 Thomas I. Hewat
Thomas I. Hewat Matthew B. Johnson
Matthew B. Johnson Sarah E. Flanagan
Sarah E. Flanagan- Institute of Biomedical and Clinical Science, University of Exeter Medical School, Exeter, United Kingdom
Congenital hyperinsulinism is characterised by the inappropriate release of insulin during hypoglycaemia. This potentially life-threatening disorder can occur in isolation, or present as a feature of syndromic disease. Establishing the underlying aetiology of the hyperinsulinism is critical for guiding medical management of this condition especially in children with diazoxide-unresponsive hyperinsulinism where the underlying genetics determines whether focal or diffuse pancreatic disease is present. Disease-causing single nucleotide variants affecting over 30 genes are known to cause persistent hyperinsulinism with mutations in the KATP channel genes (ABCC8 and KCNJ11) most commonly identified in children with severe persistent disease. Defects in methylation, changes in chromosome number, and large deletions and duplications disrupting multiple genes are also well described in congenital hyperinsulinism, further highlighting the genetic heterogeneity of this condition. Next-generation sequencing has revolutionised the approach to genetic testing for congenital hyperinsulinism with targeted gene panels, exome, and genome sequencing being highly sensitive methods for the analysis of multiple disease genes in a single reaction. It should though be recognised that limitations remain with next-generation sequencing with no single application able to detect all reported forms of genetic variation. This is an important consideration for hyperinsulinism genetic testing as comprehensive screening may require multiple investigations.
Introduction
Persistent congenital hyperinsulinism (HI) is characterised by the inappropriate secretion of insulin during hypoglycaemia which continues beyond 3 months. A prompt diagnosis of HI and effective management of glucose levels is critical to prevent adverse outcomes (1).
Persistent HI affects approximately 1 in 13,500 to 1 in 45,000 new-borns in non-consanguineous populations (2–5). In some isolated communities where founder mutations have been reported, and in populations with high rates of consanguinity, the incidence can increase to approximately 1 in 3,000 (6, 7). At least 36 different genetic causes of HI have been reported which follow recessive, dominant, X-linked, or sporadic inheritance (Table 1). The underlying genetic aetiology will determine whether the HI presents as isolated pancreatic disease or occurs as part of a rare syndrome.
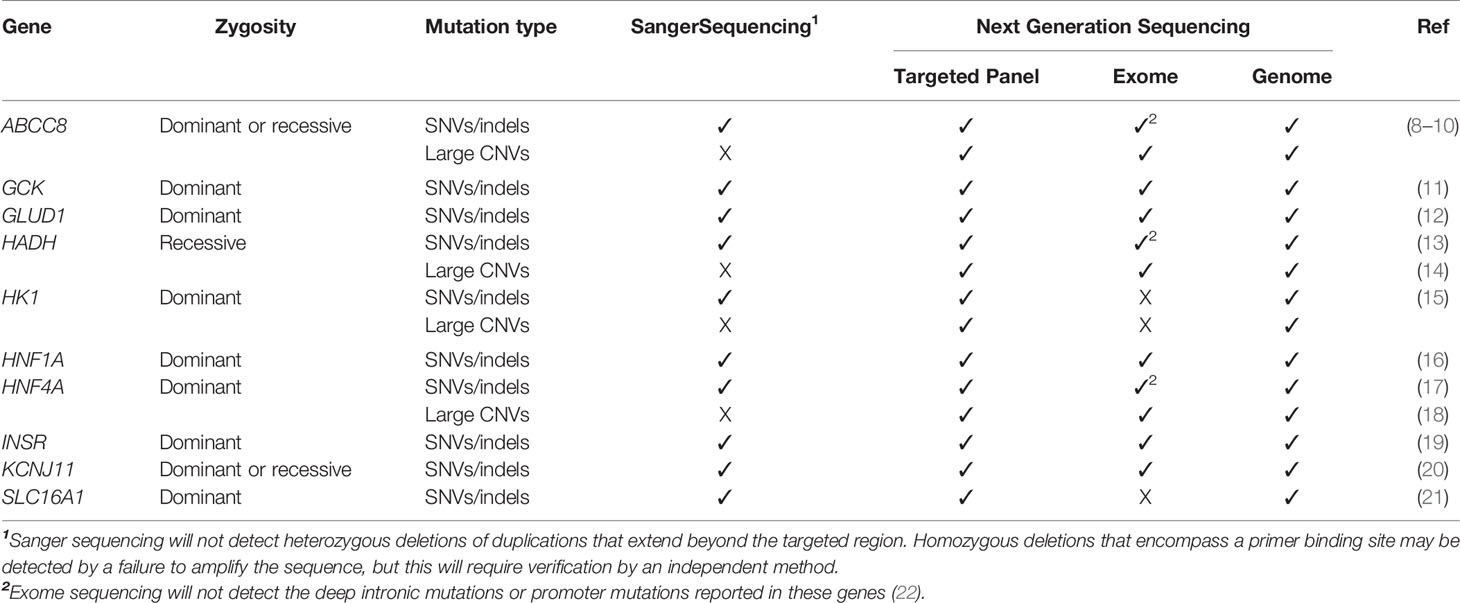
Table 1 Known genetic causes of isolated congenital hyperinsulinism and current approaches to genetic testing for this condition. A tick (✓) or cross (X) denote whether the form of genetic variation can be detected by the screening approach. None of the variants listed will be detected by methylation studies or array-CGH analysis. SNVs are single nucleotide variants, Indels are insertion/deletion variants and CNVs are copy number variants (deletions and duplications).
Many laboratories provide genetic testing for congenital HI; however, strategies vary between testing centres both in terms of the genes that are screened and the types of variation that can be detected (23–25). The different approaches to testing employed by each laboratory could help explain the differences in the percentage of mutation positive cases between cohorts which range from 45% to 79% (3, 4, 26, 27). Furthermore, the large number of genes which cause HI, the variable penetrance observed both within and between families with the same disease-causing variants, and the multiple modes of inheritance reported can hinder genetic interpretation which will also impact on the pick-up rates reported by each laboratory.
In this review, we describe the genetic causes of HI and discuss the benefits and limitations of the different methodological approaches currently used for genetic screening of this condition.
