 Iga Zendran
Iga Zendran Gabriela Gut
Gabriela Gut Marcin Kałużny
Marcin Kałużny Katarzyna Zawadzka1
Katarzyna Zawadzka1 Marek Bolanowski
Marek Bolanowski
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 09 June 2022
Sec. Pituitary Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.867965
This article is part of the Research TopicFunctional Neuroendocrine TumorsView all 11 articles
Introduction: Ectopic acromegaly is a rare condition caused most frequently by growth hormone releasing hormone (GHRH) secretion from neuroendocrine tumors. The diagnosis is often difficult to establish as its main symptoms do not differ from those of acromegaly of pituitary origin.
Objectives: To determine most common clinical features and diagnostic challenges in ectopic acromegaly.
Patients and Methods: A search for ectopic acromegaly cases available in literature was performed using PubMed, Cochrane, and MEDline database. In this article, 127 cases of ectopic acromegaly described after GHRH isolation in 1982 are comprehensively reviewed, along with a summary of current state of knowledge on its clinical features, diagnostic methods, and treatment modalities. The most important data were compiled and compared in the tables.
Results: Neuroendocrine tumors were confirmed in 119 out of 121 patients with histopathological evaluation, mostly of lung and pancreatic origin. Clinical manifestation comprise symptoms associated with pituitary hyperplasia, such as headache or visual field disturbances, as well as typical signs of acromegaly. Other endocrinopathies may also be present depending on the tumor type. Definitive diagnosis of ectopic acromegaly requires confirmation of GHRH secretion from a tumor using either histopathological methods or GHRH plasma concentration assessment. Hormonal evaluation was available for 84 patients (66%) and histopathological confirmation for 99 cases (78%). Complete tumor resection was the main treatment method for most patients as it is a treatment of choice due to its highest effectiveness. When not feasible, somatostatin receptor ligands (SRL) therapy is the preferred treatment option. Prognosis is relatively favorable for neuroendocrine GHRH-secreting tumors with high survival rate.
Conclusion: Although ectopic acromegaly remains a rare disease, one should be aware of it as a possible differential diagnosis in patients presenting with additional symptoms or those not responding to classic treatment of acromegaly.
Acromegaly is a rare disease with prevalence ranging between 2.8 and 13.7/100,000 persons, mostly caused by a benign pituitary growth hormone (GH) secreting adenoma (1). The term ‘ectopic acromegaly’ refers to a syndrome caused by secretion of growth hormone releasing hormone (GHRH), or occasionally, by an extra-pituitary source of GH and accounts for less than 1% of all acromegaly cases (2, 3). Ectopic GHRH derives most commonly from functional neuroendocrine tumors (NETs) originating in the lung or the pancreas and results in pituitary hyperplasia and excess GH secretion (4). The term ‘ectopic’ is used in a broader sense in this review, applying not only to its most common meaning of an abnormal localization, but also basically to the secretion of a hormone by a tissue that does not produce it under physiological circumstances (5). Therefore, GHRH-secreting pituitary gangliocytoma will also be included in the review, as they fall under given definition (6). Ectopic acromegaly clinical features are indistinguishable from those of acromegaly caused by a pituitary somatotropinoma (7). The suspicion of ectopic acromegaly is most commonly drawn when no discrete adenoma is shown in magnetic resonance (MR) imaging of the pituitary gland. Additional manifestations of an extracranial tumor, such as cough or dyspnea for the lung neoplasm or other endocrinopathies for a pancreatic NET, as well as lack of remission of the disease after transsphenoidal adenomectomy may also be suggestive of the disease (3, 8).
An extensive search was performed for ectopic acromegaly cases described in literature between 1982 and 2021. Research was conducted in PubMed, Cochrane, and MEDline databases using the particular keywords acromegaly, GHRH, ectopic, and neuroendocrine tumors. More than 300 articles were screened. Duplicates, unusable records, and those with partial information were rejected. According to the specific criteria, 127 cases were selected containing confirmation of ectopic GHRH secretion by biochemical and/or histopathological examination, some of them described as a part of case series. GHRH-secreting pituitary gangliocytoma was also included.
Since the initial report of the possible association between the neuroendocrine functional tumor and acromegaly, suggested by Altmann et al. in 1959, around 170 cases of suspected ectopic acromegaly have been described in literature to date, mostly as case reports (4, 6, 9–21). To our best knowledge, only 19 cases were described as caused by an ectopic source of GH (22–27). Consequently, the vast majority of the cases were reports of acromegaly due to ectopic GHRH secretion. However, until 1982, when the isolation of GHRH from pancreatic tumors was achieved simultaneously by two research groups, the underlying cause of ectopic acromegaly could only have been suspected (28, 29). To date, 10 cases of possible GHRH secretion by neuroendocrine neoplasms have been reported (7 of the lung, 2 of the pancreas, and one of the foregut origin) and will not be covered in this review (4). Also, 13 cases described between 1984 and 2002 and mentioned in previous reviews will also not be included here due to the lack of records. In this review, 127 cases of acromegaly caused by ectopic GHRH secretion confirmed by biochemical or histopathological examination will be discussed, comprehensively summarizing current knowledge on this rare condition.
