 Ellen Conceição-Furber
Ellen Conceição-Furber Tamer Coskun
Tamer Coskun Ricardo J. Samms
Ricardo J. Samms
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 25 April 2022
Sec. Gut Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.868037
This article is part of the Research Topic Targeting GPCRs and their Ligands in the Gut Establishing Glucose Homeostasis and Appetite Regulation View all 6 articles
A major challenge of obesity therapy is to sustain clinically relevant weight loss over time. Achieving this goal likely requires both reducing daily caloric intake and increasing caloric expenditure. Over the past decade, advances in pharmaceutical engineering of ligands targeting G protein-coupled receptors have led to the development of highly effective anorectic agents. These include mono-agonists of the GLP-1R and dual GIPR/GLP-1R co-agonists that have demonstrated substantial weight loss in experimental models and in humans. By contrast, currently, there are no medicines available that effectively augment metabolic rate to promote weight loss. Here, we present evidence indicating that activation of the GCGR may provide a solution to this unmet therapeutic need. In adult humans, GCGR agonism increases energy expenditure to a magnitude sufficient for inducing a negative energy balance. In preclinical studies, the glucagon-GCGR system affects key metabolically relevant organs (including the liver and white and brown adipose tissue) to boost whole-body thermogenic capacity and protect from obesity. Further, activation of the GCGR has been shown to augment both the magnitude and duration of weight loss that is achieved by either selective GLP-1R or dual GIPR/GLP-1R agonism in rodents. Based on the accumulation of such findings, we propose that the thermogenic activity of GCGR agonism will also complement other anti-obesity agents that lower body weight by suppressing appetite.
The metabolic actions of the hormone glucagon are transduced via the glucagon receptor (GCGR), a 477 amino acid, cell membrane-spanning protein (1, 2) belonging to the diverse superfamily of G protein-coupled receptors (GPCRs). In addition to possessing a signature transmembrane region consisting of seven membrane spanning alpha helices, the GCGR contains a large N-terminal extracellular domain that aides in glucagon recognition for receptor binding. Phylogenetically, this unique structural feature places the GCGR in the ‘Secretin’ sub-family of GPCRs (3), a small group of 15 peptide hormone receptors named in recognition of the secretin receptor, as its sequence was the first member determined (4). The GCGR is coupled to the GS heterotrimeric G protein, and upon glucagon binding, the receptor catalyzes the exchange of GDP for GTP, leading to the dissociation of GαS from Gβγ and activation of adenylyl cyclase. This then catalyzes the conversion of ATP to cAMP, the primary second messenger that mediates glucagon signaling. The GCGR is abundantly expressed in hepatocytes of the liver, but its mRNA is also detected in cell types of the brain, pancreatic islets, adipose tissue, the kidney, and intestinal smooth muscle (5).
Historically, the most widely studied function of the glucagon-GCGR axis has been its role in maintaining euglycemia in response to an overnight fast. This is largely a protective function, where in response to a decrease in blood glucose, glucagon is released from pancreatic alpha cells into the hepatic portal vein, thereby quickly reaching hepatocytes and stimulating endogenous glucose production. This occurs through the binding of glucagon to the GCGR, stimulation of the adenylyl cyclase-cAMP system (6), and a subsequent activation of the protein kinase A pathway to stimulate glycogenolysis and gluconeogenesis while simultaneously inhibiting glycogen synthesis (7). Thus, due to its fundamental role in promoting hepatic glucose production, therapeutic strategies aimed at both activating the GCGR to acutely rescue from hypoglycemia and at blocking glucagon-mediated hyperglycemia for the treatment of type 2 diabetes (T2D) have been pursued (8).
Ground-breaking work from Roger Unger and colleagues showed that the disruption of the glucagon-insulin bi-hormonal relationship may contribute to hyperglycemia in the diabetic condition (9). The concept was further supported by the discovery that patients with T2D often have higher concentrations of circulating glucagon compared with normo-glycemic individuals (10). Work characterizing the phenotype of Gcgr knockout mice (11, 12) and various therapeutic modalities targeting the glucagon-GCGR system to lower glucose in an array of preclinical rodent models (13–15) produced results that supported discovering agents to block glucagon action as a way to reduce hyperglycemia. However, although several GCGR antagonists have entered clinical development for the treatment of T2D, to date, none have advanced to regulatory approval (16).
Intriguingly, the glucagon-GCGR axis is also subject to investigational efforts aimed at exploiting the long-term effects of activating the GCGR. The potential therapeutic advantage of GCGR agonism is supported by other foundational studies showing that the infusion of glucagon can have beneficial effects on lipid and bile acid metabolism, and most importantly, on increasing energy expenditure in adult humans. Thus, the GCGR agonist approach may have utility in treating obesity and possibly other metabolic conditions such as non-alcoholic steato-hepatitis (NASH). To reduce the risk of inducing hyperglycemia by GCGR signaling, GCGR agonism has been combined with other mechanisms, such as glucagon-like peptide-1 receptor (GLP-1R) agonism (17–19) and glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism (20), both of which stimulate insulin secretion upon the elevation of blood glucose. In addition to controlling glycemia, since both GLP-1R mono- and GIPR/GLP-1R dual agonism reduce body weight largely by decreasing caloric intake, the combination with an energy expenditure agent like a GCGR agonist should offer complementary metabolic benefits. The article herein discusses the key attributes of GCGR activation to promote and possibly maintain body weight loss, highlighting key mechanisms of GCGR agonism that make it an attractive partner for pairing with other therapeutic approaches in the complex treatment of obesity.
The prevalence of obesity has dramatically increased in the past 50 years (21, 22), placing major economic and operational strains on healthcare systems worldwide (23, 24). Driven by increased caloric intake relative to expenditure [see Box 1 (25)], the management of obesity is often stigmatized due to the notion that excess body weight is caused by gluttony and sloth (26). However, obesity is a chronic disease that occurs frequently in an obesogenic environment, in genetically susceptible individuals, likely due to a dysregulation of the neuronal circuits that regulate body weight at a pre-defined healthy set-point (27–29). Yet, although our understanding of the central and peripheral pathways that regulate energy homeostasis and control metabolic rate have substantially increased (29, 30), current approaches employed to combat excess adiposity are focused primarily on reducing daily caloric intake (31–34). However, although effective in the short-term (35), inducing a negative energy balance by reducing daily food intake faces a significant challenge posed by a natural physiological defense system (see Box 1) that has evolved to protect against major weight loss (35–37). Specifically, reducing body weight by decreasing caloric intake often leads to increased hunger, lowered sensitivity to satiety factors (increasing meal frequency and/or meal size), and a reduced resting metabolic rate (metabolic adaptation) that is greater than expected for the amount of fat and fat-free mass that is lost (38–41). Together, this increased drive to feed in a situation of reduced caloric expenditure plays a key role in driving the body weight regain that often occurs in response to dietary intervention programs (36). Therefore, it is imperative to identify agents that both suppress appetite and increase whole-body metabolic rate, both in a state of energy surplus, and in the face of a negative energy balance (see Box 1). Taking this approach should not only maximize the magnitude of weight loss, but more importantly the duration of reduced body weight.