Genetic Types of Congenital Hyperinsulinism
Disease-causing variants in 10 genes have been reported to cause isolated, persistent HI (Table 1). Loss-of-function variants in the ABCC8 and KCNJ11 genes, which encode the two subunits of the pancreatic beta-cell ATP-sensitive potassium (KATP) channel, are most common and reported in 30-66% of cases referred for genetic testing (3, 4, 26, 27). A wide range of clinical severity is associated with KATP-HI with the functionally mildest variants causing transient disease which responds well to diazoxide treatment (the frontline drug for HI), whilst the most functionally severe variants cause diazoxide-unresponsive HI that persists throughout childhood (8, 28, 29). For individuals with diazoxide-unresponsive HI, pancreatic resection may be required to prevent life-threatening hypoglycaemia. For these infants, rapid genetic testing of the KATP channel genes is critical as it will determine the histological subtype of disease. Identifying biallelic (two disease-causing variants on opposite alleles) or a single dominant KATP channel disease-causing variant confirms diffuse pancreatic disease. In contrast finding a paternally inherited, recessive KATP channel variant, predicts focal disease with a sensitivity of 97% (27, 30). In these individuals the variant is rendered homozygous by a second somatic genetic event within the pancreas (uniparental isodisomy) (31, 32). This can be genetically confirmed by testing the pancreatic tissue following a lesionectomy, which proves curative in most cases.
Clinical characteristics can help to predict some genetic forms of isolated HI. For example, high ammonia concentrations are a consistent feature of GLUD1-HI (12), a family history of Maturity-Onset Diabetes of the Young (MODY) can predict HNF4A or HNF1A- HI (16, 17), and exercise-induced HI suggests a role for the beta-cell disallowed gene, SLC16A1 in disease pathogenesis (21).
Over 28 different syndromes which feature HI have been reported with the most common being Beckwith-Wiedemann syndrome (BWS) and Kabuki syndrome (33) (Table 2). The proportion of individuals with syndromic disease who present with HI varies between genetic subgroups. In some conditions HI is reported as a cardinal feature [e.g. Beckwith-Wiedemann syndrome (66)] whilst for others it is reported as a rare feature of the disease [e.g. Chromosome 9p deletions (40)]. Without genetic testing it can be hard to accurately diagnose syndromic disease, especially when HI is the presenting feature and dysmorphisms develop after birth, or when the clinical features are not specific to a genetic syndrome (67). For individuals with syndromic HI a genetic diagnosis is important as it will inform on prognosis and allow for the effective monitoring of new features of the disease.
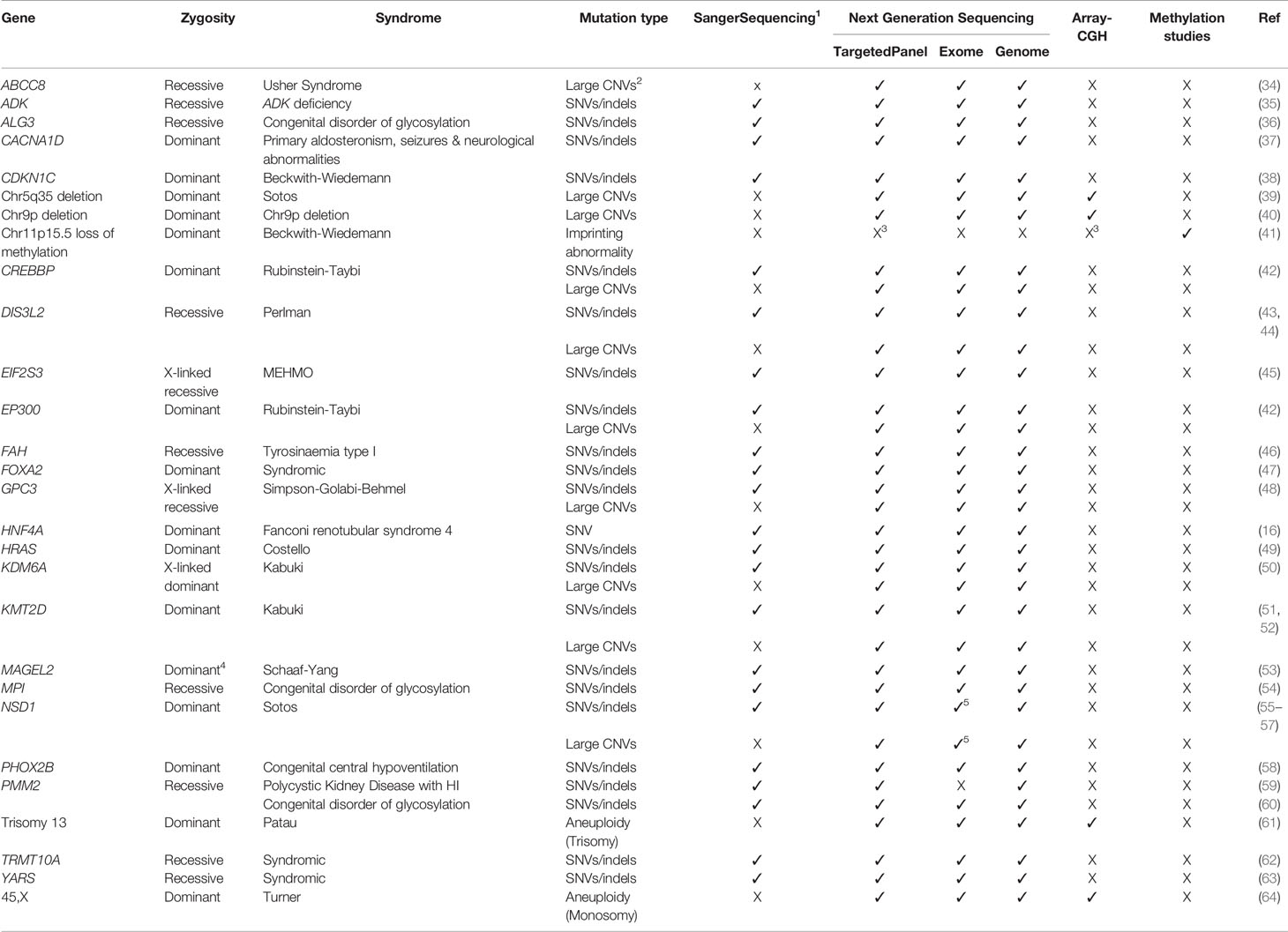
Table 2 Known genetic causes of syndromic disease in which congenital hyperinsulinism can be a rare or common feature and the current approaches to genetic testing for this condition. A tick (✓) or cross (X) denote whether the form of genetic variation can be detected by the screening approach. Methylation studies refer to methodologies that can detect changes in DNA methylation patterns (e.g. Epic array analysis, Methylation-specific MLPA). SNVs are single nucleotide variants, Indels are insertion/deletion variants and CNVs are copy number variants (deletions and duplications).
