 Yang Yang
Yang Yang Gaosi Xu*
Gaosi Xu*- Department of Nephrology, The Second Affiliated Hospital of Nanchang University, Nanchang, China
In the existing stages of diabetic kidney disease (DKD), the first stage of DKD is called the preclinical stage, characterized by glomerular hyperfiltration, an abnormally elevated glomerular filtration rate. Glomerular hyperfiltration is an independent risk factor for accelerated deterioration of renal function and progression of nephropathy, which is associated with a high risk for metabolic and cardiovascular disease. It is imperative to understand hyperfiltration and identify potential treatments to delay DKD progress. This paper summarizes the current mechanisms of hyperfiltration in early DKD. We pay close attention to the effect of glucose reabsorption mediated by sodium–glucose cotransporters and renal growth on hyperfiltration in DKD patients, as well as the mechanisms of nitric oxide and adenosine actions on renal afferent arterioles via tubuloglomerular feedback. Furthermore, we also focus on the contribution of the atrial natriuretic peptide, cyclooxygenase, renin–angiotensin–aldosterone system, and endothelin on hyperfiltration. Proposing potential treatments based on these mechanisms may offer new therapeutic opportunities to reduce the renal burden in this population.
Introduction
Diabetic kidney disease (DKD) is the most common cause of end-stage kidney disease, and its diagnostic standards include a decline in renal function, diabetic retinopathy, urine albumin-to-creatinine ratio increases, and a reduction in the glomerular filtration rate (GFR) (1). Due to the rising incidence of diabetes, the dominating cause of chronic kidney disease (CKD) in China has changed from chronic glomerulonephritis to diabetes-related CKD (2), with most developing from type 2 diabetes (T2DM). DKD is a heavy societal burden on society since it is detrimental to the health of affected patients. Despite lifestyle modification and the use of hypoglycemic drugs, the prevention and treatment of DKD still face severe challenges (2). One challenge to overcome is the difficulty of diagnosing DKD in its initial stages. Glomerular hyperfiltration usually occurs in an early stage of renal damage before the appearance of proteinuria (3, 4), without clinical manifestations. It is considered to be a risk factor for DKD (5). Of course, not all people with diabetes develop DKD; not everyone follows the established DKD phasing either. A Thailand study shows that nearly 40% of T2DM are already at stage 2 or worse when diagnosed with DKD (6).
Glomerular hyperfiltration is observed in type 1 diabetes mellitus (T1DM) and T2DM (7). At present, there is no clear cut-off value to define hyperfiltration (8, 9). This paper summarizes the prevalence of diabetic hyperfiltration in recent years (Table 1). On one hand, different methods get different results. Researchers usually used 51Cr-labeled ethylenediaminetetraacetic acid (51Cr-EDTA) to measure GFR in the 1990s (7), while its accuracy and precision are lower than technetium-99m-diethylenetriaminepentaacetic acid (99mTc-DTPA) clearance (22). On the other hand, biological factors, such as glycemic control, body mass index, age, sex, and ethnicity, can all influence GFR (8, 23). Notably, hyperfiltration can also occur in prediabetics whose glucose is impaired (4). In T1DM and T2DM, hyperfiltration may further exacerbate the decline in renal function, causing a faster decrease in GFR compared to non-hyperfiltration and ultimately resulting in CKD (5, 16, 19, 24). A prospective study based on 1,014 T2DM patients showed that baseline hyperfiltration is significantly associated with the odds of rapid renal decline (16). Therefore, by improving our existing knowledge about hyperfiltration, we can discover new strategies to delay the occurrence of DKD.
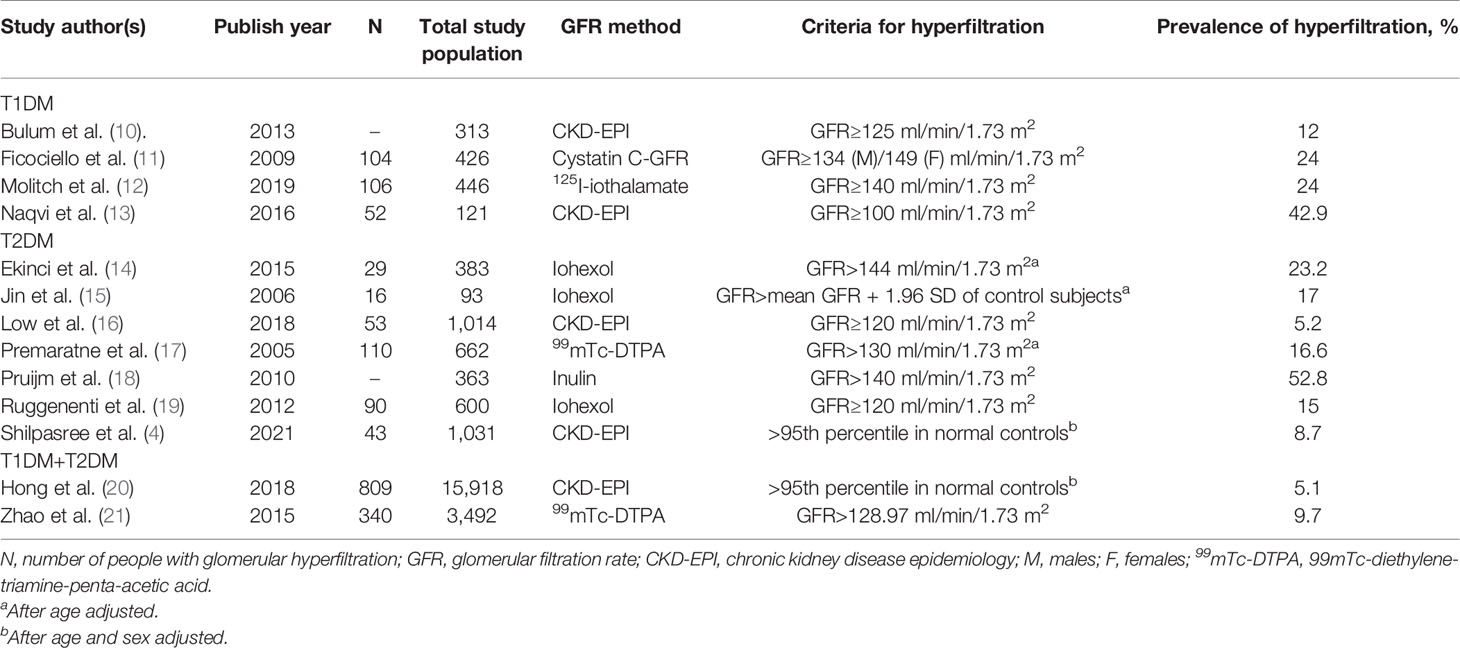
Table 1 Prevalence of diabetic hyperfiltration in recent years.
To date, the most common hypothesis about the mechanisms of diabetic hyperfiltration is the tubular event (7, 25–34). This event refers to any process that affects the increase in proximal tubule glucose reabsorption and involves the upregulation of sodium–glucose cotransporters (SGLTs) and the growth of renal tubule, then through tubuloglomerular feedback (TGF) to alter GFR. Furthermore, some level of hormones and vasoactive substances are increased in people with diabetes; these substances control the contraction and dilation of the pre-glomerular and post-glomerular arteriolar, causing hyperfiltration by altering vascular resistance (7, 9, 28, 30, 35, 36). This review aims at elaborating on the interaction of the above views on hyperfiltration and discussing the potential therapies of DKD.
Effect of Sodium–Glucose Cotransporter on Renal Glucose Reabsorption
Nearly 180 g of glucose is filtrated by the renal glomerulus every day, and little-to-no concentration of glucose is present in the urine, which is related to the reabsorption of the renal tubule. Glucose appears in urine when filtered glucose surpasses the maximum renal absorptive capacity. SGLTs are present in proximal tubules (37) where sodium ions traveling intracellularly along the electrochemical gradient carry glucose molecules in the same direction. Next, glucose is transported to the renal interstitial fluid through glucose transoporters (GLUTs) by the way of facilitated diffusion and eventually backflow into the bloodstream (Figure 1).
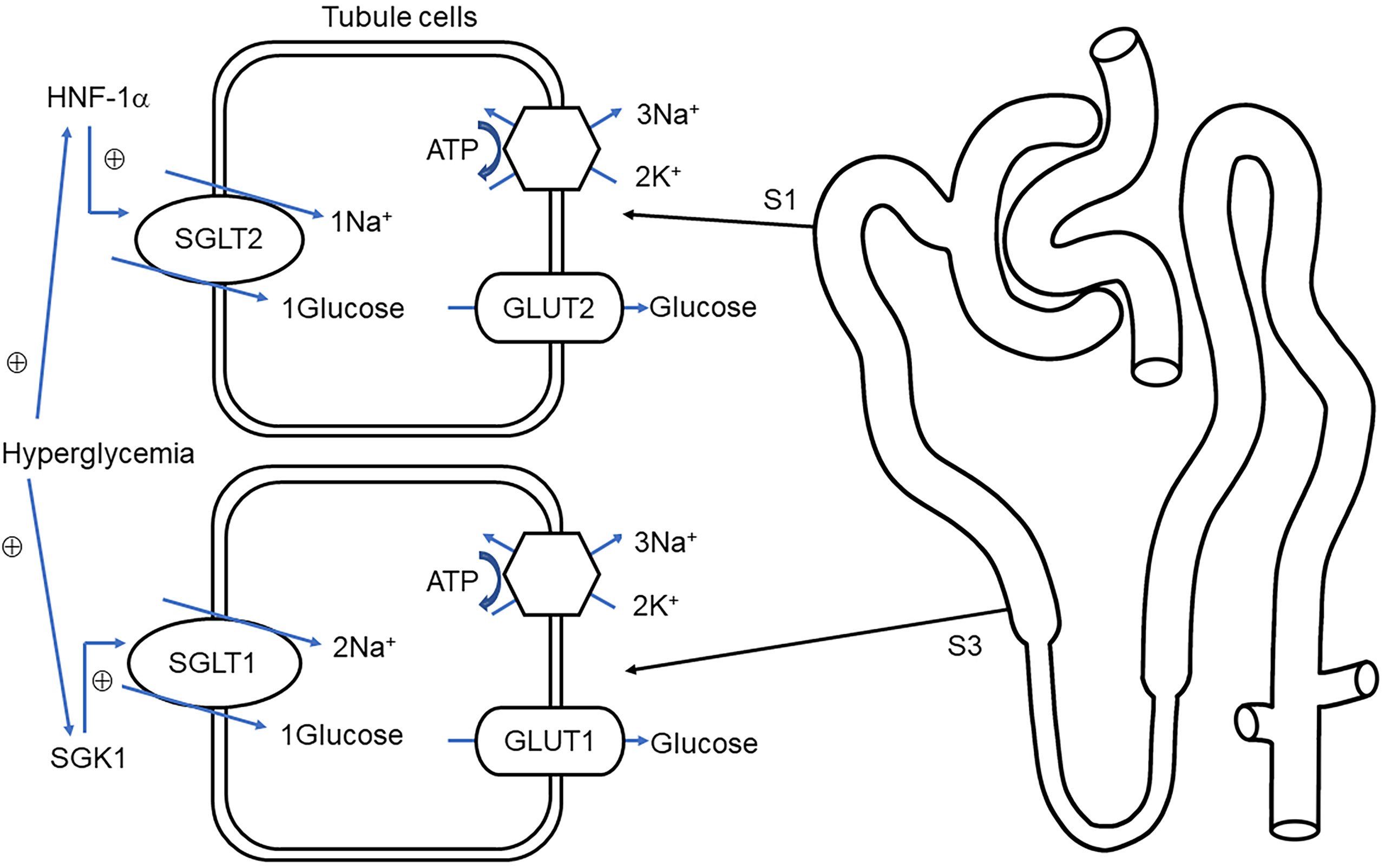
Figure 1 Overview of glucose transport in proximal tubules. Sodium–glucose cotransporter 1/2 (SGLT1, SGLT2) protein located on the brush border membrane is responsible for reabsorption of glucose and sodium in the early renal proximal tubule (S1) and distal proximal tubule (S3), respectively. The glucose on the S1 and S3 correspondingly combined with glucose transporter 2/1 (GLUT2, GLUT1) into the renal interstitial (38). Na+-K+-ATPase, located in the basolateral membrane maintains the potential gradient and concentration gradient of sodium ions and promotes the passive transport of sodium ions. Under hyperglycemia conditions, hepatocyte nuclear factor 1 alpha (HNF-1α), serum, and glucocorticoid-induced kinase-1 (SGK1) stimulate the upregulation of SGLT2 and SGLT1, respectively, thereby increasing glucose reabsorption (39, 40).