Box 1. Energy balance
The first law in thermodynamics states that energy is neither created nor destroyed, but it can be converted into different forms. This applies to human physiology, energy intake must equal energy expended for body weight to remain stable (42). The term ‘energy balance’ is used to describe this metabolic equilibrium (42), and since humans have a low capacity to store adenosine triphosphate (ATP), regulatory systems have evolved to regulate body weight and control energy intake, and expenditure (29). Following the consumption of a meal (postprandial state), ingested energy is stored as glycogen primarily in the liver and skeletal muscle and as triglyceride in subcutaneous white adipose tissue (WAT). The oxidation of glucose and lipids occurs primarily in mitochondria, where upon entry of acetyl-CoA into the TCA cycle, energy substrates (NADH and FADH) carry protons/electrons to the electron transport system for generating an electrochemical gradient that is utilized by ATP synthase in the presence of oxygen to convert adenosine diphosphate (ADP, signals energetic need) into ATP (the energy currency of the cell). Total daily energy expenditure can vary greatly between individuals, depending on differences in resting metabolic rate (amount of energy needed to fuel the body at rest), the thermic effect of food (energetic cost of absorbing and metabolizing nutrients), and differences in levels of physical activity (42). Thus, if nutrient intake exceeds that of caloric expenditure, excess energy is stored as fat, while prolonged periods of energy restriction result in weight loss. To maintain energy balance (body weight) within healthy limits, the brain senses, monitors, and integrates circulating signals (metabolic, hormonal, and neuronal) of short- and long-term energy levels and adjusts energy intake and expenditure accordingly (42).
Although GLP-1R agonist based technologies have expanded the obesity medication toolbox over the past decade (43–45), it is important to note that anorectic agents are governed by the same laws of energy balance (see Box 1) as dietary intervention induced weight loss, such that metabolic adaptation (reduced resting metabolic rate) may still present a major barrier to achieving prolonged weight loss (37, 46–48). Thus, although there is some evidence that the activation of the GCGR can curb appetite (49, 50), the predominant benefit of adding this mechanism to either GLP-1R agonism or dual GIPR/GLP-1R agonism likely lies in its ability to increase energy utilization (20, 51–54).
Over the past 60 years (see Figure 1), a considerable amount of evidence has accumulated to suggest that glucagon is a highly effective thermogenic agent (55), capable of inducing a negative energy balance by rapidly activating energy wasting pathways and enhancing thermogenic capacity to increase whole-body caloric expenditure in both states of energy surplus and deficit. Clinically, infusion of glucagon (45 min to 13 hours, 6-50 ng/kg/min) increases energy expenditure in the fed state in lean, overweight, and obese participants (56, 57), although it has also been reported that sub-chronic administration of glucagon (72-hours, 25 ng/kg/min) failed to impact energy utilization (58). Preclinically, the robustness of glucagon’s thermogenic activity is exemplified by its ability to increase energy expenditure across multiple species, including mice, rats, penguins, pigs, and dogs [see Figure 1 (59–62)], and by findings showing that the administration of glucagon reduces body weight in already obese animals and protects from diet-induced obesity in mice and rats (17, 20, 54, 63). Further, the therapeutic potential of the thermogenic activity of glucagon is validated by studies showing that GCGR activation boosts the magnitude of weight loss achieved by both selective GLP-1R and dual GIPR/GLP-1R agonism in obese animals due to an induction of whole-body metabolic rate (20). Thus, glucagon is a highly effective thermogenic agent that increases energy expenditure across multiple species including adult humans.

Figure 1 Key Discoveries Highlighting the Thermogenic Activity of Glucagon Receptor Agonism Preclinical Models and Man. Glucagon (GCG), energy expenditure (EE), brown adiose tissue (BAT, white adipose tissue (WAT), glucagon-like peptide 1 receptor (GLP-1R).
In response to weight loss, resting energy expenditure is reduced on average by 30 kcal/kg/day or 300 kcal/10 kg (10% weight loss in a 100 kg individual) (40, 41, 64), presenting a major challenge to achieving prolonged weight loss. Importantly, the administration of glucagon is still capable of augmenting metabolic rate following an overnight fast (or state of energy deficit) in adult humans and experimental models of obesity (51, 60, 65–67). In humans, acute infusion of glucagon (45 min to 210 min, 3-50 ng/kg/min) raises caloric expenditure on average by 200 kcal/day in lean and obese fasted subjects (51, 52, 68), and administration of glucagon increases metabolic rate in fasted preclinical models (60, 65, 66). Thus, with rodent studies indicating that the effect of glucagon on energy utilization progressively increases over time due to an enhancement of thermogenic capacity (54, 69), it is hypothesized that chronic GCGR agonism is sufficient to counter the reduced metabolic rate that occurs following weight loss. Indeed, administration of glucagon increases energy expenditure in the presence of GLP-1R agonism in obese humans (57), and glucagon activation augments metabolic rate and stimulates a right-shift in the weight loss curve induced by selective GLP-1R and dual GIP and GLP-1 receptor agonism in obese mice (17, 20). Further, there is evidence supporting the translation of this pharmacology in early clinical trials (70–72).
In summary, the studies presented above highlight the ability of GCGR activation to raise metabolic rate in both fed and fasted conditions, and further validate the glucagon-GCGR axis as an attractive therapeutic mechanism to pair with obesity medications that reduce body weight by suppressing caloric intake. Importantly, GCGR agonism appears to stimulate weight loss by rapidly activating pathways that function to waste energy and by targeting key metabolically relevant organs to augment thermogenic capacity. Together, these effects not only boost the magnitude of weight loss achieved but also prolong the duration of reduced body weight.
A prerequisite for a therapeutic agent that effectively increases whole-body metabolic rate is the ability to both activate existing thermogenic machinery and increase thermogenic capacity (73). In line with these criteria, glucagon targets several metabolically relevant organs to both activate pathways that function to waste energy and to stimulate the production of thermogenic machinery (see Figure 2). Below we outline the proposed target organs and mechanisms by which glucagon action increases caloric expenditure.