Sanger Sequencing
Causative genes for HI were historically screened by Sanger sequencing; an approach that allows a few hundred nucleotides (typically a single exon) to be rapidly sequenced in a single reaction. This is followed by semi-automated analysis by alignment and inspection of the DNA sequence. These constraints force laboratories to screen genes sequentially in descending order of prior probability based on clinical characteristics and how commonly disease-causing variants in the gene are identified. Whilst this phenotype-driven approach works well in many scenarios [for example in the rapid screening of KATP channel genes in individuals with diazoxide-unresponsive disease (68, 69)], the reliance of clinical features to guide testing can delay a genetic diagnosis for individuals with an atypical presentation. This is an important consideration for HI, as phenotypic variability is described within most genetic subgroups, for example the presence of normal ammonia levels in some children with GLUD1-HI (70, 71). Using the clinical characteristics to guide genetic testing in syndromic HI should also be applied with caution as additional features may develop after the diagnosis of HI (72).
A further major limitation of Sanger sequencing is its inability to detect heterozygous deletions and duplications that extend beyond the targeted region, changes in the number of chromosomes (aneuploidies), and defects in methylation, all of which have been reported to cause HI (Table 1).
Despite its limitations, Sanger sequencing remains a highly sensitive test for the rapid detection of single-nucleotide variants and small insertion/deletion variants (indels) in both the coding and non-coding regions of the genome. It can also detect mosaic variants (i.e. a genetic variant that is introduced during cell division that does not affect every cell within the body) that are present in the sampled tissue at a level of >8% (73). This is important, as disease-causing mosaic variants have been reported in the known HI genes including KMT2D, KDM6A, NSD1, and CREBBP (74–76).
Next-Generation Sequencing
Since 2005, next-generation sequencing has provided a method to allow for the simultaneous analysis of multiple genes in a single assay (77). This technology revolutionised diagnostic testing for genetically heterogeneous disorders such as HI by allowing for the parallel screening of all known disease-causing genes/genomic regions in a single assay at a much lower cost than Sanger sequencing. This led to a paradigm shift for conditions like syndromic HI where genetic testing can precede the development of the full clinical spectrum of disease, serving to make, rather than confirm, the clinical diagnosis (67).
Targeted Gene Panel Analysis by Next-Generation Sequencing
A targeted gene panel typically includes all known genetic causes of a disease and DNA samples are enriched for DNA in these loci prior to next-generation sequencing. For most targeted gene panels, the average coverage achieved often reaches many hundreds of reads over each base (78). This high-depth sequencing data can be exploited to detect changes in copy number over targeted regions and allows for the accurate detection of mosaic variants occurring at a level of >1% (73). Recent studies have shown that off-target reads generated during the sequencing process can be analysed to assess read-depth across the entire genome allowing for the detection of large deletions and duplications outside of targeted regions (79). These off-target reads have been used successfully to detect disease-causing deletions on chromosome 9p in individuals with HI (40). The potential to identify large deletions and duplications from off-target reads will though depend on the methodology used for the targeted next-generation sequencing; amplicon-based approaches that sequence PCR products will not generate the off-target sequencing data.
The major limitation of targeted next-generation sequencing is that it only allows screening of a predetermined list of genomic regions, and this list often differs between laboratories. For genetically heterogenous conditions such as HI, it is therefore important that clinicians who order panel testing are aware of which genes are included on the targeted panels and whether copy number analysis has been performed as this requires a separate bioinformatic analysis.
Exome and Genome Sequencing
The introduction of next-generation sequencing has enabled the rapid sequencing of the coding regions of all genes (the exome) or the entire human genome (coding and non-coding regions) at much lower cost than previous methods. The approach to the interpretation of exome and genome sequencing data will differ between centres with some analysing variants called within a pre-defined set of known disease-causing genes whilst other laboratories will perform a gene-agnostic analysis. The latter approach has the advantage of being able to identify new genes for HI, with recent successes including the discovery of the syndromic HI genes CACNA1D, PMM2, FOXA2, TRMT10A, EIF2S3, YARS, and KMT2D by exome sequencing and more recently the finding of regulatory variants deep within intron 2 of the beta-cell disallowed gene, HK1, by genome sequencing in individuals with isolated hyperinsulinism (15, 37, 45, 47, 51, 59, 62, 63). The ability of a laboratory to utilise next-generation sequencing data for genetic discovery will largely depend on their ability to perform robust genetic and functional studies to assess novel variation.
Exome sequencing targets the ~2% of the genome which codes for protein, making it a cheaper alternative to genome sequencing. This, together with the knowledge that 85% of known disease-causing mutations reside within coding regions, has led to exome sequencing being widely adopted within the clinical setting (80). For example, in the UK, rapid exome sequencing for acutely unwell neonates is available through the country’s National Health Service with 38% of patients tested receiving a rapid diagnosis (81). Unlike targeted next-generation sequencing, which screens a predetermined list of genes, exome sequencing provides an extremely effective method to comprehensively analyse the coding regions and intron/exon boundaries of all known HI genes and to assess copy number status. The major limitation of the approach is that it will not detect non-coding mutations such as the deep intronic mutations reported in ABCC8, HADH and HK1 or promoter variants in genes such as HNF4A, PMM2, and SLC16A1 (15, 21, 22, 59, 82).
Genome sequencing represents the gold standard approach to genetic testing given its ability to detect the largest range of genetic variation. As well as providing data on coding and non-coding regions, genome sequencing can be used to search for structural changes, copy number variants (large deletions, duplications, and aneuploidies) and mosaic variants although the lower read depth achieved makes this a less sensitive approach for detecting low-level mosaic variants compared to targeted next-generation sequencing.
The costs associated with sequencing the entire genome and the large amount of data produced (approximately 200GB of processed data per sample versus 11GB per sample for exome sequencing) had prohibited the adoption of routine genome sequencing. Until recently it had been largely reserved for genetic screening when a disease-causing variant had not been detected by targeted next-generation sequencing or exome sequencing. This approach successful resulted in an increase in diagnostic yield for many rare genetic diseases (83, 84).
Improvements in sequencing capabilities leading to reduced costs are though now leading to the emergence of genome sequencing as a first line diagnostic test in specific healthcare settings, for example in the screening of some rare developmental disorders in the UK National Health Service (85). While genome sequencing is not the current approach for investigating the genetic cause of HI in many centres, it seems likely that this will become the first line test in the coming years.
Non Sequencing Based Methods to Detect Copy Number Variants and Methylation Defects
Aneuploidies and large deletions and duplications (copy number variants) are a rare but important cause of HI (Tables 1 and 2). Unlike Sanger sequencing, next-generation sequencing can detect these forms of genetic variation, but many laboratories will not routinely screen for them as a separate analysis pipeline is required. This is an important consideration when disease-causing variants are not detected in children with HI and particularly for those where there are additional syndromic features (Table 2).