There are two dominating SGLTs in the proximal tubule that are responsible for glucose reabsorption, SGLT2 and SGLT1 (41). Evidence indicates that the renal reabsorption of glucose was significantly lower in SGLT2 knockout mice (42) and their urinary glucose excretion is significantly higher (43). David et al. demonstrated that the 24 h urinary glucose excretion of SGLT1 and SGLT2 knockout mice accounted for ~2% and ~30% of the double gene knockout mice, respectively (44). Further evidence suggests that familial renal glycosuria is caused by the individuals’ mutations of the SGLT2 gene, which is featured with persistent glycosuria without hyperglycemia or any symptom of renal tubular dysfunction (45). On the contrary, SGLT1 mainly mediates intestinal glucose absorption, so genetic defects in SGLT1 are more likely to cause neonatal diarrhea and rarely with glycosuria (38, 46). In summary, low-affinity, high-capacity SGLT2 localized at the brush border of the early proximal tubule plays a major role in glucose reabsorption (43, 47) while high-affinity, low-capacity SGLT1 plays an indispensable role in complementing SGLT2 in distal proximal tubule-mediated glucose transport (42).
Different from SGLT1 and SGLT2, it has been shown earlier that SGLT3 is not a glucose transporter but a glucosensor (48). One study showed SGLT3 overexpression in HK-2 cells in diabetic patients, which increased intracellular Na+ levels and induced diabetic hyperfiltration and kidney injury (49). Both SGLT4 and SGLT5 are thought to be kidney mannose and fructose transporters. Meanwhile, SGLT5, also involved in controlling glucose reabsorption, may be related to the level of 1,5-anhydroglucitol (50). The location of SGLT6 in the kidney is not clear; it primarily transports myo-inositol rather than glucose (51). SLC5A11, encoding SGLT6, may play a role in human autoimmune diseases by interacting with immune genes (52).
Tubular Mechanisms for Hyperfiltration in Early Diabetic Kidney Disease
Increased Reabsorption
People with diabetes typically have higher blood glucose in comparison to normal people, and to balance homeostasis, more glucose needs to be reabsorbed to ensure the absence of glycosuria, which primarily depends on the upregulation of SGLTs and GLUTs (53–55). When comparing the diabetic rats to the normal ones, the mRNA of SGLT2 and SGLT1 was increased by 36% and 20%, respectively (56). Other evidence demonstrates that more expression of SGLT2 is present within the urine from a human who has non-insulin-dependent diabetes (57). With the increases of renal tubule reabsorption, the sodium and chloride delivered to the macula densa decreases; reducing the signal for TGF leads to the elevation of the single nephron glomerular filtration rate (SNGFR) (25, 32). The published concentration of sodium chloride on the macula densa of the diabetic nephron is less than normal (25, 29), which shows an evidence of hyperreabsorption.
Excessive glucose stimulates the transcription level of serum and glucocorticoid-induced kinase-1 (SGK-1), which, in turn, excites many carriers including the Na+/H+ exchanger NHE3, SGLT1, GLUT1 to decrease glycosuria (39, 58). In the SGK-1-knockout mice, the excretion of urinary glucose is higher (58). Moreover, SGK1 is also involved in promoting the fibrosis of diabetic kidneys (39). Similarly, enhanced SGLT2 mRNA in diabetes is positively correlated to the activation of hepatocyte nuclear factor 1 alpha (HNF-1α) (40). This observation was supported, when subsequent data showed that the expression of SGLT2 is lower in HNF-1α-null mice (59). Hence, there is reason to believe that SGK-1 and HNF-1α have a certain impact on the development of DKD. At the same time, excessive filtration increases oxygen consumption; continuous reabsorption further worsens tubular hypoxia and tubulointerstitial fibrosis (60).
The contribution of a newly oral antidiabetic drug, SGLT2 inhibitors (SGLT2is), has been well established in T1DM and T2DM. It recovers the signal of TGF by increasing the concentration of sodium in macula densa, ameliorating glomerular hypertension and hyperfiltration to the same extent as the tubular hypothesis (61). Reducing the glucose levels is the vital pharmacologic action that it provides. The reduction of hyperglycemia mitigates renal tubular transport; it also reduces renal oxygenation at the corticomedullary junction, resulting in the accumulation of hypoxia-inducible factors, which is beneficial for the production of serum erythropoietin (34). In addition, SGLT2is also lead to weight loss and a drop in blood pressure, thereby significantly reducing the incidence of cardiovascular events (34). Existing clinical trials have demonstrated that SGLT2is reduce the risk of renal failure and cardiovascular outcomes (62–65). A multinational observational cohort study showed 114 and 237 cases of eventual renal outcomes after the application of SGLT2is and other hypoglycemic agents, respectively, during the follow-up period, with the former showing a significantly lower risk (65). Notably, most trails enrolled patients whose GFR is low, with few early-stage patients (62–64). Future studies of SGLT2is should target more patients with an early stage of DKD; after all, hyperfiltration is a cause of DKD progress.
While SGLT1, as a main protein carrier of intestinal glucose absorption, also has great therapeutic potential (66), a dual SGLT1/2 inhibitor, licogliflozin, reduces blood glucose by changing urinary glucose excretion and hemodynamics decline (67). Larger and longer clinical trials are needed to investigate the long-term safety, efficacy, and potential beneficial effects of this drug.
Kidney Growth and Salt Paradox
At the onset of diabetes, kidneys grow due to expanded nephron size; renal hypertrophy occurs mainly in the cortex of the diabetic kidney and is linked to subsequent proximal tubular hyperreabsorption (25, 55). Uehara−Watanabe et al. showed that the kidney weight, proximal tubules, and glomeruli size were significantly higher in streptozotocin-induced rats (55). Greater glomerular volume and glomerular basement membrane (GBM) width were also demonstrated in patients with early DKD (68). The elevated surface density of peripheral GBM and glomerular filtration surface area are closely related to glomerular hyperfiltration (68). Kidney growth is most likely caused by various cytokines and growth factors stimulating various signal pathways to respond to hyperglycemia, including the transforming growth factor-β (TGF-β) system, which is crucial for mesangial cell hypertrophy, fibrosis, and glomerulosclerosis (69). The vascular factor and protein glycation products amplify the expression of TGF-β on diabetes (69), from cell proliferation to hypertrophy to cellular senescence (28, 29, 31). It is well known that hyperglycemia plays a key role in the progression of DKD by activating advanced glycation end products (AGEs), protein kinase C (PKC), and the aldose reductase pathway (70). Cell growth, fibrosis, and tissue damage are related to increased PKC activity, especially in diabetes (71). In a diabetic mice model, the selective inhibitor of PKC-β, ruboxistaurin, amelioration overexpression of TGF-β improved glomerular hyperfiltration and reduced albuminuria (71, 72). Ruboxistaurin has been certified as beneficial to DKD therapy in a short time of clinical studies but not in long-term studies (72).
Based on a renal tubule growth phenotype, researchers found an inverse relationship between dietary NaCl intake and GFR, which is called salt paradox (73). In other words, the lower the NaCl intake of diabetics, the higher their GFR. The possible explanation is that after high salt intake, the concentration of sodium chloride reaching the macula densa is increased; the TGF signal is reactivated, which reduces SNGFR (26, 73). Persistent hyperglycemia makes reabsorption increase sensitivity to dietary NaCl (73). The activity of ornithine decarboxylase (ODC) is increased many times in diabetic rats (32, 74); it is required for the salt paradox (75). After using an ODC inhibitor, the proximal tubule hyperresponsiveness to salt was remedied, tubular growth was limited, and proximal reabsorption was attenuated, all of which improved hyperfiltration (32, 75). Therefore, ODC is necessary for renal growth and salt paradox.
Adenosine–Tubuloglomerular Feedback
TGF is a negative feedback system, which is modulated by sensing the concentration of sodium chloride in the macula densa (76, 77). TGF is a major controller of GFR changes in the tubular hypothesis (27, 30, 31). The physiological function of TGF is reliant on the adenosine 5’-triphosphate (ATP) and adenosine (76, 77). When the macula densa detects an increase in sodium chloride concentration in the tubule fluid, it stimulates ATP hydrolysis to adenosine and extracellular release, which, in turn, acts on A1 adenosine receptors (A1AR) in afferent arterioles. TGF can then be activated, leading to the contraction of the afferent arteriole followed by decreased GFR (31, 76). Therefore, the role of adenosine cannot be ignored, but the exact effect of A1AR for TGF on diabetic hyperfiltration has yet to be clarified. In some A1AR knockout diabetic mice, the degree of diabetic hyperfiltration was the same as the control groups (78, 79), which does not support TGF-mediated increase in GFR. Another point of view is that A1AR knockout diabetic mice reduce the activity of TGF (80, 81) and ameliorates hyperfiltration (82). The difference between two viewpoints is the interference of confounding factors like blood pressure and severe hyperglycemia (29). The use of empagliflozin validates the response of A1AR to TGF (83).