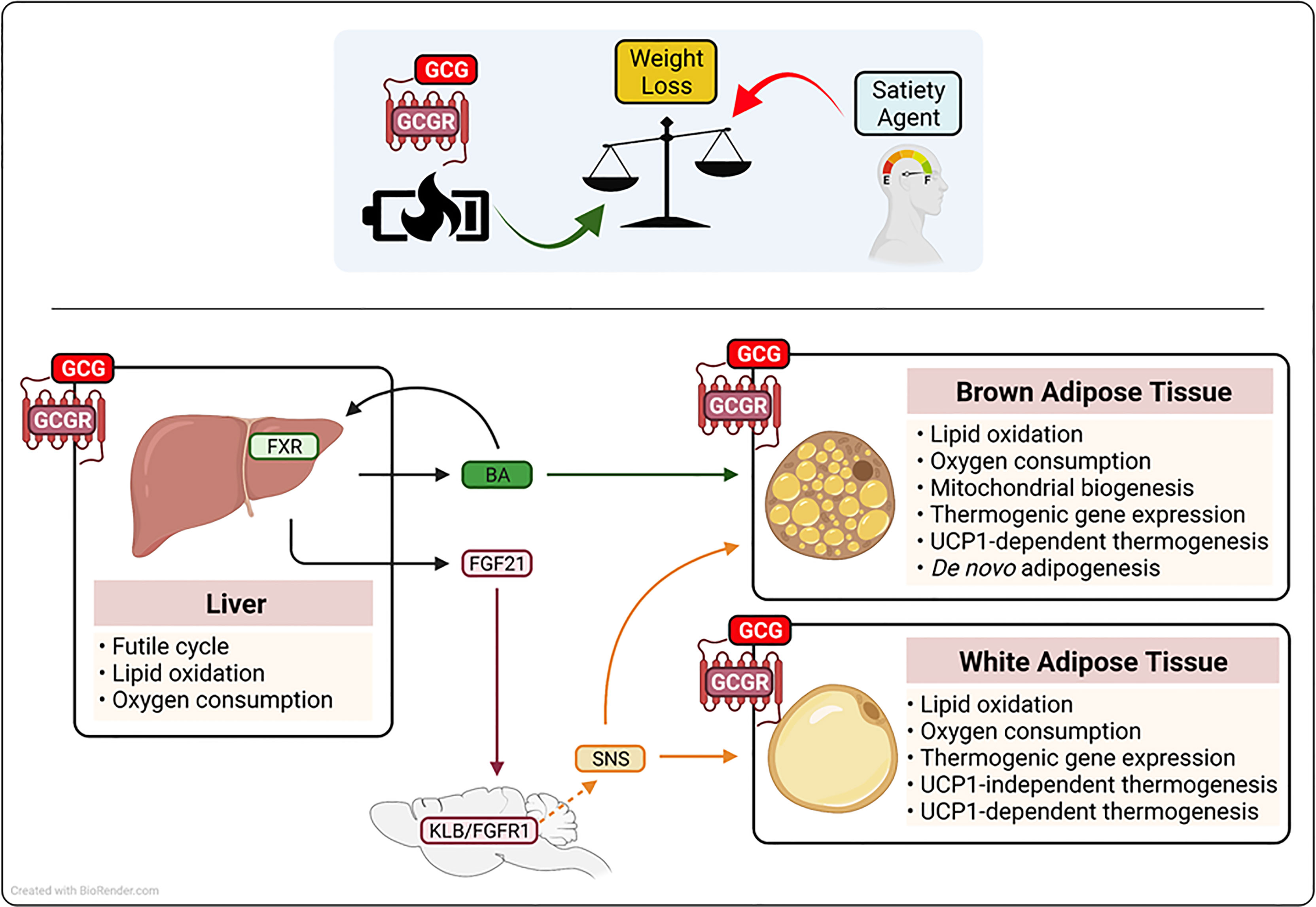
Figure 2 Schematic representation of the proposed mechanism(s) by which glucagon receptor (GCGR) activation augments metabolic rate and drive weight loss. Glucagon (GCGR)-GCGR agonism contributes to anti-obesity strategies that employ low caloric intake (satiety agents) by augmenting of metabolic rate. Glucagon-GCGR activation increases whole-body energy expenditure by the activation of hepatic futile cycling, and the secretion of thermogenic agents fibrolast growth factor 21 (FGF21) and bile acids (BA) from the liver. Further, GCG-GCGR agonism increases caloric expenditure to protect from obesity, by leveraging the energy wasting activity of uncoupling protein 1 (UCP1) in brown adipose tissue and UCP1-dependent and-independent futile cycling in white adipose tissue. Beta-klotho(KLB), fibroblast growth factor receptor 1 (FGFR1), farnesoid X receptor (FXR), sympahetic nervous system (SNS).
The liver plays an essential role in the regulation of glycemic control, lipid homeostasis, and energy balance (74). The hepatocyte is the major metabolic cell type in the liver and is characterized by high expression of enzymes associated with glucose, lipid and amino acid metabolism, the dense presence of mitochondria, and the production of hormones (hepatokines) that impact systemic energy homeostasis (74, 75). Due to its high metabolic activity, the liver accounts for approximately 17% of basal metabolic rate, and as highlighted by liver-specific uncouplers, it has the capacity to further impact total energy expenditure to induce a negative energy balance (76, 77). The GCGR is expressed by hepatocytes (5), where in addition to regulating hepatic glucose production, GCGR signaling reduces liver fat content by inducing lipid oxidation, augmenting metabolic enzyme activity, enhancing mitochondrial function, and increasing liver-specific metabolic rate (54, 78–80). The importance of the liver in mediating the anti-obesity action of glucagon administration is exemplified by findings in liver-specific knockout models, where the absence of the GCGR ablates the ability of glucagon to induce weight loss (54, 81). Together, these studies demonstrate that glucagon action augments energy expenditure and drives weight loss by GCGR activation in the liver; mechanistically, this is due to farnesoid X receptor (FXR)-mediated hepatic futile cycling (see Box 2), the secretion of the hepatokine fibroblast growth factor 21 (FGF21) and an induction of plasma levels of bile acid (BA) species known to impact energy homeostasis (54, 63). Treatment of obese mice with a long-acting GCGR agonist increased systemic levels of cholic acid, a BA species that elevates caloric expenditure through brown-fat thermogenesis (54, 82). Further, BAs are ligands for the FXR, a nuclear receptor known to regulate both adipogenesis and adaptive thermogenesis in response to both fasting and cold exposure in mice (83). Importantly, absence of hepatic FXR nullifies the effect of GCGR agonism on metabolic rate, fatty acid oxidation, and weight loss (54). In addition to its effect on BAs/FXR, glucagon rapidly and dose-dependently increases hepatic mRNA expression and circulating levels of the thermogenic hormone FGF21 in mice and adult humans (63, 84). Notably, FGF21 and the FGF21 receptor complex (FGFR1c and KLB) knockout mouse models indicate that glucagon requires the FGF21 pathway to protect from obesity (54, 63, 81). Mechanistically, FGF21 acts via both central and peripheral mechanisms to leverage the energy-burning power of white and brown adipose tissue to augment metabolic rate in both a UCP1-dependent and -independent manner [see Box 2 (85–88)].
Box 2. The thermogenic adipocyte
Beyond exercise, there are two primary ways of augmenting energetic demand (by increasing ADP availability) and increasing whole-body metabolic rate; 1) direct induction of mitochondrial proton leak, and 2) stimulation of metabolic futile cycling (89, 90). Although exercise is effective in the short-term as a weight loss approach, adherence is problematic, and therefore, exercise programs are often ineffective. In addition to the energy storage reservoir of WAT, there are two types of thermogenic adipose tissue in mammals: classical brown and inducible beige fat (91). Brown adipocytes are highly metabolically active cells characterized by multi-locular lipid droplets, high thermogenic capacity, the expression of uncoupling protein 1 (UCP1), an enriched number of mitochondria, and the presence of highly expressed metabolic and thermogenic genes (92). The primary physiological role of brown adipose tissue (BAT) is to defend body temperature in response to cold exposure, where UCP1 uncouples the mitochondrial electrochemical protein gradient, bypassing ATP synthase and releasing energy as heat (92). In rodents and newborn humans, BAT is located in defined anatomical regions, including the interscapular and perirenal BAT depots, while in adult humans, interscapular BAT is replaced by brown fat depots located in the cervical, supraclavicular, axillary, and paravertebral regions (93). In addition to classic BAT, a second type of thermogenic adipocyte can emerge in subcutaneous white adipose tissue (WAT); this has been demonstrated to occur in response to cold exposure, β3 adrenergic agonist treatment, and several metabolic hormones (93). These so-called “inducible,” “beige,” or “brown-in-white” (BRITE) adipocytes arise from a unique developmental origin versus that of the classical brown fat and are recruited via a process known as the browning of WAT (93). Here, beige preadipocyte/progenitor cells differentiate and/or mature white adipocytes trans-differentiate into thermogenically competent fat cells (94). Like brown adipocytes, beige adipocytes are exemplified by a high oxidative/thermogenic capacity, the expression of UCP1, a high mitochondrial density, and the presence of highly expressed metabolic/thermogenic genes (89). However, in addition to utilizing the thermogenic activity of UCP1, beige adipocytes can bypass the mitochondrial electrochemical proton gradient and waste energy as heat through the induction of metabolic futile cycles (when ATP consuming pathways run simultaneously in opposite directions, releasing energy as heat), including the creatine, succinate, and lipid dependent substrate cycles (89). Importantly, leveraging the thermogenic action of brown and beige fat offers potential for the treatment of obesity and its associated comorbidities. Indeed, a recent study demonstrated that the presence of BAT in adult humans is associated with protection from metabolic diseases (95). Of further note, in both preclinical models and adult humans, cold-induced recruitment of BAT activity lowers body weight (96).