Multiplex-ligation dependent probe amplification (MLPA) can detect disease-causing deletions and duplications in individuals with HI. This approach is commonly used to screen for deletions in the ABCC8 gene and can detect mosaicism (9). The usefulness of MLPA is limited by its ability to analyse a maximum of 60 different small genomic regions (generally single exons) in a single assay thus preventing the simultaneous analysis of all HI genes in which copy number changes have been reported.
Microarray-based comparative genomic hybridization (array CGH) is a well-established method that is used to detect large deletions/duplications and aneuploidies in individuals with HI. Unlike MLPA, array GCH is not able to detect low level mosaicism (<30% mosaicism for deletions and duplications and <10% for aneuploidies). The approach does however allow for the analysis of copy number variation across a greater percentage of the genome although the targeted region will vary across arrays and will not always target the regions known to cause HI with enough precision.
Current diagnostic sequencing approaches are also unable to detect changes in DNA methylation. Individuals with clinical suspicion of an imprinting disorder such as Beckwith-Wiedemann syndrome may therefore require additional methylation studies, such as methylation-specific MLPA (MS-MLPA) (86) or Infinium Methylation EPIC array analysis (87). Emerging technologies, such as Oxford Nanopore sequencing, may allow for the simultaneous detection of sequence variation and DNA methylation status but have not been widely used clinically. This technology does offer the hope of a single comprehensive test for genetically heterogeneous disorders like HI although to date it has mainly been used for genes that are hard to sequence by other methodologies (88–91).
Further Considerations and Concluding Remarks
Diagnostic testing for HI is routinely performed on DNA extracted from peripheral blood leukocytes, saliva, or buccal samples. For conditions such as HI it is important to consider the source of DNA being screened, given that somatic mutations which are only present in the pancreatic tissue have been reported (27, 92). Therefore, when a mutation is not identified in the blood, and a pancreatectomy has been performed, re-testing the known HI genes to search for a variant present only within the pancreatic DNA should be considered.
In conclusion, several different genetic approaches exist for routine diagnostic screening in HI with genome sequencing representing the gold standard approach to testing. For healthcare professionals managing this genetically heterogenous disorder it is important that the limitations of each approach including genome sequencing, are recognised as no single test can detect all known types of genetic variation reported in HI. This is particularly important when managing syndromic disease, where copy number variants or defects in methylation are common. Despite there being a broad range of genetic screening approaches that are available for HI, in reality the testing strategy is most likely to be influenced by the capabilities of the local genetic diagnostic laboratory, affordability and importantly how quickly the tests can be performed and results reported back. This is especially critical for children with diazoxide-unresponsive disease as identifying a paternally inherited KATP disease-causing variant suggests focal pancreatic disease which can be cured by lesionectomy.
Author Contributions
TH and SF performed the literature searches and reviews. TH, MJ, and SF drafted and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
SF is a Wellcome Trust Senior Research Fellow (Grant Number 223187/Z/21/Z). TH is the recipient of a PhD studentship and MJ has an Independent Fellowship from the Exeter Diabetes Centre of Excellence funded by Research England’s Expanding Excellence in England (E3) fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Dr Thomas Laver and Dr Matthew Wakeling for their contributions to this review.
References
1. Helleskov A, Melikyan M, Globa E, Shcherderkina I, Poertner F, Larsen AM, et al. Both Low Blood Glucose and Insufficient Treatment Confer Risk of Neurodevelopmental Impairment in Congenital Hyperinsulinism: A Multinational Cohort Study. Front Endocrinol (Lausanne) (2017) 8:156. doi: 10.3389/fendo.2017.00156
2. Yau D, Laver TW, Dastamani A, Senniappan S, Houghton JAL, Shaikh G, et al. Using Referral Rates for Genetic Testing to Determine the Incidence of a Rare Disease: The Minimal Incidence of Congenital Hyperinsulinism in the UK is 1 in 28,389. PloS One (2020) 15(2):e0228417. doi: 10.1371/journal.pone.0228417
3. Rozenkova K, Malikova J, Nessa A, Dusatkova L, Bjorkhaug L, Obermannova B, et al. High Incidence of Heterozygous ABCC8 and HNF1A Mutations in Czech Patients With Congenital Hyperinsulinism. J Clin Endocrinol Metab (2015) 100(12):E1540–9. doi: 10.1210/jc.2015-2763
4. Mannisto JME, Jaaskelainen J, Otonkoski T, Huopio H. Long-Term Outcome and Treatment in Persistent and Transient Congenital Hyperinsulinism: A Finnish Population-Based Study. J Clin Endocrinol Metab (2021) 106(4):e1542–e51. doi: 10.1210/clinem/dgab024
5. Kawakita R, Sugimine H, Nagai S, Kawai M, Kusuda S, Yorifuji T. Clinical Characteristics of Congenital Hyperinsulinemic Hypoglycemia in Infant: A Nationwide Epidemiological Survey in Japan. Nihon Shonika Gakkai Zasshi (2011) 115:563–9.