Except for A1AR-mediated vasoconstriction in the low concentration range, the adenosine diphasic response has been shown in the isolation and perfusion of renal glomerular arterioles (84). Another subtype of adenosine receptor A2, involving A2aAR and A2bAR, mediates vasodilation at higher concentrations (76). Under normal circumstances, compared to A2bAR, A2aAR has a higher affinity for efferent arterioles and preferentially dilates the efferent arterioles, maintaining GFR in a normal range (85, 86). Interestingly, in diabetes, the effect of adenosine A2aAR is diminished, increasing the resistance of efferent arterioles leading to the elevation of the filtration fraction and GFR (86, 87). Recently, Patinha et al. demonstrated that the decrease in plasma glucose, reduction in proteinuria, and improvement in renal fibrosis in diabetic mice may be associated with the upregulation of A2aAR, which may serve as a promising therapeutic target for hypertension-DKD (88). Furthermore, the absence of A1AR does not influence the effect of TGF changes on the activation of the A2aAR on the efferent arterioles (31). Whether hyperfiltration can be controlled by stimulating A2aAR in the clinic, it may be a new alternative treatment that can delay the progression of DKD; more experiments must be conducted to confirm this.
In addition, A2bAR has a pathogenic effect in early glomerular dysfunction in diabetic rats (89). An experiment model shows the A2bAR-mediated overproduction of the vascular endothelial growth factor (VEGF) in early DKD rats (89), which is associated with hyperfiltration, proteinuria, and the ultrastructural changes of glomerulus (90). VEGF also promotes endothelial cell damage, which is the first barrier to the glomerular filtration membrane and may lead to the production of microalbuminuria (91). However, VEGF is an important mediator in the recovery from other kidney diseases (92). A2bAR blockers may be a novel alternative for the treatment of DKD patients.
SGLT1-NO Synthesis1 Pathway
NO was thought to be a powerful vasodilator (93). The increased expression of NO, which is associated with hyperfiltration, was reported in diabetic kidneys (94–96). Zhang et al. used the selective GLUT1 antagonist and NOS inhibitor, respectively, to block the response of the vascular and afferent arterioles to hyperglycemia successively; it indicated that vasodilation induced by hyperglycemia is achieved through the GLUT1-mediated endothelium-dependent production of NO (97).
Recent research has proposed a novel idea that increased production of NOS-dependent NO in the macula densa results from sensing elevated glucose concentration in the luminal of macula densa through SGLT1, thereby reducing TGF activity resulting in hyperfiltration (98–100). Raising GFR through the SGLT1-NOS1-GFR pathway maintains the urinary sodium and fluid excretion and volume balance (31). Normally, the concentration of glucose in luminal can be ignored; however, in a high-sugar environment or the use of the inhibitor of SGLT2, there is an elevated glucose concentration at the lumen of the macula densa when filtrated glucose exceeds the maximum reabsorption capacity. One study found that elevated GFR induced by acute hyperglycemia was significantly attenuated in mice without NOS1 (99). There was another experiment with SGLT1 knockout mice, whose GFR, kidney weight, glomerular size, and proteinuria were reduced (98). Those discoveries established the decisive factor of macula densa NOS1 and SGLT1 in glomerular hyperfiltration related to high glucose and may provide evidence for potential new therapeutic targets, but whether it exists in chronic mild hyperglycemia is not clear; after all, increased glucose delivery to macula densa is generally found only in the medium-to-high blood glucose situations.
Meanwhile, this pathway can also be activated after a high-protein meal (101). According to a retrospective cohort study of forty-three kidney donors in Tokyo, Oba et al. found that high protein intake is positively related to SNGFR and leads to hyperfiltration (102). They measured GFR at the level of a single nephron as a representative parameter on renal hemodynamic change (102), avoiding the number of functioning glomeruli affecting total renal filtration. Compared to animal protein, plant-sourced protein shows a strong beneficial effect in DKD (103). Thus, diabetic patients should not only strictly limit their sugar intake but also animal protein intake to avoid accelerating the progression of this disease (Figure 2).
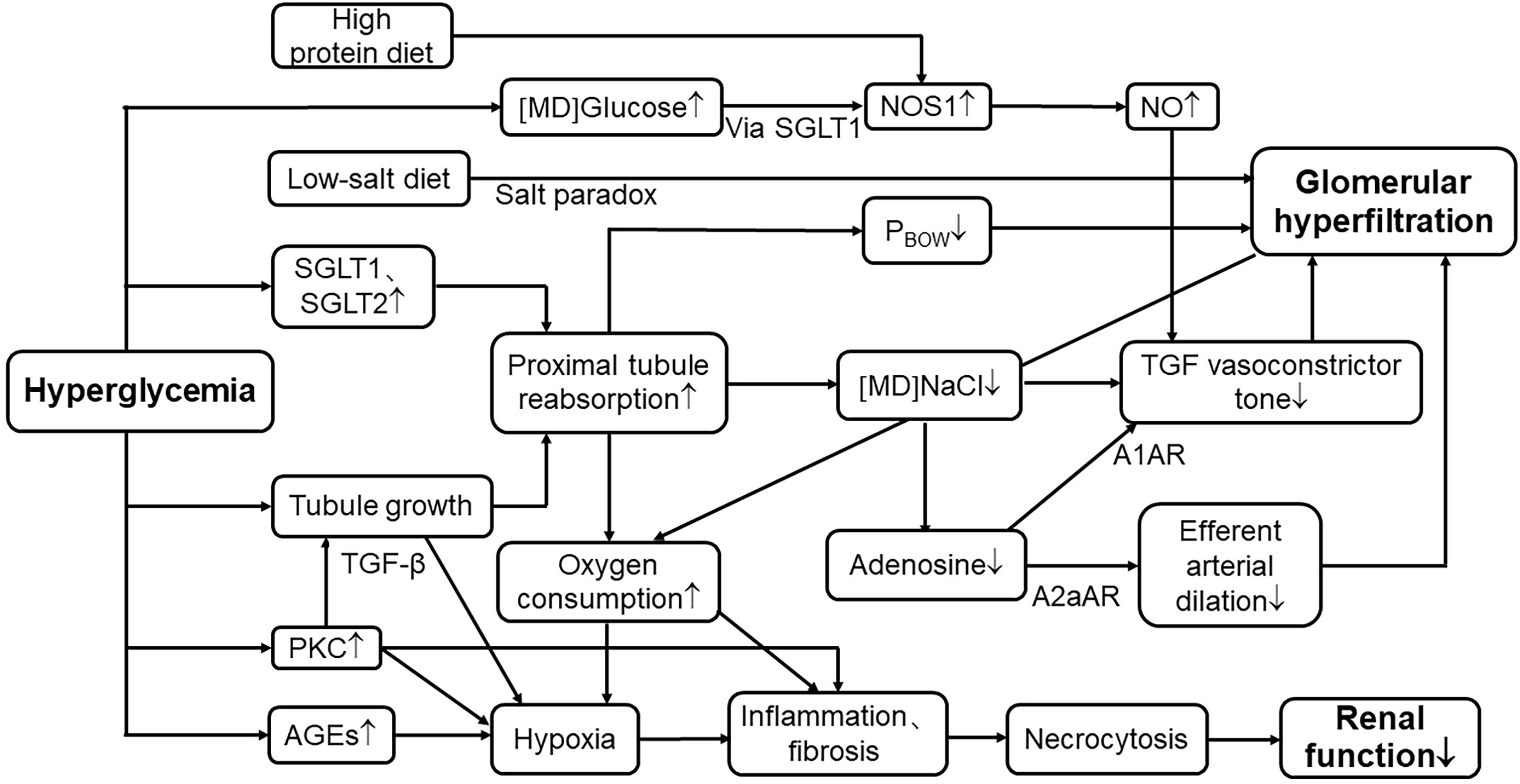
Figure 2 Mechanisms of hyperglycemia in renal tubular events leading to hyperfiltration and progression of nephropathy. SGLT1, sodium–glucose cotransporter1; SGLT2, sodium–glucose cotransporter2; [MD]NaCl, the concentration of sodium chloride in macula densa; TGF, tubuloglomerular feedback; PBOW, hydraulic pressure in Bowman’s space; [MD]Glucose, the concentration of glucose in macula densa; NOS1, nitric oxide synthase1; NO, nitric oxide; A1AR, A1 adenosine receptor; A2aAR, A2 adenosine receptor; PKC, protein kinase C; AGE, advanced glycation end products; TGF-β, transforming growth factor-β; ↑, elevate or increase; ↓, decrease or reduce.
Vascular Mechanisms for Hyperfiltration in Early Diabetic Kidney Disease
Cyclooxygenase
The COX-metabolite of arachidonic acid is prostaglandins (PGs); it plays a pivotal role in regulating the renal blood flow and GFR. Several observations in the diabetic mice model have verified that renal PGs are increased, including prostaglandin E2 (PGE2), prostaglandin I2 (PGI2), prostaglandin F2 alpha (PGF2α), and thromboxane B2 (104, 105). Increased PGs alters diabetic renal hemodynamics in a way of dilating renal afferent arterioles. Increased glomerular capillary pressure heightens the tensile stress on the capillary wall, resulting in an increase of the length and area of GBM, while an increased ultrafiltrate, in turn, elevates the shear force on the podocyte foot processes and body surface (106). Both mechanical stresses eventually lead to podocyte shedding, extensive GBM exfoliation, and segmental sclerosis, exacerbating kidney function damage (106).
Indomethacin, a nonselective COX inhibitor, partially reduced GFR (107). Further experimental demonstrated that COX2 inhibitors significantly reduce GFR; despite the lack of normalization (108), but selective COX1 inhibitors did not affect renal hemodynamics in diabetic rats (109). Nevertheless, Craven et al. considered that the hyperfiltration mediated by PGs occurred within 2 weeks of streptozotocin-induced diabetic mice; PG production did not increase after 4 weeks, so persistent hyperfiltration was not mediated by PGs, suggesting that sustained effects may be mediated by other factors (105). COX2 is a pivotal component in stimulating renin release by the macula densa, possibly through the release of renin-stimulating PGE2 and PGI2 (110). Consistent with this view, the level of renin decreased in COX2 knockout mice, suggesting that the PGs produced by COX-2 could affect the renal hemodynamic balance by regulating the activity of the renin–angiotensin–aldosterone system (RAAS) (111).