Brown adipose tissue (BAT) is a highly metabolically active organ characterized by an abundance of the thermogenic protein uncoupling protein 1 (UCP1), which uncouples the mitochondrial electrochemical proton gradient, thereby releasing energy as heat (see Box 2), (92). To augment metabolic rate, BAT combusts both stored and circulating energy substrates, including glucose, lipids, and amino acids (97, 98). Therefore, with the re-discovery of BAT in adult humans (73), and clinical studies highlighting the importance of BAT to metabolic health (99), harnessing the energy-wasting capacity of BAT has potential for the treatment of obesity and its associated comorbidities in adult humans (100). In addition to utilizing the thermogenic activity of BAT via the action of FGF21, studies performed since the late 1960s have shown that glucagon has the potential to directly signal in brown fat to augment metabolic rate (101–103). The GCGR is expressed in BAT (104), and the thermogenic capacity of GCGR activation is highlighted by some (105), but not other (104) loss of function mouse models, where absence of glucagon activity impairs adaptive thermogenesis (104, 105). Pharmacologically, the ability of glucagon to utilize the thermogenic capacity of BAT is demonstrated by studies showing that it promotes brown fat respiration and heat production (101, 103, 104). Further, in BAT explants, glucagon stimulates free fatty acid (FFA) release, augments lipid oxidation, and increases oxygen consumption [see Figures 1, 2 (101, 104)]. In vivo, in preclinical models, the administration of glucagon augments BAT blood flow (helping ensure optimal nutrient and oxygen delivery), stimulates BAT heat production, increases core body temperature, and rapidly increases energy expenditure in mice housed in thermal neutral (27-30°C) conditions (103, 104, 106). Mechanistically, glucagon recruits BAT-induced non-shivering thermogenesis via both the activation of UCP1 in existing thermogenic adipocytes, and the generation of new brown adipocytes and/or the production of new thermogenic machinery (69, 104, 105). Under non-stimulated conditions, UCP1 is inhibited by purine nucleotides (92). However, in response to cold exposure (or thermogenic stress), this inhibition is overcome and UCP1 is activated by long-chain fatty acids that are released through norepinephrine induced lipolysis (92, 107). In accordance, the administration of glucagon rapidly increases whole-body metabolic rate in vivo, and stimulates lipid breakdown, fatty acid oxidation, and oxygen consumption rates in brown-fat explants (101, 102). Thus, glucagon may leverage the classical adrenergic pathway to activate UCP1 activity and stimulate whole-body energy expenditure. Further, glucagon appears to increase the thermogenic capacity of BAT by promoting de novo adipogenesis, driving mitochondrial biogenesis, and stimulating the expression of metabolic and thermogenic genes [see Figure 2 (69, 104, 108)].
In addition to classical BAT, brown-like (or beige) adipocytes can develop in WAT, via a process known as the browning of WAT [see Box 2 (109)]. These thermogenically competent adipocytes arise from beige adipocyte progenitor cells via de novo adipogenesis and/or through the transdifferentiation of exiting white adipocytes (109). Importantly, activation of beige adipocytes expends energy by both UCP1 non-shivering thermogenesis and the induction of metabolic futile cycling [see Box 2 (89)]. Interestingly, glucagon has been reported to induce metabolic rate in the absence of classical BAT recruitment in preclinical models and man (52, 62, 66). Specifically, the administration of glucagon increases metabolic rate in adult humans without activating brown fat (52), and in experimental models (pigs and dogs) without functional BAT (62, 66). In addition, glucagon treatment increases energy expenditure in UCP1 knockout mice and in BAT-specific glucagon receptor null mice (104). These findings have led to the suggestion that glucagon may not require brown fat to augment metabolic rate. However, classical brown adipocytes derive from a distinct progenitor cell [myogen factor 5 positive (Myf5-positive)] from that of most beige adipocytes (Myf5-negative) (110). Thus, although glucagon may not require brown fat per se, it may still leverage the thermogenic capacity of beige adipose tissue. Indeed, the GCGR is expressed in WAT, and it is noteworthy that glucagon stimulates lipid breakdown, fatty acid oxidation, and induces oxygen consumption rates in WAT explants, and it promotes the expression of thermogenic genes in WAT (108, 111). Together, these findings lead to the intriguing hypothesis that glucagon augments metabolic rate by promoting the browning of WAT and the induction of UCP1-independent metabolic futile cycles (see Figure 2).
The primary objective of an effective weight loss program is to deliver clinically meaningful weight loss over the long-term (112). To achieve this goal there is a need to target both sides of the energy balance equation to reduce energy intake and increase caloric expenditure (42). Over the past 20 years, the impressive weight loss induced by bariatric surgery (113), the effectiveness of the GLP-1R agonist drug class (43, 44), and the emerging benefits of dual GIPR/GLP-1R agonism (114) have helped fuel major interest in understanding how the periphery of the body communicates with the brain to suppress appetite (113). Together, this has led to an increase in the number of potential anorectic agents (e.g., analogues of PYY, GDF-15, Amylin, Amylin/Calcitonin dual agonists, etc.) under investigation for the treatment of obesity and T2D (44). By contrast, despite seminal discoveries in the field of adipocyte bioenergetics in particular (73, 89, 109), there are currently no effective thermogenic-based medications approved for the treatment of obesity. Here, we have presented GCGR activation as a potential solution to this unmet therapeutic need. Firstly, GCGR agonism rapidly activates energy expenditure in adult humans, and it increases thermogenic capacity and metabolic rate to drive weight loss in preclinical models of obesity. Secondly, glucagon targets several key metabolic organs to mediate its whole-body thermogenic activity. And finally, activation of the GCGR both increases the magnitude and duration of the weight loss achieved by selective GLP-1R and dual GIPR/GLP-1R agonism in rodents. Thus, it is anticipated that GCGR activation may become a trail blazer in the field of thermogenic therapeutics, not only enhancing the weight loss profile of current therapies, but also that of other anti-obesity medications that function by reducing daily caloric intake.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
All authors are current or past employees of Eli Lilly and Company.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Ana Bueno, Matthew Coghlan, Julie Moyers, and Francis Willard for review and helpful discussion of the manuscript.