Acromegaly due to ectopic GHRH secretion is more common in women who represent 70% of reported cases (Table 1). Patients ranged in age from 14 to 77, with mean age of 43.3 years at the time of diagnosis. The age distribution was comparable for men and women, with mean age of 43.9 and 41.7 years, respectively. The duration of the disease before diagnosis was approximately 7.4 years, which is consistent with the course of acromegaly caused by somatotropinoma (30, 31). Although the age at diagnosis was similar for both men and women, there has been a sex disparity from the time of the onset of symptoms until diagnosis. The diagnostic delay was longer in men by approximately 3.5 years with mean duration of 9.9 years in comparison to 6.2 years in women, which is also in line with pituitary acromegaly characteristics (32).
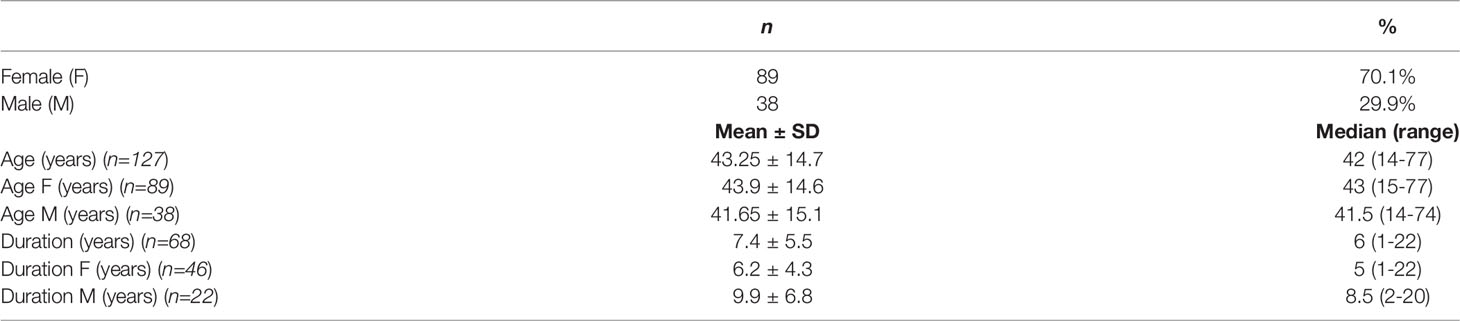
Table 1 Clinical features of 127 patients with GHRH-secreting tumors.
Extracranial tumors constituted 78.7% of the cases and the remaining 27 cases presented with GHRH-secreting tumors within the sellar region are shown in Table 2. Histopathological evaluation was available for 121 tumors, indicating that 119 of them were neuroendocrine tumors with only 2 exceptions for a pituitary diffuse large B-cell lymphoma (DLBCL) and an adenoid cystic carcinoma of the lung (18, 33). Most common histopathological assessment for tumors of the sellar region was mixed gangliocytoma-pituitary adenoma, representing 77.8% of all intracranial cases. As for the extracranial tumors, the majority of them originated in the lung and pancreas (50% and 35%, respectively), with typical bronchial carcinoid as the most common histopathological diagnosis. Albeit rare, GHRH secretion in other tumors, including pheochromocytoma, lymphoma, paraganglioma, or thymoma has also been reported (4, 11, 15, 34–37). In some cases, tumor resection preceded possible acromegaly development (37–39). According to gross pathology data available for 72 patients, extracranial tumors measured 6.6 cm on average, ranging from 1 to 25 cm. Consequently, most of them were apparent in conventional imaging, with an estimated 86% sensitivity of computed tomography (CT) scan described in the French series of 21 cases (40). The largest pituitary mass found in a case of a mixed gangliocytoma-pituitary adenoma measured 6.5 cm (41). In a few cases, multiple pancreatic tumors were identified (13, 42–44). Gangliocytomas may present a characteristic MRI appearance. Usually these tumors consist of cystic and solid components. Gangliocytomas are hypo- to isointense relative to cortex on T1-weighted images and hyperintense on T2/FLAIR images. The solid portions of the tumors show variable enhancement after gadolinium administration: from none to striking homogeneous enhancement. About one-third of cases contain calcifications, which can be seen on susceptibility weighted imaging (SWI) sequence as low signal structures. There are no signs of hemorrhage or necrosis within the tumor, which may be present in common somatotropinomas. Gangliocytomas can occur anywhere in the central nervous system, however the most common and typical location is the temporal lobe. Other reported sites include the brainstem, sellar region, and spinal cord. At diagnosis, lymph node or distant metastases were present in 42 patients with liver, bones, and lung as the most frequent metastatic site. Presence of metastatic cancerous cells was also reported in the breast, spleen, central nervous system, or heart tissue in isolated cases (33, 40, 45–48). Since lung and pancreas tumors constituted the majority of GHRH-secreting tumors, an additional comparison of their main features and treatment results has been performed in Table 3, indicating that pancreatic tumors were associated with multiple endocrine neoplasia 1 (MEN 1) syndrome in over half of the cases and were more inclined to produce other hormones, especially insulin, gastrin, and pancreatic polypeptide (40, 42, 44, 49–52).