Atrial Natriuretic Peptide
ANP can cause the dilation of renal afferent arteriolar and constriction of efferent arteriolar (112) and was hypothesized to be a potential mediator of diabetic hyperfiltration as early as the 1980s (113). In diabetic mice, afferent arteriolar resistance is reduced more than the efferent to increase intraglomerular pressure and GFR (107). ANP contributes to the hyperfiltration as documented by the elevated level of plasma concentration in diabetic mice and patients and by the reduction of GFR that followed the injection of the ANP-specific antiserum or antagonist (107, 114, 115). Liu et al. showed that the level of plasma ANP is significantly associated with the secretion of cytokines, which promotes the progression of DKD (115). Moreover, ANP also increases urinary albumin excretion in normoalbuminuric T2DM (116). The dual effect of ANP on hyperfiltration and proteinuria may be a predictor of the development of DKD in the long term.
However, on the other hand, ANP showed benefit in preventing and reversing kidney injury. Sacubitril/valsartan, as a dual inhibitor of neprilysin and the angiotensin II (Ang II) receptor, strengthens the effect of ANP and mitigates the hyperfiltration and renal tubular injury in the animal models of early DKD (117). This may counteract the dilated effect of ANP due to the inhibition of the vasoconstrictive effect of Ang II, resulting in a slight decrease in intraglomerular pressure and thus a protective effect on the kidney. In addition, the effect of this drug is limited to patients with heart failure (118). Therefore, more studies are needed in diabetic patients without heart failure or varying stages of DKD to explore the prognosis of this drug on the kidney.
Renin–Angiotensin–Aldosterone System
Regarding the intrarenal hemodynamic alterations, the role of RAAS cannot be ignored (119–121). Ang II, which is the key substance in RAAS, mediates the contraction of the afferent and efferent arteries. When RAAS is activated, it causes intraglomerular pressure rises, damages tissue, stimulates fibrosis, promotes the mesangial matrix increases, and ultimately leads to diabetic glomerulosclerosis (121, 122). The SGLT2is also decreased the activation of RAAS to reduce hyperfiltration (61). Notably, after the use of the RAAS inhibitor, GFR did not fall to normal levels. This was due to the vasoconstriction effect of Ang II that may be regulated by a vasodilator, including NO and PGs, which also have an influence on hyperfiltration (123). One of the effects of sacubitril/valsartan described above is the inhibition of the Ang II receptor, which influences hemodynamic changes by dilating the efferent arterioles. It has become a hot topic of research due to its reduced risk of renal insufficiency compared with RAS inhibitors alone (124).
Furthermore, angiotensin 1-7 (Ang1-7)-mediated production by angiotensin- converting enzyme2 is also associated with hyperfiltration (125). Ren et al. found that Ang1-7 has vasodilation on afferent arteriole in isolated rabbits, further increases the intraglomerular pressure as well as stimulus production of PGs and NO (126). This function eliminated by the NOS inhibitor rather than COX means that the vasodilatory effect of Ang1-7 is dependent on the production of endothelial NO (126). Until now, RAAS inhibitors have been the gold standard therapy in DKD, since on one hand, they reduce efferent arterial contraction and blood pressure and thereby improve hyperfiltration (127). On the other hand, they prevent fibroblast activation, which delays the development of nephropathy (128).
Endothelin
Another endodermal material, endothelin-1 (ET-1), is vital in maintaining the homeostasis of sodium and water, as well as controlling the glomerular vascular tone and hemodynamics (129). Like Ang II, ET-1 can induce inflammation and prompt fibrosis (129); both factors are involved in diabetic kidney growth that is associated with the activation of TGF-β (69). ET-1 combined with ETA receptor located on the vascular smooth muscle stimulates vascular constriction, whereas the release of NO and PG triggers vasodilation through the ETB receptor (129, 130). Under normal conditions, the small amount of ET produced by vascular endothelium has little effect on systemic hemodynamics (131). Elevated plasma ET-1 levels have been found in T2DM, which leads to endothelial dysfunction (132). Recently, in a three-clinical-trial analysis, the kidney hemodynamic profile in adults with type 2 diabetes showed that endothelial dysfunction is associated with glomerular hyperfiltration (133). Evidence suggests that the vasoconstrictive effect of endothelin is amplified in DKD (129, 130). Renal vascular resistance and filtration fraction increased compared with the control group after exogenous ET administration, which indicates the presence of renal hyperfiltration (35, 131). Endothelin-receptor antagonists have been shown in several clinical trials and experimental models to prevent diabetic hyperfiltration, reduce proteinuria, and delay the progression of renal damage (134–136). Despite some complications of this antagonist, more trials are being explored with the promise of this new drug treating DKD (121) and whether it has greater pharmacological benefits in combination with drugs such as SGLT2is (Figure 3).
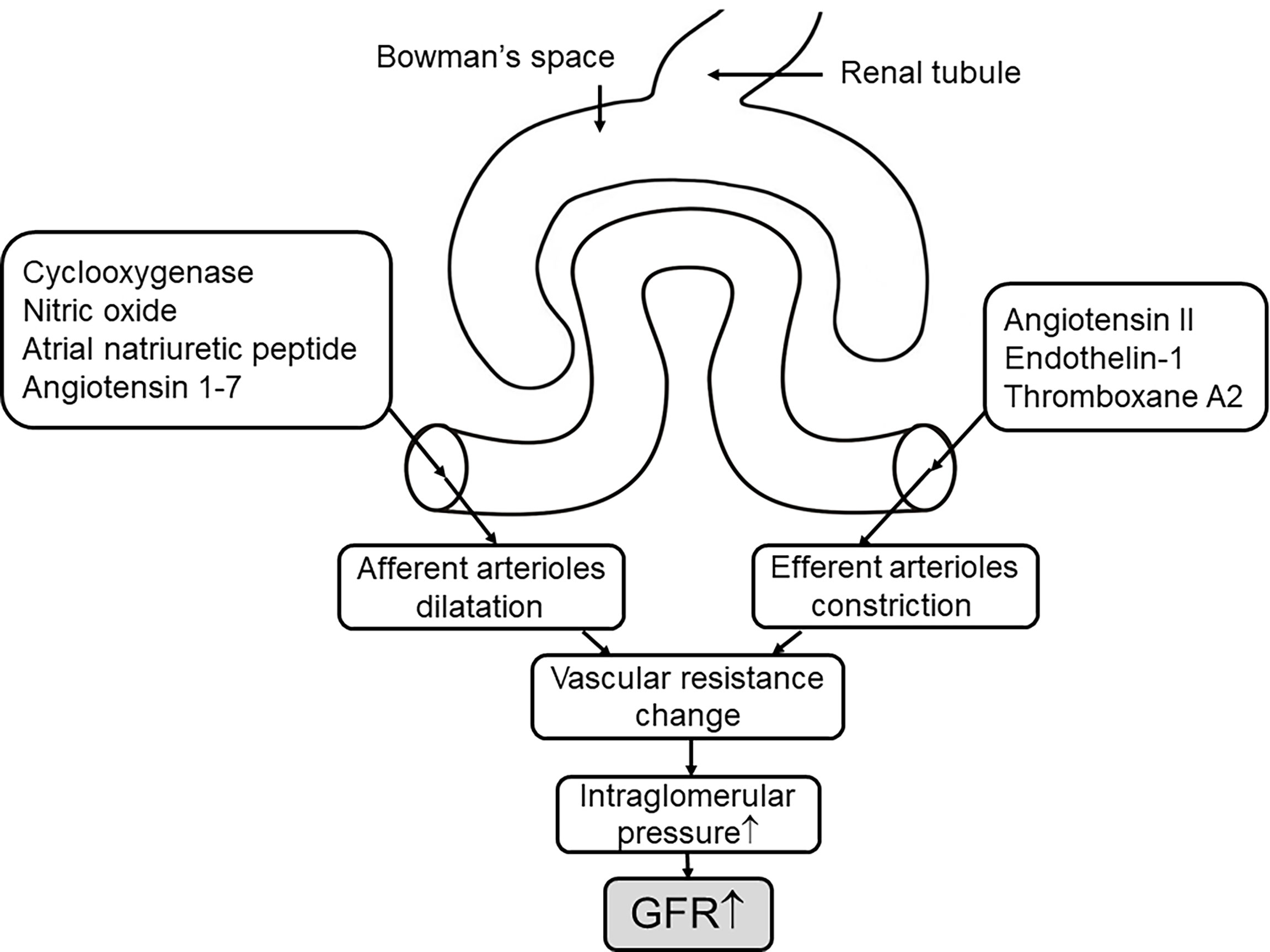
Figure 3 The mechanisms of the vascular events on hyperfiltration. The concentration of hormones and vasoactive mediators that mediate afferent arteriole dilation and efferent arteriole constriction increase in response to hyperglycemia (7) through altering the vascular resistance to elevate the pressure of intraglomerular, thus leading to increased GFR. ↑, elevate or increase.
Conclusion
With increasing attention to the DKD, there is growing evidence that hyperfiltration affects the progression of DKD. It occurs through a variety of mechanisms, including glucose reabsorption mediated by SGLTs, renal growth, the adenosine signal, and SGLT1-NOS1-pathway; all of these elevate GFR through weakened TGF signaling. While vasoactive mediators cause changes in GFR by altering the vascular resistance of the afferent and efferent arterioles, we attempt to propose underlying therapies to improve hyperfiltration. Clinical trials have demonstrated the renal protective effect of SGLT2is and RAAS inhibition. Upregulated A2aAR, PKC-β inhibitors, and endothelin-receptor antagonists provide new ideas for delaying the progression of DKD, but their limitations also need to be considered. Further studies of renal prognosis are needed to assess the long-term effectiveness and safety of these strategies.
Author Contributions
YY: conceptualization, software, writing—original draft. GX: idea, funds, and paper revision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81970583 & 82060138), the Nature Science Foundation of Jiangxi Province (No. 20202BABL206025), and the Projects in the Second Affiliated Hospital of Nanchang University (No. 2019YNLZ12008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
A1AR, A1 adenosine receptors; A2aAR, A2bAR, A2 adenosine receptors; AGE, advanced glycation end product; Ang II, angiotensin II; Ang1-7, angiotensin 1-7; ANP, atrial natriuretic peptide; ATP, adenosine 5’-triphosphate; CKD, chronic kidney disease; COX, cyclooxygenase; 51Cr-EDTA, 51Cr-labeled ethylenediaminetetraacetic acid; DKD, diabetic kidney disease; ET-1, endothelin-1; GBM, glomerular basement membrane; GFR, glomerular filtration rate; GLUT, glucose transporter; GLUT1, glucose transporter 1; HNF-1α, hepatocyte nuclear factor 1 alpha; 99mTc-DTPA, technetium-99m-diethylenetriaminepentaacetic acid; NO, nitric oxide; NOS1, NO synthesis 1; Nrf2, nuclear factor erythroid 2-related factor 2; ODC, ornithine decarboxylase; PGE2, prostaglandin E2; PGF2α, prostaglandin F2 alpha; PGI2. prostaglandin I2; PG, prostaglandin; PKC, protein kinase C; RAAS, renin–angiotensin–aldosterone system; SGK-1, serum and glucocorticoid-induced kinase-1; SGLT1, sodium–glucose cotransporter 1; SGLT2, sodium–glucose cotransporter 2; SGLT2i, SGLT2 inhibitor; SGLT3, sodium–glucose cotransporter 3; SGLT4, sodium–glucose cotransporter 4; SGLT5, sodium–glucose cotransporter 5; SGLT6, sodium–glucose cotransporter 6; SGLT, sodium–glucose cotransporter; SNGFR, single nephron glomerular filtration rate; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; TGF, tubuloglomerular feedback; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor.
