. J Cell Physiol (2012) 227(2):639–48. doi: 10.1002/jcp.22768
60. Sambandam Y, Townsend MT, Pierce JJ, Lipman CM, Haque A, Bateman TA, et al. Microgravity Control of Autophagy Modulates Osteoclastogenesis. Bone (2014) 61:125–31. doi: 10.1016/j.bone.2014.01.004
61. Arai A, Kim S, Goldshteyn V, Kim T, Park NH, Wang CY, et al. Beclin1 Modulates Bone Homeostasis by Regulating Osteoclast and Chondrocyte Differentiation. J Bone Miner Res (2019) 34(9):1753–66. doi: 10.1002/jbmr.3756
62. Lin NY, Stefanica A, Distler JH. Autophagy: A Key Pathway of TNF-Induced Inflammatory Bone Loss. Autophagy (2013) 9(8):1253–5. doi: 10.4161/auto.25467
63. Chung YH, Yoon SY, Choi B, Sohn DH, Yoon KH, Kim WJ, et al. Microtubule-Associated Protein Light Chain 3 Regulates Cdc42-Dependent Actin Ring Formation in Osteoclast. Int J Biochem Cell Biol (2012) 44(6):989–97. doi: 10.1016/j.biocel.2012.03.007
64. DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, et al. Autophagy Proteins Regulate the Secretory Component of Osteoclastic Bone Resorption. Dev Cell (2011) 21(5):966–74. doi: 10.1016/j.devcel.2011.08.016
65. Muthusami S, Ramachandran I, Muthusamy B, Vasudevan G, Prabhu V, Subramaniam V, et al. Ovariectomy Induces Oxidative Stress and Impairs Bone Antioxidant System in Adult Rats. Clin Chim Acta (2005) 360(1-2):81–6. doi: 10.1016/j.cccn.2005.04.014
66. Bartolomé A, López-Herradón A, Portal-Núñez S, García-Aguilar A, Esbrit P, Benito M, et al. Autophagy Impairment Aggravates the Inhibitory Effects of High Glucose On Osteoblast Viability and Function. Biochem J (2013) 455(3):329–37. doi: 10.1042/BJ20130562
67. Zhang S, Liu Y, Liang Q. Low-Dose Dexamethasone Affects Osteoblast Viability by Inducing Autophagy Via Intracellular Ros. Mol Med Rep (2018) 17(3):4307–16. doi: 10.3892/mmr.2018.8461
68. Camuzard O, Santucci-Darmanin S, Breuil V, Cros C, Gritsaenko T, Pagnotta S, et al. Sex-Specific Autophagy Modulation in Osteoblastic Lineage: A Critical Function to Counteract Bone Loss in Female. Oncotarget (2016) 7(41):66416–28. doi: 10.18632/oncotarget.12013
69. Yang L, Meng H, Yang M. Autophagy Protects Osteoblasts From Ages Induced Apoptosis Through. J Mol Endocrinol (2016) 56(4):291–300. doi: 10.1530/JME-15-0267
70. Yang YH, Chen K, Li B, Chen JW, Zheng XF, Wang YR, et al. Estradiol Inhibits Osteoblast Apoptosis Via Promotion of Autophagy Through the ER-ERK-mTOR Pathway. Apoptosis (2013) 18(11):1363–75. doi: 10.1007/s10495-013-0867-x
71. Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, et al. Forkhead Transcription Factor Foxo3a Protects Quiescent Cells From Oxidative Stress. Nature (2002) 419(6904):316–21. doi: 10.1038/nature01036
72. Ho KK, McGuire VA, Koo CY, Muir KW, de Olano N, Maifoshie E, et al. Phosphorylation of Foxo3a On Ser-7 by P38 Promotes Its Nuclear Localization in Response to Doxorubicin. J Biol Chem (2012) 287(2):1545–55. doi: 10.1074/jbc.M111.284224
73. Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, et al. Foxo Transcription Factor Activation by Oxidative Stress Mediated by the Small Gtpase Ral and Jnk. EMBO J (2004) 23(24):4802–12. doi: 10.1038/sj.emboj.7600476
74. Gómez-Puerto MC, Verhagen LP, Braat AK, Lam EWF, Coffer PJ, Lorenowicz MJ. Activation of Autophagy by Foxo3 Regulates Redox Homeostasis During Osteogenic Differentiation. Autophagy (2016) 12(10):1804–16. doi: 10.1080/15548627.2016.1203484
75. Gu X, Han D, Chen W, Zhang L, Lin Q, Gao J, et al. Sirt1-Mediated Foxos Pathways Protect Against Apoptosis by Promoting Autophagy in Osteoblast-Like Mc3T3-E1 Cells Exposed to Sodium Fluoride. Oncotarget (2016) 7(40):65218–30. doi: 10.18632/oncotarget.11573
76. Hardie DG. Amp-Activated/Snf1 Protein Kinases: Conserved Guardians of Cellular Energy. Nat Rev Mol Cell Biol (2007) 8(10):774–85. doi: 10.1038/nrm2249
