 Lei Wei
Lei Wei Jianjian Shi
Jianjian Shi- Herman B Wells Center for Pediatric Research, Department of Pediatrics, Indiana University, School of Medicine, Indianapolis, IN, United States
Obesity and associated complications increasingly jeopardize global health and contribute to the rapidly rising prevalence of type 2 diabetes mellitus and obesity-related diseases. Developing novel methods for the prevention and treatment of excess body adipose tissue expansion can make a significant contribution to public health. Rho kinase is a Rho-associated coiled-coil-containing protein kinase (Rho kinase or ROCK). The ROCK family including ROCK1 and ROCK2 has recently emerged as a potential therapeutic target for the treatment of metabolic disorders. Up-regulated ROCK activity has been involved in the pathogenesis of all aspects of metabolic syndrome including obesity, insulin resistance, dyslipidemia and hypertension. The RhoA/ROCK-mediated actin cytoskeleton dynamics have been implicated in both white and beige adipogenesis. Studies using ROCK pan-inhibitors in animal models of obesity, diabetes, and associated complications have demonstrated beneficial outcomes. Studies via genetically modified animal models further established isoform-specific roles of ROCK in the pathogenesis of metabolic disorders including obesity. However, most reported studies have been focused on ROCK1 activity during the past decade. Due to the progress in developing ROCK2-selective inhibitors in recent years, a growing body of evidence indicates more attention should be devoted towards understanding ROCK2 isoform function in metabolism. Hence, studying individual ROCK isoforms to reveal their specific roles and principal mechanisms in white and beige adipogenesis, insulin sensitivity, energy balancing regulation, and obesity development will facilitate significant breakthroughs for systemic treatment with isoform-selective inhibitors. In this review, we give an overview of ROCK functions in the pathogenesis of obesity and insulin resistance with a particular focus on the current understanding of ROCK isoform signaling in white and beige adipogenesis, obesity and thermogenesis in adipose tissue and other major metabolic organs involved in energy homeostasis regulation.
Introduction
Obesity is known as a major risk factor for type 2 diabetes, cardiovascular disease and several cancers, with obvious and significant impacts on public health. In 2016, over 1.9 billion adults and 340 million minors worldwide aged 5-19 were overweight or obese (1–7). In 2020, 39 million children under the age of 5 were overweight or obese. As the result of excess body mass, 2.8 million people die each year, and an additional estimated 35.8 million (2.3%) of global disability-adjusted life years (DALYs) are affected (7). More recently, during the COVID-19 pandemic, there has been a sharp increase in BMI among young school-aged children aged 2–19 years described in a recent longitudinal cohort study (8). These findings highlight the importance of integrating obesity management effort into public health interventions in present day. On the other hand, we must clearly appreciate that obesity is a preventable global health issue; therefore developing novel therapies to stop excessive adipose tissue expansion can significantly contribute to public health.
White adipose tissue (WAT) not only stores energy in the form of fat, but also displays important endocrine function in metabolic homeostasis (9). In contrast, brown adipose tissue (BAT) and beige adipocytes are thermogenic and can convert glucose and fatty acids into heat, thereby acting as thermogenic tissue to increase energy expenditure (10). BAT is mainly found in the dorsal interscapulum region in rodents and characterized by high constitutive expression of thermogenic genes, most notably: uncoupling protein-1 (UCP1), which resides in the inner mitochondrial membrane of brown adipocytes (11). UCP1 mediates heat generation in brown adipocytes by uncoupling respiratory chain, thus causing a fast substrate oxidation without efficient ATP production. In addition, clusters of “brown-like” UCP1-positive cells, also known as beige adipocytes, develop in WAT in response to various activators including cold exposure and β-adrenergic stimulation. BAT is also found in adult humans, but it is mainly characterized as inducible beige fat described in rodents (12, 13). Various factors may apply negative effects on brown adipocytes regeneration leading to its decreased prevalence as humans age (14). It is worth noting that increased thermogenic activities in either brown or beige adipocytes have been linked to obesity resistance which were reported in numerous studies involving mouse models (9, 15–17) and humans (18–24). Increasing energy expenditure through the formation and activation of brown and beige adipocytes is an electrifying pathway with great potential in reducing obesity.
One strategy is to target molecules involved in the adipocyte formation and differentiation process. Rho kinases (hereafter referred to as ROCKs) are major downstream effectors of the small GTPase RhoA (25–28). The ROCK family has recently emerged as a potential therapeutic target for metabolic disorders. In human and mouse, both ROCK1 and ROCK2 are ubiquitously expressed across tissues (27). ROCKs play central roles in the organization of the actin cytoskeleton and is involved in a wide range of fundamental cellular functions such as smooth muscle cell contraction, cell proliferation, adhesion, migration, polarity, cytokinesis, differentiation and survival in many cell types. ROCK activity change has been demonstrated in pathogenesis of various diseases. Up-regulated ROCK activity is implicated in the pathogenesis of all aspects of metabolic syndrome including obesity, insulin resistance, dyslipidemia and hypertension (29–33). RhoA/ROCK-mediated cytoskeleton changes are identified as important regulators in adipogenesis (29, 34–37) and insulin sensitivity (30, 38–42). Studies using ROCK inhibitors in animal models of obesity, diabetes, and associated complications have demonstrated beneficial outcomes (29, 43–53). However, most ROCK inhibitors in these studies are non-isoform selective, thereby limiting their therapeutic potential due to observed dose-dependent smooth muscle relaxation and vascular hypotension in systemic treatment, which obviously hampers ROCK inhibition as a novel treatment (54–56). Thus, development of isoform-selective inhibitors can help speed up a new strategy breakthrough.
In addition to previous metabolic studies utilizing non-selective, exogenously administered chemical inhibitors of ROCK, studies which utilize genetically modified animal models to identify isoform selective roles of ROCK in the pathogenesis of metabolic disorders including obesity, have also been of particular interest to medical scientific community. Although, earlier published studies in the past decade were focused on ROCK1 activity (33, 57–59), a growing interest in understanding ROCK2 isoform function has emerged more recently with the development of ROCK2-selective inhibitors (54–56). For example, KD025 (also known as SLx-2119), a selective ROCK2 inhibitor, shows no major side effects in various clinical trials focusing on non-metabolic diseases (60–63). Hence, studying individual ROCK isoforms to elucidate their unique roles and mechanisms in white and beige adipogenesis, insulin sensitivity, and energy balance will facilitate significant breakthroughs in systemic application of isoform-selective inhibitors for obesity prevention and treatment. In this review, we give an overview of ROCK functions in the pathogenesis of obesity with a particular focus on the current understanding of ROCK isoform signaling involved in white and beige adipogenesis, obesity development and thermogenesis in adipose tissue and other major metabolic organs involved in energy homeostasis regulation.
Overview of Major ROCK Signaling Pathways in Regulating Metabolic Function
Regulation of ROCK Activity in Metabolic Tissues
ROCK1 and ROCK2 are downstream targets of the small GTP-binding protein RhoA, they work as mediators in the RhoA-dependent signaling pathway. Stimulation of tyrosine kinase and G-protein-coupled receptors leads to activation of RhoA via the recruitment and activation of guanine nucleotide exchange factors (GEFs) (64, 65). Activated RhoA directly interacts with ROCK at the C-terminal portion of the coiled-coil domain and induces a conformational change, leading to activation of the serine/threonine kinase toward selective substrates (25–28). ROCK activity can also be modulated through interaction of C-terminal pleckstrin-homology domain with lipid mediators and with the plasma membrane (66–68), auto-phosphorylation through dimerization (69), and proteolytic cleavage of the inhibitory C-terminal domain (70–72). In addition, recent microRNA (miRNA) research has identified numerous miRNAs which are involved in regulating RhoA and ROCK expression and kinase activity (73, 74), suggesting that miRNAs have a very important position in RhoA/ROCKs gene regulation.
Both experimental (29, 43–45) and clinical studies (31, 41, 75) have established a clear link between aberrant ROCK activity and metabolic disease including obesity and insulin resistance (Figure 1). The factors that can change kinase activity and/or gene expression are context dependent, as indicated in hyperglycemia, hyperlipidemia, mechanical stress, oxidative stress and increased production of inflammatory cytokines etc. (29, 31, 41, 43–45, 75) (Figure 1). Metabolic stresses including hyperglycemia and hyperlipidemia mainly stimulate RhoA/ROCK activity via oxidative stress-dependent pathways and through activation of GEFs by tyrosine kinase and G-protein-coupled receptors; moreover, other protein kinases including adenosine monophosphate-activated protein kinase (AMPK) can activate RhoA (29–33, 76). Recent mechanistic studies which comprehensively link hyperglycemia and hyperlipidemia with aberrant RhoA/ROCK activity have shown that the increased production of proinflammatory cytokines and oxidative stress can disturb miRNA expression which subsequently change RhoA/ROCK expression and activity as well (77, 78). miRNAs were found in metabolic tissues including adipose tissue (79), liver (80, 81), skeletal muscle (82) and kidney (83), et al., where they regulate RhoA, ROCK1 and ROCK2 expression and activity (Table 1). Although miRNAs are known as a class of regulatory molecules, elucidating their biological relevance of individual miRNAs is challenging. Their up- or down-regulations can be either protective or detrimental to insulin resistance (82), diabetes (83) and obesity (79–81). Despite their elusive role, miRNAs modify RhoA/ROCK signaling pathway and obviously contribute to animal development, metabolic homeostasis, and disease pathogenesis.
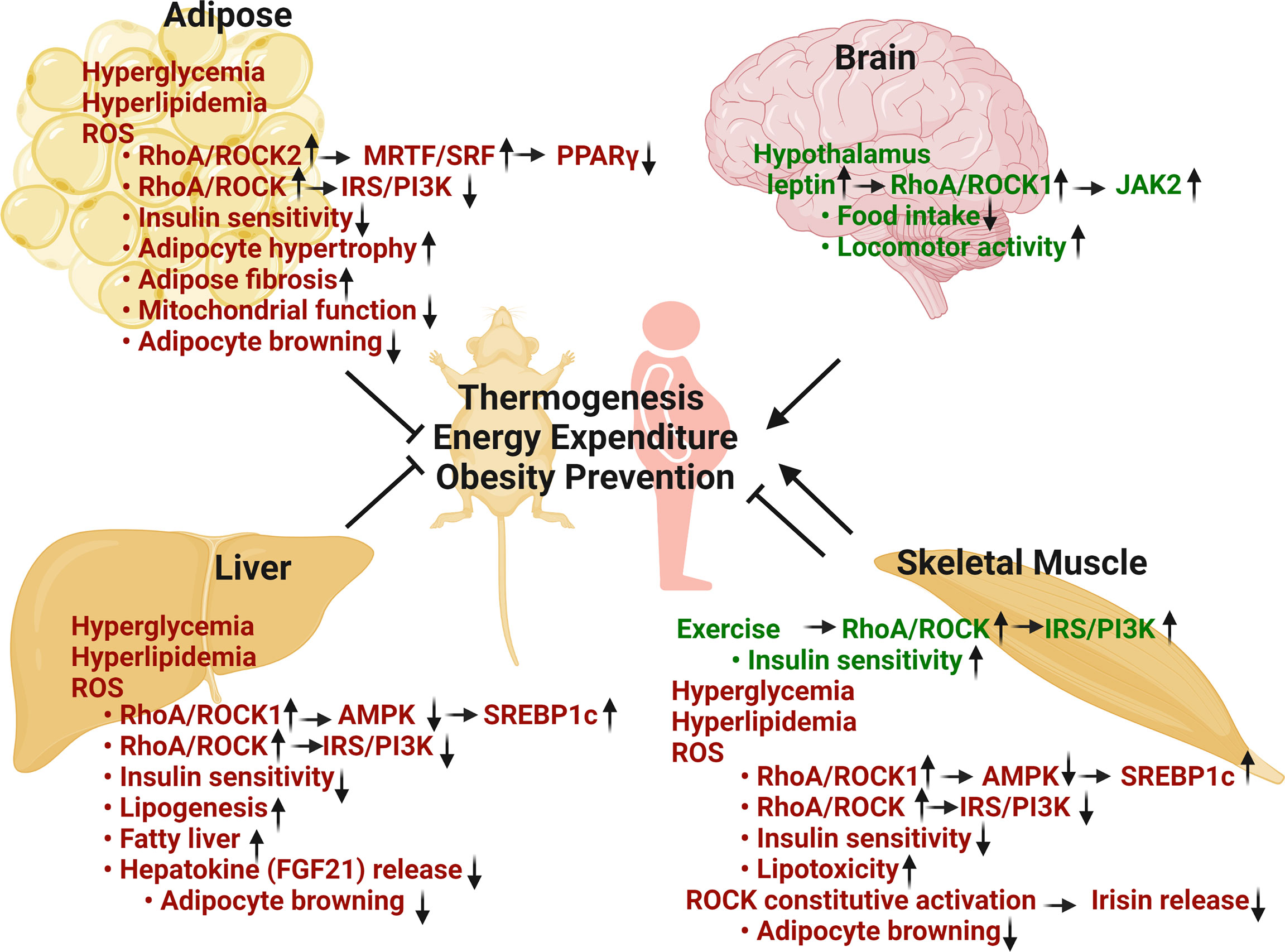
Figure 1 Metabolic regulation of RhoA/ROCK pathway in thermogenesis, energy balance, obesity prevention and insulin sensitivity. Adipose RhoA/ROCK activation, caused by metabolic stresses including hyperglycemia, hyperlipidemia and ROS, is associated with disease phenotypes. Total ROCK activity and ROCK2 activity are negative regulators of thermogenic, adipogenic, and mitochondrial gene expression via stimulation of F-actin/MRTF/SRF signaling pathway and inhibition of insulin signaling pathway. Hepatic RhoA/ROCK activation by risk-factors is also associated with aberrant regulation. Total ROCK activity and ROCK1 activity are positive regulators of lipogenic pathway through inhibition of AMPK and activation of SREBP1c. Hepatic RhoA/ROCK activities are also negative regulators of thermogenesis and insulin signaling pathway. Skeletal muscle RhoA/ROCK activation shows both beneficial and detrimental outcomes. ROCK activation under basal or exercise condition is critical to glucose regulation via promoting insulin signaling pathway. Conversely, ROCK activation by metabolic stresses is associated with disease through inhibition of AMPK and insulin signaling. Hypothalamic RhoA/ROCK1 activation by circulating leptin decreases body weight and adiposity through reducing food intake, increasing energy expenditure and locomotor activity (Green texts indicate beneficial effects in metabolism; red texts indicate detrimental effects in metabolism) (Figure is created with BioRender.com).
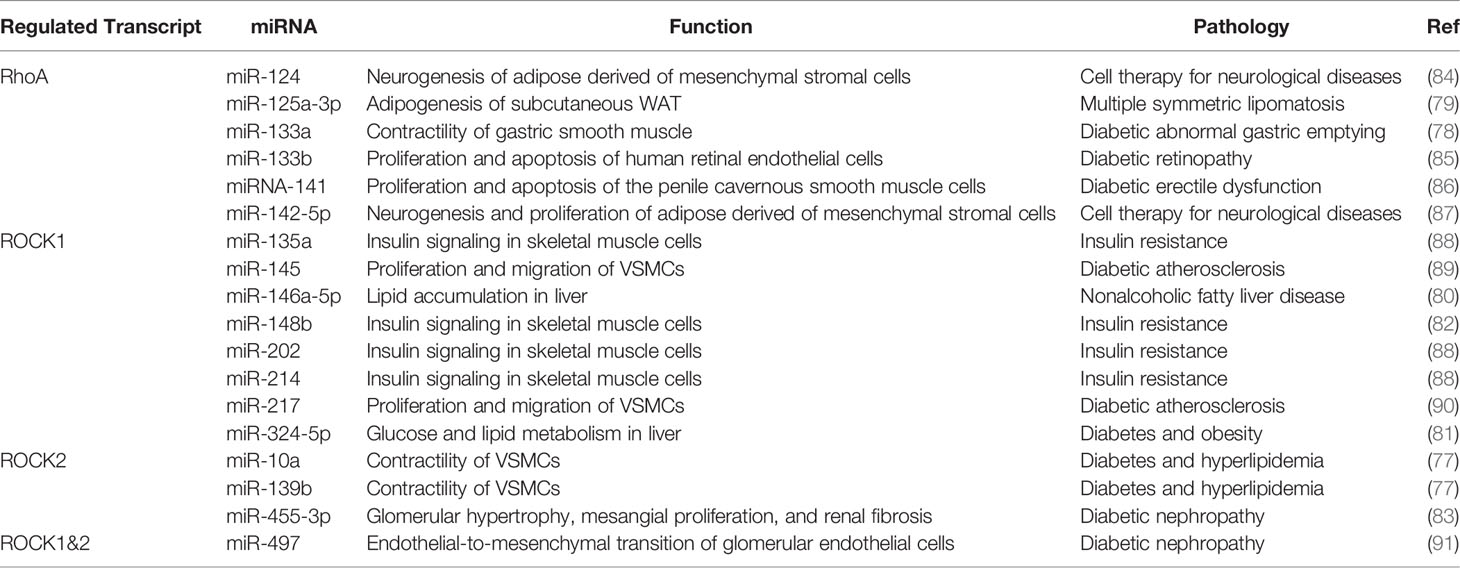
Table 1 Down-regulation of RhoA, ROCK1 and ROCK2 by miRNAs in metabolic tissues.