6. Otonkoski T, Ammala C, Huopio H, Cote GJ, Chapman J, Cosgrove K, et al. A Point Mutation Inactivating the Sulfonylurea Receptor Causes the Severe Form of Persistent Hyperinsulinemic Hypoglycemia of Infancy in Finland. Diabetes. (1999) 48(2):408–15. doi: 10.2337/diabetes.48.2.408
7. Mathew PM, Young JM, Abu-Osba YK, Mulhern BD, Hammoudi S, Hamdan JA, et al. Persistent Neonatal Hyperinsulinism. Clin Pediatr (Phila) (1988) 27(3):148–51. doi: 10.1177/000992288802700307
8. Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, et al. Mutations in the Sulfonylurea Receptor Gene in Familial Persistent Hyperinsulinemic Hypoglycemia of Infancy. Science (1995) 268(5209):426–9. doi: 10.1126/science.7716548
9. Flanagan S, Damhuis A, Banerjee I, Rokicki D, Jefferies C, Kapoor R, et al. Partial ABCC8 Gene Deletion Mutations Causing Diazoxide-Unresponsive Hyperinsulinaemic Hypoglycaemia. Pediatr Diabetes (2012) 13(3):285–9. doi: 10.1111/j.1399-5448.2011.00821.x
10. De Franco E, Saint-Martin C, Brusgaard K, Knight Johnson AE, Aguilar-Bryan L, Bowman P, et al. Update of Variants Identified in the Pancreatic Beta-Cell KATP Channel Genes KCNJ11 and ABCC8 in Individuals With Congenital Hyperinsulinism and Diabetes. Hum Mutat (2020) 41(5):884–905. doi: 10.1002/humu.23995
11. Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, et al. Familial Hyperinsulinism Caused by an Activating Glucokinase Mutation. N Engl J Med (1998) 338(4):226–30. doi: 10.1056/NEJM199801223380404
12. Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, et al. Hyperinsulinism and Hyperammonemia in Infants With Regulatory Mutations of the Glutamate Dehydrogenase Gene. N Engl J Med (1998) 338(19):1352–7. doi: 10.1056/NEJM199805073381904
13. Clayton PT, Eaton S, Aynsley-Green A, Edginton M, Hussain K, Krywawych S, et al. Hyperinsulinism in Short-Chain L-3-Hydroxyacyl-CoA Dehydrogenase Deficiency Reveals the Importance of Beta-Oxidation in Insulin Secretion. J Clin Invest (2001) 108(3):457–65. doi: 10.1172/JCI200111294
14. Flanagan SE, Patch AM, Locke JM, Akcay T, Simsek E, Alaei M, et al. Genome-Wide Homozygosity Analysis Reveals HADH Mutations as a Common Cause of Diazoxide-Responsive Hyperinsulinemic-Hypoglycemia in Consanguineous Pedigrees. J Clin Endocrinol Metab (2011) 96(3):E498–502. doi: 10.1210/jc.2010-1906
15. Wakeling MN, Owens NDL, Hopkinson JR, Johnson MB, Houghton JAL, Dastamani A, et al. A Novel Disease Mechanism Leading to the Expression of a Disallowed Gene in the Pancreatic Beta-Cell Identified by non-Coding, Regulatory Mutations Controlling HK1. medRxiv (2021) 2021.12.03.21267240. doi: 10.1101/2021.12.03.21267240
16. Stanescu DE, Hughes N, Kaplan B, Stanley CA, De Leon DD. Novel Presentations of Congenital Hyperinsulinism Due to Mutations in the MODY Genes: HNF1A and HNF4A. J Clin Endocrinol Metab (2012) 97(10):E2026–30. doi: 10.1210/jc.2012-1356
17. Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, et al. Macrosomia and Hyperinsulinaemic Hypoglycaemia in Patients With Heterozygous Mutations in the HNF4A Gene. PloS Med (2007) 4(4):e118. doi: 10.1371/journal.pmed.0040118
18. Tung JY, Boodhansingh K, Stanley CA, De Leon DD. Clinical Heterogeneity of Hyperinsulinism Due to HNF1A and HNF4A Mutations. Pediatr Diabetes (2018) 19(5):910–6. doi: 10.1111/pedi.12655
19. Hojlund K, Hansen T, Lajer M, Henriksen JE, Levin K, Lindholm J, et al. A Novel Syndrome of Autosomal-Dominant Hyperinsulinemic Hypoglycemia Linked to a Mutation in the Human Insulin Receptor Gene. Diabetes (2004) 53(6):1592–8. doi: 10.2337/diabetes.53.6.1592
20. Thomas P, Ye Y, Lightner E. Mutation of the Pancreatic Islet Inward Rectifier Kir6.2 Also Leads to Familial Persistent Hyperinsulinemic Hypoglycemia of Infancy. Hum Mol Genet (1996) 5(11):1809–12. doi: 10.1093/hmg/5.11.1809
21. Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, Parton LE, et al. Physical Exercise-Induced Hypoglycemia Caused by Failed Silencing of Monocarboxylate Transporter 1 in Pancreatic Beta Cells. Am J Hum Genet (2007) 81(3):467–74. doi: 10.1086/520960
22. Flanagan SE, Xie W, Caswell R, Damhuis A, Vianey-Saban C, Akcay T, et al. Next-Generation Sequencing Reveals Deep Intronic Cryptic ABCC8 and HADH Splicing Founder Mutations Causing Hyperinsulinism by Pseudoexon Activation. Am J Hum Genet (2013) 92(1):131–6. doi: 10.1016/j.ajhg.2012.11.017
23. Novoa-Medina Y, Dominguez Garcia A, Quinteiro Gonzalez S, Garcia Cruz LM, Santana Rodriguez A. Congenital Hyperinsulinism in Gran Canaria, Canary Isles. Pediatr (Engl Ed) (2021) 95(2):93–100. doi: 10.1016/j.anpede.2020.08.006
24. Casertano A, Rossi A, Fecarotta S, Rosanio FM, Moracas C, Di Candia F, et al. An Overview of Hypoglycemia in Children Including a Comprehensive Practical Diagnostic Flowchart for Clinical Use. Front Endocrinol (Lausanne) (2021) 12:684011. doi: 10.3389/fendo.2021.684011
25. Razzaghy-Azar M, Saeedi S, Dayani SB, Enayati S, Abbasi F, Hashemian S, et al. Investigating Genetic Mutations in a Large Cohort of Iranian Patients With Congenital Hyperinsulinism. J Clin Res Pediatr Endocrinol (2022) 14(1):87–95. doi: 10.4274/jcrpe.galenos.2021.2021.0071
26. Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and Molecular Characterisation of 300 Patients With Congenital Hyperinsulinism. Eur J Endocrinol (2013) 168(4):557–64. doi: 10.1530/EJE-12-0673
27. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and Phenotype Correlations in 417 Children With Congenital Hyperinsulinism. J Clin Endocrinol Metab (2013) 98(2):E355–63. doi: 10.1210/jc.2012-2169
28. Taschenberger G, Mougey A, Shen S, Lester LB, LaFranchi S, Shyng SL. Identification of a Familial Hyperinsulinism-Causing Mutation in the Sulfonylurea Receptor 1 That Prevents Normal Trafficking and Function of KATP Channels. J Biol Chem (2002) 277(19):17139–46. doi: 10.1074/jbc.M200363200
29. Kumaran A, Kapoor RR, Flanagan SE, Ellard S, Hussain K. Congenital Hyperinsulinism Due to a Compound Heterozygous ABCC8 Mutation With Spontaneous Resolution at Eight Weeks. Horm Res Paediatr (2010) 73(4):287–92. doi: 10.1159/000284394
30. Mohnike K, Wieland I, Barthlen W, Vogelgesang S, Empting S, Mohnike W, et al. Clinical and Genetic Evaluation of Patients With KATP Channel Mutations From the German Registry for Congenital Hyperinsulinism. Horm Res Paediatr (2014) 81(3):156–68. doi: 10.1159/000356905
31. de Lonlay P, Fournet JC, Rahier J, Gross-Morand MS, Poggi-Travert F, Foussier V, et al. Somatic Deletion of the Imprinted 11p15 Region in Sporadic Persistent Hyperinsulinemic Hypoglycemia of Infancy is Specific of Focal Adenomatous Hyperplasia and Endorses Partial Pancreatectomy. J Clin Invest (1997) 100(4):802–7. doi: 10.1172/JCI119594
32. Damaj L, le Lorch M, Verkarre V, Werl C, Hubert L, Nihoul-Fekete C, et al. Chromosome 11p15 Paternal Isodisomy in Focal Forms of Neonatal Hyperinsulinism. J Clin Endocrinol Metab (2008) 93(12):4941–7. doi: 10.1210/jc.2008-0673
33. Kostopoulou E, Dastamani A, Guemes M, Clement E, Caiulo S, Shanmugananda P, et al. Syndromic Forms of Hyperinsulinaemic Hypoglycaemia-A 15-Year Follow-Up Study. Clin Endocrinol (Oxf) (2021) 94(3):399–412. doi: 10.1111/cen.14393
34. Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, et al. A Recessive Contiguous Gene Deletion Causing Infantile Hyperinsulinism, Enteropathy and Deafness Identifies the Usher Type 1C Gene. Nat Genet (2000) 26(1):56–60. doi: 10.1038/79178
35. Staufner C, Lindner M, Dionisi-Vici C, Freisinger P, Dobbelaere D, Douillard C, et al. Adenosine Kinase Deficiency: Expanding the Clinical Spectrum and Evaluating Therapeutic Options. J Inherit Metab Dis (2016) 39(2):273–83. doi: 10.1007/s10545-015-9904-y
36. Sun L, Eklund EA, Chung WK, Wang C, Cohen J, Freeze HH. Congenital Disorder of Glycosylation Id Presenting With Hyperinsulinemic Hypoglycemia and Islet Cell Hyperplasia. J Clin Endocrinol Metab (2005) 90(7):4371–5. doi: 10.1210/jc.2005-0250
37. Flanagan SE, Vairo F, Johnson MB, Caswell R, Laver TW, Lango Allen H, et al. A CACNA1D Mutation in a Patient With Persistent Hyperinsulinaemic Hypoglycaemia, Heart Defects, and Severe Hypotonia. Pediatr Diabetes (2017) 18(4):320–3. doi: 10.1111/pedi.12512
38. Hatada I, Ohashi H, Fukushima Y, Kaneko Y, Inoue M, Komoto Y, et al. An Imprinted Gene P57kip2 is Mutated in Beckwith-Wiedemann Syndrome. Nat Genet (1996) 14(2):171–3. doi: 10.1038/ng1096-171
39. Matsuo T, Ihara K, Ochiai M, Kinjo T, Yoshikawa Y, Kojima-Ishii K, et al. Hyperinsulinemic Hypoglycemia of Infancy in Sotos Syndrome. Am J Med Genet A (2013) 161A(1):34–7. doi: 10.1002/ajmg.a.35657
40. Banerjee I, Senniappan S, Laver TW, Caswell R, Zenker M, Mohnike K, et al. Refinement of the Critical Genomic Region for Congenital Hyperinsulinism in the Chromosome 9p Deletion Syndrome. Wellcome Open Res (2019) 4:149. doi: 10.12688/wellcomeopenres.15465.1
41. Munns CF, Batch JA. Hyperinsulinism and Beckwith-Wiedemann Syndrome. Arch Dis Child Fetal Neonatal Ed (2001) 84(1):F67–9. doi: 10.1136/fn.84.1.F67
42. Welters A, El-Khairi R, Dastamani A, Bachmann N, Bergmann C, Gilbert C, et al. Persistent Hyperinsulinaemic Hypoglycaemia in Children With Rubinstein-Taybi Syndrome. Eur J Endocrinol (2019) 181(2):121–8. doi: 10.1530/EJE-19-0119
43. Henneveld HT, van Lingen RA, Hamel BC, Stolte-Dijkstra I, van Essen AJ. Perlman Syndrome: Four Additional Cases and Review. Am J Med Genet (1999) 86(5):439–46. doi: 10.1002/(SICI)1096-8628(19991029)86:5<439::AID-AJMG9>3.0.CO;2-4
44. Astuti D, Morris MR, Cooper WN, Staals RH, Wake NC, Fews GA, et al. Germline Mutations in DIS3L2 Cause the Perlman Syndrome of Overgrowth and Wilms Tumor Susceptibility. Nat Genet (2012) 44(3):277–84. doi: 10.1038/ng.1071
45. Gregory LC, Ferreira CB, Young-Baird SK, Williams HJ, Harakalova M, van Haaften G, et al. Impaired EIF2S3 Function Associated With a Novel Phenotype of X-Linked Hypopituitarism With Glucose Dysregulation. EBioMedicine. (2019) 42:470–80. doi: 10.1016/j.ebiom.2019.03.013
46. Baumann U, Preece MA, Green A, Kelly DA, McKiernan PJ. Hyperinsulinism in Tyrosinaemia Type I. J Inherit Metab Dis (2005) 28(2):131–5. doi: 10.1007/s10545-005-5517-1
47. Giri D, Vignola ML, Gualtieri A, Scagliotti V, McNamara P, Peak M, et al. Novel FOXA2 Mutation Causes Hyperinsulinism, Hypopituitarism With Craniofacial and Endoderm-Derived Organ Abnormalities. Hum Mol Genet (2017) 26(22):4315–26. doi: 10.1093/hmg/ddx318
48. Terespolsky D, Farrell SA, Siegel-Bartelt J, Weksberg R. Infantile Lethal Variant of Simpson-Golabi-Behmel Syndrome Associated With Hydrops Fetalis. Am J Med Genet (1995) 59(3):329–33. doi: 10.1002/ajmg.1320590310
49. Alexander S, Ramadan D, Alkhayyat H, Al-Sharkawi I, Backer KC, El-Sabban F, et al. Costello Syndrome and Hyperinsulinemic Hypoglycemia. Am J Med Genet A (2005) 139(3):227–30. doi: 10.1002/ajmg.a.31011