References
1. Samsu N. Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. BioMed Res Int (2021) 2021:1497449. doi: 10.1155/2021/1497449
2. Shilian H, Jing W, Cui C, Xinchun W. Analysis of Epidemiological Trends in Chronic Diseases of Chinese Residents. Aging Med (Milton) (2020) 3(4):226–33. doi: 10.1002/agm2.12134
3. Oh SW, Yang JH, Kim MG, Cho WY, Jo SK. Renal Hyperfiltration as a Risk Factor for Chronic Kidney Disease: A Health Checkup Cohort Study. PloS One (2020) 15(9):e0238177. doi: 10.1371/journal.pone.0238177
4. Shilpasree AS, Patil VS, Revanasiddappa M, Patil VP, Ireshnavar D. Renal Dysfunction in Prediabetes: Confirmed by Glomerular Hyperfiltration and Albuminuria. J Lab Physicians (2021) 13(3):257–62. doi: 10.1055/s-0041-1731107
5. Magee GM, Bilous RW, Cardwell CR, Hunter SJ, Kee F, Fogarty DG. Is Hyperfiltration Associated With the Future Risk of Developing Diabetic Nephropathy? A Meta-Analysis. Diabetologia (2009) 52(4):691–7. doi: 10.1007/s00125-009-1268-0
6. Nata N, Rangsin R, Supasyndh O, Satirapoj B. Impaired Glomerular Filtration Rate in Type 2 Diabetes Mellitus Subjects: A Nationwide Cross-Sectional Study in Thailand. J Diabetes Res (2020) 2020:6353949. doi: 10.1155/2020/6353949
7. Tonneijck L, Muskiet MH, Smits MM, van Bommel EJ, Heerspink HJ, van Raalte DH, et al. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J Am Soc Nephrol (2017) 28(4):1023–39. doi: 10.1681/ASN.2016060666
8. Cachat F, Combescure C, Cauderay M, Girardin E, Chehade H. A Systematic Review of Glomerular Hyperfiltration Assessment and Definition in the Medical Literature. Clin J Am Soc Nephrol (2015) 10(3):382–9. doi: 10.2215/CJN.03080314
9. Helal I, Fick-Brosnahan GM, Reed-Gitomer B, Schrier RW. Glomerular Hyperfiltration: Definitions, Mechanisms and Clinical Implications. Nat Rev Nephrol (2012) 8(5):293–300. doi: 10.1038/nrneph.2012.19
10. Bulum T, Kolarić B, Prkacin I, Duvnjak L. Hyperfiltration in Normoalbuminuric Type 1 Diabetic Patients: Relationship With Urinary Albumin Excretion Rate. Coll Antropol (2013) 37(2):471–6.
11. Ficociello LH, Perkins BA, Roshan B, Weinberg JM, Aschengrau A, Warram JH, et al. Renal Hyperfiltration and the Development of Microalbuminuria in Type 1 Diabetes. Diabetes Care (2009) 32(5):889–93. doi: 10.2337/dc08-1560
12. Molitch ME, Gao X, Bebu I, de Boer IH, Lachin J, Paterson A, et al. Early Glomerular Hyperfiltration and Long-Term Kidney Outcomes in Type 1 Diabetes: The Dcct/Edic Experience. Clin J Am Soc Nephrol (2019) 14(6):854–61. doi: 10.2215/CJN.14831218
13. Naqvi SA, Ahsan S, Fawwad A, Basit A, Shera AS. Effect of Angiotensin Converting Enzyme Inhibitor on Glomerular Hyperfiltration in Patients With Type 1 Diabetes. Pak J Med Sci (2016) 32(3):559–64. doi: 10.12669/pjms.323.9399
14. Ekinci EI, Hughes JT, Chatfield MD, Lawton PD, Jones GR, Ellis AG, et al. Hyperfiltration in Indigenous Australians With and Without Diabetes. Nephrol Dial Transplant (2015) 30(11):1877–84. doi: 10.1093/ndt/gfv230
15. Jin Y, Moriya T, Tanaka K, Matsubara M, Fujita Y. Glomerular Hyperfiltration in Non-Proteinuric and Non-Hypertensive Japanese Type 2 Diabetic Patients. Diabetes Res Clin Pract (2006) 71(3):264–71. doi: 10.1016/j.diabres.2005.06.014
16. Low S, Zhang X, Wang J, Yeoh LY, Liu YL, Ang KKL, et al. Long-Term Prospective Observation Suggests That Glomerular Hyperfiltration Is Associated With Rapid Decline in Renal Filtration Function: A Multiethnic Study. Diabetes Vasc Dis Res (2018) 15(5):417–23. doi: 10.1177/1479164118776465
17. Premaratne E, Macisaac RJ, Tsalamandris C, Panagiotopoulos S, Smith T, Jerums G. Renal Hyperfiltration in Type 2 Diabetes: Effect of Age-Related Decline in Glomerular Filtration Rate. Diabetologia (2005) 48(12):2486–93. doi: 10.1007/s00125-005-0002-9
18. Pruijm M, Wuerzner G, Maillard M, Bovet P, Renaud C, Bochud M, et al. Glomerular Hyperfiltration and Increased Proximal Sodium Reabsorption in Subjects With Type 2 Diabetes or Impaired Fasting Glucose in a Population of the African Region. Nephrol Dial Transplant (2010) 25(7):2225–31. doi: 10.1093/ndt/gfq008
19. Ruggenenti P, Porrini EL, Gaspari F, Motterlini N, Cannata A, Carrara F, et al. Glomerular Hyperfiltration and Renal Disease Progression in Type 2 Diabetes. Diabetes Care (2012) 35(10):2061–8. doi: 10.2337/dc11-2189
20. Hong S, Choi YM, Ihm SH, Kim D, Choi MG, Yu JM, et al. Association Between Metabolic Parameters and Glomerular Hyperfiltration in a Representative Korean Population Without Chronic Kidney Disease. PloS One (2018) 13(12):e0207843. doi: 10.1371/journal.pone.0207843
21. Zhao F, Zhang L, Lu J, Guo K, Wu M, Yu H, et al. The Chronic Kidney Disease Epidemiology Collaboration Equation Improves the Detection of Hyperfiltration in Chinese Diabetic Patients. Int J Clin Exp Med (2015) 8(12):22084–97.
22. Vidal-Petiot E, Courbebaisse M, Livrozet M, Corrégé G, Rusu T, Montravers F, et al. Comparison of (51)Cr-Edta and (99m)Tc-Dtpa for Glomerular Filtration Rate Measurement. J Nephrol (2021) 34(3):729–37. doi: 10.1007/s40620-020-00932-9
23. Jerums G, Premaratne E, Panagiotopoulos S, MacIsaac RJ. The Clinical Significance of Hyperfiltration in Diabetes. Diabetologia (2010) 53(10):2093–104. doi: 10.1007/s00125-010-1794-9
24. Bjornstad P, Cherney DZ, Snell-Bergeon JK, Pyle L, Rewers M, Johnson RJ, et al. Rapid Gfr Decline Is Associated With Renal Hyperfiltration and Impaired Gfr in Adults With Type 1 Diabetes. Nephrol Dial Transplant (2015) 30(10):1706–11. doi: 10.1093/ndt/gfv121
25. Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular Hyperfiltration in Experimental Diabetes Mellitus: Potential Role of Tubular Reabsorption. J Am Soc Nephrol (1999) 10(12):2569–76. doi: 10.1681/asn.V10122569
26. Vallon V, Blantz RC, Thomson S. Glomerular Hyperfiltration and the Salt Paradox in Early [Corrected] Type 1 Diabetes Mellitus: A Tubulo-Centric View. J Am Soc Nephrol (2003) 14(2):530–7. doi: 10.1097/01.asn.0000051700.07403.27
27. Thomson SC, Vallon V, Blantz RC. Kidney Function in Early Diabetes: The Tubular Hypothesis of Glomerular Filtration. Am J Physiol Renal Physiol (2004) 286(1):F8–15. doi: 10.1152/ajprenal.00208.2003
28. Vallon V, Komers R. Pathophysiology of the Diabetic Kidney. Compr Physiol (2011) 1(3):1175–232. doi: 10.1002/cphy.c100049
29. Vallon V, Thomson SC. Renal Function in Diabetic Disease Models: The Tubular System in the Pathophysiology of the Diabetic Kidney. Annu Rev Physiol (2012) 74:351–75. doi: 10.1146/annurev-physiol-020911-153333
30. Takenaka T, Inoue T, Watanabe Y. How the Kidney Hyperfiltrates in Diabetes: From Molecules to Hemodynamics. World J Diabetes (2015) 6(4):576–82. doi: 10.4239/wjd.v6.i4.576
31. Vallon V, Thomson SC. The Tubular Hypothesis of Nephron Filtration and Diabetic Kidney Disease. Nat Rev Nephrol (2020) 16(6):317–36. doi: 10.1038/s41581-020-0256-y
32. Thomson SC, Deng A, Bao D, Satriano J, Blantz RC, Vallon V. Ornithine Decarboxylase, Kidney Size, and the Tubular Hypothesis of Glomerular Hyperfiltration in Experimental Diabetes. J Clin Invest (2001) 107(2):217–24. doi: 10.1172/jci10963
33. Hallow KM, Gebremichael Y, Helmlinger G, Vallon V. Primary Proximal Tubule Hyperreabsorption and Impaired Tubular Transport Counterregulation Determine Glomerular Hyperfiltration in Diabetes: A Modeling Analysis. Am J Physiol Renal Physiol (2017) 312(5):F819–F35. doi: 10.1152/ajprenal.00497.2016
34. Provenzano M, Pelle MC, Zaffina I, Tassone B, Pujia R, Ricchio M, et al. Sodium–glucose Co-Transporter-2 Inhibitors and Nephroprotection in Diabetic Patients: More Than a Challenge. Front Med (Lausanne) (2021) 8:654557. doi: 10.3389/fmed.2021.654557
35. Sasson AN, Cherney DZ. Renal Hyperfiltration Related to Diabetes Mellitus and Obesity in Human Disease. World J Diabetes (2012) 3(1):1–6. doi: 10.4239/wjd.v3.i1.1
36. Carmines PK. The Renal Vascular Response to Diabetes. Curr Opin Nephrol Hypertens (2010) 19(1):85–90. doi: 10.1097/MNH.0b013e32833240fc