ROCKs Regulate Actin Cytoskeleton Function in all Metabolic Tissues
ROCKs are downstream of the small GTPase RhoA and regulate actin cytoskeleton function as central regulators (25–27). The two ROCK isoforms are highly homologous with 92% amino acid sequence identity in the kinase domain and an overall shared identity of 65% (25–27). ROCK1 and ROCK2 share more than 30 immediate downstream substrates (reviewed in refs (73, 92–94)) which share the consensus amino acid sequences: R/K-X-S/T or R/K-X-X-S/T (R, arginine; K, lysine; S, serine; T, threonine, all potential phosphorylation sites by ROCK) (95, 96). The major downstream substrates of ROCKs include the myosin binding subunit of myosin light chain (MLC) phosphatase-1 (MYPT1) (95, 97, 98), MLC2 (97, 99) and LIM kinases (LIMK) (96, 100–103). ROCKs/MYPT1/MLC2 pathway promotes actomyosin contractility through MLC2 phosphorylation, and ROCKs/LIMK/cofilin pathway stabilizes actin filaments (F-actin) through cofilin phosphorylation, which inhibits actin-depolymerization activity (Figure 2A). These two signaling pathways modulate actin cytoskeleton organization, stress fiber formation and smooth muscle cell contraction, and remain the best characterized mechanism presenting in all metabolic tissues.
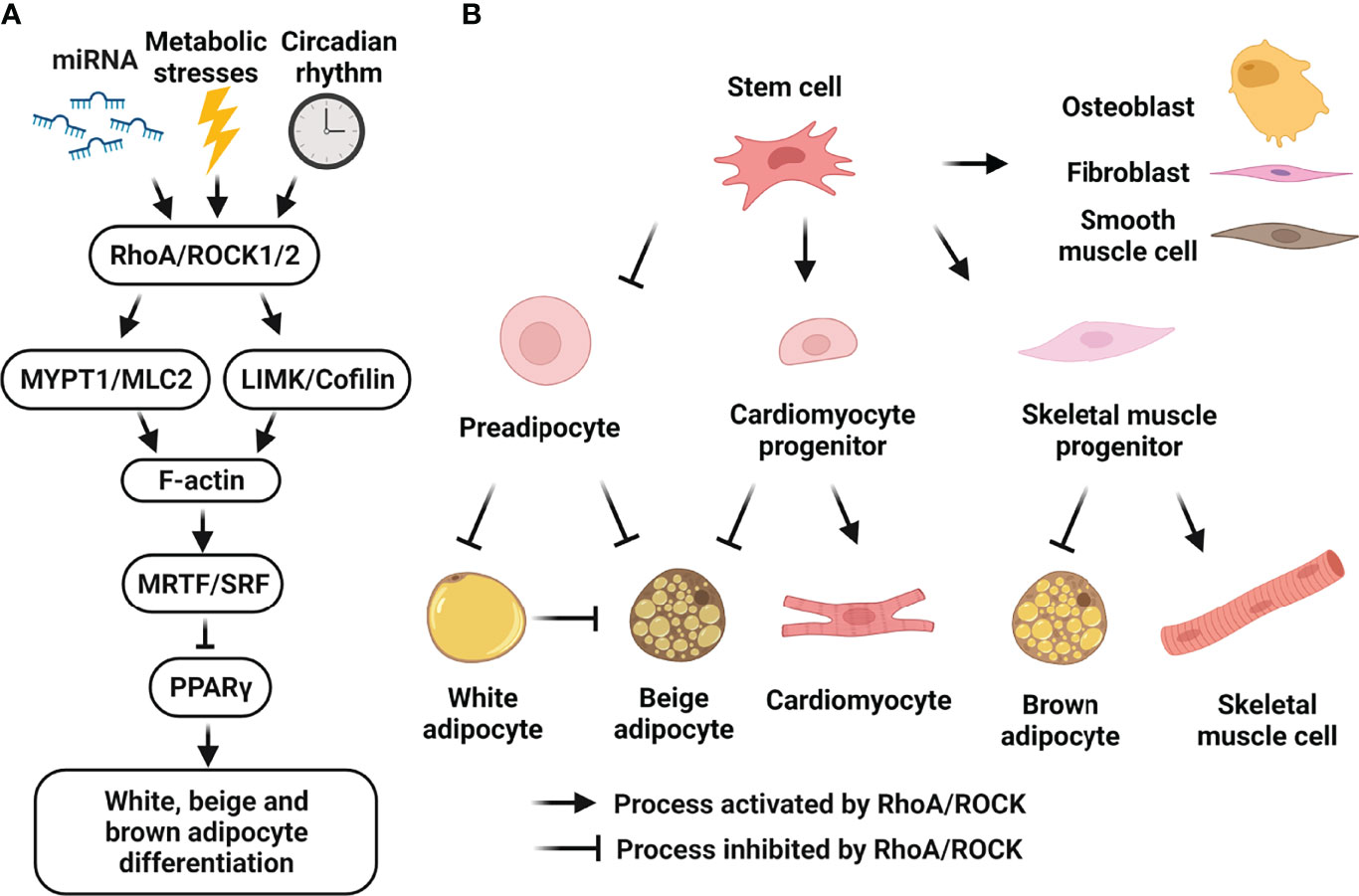
Figure 2 Cellular roles of RhoA/ROCK signaling in white, beige and brown adipocyte differentiation. (A) Schematic representation of RhoA/ROCK pathway which negatively regulates adipocyte differentiation. RhoA/ROCK signaling-cascade activation can be induced by many risk-factors including mechanical stress, mitochondrial dysfunction, inflammation, ROS, aging, hyperglycemia and dyslipidemia, etc. Activated pathway causes actin polymerization and formation of stress fibers, leading to nuclear translocation of MRTF and suppression of PPARγ activity. (B) Schematic representation of the regulation of cell fate determination of mesenchymal stem cells by RhoA/ROCK pathway. Activation of RhoA/ROCK signaling by local mechanical stresses and environment factors promotes the commitment to osteogenic versus adipogenic lineages of bone marrow mesenchymal stem cells and adipose tissue-derived stem cells. RhoA/ROCK activation also promotes the commitment to smooth muscle cell-like and fibrogenic lineages versus adipogenic lineage of perivascular progenitors. Moreover, RhoA/ROCK activation favors a switch toward myogenesis versus brown adipogenesis of common mesenchymal precursors, and a switch toward cardiomyocytes versus beige adipocytes of embryonic cardiac progenitors. Finally, RhoA/ROCK activation negatively regulates preadipocyte differentiation towards white and beige adipocytes, and transdifferentiation of white adipocytes to beige adipocytes. (Figure is created with BioRender.com).
MRTF and SRF are Downstream Mediators Mainly in Adipose and Vascular Tissues
The prominent effects of RhoA/ROCK on cytoskeleton dynamics are not only limited to cell contraction, adhesion, morphology and motility, but also include transcriptional regulation. For instance, serum response factor (SRF) activity is regulated by RhoA/ROCK signaling on actin polymerization (104–107). Myocardin, the myocardin-related transcription factors A and B (MRTF-A/MKL1 and MRTF-B/MKL2) and MASTR constitute an SRF coactivator family, and their activity depends on actin dynamics (106–109). Association of MRTF-A with monomeric globular actin (G-actin) results in its sequestration in the cytoplasm, and actin polymerization leads to MRTF-A translocation into the nucleus and SRF target gene activation (106, 107). The RhoA/ROCK/F-actin/MRTF-A/SRF pathway has been well-established in smooth muscle cells and regulates transcription of genes involved in the contractile function of smooth muscle cells (110).
Recent studies also demonstrate the role of this pathway in regulating lipid and glucose metabolisms (111, 112). In adipose tissue, MRTF-A/SRF pathway suppresses peroxisome proliferator-activated receptor γ (PPARγ) expression/activity and negatively regulates adipocyte differentiation including white (37, 113), beige (16, 114) and brown adipogenesis (115) (Figure 2). Furthermore, activation of this pathway was found to reduce expression of insulin receptor substrate-1 (IRS1) and PPARγ in mature human adipocytes and in hypertrophic adipocytes from ob/ob mice, thus contributing to adipocyte dysfunction and insulin resistance (116). This pathway also promotes fibrosis, inflammation and insulin resistance in diet-induced obesity, since perivascular progenitor cells will preferentially differentiate into a fibroblast lineage as opposed to a normal adipocyte lineage (117). Even in mature adipocytes, this pathway promotes their fibroblast-like phenotype contributing to adipocyte dysfunction and mitochondrial dysfunction (118). In addition to adipose tissue, MRTF-A/SRF pathway is involved in hepatic fibrosis (119) and diabetic nephropathy (120): MRTF-A knockout reduced macrophage infiltration, and ameliorated inflammation and fibrosis of liver tissues in mice with nonalcoholic steatohepatitis induced by high-fat diet (HFD) (119); through an epigenetic mechanism, MRTF-A deficiency also attenuated connective tissue growth factor in diabetic nephropathy (120). It should be noted some metabolic studies cited above were interested in the connection between RhoA/ROCK/F-actin and MRTF-A/SRF pathways (16, 37, 114, 116), but some mainly focused on MRTF-A and its downstream effectors (113, 115, 117). Together, besides the well-established role for the RhoA/ROCK/F-actin/MRTF-A/SRF pathway in regulating vascular cell function, abovementioned studies support that the RhoA/ROCK/F-actin/MRTF-A/SRF pathway negatively regulates adipocyte differentiation, plays a negative role in maintaining adipose tissue health, and contributes to liver fibrosis and diabetic nephropathy.
Impacts of ROCKs on Insulin Signaling
The relationship between ROCK activity and IRS1 phosphorylation has been documented in various cell types and organs including adipose tissues, liver, skeletal muscle and vascular system (Figure 1). It is widely believed that serine phosphorylation of IRS1 by ROCK activity in WAT and vascular cells leads to reduced IRS1-mediated phosphatidylinositol 3-kinase (PI3K) activation, resulting in decreased insulin sensitivity (30, 34, 42, 43, 121, 122). However, ROCK-mediated IRS1 phosphorylation can also positively impact on insulin signaling such as in skeletal muscle, and in cultured adipocytes and muscle cells (38–41). Although it has been consistently reported that ROCK activation increases serine phosphorylation levels of IRS1 (Ser307 and Ser632/635) and IRS2 (39, 122–124), the effects on insulin-stimulated tyrosine phosphorylation of IRS1 and IRS2, and on PI3K activation appear to be complex and context-dependent (38). It is also noted that ROCK-mediated actin cytoskeleton dynamics and insulin signaling can be inter-regulated; for instance, the changes of actin dynamics in ROCK1 deficient MEFs could be linked to improved insulin signaling through increased insulin receptor (IR) activation (30). Therefore, these studies support the concept that ROCK activity has both positive and negative actions on insulin signaling in a cell-type and tissue-dependent manner.
AMPK and SREBP are Downstream Mediators Mainly in Liver and Skeletal Muscle
AMPK is a key regulator of glucose and fatty acid metabolism and plays a major role in obesity and type 2 diabetes. The relationship between ROCK and AMPK has been documented in various cell types and organs, although physical interaction of these molecules has not been identified (125). ROCK1 has been identified as an upstream negative regulator of AMPK activity through an unknown mechanism in liver (47, 48, 59, 80, 125), skeletal muscle (47, 48, 126) and pancreatic β-cells (127). In liver tissue, ROCK1/AMPK/sterol regulatory element-binding protein-1c (SREBP1c) axis was reported to regulate hepatic lipogenesis and contribute to nonalcoholic fatty liver diseases (59, 125) (Figure 1). In skeletal muscle cells, ROCK1/AMPK/SREBP1c axis was involved in lipotoxicity and insulin resistance (126) (Figure 1). In pancreatic β-cells, ROCK1/AMPK axis contributed to β-cell dysfunction under lipotoxic stress (127). Moreover, systemic ROCK inhibition increased whole body energy consumption and ameliorated metabolic disorders through AMPK activation in the skeletal muscle and liver (47, 48). Overall, numerous studies have suggested a detrimental role of ROCK1/AMPK signaling in nutrient metabolism in liver, skeletal muscle and pancreatic β-cells.
Different from ROCK1/AMPK axis mentioned above, AMPK has been reported as an upstream positive or negative regulator of RhoA/ROCK pathway through various context dependent mechanisms in smooth muscle cells (76, 128, 129), endothelial cells (130) and podocytes (131). For instance, AMPK activation by metabolic stress reduced smooth muscle contractility attributed to phosphorylating RhoA and inhibiting the RhoA/ROCK/MYPT1/MLC2 pathway (76). In contrast, Toll-like receptor 9-dependent AMPK activation contributed to RhoA/ROCK activation and actin polymerization in smooth muscle cells (128). It is worth noting that whether ROCK1/AMPK or AMPK/RhoA/ROCK/MYPT1/MLC2 pathways could play a significant role in metabolic functions of adipose tissues has not yet been documented.
ROCK Isoforms Differentially Function in Various Cell Types
Though the two ROCK isoforms are very similar and are possibly somewhat redundant, a solid body of evidence indicates that they also have some unique functions. For instance, although both ROCK1 and ROCK2 control assembly of the actin cytoskeleton and cell contractility via phosphorylation of MYPT1, the mechanism may vary between the two. Only ROCK2 binds directly to and phosphorylates MYPT1 (132), suggesting that intermediate proteins are involved in ROCK1 binding to MYPT1. Moreover, these two ROCK isoforms differ in their binding capacities for IRS1 and only ROCK2 was found to bind to IRS1 although both isoforms are involved in insulin-stimulated phosphorylation of IRS1 at serine 632/635 (40). The in vivo functional similarity and differences of ROCK1 and ROCK2 have been shown in mouse genetic studies during embryonic development and under pathological conditions (133). For instance, genetic approaches have revealed pleiotropic actions of ROCKs in regulating insulin signaling and obesity, and the ultimate phenotype depends on ROCK isoforms and metabolic organs (30, 38, 41, 42, 121, 125, 134). Their functional differences could be explained by the facts that both isoforms are expressed at different levels and/or they have different interacting partners in individual cell types.
Recent Advances in Metabolic Study of ROCK2 Isoform-Specific Inhibitors
The high sequence identity between ROCK1 and ROCK2, particularly in their kinase domain, makes the design of isoformselective inhibitors very challenging (55, 56, 135). KD025 is the first highly selective ROCK2 inhibitor, and it achieved a good isoform selectivity of >200-fold for ROCK2 versus ROCK1 (60). Its therapeutic potential has been explored in fibrotic disease (60), focal cerebral ischemia (61, 136, 137), brain injury related to blood-brain barrier disruption (137), pulmonary arterial hypertension (138), and auto-immune disease (62, 139–144). Notably, the hypotensive phenotype was not observed when KD025 was tested in systemic application (61), therefore, emerging as an important breakthrough in systemic application. As a potential new drug candidate, KD025 is currently undergoing several phase 2 clinical trials for psoriasis, idiopathic pulmonary fibrosis, chronic graft-versus-host disease, and systemic sclerosis (142, 145, 146).
Recent studies explored therapeutic potential of KD025 in metabolic disease including renal fibrosis in the unilateral ureteral obstruction mice (147), arteriosclerosis and vascular fibrosis in a diabetic mouse model (148). Their therapeutic effects are partly due to the inhibition of profibrotic pathways and fat metabolism in both ROCK2-dependent and independent manners (149). Moreover, KD025 has been shown to inhibit adipogenesis in human adipose-derived stem cells, 3T3-L1 preadipocytes and human orbital fibroblasts through a ROCK2-independent mechanism (150–152), by inhibiting casein kinase 2 (153). In stromal vascular (SV) cells isolated from mouse subcutaneous WAT, we observed both pro-beige adipogenic (through ROCK2 inhibition) and anti-adipogenic (targets not clear) actions of KD025, which can be dissociated by dose- and time-dependent analyses during the in vitro differentiation process (154). A recent bioinformatics analysis confirmed the function of KD025 in regulating inflammation and adipogenesis pathways and revealed several novel regulatory functions, including oxidative phosphorylation, Wnt signaling, angiogenesis, and KRAS signaling (155). These studies revealed the presence of on-target (ROCK2-dependent) and off-target (ROCK2-independent) effects of KD025.
Owing to the off-target effects such as anti-adipogenic activity through inhibiting casein kinase 2 (153), KD025 may produce combinatorial effects on adipogenesis resulting from the dual inhibition of ROCK2 (promoting adipocyte differentiation) and CK2 (inhibiting adipocyte differentiation). Although the effects of KD025 on adipogenesis has been extensively studied in cell culture models, its therapeutic potential in reducing adiposity and improving insulin sensitivity has not yet been evaluated in experimental animals such as diet-induced obese mice or genetically obese mice. Furthermore, in a phase 2 study of patients with psoriasis vulgaris who were mostly obese with BMI > 30, giving oral administration of three different doses of KD025 for 3 months, didn’t see significant differences in the body weight among the dose-different groups (142), suggesting that specific studies focusing on obese and diabetic patients should be designed in order to rigorously evaluate its anti-obesity potential. Therefore, both experimental and clinical studies are required to evaluate the prospect of KD025 in prevention and treatment of obesity.