50. Gole H, Chuk R, Coman D. Persistent Hyperinsulinism in Kabuki Syndrome 2: Case Report and Literature Review. Clin Pract (2016) 6(3):848. doi: 10.4081/cp.2016.848
51. Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. Exome Sequencing Identifies MLL2 Mutations as a Cause of Kabuki Syndrome. Nat Genet (2010) 42(9):790–3. doi: 10.1038/ng.646
52. White SM, Thompson EM, Kidd A, Savarirayan R, Turner A, Amor D, et al. Growth, Behavior, and Clinical Findings in 27 Patients With Kabuki (Niikawa-Kuroki) Syndrome. Am J Med Genet A (2004) 127A(2):118–27. doi: 10.1002/ajmg.a.20674
53. Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of Exome and Genome Sequencing Guided by Acuity of Illness for Diagnosis of Neurodevelopmental Disorders. Sci Transl Med (2014) 6(265):265ra168. doi: 10.1126/scitranslmed.3010076
54. Deeb A, Al Amoodi A. A Novel Homozygous Mutation in the Mannose Phosphate Isomerase Gene Causing Congenital Disorder of Glycation and Hyperinsulinemic Hypoglycemia in an Infant. Clin Case Rep (2018) 6(3):479–83. doi: 10.1002/ccr3.1387
55. Carrasco Salas P, Palma Milla C, Lezana Rosales JM, Benito C, Franco Freire S, Lopez Siles J. Hyperinsulinemic Hypoglycemia in a Patient With an Intragenic NSD1 Mutation. Am J Med Genet A (2016) 170A(2):544–6. doi: 10.1002/ajmg.a.37440
56. Grand K, Gonzalez-Gandolfi C, Ackermann AM, Aljeaid D, Bedoukian E, Bird LM, et al. Hyperinsulinemic Hypoglycemia in Seven Patients With De Novo NSD1 Mutations. Am J Med Genet A (2019) 179(4):542–51. doi: 10.1002/ajmg.a.61062
57. Douglas J, Tatton-Brown K, Coleman K, Guerrero S, Berg J, Cole TR, et al. Partial NSD1 Deletions Cause 5% of Sotos Syndrome and are Readily Identifiable by Multiplex Ligation Dependent Probe Amplification. J Med Genet (2005) 42(9):e56. doi: 10.1136/jmg.2005.031930
58. Hennewig U, Hadzik B, Vogel M, Meissner T, Goecke T, Peters H, et al. Congenital Central Hypoventilation Syndrome With Hyperinsulinism in a Preterm Infant. J Hum Genet (2008) 53(6):573–7. doi: 10.1007/s10038-008-0275-1
59. Cabezas OR, Flanagan SE, Stanescu H, Garcia-Martinez E, Caswell R, Lango-Allen H, et al. Polycystic Kidney Disease With Hyperinsulinemic Hypoglycemia Caused by a Promoter Mutation in Phosphomannomutase 2. J Am Soc Nephrol (2017) 28(8):2529–39. doi: 10.1681/ASN.2016121312
60. Bohles H, Sewell AA, Gebhardt B, Reinecke-Luthge A, Kloppel G, Marquardt T. Hyperinsulinaemic Hypoglycaemia–Leading Symptom in a Patient With Congenital Disorder of Glycosylation Ia (Phosphomannomutase Deficiency). J Inherit Metab Dis (2001) 24(8):858–62. doi: 10.1023/A:1013944308881
61. Tamame T, Hori N, Homma H, Yoshida R, Inokuchi M, Kosaki K, et al. Hyperinsulinemic Hypoglycemia in a Newborn Infant With Trisomy 13. Am J Med Genet A (2004) 129A(3):321–2. doi: 10.1002/ajmg.a.30147
62. Gillis D, Krishnamohan A, Yaacov B, Shaag A, Jackman JE, Elpeleg O. TRMT10A Dysfunction is Associated With Abnormalities in Glucose Homeostasis, Short Stature and Microcephaly. J Med Genet (2014) 51(9):581–6. doi: 10.1136/jmedgenet-2014-102282
63. Zeiad R, Ferren EC, Young DD, De Lancy SJ, Dedousis D, Schillaci LA, et al. A Novel Homozygous Missense Mutation in the YARS Gene: Expanding the Phenotype of YARS Multisystem Disease. J Endocr Soc (2021) 5(2):bvaa196. doi: 10.1210/jendso/bvaa196
64. Alkhayyat H, Christesen HB, Steer J, Stewart H, Brusgaard K, Hussain K. Mosaic Turner Syndrome and Hyperinsulinaemic Hypoglycaemia. J Pediatr Endocrinol Metab (2006) 19(12):1451–7. doi: 10.1515/JPEM.2006.19.12.1451
65. Brioude F, Lacoste A, Netchine I, Vazquez MP, Auber F, Audry G, et al. Beckwith-Wiedemann Syndrome: Growth Pattern and Tumor Risk According to Molecular Mechanism, and Guidelines for Tumor Surveillance. Horm Res Paediatr (2013) 80(6):457–65. doi: 10.1159/000355544
66. Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert Consensus Document: Clinical and Molecular Diagnosis, Screening and Management of Beckwith-Wiedemann Syndrome: An International Consensus Statement. Nat Rev Endocrinol (2018) 14(4):229–49. doi: 10.1038/nrendo.2017.166
67. Hoermann H, El-Rifai O, Schebek M, Lodefalk M, Brusgaard K, Bachmann N, et al. Comparative Meta-Analysis of Kabuki Syndrome With and Without Hyperinsulinaemic Hypoglycaemia. Clin Endocrinol (Oxf) (2020) 93(3):346–54. doi: 10.1111/cen.14267
68. Hewat TI, Yau D, Jerome JCS, Laver TW, Houghton JAL, Shields BM, et al. Birth Weight and Diazoxide Unresponsiveness Strongly Predict the Likelihood of Congenital Hyperinsulinism Due to a Mutation in ABCC8 or KCNJ11. Eur J Endocrinol (2021) 185(6):813–8. doi: 10.1530/EJE-21-0476
69. Banerjee I, Skae M, Flanagan SE, Rigby L, Patel L, Didi M, et al. The Contribution of Rapid KATP Channel Gene Mutation Analysis to the Clinical Management of Children With Congenital Hyperinsulinism. Eur J Endocrinol (2011) 164(5):733–40. doi: 10.1530/EJE-10-1136
70. Brandt A, Agarwal N, Giri D, Yung Z, Didi M, Senniappan S. Hyperinsulinism Hyperammonaemia (HI/HA) Syndrome Due to GLUD1 Mutation: Phenotypic Variations Ranging From Late Presentation to Spontaneous Resolution. J Pediatr Endocrinol Metab (2020) 33(5):675–9. doi: 10.1515/jpem-2019-0416
71. Kapoor RR, Flanagan SE, Fulton P, Chakrapani A, Chadefaux B, Ben-Omran T, et al. Hyperinsulinism-Hyperammonaemia Syndrome: Novel Mutations in the GLUD1 Gene and Genotype-Phenotype Correlations. Eur J Endocrinol (2009) 161(5):731–5. doi: 10.1530/EJE-09-0615