1. Jelinek LJ, Lok S, Rosenberg GB, Smith RA, Grant FJ, Biggs S, et al. Expression Cloning and Signaling Properties of the Rat Glucagon Receptor. Science (1993) 259(5101):1614–6. doi: 10.1126/science.8384375
2. Lok S, Kuijper JL, Jelinek LJ, Kramer JM, Whitmore TE, Sprecher CA, et al. The Human Glucagon Receptor Encoding Gene: Structure, Cdna Sequence and Chromosomal Localization. Gene (1994) 140(2):203–9. doi: 10.1016/0378-1119(94)90545-2
3. Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol Pharmacol (2003) 63(6):1256–72. doi: 10.1124/mol.63.6.1256
4. Ishihara T, Nakamura S, Kaziro Y, Takahashi T, Takahashi K, Nagata S. Molecular Cloning and Expression of a Cdna Encoding the Secretin Receptor. EMBO J (1991) 10(7):1635–41. doi: 10.1002/j.1460-2075.1991.tb07686.x
5. Svoboda M, Tastenoy M, Vertongen P, Robberecht P. Relative Quantitative Analysis of Glucagon Receptor Mrna in Rat Tissues. Mol Cell Endocrinol (1994) 105(2):131–7. doi: 10.1016/0303-7207(94)90162-7
6. Rodbell M, Krans HM, Pohl SL, Birnbaumer L. The Glucagon-Sensitive Adenyl Cyclase System in Plasma Membranes of Rat Liver. IV. Effects of Guanylnucleotides on Binding of 125I-Glucagon. J Biol Chem (1971) 246(6):1872–6.
7. Stevenson RW, Steiner KE, Davis MA, Hendrick GK, Williams PE, Lacy WW, et al. Similar Dose Responsiveness of Hepatic Glycogenolysis and Gluconeogenesis to Glucagon In Vivo. Diabetes (1987) 36(3):382–9. doi: 10.2337/diab.36.3.382
8. Scheen AJ, Paquot N, Lefèbvre PJ. Investigational Glucagon Receptor Antagonists in Phase I and II Clinical Trials for Diabetes. Expert Opin Investig Drugs (2017) 26(12):1373–89. doi: 10.1080/13543784.2017.1395020
9. Unger RH. Letter: Glucagon in Pathogenesis of Diabetes. Lancet (1975) 1(7914):1036–2. doi: 10.1016/S0140-6736(75)91985-6
10. Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. Documentation of Hyperglucagonemia Throughout the Day in Nonobese and Obese Patients With Noninsulin-Dependent Diabetes Mellitus. J Clin Endocrinol Metab (1987) 64(1):106–10. doi: 10.1210/jcem-64-1-106
11. Parker JC, Andrews KM, Allen MR, Stock JL, McNeish JD. Glycemic Control in Mice With Targeted Disruption of the Glucagon Receptor Gene. Biochem Biophys Res Commun (2002) 290(2):839–43. doi: 10.1006/bbrc.2001.6265
12. Gelling RW, Du XQ, Dichmann DS, Romer J, Huang H, Cui L, et al. Lower Blood Glucose, Hyperglucagonemia, and Pancreatic Alpha Cell Hyperplasia in Glucagon Receptor Knockout Mice. Proc Natl Acad Sci USA. (2003) 100(3):1438–43. doi: 10.1073/pnas.0237106100
13. Sloop KW, Cao JX, Siesky AM, Zhang HY, Bodenmiller DM, Cox AL, et al. Hepatic and Glucagon-Like Peptide-1-Mediated Reversal of Diabetes by Glucagon Receptor Antisense Oligonucleotide Inhibitors. J Clin Invest (2004) 113(11):1571–81. doi: 10.1172/JCI20911
14. Qureshi SA, Rios Candelore M, Xie D, Yang X, Tota LM, Ding VD, et al. A Novel Glucagon Receptor Antagonist Inhibits Glucagon-Mediated Biological Effects. Diabetes (2004) 53(12):3267–73. doi: 10.2337/diabetes.53.12.3267
15. Brand CL, Rolin B, Jorgensen PN, Svendsen I, Kristensen JS, Holst JJ. Immunoneutralization of Endogenous Glucagon With Monoclonal Glucagon Antibody Normalizes Hyperglycaemia in Moderately Streptozotocin-Diabetic Rats. Diabetologia (1994) 37(10):985–93. doi: 10.1007/BF00400461
16. Scheen AJ, Paquot N, Lefebvre PJ. Investigational Glucagon Receptor Antagonists in Phase I and II Clinical Trials for Diabetes. Expert Opin Investig Drugs (2017) 26(12):1373–89. doi: 10.1080/13543784.2017.1395020
17. Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, et al. A New Glucagon and GLP-1 Co-Agonist Eliminates Obesity in Rodents. Nat Chem Biol (2009) 5(10):749–57. doi: 10.1038/nchembio.209
18. Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, et al. Glucagon-Like Peptide 1/Glucagon Receptor Dual Agonism Reverses Obesity in Mice. Diabetes (2009) 58(10):2258–66. doi: 10.2337/db09-0278
19. Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M, et al. Robust Anti-Obesity and Metabolic Effects of a Dual GLP-1/Glucagon Receptor Peptide Agonist in Rodents and Non-Human Primates. Diabetes Obes Metab (2016) 18(12):1176–90. doi: 10.1111/dom.12735
20. Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, et al. A Rationally Designed Monomeric Peptide Triagonist Corrects Obesity and Diabetes in Rodents. Nat Med (2015) 21(1):27–36. doi: 10.1038/nm.3761
21. Ward ZJ, Bleich SN, Cradock AL, Barrett JL, Giles CM, Flax C, et al. Projected U.S. State-Level Prevalence of Adult Obesity and Severe Obesity. N Engl J Med (2019) 381(25):2440–50. doi: 10.1056/NEJMsa1909301
22. Hruby A, Hu FB. The Epidemiology of Obesity: A Big Picture. PharmacoEconomics (2015) 33(7):673–89. doi: 10.1007/s40273-014-0243-x
23. Tremmel M, Gerdtham U-G, Nilsson PM, Saha S. Economic Burden of Obesity: A Systematic Literature Review. Int J Environ Res Public Health (2017) 14(4):435. doi: 10.3390/ijerph14040435
24. Cawley J, Meyerhoefer C. The Medical Care Costs of Obesity: An Instrumental Variables Approach. J Health Econ (2012) 31(1):219–30. doi: 10.1016/j.jhealeco.2011.10.003
25. Romieu I, Dossus L, Barquera S, Blottière HM, Franks PW, Gunter M, et al. Energy Balance and Obesity: What Are the Main Drivers? Cancer Causes Control: CCC (2017) 28(3):247–58. doi: 10.1007/s10552-017-0869-z
26. Rubino F, Puhl RM, Cummings DE, Eckel RH, Ryan DH, Mechanick JI, et al. Joint International Consensus Statement for Ending Stigma of Obesity. Nat Med (2020) 26(4):485–97. doi: 10.1038/s41591-020-0803-x
27. Müller MJ, Geisler C, Heymsfield SB, Bosy-Westphal A. Recent Advances in Understanding Body Weight Homeostasis in Humans. F1000Research (2018) 7:F1000 Faculty Rev–1025. doi: 10.12688/f1000research.14151.1
28. Blüher M. Obesity: Global Epidemiology and Pathogenesis. Nat Rev Endocrinol (2019) 15(5):288–98. doi: 10.1038/s41574-019-0176-8