Cellular Actions of ROCK Signaling Pathways in Adipose Tissue
Healthy white adipocytes maintain a remarkably high degree of plasticity, quickly responding to whole body energy demands either through the release of fatty acids and glycerol, storage of excess calories as triglycerides and secretion of adipokines. Unhealthy remodeling of adipose tissue, especially visceral WAT from overfeeding leads to adipocyte dysfunction, reduced adipogenic capacity, and tissue fibrosis, all of which can contribute to whole body metabolic dysfunction, including type 2 diabetes and cardiovascular complications. BAT has significant effects on whole-body energy homeostasis. Promoting BAT function and the induction of beige adipocyte formation in WAT, which is a hallmark feature of adipose tissue browning, have emerged as promising therapeutic targets to increase energy expenditure and counteract unhealthy remodeling of adipose tissue. In response to certain environmental, genetic or pharmacological stimuli, the newly formed beige adipocytes in WAT are derived from 1) the recruitment of beige adipocyte progenitors from perivascular progenitors, 2) differentiation of preadipocytes, and 3) the transdifferentiation from mature white adipocytes (Figure 2). It is worthy to note that recent lineage-tracing studies have revealed that instead of the transdifferentiation from mature white adipocytes, beige adipocytes can arise from reactivation of dormant beige adipocytes which acquire white adipocyte-like unilocular morphology after warm adaptation (156). Here, we focus on the current status of RhoA/ROCK research in regulating the recruitment of adipocyte progenitors and adipocyte differentiation for all three types of adipocytes, WAT browning, and WAT remodeling due to overfeeding.
Lineage Commitment and Recruitment of Adipocyte Progenitors
Early studies support the contribution of RhoA/ROCK pathway to regulating a switch between adipogenesis and osteogenesis of mesenchymal stem cells (36) (Figure 2). Many local mechanical stresses and environment factors influence stem cells in developmental and adult contexts, including changes in cell shape, cytoskeletal tension and RhoA/ROCK signaling which are essential to the commitment of stem cell fate (35, 36, 157–159). Inhibition of RhoA/ROCK activity enhances adipogenic differentiation of human and murine bone marrow mesenchymal stem cells (36, 157, 158) and of human adipose tissue-derived stem cells (160). More recent studies support the contribution of MRTF-A/SRF to acting at downstream of the RhoA/ROCK pathway to promote murine bone marrow mesenchymal stem cell commitment (113) and human adipose tissue-derived stem cell commitment (161) to osteogenic versus adipogenic lineages. As mentioned above, by controlling expression of adipogenesis-related genes as well as cytoskeleton, focal adhesion, and extracellular matrix genes, MRTF-A/SRF inhibits adipogenesis. In addition, RhoA/ROCK-mediated actin cytoskeleton formation also promotes nuclear translocation of transcription factors yes-associated protein 1 (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), which promote osteogenesis and suppress adipogenesis (162). Moreover, a reciprocal enhancement of cell adhesion mediated by focal adhesion kinase activity and actin dynamics mediated by RhoA/ROCK signaling is involved in the commitment towards osteogenic versus adipogenic lineages of human adipose stem cells (163). In addition, RhoA/ROCK/AMPK/SREBP axis is involved in inhibiting adipogenesis and promoting osteogenesis during cell sheet formation of human adipose tissue-derived stem cells, which represents a cell culture model of spontaneous self-organization of multipotent mesenchymal stem/stromal cells (164). These studies support a negative role of RhoA/ROCK/F-actin/MRTF-A/SRF, RhoA/ROCK/F-actin/YAP/TAZ, or RhoA/ROCK/AMPK/SREBP pathway in the commitment of mesenchymal stem cells to the adipogenic lineage.
RhoA/ROCK pathway has also been shown to control a switch between adipogenesis and myogenesis of common mesenchymal precursors in vivo and in vitro (35) (Figure 2). In embryonic heart, ROCK inhibition could lead to fibrofatty replacement of cardiomyocytes in the right ventricle of adult mice resulting in arrhythmogenic right ventricular cardiomyopathy (165). Mechanistically, both Wnt/β-catenin and RhoA/ROCK pathways must be inactive for a significant increase of PPARγ expression, which is responsible for mediating arrhythmogenic right ventricular cardiomyopathy pathogenesis (166). Moreover, in embryonic heart, the RhoA/ROCK/F-actin/MRTF-A/SRF pathway is essential to maintaining cell-cell contacts of developing cardiomyocytes and inhibiting this pathway during cardiomyocyte differentiation primes cardiac progenitors to switch to the brown/beige adipocyte lineage in response to adipogenesis-inducing signals (114). Mechanistically, inhibition of RhoA/ROCK/F-actin/MRTF-A/SRF pathway due to impaired cell-cell contacts at the intercalated disc represses the MRTF-A/SRF‐regulated myogenic lineage commitment while increasing PPARγ gene expression and activation of beige adipogenic program. These studies support a negative role of the RhoA/ROCK/F-actin/MRTF-A/SRF pathway in directing the commitment of skeletal muscle progenitors and cardiomyocyte progenitors to the brown/beige adipocyte lineage.
In agreement with the contribution of RhoA/ROCK/F-actin/MRTF-A/SRF cascade in modulating recruitment of white as well as beige and brown adipocyte progenitors, a bone morphogenetic protein-7 (BMP7)-induced inhibition of RhoA/ROCK/F-actin/MRTF-A/SRF axis directs the commitment of adipose stromal progenitors to beige adipocyte over vascular lineages (16); knocking down SRF or MRTF-A/B in multipotent mesenchymal stem cells enhanced lineage commitment of mesenchymal progenitors to brown adipocyte lineage (115). Mechanistically, inhibition of SRF or MRTF-A/B attenuated TGF-β signaling pathway and increased BMP signaling pathway, suggesting a potential regulatory feedforward loop between MRTF/SRF and TGF-β/BMP for lineage specification in thermogenic adipocyte progenitors.
Adipocyte Differentiation
Inhibition of RhoA/ROCK pathway not only promotes recruitment of adipocyte progenitors, but also facilitates preadipocyte differentiation into mature adipocytes (34, 37, 152, 167–170) (Figure 2). Similar downstream pathways and crosstalk pathways with RhoA/ROCK are involved in both lineage commitment and differentiation of adipocytes. For instance, a cross-talk between the Rho/ROCK pathway and the Wnt/β-catenin pathway controls differentiation of preadipocytes (168); MRTF/SRF signaling suppresses preadipocyte differentiation (37, 167). It should be mentioned that adipocyte differentiation is accompanied by a pronounced change in the actin cytoskeleton, which is characterized by the disruption of the F-actin stress fibers and their reorganization into cortical F-actin structures which are essential for the completion of adipocyte differentiation. Both the MRTF/SRF signaling pathway (negative regulator) and the insulin/PI3K/Rac1 pathway (positive regulator) control the formation of adipocyte-associated cortical actin structures (170).
There is also mounting evidence supporting a negative impact of ROCK activity on brown and beige adipocyte differentiation by multiple mechanisms. Early studies support the contribution of RhoA/ROCK/IRS pathway to the suppression of brown adipogenesis through inhibiting insulin signaling (171–173). More recent studies support the concept that the RhoA/ROCK/F-actin/MRTF/SRF signaling pathway inhibits brown and beige adipogenesis and PPARγ expression (16, 115, 154). Knocking down SRF or MRTF-A/B in brown preadipocytes enhanced brown preadipocyte terminal differentiation associated with attenuated TGF-β signaling pathway and increased BMP signaling pathway (115).
During adipogenesis of white, beige and brown adipocytes, the protein expression and activity of RhoA, ROCK, MRTF-A/B and SRF are down-regulated (16, 115, 154). Both cyclic AMP and cyclic guanosine monophosphate contribute to the downregulation of RhoA/ROCK signaling (171–173). Moreover, among other upstream regulators, the regulator of G protein signaling 2 suppresses Gq/RhoA/ROCK signaling and promotes brown adipogenesis (174). Furthermore, PPARγ increases Rho GTPase-activating protein (GAP) DLC1 expression leading to suppression of RhoA/ROCK signaling, this provides a regulatory feedback loop between PPARγ and RhoA/ROCK pathways during white and brown adipocyte differentiation (175).
As mentioned above, the established general concept is that the inhibition of RhoA/ROCK-mediated actin cytoskeleton dynamics is required for the recruitment of progenitors and differentiation of white, brown and beige adipocytes. Recent studies support the concept that the grade of RhoA/ROCK inhibition can differently regulate these three types of adipocyte differentiation. For instance, there is an inverse relationship between RhoA/ROCK activity and the abundance of brown/beige adipocytes in fat depots (154). Specifically, ROCK1 and ROCK2 protein expression, total ROCK activity, especially ROCK2 activity were the lowermost in BAT, in addition, they were lower in subcutaneous WAT compared to epididymal WAT (154). In addition, β-adrenergic stimulation, which enhances beige adipocyte formation in subcutaneous WAT, down-regulates RhoA/ROCK protein expression and activity, including ROCK2 protein expression and total ROCK activity without affecting ROCK1 protein expression (154).
A negative role for ROCK2 in regulating beige adipocyte formation has been supported by the observations that reduction in ROCK2 signaling poises differentiating adipocytes towards beige adipocytes in vivo and in vitro (154). Indeed, acquired data from either systemic ROCK2+/- mouse model, or from ROCK2+/KD mouse model in which mouse harboring an allele with a kinase-dead (KD) mutation, showed a lean body mass phenotype during aging and was associated with increased amounts of beige cells in subcutaneous WAT and increased thermogenic gene expression, including UCP1 in WAT and BAT (154). ROCK2+/- mice also exhibited increased sensitivity to the browning effects of β-adrenergic stimulation resulting in increased beige cell formation in subcutaneous WAT, and increased BAT mass. In vitro differentiated ROCK2+/- SV cells isolated from subcutaneous WAT or BAT exhibited increased beige or brown adipogenesis, respectively (154). It remains to be determined through cell type-specific knockout approach if ROCK2 activity inhibits the recruitment of beige adipocyte progenitors, differentiation of preadipocytes to beige adipocytes, and the transdifferentiation from mature white adipocytes in vivo.
Adipose RhoA/ROCK Activation Contributes to Adipocyte Hypertrophy, Inflammation and Fibrosis
WAT undergoes expansion and extensive remodeling during diet-induced obesity, including increased adipose cell size (hypertrophy) and cell number (hyperplasia) after short-term overfeeding. This process is accompanied by the development of a chronic low-grade inflammation in adipose tissue, presenting as infiltration of immune cells and increased levels of pro-inflammatory cytokines, then followed by the development of tissue fibrosis after chronic overfeeding. Hypertrophic adipocytes are less responsive to insulin, and adipocyte size serves as a predictor for development of type 2 diabetes.
Mouse on HFD is linked with augmented RhoA/ROCK activity in adipose tissue where increased F-actin assembly and changed expression of actin-regulating proteins favor actin polymerization (29, 30, 154, 176), which further enhances adipocyte stretch due to excess lipid accumulation and creates a vicious cycle of adipocyte hypertrophy and dysfunction (29). Although there is a unanimous agreement on the increased total ROCK activity in adipose tissue from HFD-fed mice (29, 176), there are also discrepancies on increased isoform-specific activity (30, 121). For instance, in one study, a significant increase in ROCK2 activity without changes in its protein expression was reported in adipose tissue from HFD-fed mice, while ROCK1 protein expression and activity remained unchanged (121). But another study observed increased activity of both isoforms, ROCK1 and ROCK2 in adipose tissue of HFD-fed mice, however, no change was found in protein expression of either isoform (30).
Transgenic mice that overexpress an adipocyte-specific, dominant-negative form of RhoA showed decreased ROCK activity in adipocytes, decreased adipocyte hypertrophy and dysfunction, and reduced macrophages in adipose tissue (29). Adipocyte-specific ROCK1 knockout mice showed modest amelioration in insulin sensitivity and insulin signaling, but no significant effects on adipocyte hypertrophy and inflammation under HFD (30), suggesting that the ROCK2 isoform in adipose tissue may play a dominant role in controlling adiposity and insulin sensitivity. Indeed, partial deletion of ROCK2 in mice demonstrated diminished adipocyte hypertrophy, inflammation and fibrosis under HFD (121, 154). Future animal study with adipocyte specific ROCK2 knockout should help determine adipocyte autonomous function of ROCK2 in vivo. Mechanistically, downstream of RhoA/ROCK-mediated F-actin assembly, nuclear translocation of MRTF-A suppresses the expression of IRS1 and PPARγ in hypertrophic adipocytes, contributing to adipocyte dysfunction and insulin resistance (116). Moreover, RhoA/ROCK-mediated actomyosin contractility promotes nuclear translocation of the transcriptional co-activators YAP/TAZ which suppress expression of pro-apoptotic factor Bim and protect against white adipocyte cell death during obesity (177).
In addition to increasing adipocyte size, visceral WAT adipocytes respond to chronic overfeeding by adopting a fibroblast-like phenotype, which is characterized by enhanced expression of extracellular matrix proteins, focal adhesion and cytoskeletal genes, and suppression of many adipocyte programs most notably those associated with mitochondria (118). In obesity, visceral WAT adipocytes progressively become metabolically flawed due to the acquirement of fibrogenic functions. During this process, RhoA/ROCK/MRTF-A/SRF signaling within the adipocytes contributes to both upregulation of morphological genes as well as suppression of mitochondrial programs (118). Moreover, outside of adipocytes, RhoA/ROCK/MRTF-A/SRF activation within visceral WAT shifts the fate of perivascular progenitors to fibrogenic versus adipogenic progenitors (117). Therefore, both cellular mechanisms of RhoA/ROCK/MRTF-A/SRF signaling contribute to chronic obesity-induced fibrosis and metabolic dysfunction of adipose tissue.
Adipose RhoA/ROCK Activation Contributes to Obesity and Insulin Resistance, and Inhibits Energy Expenditure
As mentioned above, transgenic mice overexpressing an adipocyte-specific, dominant-negative form of RhoA exhibited decreased visceral adipose tissue remodeling associated with decreased HFD-induced weight gain and improved glucose metabolism (29). Partial deletion of ROCK2 in mice also decreased adipose tissue remodeling associated with decreased HFD-induced adipose and systemic insulin resistance (121, 154). However, there is still discrepancy on the effects of partial deletion of ROCK2 on HFD-induced weight gain from studies using mice of different genetic background (42, 121, 154).
In addition to visceral WAT remodeling, we recently revealed a role for ROCK2 as a negative regulator of thermogenic programs in WAT and BAT through facilitating actin cytoskeleton assembly (154). ROCK2+/- mice on HFD demonstrated increased sensitivity to the browning effects of beta-adrenergic stimulation, increased energy expenditure with reduced obesity, and improved insulin sensitivity. In contrast to ROCK2+/- and ROCK2+/KD mice, ROCK1+/- and ROCK1+/KD mouse models didn’t exhibit a lean body mass phenotype (154) which is consistent with adipocyte-specific ROCK1 knockout mice with modest amelioration in systemic insulin sensitivity and adipose insulin signaling, but with no significant effects on body weight gain under HFD (30). Thus, inhibition of RhoA/ROCK2 in adipose tissue may provide a potential therapeutic strategy to combat obesity and insulin resistance (Figure 1).
Metabolic Roles of Liver, Skeletal Muscle, Brain and Vascular RhoA/ROCK Signaling Pathways in Energy Expenditure, Diet-Induced Obesity and Insulin Resistance
Aforementioned both ROCK1 and ROCK2 are ubiquitously expressed across tissues in human and mouse (27). Importantly, up-regulated ROCK activity has been implicated in the pathogenesis of all aspects of metabolic syndrome including obesity, insulin resistance, dyslipidemia and hypertension (29–33). Here, we focus on the current status of RhoA/ROCK research in major metabolic organs that play vital roles in the development of obesity and insulin resistance. We also devoted special attention to the impacts of RhoA/ROCK activation in liver, skeletal muscle and brain on adipose tissue remodeling and energy expenditure through paracrine mechanisms (Figure 1).
Metabolic Roles of Liver RhoA/ROCK Signaling
Both liver and skeletal muscle are major metabolic tissues involved in the regulation of whole-body glucose, lipid and energy homeostasis. Systemic overexpression of dominant-negative ROCK in mice or systemic ROCK inhibition with fasudil improved HFD-induced metabolic disorders including obesity, hypercholesterolemia and glucose intolerance in mice (47, 48). These metabolic improvements were mediated through the activation of AMPK pathway in liver and skeletal muscle which contributed to increased whole body energy expenditure (47, 48). In addition to systemic metabolic improvements, treatment with ROCK inhibitors ameliorates liver fibrosis in diabetic non-alcoholic steatohepatitis (178–180).