72. Yap KL, Johnson AEK, Fischer D, Kandikatla P, Deml J, Nelakuditi V, et al. Congenital Hyperinsulinism as the Presenting Feature of Kabuki Syndrome: Clinical and Molecular Characterization of 9 Affected Individuals. Genet Med (2019) 21(1):233–42. doi: 10.1038/s41436-018-0013-9
73. Brewer CJ, Gillespie M, Fierro J, Scaringe WA, Li JM, Lee CY, et al. The Value of Parental Testing by Next-Generation Sequencing Includes the Detection of Germline Mosaicism. J Mol Diagn (2020) 22(5):670–8. doi: 10.1016/j.jmoldx.2020.02.001
74. Murakami H, Tsurusaki Y, Enomoto K, Kuroda Y, Yokoi T, Furuya N, et al. Update of the Genotype and Phenotype of KMT2D and KDM6A by Genetic Screening of 100 Patients With Clinically Suspected Kabuki Syndrome. Am J Med Genet A (2020) 182(10):2333–44. doi: 10.1002/ajmg.a.61793
75. Castronovo C, Rusconi D, Crippa M, Giardino D, Gervasini C, Milani D, et al. A Novel Mosaic NSD1 Intragenic Deletion in a Patient With an Atypical Phenotype. Am J Med Genet A (2013) 161A(3):611–8. doi: 10.1002/ajmg.a.35814
76. Lin S, He Z, Huang L, Liu J, Lei T, Wu J, et al. Case Report: Low-Level Maternal Mosaicism of a Novel CREBBP Variant Causes Recurrent Rubinstein-Taybi Syndrome in Two Siblings of a Chinese Family. Front Genet (2021) 12:640992. doi: 10.3389/fgene.2021.640992
77. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome Sequencing in Microfabricated High-Density Picolitre Reactors. Nature. (2005) 437(7057):376–80. doi: 10.1038/nature03959
78. Cleveland MH, Zook JM, Salit M, Vallone PM. Determining Performance Metrics for Targeted Next-Generation Sequencing Panels Using Reference Materials. J Mol Diagn (2018) 20(5):583–90. doi: 10.1016/j.jmoldx.2018.04.005
79. Laver TW, De Franco E, Johnson MB, Patel KA, Ellard S, Weedon MN, et al. SavvyCNV: Genome-Wide CNV Calling From Off-Target Reads. PloS Comput Biol (2022) 18(3):e1009940. doi: 10.1371/journal.pcbi.1009940
80. Scacheri CA, Scacheri PC. Mutations in the Noncoding Genome. Curr Opin Pediatr (2015) 27(6):659–64. doi: 10.1097/MOP.0000000000000283
81. McDermott H, Baple E, Ellard S, Naik S. 1435 Rapid Exome Sequencing in Acutely Unwell Children – Providing New Diagnostic Options in Intensive Care Settings. Arch Dis Child (2021), :106(Suppl 1):A1–A514. doi: 10.1136/archdischild-2021-rcpch.641
82. Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S. Mutations in the Genes Encoding the Transcription Factors Hepatocyte Nuclear Factor 1 Alpha and 4 Alpha in Maturity-Onset Diabetes of the Young and Hyperinsulinemic Hypoglycemia. Hum Mutat (2013) 34(5):669–85. doi: 10.1002/humu.22279
83. Mattick JS, Dinger M, Schonrock N, Cowley M. Whole Genome Sequencing Provides Better Diagnostic Yield and Future Value Than Whole Exome Sequencing. Med J Aust (2018) 209(5):197–9. doi: 10.5694/mja17.01176
84. Investigators GPP, Smedley D, Smith KR, Martin A, Thomas EA, McDonagh EM, et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care - Preliminary Report. N Engl J Med (2021) 385(20):1868–80. doi: 10.1056/NEJMoa2035790
85. Caulfield M, Davies J, Dennys M, Elbahy L, Fowler T, Hill S, et al. The National Genomics Research and Healthcare Knowledgebase. (2020).
86. Scott RH, Douglas J, Baskcomb L, Nygren AO, Birch JM, Cole TR, et al. Methylation-Specific Multiplex Ligation-Dependent Probe Amplification (MS-MLPA) Robustly Detects and Distinguishes 11p15 Abnormalities Associated With Overgrowth and Growth Retardation. J Med Genet (2008) 45(2):106–13. doi: 10.1136/jmg.2007.053207
87. Aref-Eshghi E, Kerkhof J, Pedro VP, France GD, Barat-Houari M, Ruiz-Pallares N, et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am J Hum Genet (2021) 108(6):1161–3. doi: 10.1016/j.ajhg.2021.04.022
88. Nowak A, Murik O, Mann T, Zeevi DA, Altarescu G. Detection of Single Nucleotide and Copy Number Variants in the Fabry Disease-Associated GLA Gene Using Nanopore Sequencing. Sci Rep (2021) 11(1):22372. doi: 10.1038/s41598-021-01749-7
89. Leija-Salazar M, Sedlazeck FJ, Toffoli M, Mullin S, Mokretar K, Athanasopoulou M, et al. Evaluation of the Detection of GBA Missense Mutations and Other Variants Using the Oxford Nanopore MinION. Mol Genet Genomic Med (2019) 7(3):e564. doi: 10.1002/mgg3.564
90. Mahaweni NM, Olieslagers TI, Rivas IO, Molenbroeck SJJ, Groeneweg M, Bos GMJ, et al. A Comprehensive Overview of FCGR3A Gene Variability by Full-Length Gene Sequencing Including the Identification of V158F Polymorphism. Sci Rep (2018) 8(1):15983. doi: 10.1038/s41598-018-34258-1
91. Minervini CF, Cumbo C, Orsini P, Brunetti C, Anelli L, Zagaria A, et al. TP53 Gene Mutation Analysis in Chronic Lymphocytic Leukemia by Nanopore MinION Sequencing. Diagn Pathol (2016) 11(1):96. doi: 10.1186/s13000-016-0550-y
Keywords: hyperinsulinism, hypoglycaemia, genetic screening, genetics, next generation sequencing - NGS
Citation: Hewat TI, Johnson MB and Flanagan SE (2022) Congenital Hyperinsulinism: Current Laboratory-Based Approaches to the Genetic Diagnosis of a Heterogeneous Disease. Front. Endocrinol. 13:873254. doi: 10.3389/fendo.2022.873254
Received: 10 February 2022; Accepted: 14 June 2022;
Published: 07 July 2022.
Edited by:
Klaus Mohnike, University Hospital Magdeburg, GermanyReviewed by:
Charles Alfred Stanley, Children’s Hospital of Philadelphia, United StatesTimo Otonkoski, University of Helsinki, Finland
Copyright © 2022 Hewat, Johnson and Flanagan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah E. Flanagan, Uy5GbGFuYWdhbkBleGV0ZXIuYWMudWs=