Table 2 Origin of the tumor.
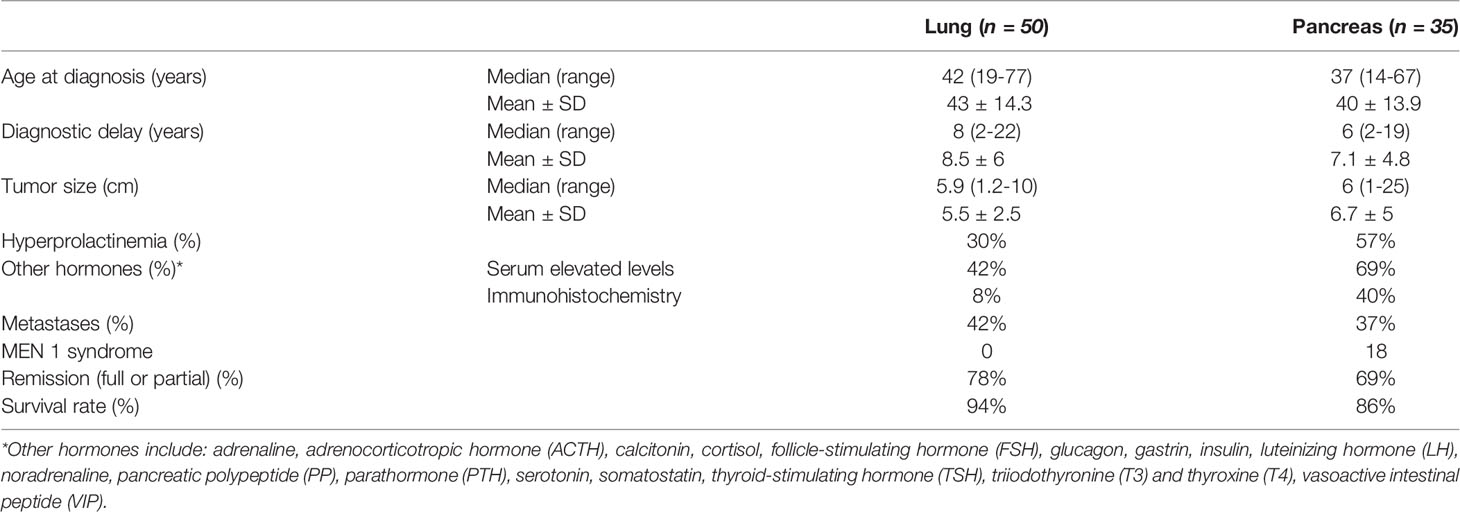
Table 3 Lung and pancreatic tumor comparison.