37. Kanai Y, Lee WS, You G, Brown D, Hediger MA. The Human Kidney Low Affinity Na+/Glucose Cotransporter Sglt2. Delineation of the Major Renal Reabsorptive Mechanism for D-Glucose. J Clin Invest (1994) 93(1):397–404. doi: 10.1172/jci116972
38. Wright EM, Loo DD, Hirayama BA. Biology of Human Sodium Glucose Transporters. Physiol Rev (2011) 91(2):733–94. doi: 10.1152/physrev.00055.2009
39. Lang F, Gorlach A, Vallon V. Targeting Sgk1 in Diabetes. Expert Opin Ther Targets (2009) 13(11):1303–11. doi: 10.1517/14728220903260807
40. Freitas HS, Anhe GF, Melo KF, Okamoto MM, Oliveira-Souza M, Bordin S, et al. Na(+) -Glucose Transporter-2 Messenger Ribonucleic Acid Expression in Kidney of Diabetic Rats Correlates With Glycemic Levels: Involvement of Hepatocyte Nuclear Factor-1alpha Expression and Activity. Endocrinology (2008) 149(2):717–24. doi: 10.1210/en.2007-1088
41. Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, Wright EM. Glucose Transport by Human Renal Na+/D-Glucose Cotransporters Sglt1 and Sglt2. Am J Physiol Cell Physiol (2011) 300(1):C14–21. doi: 10.1152/ajpcell.00388.2010
42. Rieg T, Masuda T, Gerasimova M, Mayoux E, Platt K, Powell DR, et al. Increase in Sglt1-Mediated Transport Explains Renal Glucose Reabsorption During Genetic and Pharmacological Sglt2 Inhibition in Euglycemia. Am J Physiol Renal Physiol (2014) 306(2):F188–93. doi: 10.1152/ajprenal.00518.2013
43. Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, et al. Knockout of Na-Glucose Transporter Sglt2 Attenuates Hyperglycemia and Glomerular Hyperfiltration But Not Kidney Growth or Injury in Diabetes Mellitus. Am J Physiol Renal Physiol (2013) 304(2):F156–67. doi: 10.1152/ajprenal.00409.2012
44. Powell DR, DaCosta CM, Gay J, Ding ZM, Smith M, Greer J, et al. Improved Glycemic Control in Mice Lacking Sglt1 and Sglt2. Am J Physiol Endocrinol Metab (2013) 304(2):E117–30. doi: 10.1152/ajpendo.00439.2012
45. Santer R, Kinner M, Lassen CL, Schneppenheim R, Eggert P, Bald M, et al. Molecular Analysis of the Sglt2 Gene in Patients With Renal Glucosuria. J Am Soc Nephrol (2003) 14(11):2873–82. doi: 10.1097/01.asn.0000092790.89332.d2
46. Martín MG, Turk E, Lostao MP, Kerner C, Wright EM. Defects in Na+/Glucose Cotransporter (Sglt1) Trafficking and Function Cause Glucose-Galactose Malabsorption. Nat Genet (1996) 12(2):216–20. doi: 10.1038/ng0296-216
47. Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, et al. Sglt2 Mediates Glucose Reabsorption in the Early Proximal Tubule. J Am Soc Nephrol (2011) 22(1):104–12. doi: 10.1681/ASN.2010030246
48. Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C, et al. A Glucose Sensor Hiding in a Family of Transporters. Proc Natl Acad Sci USA (2003) 100(20):11753–8. doi: 10.1073/pnas.1733027100
49. Kothinti RK, Blodgett AB, North PE, Roman RJ, Tabatabai NM. A Novel Sglt Is Expressed in the Human Kidney. Eur J Pharmacol (2012) 690(1-3):77–83. doi: 10.1016/j.ejphar.2012.06.033
50. Gyimesi G, Pujol-Gimenez J, Kanai Y, Hediger MA. Sodium-Coupled Glucose Transport, the Slc5 Family, and Therapeutically Relevant Inhibitors: From Molecular Discovery to Clinical Application. Pflugers Arch (2020) 472(9):1177–206. doi: 10.1007/s00424-020-02433-x
51. Coady MJ, Wallendorff B, Gagnon DG, Lapointe JY. Identification of a Novel Na+/Myo-Inositol Cotransporter. J Biol Chem (2002) 277(38):35219–24. doi: 10.1074/jbc.M204321200
52. Tsai LJ, Hsiao SH, Tsai LM, Lin CY, Tsai JJ, Liou DM, et al. The Sodium-Dependent Glucose Cotransporter Slc5a11 as an Autoimmune Modifier Gene in Sle. Tissue Antigens (2008) 71(2):114–26. doi: 10.1111/j.1399-0039.2007.00975.x
53. Gorboulev V, Schurmann A, Vallon V, Kipp H, Jaschke A, Klessen D, et al. Na(+)-D-Glucose Cotransporter Sglt1 Is Pivotal for Intestinal Glucose Absorption and Glucose-Dependent Incretin Secretion. Diabetes (2012) 61(1):187–96. doi: 10.2337/db11-1029
54. Bakris GL, Fonseca VA, Sharma K, Wright EM. Renal Sodium–glucose Transport: Role in Diabetes Mellitus and Potential Clinical Implications. Kidney Int (2009) 75(12):1272–7. doi: 10.1038/ki.2009.87
55. Uehara-Watanabe N, Okuno-Ozeki N, Minamida A, Nakamura I, Nakata T, Nakai K, et al. Direct Evidence of Proximal Tubular Proliferation in Early Diabetic Nephropathy. Sci Rep (2022) 12(1):778. doi: 10.1038/s41598-022-04880-1
56. Vestri S, Okamoto MM, de Freitas HS, Aparecida Dos Santos R, Nunes MT, Morimatsu M, et al. Changes in Sodium or Glucose Filtration Rate Modulate Expression of Glucose Transporters in Renal Proximal Tubular Cells of Rat. J Membr Biol (2001) 182(2):105–12. doi: 10.1007/s00232-001-0036-y
57. Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose Transporters in Human Renal Proximal Tubular Cells Isolated From the Urine of Patients With Non-Insulin-Dependent Diabetes. Diabetes (2005) 54(12):3427–34. doi: 10.2337/diabetes.54.12.3427
58. Ackermann TF, Boini KM, Volkl H, Bhandaru M, Bareiss PM, Just L, et al. Sgk1-Sensitive Renal Tubular Glucose Reabsorption in Diabetes. Am J Physiol Renal Physiol (2009) 296(4):F859–66. doi: 10.1152/ajprenal.90238.2008
59. Pontoglio M, Prié D, Cheret C, Doyen A, Leroy C, Froguel P, et al. Hnf1alpha Controls Renal Glucose Reabsorption in Mouse and Man. EMBO Rep (2000) 1(4):359–65. doi: 10.1093/embo-reports/kvd071
60. Chang J, Yan J, Li X, Liu N, Zheng R, Zhong Y. Update on the Mechanisms of Tubular Cell Injury in Diabetic Kidney Disease. Front Med (Lausanne) (2021) 8:661076. doi: 10.3389/fmed.2021.661076
61. Ravindran S, Munusamy S. Renoprotective Mechanisms of Sodium-Glucose Co-Transporter 2 (Sglt2) Inhibitors Against the Progression of Diabetic Kidney Disease. J Cell Physiol (2021) 237(2):1182–205. doi: 10.1002/jcp.30621
62. Heerspink HJL, Stefansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in Patients With Chronic Kidney Disease. N Engl J Med (2020) 383(15):1436–46. doi: 10.1056/NEJMoa2024816
63. Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med (2019) 380(24):2295–306. doi: 10.1056/NEJMoa1811744
64. Perkovic V, de Zeeuw D, Mahaffey KW, Fulcher G, Erondu N, Shaw W, et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes: Results From the Canvas Program Randomised Clinical Trials. Lancet Diabetes Endocrinol (2018) 6(9):691–704. doi: 10.1016/s2213-8587(18)30141-4
65. Heerspink HJL, Karasik A, Thuresson M, Melzer-Cohen C, Chodick G, Khunti K, et al. Kidney Outcomes Associated With Use of Sglt2 Inhibitors in Real-World Clinical Practice (Cvd-Real 3): A Multinational Observational Cohort Study. Lancet Diabetes Endocrinol (2020) 8(1):27–35. doi: 10.1016/s2213-8587(19)30384-5
66. Dominguez Rieg JA, Rieg T. What Does Sodium-Glucose Co-Transporter 1 Inhibition Add: Prospects for Dual Inhibition. Diabetes Obes Metab (2019) 21 Suppl 2:43–52. doi: 10.1111/dom.13630
67. He Y, Pachori A, Chen P, Ma S, Mendonza AE, Amer A, et al. Glucosuric, Renal and Haemodynamic Effects of Licogliflozin, a Dual Inhibitor of Sodium-Glucose Co-Transporter-1 and Sodium-Glucose Co-Transporter-2, in Patients With Chronic Kidney Disease: A Randomized Trial. Diabetes Obes Metab (2021) 23(5):1182–90. doi: 10.1111/dom.14327
68. Moriya T, Tsuchiya A, Okizaki S, Hayashi A, Tanaka K, Shichiri M. Glomerular Hyperfiltration and Increased Glomerular Filtration Surface Are Associated With Renal Function Decline in Normo- and Microalbuminuric Type 2 Diabetes. Kidney Int (2012) 81(5):486–93. doi: 10.1038/ki.2011.404
69. Wolf G, Ziyadeh FN. Molecular Mechanisms of Diabetic Renal Hypertrophy. Kidney Int (1999) 56(2):393–405. doi: 10.1046/j.1523-1755.1999.00590.x
70. Ohshiro Y, Ma RC, Yasuda Y, Hiraoka-Yamamoto J, Clermont AC, Isshiki K, et al. Reduction of Diabetes-Induced Oxidative Stress, Fibrotic Cytokine Expression, and Renal Dysfunction in Protein Kinase Cbeta-Null Mice. Diabetes (2006) 55(11):3112–20. doi: 10.2337/db06-0895
71. Tuttle KR, Bakris GL, Toto RD, McGill JB, Hu K, Anderson PW. The Effect of Ruboxistaurin on Nephropathy in Type 2 Diabetes. Diabetes Care (2005) 28(11):2686–90. doi: 10.2337/diacare.28.11.2686