77. Mihaylova MM, Shaw RJ. The Ampk Signalling Pathway Coordinates Cell Growth, Autophagy and Metabolism. Nat Cell Biol (2011) 13(9):1016–23. doi: 10.1038/ncb2329
78. Wang S, Song P, Zou MH. Amp-Activated Protein Kinase, Stress Responses and Cardiovascular Diseases. Clin Sci (Lond) (2012) 122(12):555–73. doi: 10.1042/CS20110625
79. Kim J, Kundu M, Viollet B, Guan KL. Ampk and Mtor Regulate Autophagy Through Direct Phosphorylation of Ulk1. Nat Cell Biol (2011) 13(2):132–41. doi: 10.1038/ncb2152
80. She C, Zhu L, Zhen Y, Wang X, Dong Q. Activation of Ampk Protects Against Hydrogen Peroxide-Induced Osteoblast Apoptosis Through Autophagy Induction and Nadph Maintenance: New Implications for Osteonecrosis Treatment? Cell Signal (2014) 26(1):1–8. doi: 10.1016/j.cellsig.2013.08.046
81. Liu W, Mao L, Ji F, Chen F, Hao Y, Liu G. Targeted Activation of Ampk by Gsk621 Ameliorates H2O2-Induced Damages in Osteoblasts. Oncotarget (2017) 8(6):10543–52. doi: 10.18632/oncotarget.14454
82. Zhu Y, Zhou J, Ao R, Yu B. A-769662 Protects Osteoblasts From Hydrogen Dioxide-Induced Apoptosis Through Activating of Amp-Activated Protein Kinase (Ampk). Int J Mol Sci (2014) 15(6):11190–203. doi: 10.3390/ijms150611190
83. Zhao D, Yang J, Yang L. Insights for Oxidative Stress and mTOR Signaling in Myocardial Ischemia/Reperfusion Injury Under Diabetes. Oxid Med Cell Longev (2017) 2017:6437467. doi: 10.1155/2017/6437467
84. Hay N, Sonenberg N. Upstream and Downstream of mTOR. Genes Dev (2004) 18(16):1926–45. doi: 10.1101/gad.1212704
85. Zhai C, Cheng J, Mujahid H, Wang H, Kong J, Yin Y, et al. Selective Inhibition of PI3K/Akt/ mTOR Signaling Pathway Regulates Autophagy of Macrophage and Vulnerability of Atherosclerotic Plaque. PLoS One (2014) 9(3):e90563. doi: 10.1371/journal.pone.0090563
86. Koundouros N, Poulogiannis G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front Oncol (2018) 8. doi: 10.3389/fonc.2018.00160
87. Wang X, Feng Z, Li J, Chen L, Tang W. High Glucose Induces Autophagy of Mc3T3-E1 Cells Via Ros-Akt- mTOR Axis. Mol Cell Endocrinol (2016) 429:62–72. doi: 10.1016/j.mce.2016.03.036
88. Leslie NR, Downes CP. Pten: The Down Side of PI3-Kinase Signalling. Cell Signal (2002) 14(4):285–95. doi: 10.1016/s0898-6568(01)00234-0
89. Shi Y, Liu XY, Jiang YP, Zhang JB, Zhang QY, Wang NN, et al. Monotropein Attenuates Oxidative Stress Via Akt/mTOR-Mediated Autophagy in Osteoblast Cells. BioMed Pharmacother (2020) 121:109566. doi: 10.1016/j.biopha.2019.109566
90. Suzuki M, Bandoski C, Bartlett JD. Fluoride Induces Oxidative Damage and Sirt1/Autophagy Through Ros-Mediated JNK Signaling. Free Radic Biol Med (2015) 89:369–78. doi: 10.1016/j.freeradbiomed.2015.08.015
91. Peng P, Nie Z, Sun F, Peng H. Glucocorticoids Induce Femoral Head Necrosis in Rats Through the ROS/JNK/c-Jun Pathway. FEBS Open Bio (2021) 11(1):312–21. doi: 10.1002/2211-5463.13037
92. Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-Xl Play Important Roles in the Crosstalk Between Autophagy and Apoptosis. FEBS J (2011) 278(3):403–13. doi: 10.1111/j.1742-4658.2010.07965.x
93. Luo S, Rubinsztein DC. Bcl2L11/Bim: A Novel Molecular Link Between Autophagy and Apoptosis. Autophagy (2013) 9(1):104–5. doi: 10.4161/auto.22399
94. Wei Y, Sinha S, Levine B. Dual Role of JNK1-Mediated Phosphorylation of Bcl-2 in Autophagy and Apoptosis Regulation. Autophagy (2008) 4(7):949–51. doi: 10.4161/auto.6788
95. Shimizu S, Konishi A, Nishida Y, Mizuta T, Nishina H, Yamamoto A, et al. Involvement of JNK in the Regulation of Autophagic Cell Death. Oncogene (2010) 29(14):2070–82. doi: 10.1038/onc.2009.487
96. Yang Y, Zheng X, Li B, Jiang S, Jiang L. Increased Activity of Osteocyte Autophagy in Ovariectomized Rats and Its Correlation With Oxidative Stress Status and Bone Loss. Biochem Biophys Res Commun (2014) 451(1):86–92. doi: 10.1016/j.bbrc.2014.07.069