Overt acromegaly at different stages was presented by 124 patients, ranging from mild acral enlargement to advanced metabolic complications such as hypertension, diabetes, or hyperparathyroidism, considerably reducing the quality of life (53). Acromegaly symptoms did not differ from those of the classic form of the disease (30, 31, 54). In almost half of the cases, elevated levels of other hormones were observed (Table 3). Hyperprolactinemia was the most common symptom associated endocrinopathy which was documented in 44 patients (34.7%) and though usually asymptomatic, it manifested with amenorrhea-galactorrhea syndrome in some patients (36, 55–62). Prolactin hypersecretion derived rather from GHRH-induced pituitary hyperplasia or stalk compression than from the ectopic tumor itself, as its expression was documented immunohistochemically only in intracranial tumor cases. Elevated prolactin levels occur more often in patients with acromegaly caused by an ectopic GHRH source than in those with pituitary acromegaly (30). Less common manifestations included diabetes insipidus (63), Zollinger-Ellison syndrome (49, 51), Cushing syndrome (37, 59, 64), carcinoid syndrome (55), and typical pheochromocytoma symptoms (34), as appropriate for corresponding tumors. MEN 1 syndrome was highly probable in 23 patients based on clinical features. Genetic confirmation of MEN1 mutation was available for 19 tumors, including 18 pancreatic NETs and one thymic carcinoid (13, 36, 40, 42–44, 52, 57, 61, 65, 66). In 4 patients, genetic testing had not been performed. Apart from acromegaly and other features caused by pancreatic tumors, the syndromes included hyperparathyroidism in almost all MEN1 cases (13, 36, 40, 42–44, 52, 57, 61, 65), as well as gonadotroph (42) or mixed PRL-GH (40) secreting pituitary adenomas. A null cell pituitary tumor was also detected in one case (65). Some patients suffered from visual field disturbances and severe headaches due to large masses located in the sellar region, mostly macroadenoma or somatotroph hyperplasia (41, 47, 59, 67–72). In 3 cases, acromegaly signs have not been observed despite elevated growth hormone levels (37–39). Taking the typical long course of acromegaly into consideration, it is possible that the disease was diagnosed in its initial stages based on hormone and imaging results, enabling prompt and effective treatment. In some patients, acromegaly symptoms manifested for a long time after surgical treatment of an initially asymptomatic bronchial or pancreatic tumor, due to relapse or remaining metastasis. The longest latency period in these cases lasted as many as 30 years (42, 43, 45, 47, 48, 73–75).
The most described symptom in cases at preliminary diagnosis was acromegaly, recognized by clinical signs and confirmed with GH level not suppressed after oral glucose tolerance test (OGTT) and/or elevated insulin-like growth factor 1 (IGF-1) concentration. Ectopic source should be suspected when no improvement is observed after pituitary surgery, which was the case in 21 patients (17, 19, 35, 40, 42, 56, 57, 61, 66, 73, 76–82). Other findings leading to ectopic GHRH secretion diagnosis were acromegalic features without any pituitary lesion in magnetic resonance imaging or conversely, acromegaly in the setting of a previously known non-pituitary tumor, sometimes accompanied by other manifestations mentioned above (20.5% and 39.3% of the patients, respectively). In 24 cases of intracranial tumors, ectopic acromegaly was not suspected until the histopathological examination revealed unphysiological expression of GHRH, mostly by gangliocytoma cells (6). Diagnostic criteria of acromegaly, due to GHRH-producing tumor, are met when hypersecretion of GHRH is substantiated in a patient with overt acromegaly and recovery following the resection of corresponding tumor is observed (3, 7). GHRH tumoral secretion can be proved by measuring its plasmatic levels and by positive immunostaining or radioimmunoassay with antibodies anti-GHRH 1-40 and anti-GHRH 1-44 in histopathological examination (3, 83). However, though useful,the immunohistochemical technique is not widely available due to limited access to reagents and tumor tissue which may not always be obtained in the required amount (19). Other less common laboratory methods include hormone extraction (50, 56, 76), measurement of arterio-venous gradient of GHRH across tumoral tissue (57, 84), high performance liquid chromatography (HPLC), or ion exchange chromatography (73, 85) and GHRH mRNA detection with polymerase chain reaction (PCR) (63, 86, 87). Before GHRH isolation in 1982, its possible secretion was tested mostly with bioassay, which is an indirect examination based on GH production by rat pituitary cells triggered by the substance obtained from tumor extracts (88). This method may still serve as an additional test and was recently performed on cultured human pituitary cells (16, 60). As shown in Table 4, GHRH expression was documented histopathologically in 99 tumors, mostly by means of immunostaining which was performed in 83 cases. Although GHRH expression by various tumors may be considered more often than it was initially believed, only a small number of patients present overt clinical acromegaly. In vitro tests with immunohistochemistry (IHC) and radioimmunoassay (RIA) using anti-GHRH antibodies revealed GHRH expression in up to 14% and 56% (IHC and RIA, respectively) of all type tumors, mostly small cell lung carcinoma, breast carcinoma, and pheochromocytoma, however, acromegalic features were rarely observed (8, 30, 89). This may be due to different reasons, beginning with the possibility that produced GHRH is either not released from the tumor or its amount is not enough to stimulate pituitary cells to produce GH. Moreover, ectopic GHRH activity might be reduced due to abnormal chemical structure or concurrent secretion of somatostatin from tumoral tissue (4, 81). Positive immunostaining for hormones other than GHRH was documented in 47 neoplasms; 23 of them were intracranial tumors, mostly mixed gangliocytoma-pituitary adenoma positive for GH and sometimes also PRL. Somatostatin expression has been found in both intra- and extracranial tumor tissues (6 and 9 documented cases, respectively). Other marked substances included ACTH, calcitonin, insulin, gastrin, glucagon, PP, serotonin, and VIP, produced one or more at a time by several tumors (16, 33, 40, 77, 90). As above mentioned, tumoral hormone expression has not always translated to elevated plasmatic levels, let alone the symptoms.