72. Koya D. Dual Protein Kinase C Alpha and Beta Inhibitors and Diabetic Kidney Disease: A Revisited Therapeutic Target for Future Clinical Trials. J Diabetes Investig (2014) 5(2):147–8. doi: 10.1111/jdi.12154
73. Vallon V, Huang DY, Deng A, Richter K, Blantz RC, Thomson S. Salt-Sensitivity of Proximal Reabsorption Alters Macula Densa Salt and Explains the Paradoxical Effect of Dietary Salt on Glomerular Filtration Rate in Diabetes Mellitus. J Am Soc Nephrol (2002) 13(7):1865–71. doi: 10.1097/01.asn.0000016441.41118.57
74. Pedersen SB, Flyvbjerg A, Richelsen B. Inhibition of Renal Ornithine Decarboxylase Activity Prevents Kidney Hypertrophy in Experimental Diabetes. Am J Physiol (1993) 264(2 Pt 1):C453–6. doi: 10.1152/ajpcell.1993.264.2.C453
75. Miracle CM, Rieg T, Mansoury H, Vallon V, Thomson SC. Ornithine Decarboxylase Inhibitor Eliminates Hyperresponsiveness of the Early Diabetic Proximal Tubule to Dietary Salt. Am J Physiol Renal Physiol (2008) 295(4):F995–F1002. doi: 10.1152/ajprenal.00491.2007
76. Castrop H. Mediators of Tubuloglomerular Feedback Regulation of Glomerular Filtration: Atp and Adenosine. Acta Physiol (Oxf) (2007) 189(1):3–14. doi: 10.1111/j.1748-1716.2006.01610.x
77. Thomson S, Bao D, Deng A, Vallon V. Adenosine Formed by 5’-Nucleotidase Mediates Tubuloglomerular Feedback. J Clin Invest (2000) 106(2):289–98. doi: 10.1172/jci8761
78. Sallstrom J, Carlsson PO, Fredholm BB, Larsson E, Persson AE, Palm F. Diabetes-Induced Hyperfiltration in Adenosine a(1)-Receptor Deficient Mice Lacking the Tubuloglomerular Feedback Mechanism. Acta Physiol (Oxf) (2007) 190(3):253–9. doi: 10.1111/j.1748-1716.2007.01705.x
79. Faulhaber-Walter R, Chen L, Oppermann M, Kim SM, Huang Y, Hiramatsu N, et al. Lack of A1 Adenosine Receptors Augments Diabetic Hyperfiltration and Glomerular Injury. J Am Soc Nephrol (2008) 19(4):722–30. doi: 10.1681/ASN.2007060721
80. Brown R, Ollerstam A, Johansson B, Skøtt O, Gebre-Medhin S, Fredholm B, et al. Abolished Tubuloglomerular Feedback and Increased Plasma Renin in Adenosine A1 Receptor-Deficient Mice. Am J Physiol Regul Integr Comp Physiol (2001) 281(5):R1362–7. doi: 10.1152/ajpregu.2001.281.5.R1362
81. Li L, Lai EY, Huang Y, Eisner C, Mizel D, Wilcox CS, et al. Renal Afferent Arteriolar and Tubuloglomerular Feedback Reactivity in Mice With Conditional Deletions of Adenosine 1 Receptors. Am J Physiol Renal Physiol (2012) 303(8):F1166–75. doi: 10.1152/ajprenal.00222.2012
82. Vallon V, Schroth J, Satriano J, Blantz RC, Thomson SC, Rieg T. Adenosine a(1) Receptors Determine Glomerular Hyperfiltration and the Salt Paradox in Early Streptozotocin Diabetes Mellitus. Nephron Physiol (2009) 111(3):p30–8. doi: 10.1159/000208211
83. Kidokoro K, Cherney DZI, Bozovic A, Nagasu H, Satoh M, Kanda E, et al. Evaluation of Glomerular Hemodynamic Function by Empagliflozin in Diabetic Mice Using in Vivo Imaging. Circulation (2019) 140(4):303–15. doi: 10.1161/circulationaha.118.037418
84. Carlstrom M, Wilcox CS, Welch WJ. Adenosine A2a Receptor Activation Attenuates Tubuloglomerular Feedback Responses by Stimulation of Endothelial Nitric Oxide Synthase. Am J Physiol Renal Physiol (2011) 300(2):F457–64. doi: 10.1152/ajprenal.00567.2010
85. Persson P, Hansell P, Palm F. Adenosine A2 Receptor-Mediated Regulation of Renal Hemodynamics and Glomerular Filtration Rate Is Abolished in Diabetes. Adv Exp Med Biol (2013) 765:225–30. doi: 10.1007/978-1-4614-4989-8_31
86. Persson P, Hansell P, Palm F. Reduced Adenosine A2a Receptor-Mediated Efferent Arteriolar Vasodilation Contributes to Diabetes-Induced Glomerular Hyperfiltration. Kidney Int (2015) 87(1):109–15. doi: 10.1038/ki.2014.219
87. Patinha D, Carvalho C, Abreu C, Cunha OM, Mota MC, Afonso J, et al. Diabetes Downregulates Renal Adenosine A2a Receptors in an Experimental Model of Hypertension. PloS One (2019) 14(5):e0217552. doi: 10.1371/journal.pone.0217552
88. Patinha D, Abreu C, Carvalho C, Cunha OM, Mota M, Afonso J, et al. Adenosine A2a and A3 Receptors as Targets for the Treatment of Hypertensive-Diabetic Nephropathy. Biomedicines (2020) 8(11):529. doi: 10.3390/biomedicines8110529
89. Cardenas A, Toledo C, Oyarzun C, Sepulveda A, Quezada C, Guillen-Gomez E, et al. Adenosine a(2b) Receptor-Mediated Vegf Induction Promotes Diabetic Glomerulopathy. Lab Invest (2013) 93(1):135–44. doi: 10.1038/labinvest.2012.143
90. Dessapt-Baradez C, Woolf AS, White KE, Pan J, Huang JL, Hayward AA, et al. Targeted Glomerular Angiopoietin-1 Therapy for Early Diabetic Kidney Disease. J Am Soc Nephrol (2014) 25(1):33–42. doi: 10.1681/ASN.2012121218
91. Chen SJ, Lv LL, Liu BC, Tang RN. Crosstalk Between Tubular Epithelial Cells and Glomerular Endothelial Cells in Diabetic Kidney Disease. Cell Prolif (2020) 53(3):e12763. doi: 10.1111/cpr.12763
92. Schrijvers BF, Flyvbjerg A, De Vriese AS. The Role of Vascular Endothelial Growth Factor (Vegf) in Renal Pathophysiology. Kidney Int (2004) 65(6):2003–17. doi: 10.1111/j.1523-1755.2004.00621.x
93. Edwards RM, Trizna W. Modulation of Glomerular Arteriolar Tone by Nitric Oxide Synthase Inhibitors. J Am Soc Nephrol (1993) 4(5):1127–32. doi: 10.1681/asn.V451127
94. Sugimoto H, Shikata K, Matsuda M, Kushiro M, Hayashi Y, Hiragushi K, et al. Increased Expression of Endothelial Cell Nitric Oxide Synthase (Ecnos) in Afferent and Glomerular Endothelial Cells Is Involved in Glomerular Hyperfiltration of Diabetic Nephropathy. Diabetologia (1998) 41(12):1426–34. doi: 10.1007/s001250051088
95. Chiarelli F, Cipollone F, Romano F, Tumini S, Costantini F, di Ricco L, et al. Increased Circulating Nitric Oxide in Young Patients With Type 1 Diabetes and Persistent Microalbuminuria: Relation to Glomerular Hyperfiltration. Diabetes (2000) 49(7):1258–63. doi: 10.2337/diabetes.49.7.1258
96. Hiragushi K, Sugimoto H, Shikata K, Yamashita T, Miyatake N, Shikata Y, et al. Nitric Oxide System Is Involved in Glomerular Hyperfiltration in Japanese Normo- and Micro-Albuminuric Patients With Type 2 Diabetes. Diabetes Res Clin Pract (2001) 53(3):149–59. doi: 10.1016/s0168-8227(01)00260-1
97. Zhang J, Jiang S, Wei J, Yip KP, Wang L, Lai EY, et al. Glucose Dilates Renal Afferent Arterioles Via Glucose Transporter-1. Am J Physiol Renal Physiol (2018) 315(1):F123–F9. doi: 10.1152/ajprenal.00409.2017
98. Song P, Huang W, Onishi A, Patel R, Kim YC, van Ginkel C, et al. Knockout of Na(+)-Glucose Cotransporter Sglt1 Mitigates Diabetes-Induced Upregulation of Nitric Oxide Synthase Nos1 in the Macula Densa and Glomerular Hyperfiltration. Am J Physiol Renal Physiol (2019) 317(1):F207–F17. doi: 10.1152/ajprenal.00120.2019
99. Zhang J, Wei J, Jiang S, Xu L, Wang L, Cheng F, et al. Macula Densa Sglt1-Nos1-Tubuloglomerular Feedback Pathway, a New Mechanism for Glomerular Hyperfiltration During Hyperglycemia. J Am Soc Nephrol (2019) 30(4):578–93. doi: 10.1681/ASN.2018080844
100. Zhang J, Cai J, Cui Y, Jiang S, Wei J, Kim YC, et al. Role of the Macula Densa Sodium Glucose Cotransporter Type 1-Neuronal Nitric Oxide Synthase-Tubuloglomerular Feedback Pathway in Diabetic Hyperfiltration. Kidney Int (2022) 101(3):541–50. doi: 10.1016/j.kint.2021.10.037
101. Wei J, Zhang J, Jiang S, Wang L, Persson AEG, Liu R. High-Protein Diet-Induced Glomerular Hyperfiltration Is Dependent on Neuronal Nitric Oxide Synthase Beta in the Macula Densa Via Tubuloglomerular Feedback Response. Hypertension (2019) 74(4):864–71. doi: 10.1161/HYPERTENSIONAHA.119.13077
102. Oba R, Kanzaki G, Sasaki T, Okabayashi Y, Haruhara K, Koike K, et al. Dietary Protein Intake and Single-Nephron Glomerular Filtration Rate. Nutrients (2020) 12(9):2549. doi: 10.3390/nu12092549
103. Adeva-Andany MM, Fernández-Fernández C, Carneiro-Freire N, Vila-Altesor M, Ameneiros-Rodríguez E. The Differential Effect of Animal Versus Vegetable Dietary Protein on the Clinical Manifestations of Diabetic Kidney Disease in Humans. Clin Nutr ESPEN (2022) 48:21–35. doi: 10.1016/j.clnesp.2022.01.030