It is noteworthy that different from adipose tissues where ROCK2 appears to the major isoform contributing to ROCK activity, ROCK1 is reportedly the major isoform in liver and mediating diet-induced obesity and insulin resistance through the ROCK1/AMPK signaling pathway (47, 48, 59, 125, 181) (Figure 1). Liver-specific knockout of ROCK1 mice were resistant to diet-induced obesity due to increased energy expenditure and thermogenic gene expression in adipose tissue, whereas hepatic overexpression of ROCK1 was sufficient to promote adiposity, insulin resistance, and hepatic lipid accumulation in mice fed with a HFD (125). In addition, ROCK1/AMPK/SREBP1c axis was reported to regulate hepatic lipogenesis and contribute to nonalcoholic fatty liver diseases (125). Furthermore, a paracrine mechanism through liver-released hepatokines including FGF21, increased in liver of hepatic ROCK1 knockout mice, potentially mediating the effects of hepatic ROCK1 knockout on inducing thermogenic gene expression in adipose tissue and increasing energy expenditure (125). Although these studies established a role for hepatic ROCK1 in promoting adiposity, insulin resistance, and hepatic lipid accumulation, the role for hepatic ROCK2 in regulating these metabolic functions remains to be explored.
Metabolic Roles of Skeletal Muscle RhoA/ROCK Signaling
In skeletal muscle, ROCK1 and total ROCK activity have been reported to play either positive or negative metabolic roles depending on metabolic conditions (38, 41, 126, 182–185) (Figure 1). On one hand, ROCK-mediated IRS1 phosphorylation in skeletal muscle can positively impact insulin signaling under normal diet or in exercised animals (38, 41, 183, 184). On the other hand, ROCK1/AMPK/SREBP1c and ROCK/IRS1 pathways have been found involved in lipotoxicity and insulin resistance on HFD (126, 185). A finding indicated that increased ROCK1 activity in skeletal muscle through muscle-specific expression of constitutive ROCK1 reduced irisin expression in muscle resulting in a low level of irisin in circulation. Low level of irisin suppressed UCP1 expression in BAT and subcutaneous WAT through a paracrine mechanism (134). Irisin administration to these mice partially reversed insulin resistance and obesity, and these changes were associated with increased expression of UCP1 in subcutaneous WAT (134). Hence, the reduced adipocyte browning contributes to reduced heat production, development of obesity and insulin resistance in transgenic mice. It remains to be determined if ROCK2 activation in skeletal muscle also regulate these metabolic functions.
Metabolic Roles of Central Nervous System RhoA/ROCK Signaling
Different from liver where ROCK1 activation plays detrimental metabolic roles, ROCK1 plays a positive role in leptin signaling in the brain which controls feeding behavior, energy expenditure, and glucose metabolism (58, 186, 187) (Figure 1). Diet-induced and genetic forms (db/db and ob/ob) of obesity were associated with reduced ROCK1 activity in murine hypothalamic neurons (187). In addition, mice lacking ROCK1 in pro-opiomelanocortin and agouti-related protein neurons, which are mediators of leptin action, displayed obesity and impaired leptin sensitivity associated with increased food intake, reduced energy expenditure and locomotor activity (186, 187). Mechanistically, the leptin/ROCK1/JAK2 pathway promotes downstream signaling pathways of leptin including Stat3 and PI3K signaling in hypothalamic neurons (186, 187). Moreover, mice lacking RhoA in hypothalamic tyrosine hydroxylase neurons showed increased sensitivity to ghrelin and decreased sensitivity to leptin, resulting in increased food intake and development of obesity (188). It must be noted that systemic ROCK1-/-, ROCK1+/- and ROCK1+/KD mouse models did not reveal overeating behavior and increasingly gained body weight compared to control mice (38, 154), suggesting that the central effects of ROCK1 deficiency in hypothalamic neurons are compensated by opposite metabolic effects of ROCK1 deficiency in other cells and organs. It remains to be determined through cell type-specific knockout approach as performed for ROCK1 if ROCK2 in central nervous system regulates food intake and energy expenditure.
Metabolic Roles of Vascular RhoA/ROCK Signaling and Metabolic Effects Of Systemic Inhibition of RhoA/ROCK Signaling
Fasudil is a non-isoform selective inhibitor approved for human use in Japan for cerebral vasospasm after surgery of subarachnoid hemorrhage (189, 190). Studies using ROCK inhibitors in animal models of obesity, diabetes, and associated complications have demonstrated beneficial outcomes (29, 43–53). However, ROCK inhibitors have not yet been accepted for treating metabolic disorders due to potential systemic side effects, including hypotension caused by smooth muscle relaxation due to both ROCK1 and ROCK2 are similarly inhibited (54–56, 191).
In recent studies described above, the major organs mediating the beneficial metabolic effects of systemic ROCK inhibition includes adipose tissues (29, 121, 154), liver (47, 48, 178, 180), skeletal muscle (47, 48), hearts (42, 192) and intestine (193). In earlier studies, the beneficial effects have been focused on the vascular systems. For instance, ROCK inhibitor treatment reduced atherosclerotic lesion formation and vascular dysfunction in diabetic mice (49, 194–196). Mechanistically, perivascular adipose tissue in HFD obese mice releases reactive oxygen species (ROS), vasoconstrictors and proinflammatory factors, which in turn increase RhoA/ROCK-mediated vascular smooth muscle cell (VSMC) contractility and vascular remodeling (197–199). In addition, hyperglycemia, hyperlipidemia and advanced glycation endproducts activate RhoA/ROCK pathway in endothelial cells and VSMCs. This results in endothelial dysfunction and increased contractility, proliferation, migration and production of extracellular matrix proteins of VSMCs leading to obesity-induced hypertension and atherosclerosis (148, 200–204).
Growing Research Areas in RhoA/ROCK Signaling With Metabolic Implications
In addition to the established research areas described above, there are several rapid growing research areas on RhoA/ROCK signaling with noticeable implications on metabolic regulation. Here, we focus on the current understanding of RhoA/ROCK research on circadian rhythm, mitochondrial function, mitophagy, and miRNAs. Future studies are expected to fully explore their regulation and function in metabolic tissues.
Metabolic Association of RhoA/ROCK Signaling With Circadian Rhythm
Circadian rhythm exhibits 24-h cycles in the body, and metabolic processes are under circadian regulation. Disruption of the circadian rhythms can disrupt clock gene expressions in both central (e.g., suprachiasmatic nucleus) and peripheral tissues (e.g., adipose tissue, liver and vasculature) causing dysfunction in energy metabolism and nutrient metabolic homeostasis, and leading to development of metabolic disorders including obesity, diabetes, dyslipidemia and hypertension (205). Numerous studies support a role for ROCKs in regulating circadian rhythm of vascular contractility, giving rise to blood pressure circadian rhythm, which is essential for cardiovascular health (206, 207). For instance, the circadian rhythm of ROCK1 and ROCK2 expression in vascular smooth muscle was disrupted in db/db obese mice (208). In addition, ROCK2 played a pivotal role in generating the intrinsic circadian rhythm of vascular contractility in the mouse aorta as ROCK2 expression exhibited the circadian oscillation in phase with that of MLC2 phosphorylation (209, 210). Moreover, an altered circadian ROCK activity in circulating leukocytes was associated with alterations in coronary vasomotor responses and autonomic activity in vasospastic angina patients (211). Mechanistically, the circadian ROCK2 expression in vasculature is under the control of clock genes RORα and Bmal1 as both activate the transcription of ROCK2 (209, 212). ROCK1 expression was found negatively regulated by clock gene CLOCK in human umbilical vein endothelial cells (213). In addition to being transcriptionally regulated by clock genes, RhoA/ROCK signaling can act upstream and regulate clock gene activity. For instance, RhoA/ROCK-mediated mechano-sensing to extracellular stiffness regulates the activity of the core circadian clock complex in primary breast epithelial cells (214). The role RhoA/ROCK signaling in controlling or mediating circadian rhythm in other metabolic tissues currently remains to be explored.
Impacts of RhoA/ROCK Signaling on Mitochondrial Function and Mitophagy
The mitochondria play critical roles in oxidative phosphorylation and energy metabolism in all metabolic tissues. As described above, RhoA/ROCK signaling contributes to the regulation of thermogenic gene expression and mitochondrial biogenesis in adipose tissue. Inhibition of RhoA/ROCK signaling results in increased PPARγ expression, which promotes thermogenic gene expressions and energy expenditure in adipose tissue (Figure 1). In liver and skeletal muscle, inhibition of RhoA/ROCK signaling induces thermogenic gene expression in adipose tissue through a paracrine mechanism (Figure 2). Due to the critical roles of mitochondria in regulating cellular and systemic energy homeostasis, it is necessary to discuss some recent advances in our understanding on the impacts of RhoA/ROCK signaling to mitochondrial biology.
RhoA/ROCK signaling has been reported to function both upstream and downstream of mitochondria. On one hand, the mitochondria/ROS/RhoA/ROCK pathway is well-documented, which is trigged by mitochondrial dysfunction leading to the production of excessive ROS and subsequently RhoA/ROCK activation. For example, impaired mitochondrial function in the perivascular adipocytes of obese mice produced excessive ROS which triggered RhoA/ROCK activation resulting in increased vascular contractility (198, 215). Similarly, a critical mechanism contributing to the development of pulmonary hypertension in vascular hypercontraction is hypoxia. Under this condition, excessive ROS was produced by affected mitochondria that triggered activation of RhoA/ROCK signaling (216, 217). On the other hand, the RhoA/ROCK/dynamin-related protein-1 (Drp1) and F-actin/mitochondrial fission/ROS/apoptosis pathway has been reported in various diseases including diabetes, hypertension and Parkinson’s disease. For example, hyperglycemia-induced mitochondrial fission and dysfunction in endothelial cells is mediated by activating the RhoA/ROCK1/Drp1 signaling pathway, thus contributing to vascular complications of diabetes (218–220). This pathway also contributes to hypercontraction of VSMCs involved in artery constriction (221), cardiac inflammatory injury (222, 223), and dopaminergic nerve cell apoptosis in Parkinson’s disease (224).
Interestingly, inhibition of RhoA/ROCK pathways can also improve mitochondrial quality control by activating mitophagy. In the context of Parkinson’s disease, recent evidence supports beneficial effects of ROCK inhibition in promoting the Parkin-mediated mitophagy pathway which removes damaged mitochondria in dopaminergic neurons (225, 226). Mechanistically, ROCK2 inhibition promotes the activity of this pathway by increasing the recruitment of hexokinase 2, a positive regulator of Parkin, to mitochondria. This recruitment enables increased docking of impaired mitochondria to lysosomes, subsequently enhancing the removal of damaged mitochondria from cells (225, 226). In addition to the neuronal ROCK2/Parkin/mitophagy axis, the ROCK1/Parkin/mitophagy pathway was reported to mediate cisplatin-related resistance in lung cancer cells (227). It will be interesting to further explore if the ROCK/Parkin/mitophagy axis plays a role in regulating mitochondrial quality control in metabolic diseases like obesity and diabetes in future studies.
miRNAs Contribute to Isoform-Selective Activation and/or Inhibition of RhoA/ROCK Signaling in Metabolic Tissues
Aforesaid recent research has identified miRNAs functioning as endogenous physiologic activators and/or inhibitors of ROCK1 and ROCK2 to achieve isoform-selective activation or inhibition in metabolic tissues (Table 1). The upstream activators of ROCK are affected by changing miRNAs expression levels (Table 1). For instance, when miR-125a-3p expression was up-regulated which then targeted RhoA leading to a down-regulation of RhoA/ROCK activity and followed by increased adipogenesis in subcutaneous WAT in patients with multiple symmetric lipomatosis (79). On the other hand, when miR-133a expression in gastric smooth muscle of ob/ob mice was down-regulated, as the result of RhoA was targeted, there was an increase in RhoA/ROCK signaling and muscle contraction (78). Furthermore, down-regulation of miRNA-141 was observed in penile cavernous smooth muscle cells of rats with diabetic erectile dysfunction, and the decreased expression of miRNA-141 was also associated with up-regulation of RhoA/ROCK signaling (86). Even though miRNAs expression changes are frequently linked to up- or down-regulation changes in RhoA/ROCK signaling pathway, the metabolic outcome is detrimental in most cases.
Known that ROCK1 and ROCK2 have different miRNA-binding sites, therefore, each isoform is individually up- and down-regulated based on available data (Table 1). ROCK1 was found to be a target of miR-146a-5p which was down-regulated in liver tissue taken from models of a nonalcoholic fatty liver; miR-146a-5p down-regulation in these models promoted lipid accumulation in hepatocytes through inhibiting adenosine monophosphate-activated protein kinase (AMPK) pathway (80). Down-regulation of miR-145 was also observed in cultured human vascular smooth muscle cells (VSMCs) exposed to high glucose; miR-145 down-regulation promoted ROCK1 expression and increased proliferation and migration of VSMCs which are important steps in diabetic atherosclerosis (89). In circulating peripheral blood and liver tissue of db/db mice, raised miR-324-5p expression was associated with hyperglycemia or hyperlipidemia leading to impaired glucose and lipid metabolism due to suppressed ROCK1 expression (81). During period of physical inactivity, the increased skeletal muscle miR-148b expression triggered the downregulation of ROCK1 expression and insulin resistance in humans and mice (82). In contrast to the detrimental effects of miRNA expression fluctuations in the metabolic tissues, up-regulation of miR-217 was found to be beneficial in both VSMCs treated by high glucose and in aorta VSMCs of diabetic rats, miR-217 up-regulation suppressed ROCK1expression in these cases and inhibited excessive proliferation and migration of VSMCs (90).
ROCK2, instead, was found to be a target of miR-455-3p which was down-regulated in the human mesangial cells and human proximal tubule epithelial cells when stimulating with high glucose or transforming growth factor beta 1 (TGF-β1); restoring miR-455-3p expression in a rat diabetic nephropathy model decreased ROCK2 expression and suppressed renal fibrosis (83). Down-regulation of miR-10a and miR-139b in VSMCs induced ROCK2 expression and contributed to diabetes and hyperlipidemia-induced vascular hyperreactivity (77). Lastly, some miRNAs target both ROCK1 and ROCK2, such as miR-497, which was increased in glomerular endothelial cells of diabetic rats and contributed to endothelial-to-mesenchymal transition and albuminuria through suppressing ROCK1 and ROCK2 (91). Integrative analysis of gene expression profiles including miRNAs and their target gene network in metabolic tissues has contributed significantly to our understanding of insulin resistance and obesity (228). Future studies in this area are expected to further expand the list of miRNAs targeting RhoA/ROCK pathways in metabolic diseases (Table 1) and elucidate their biological role at mechanistic level.
Concluding Remarks
Studies using chemical inhibitors, whole-body and tissue-specific genetic manipulations in animal models have demonstrated that abnormal RhoA/ROCK signaling contributes to the pathogenesis of obesity and type 2 diabetes. These studies have identified that the ROCK isoforms have critical functions in various metabolic tissues, including adipose tissue, liver, skeletal muscle, and hypothalamus where they regulate thermogenesis, glucose and lipid metabolism, food intake, and locomotor activity (Figure 1). Significant progress has been made in dissecting the molecular and cellular mechanisms underlying these aberrant global or tissue-specific metabolism in which ROCK inhibition mainly shows beneficial outcome. In adipose tissue, ROCK inhibition provides favorable effects through reducing WAT remodeling under metabolic stress conditions, promoting BAT thermogenic function and browning process of WAT, leading to increased energy expenditure and reduced adiposity. Metabolic beneficial effects of ROCK inhibition are consistently observed in liver, skeletal muscle, vascular system and kidney under metabolic stress conditions. In contrast, ROCK inhibition in skeletal muscle and hypothalamus is metabolically detrimental under basal condition. These tissue-specific beneficial and detrimental effects of ROCK inhibition highlight the importance of tissue-specific targeting of ROCK activity via innovative delivery strategies (229–232).
Considering recent advances in understanding RhoA/ROCK signaling in regulating metabolic functions, promoting brown adipogenesis and browning process of WAT by ROCK inhibition, particularly ROCK2 inhibition, have emerged as promising strategies to treat obesity and its related metabolic disorders. However, a ROCK2-selelective inhibitor KD025, initially bearing great therapeutic potential, has been discovered to exhibit an anti-adipogenic activity through inhibiting casein kinase 2, a ROCK2-independent mechanism. With this off-target effect, it makes KD025 possibly unsuitable as a molecular tool to enhance thermogenesis.
We anticipate that future research in the field has three main directions (1): continuous dissection of molecular mechanisms and cellular functions of RhoA/ROCK signaling in the pathogenesis of obesity and type 2 diabetes with increased insights into mitochondrial dysfunction and circadian rhythm disruption; (2) continuous development of ROCK isoform-selective inhibitors and exploration of the endogenous physiologic activators and inhibitors of ROCK1 and ROCK2 such as miRNAs to achieve isoform-selective activation or inhibition in metabolic tissues; and (3) increasing efforts on studying RhoA/ROCK pathways in human obesity and other related metabolic disease, which is pathophysiological more complex than rodent models. Together, all these research efforts will improve our understanding of RhoA/ROCK signaling in thermogenesis and energy expenditure with the translational goal of developing new treatment for obesity and related disease.