Table 4 Diagnostic methods used to confirm GHRH-induced acromegaly.
GHRH plasma levels were available for 84 patients with a median concentration of 1,273 ng/L, as shown in Table 5, along with other growth hormone axis levels. The cutoff suggesting ectopic acromegaly (triggered by GHRH ectopic secretion) is a GHRH level of 300 ng/L according to Scheithauer et al. (91). However, in 2012 Garby et al. proposed the threshold of 250 ng/L as a highly specific marker of an ectopic release of GHRH causing acromegaly based on a series of 21 cases (40). In reviewed cases, 76 out of 81 patients with data available in SI values (93.8%) had GHRH levels above the 250 ng/L cutoff value. Elevated GHRH plasma level (especially >250-300 ng/L), may enable differentiation between ectopic and eutopic acromegaly which usually presents with undetectable GHRH serum concentration (10). Interestingly, in patients with hypothalamic GHRH-secreting tumors, plasma level of this hormone is also low (92, 93). The same laboratory criteria are used to diagnose both pituitary and ectopic acromegaly, namely GH hypersecretion, lack of suppression of GH levels during oral glucose tolerance test (OGTT), and elevated IGF-1 levels (3, 7) and values are shown in Table 5. It has been described that ectopic acromegaly is more frequently related with paradoxical serum GH rise (>50% of baseline) after TRH or glucose administration. Another noticed difference was that after GHRH administration, GH rise was attenuated in ectopic acromegaly (7, 30). The most popular commercially available GHRH tests are the enzyme-linked immunosorbent assays (ELISA), which use highly specific antibody-antigen interactions. There are GHRH tests with different sensitivities available for various types of samples. ELISA is the gold standard of immunoassays. Another sensitive but less commercial method of quantifying GHRH in liquid sample is RIA, a method based on radiolabeled antibodies (I125). Some examples for the most commercially available ELISA kits are: Human GHRH ELISA Kit (sandwich ELISA), detecting GHRH in plasma, tissue homogenates, and other biology fluids, with sensitivity < 0.75 pg/mL (Antibodies.com8, United Kingdom; Biorbyt, United Kingdom). Human GHRH ELISA Kit (sandwich ELISA), detecting GHRH in serum, plasma and other biological fluids with sensivity 9.38 pg/mL (Novus Biologicals, LLC, USA; LSBio, USA; Assaygenie, Ireland). Human GHRH ELISA Kit (sandwich ELISA), detecting GHRH in serum, plasma, cell culture supernatants, body fluid, and tissue homogenate with sensitivity 1.0 pg/mL (MyBioSource, Inc.USA). Taking into consideration the poor accessibility of GHRH plasma measurement in some areas of the world, the most common side effect, and relatively large dimensions of the related extracranial tumors, the authors suggest that a chest radiograph and an abdomen ultrasound should be performed in every acromegaly case as a minimum diagnostic procedures in order not to overlook a source of an ectopic production. In some cases abdomen and chest CT should be considered, especially in cases of unequivocal acromegaly diagnosis and the absence of a pituitary tumor in MR imaging.
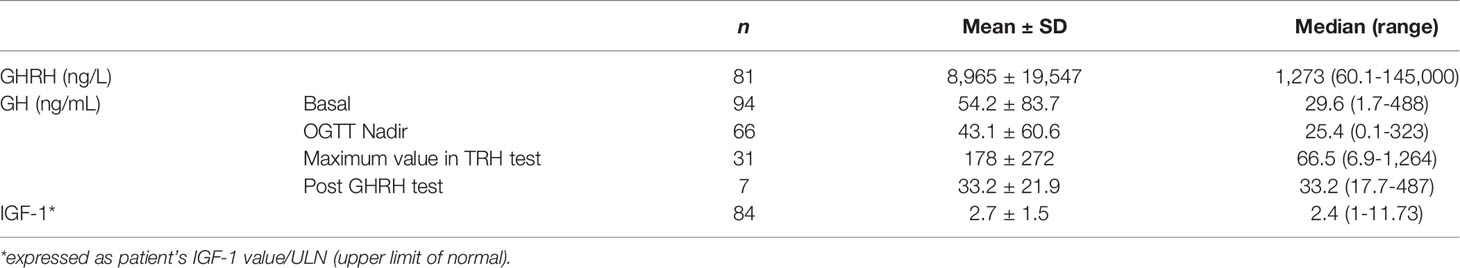
Table 5 Serum/plasma concentrations of hormones in the hypothalamic–pituitary–somatotropic axis (HPS axis) in patients with ectopic acromegaly.