29. Loos RJF, Yeo GSH. The Genetics of Obesity: From Discovery to Biology. Nat Rev Genet (2021) 1–14. doi: 10.1038/s41576-021-00414-z
30. Farooqi IS. Defining the Neural Basis of Appetite and Obesity: From Genes to Behaviour. Clin Med (Lond) (2014) 14(3):286–9. doi: 10.7861/clinmedicine.14-3-286
31. Müller TD, Clemmensen C, Finan B, DiMarchi RD, Tschöp MH. Anti-Obesity Therapy: From Rainbow Pills to Polyagonists. Pharmacol Rev (2018) 70(4):712–46. doi: 10.1124/pr.117.014803
32. American Diabetes A. Standards of Medical Care in Diabetes-2020 Abridged for Primary Care Providers. Clin diabetes: Publ Am Diabetes Assoc (2020) 38(1):10–38. doi: 10.2337/cd20-as01
33. Apovian CM, Aronne LJ, Bessesen DH, McDonnell ME, Murad MH, Pagotto U, et al. Pharmacological Management of Obesity: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2015) 100(2):342–62. doi: 10.1210/jc.2014-3415
34. Srivastava G, Apovian CM. Current Pharmacotherapy for Obesity. Nat Rev Endocrinol (2018) 14(1):12–24. doi: 10.1038/nrendo.2017.122
35. Maclean PS, Bergouignan A, Cornier M-A, Jackman MR. Biology’s Response to Dieting: The Impetus for Weight Regain. Am J Physio Regulatory Integr Comp Physiol (2011) 301(3):R581–600. doi: 10.1152/ajpregu.00755.2010
36. Piaggi P. Metabolic Determinants of Weight Gain in Humans. Obes (Silver Spring) (2019) 27(5):691–9. doi: 10.1002/oby.22456
37. Ravussin E, Smith SR, Ferrante AW Jr. Physiology of Energy Expenditure in the Weight-Reduced State. Obes (Silver Spring) (2021) 29 Suppl 1:S31–s38. doi: 10.1002/oby.23095
38. Greenway FL. Physiological Adaptations to Weight Loss and Factors Favouring Weight Regain. Int J Obes (Lond) (2015) 39(8):1188–96. doi: 10.1038/ijo.2015.59
39. Ravussin E, Ryan DH. Energy Expenditure and Weight Control: Is the Biggest Loser the Best Loser? Obes (Silver Spring) (2016) 24(8):1607–8. doi: 10.1002/oby.21586
40. Polidori D, Sanghvi A, Seeley RJ, Hall KD. How Strongly Does Appetite Counter Weight Loss? Quantification of the Feedback Control of Human Energy Intake. Obes (Silver Spring) (2016) 24(11):2289–95. doi: 10.1002/oby.21653
41. Fothergill E, Guo J, Howard L, Kerns JC, Knuth ND, Brychta R, et al. Persistent Metabolic Adaptation 6 Years After “the Biggest Loser” Competition. Obes (Silver Spring) (2016) 24(8):1612–9. doi: 10.1002/oby.21538
42. Hall KD, Heymsfield SB, Kemnitz JW, Klein S, Schoeller DA, Speakman JR. Energy Balance and Its Components: Implications for Body Weight Regulation. Am J Clin Nutr (2012) 95(4):989–94. doi: 10.3945/ajcn.112.036350
43. Müller TD, Finan B, Bloom SR, D'Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-Like Peptide 1 (GLP-1). Mol Metab (2019) 30:72–130. doi: 10.1016/j.molmet.2019.09.010
44. Müller TD, Blüher M, Tschöp MH, DiMarchi RD. Anti-Obesity Drug Discovery: Advances and Challenges. Nat Rev Drug Discov (2021) p:1–23. doi: 10.1038/s41573-021-00337-8
45. Moller DE. Metabolic Disease Drug Discovery- “Hitting the Target” is Easier Said Than Done. Cell Metab (2012) 15(1):19–24. doi: 10.1016/j.cmet.2011.10.012
46. van Eyk HJ, Paiman EHM, Bizino MB, Ijzermans SL, Kleiburg F, Boers TGW, et al. Liraglutide Decreases Energy Expenditure and Does Not Affect the Fat Fraction of Supraclavicular Brown Adipose Tissue in Patients With Type 2 Diabetes. Nutrition Metab Cardiovasc Dis (2020) 30(4):616–24. doi: 10.1016/j.numecd.2019.12.005
47. Ladenheim EE. Liraglutide and Obesity: A Review of the Data So Far. Drug Des Devel Ther (2015) 9:1867–75. doi: 10.2147/DDDT.S58459
48. Flint A, Raben A, Rehfeld JF, Holst JJ, Astrup A. The Effect of Glucagon-Like Peptide-1 on Energy Expenditure and Substrate Metabolism in Humans. Int J Obes Relat Metab Disord (2000) 24(3):288–98. doi: 10.1038/sj.ijo.0801126
49. Penick SB, Hinkle LE Jr. Depression of Food Intake Induced in Healthy Subjects by Glucagon. N Engl J Med (1961) 264:893–7. doi: 10.1056/NEJM196105042641801
50. Schulman JL, Carleton JL, Whitney G, Whitehorn JC. Effect of Glucagon on Food Intake and Body Weight in Man. J Appl Physiol (1957) 11(3):419–21. doi: 10.1152/jappl.1957.11.3.419
51. Nair KS. Hyperglucagonemia Increases Resting Metabolic Rate in Man During Insulin Deficiency. J Clin Endocrinol Metab (1987) 64(5):896–901. doi: 10.1210/jcem-64-5-896
52. Salem V, Izzi-Engbeaya C, Coello C, Thomas DB, Chambers ES, Comninos AN, et al. Glucagon Increases Energy Expenditure Independently of Brown Adipose Tissue Activation in Humans. Diabetes Obes Metab (2016) 18(1):72–81. doi: 10.1111/dom.12585
53. Tan TM, Field BCT, McCullough KA, Troke RC, Chambers ES, Salem V, et al. Coadministration of Glucagon-Like Peptide-1 During Glucagon Infusion in Humans Results in Increased Energy Expenditure and Amelioration of Hyperglycemia. Diabetes (2013) 62(4):1131–8. doi: 10.2337/db12-0797
54. Kim T, Nason S, Holleman C, Pepin M, Wilson L, Berryhill TF, et al. Glucagon Receptor Signaling Regulates Energy Metabolism via Hepatic Farnesoid X Receptor and Fibroblast Growth Factor 21. Diabetes (2018) 67(9):1773–82. doi: 10.2337/db17-1502
55. Kleinert M, Sachs S, Habegger KM, Hofmann SM, Müller TD. Glucagon Regulation of Energy Expenditure. Int J Mol Sci (2019) 20(21):5407. doi: 10.3390/ijms20215407
56. Calles-Escandón J. Insulin Dissociates Hepatic Glucose Cycling and Glucagon-Induced Thermogenesis in Man. Metabolism (1994) 43(8):1000–5. doi: 10.1016/0026-0495(94)90180-5
57. Tan TM, et al. Coadministration of Glucagon-Like Peptide-1 During Glucagon Infusion in Humans Results in Increased Energy Expenditure and Amelioration of Hyperglycemia. Diabetes (2013) 62(4):1131–8. doi: 10.2337/db12-0797
58. Whytock KL, Carnero EA, Vega RB, Tillner J, Bock C, Chivukula K, et al. Prolonged Glucagon Infusion Does Not Affect Energy Expenditure in Individuals With Overweight/Obesity: A Randomized Trial. Obes (Silver Spring) (2021) 29(6):1003–13. doi: 10.1002/oby.23141