Author Contributions
JS and LW contributed to the idea for the article, performed the literature search and data analysis, drafted and revised the article. JS and LW agreed with the content of this manuscript and have approved the manuscript before submission.
Funding
This work was supported by National Institutes of Health grants HL151480 (to LW and JS), HL107537 (to LW), HL134599 (to LW), Riley Children’s Foundation (RCF) (to JS and to LW), the Indiana Clinical and Translational Sciences Institute (CTSI) including Biomedical Research Grant of Indiana University School of Medicine 2286128 (to JS) and the Pilot Funding for Research Use of Core Facilities 2286118 (to JS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
Actin filaments, F-actin; AMPK, adenosine monophosphate -activated protein kinase; BAT, brown adipose tissue; BMP, bone morphogenetic protein; Drp1, dynamin-related protein-1; G-actin, globular actin; GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; HFD, high-fat diet; IR, insulin receptor; IRS1, insulin receptor substrate-1; KD, kinase-dead; miRNA, microRNAs; LIMK, LIM kinases; MRTF, myocardin-related transcription factor; MLC, myosin light chain; MYPT1, myosin binding subunit of myosin light chain phosphatase-1; PI3K, phosphatidylinositol 3-kinase; PPARγ, peroxisome proliferator-activated receptor γ; ROCK, Rho-associated coiled-coil-containing protein kinase; ROS, reactive oxygen species; SRF, serum response factor; SREBP1c, sterol regulatory element-binding protein-1c; SV, stromal vascular; TGF-β1, transforming growth factor beta 1; TAZ, transcriptional coactivator with PDZ-binding motif; UCP1, uncoupling protein-1; VSMCs, vascular smooth muscle cells; WAT, white adipose tissue; YAP, yes-associated protein 1.
References
1. O’Neill S, O’Driscoll L. Metabolic Syndrome: A Closer Look at the Growing Epidemic and its Associated Pathologies. Obes Rev (2015) 16:1–12. doi: 10.1111/obr.12229
2. Pedersen SD. Metabolic Complications of Obesity. Best Pract Res Clin Endocrinol Metab (2013) 27:179–93. doi: 10.1016/j.beem.2013.02.004
3. Vucenik I, Stains JP. Obesity and Cancer Risk: Evidence, Mechanisms, and Recommendations. Ann NY Acad Sci (2012) 1271:37–43. doi: 10.1111/j.1749-6632.2012.06750.x
4. Fonseca VA, Kirkman MS, Darsow T, Ratner RE. The American Diabetes Association Diabetes Research Perspective. Diabetes (2012) 61:1338–45. doi: 10.2337/db12-0435
5. Cowie CC, Rust KF, Ford ES, Eberhardt MS, Byrd-Holt DD, Li C, et al. Full Accounting of Diabetes and Pre-Diabetes in the U.S. Population in 1988-1994 and 2005-2006. Diabetes Care (2009) 32:287–94. doi: 10.2337/dc08-1296
6. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of Childhood and Adult Obesity in the United States, 2011-2012. JAMA (2014) 311:806–14. doi: 10.1001/jama.2014.732
7. World Health Organization. Fact Sheet: Obesity and Overweight (2021). Available at: https://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight.
8. Centers of Disease and Prevention. Prevalence of Overweight, Obesity, and Severe Obesity Among Children and Adolescents Aged 2-19 Years: United States, 1963-1965 Through 2017-2018 (2020). Available at: https://www.cdc.gov/nchs/data/hestat/obesity-child-17-18/overweight-obesity-child-H.pdf.
9. Harms M, Seale P. Brown and Beige Fat: Development, Function and Therapeutic Potential. Nat Med (2013) 19:1252–63. doi: 10.1038/nm.3361
10. Kajimura S, Spiegelman BM, Seale P. Brown and Beige Fat: Physiological Roles Beyond Heat Generation. Cell Metab (2015) 22:546–59. doi: 10.1016/j.cmet.2015.09.007
11. Nedergaard J, Ricquier D, Kozak LP. Uncoupling Proteins: Current Status and Therapeutic Prospects. EMBO Rep (2005) 6:917–21. doi: 10.1038/sj.embor.7400532
12. Leitner BP, Huang S, Brychta RJ, Duckworth CJ, Baskin AS, McGehee S, et al. Mapping of Human Brown Adipose Tissue in Lean and Obese Young Men. Proc Natl Acad Sci USA (2017) 114:8649–54. doi: 10.1073/pnas.1705287114
13. Mulya A, Kirwan JP. Brown and Beige Adipose Tissue: Therapy for Obesity and Its Comorbidities? Endocrinol Metab Clin North Am (2016) 45:605–21. doi: 10.1016/j.ecl.2016.04.010
14. Graja A, Schulz TJ. Mechanisms of Aging-Related Impairment of Brown Adipocyte Development and Function. Gerontology (2015) 61:211–7. doi: 10.1159/000366557
15. Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, et al. Prdm16 Determines the Thermogenic Program of Subcutaneous White Adipose Tissue in Mice. J Clin Invest (2011) 121:96–105. doi: 10.1172/JCI44271
16. McDonald ME, Li C, Bian H, Smith BD, Layne MD, Farmer SR. Myocardin-Related Transcription Factor A Regulates Conversion of Progenitors to Beige Adipocytes. Cell (2015) 160:105–18. doi: 10.1016/j.cell.2014.12.005
17. Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection From Obesity and Diabetes by Blockade of TGF-Beta/Smad3 Signaling. Cell Metab (2011) 14:67–79. doi: 10.1016/j.cmet.2011.04.013
18. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and Importance of Brown Adipose Tissue in Adult Humans. N Engl J Med (2009) 360:1509–17. doi: 10.1056/NEJMoa0810780
19. Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, et al. High Incidence of Metabolically Active Brown Adipose Tissue in Healthy Adult Humans: Effects of Cold Exposure and Adiposity. Diabetes (2009) 58:1526–31. doi: 10.2337/db09-0530
20. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold-Activated Brown Adipose Tissue in Healthy Men. N Engl J Med (2009) 360:1500–8. doi: 10.1056/NEJMoa0808718
21. Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, et al. Recruited Brown Adipose Tissue as an Antiobesity Agent in Humans. J Clin Invest (2013) 123:3404–8. doi: 10.1172/JCI67803
22. Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elia E, Kessler SH, Kahn PA, et al. Activation of Human Brown Adipose Tissue by a Beta3-Adrenergic Receptor Agonist. Cell Metab (2015) 21:33–8. doi: 10.1016/j.cmet.2014.12.009
23. Betz MJ, Enerback S. Human Brown Adipose Tissue: What We Have Learned So Far. Diabetes (2015) 64:2352–60. doi: 10.2337/db15-0146
24. Lidell ME, Betz MJ, Enerback S. Brown Adipose Tissue and its Therapeutic Potential. J Intern Med (2014) 276:364–77. doi: 10.1111/joim.12255
25. Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, et al. Rho-Associated Kinase, a Novel Serine/Threonine Kinase, as a Putative Target for Small GTP Binding Protein Rho. EMBO J (1996) 15:2208–16. doi: 10.1002/j.1460-2075.1996.tb00574.x
26. Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, et al. The Small GTP-Binding Protein Rho Binds to and Activates a 160 kDa Ser/Thr Protein Kinase Homologous to Myotonic Dystrophy Kinase. EMBO J (1996) 15:1885–93. doi: 10.1002/j.1460-2075.1996.tb00539.x
27. Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, Two Isoforms of Rho-Associated Coiled-Coil Forming Protein Serine/Threonine Kinase in Mice. FEBS Lett (1996) 392:189–93. doi: 10.1016/0014-5793(96)00811-3
28. Leung T, Chen XQ, Manser E, Lim L. The P160 RhoA-Binding Kinase ROK Alpha is a Member of a Kinase Family and is Involved in the Reorganization of the Cytoskeleton. Mol Cell Biol (1996) 16:5313–27. doi: 10.1128/MCB.16.10.5313
29. Hara Y, Wakino S, Tanabe Y, Saito M, Tokuyama H, Washida N, et al. Rho and Rho-Kinase Activity in Adipocytes Contributes to a Vicious Cycle in Obesity That may Involve Mechanical Stretch. Sci Signal (2011) 4:ra3. doi: 10.1126/scisignal.2001227
30. Lee SH, Huang H, Choi K, Lee DH, Shi J, Liu T, et al. ROCK1 Isoform-Specific Deletion Reveals a Role for Diet-Induced Insulin Resistance. Am J Physiol Endocrinol Metab (2014) 306:E332–43. doi: 10.1152/ajpendo.00619.2013
31. Liu PY, Chen JH, Lin LJ, Liao JK. Increased Rho Kinase Activity in a Taiwanese Population With Metabolic Syndrome. J Am Coll Cardiol (2007) 49:1619–24. doi: 10.1016/j.jacc.2006.12.043
32. Liu L, Tan L, Lai J, Li S, Wang DW. Enhanced Rho-Kinase Activity: Pathophysiological Relevance in Type 2 Diabetes. Clin Chim Acta (2016) 462:107–10. doi: 10.1016/j.cca.2016.09.003
33. Jahani V, Kavousi A, Mehri S, Karimi G. Rho Kinase, a Potential Target in the Treatment of Metabolic Syndrome. BioMed Pharmacother (2018) 106:1024–30. doi: 10.1016/j.biopha.2018.07.060
34. Noguchi M, Hosoda K, Fujikura J, Fujimoto M, Iwakura H, Tomita T, et al. Genetic and Pharmacological Inhibition of Rho-Associated Kinase II Enhances Adipogenesis. J Biol Chem (2007) 282:29574–83. doi: 10.1074/jbc.M705972200
35. Sordella R, Jiang W, Chen GC, Curto M, Settleman J. Modulation of Rho GTPase Signaling Regulates a Switch Between Adipogenesis and Myogenesis. Cell (2003) 113:147–58. doi: 10.1016/S0092-8674(03)00271-X
36. McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell Shape, Cytoskeletal Tension, and RhoA Regulate Stem Cell Lineage Commitment. Dev Cell (2004) 6:483–95. doi: 10.1016/S1534-5807(04)00075-9
37. Nobusue H, Onishi N, Shimizu T, Sugihara E, Oki Y, Sumikawa Y, et al. Regulation of MKL1 via Actin Cytoskeleton Dynamics Drives Adipocyte Differentiation. Nat Commun (2014) 5:3368. doi: 10.1038/ncomms4368
38. Lee DH, Shi J, Jeoung NH, Kim MS, Zabolotny JM, Lee SW, et al. Targeted Disruption of ROCK1 Causes Insulin Resistance in vivo. J Biol Chem (2009) 284:11776–80. doi: 10.1074/jbc.C900014200
39. Furukawa N, Ongusaha P, Jahng WJ, Araki K, Choi CS, Kim HJ, et al. Role of Rho-Kinase in Regulation of Insulin Action and Glucose Homeostasis. Cell Metab (2005) 2:119–29. doi: 10.1016/j.cmet.2005.06.011
40. Chun KH, Araki K, Jee Y, Lee DH, Oh BC, Huang H, et al. Regulation of Glucose Transport by ROCK1 Differs From That of ROCK2 and is Controlled by Actin Polymerization. Endocrinology (2012) 153:1649–62. doi: 10.1210/en.2011-1036
41. Chun KH, Choi KD, Lee DH, Jung Y, Henry RR, Ciaraldi TP, et al. In Vivo Activation of ROCK1 by Insulin is Impaired in Skeletal Muscle of Humans With Type 2 Diabetes. Am J Physiol Endocrinol Metab (2011) 300:E536–42. doi: 10.1152/ajpendo.00538.2010
42. Soliman H, Nyamandi V, Garcia-Patino M, Varela JN, Bankar G, Lin G, et al. Partial Deletion of ROCK2 Protects Mice From High-Fat Diet-Induced Cardiac Insulin Resistance and Contractile Dysfunction. Am J Physiol Heart Circ Physiol (2015) 309:H70–81. doi: 10.1152/ajpheart.00664.2014
43. Kanda T, Wakino S, Homma K, Yoshioka K, Tatematsu S, Hasegawa K, et al. Rho-Kinase as a Molecular Target for Insulin Resistance and Hypertension. FASEB J (2006) 20:169–71. doi: 10.1096/fj.05-4197fje
44. Kikuchi Y, Yamada M, Imakiire T, Kushiyama T, Higashi K, Hyodo N, et al. A Rho-Kinase Inhibitor, Fasudil, Prevents Development of Diabetes and Nephropathy in Insulin-Resistant Diabetic Rats. J Endocrinol (2007) 192:595–603. doi: 10.1677/JOE-06-0045
45. Peng F, Wu D, Gao B, Ingram AJ, Zhang B, Chorneyko K, et al. RhoA/Rho-Kinase Contribute to the Pathogenesis of Diabetic Renal Disease. Diabetes (2008) 57:1683–92. doi: 10.2337/db07-1149
46. Li CB, Li XX, Chen YG, Gao HQ, Bao CM, Liu XQ, et al. Myocardial Remodeling in Rats With Metabolic Syndrome: Role of Rho-Kinase Mediated Insulin Resistance. Acta Biochim Pol (2012) 59:249–54. doi: 10.18388/abp.2012_2146
47. Noda K, Nakajima S, Godo S, Saito H, Ikeda S, Shimizu T, et al. Rho-Kinase Inhibition Ameliorates Metabolic Disorders Through Activation of AMPK Pathway in Mice. PLoS One (2014) 9:e110446. doi: 10.1371/journal.pone.0110446
48. Noda K, Godo S, Saito H, Tsutsui M, Shimokawa H. Opposing Roles of Nitric Oxide and Rho-Kinase in Lipid Metabolism in Mice. Tohoku J Exp Med (2015) 235:171–83. doi: 10.1620/tjem.235.171
49. Shin HK, Huang PL, Ayata C. Rho-Kinase Inhibition Improves Ischemic Perfusion Deficit in Hyperlipidemic Mice. J Cereb Blood Flow Metab (2014) 34:284–7. doi: 10.1038/jcbfm.2013.195
50. Sezen SF, Lagoda G, Musicki B, Burnett AL. Hydroxyl Fasudil, an Inhibitor of Rho Signaling, Improves Erectile Function in Diabetic Rats: A Role for Neuronal ROCK. J Sex Med (2014) 11:2164–71. doi: 10.1111/jsm.12613
51. Matoba K, Kawanami D, Okada R, Tsukamoto M, Kinoshita J, Ito T, et al. Rho-Kinase Inhibition Prevents the Progression of Diabetic Nephropathy by Downregulating Hypoxia-Inducible Factor 1alpha. Kidney Int (2013) 84:545–54. doi: 10.1038/ki.2013.130
52. Pearson JT, Jenkins MJ, Edgley AJ, Sonobe T, Joshi M, Waddingham MT, et al. Acute Rho-Kinase Inhibition Improves Coronary Dysfunction In Vivo, in the Early Diabetic Microcirculation. Cardiovasc Diabetol (2013) 12:111. doi: 10.1186/1475-2840-12-111
53. Zhou L, Liu F, Huang XR, Chen H, Chung AC, Shi J, et al. Amelioration of Albuminuria in ROCK1 Knockout Mice With Streptozotocin-Induced Diabetic Kidney Disease. Am J Nephrol (2011) 34:468–75. doi: 10.1159/000332040
54. Feng Y, LoGrasso PV. Rho Kinase Inhibitors: A Patent Review (2012 - 2013). Expert Opin Ther Pat (2014) 24:295–307. doi: 10.1517/13543776.2014.863279
55. Feng Y, LoGrasso PV, Defert O, Li R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J Med Chem (2016) 59:2269–300. doi: 10.1021/acs.jmedchem.5b00683
56. Defert O, Boland S. Rho Kinase Inhibitors: A Patent Review (2014 - 2016). Expert Opin Ther Pat (2017) 27:507–15. doi: 10.1080/13543776.2017.1272579
57. Landry T, Shookster D, Huang H. Tissue-Specific Approaches Reveal Diverse Metabolic Functions of Rho-Kinase 1. Front Endocrinol (2020) 11:622581. doi: 10.3389/fendo.2020.622581
58. Huang H, Lee DH, Zabolotny JM, Kim YB. Metabolic Actions of Rho-Kinase in Periphery and Brain. Trends Endocrinol Metab (2013) 24:506–14. doi: 10.1016/j.tem.2013.06.003