Pituitary imaging was available for 114 patients (89.8%), with MR imaging being the most commonly used method. As shown in Tables 6, 7, its interpretation might be difficult and misleading in GHRH-induced acromegaly. Exposure to GHRH hypersecretion often leads to pituitary somatotroph hyperplasia, which is considered to be a characteristic feature of ectopic acromegaly. Apart from this, somatotroph hyperplasia has only been found in patients with McCune-Albright syndrome (4). However, studies show that pituitary adenoma may also occur in the course of ectopic GHRH secretion when the exposure is prolonged which was proved in laboratory transgenic mice (94). Although rare, such cases have also been described in patients, however it has not been proved that the adenoma was due to GHRH secretion and not incidental (40, 65, 77). The association has also been shown for sellar region gangliocytoma where the proximity of GHRH source may result in paracrine stimulation of pituitary cells eventually leading to adenomatous transformation (41, 62, 95). It has been suggested that somatotroph transformation in response to ectopic GHRH may exhibit a continuum model of transformation rather than a surge character of changes and therefore both hyperplasia and adenomatous cells may be present at the same time (57, 96, 97). Hence, proper distinction between pituitary adenoma and hyperplasia is not always achievable by means of imaging methods. This may, in consequence, lead to unnecessary surgical resection, especially when there is no other indication of an extracranial neoplasm as an underlying cause of acromegaly. Pituitary surgery via transsphenoidal approach or craniotomy was performed in 54 patients. In 28 cases it appeared to be curative, as the pituitary tumor has been found to cause acromegaly. In an additional 5 patients it served as symptomatic treatment of bitemporal hemianopsia or headache induced by the pituitary mass. However, in 21 cases (38.9% of performed procedures), resection was proved not to be necessary, revealing hyperplasia or normal pituitary tissue in histopathological examination and no improvement afterwards. This stresses the need for a cautious approach while interpreting pituitary images and proves there is necessity for accurate hormonal evaluation, especially in ambiguous cases, in order to avoid unnecessary surgery. In patients with extracranial ectopic source of GHRH, sellar enlargement proved to be reversible in many cases and pituitary size diminished significantly after resection of the primary tumor or somatostatin receptor ligands (SRL) therapy (21, 49, 52, 58, 68, 98).

Table 6 Pituitary imaging and histopathology of intracranial GHRH-secreting tumors.

Table 7 Pituitary imaging and histopathology of extracranial GHRH-secreting tumors.
Despite various treatment modalities being used in ectopic acromegaly management, tumor surgical resection remains a method of choice due to its highest effectiveness and should be performed when feasible (3). Complete tumor resection was the main treatment method for 85 patients, including adenomectomy in 26 of 27 intracranial tumors cases. In a few cases, surgical resection of liver metastases was performed with satisfactory results (40). SRL therapy is the preferred treatment option for patients with inoperable tumors or disseminated metastatic disease, yet, it may also serve successfully as an adjuvant therapy for patients who undergo surgical procedures (3). Therapy with SRL was administered to 37.8% of patients at different stages of treatment, being the main method in 22 cases (9, 40, 45, 47, 50, 68, 75, 99, 100). However, in some cases it was proved to not be sufficiently effective, presumably due to the lack of somatostatin receptor 2 (SSTR-2) in tumor tissue, which is also uncommon, in hyperplastic somatotroph cells (61). In other cases, even if somatostatin receptors were not found in tumor tissue, SRL therapy showed at least partial efficacy due to typical SSTR expression in somatotroph cells (61). In the other cases it appeared to be the only successful method even after surgical treatment (78). The efficacy of surgical resection and SRL therapy cannot be righteously compared in this condition, as the second method has usually served as basic treatment in more advanced cases, often with metastatic disease which itself is a factor of worse prognosis (36). Along with SRL, other supportive methods that may be mentioned are chemotherapy, radiotherapy, immunotherapy, metastases embolization, and other hormonal treatments such as bromocriptine or pegvisomant (9, 18, 33, 36, 40, 42, 43, 46, 49, 51, 73, 78, 90, 96). Altogether, 41 patients received one of these adjuvant treatment options, with pituitary radiation being the most frequent modality (18, 19, 40, 59, 64, 67, 70, 77, 78, 96). Therapeutic results were available for 115 patients with an overall survival rate of 88.7%. Complete and partial remission following treatment was documented in 47.8% and 28.7%, respectively. As for patients with metastatic tumors, their overall survival rate did not differ much from patients with non-metastatic tumors, with outcomes of 83% and 92%, respectively. Nevertheless, metastatic disease appears to be a factor of treatment with worse results, as full or partial recovery was documented for 87.8% of patients without metastases and only 56.1% with disseminated tumors. Relapse and progression were also observed much more frequently within the latter (40, 57, 91). Mean follow-up was 4.2 ± 5.2 years for all 81 patients with available data. This outcome is in accordance with the literature data indicating that prognosis in neuroendocrine tumors is rather favorable even if diagnosed at a metastatic stage and the course of disease remains indolent in most cases (101, 102). Recovery was defined by improvement of symptoms as well as normalization in GH and IGF-1 serum levels. Full GHRH normalization is achievable after surgical treatment but not with SRL therapy. Hormonal treatment appears to reduce GHRH level but not below the detection limit (10). Evaluation of plasma GHRH may be considered useful when anticipating relapse as the rise of GHRH concentration can occur before recurrence of clinical manifestations (11).