59. Davidson IWF, Salter JM, Best CH. Calorigenic Action of Glucagon. Nature (1957) 180(4595):1124–4. doi: 10.1038/1801124a0
60. Davidson IWF, Salter JM, Best CH. The Effect of Glucagon on the Metabolic Rate of Rats. Am J Clin Nutr (1960) 8(5):540–6. doi: 10.1093/ajcn/8.5.540
61. Barre H, Rouanet JL. Calorigenic Effect of Glucagon and Catecholamines in King Penguin Chicks. Am J Physiol (1983) 244(6):R758–63. doi: 10.1152/ajpregu.1983.244.6.R758
62. Ingram DL, Kaciuba-Uscilko H. Metabolic Effects of Glucagon in the Young Pig. Horm Metab Res (1980) 12(9):430–3. doi: 10.1055/s-2007-999167
63. Habegger KM, Stemmer K, Cheng C, Muller TD, Heppner KM, Ottaway N, et al. Fibroblast Growth Factor 21 Mediates Specific Glucagon Actions. Diabetes (2013) 62(5):1453–63. doi: 10.2337/db12-1116
64. Leibel RL, Rosenbaum M, Hirsch J. Changes in Energy Expenditure Resulting From Altered Body Weight. N Engl J Med (1995) 332(10):621–8. doi: 10.1056/NEJM199503093321001
65. Davidson IW, Salter JM, Best CH. Calorigenic Action of Glucagon. Nature (1957) 180(4595):1124. doi: 10.1038/1801124a0
66. Weiser PC, Grande F. Calorigenic Effects of Glucagon and Epinephrine in Anesthetized Dogs. Proc Soc Exp Biol Med (1974) 145(3):912–7. doi: 10.3181/00379727-145-37923
67. Salem V, Izzi-Engbeaya C, Coello C, Thomas DB, Chambers ES, Comninos AN, et al. Glucagon Increases Energy Expenditure Independently of Brown Adipose Tissue Activation in Humans. Diabetes Obes Metab (2016) 18(1):72–81. doi: 10.1111/dom.12585
68. Stahel P, Lee SJ, Sud SK, Floh A, Dash S. Intranasal Glucagon Acutely Increases Energy Expenditure Without Inducing Hyperglycaemia in Overweight/Obese Adults. Diabetes Obes Metab (2019) 21(6):1357–64. doi: 10.1111/dom.13661
69. Billington CJ, Bartness TJ, Briggs J, Levine AS, Morley JE. Glucagon Stimulation of Brown Adipose Tissue Growth and Thermogenesis. Am J Physiol (1987) 252(1 Pt 2):R160–5. doi: 10.1152/ajpregu.1987.252.1.R160
70. Ambery P, Parker VE, Stumvoll M, Posch MG, Heise T, Plum-Moerschel L, et al. MEDI0382, a GLP-1 and Glucagon Receptor Dual Agonist, in Obese or Overweight Patients With Type 2 Diabetes: A Randomised, Controlled, Double-Blind, Ascending Dose and Phase 2a Study. Lancet (2018) 391(10140):2607–18. doi: 10.1016/S0140-6736(18)30726-8
71. Di Prospero NA, Yee J, Frustaci ME, Samtani MN, Alba M, Fleck P. Efficacy and Safety of Glucagon-Like Peptide-1/Glucagon Receptor Co-Agonist JNJ-64565111 in Individuals With Type 2 Diabetes Mellitus and Obesity: A Randomized Dose-Ranging Study. Clin Obes (2021) 11(2):e12433. doi: 10.1111/cob.12433
72. Bossart M, Wagner M, Elvert R, Evers A, Hübschle T, Kloeckener T, et al. Effects on Weight Loss and Glycemic Control With SAR441255, a Potent Unimolecular Peptide GLP-1/GIP/GCG Receptor Triagonist. Cell Metab (2022) 34(1):59–74.e10. doi: 10.1016/j.cmet.2021.12.005
73. Nedergaard J, Cannon B. The Changed Metabolic World With Human Brown Adipose Tissue: Therapeutic Visions. Cell Metab (2010) 11(4):268–72. doi: 10.1016/j.cmet.2010.03.007
74. Rui L. Energy Metabolism in the Liver. Compr Physiol (2014) 4(1):177–97. doi: 10.1002/cphy.c130024
75. Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr Rev (2019) 40(5):1367–93. doi: 10.1210/er.2019-00034
76. Goedeke L, Shulman GI. Therapeutic Potential of Mitochondrial Uncouplers for the Treatment of Metabolic Associated Fatty Liver Disease and NASH. Mol Metab (2021) 46:101178. doi: 10.1016/j.molmet.2021.101178
77. Löffler MC, Betz MJ, Blondin DP, Augustin R, Sharma AK, Tseng YH, et al. Challenges in Tackling Energy Expenditure as Obesity Therapy: From Preclinical Models to Clinical Application. Mol Metab (2021) 51:101237. doi: 10.1016/j.molmet.2021.101237
78. Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschöp MH. The Metabolic Actions of Glucagon Revisited. Nat Rev Endocrinol (2010) 6(12):689–97. doi: 10.1038/nrendo.2010.187
79. Nason SR, Kim T, Antipenko JP, Finan B, DiMarchi R, Hunter CS, et al. Glucagon-Receptor Signaling Reverses Hepatic Steatosis Independent of Leptin Receptor Expression. Endocrinology (2020) 161(1). doi: 10.1210/endocr/bqz013
80. Breton L, Clot JP, Baudry M. Effects of Glucagon on Basal Metabolic Rate and Oxidative Phosphorylation of Rat Liver Mitochondria. Horm Metab Res (1983) 15(9):429–32. doi: 10.1055/s-2007-1018747
81. Nason SR, Antipenko J, Presedo N, Cunningham SE, Pierre TH, Kim T, et al. Glucagon Receptor Signaling Regulates Weight Loss via Central KLB Receptor Complexes. JCI Insight (2021) 6(4). doi: 10.1172/jci.insight.141323
82. Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile Acids Induce Energy Expenditure by Promoting Intracellular Thyroid Hormone Activation. Nature (2006) 439(7075):484–9. doi: 10.1038/nature04330
83. Cariou B, Bouchaert E, Abdelkarim M, Dumont J, Caron S, Fruchart JC, et al. FXR-Deficiency Confers Increased Susceptibility to Torpor. FEBS Lett (2007) 581(27):5191–8. doi: 10.1016/j.febslet.2007.09.064
84. Arafat AM, Kaczmarek P, Skrzypski M, Pruszyńska-Oszmalek E, Kołodziejski P, Szczepankiewicz D, et al. Glucagon Increases Circulating Fibroblast Growth Factor 21 Independently of Endogenous Insulin Levels: A Novel Mechanism of Glucagon-Stimulated Lipolysis? Diabetologia (2013) 56(3):588–97. doi: 10.1007/s00125-012-2803-y
85. Lewis JE, Ebling FJP, Samms RJ, Tsintzas K. Going Back to the Biology of FGF21: New Insights. Trends Endocrinol Metab (2019) 30(8):491–504. doi: 10.1016/j.tem.2019.05.007
86. Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, et al. FGF21 Regulates PGC-1α and Browning of White Adipose Tissues in Adaptive Thermogenesis. Genes Dev (2012) 26(3):271–81. doi: 10.1101/gad.177857.111
87. Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, et al. FGF21 Acts Centrally to Induce Sympathetic Nerve Activity, Energy Expenditure, and Weight Loss. Cell Metab (2014) 20(4):670–7. doi: 10.1016/j.cmet.2014.07.012
88. Véniant MM, Sivits G, Helmering J, Komorowski R, Lee J, Fan W, et al. Pharmacologic Effects of FGF21 Are Independent of the “Browning” of White Adipose Tissue. Cell Metab (2015) 21(5):731–8. doi: 10.1016/j.cmet.2015.04.019
89. Chouchani ET, Kazak L, Spiegelman BM. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab (2019) 29(1):27–37. doi: 10.1016/j.cmet.2018.11.002
90. Geisler JG. Targeting Energy Expenditure via Fuel Switching and Beyond. Diabetologia (2011) 54(2):237–44. doi: 10.1007/s00125-010-1932-4
91. Rosen ED, Spiegelman BM. What We Talk About When We Talk About Fat. Cell (2014) 156(1-2):20–44. doi: 10.1016/j.cell.2013.12.012
92. Cannon B, Nedergaard J. Brown Adipose Tissue: Function and Physiological Significance. Physiol Rev (2004) 84(1):277–359. doi: 10.1152/physrev.00015.2003
93. Ikeda K, Maretich P, Kajimura S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol Metab (2018) 29(3):191–200. doi: 10.1016/j.tem.2018.01.001
94. Kusminski CM, Bickel PE, Scherer PE. Targeting Adipose Tissue in the Treatment of Obesity-Associated Diabetes. Nat Rev Drug Discov (2016) 15(9):639–60. doi: 10.1038/nrd.2016.75
95. Becher T, Palanisamy S, Kramer DJ, Eljalby M, Marx SJ, Wibmer AG, et al. Brown Adipose Tissue Is Associated With Cardiometabolic Health. Nat Med (2021). doi: 10.1101/2020.02.08.933754
96. Sidossis L, Kajimura S. Brown and Beige Fat in Humans: Thermogenic Adipocytes That Control Energy and Glucose Homeostasis. J Clin Invest (2015) 125(2):478–86. doi: 10.1172/JCI78362
97. Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, et al. Brown Adipose Tissue Activity Controls Triglyceride Clearance. Nat Med (2011) 17(2):200–5. doi: 10.1038/nm.2297
98. Yoneshiro T, Wang Q, Tajima K, Matsushita M, Maki H, Igarashi K, et al. BCAA Catabolism in Brown Fat Controls Energy Homeostasis Through SLC25A44. Nature (2019) 572(7771):614–9. doi: 10.1038/s41586-019-1503-x
99. Becher T, Palanisamy S, Kramer DJ, Eljalby M, Marx SJ, Wibmer AG, et al. Brown Adipose Tissue Is Associated With Cardiometabolic Health. Nat Med (2021) 27(1):58–65. doi: 10.1038/s41591-020-1126-7
100. Chen KY, Brychta RJ, Abdul Sater Z, Cassimatis TM, Cero C, Fletcher LA, et al. Opportunities and Challenges in the Therapeutic Activation of Human Energy Expenditure and Thermogenesis to Manage Obesity. J Biol Chem (2020) 295(7):1926–42. doi: 10.1074/jbc.REV119.007363
101. Joel CD. Stimulation of Metabolism of Rat Brown Adipose Tissue by Addition of Lipolytic Hormones In Vitro. J Biol Chem (1966) 241(4):814–21. doi: 10.1016/S0021-9258(18)96838-9
102. Cockburn F, Hull D, Walton I. The Effect of Lipolytic Hormones and Theophylline on Heat Production in Brown Adipose Tissue In Vivo. Br J Pharmacol Chemother (1967) 31(3):568–77. doi: 10.1111/j.1476-5381.1967.tb00421.x
103. Heim T, Hull D. The Effect of Propranalol on the Calorigenic Response in Brown Adipose Tissue of New-Born Rabbits to Catecholamines, Glucagon, Corticotrophin and Cold Exposure. J Physiol (1966) 187(2):271–83. doi: 10.1113/jphysiol.1966.sp008088
104. Beaudry JL, Kaur KD, Varin EM, Baggio LL, Cao X, Mulvihill EE, et al. The Brown Adipose Tissue Glucagon Receptor Is Functional But Not Essential for Control of Energy Homeostasis in Mice. Mol Metab (2019) 22:37–48. doi: 10.1016/j.molmet.2019.01.011
105. Kinoshita K, Ozaki N, Takagi Y, Murata Y, Oshida Y, Hayashi Y. Glucagon is Essential for Adaptive Thermogenesis in Brown Adipose Tissue. Endocrinology (2014) 155(9):3484–92. doi: 10.1210/en.2014-1175
106. Yahata T, Habara Y, Kuroshima A. Effects of Glucagon and Noradrenaline on the Blood Flow Through Brown Adipose Tissue in Temperature-Acclimated Rats. Jpn J Physiol (1983) 33(3):367–76. doi: 10.2170/jjphysiol.33.367
107. Fedorenko A, Lishko PV, Kirichok Y. Mechanism of Fatty-Acid-Dependent UCP1 Uncoupling in Brown Fat Mitochondria. Cell (2012) 151(2):400–13. doi: 10.1016/j.cell.2012.09.010
108. Townsend LK, Medak KD, Knuth CM, Peppler WT, Charron MJ, Wright DC. Loss of Glucagon Signaling Alters White Adipose Tissue Browning. FASEB J (2019) 33(4):4824–35. doi: 10.1096/fj.201802048RR
109. Wu J, Cohen P, Spiegelman BM. Adaptive Thermogenesis in Adipocytes: Is Beige the New Brown? Genes Dev (2013) 27(3):234–50. doi: 10.1101/gad.211649.112
110. Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, et al. PRDM16 Controls a Brown Fat/Skeletal Muscle Switch. Nature (2008) 454(7207):961–7. doi: 10.1038/nature07182
111. Hagen JH. Effect of Glucagon on the Metabolism of Adipose Tissue. J Biol Chem (1961) 236:1023–7. doi: 10.1016/S0021-9258(18)64236-X
112. 8. Obesity Management for the Treatment of Type 2 Diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care (2021) 44(Suppl 1):S100–s110. doi: 10.2337/dc21-S008
113. Gimeno RE, Briere DA, Seeley RJ. Leveraging the Gut to Treat Metabolic Disease. Cell Metab (2020) 31(4):679–98. doi: 10.1016/j.cmet.2020.02.014
Keywords: glucagon-receptor (GCGR), G protein-coupled receptor (GPCR), energy balance, obesity, weight loss
Citation: Conceição-Furber E, Coskun T, Sloop KW and Samms RJ (2022) Is Glucagon Receptor Activation the Thermogenic Solution for Treating Obesity? Front. Endocrinol. 13:868037. doi: 10.3389/fendo.2022.868037
Received: 02 February 2022; Accepted: 07 March 2022;
Published: 25 April 2022.
Edited by:
Aylin Carla Hanyaloglu, Imperial College London, United KingdomReviewed by:
Richard Ijzerman, VU Medical Center, NetherlandsCopyright © 2022 Conceição-Furber, Coskun, Sloop and Samms. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ricardo J. Samms, c2FtbXNfcmljYXJkb19qQGxpbGx5LmNvbQ==; Kyle W. Sloop, c2xvb3Bfa3lsZV93QGxpbGx5LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.