59. Sousa-Lima I, Kim HJ, Jones J, Kim YB. Rho-Kinase as a Therapeutic Target for Nonalcoholic Fatty Liver Diseases. Diabetes Metab J (2021) 45:655–74. doi: 10.4093/dmj.2021.0197
60. Boerma M, Fu Q, Wang J, Loose DS, Bartolozzi A, Ellis JL, et al. Comparative Gene Expression Profiling in Three Primary Human Cell Lines After Treatment With a Novel Inhibitor of Rho Kinase or Atorvastatin. Blood Coagul Fibrinolysis (2008) 19:709–18. doi: 10.1097/MBC.0b013e32830b2891
61. Lee JH, Zheng Y, von Bornstadt D, Wei Y, Balcioglu A, Daneshmand A, et al. Selective ROCK2 Inhibition in Focal Cerebral Ischemia. Ann Clin Transl Neurol (2014) 1:2–14. doi: 10.1002/acn3.19
62. Zanin-Zhorov A, Weiss JM, Nyuydzefe MS, Chen W, Scher JU, Mo R, et al. Selective Oral ROCK2 Inhibitor Down-Regulates IL-21 and IL-17 Secretion in Human T Cells via STAT3-Dependent Mechanism. Proc Natl Acad Sci USA (2014) 111:16814–9. doi: 10.1073/pnas.1414189111
63. Sharma P, Roy K. ROCK-2-Selective Targeting and its Therapeutic Outcomes. Drug Discov Today (2020) 25:446–55. doi: 10.1016/j.drudis.2019.11.017
64. Suzuki N, Hajicek N, Kozasa T. Regulation and Physiological Functions of G12/13-Mediated Signaling Pathways. Neurosignals (2009) 17:55–70. doi: 10.1159/000186690
65. Aittaleb M, Boguth CA, Tesmer JJ. Structure and Function of Heterotrimeric G Protein-Regulated Rho Guanine Nucleotide Exchange Factors. Mol Pharmacol (2010) 77:111–25. doi: 10.1124/mol.109.061234
66. Fu X, Gong MC, Jia T, Somlyo AV, Somlyo AP. The Effects of the Rho-Kinase Inhibitor Y-27632 on Arachidonic Acid-, GTPgammaS-, and Phorbol Ester-Induced Ca2+-Sensitization of Smooth Muscle. FEBS Lett (1998) 440:183–7. doi: 10.1016/S0014-5793(98)01455-0
67. Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, et al. Rho-Associated Kinase of Chicken Gizzard Smooth Muscle. J Biol Chem (1999) 274:3744–52. doi: 10.1074/jbc.274.6.3744
68. Shirao S, Kashiwagi S, Sato M, Miwa S, Nakao F, Kurokawa T, et al. Sphingosylphosphorylcholine is a Novel Messenger for Rho-Kinase-Mediated Ca2+ Sensitization in the Bovine Cerebral Artery: Unimportant Role for Protein Kinase C. Circ Res (2002) 91:112–9. doi: 10.1161/01.RES.0000026057.13161.42
69. Doran JD, Liu X, Taslimi P, Saadat A, Fox T. New Insights Into the Structure-Function Relationships of Rho-Associated Kinase: A Thermodynamic and Hydrodynamic Study of the Dimer-to-Monomer Transition and its Kinetic Implications. Biochem J (2004) 384:255–62. doi: 10.1042/BJ20040344
70. Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane Blebbing During Apoptosis Results From Caspase-Mediated Activation of ROCK I. Nat Cell Biol (2001) 3:339–45. doi: 10.1038/35070009
71. Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-Mediated Cleavage of ROCK I Induces MLC Phosphorylation and Apoptotic Membrane Blebbing. Nat Cell Biol (2001) 3:346–52. doi: 10.1038/35070019
72. Sebbagh M, Hamelin J, Bertoglio J, Solary E, Breard J. Direct Cleavage of ROCK II by Granzyme B Induces Target Cell Membrane Blebbing in a Caspase-Independent Manner. J Exp Med (2005) 201:465–71. doi: 10.1084/jem.20031877
73. Wei L, Surma M, Shi S, Lambert-Cheatham N, Shi J. Novel Insights Into the Roles of Rho Kinase in Cancer. Arch Immunol Ther Exp (Warsz) (2016) 64:259–78. doi: 10.1007/s00005-015-0382-6
74. Uray K, Major E, Lontay B. MicroRNA Regulatory Pathways in the Control of the Actin-Myosin Cytoskeleton. Cells (2020) 9:1649. doi: 10.3390/cells9071649
75. O’Brien M, Carbin S, Morrison JJ, Smith TJ. Decreased Myometrial P160 ROCK-1 Expression in Obese Women at Term Pregnancy. Reprod Biol Endocrinol (2013) 11:79. doi: 10.1186/1477-7827-11-79
76. Smith CA, Miner AS, Barbee RW, Ratz PH. Metabolic Stress-Induced Activation of AMPK and Inhibition of Constitutive Phosphoproteins Controlling Smooth Muscle Contraction: Evidence for Smooth Muscle Fatigue? Front Physiol (2017) 8:681. doi: 10.3389/fphys.2017.00681
77. Li T, Yang GM, Zhu Y, Wu Y, Chen XY, Lan D, et al. Diabetes and Hyperlipidemia Induce Dysfunction of VSMCs: Contribution of the Metabolic Inflammation/miRNA Pathway. Am J Physiol Endocrinol Metab (2015) 308:E257–69. doi: 10.1152/ajpendo.00348.2014
78. Mahavadi S, Sriwai W, Manion O, Grider JR, Murthy KS. Diabetes-Induced Oxidative Stress Mediates Upregulation of RhoA/Rho Kinase Pathway and Hypercontractility of Gastric Smooth Muscle. PLoS One (2017) 12:e0178574. doi: 10.1371/journal.pone.0178574
79. Chen K, He H, Xie Y, Zhao L, Zhao S, Wan X, et al. miR-125a-3p and miR-483-5p Promote Adipogenesis via Suppressing the RhoA/ROCK1/ERK1/2 Pathway in Multiple Symmetric Lipomatosis. Sci Rep (2015) 5:11909. doi: 10.1038/srep11909
80. Chen X, Tan XR, Li SJ, Zhang XX. LncRNA NEAT1 Promotes Hepatic Lipid Accumulation via Regulating miR-146a-5p/ROCK1 in Nonalcoholic Fatty Liver Disease. Life Sci (2019) 235:116829. doi: 10.1016/j.lfs.2019.116829
81. Guo J, Yang C, Lin Y, Hu G, Wei J, Zhang X, et al. Enhanced Peripheral Blood miR-324-5p is Associated With the Risk of Metabolic Syndrome by Suppressing ROCK1. Biochim Biophys Acta Mol Cell Biol Lipids (2020) 1865:158727. doi: 10.1016/j.bbalip.2020.158727
82. Gastebois C, Chanon S, Rome S, Durand C, Pelascini E, Jalabert A, et al. Transition From Physical Activity to Inactivity Increases Skeletal Muscle miR-148b Content and Triggers Insulin Resistance. Physiol Rep (2016) 4:e12902. doi: 10.14814/phy2.12902
83. Wu J, Liu J, Ding Y, Zhu M, Lu K, Zhou J, et al. MiR-455-3p Suppresses Renal Fibrosis Through Repression of ROCK2 Expression in Diabetic Nephropathy. Biochem Biophys Res Commun (2018) 503:977–83. doi: 10.1016/j.bbrc.2018.06.105
84. Wang Y, Wang D, Guo D. MiR-124 Promote Neurogenic Transdifferentiation of Adipose Derived Mesenchymal Stromal Cells Partly Through RhoA/ROCK1, But Not ROCK2 Signaling Pathway. PLoS One (2016) 11:e0146646. doi: 10.1371/journal.pone.0146646
85. Yao J, Wang J, Yao Y, Wang K, Zhou Q, Tang Y. Mir133b Regulates Proliferation and Apoptosis in Highglucoseinduced Human Retinal Endothelial Cells by Targeting Ras Homolog Family Member a. Int J Mol Med (2018) 42:839–50. doi: 10.3892/ijmm.2018.3694
86. Zhang Y, Jia L, Ji W, Li H. MicroRNA-141 Inhibits the Proliferation of Penile Cavernous Smooth Muscle Cells Associated With Down-Regulation of the RhoA/Rho Kinase Signaling Pathway. Cell Physiol Biochem (2018) 48:348–60. doi: 10.1159/000491741
87. Yang L, Wang ZF, Wu H, Wang W. miR-142-5p Improves Neural Differentiation and Proliferation of Adipose-Derived Stem Cells. Cell Physiol Biochem (2018) 50:2097–107. doi: 10.1159/000495054
88. Honardoost M, Keramati F, Arefian E, Mohammadi Yeganeh S, Soleimani M. Network of Three Specific microRNAs Influence Type 2 Diabetes Through Inducing Insulin Resistance in Muscle Cell Lines. J Cell Biochem (2019) 120:1532–38. doi: 10.1002/jcb.27381
89. Chen M, Zhang Y, Li W, Yang J. MicroRNA-145 Alleviates High Glucose-Induced Proliferation and Migration of Vascular Smooth Muscle Cells Through Targeting ROCK1. BioMed Pharmacother (2018) 99:81–6. doi: 10.1016/j.biopha.2018.01.014
90. Zhou W, Ye S, Wang W. miR-217 Alleviates High-Glucose-Induced Vascular Smooth Muscle Cell Dysfunction via Regulating ROCK1. J Biochem Mol Toxicol (2021) 35:e22668. doi: 10.1002/jbt.22668
91. Liu F, Zhang S, Xu R, Gao S, Yin J. Melatonin Attenuates Endothelial-To-Mesenchymal Transition of Glomerular Endothelial Cells via Regulating miR-497/ROCK in Diabetic Nephropathy. Kidney Blood Press Res (2018) 43:1425–36. doi: 10.1159/000493380
92. Amano M, Nakayama M, Kaibuchi K. Rho-Kinase/ROCK: A Key Regulator of the Cytoskeleton and Cell Polarity. Cytoskeleton (Hoboken) (2010) 67:545–54. doi: 10.1002/cm.20472
93. Shi J, Wei L. Rho Kinase in the Regulation of Cell Death and Survival. Arch Immunol Ther Exp (Warsz) (2007) 55:61–75. doi: 10.1007/s00005-007-0009-7
94. Surma M, Wei L, Shi J. Rho Kinase as a Therapeutic Target in Cardiovascular Disease. Future Cardiol (2011) 7:657–71. doi: 10.2217/fca.11.51
95. Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, et al. Phosphorylation of Myosin-Binding Subunit (MBS) of Myosin Phosphatase by Rho-Kinase. vivo J Cell Biol (1999) 147:1023–38. doi: 10.1083/jcb.147.5.1023
96. Sumi T, Matsumoto K, Nakamura T. Specific Activation of LIM Kinase 2 via Phosphorylation of Threonine 505 by ROCK, a Rho-Dependent Protein Kinase. J Biol Chem (2001) 276:670–6. doi: 10.1074/jbc.M007074200
97. Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, et al. Phosphorylation and Activation of Myosin by Rho-Associated Kinase (Rho-Kinase). J Biol Chem (1996) 271:20246–9. doi: 10.1074/jbc.271.34.20246
98. Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of Myosin Phosphatase by Rho and Rho-Associated Kinase (Rho-Kinase). Science (1996) 273:245–8. doi: 10.1126/science.273.5272.245
99. Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, et al. Rho-Associated Kinase Directly Induces Smooth Muscle Contraction Through Myosin Light Chain Phosphorylation. J Biol Chem (1997) 272:12257–60. doi: 10.1074/jbc.272.19.12257
100. Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, et al. Signaling From Rho to the Actin Cytoskeleton Through Protein Kinases ROCK and LIM-Kinase. Science (1999) 285:895–8. doi: 10.1126/science.285.5429.895
101. Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-Associated Kinase ROCK Activates LIM-Kinase 1 by Phosphorylation at Threonine 508 Within the Activation Loop. J Biol Chem (2000) 275:3577–82. doi: 10.1074/jbc.275.5.3577
102. Amano T, Tanabe K, Eto T, Narumiya S, Mizuno K. LIM-Kinase 2 Induces Formation of Stress Fibres, Focal Adhesions and Membrane Blebs, Dependent on Its Activation by Rho-Associated Kinase-Catalysed Phosphorylation at Threonine-505. Biochem J (2001) 354:149–59. doi: 10.1042/bj3540149
103. Lin T, Zeng L, Liu Y, DeFea K, Schwartz MA, Chien S, et al. Rho-ROCK-LIMK-Cofilin Pathway Regulates Shear Stress Activation of Sterol Regulatory Element Binding Proteins. Circ Res (2003) 92:1296–304. doi: 10.1161/01.RES.0000078780.65824.8B
104. Sotiropoulos A, Gineitis D, Copeland J, Treisman R. Signal-Regulated Activation of Serum Response Factor is Mediated by Changes in Actin Dynamics. Cell (1999) 98:159–69. doi: 10.1016/S0092-8674(00)81011-9
105. Posern G, Treisman R. Actin’ Together: Serum Response Factor, its Cofactors and the Link to Signal Transduction. Trends Cell Biol (2006) 16:588–96. doi: 10.1016/j.tcb.2006.09.008
106. Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin Dynamics Control SRF Activity by Regulation of its Coactivator MAL. Cell (2003) 113:329–42. doi: 10.1016/S0092-8674(03)00278-2
107. Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear Actin Regulates Dynamic Subcellular Localization and Activity of the SRF Cofactor MAL. Science (2007) 316:1749–52. doi: 10.1126/science.1141084
108. Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, et al. Activation of Cardiac Gene Expression by Myocardin, a Transcriptional Cofactor for Serum Response Factor. Cell (2001) 105:851–62. doi: 10.1016/S0092-8674(01)00404-4
109. Wang DZ, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, et al. Potentiation of Serum Response Factor Activity by a Family of Myocardin-Related Transcription Factors. Proc Natl Acad Sci USA (2002) 99:14855–60. doi: 10.1073/pnas.222561499
110. Olson EN, Nordheim A. Linking Actin Dynamics and Gene Transcription to Drive Cellular Motile Functions. Nat Rev Mol Cell Biol (2010) 11:353–65. doi: 10.1038/nrm2890
111. Sward K, Stenkula KG, Rippe C, Alajbegovic A, Gomez MF, Albinsson S. Emerging Roles of the Myocardin Family of Proteins in Lipid and Glucose Metabolism. J Physiol (2016) 594:4741–52. doi: 10.1113/JP271913
112. Lin JZ, Farmer SR. Morphogenetics in Brown, Beige and White Fat Development. Adipocyte (2016) 5:130–5. doi: 10.1080/21623945.2016.1140708
113. Bian H, Lin JZ, Li C, Farmer SR. Myocardin-Related Transcription Factor A (MRTFA) Regulates the Fate of Bone Marrow Mesenchymal Stem Cells and its Absence in Mice Leads to Osteopenia. Mol Metab (2016) 5:970–9. doi: 10.1016/j.molmet.2016.08.012
114. Dorn T, Kornherr J, Parrotta EI, Zawada D, Ayetey H, Santamaria G, et al. Interplay of Cell-Cell Contacts and RhoA/MRTF-A Signaling Regulates Cardiomyocyte Identity. EMBO J (2018) 37:e98133. doi: 10.15252/embj.201798133
115. Liu R, Xiong X, Nam D, Yechoor V, Ma K. SRF-MRTF Signaling Suppresses Brown Adipocyte Development by Modulating TGF-Beta/BMP Pathway. Mol Cell Endocrinol (2020) 515:110920. doi: 10.1016/j.mce.2020.110920
116. Hansson B, Schumacher S, Fryklund C, Moren B, Bjorkqvist M, Sward K, et al. A Hypothesis for Insulin Resistance in Primary Human Adipocytes Involving MRTF-A and Suppression of PPARgamma. Biochem Biophys Res Commun (2020) 533:64–9. doi: 10.1016/j.bbrc.2020.08.105
117. Lin JZ, Rabhi N, Farmer SR. Myocardin-Related Transcription Factor a Promotes Recruitment of ITGA5+ Profibrotic Progenitors During Obesity-Induced Adipose Tissue Fibrosis. Cell Rep (2018) 23:1977–87. doi: 10.1016/j.celrep.2018.04.057
118. Jones JEC, Rabhi N, Orofino J, Gamini R, Perissi V, Vernochet C, et al. The Adipocyte Acquires a Fibroblast-Like Transcriptional Signature in Response to a High Fat Diet. Sci Rep (2020) 10:2380. doi: 10.1038/s41598-020-59284-w
119. Zhang L, Li HL, Zhang DD, Cui XC. Therapeutic Effects of Myocardin-Related Transcription Factor A (MRTF-A) Knockout on Experimental Mice With Nonalcoholic Steatohepatitis Induced by High-Fat Diet. Hum Exp Toxicol (2021) 40:1634–45. doi: 10.1177/09603271211002886