Acromegaly caused by GHRH release is very rare disease, although one should be aware it exists. Signs, symptoms, and common hormonal evaluation do not differentiate clearly between tumors secreting GH and GHRH. Pituitary imaging does not provide proper diagnosis between pituitary adenoma and hypertrophy and could result in unnecessary surgery. GHRH tumor secretion can be proved by measuring its plasmatic levels and using histopathological methods with immunohistochemical techniques, however, these are still hardly accessible procedures. GHRH level above 250 ng/L indicates a high probability of an ectopic cause of acromegaly. Vast majority of GHRH-producing tumors are neuroendocrine tumors, quite often associated with MEN-1 syndrome.
Idea for the article: MB, MK, and KZ; literature search and data analysis: IZ and GG, drafting the manuscript: MK, IZ, and GG; supervision and critical revision of the work: MB, MK, and KZ. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lavrentaki A, Paluzzi A, Wass JA, Karavitaki N. Epidemiology of Acromegaly: Review of Population Studies. Pituitary (2017) 20:4–9. doi: 10.1007/s11102-016-0754-x
2. Thorner MO, Frohman LA, Leong DA, Thominet J, Downs T, Hellmann P, et al. Extrahypothalamic Growth-Hormone-Releasing Factor (GRF) Secretion is a Rare Cause of Acromegaly: Plasma GRF Levels in 177 Acromegalic Patients. J Clin Endocrinol Metab (1984) 59:846–9. doi: 10.1210/jcem-59-5-846
3. Faglia G, Arosio M, Bazzoni N. Ectopic Acromegaly. Endocrinol Metab Clin North Am (1992) 21:575–95. doi: 10.1016/S0889-8529(18)30203-2
4. Sano T, Asa SL, Kovacs K. Growth Hormone-Releasing Hormone-Producing Tumors: Clinical, Biochemical, and Morphological Manifestations. Endocr Rev (1988) 9:357–73. doi: 10.1210/edrv-9-3-357
5. Yeung SCJ, Gagel RF. Endocrine Paraneoplastic Syndromes (“Ectopic” Hormone Production). In: Kufe DW, Pollock RE, Weichselbaum RR, editors. Holland-Frei Cancer Medicine, 6th edition. Hamilton (ON: BC Decker (2003). Available at: https://www.ncbi.nlm.nih.gov/books/NBK12609/.