104. Kreisberg JI, Patel PY. The Effects of Insulin, Glucose and Diabetes on Prostaglandin Production by Rat Kidney Glomeruli and Cultured Glomerular Mesangial Cells. Prostaglandins Leukot Med (1983) 11(4):431–42. doi: 10.1016/0262-1746(83)90097-5
105. Craven PA, Caines MA, DeRubertis FR. Sequential Alterations in Glomerular Prostaglandin and Thromboxane Synthesis in Diabetic Rats: Relationship to the Hyperfiltration of Early Diabetes. Metabolism (1987) 36(1):95–103. doi: 10.1016/0026-0495(87)90070-9
106. Chagnac A, Zingerman B, Rozen-Zvi B, Herman-Edelstein M. Consequences of Glomerular Hyperfiltration: The Role of Physical Forces in the Pathogenesis of Chronic Kidney Disease in Diabetes and Obesity. Nephron (2019) 143(1):38–42. doi: 10.1159/000499486
107. Perico N, Benigni A, Gabanelli M, Piccinelli A, Ròg M, De Riva C, et al. Atrial Natriuretic Peptide and Prostacyclin Synergistically Mediate Hyperfiltration and Hyperperfusion of Diabetic Rats. Diabetes (1992) 41(4):533–8. doi: 10.2337/diab.41.4.533
108. Cherney DZ, Miller JA, Scholey JW, Bradley TJ, Slorach C, Curtis JR, et al. The Effect of Cyclooxygenase-2 Inhibition on Renal Hemodynamic Function in Humans With Type 1 Diabetes. Diabetes (2008) 57(3):688–95. doi: 10.2337/db07-1230
109. Komers R, Lindsley JN, Oyama TT, Schutzer WE, Reed JF, Mader SL, et al. Immunohistochemical and Functional Correlations of Renal Cyclooxygenase-2 in Experimental Diabetes. J Clin Invest (2001) 107(7):889–98. doi: 10.1172/jci10228
110. Jensen BL, Schmid C, Kurtz A. Prostaglandins Stimulate Renin Secretion and Renin Mrna in Mouse Renal Juxtaglomerular Cells. Am J Physiol (1996) 271(3 Pt 2):F659–69. doi: 10.1152/ajprenal.1996.271.3.F659
111. Yang T, Endo Y, Huang YG, Smart A, Briggs JP, Schnermann J. Renin Expression in Cox-2-Knockout Mice on Normal or Low-Salt Diets. Am J Physiol Renal Physiol (2000) 279(5):F819–25. doi: 10.1152/ajprenal.2000.279.5.F819
112. Choi MR, Fernandez BE. Protective Renal Effects of Atrial Natriuretic Peptide: Where Are We Now? Front Physiol (2021) 12:680213. doi: 10.3389/fphys.2021.680213
113. Ortola FV, Ballermann BJ, Anderson S, Mendez RE, Brenner BM. Elevated Plasma Atrial Natriuretic Peptide Levels in Diabetic Rats. Potential Mediator of Hyperfiltration. J Clin Invest (1987) 80(3):670–4. doi: 10.1172/jci113120
114. Zhang PL, Mackenzie HS, Troy JL, Brenner BM. Effects of an Atrial Natriuretic Peptide Receptor Antagonist on Glomerular Hyperfiltration in Diabetic Rats. J Am Soc Nephrol (1994) 4(8):1564–70. doi: 10.1681/asn.V481564
115. Liu C, Li Q, Feng X, Zhu J, Li Q. Deterioration of Diabetic Nephropathy Via Stimulating Secretion of Cytokines by Atrial Natriuretic Peptide. BMC Endocr Disord (2021) 21(1):204. doi: 10.1186/s12902-021-00867-7
116. Moore KB, McKenna K, Osman M, Tormey WP, McDonald D, Thompson CJ. Atrial Natriuretic Peptide Increases Urinary Albumin Excretion in People With Normoalbuminuric Type-2 Diabetes. Ir J Med Sci (2007) 176(2):67–73. doi: 10.1007/s11845-007-0030-1
117. Habibi J, Aroor AR, Das NA, Manrique-Acevedo CM, Johnson MS, Hayden MR, et al. The Combination of a Neprilysin Inhibitor (Sacubitril) and Angiotensin-Ii Receptor Blocker (Valsartan) Attenuates Glomerular and Tubular Injury in the Zucker Obese Rat. Cardiovasc Diabetol (2019) 18(1):40. doi: 10.1186/s12933-019-0847-8
118. Spannella F, Giulietti F, Filipponi A, Sarzani R. Effect of Sacubitril/Valsartan on Renal Function: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. ESC Heart Fail (2020) 7(6):3487–96. doi: 10.1002/ehf2.13002
119. Hollenberg NK, Price DA, Fisher ND, Lansang MC, Perkins B, Gordon MS, et al. Glomerular Hemodynamics and the Renin-Angiotensin System in Patients With Type 1 Diabetes Mellitus. Kidney Int (2003) 63(1):172–8. doi: 10.1046/j.1523-1755.2003.00701.x
120. Gurley SB, Coffman TM. The Renin-Angiotensin System and Diabetic Nephropathy. Semin Nephrol (2007) 27(2):144–52. doi: 10.1016/j.semnephrol.2007.01.009
121. Lytvyn Y, Bjornstad P, van Raalte DH, Heerspink HL, Cherney DZI. The New Biology of Diabetic Kidney Disease-Mechanisms and Therapeutic Implications. Endocr Rev (2020) 41(2):202–31. doi: 10.1210/endrev/bnz010
122. Lovshin JA, Boulet G, Lytvyn Y, Lovblom LE, Bjornstad P, Farooqi MA, et al. Renin-Angiotensin-Aldosterone System Activation in Long-Standing Type 1 Diabetes. JCI Insight (2018) 3(1):e96968. doi: 10.1172/jci.insight.96968
123. Sochett EB, Cherney DZ, Curtis JR, Dekker MG, Scholey JW, Miller JA. Impact of Renin Angiotensin System Modulation on the Hyperfiltration State in Type 1 Diabetes. J Am Soc Nephrol (2006) 17(6):1703–9. doi: 10.1681/ASN.2005080872
124. Peikert A, Vaduganathan M, Mc Causland F, Claggett BL, Chatur S, Packer M, et al. Effects of Sacubitril/Valsartan Versus Valsartan on Renal Function in Patients With and Without Diabetes and Heart Failure With Preserved Ejection Fraction: Insights From Paragon-Hf. Eur J Heart Fail (2022). doi: 10.1002/ejhf.2450
125. Tikellis C, Brown R, Head GA, Cooper ME, Thomas MC. Angiotensin-Converting Enzyme 2 Mediates Hyperfiltration Associated With Diabetes. Am J Physiol Renal Physiol (2014) 306(7):F773–80. doi: 10.1152/ajprenal.00264.2013
126. Ren Y, Garvin JL, Carretero OA. Vasodilator Action of Angiotensin-(1-7) on Isolated Rabbit Afferent Arterioles. Hypertension (2002) 39(3):799–802. doi: 10.1161/hy0302.104673
127. Lytvyn Y, Bjornstad P, Lovshin JA, Boulet G, Farooqi MA, Lai V, et al. Renal Hemodynamic Function and Raas Activation Over the Natural History of Type 1 Diabetes. Am J Kidney Dis (2019) 73(6):786–96. doi: 10.1053/j.ajkd.2018.12.034
128. Koszegi S, Molnar A, Lenart L, Hodrea J, Balogh DB, Lakat T, et al. Raas Inhibitors Directly Reduce Diabetes-Induced Renal Fibrosis Via Growth Factor Inhibition. J Physiol (2019) 597(1):193–209. doi: 10.1113/JP277002
129. Raina R, Chauvin A, Chakraborty R, Nair N, Shah H, Krishnappa V, et al. The Role of Endothelin and Endothelin Antagonists in Chronic Kidney Disease. Kidney Dis (Basel) (2020) 6(1):22–34. doi: 10.1159/000504623
130. Benz K, Amann K. Endothelin in Diabetic Renal Disease. Contrib Nephrol (2011) 172:139–48. doi: 10.1159/000328695
131. Rabelink TJ, Kaasjager KA, Boer P, Stroes EG, Braam B, Koomans HA. Effects of Endothelin-1 on Renal Function in Humans: Implications for Physiology and Pathophysiology. Kidney Int (1994) 46(2):376–81. doi: 10.1038/ki.1994.284
132. Jung C, Rafnsson A, Brismar K, Pernow J. Endothelial Progenitor Cells in Relation to Endothelin-1 and Endothelin Receptor Blockade: A Randomized, Controlled Trial. Int J Cardiol (2013) 168(2):1017–22. doi: 10.1016/j.ijcard.2012.10.032
133. Hesp AC, Smits MM, van Bommel EJM, Muskiet MHA, Tonneijck L, Nieuwdorp M, et al. Kidney Hemodynamic Profile and Systemic Vascular Function in Adults With Type 2 Diabetes: Analysis of Three Clinical Trials. J Diabetes Complications (2022) 36(3):108127. doi: 10.1016/j.jdiacomp.2022.108127
134. Zhang L, Xue S, Hou J, Chen G, Xu ZG. Endothelin Receptor Antagonists for the Treatment of Diabetic Nephropathy: A Meta-Analysis and Systematic Review. World J Diabetes (2020) 11(11):553–66. doi: 10.4239/wjd.v11.i11.553
135. Yuan W, Li Y, Wang J, Li J, Gou S, Fu P. Endothelin-Receptor Antagonists for Diabetic Nephropathy: A Meta-Analysis. Nephrol (Carlton) (2015) 20(7):459–66. doi: 10.1111/nep.12442
Keywords: diabetic kidney disease, glomerular hyperfiltration, sodium–glucose cotransporter, tubuloglomerular feedback, renal hemodynamics
Citation: Yang Y and Xu G (2022) Update on Pathogenesis of Glomerular Hyperfiltration in Early Diabetic Kidney Disease. Front. Endocrinol. 13:872918. doi: 10.3389/fendo.2022.872918
Received: 10 February 2022; Accepted: 11 April 2022;
Published: 19 May 2022.
Edited by:
Francisco Westermeier, FH Joanneum, AustriaReviewed by:
Akira Sugawara, Tohoku University, JapanTatsuo Shimosawa, International University of Health and Welfare (IUHW), Japan
Sebastián Alarcón, Austral University of Chile, Chile
Copyright © 2022 Yang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaosi Xu, Z2Fvc2l4dUAxNjMuY29t