120. Shao J, Xu H, Wu X, Xu Y. Epigenetic Activation of CTGF Transcription by High Glucose in Renal Tubular Epithelial Cells is Mediated by Myocardin-Related Transcription Factor a. Cell Tissue Res (2020) 379:549–59. doi: 10.1007/s00441-019-03124-5
121. Soliman H, Varela JN, Nyamandi V, Garcia-Patino M, Lin G, Bankar GR, et al. Attenuation of Obesity-Induced Insulin Resistance in Mice With Heterozygous Deletion of ROCK2. Int J Obes (Lond) (2016) 40:1435–43. doi: 10.1038/ijo.2016.89
122. Begum N, Sandu OA, Ito M, Lohmann SM, Smolenski A. Active Rho Kinase (ROK-Alpha) Associates With Insulin Receptor Substrate-1 and Inhibits Insulin Signaling in Vascular Smooth Muscle Cells. J Biol Chem (2002) 277:6214–22. doi: 10.1074/jbc.M110508200
123. Lim MJ, Choi KJ, Ding Y, Kim JH, Kim BS, Kim YH, et al. RhoA/Rho Kinase Blocks Muscle Differentiation via Serine Phosphorylation of Insulin Receptor Substrate-1 and -2. Mol Endocrinol (2007) 21:2282–93. doi: 10.1210/me.2007-0114
124. Farah S, Agazie Y, Ohan N, Ngsee JK, Liu XJ. A Rho-Associated Protein Kinase, ROKalpha, Binds Insulin Receptor Substrate-1 and Modulates Insulin Signaling. J Biol Chem (1998) 273:4740–6. doi: 10.1074/jbc.273.8.4740
125. Huang H, Lee SH, Sousa-Lima I, Kim SS, Hwang WM, Dagon Y, et al. Rho Kinase/AMPK Axis Regulates Hepatic Lipogenesis During Overnutrition. J Clin Invest (2018) 128:5335–50. doi: 10.1172/JCI63562
126. Tang S, Wu W, Tang W, Ge Z, Wang H, Hong T, et al. Suppression of Rho-Kinase 1 Is Responsible for Insulin Regulation of the AMPK/SREBP-1c Pathway in Skeletal Muscle Cells Exposed to Palmitate. Acta Diabetol (2017) 54:635–44. doi: 10.1007/s00592-017-0976-z
127. Kim HI, Lee JS, Kwak BK, Hwang WM, Kim MJ, Kim YB, et al. Metformin Ameliorates Lipotoxic Beta-Cell Dysfunction Through a Concentration-Dependent Dual Mechanism of Action. Diabetes Metab J (2019) 43:854–66. doi: 10.4093/dmj.2018.0179
128. McCarthy CG, Wenceslau CF, Ogbi S, Szasz T, Webb RC. Toll-Like Receptor 9-Dependent AMPKalpha Activation Occurs via TAK1 and Contributes to RhoA/ROCK Signaling and Actin Polymerization in Vascular Smooth Muscle Cells. J Pharmacol Exp Ther (2018) 365:60–71. doi: 10.1124/jpet.117.245746
129. Sun GQ, Li YB, Du B, Meng Y. Resveratrol via Activation of AMPK Lowers Blood Pressure in DOCA-Salt Hypertensive Mice. Clin Exp Hypertens (2015) 37:616–21. doi: 10.3109/10641963.2015.1036060
130. Wang Y, Wang B, Guerram M, Sun L, Shi W, Tian C, et al. Deoxypodophyllotoxin Suppresses Tumor Vasculature in HUVECs by Promoting Cytoskeleton Remodeling Through LKB1-AMPK Dependent Rho A Activation. Oncotarget (2015) 6:29497–512. doi: 10.18632/oncotarget.4985
131. Szrejder M, Rachubik P, Rogacka D, Audzeyenka I, Rychlowski M, Kreft E, et al. Metformin Reduces TRPC6 Expression Through AMPK Activation and Modulates Cytoskeleton Dynamics in Podocytes Under Diabetic Conditions. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165610. doi: 10.1016/j.bbadis.2019.165610
132. Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, et al. ROCK Isoform Regulation of Myosin Phosphatase and Contractility in Vascular Smooth Muscle Cells. Circ Res (2009) 104:531–40. doi: 10.1161/CIRCRESAHA.108.188524
133. Shi J, Zhang L, Wei L. Rho-Kinase in Development and Heart Failure: Insights From Genetic Models. Pediatr Cardiol (2011) 32:297–304. doi: 10.1007/s00246-011-9920-0
134. Zhou X, Li R, Liu X, Wang L, Hui P, Chan L, et al. ROCK1 Reduces Mitochondrial Content and Irisin Production in Muscle Suppressing Adipocyte Browning and Impairing Insulin Sensitivity. Sci Rep (2016) 6:29669. doi: 10.1038/srep29669
135. Zhu L, Yang Y, Lu X. The Selectivity and Promiscuity of Brain-Neuroregenerative Inhibitors Between ROCK1 and ROCK2 Isoforms: An Integration of SB-QSSR Modelling, QM/MM Analysis and In Vitro Kinase Assay. SAR QSAR Environ Res (2016) 27:47–65. doi: 10.1080/1062936X.2015.1132765
136. Akhter M, Qin T, Fischer P, Sadeghian H, Kim HH, Whalen MJ, et al. Rho-Kinase Inhibitors do Not Expand Hematoma Volume in Acute Experimental Intracerebral Hemorrhage. Ann Clin Transl Neurol (2018) 5:769–76. doi: 10.1002/acn3.569
137. Sadeghian H, Lacoste B, Qin T, Toussay X, Rosa R, Oka F, et al. Spreading Depolarizations Trigger Caveolin-1-Dependent Endothelial Transcytosis. Ann Neurol (2018) 84:409–23. doi: 10.1002/ana.25298
138. Yamamura A, Nayeem MJ, Sato M. The Rho Kinase 2 (ROCK2)-Specific Inhibitor KD025 Ameliorates the Development of Pulmonary Arterial Hypertension. Biochem Biophys Res Commun (2021) 534:795–801. doi: 10.1016/j.bbrc.2020.10.106
139. Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. Targeted Rho-Associated Kinase 2 Inhibition Suppresses Murine and Human Chronic GVHD Through a Stat3-Dependent Mechanism. Blood (2016) 127:2144–54. doi: 10.1182/blood-2015-10-678706
140. Weiss JM, Chen W, Nyuydzefe MS, Trzeciak A, Flynn R, Tonra JR, et al. ROCK2 Signaling is Required to Induce a Subset of T Follicular Helper Cells Through Opposing Effects on STATs in Autoimmune Settings. Sci Signal (2016) 9:ra73. doi: 10.1126/scisignal.aad8953
141. Zanin-Zhorov A, Flynn R, Waksaland SD, Blazar BR. Isoform-Specific Targeting of ROCK Proteins in Immune Cells. Small GTPases (2016) 7:173–7. doi: 10.1080/21541248.2016.1181698
142. Zanin-Zhorov A, Weiss JM, Trzeciak A, Chen W, Zhang J, Nyuydzefe MS, et al. Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients With Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10. J Immunol (2017) 198:3809–14. doi: 10.4049/jimmunol.1602142
143. Rozo C, Chinenov Y, Maharaj RK, Gupta S, Leuenberger L, Kirou KA, et al. Targeting the RhoA-ROCK Pathway to Reverse T-Cell Dysfunction in SLE. Ann Rheum Dis (2017) 76:740–7. doi: 10.1136/annrheumdis-2016-209850
144. Tengesdal IW, Kitzenberg D, Li S, Nyuydzefe MS, Chen W, Weiss JM, et al. The Selective ROCK2 Inhibitor KD025 Reduces IL-17 Secretion in Human Peripheral Blood Mononuclear Cells Independent of IL-1 and IL-6. Eur J Immunol (2018) 48:1679–86. doi: 10.1002/eji.201847652
145. Jagasia M, Lazaryan A, Bachier CR, Salhotra A, Weisdorf DJ, Zoghi B, et al. ROCK2 Inhibition With Belumosudil (KD025) for the Treatment of Chronic Graft-Versus-Host Disease. J Clin Oncol (2021) 39:1888–98. doi: 10.1200/JCO.20.02754
146. Yiu ZZ, Warren RB. Novel Oral Therapies for Psoriasis and Psoriatic Arthritis. Am J Clin Dermatol (2016) 17:191–200. doi: 10.1007/s40257-016-0179-3
147. You R, Zhou W, Li Y, Zhang Y, Huang S, Jia Z, et al. Inhibition of ROCK2 Alleviates Renal Fibrosis and the Metabolic Disorders in the Proximal Tubular Epithelial Cells. Clin Sci (Lond) (2020) 134:1357–76. doi: 10.1042/CS20200030
148. Behrmann A, Zhong D, Li L, Cheng SL, Mead M, Ramachandran B, et al. PTH/PTHrP Receptor Signaling Restricts Arterial Fibrosis in Diabetic LDLR (-/-) Mice by Inhibiting Myocardin-Related Transcription Factor Relays. Circ Res (2020) 126:1363–78. doi: 10.1161/CIRCRESAHA.119.316141
149. Lee JY, Stevens RP, Kash M, Zhou C, Koloteva A, Renema P, et al. KD025 Shifts Pulmonary Endothelial Cell Bioenergetics and Decreases Baseline Lung Permeability. Am J Respir Cell Mol Biol (2020) 63:519–30. doi: 10.1165/rcmb.2019-0435OC
150. Diep DTV, Duong KHM, Choi H, Jun HS, Chun KH. KD025 (SLx-2119) Suppresses Adipogenesis at Intermediate Stage in Human Adipose-Derived Stem Cells. Adipocyte (2019) 8:114–24. doi: 10.1080/21623945.2019.1590929
151. Diep DTV, Hong K, Khun T, Zheng M, Ul-Haq A, Jun HS, et al. Anti-Adipogenic Effects of KD025 (SLx-2119), a ROCK2-Specific Inhibitor, in 3T3-L1 Cells. Sci Rep (2018) 8:2477. doi: 10.1038/s41598-018-20821-3
152. Hikage F, Ichioka H, Watanabe M, Umetsu A, Ohguro H, Ida Y. ROCK Inhibitors Modulate the Physical Properties and Adipogenesis of 3D Spheroids of Human Orbital Fibroblasts in Different Manners. FASEB Bioadv (2021) 3:866–72. doi: 10.1096/fba.2021-00037
153. Tran NNQ, Chun KH. ROCK2-Specific Inhibitor KD025 Suppresses Adipocyte Differentiation by Inhibiting Casein Kinase 2. Molecules (2021) 26:4747. doi: 10.3390/molecules26164747
154. Wei L, Surma M, Yang Y, Tersey S, Shi J. ROCK2 Inhibition Enhances the Thermogenic Program in White and Brown Fat Tissue in Mice. FASEB J (2020) 34:474–93. doi: 10.1096/fj.201901174RR
155. Park J, Chun KH. Identification of Novel Functions of the ROCK2-Specific Inhibitor KD025 by Bioinformatics Analysis. Gene (2020) 737:144474. doi: 10.1016/j.gene.2020.144474
156. Shao M, Wang QA, Song A, Vishvanath L, Busbuso NC, Scherer PE, et al. Cellular Origins of Beige Fat Cells Revisited. Diabetes (2019) 68:1874–85. doi: 10.2337/db19-0308
157. Meyers VE, Zayzafoon M, Douglas JT, McDonald JM. RhoA and Cytoskeletal Disruption Mediate Reduced Osteoblastogenesis and Enhanced Adipogenesis of Human Mesenchymal Stem Cells in Modeled Microgravity. J Bone Miner Res (2005) 20:1858–66. doi: 10.1359/JBMR.050611
158. Arnsdorf EJ, Tummala P, Kwon RY, Jacobs CR. Mechanically Induced Osteogenic Differentiation–the Role of RhoA, ROCKII and Cytoskeletal Dynamics. J Cell Sci (2009) 122:546–53. doi: 10.1242/jcs.036293
159. Yao X, Peng R, Ding J. Effects of Aspect Ratios of Stem Cells on Lineage Commitments With and Without Induction Media. Biomaterials (2013) 34:930–9. doi: 10.1016/j.biomaterials.2012.10.052
160. Santos A, Bakker AD, de Blieck-Hogervorst JM, Klein-Nulend J. WNT5A Induces Osteogenic Differentiation of Human Adipose Stem Cells via Rho-Associated Kinase ROCK. Cytotherapy (2010) 12:924–32. doi: 10.3109/14653241003774011
161. Major LG, Holle AW, Young JL, Hepburn MS, Jeong K, Chin IL, et al. Volume Adaptation Controls Stem Cell Mechanotransduction. ACS Appl Mater Interfaces (2019) 11:45520–30. doi: 10.1021/acsami.9b19770
162. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in Mechanotransduction. Nature (2011) 474:179–83. doi: 10.1038/nature10137
163. Hyvari L, Ojansivu M, Juntunen M, Kartasalo K, Miettinen S, Vanhatupa S. Focal Adhesion Kinase and ROCK Signaling Are Switch-Like Regulators of Human Adipose Stem Cell Differentiation Towards Osteogenic and Adipogenic Lineages. Stem Cells Int (2018) 2018:2190657. doi: 10.1155/2018/2190657
164. Nimiritsky P, Novoseletskaya E, Eremichev R, Alexandrushkina N, Karagyaur M, Vetrovoy O, et al. Self-Organization Provides Cell Fate Commitment in MSC Sheet Condensed Areas via ROCK-Dependent Mechanism. Biomedicines (2021) 9:1192. doi: 10.3390/biomedicines9091192
165. Ellawindy A, Satoh K, Sunamura S, Kikuchi N, Suzuki K, Minami T, et al. Rho-Kinase Inhibition During Early Cardiac Development Causes Arrhythmogenic Right Ventricular Cardiomyopathy in Mice. Arterioscler Thromb Vasc Biol (2015) 35:2172–84. doi: 10.1161/ATVBAHA.115.305872
166. Parrotta EI, Procopio A, Scalise S, Esposito C, Nicoletta G, Santamaria G, et al. Deciphering the Role of Wnt and Rho Signaling Pathway in iPSC-Derived ARVC Cardiomyocytes by In Silico Mathematical Modeling. Int J Mol Sci (2021) 22:2004. doi: 10.3390/ijms22042004
167. Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, et al. Comparative Epigenomic Analysis of Murine and Human Adipogenesis. Cell (2010) 143:156–69. doi: 10.1016/j.cell.2010.09.006
168. Li L, Tam L, Liu L, Jin T, Ng DS. Wnt-Signaling Mediates the Anti-Adipogenic Action of Lysophosphatidic Acid Through Cross Talking With the Rho/Rho Associated Kinase (ROCK) Pathway. Biochem Cell Biol (2011) 89:515–21. doi: 10.1139/o11-048
169. Ida Y, Hikage F, Ohguro H. ROCK Inhibitors Enhance the Production of Large Lipid-Enriched 3D Organoids of 3T3-L1 Cells. Sci Rep (2021) 11:5479. doi: 10.1038/s41598-021-84955-7
170. Kunitomi H, Oki Y, Onishi N, Kano K, Banno K, Aoki D, et al. The Insulin-PI3K-Rac1 Axis Contributes to Terminal Adipocyte Differentiation Through Regulation of Actin Cytoskeleton Dynamics. Genes Cells (2020) 25:165–74. doi: 10.1111/gtc.12747
171. Haas B, Mayer P, Jennissen K, Scholz D, Berriel Diaz M, Bloch W, et al. Protein Kinase G Controls Brown Fat Cell Differentiation and Mitochondrial Biogenesis. Sci Signal (2009) 2:ra78. doi: 10.1126/scisignal.2000511
172. Amieux PS, McKnight GS. Cyclic Nucleotides Converge on Brown Adipose Tissue Differentiation. Sci Signal (2010) 3:pe2. doi: 10.1126/scisignal.3104pe2
173. Mitschke MM, Hoffmann LS, Gnad T, Scholz D, Kruithoff K, Mayer P, et al. Increased cGMP Promotes Healthy Expansion and Browning of White Adipose Tissue. FASEB J (2013) 27:1621–30. doi: 10.1096/fj.12-221580
174. Klepac K, Yang J, Hildebrand S, Pfeifer A. RGS2: A Multifunctional Signaling Hub That Balances Brown Adipose Tissue Function and Differentiation. Mol Metab (2019) 30:173–83. doi: 10.1016/j.molmet.2019.09.015
175. Sim CK, Kim SY, Brunmeir R, Zhang Q, Li H, Dharmasegaran D, et al. Regulation of White and Brown Adipocyte Differentiation by RhoGAP Dlc1. PLoS One (2017) 12:e0174761. doi: 10.1371/journal.pone.0174761
176. Hansson B, Moren B, Fryklund C, Vliex L, Wasserstrom S, Albinsson S, et al. Adipose Cell Size Changes are Associated With a Drastic Actin Remodeling. Sci Rep (2019) 9:12941. doi: 10.1038/s41598-019-49418-0
177. Wang L, Wang S, Shi Y, Li R, Gunther S, Ong YT, et al. YAP and TAZ Protect Against White Adipocyte Cell Death During Obesity. Nat Commun (2020) 11:5455. doi: 10.1038/s41467-020-19229-3
178. Zhou H, Fang C, Zhang L, Deng Y, Wang M, Meng F. Fasudil Hydrochloride Hydrate, a Rho-Kinase Inhibitor, Ameliorates Hepatic Fibrosis in Rats With Type 2 Diabetes. Chin Med J (Engl) (2014) 127:225–31. doi: 10.3760/cma.j.issn.0366-6999.20131917
179. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology (2016) 150:956–67. doi: 10.1053/j.gastro.2015.12.037
180. Wang J, Jiang W. The Effects of RKI-1447 in a Mouse Model of Nonalcoholic Fatty Liver Disease Induced by a High-Fat Diet and in HepG2 Human Hepatocellular Carcinoma Cells Treated With Oleic Acid. Med Sci Monit (2020) 26:e919220. doi: 10.12659/MSM.919220
181. Chen X, Tan QQ, Tan XR, Li SJ, Zhang XX. Circ_0057558 Promotes Nonalcoholic Fatty Liver Disease by Regulating ROCK1/AMPK Signaling Through Targeting miR-206. Cell Death Dis (2021) 12:809. doi: 10.1038/s41419-021-04090-z
182. Rodriguez-Fdez S, Lorenzo-Martin LF, Fernandez-Pisonero I, Porteiro B, Veyrat-Durebex C, Beiroa D, et al. Vav2 Catalysis-Dependent Pathways Contribute to Skeletal Muscle Growth and Metabolic Homeostasis. Nat Commun (2020) 11:5808. doi: 10.1038/s41467-020-19489-z
183. Munoz VR, Gaspar RC, Kuga GK, da Rocha AL, Crisol BM, Botezelli JD, et al. Exercise Increases Rho-Kinase Activity and Insulin Signaling in Skeletal Muscle. J Cell Physiol (2018) 233:4791–800. doi: 10.1002/jcp.26278
184. Munoz VR, Gaspar RC, Severino MB, Macedo APA, Simabuco FM, Ropelle ER, et al. Exercise Counterbalances Rho/ROCK2 Signaling Impairment in the Skeletal Muscle and Ameliorates Insulin Sensitivity in Obese Mice. Front Immunol (2021) 12:702025. doi: 10.3389/fimmu.2021.702025
185. Tao W, Wu J, Xie BX, Zhao YY, Shen N, Jiang S, et al. Lipid-Induced Muscle Insulin Resistance Is Mediated by GGPPS via Modulation of the RhoA/Rho Kinase Signaling Pathway. J Biol Chem (2015) 290:20086–97. doi: 10.1074/jbc.M115.657742
186. Huang H, Kong D, Byun KH, Ye C, Koda S, Lee DH, et al. Rho-Kinase Regulates Energy Balance by Targeting Hypothalamic Leptin Receptor Signaling. Nat Neurosci (2012) 15:1391–8. doi: 10.1038/nn.3207
187. Huang H, Lee SH, Ye C, Lima IS, Oh BC, Lowell BB, et al. ROCK1 in AgRP Neurons Regulates Energy Expenditure and Locomotor Activity in Male Mice. Endocrinology (2013) 154:3660–70. doi: 10.1210/en.2013-1343
188. Skov LJ, Ratner C, Hansen NW, Thompson JJ, Egerod KL, Burm H, et al. RhoA in Tyrosine Hydroxylase Neurones Regulates Food Intake and Body Weight via Altered Sensitivity to Peripheral Hormones. J Neuroendocrinol (2019) 31:e12761. doi: 10.1111/jne.12761
189. Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, et al. Effect of AT877 on Cerebral Vasospasm After Aneurysmal Subarachnoid Hemorrhage. Results of a Prospective Placebo-Controlled Double-Blind Trial. J Neurol Sci (1992) 76:571–7. doi: 10.3171/jns.1992.76.4.0571
190. Shi J, Wei L. Rho Kinases in Cardiovascular Physiology and Pathophysiology: The Effect of Fasudil. J Cardiovasc Pharmacol (2013) 62:341–54. doi: 10.1097/FJC.0b013e3182a3718f
191. Shaw D, Hollingworth G, Soldermann N, Sprague E, Schuler W, Vangrevelinghe E, et al. Novel ROCK Inhibitors for the Treatment of Pulmonary Arterial Hypertension. Bioorg Med Chem Lett (2014) 24:4812–7. doi: 10.1016/j.bmcl.2014.09.002
192. Chen J, Li Q, Dong R, Gao H, Peng H, Wu Y. The Effect of the Ras Homolog Gene Family (Rho), Member A/Rho Associated Coiled-Coil Forming Protein Kinase Pathway in Atrial Fibrosis of Type 2 Diabetes in Rats. Exp Ther Med (2014) 8:836–40. doi: 10.3892/etm.2014.1843
193. Petersen N, Frimurer TM, Terndrup Pedersen M, Egerod KL, Wewer Albrechtsen NJ, Holst JJ, et al. Inhibiting RHOA Signaling in Mice Increases Glucose Tolerance and Numbers of Enteroendocrine and Other Secretory Cells in the Intestine. Gastroenterology (2018) 155:1164–76.e2. doi: 10.1053/j.gastro.2018.06.039
194. Mallat Z, Gojova A, Sauzeau V, Brun V, Silvestre JS, Esposito B, et al. Rho-Associated Protein Kinase Contributes to Early Atherosclerotic Lesion Formation in Mice. Circ Res (2003) 93:884–8. doi: 10.1161/01.RES.0000099062.55042.9A
195. Steioff K, Rutten H, Busch AE, Plettenburg O, Ivashchenko Y, Lohn M. Long Term Rho-Kinase Inhibition Ameliorates Endothelial Dysfunction in LDL-Receptor Deficient Mice. Eur J Pharmacol (2005) 512:247–9. doi: 10.1016/j.ejphar.2005.03.001
196. Wu DJ, Xu JZ, Wu YJ, Jean-Charles L, Xiao B, Gao PJ, et al. Effects of Fasudil on Early Atherosclerotic Plaque Formation and Established Lesion Progression in Apolipoprotein E-Knockout Mice. Atherosclerosis (2009) 207:68–73. doi: 10.1016/j.atherosclerosis.2009.04.025
197. Nguyen Dinh Cat A, Antunes TT, Callera GE, Sanchez A, Tsiropoulou S, Dulak-Lis MG, et al. Adipocyte-Specific Mineralocorticoid Receptor Overexpression in Mice Is Associated With Metabolic Syndrome and Vascular Dysfunction: Role of Redox-Sensitive PKG-1 and Rho Kinase. Diabetes (2016) 65:2392–403. doi: 10.2337/db15-1627
198. da Costa RM, Fais RS, Dechandt CRP, Louzada-Junior P, Alberici LC, Lobato NS, et al. Increased Mitochondrial ROS Generation Mediates the Loss of the Anti-Contractile Effects of Perivascular Adipose Tissue in High-Fat Diet Obese Mice. Br J Pharmacol (2017) 174:3527–41. doi: 10.1111/bph.13687
199. Weng C, Shen Z, Li X, Jiang W, Peng L, Yuan H, et al. Effects of Chemerin/CMKLR1 in Obesity-Induced Hypertension and Potential Mechanism. Am J Transl Res (2017) 9:3096–104.
200. Stern C, Schreier B, Nolze A, Rabe S, Mildenberger S, Gekle M. Knockout of Vascular Smooth Muscle EGF Receptor in a Mouse Model Prevents Obesity-Induced Vascular Dysfunction and Renal Damage. vivo Diabetologia (2020) 63:2218–34. doi: 10.1007/s00125-020-05187-4
201. Ludvigsen TP, Olsen LH, Pedersen HD, Christoffersen BO, Jensen LJ. Hyperglycemia-Induced Transcriptional Regulation of ROCK1 and TGM2 Expression is Involved in Small Artery Remodeling in Obese Diabetic Gottingen Minipigs. Clin Sci (Lond) (2019) 133:2499–516. doi: 10.1042/CS20191066
202. Nuno DW, Lamping KG. Dietary Fatty Acid Saturation Modulates Sphingosine-1-Phosphate-Mediated Vascular Function. J Diabetes Res (2019) 2019:2354274. doi: 10.1155/2019/2354274
203. Elrashidy RA, Zhang J, Liu G. Long-Term Consumption of Western Diet Contributes to Endothelial Dysfunction and Aortic Remodeling in Rats: Implication of Rho-Kinase Signaling. Clin Exp Hypertens (2019) 41:174–80. doi: 10.1080/10641963.2018.1462375
204. Tang ST, Zhang Q, Tang HQ, Wang CJ, Su H, Zhou Q, et al. Effects of Glucagon-Like Peptide-1 on Advanced Glycation Endproduct-Induced Aortic Endothelial Dysfunction in Streptozotocin-Induced Diabetic Rats: Possible Roles of Rho Kinase- and AMP Kinase-Mediated Nuclear Factor kappaB Signaling Pathways. Endocrine (2016) 53:107–16. doi: 10.1007/s12020-015-0852-y
205. Engin A. Circadian Rhythms in Diet-Induced Obesity. Adv Exp Med Biol (2017) 960:19–52. doi: 10.1007/978-3-319-48382-5_2
206. Hou T, Guo Z, Gong MC. Circadian Variations of Vasoconstriction and Blood Pressure in Physiology and Diabetes. Curr Opin Pharmacol (2021) 57:125–31. doi: 10.1016/j.coph.2021.02.001
207. Silvani A. Sleep Disorders, Nocturnal Blood Pressure, and Cardiovascular Risk: A Translational Perspective. Auton Neurosci (2019) 218:31–42. doi: 10.1016/j.autneu.2019.02.006
208. Su W, Xie Z, Guo Z, Duncan MJ, Lutshumba J, Gong MC. Altered Clock Gene Expression and Vascular Smooth Muscle Diurnal Contractile Variations in Type 2 Diabetic Db/Db Mice. Am J Physiol Heart Circ Physiol (2012) 302:H621–33. doi: 10.1152/ajpheart.00825.2011
209. Saito T, Hirano M, Ide T, Ichiki T, Koibuchi N, Sunagawa K, et al. Pivotal Role of Rho-Associated Kinase 2 in Generating the Intrinsic Circadian Rhythm of Vascular Contractility. Circulation (2013) 127:104–14. doi: 10.1161/CIRCULATIONAHA.112.135608
210. Saito T. The Vascular Clock System Generates the Intrinsic Circadian Rhythm of Vascular Contractility. J Smooth Muscle Res (2015) 51:95–106. doi: 10.1540/jsmr.51.95
211. Nihei T, Takahashi J, Tsuburaya R, Ito Y, Shiroto T, Hao K, et al. Circadian Variation of Rho-Kinase Activity in Circulating Leukocytes of Patients With Vasospastic Angina. Circ J (2014) 78:1183–90. doi: 10.1253/circj.CJ-13-1458
212. Xie Z, Su W, Liu S, Zhao G, Esser K, Schroder EA, et al. Smooth-Muscle BMAL1 Participates in Blood Pressure Circadian Rhythm Regulation. J Clin Invest (2015) 125:324–36. doi: 10.1172/JCI76881
213. Tang H, Zhu M, Zhao G, Fu W, Shi Z, Ding Y, et al. Loss of CLOCK Under High Glucose Upregulates ROCK1-Mediated Endothelial to Mesenchymal Transition and Aggravates Plaque Vulnerability. Atherosclerosis (2018) 275:58–67. doi: 10.1016/j.atherosclerosis.2018.05.046
214. Yang N, Williams J, Pekovic-Vaughan V, Wang P, Olabi S, McConnell J, et al. Cellular Mechano-Environment Regulates the Mammary Circadian Clock. Nat Commun (2017) 8:14287. doi: 10.1038/ncomms14287
215. Lefranc C, Friederich-Persson M, Braud L, Palacios-Ramirez R, Karlsson S, Boujardine N, et al. MR (Mineralocorticoid Receptor) Induces Adipose Tissue Senescence and Mitochondrial Dysfunction Leading to Vascular Dysfunction in Obesity. Hypertension (2019) 73:458–68. doi: 10.1161/HYPERTENSIONAHA.118.11873
216. Dunham-Snary KJ, Wu D, Sykes EA, Thakrar A, Parlow LRG, Mewburn JD, et al. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. Chest (2017) 151:181–92. doi: 10.1016/j.chest.2016.09.001
217. Yan S, Resta TC, Jernigan NL. Vasoconstrictor Mechanisms in Chronic Hypoxia-Induced Pulmonary Hypertension: Role of Oxidant Signaling. Antioxidants (Basel) (2020) 9:999. doi: 10.3390/antiox9100999
218. Chen W, Xiang H, Chen R, Yang J, Yang X, Zhou J, et al. S1PR2 Antagonist Ameliorate High Glucose-Induced Fission and Dysfunction of Mitochondria in HRGECs via Regulating ROCK1. BMC Nephrol (2019) 20:135. doi: 10.1186/s12882-019-1323-0
219. Shi Y, Fan S, Wang D, Huyan T, Chen J, Chen J, et al. FOXO1 Inhibition Potentiates Endothelial Angiogenic Functions in Diabetes via Suppression of ROCK1/Drp1-Mediated Mitochondrial Fission. Biochim Biophys Acta Mol Basis Dis (2018) 1864:2481–94. doi: 10.1016/j.bbadis.2018.04.005
220. Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. Mitochondrial Fission Triggered by Hyperglycemia Is Mediated by ROCK1 Activation in Podocytes and Endothelial Cells. Cell Metab (2012) 15:186–200. DOI: 10.1016/j.cmet.2012.01.009
221. Liu MY, Jin J, Li SL, Yan J, Zhen CL, Gao JL, et al. Mitochondrial Fission of Smooth Muscle Cells Is Involved in Artery Constriction. Hypertension (2016) 68:1245–54. doi: 10.1161/HYPERTENSIONAHA.116.07974
222. Preau S, Delguste F, Yu Y, Remy-Jouet I, Richard V, Saulnier F, et al. Endotoxemia Engages the RhoA Kinase Pathway to Impair Cardiac Function by Altering Cytoskeleton, Mitochondrial Fission, and Autophagy. Antioxid Redox Signal (2016) 24:529–42. doi: 10.1089/ars.2015.6421
223. Shen YL, Shi YZ, Chen GG, Wang LL, Zheng MZ, Jin HF, et al. TNF-Alpha Induces Drp1-Mediated Mitochondrial Fragmentation During Inflammatory Cardiomyocyte Injury. Int J Mol Med (2018) 41:2317–27. doi: 10.3892/ijmm.2018.3385
224. Zhang Q, Hu C, Huang J, Liu W, Lai W, Leng F, et al. ROCK1 Induces Dopaminergic Nerve Cell Apoptosis via the Activation of Drp1-Mediated Aberrant Mitochondrial Fission in Parkinson’s Disease. Exp Mol Med (2019) 51:1–13. doi: 10.1038/s12276-019-0318-z
225. Moskal N, Riccio V, Bashkurov M, Taddese R, Datti A, Lewis PN, et al. ROCK Inhibitors Upregulate the Neuroprotective Parkin-Mediated Mitophagy Pathway. Nat Commun (2020) 11:88. doi: 10.1038/s41467-019-13781-3
226. Quadir H, Hakobyan K, Gaddam M, Ojinnaka U, Ahmed Z, Kannan A, et al. Role of Rho-Associated Protein Kinase Inhibition as Therapeutic Strategy for Parkinson’s Disease: Dopaminergic Survival and Enhanced Mitophagy. Cureus (2021) 13:e16973. doi: 10.7759/cureus.16973
227. Liu Y, Fu Y, Hu X, Chen S, Miao J, Wang Y, et al. Caveolin-1 Knockdown Increases the Therapeutic Sensitivity of Lung Cancer to Cisplatin-Induced Apoptosis by Repressing Parkin-Related Mitophagy and Activating the ROCK1 Pathway. J Cell Physiol (2020) 235:1197–208. doi: 10.1002/jcp.29033
228. Pujar MK, Vastrad B, Vastrad C. Integrative Analyses of Genes Associated With Subcutaneous Insulin Resistance. Biomolecules (2019) 9:37. doi: 10.3390/biom9020037
229. Federico C, Alhallak K, Sun J, Duncan K, Azab F, Sudlow GP, et al. Tumor Microenvironment-Targeted Nanoparticles Loaded With Bortezomib and ROCK Inhibitor Improve Efficacy in Multiple Myeloma. Nat Commun (2020) 11:6037. doi: 10.1038/s41467-020-19932-1
230. Mietzner R, Kade C, Froemel F, Pauly D, Stamer WD, Ohlmann A, et al. Fasudil Loaded PLGA Microspheres as Potential Intravitreal Depot Formulation for Glaucoma Therapy. Pharmaceutics (2020) 12:706. doi; 10.3390/pharmaceutics12080706
231. Klein S, Frohn F, Magdaleno F, Reker-Smit C, Schierwagen R, Schierwagen I, et al. Rho-Kinase Inhibitor Coupled to Peptide-Modified Albumin Carrier Reduces Portal Pressure and Increases Renal Perfusion in Cirrhotic Rats. Sci Rep (2019) 9:2256. doi: 10.1038/s41598-019-38678-5
Keywords: ROCK, beige adipogenesis, energy expenditure, obesity, ROCK isoform-selective
Citation: Wei L and Shi J (2022) Insight Into Rho Kinase Isoforms in Obesity and Energy Homeostasis. Front. Endocrinol. 13:886534. doi: 10.3389/fendo.2022.886534
Received: 28 February 2022; Accepted: 06 May 2022;
Published: 13 June 2022.
Edited by:
Takeshi Yoneshiro, The University of Tokyo, JapanReviewed by:
Ying Yang, Shanghai Jiao Tong University, ChinaShannon Reilly, Cornell University, United States
Shinsuke Nirengi, The Ohio State University, United States
Ankit Gilani, Cornell University, United States
Copyright © 2022 Wei and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Wei, bGV3ZWlAaXUuZWR1; Jianjian Shi, amlzaGlAaXUuZWR1