97. Kar R, Riquelme MA, Hua R, Jiang JX. Glucocorticoid-Induced Autophagy Protects Osteocytes Against Oxidative Stress Through Activation of Mapk/Erk Signaling. JBMR Plus (2019) 3(4):e10077. doi: 10.1002/jbm4.10077
98. Zhang Y, Yan M, Kuang S, Lou Y, Wu S, Li Y, et al. Bisphenol a Induces Apoptosis and Autophagy in Murine Osteocytes Mlo-Y4: Involvement of ROS-Mediated mTOR/ULK1 Pathway. Ecotoxicol Environ Saf (2021) 230:113119. doi: 10.1016/j.ecoenv.2021.113119
99. Lee JJ, Jain V, Amaravadi RK. Clinical Translation of Combined Mapk and Autophagy Inhibition in Ras Mutant Cancer. Int J Mol Sci (2021) 22(22):12402. doi: 10.3390/ijms222212402
100. Li Z, Miao Z, Ding L, Teng X, Bao J. Energy Metabolism Disorder Mediated Ammonia Gas-Induced Autophagy Via AMPK/mTOR/ULK1-Beclin1 Pathway in Chicken Livers. Ecotoxicol Environ Saf (2021) 217:112219. doi: 10.1016/j.ecoenv.2021.112219
101. Wang T, Xu F, Song L, Li J, Wang Q. Bisphenol a Exposure Prenatally Delays Bone Development and Bone Mass Accumulation in Female Rat Offspring Via the Erβ/Hdac5/Tgfβ Signaling Pathway. Toxicology (2021) 458:152830. doi: 10.1016/j.tox.2021.152830
102. Srinivasan S, Koenigstein A, Joseph J, Sun L, Kalyanaraman B, Zaidi M, et al. Role of Mitochondrial Reactive Oxygen Species in Osteoclast Differentiation. Ann N Y Acad Sci (2010) 1192(1):245–52. doi: 10.1111/j.1749-6632.2009.05377.x
103. Kim MS, Yang YM, Son A, Tian YS, Lee SI, Kang SW, et al. Rankl-Mediated Reactive Oxygen Species Pathway That Induces Long Lasting Ca2+ Oscillations Essential for Osteoclastogenesis. J Biol Chem (2010) 285(10):6913–21. doi: 10.1074/jbc.M109.051557
104. Sul OJ, Park HJ, Son HJ, Choi HS. Lipopolysaccharide (Lps)-Induced Autophagy Is Responsible for Enhanced Osteoclastogenesis. Mol Cells (2017) 40(11):880–7. doi: 10.14348/molcells.2017.0230
105. Shi J, Wang L, Zhang H, Jie Q, Li X, Shi Q, et al. Glucocorticoids: Dose-Related Effects On Osteoclast Formation and Function Via Reactive Oxygen Species and Autophagy. Bone (2015) 79:222–32. doi: 10.1016/j.bone.2015.06.014
106. Malhotra JD, Kaufman RJ. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle Or a Double-Edged Sword? Antioxid Redox Signal (2007) 9(12):2277–93. doi: 10.1089/ars.2007.1782
107. Fernández A, Ordóñez R, Reiter RJ, González-Gallego J, Mauriz JL. Melatonin and Endoplasmic Reticulum Stress: Relation to Autophagy and Apoptosis. J Pineal Res (2015) 59(3):292–307. doi: 10.1111/jpi.12264
108. Wang K, Niu J, Kim H, Kolattukudy PE. Osteoclast Precursor Differentiation by Mcpip Via Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy. J Mol Cell Biol (2011) 3(6):360–8. doi: 10.1093/jmcb/mjr021
109. Martini-Stoica H, Xu Y, Ballabio A, Zheng H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A Tfeb Perspective. Trends Neurosci (2016) 39(4):221–34. doi: 10.1016/j.tins.2016.02.002
110. Wang H, Wang N, Xu D, Ma Q, Chen Y, Xu S, et al. Oxidation of Multiple Mit/Tfe Transcription Factors Links Oxidative Stress to Transcriptional Control of Autophagy and Lysosome Biogenesis. Autophagy (2020) 16(9):1683–96. doi: 10.1080/15548627.2019.1704104
Keywords: autophagy, oxidative stress, osteoblast, osteoclast, osteoporosis
Citation: Zhu C, Shen S, Zhang S, Huang M, Zhang L and Chen X (2022) Autophagy in Bone Remodeling: A Regulator of Oxidative Stress. Front. Endocrinol. 13:898634. doi: 10.3389/fendo.2022.898634
Received: 17 March 2022; Accepted: 01 June 2022;
Published: 30 June 2022.
Edited by:
Guanwu Li, Shanghai University of Traditional Chinese Medicine, ChinaReviewed by:
Chen-he Zhou, Zhejiang University, ChinaCopyright © 2022 Zhu, Shen, Zhang, Huang, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Chen, Y2hlbmFiMDA0QDEyNi5jb20=; Lan Zhang, aGFuZ2xhbl9zZHR5QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.