6. Teramoto S, Tange Y, Ishii H, Goto H, Ogino I, Arai H. Mixed Gangliocytoma-Pituitary Adenoma Containing GH and GHRH Co-Secreting Adenoma Cells. Endocrinol Diabetes Metab Case Rep (2019) 2019:19–0099. doi: 10.1530/EDM-19-0099
7. Losa M, von Werder K. Pathophysiology and Clinical Aspects of the Ectopic GH-Releasing Hormone Syndrome. Clin Endocrinol (Oxf) (1997) 47:123–35. doi: 10.1046/j.1365-2265.1997.2311048.x
8. Gola M, Doga M, Bonadonna S, Mazziotti G, Vescovi PP, Giustina A. Neuroendocrine Tumors Secreting Growth Hormone-Releasing Hormone: Pathophysiological and Clinical Aspects. Pituitary (2006) 9:221–9. doi: 10.1007/s11102-006-0267-0
9. Van den Bruel A, Fevery J, Van Dorpe J, Hofland L, Bouillon R. Hormonal and Volumetric Long Term Control of a Growth Hormone-Releasing Hormone-Producing Carcinoid Tumor. J Clin Endocrinol Metab (1999) 84:3162–9. doi: 10.1210/jcem.84.9.6001
10. Borson-Chazot F, Garby L, Raverot G, Claustrat F, Raverot V, Sassolas G. Acromegaly Induced by Ectopic Secretion of GHRH: A Review 30 Years After GHRH Discovery. Ann Endocrinol (Paris) (2012) 73:497–502. doi: 10.1016/j.ando.2012.09.004
11. Ghazi AA, Amirbaigloo A, Dezfooli AA, Saadat N, Ghazi S, Pourafkari M, et al. Ectopic Acromegaly Due to Growth Hormone Releasing Hormone. Endocrine (2013) 43:293–302. doi: 10.1007/s12020-012-9790-0
12. Mnif Feki M, Mnif F, Kamoun M, Charfi N, Rekik N, Naceur BB, et al. Ectopic Secretion of GHRH by a Pancreatic Neuroendocrine Tumor Associated With an Empty Sella. Ann Endocrinol (Paris) (2011) 72:522–5. doi: 10.1016/j.ando.2011.06.002
13. Sala E, Ferrante E, Verrua E, Malchiodi E, Mantovani G, Filopanti M, et al. Growth Hormone-Releasing Hormone-Producing Pancreatic Neuroendocrine Tumor in a Multiple Endocrine Neoplasia Type 1 Family With an Uncommon Phenotype. Eur J Gastroenterol Hepatol (2013) 25:858–62. doi: 10.1097/MEG.0b013e32835f433f
14. Mai M, Tönjes A, Trantakis C, Wittekind C, Stumvoll M, Führer D. Hirsutismus Und Struma Multinodosa Bei Einer 40-Jährigen Uhrmacherin [Hirsutism and Multinodular Goiter in a 40-Year-Old Female Watchmaker]. Internist (Berl) (2013) 54:1137–40. doi: 10.1007/s00108-013-3351-3
15. Mumby C, Davis JR, Trouillas J, Higham CE. Phaeochromocytoma and Acromegaly: A Unifying Diagnosis. Endocrinol Diabetes Metab Case Rep (2014) 2014:140036. doi: 10.1530/EDM-14-0036
16. Zornitzki T, Rubinfeld H, Lysyy L, Schiller T, Raverot V, Shimon I, et al. pNET Co-Secreting GHRH and Calcitonin: Ex Vivo Hormonal Studies in Human Pituitary Cells. Endocrinol Diabetes Metab Case Rep (2016) 2016:150134. doi: 10.1530/EDM-15-0134
17. Cheung KK, Chow FC, Lo AW. 'Open-and-Close' Pituitary Surgery in an Acromegalic Man Presenting With Excessive Sweatiness. BMJ Case Rep (2016) 2016:215183. doi: 10.1136/bcr-2016-215183
18. Ravindra VM, Raheja A, Corn H, Driscoll M, Welt C, Simmons DL, et al. Primary Pituitary Diffuse Large B-Cell Lymphoma With Somatotroph Hyperplasia and Acromegaly: Case Report. J Neurosurg (2017) 126:1725–30. doi: 10.3171/2016.5.JNS16828
19. Kyriakakis N, Trouillas J, Dang MN, Lynch J, Belchetz P, Korbonits M, et al. Diagnostic Challenges and Management of a Patient With Acromegaly Due to Ectopic Growth Hormone-Releasing Hormone Secretion From a Bronchial Carcinoid Tumour. Endocrinol Diabetes Metab Case Rep (2017) 2017:16–0104. doi: 10.1530/EDM-16-0104
20. Stelmachowska-Banaś M, Głogowski M, Vasiljevic A, Raverot V, Raverot G, Zgliczyński W. Ectopic Acromegaly Due to Growth Hormone-Releasing Hormone Secretion From Bronchial Carcinoid Causing Somatotroph Hyperplasia and Partial Pituitary Insufficiency. Pol Arch Intern Med (2019) 129:208–10. doi: 10.20452/pamw.4413
21. Kałużny M, Polowczyk B, Bladowska J, Kubicka E, Bidlingmaier M, Bolanowski M. Acromegaly Due to Ectopic Growth Hormone–Releasing Hormone Secretion by Lung Carcinoid. Pol Arch Intern Med (2020) 130:685–7. doi: 10.20452/pamw.15337
22. Ramírez C, Hernández-Ramirez LC, Espinosa-de-los-Monteros AL, Franco JM, Guinto G, Mercado M. Ectopic Acromegaly Due to a GH-Secreting Pituitary Adenoma in the Sphenoid Sinus: A Case Report and Review of the Literature. BMC Res Notes (2013) 6:411. doi: 10.1186/1756-0500-6-411
23. Melmed S, Ezrin C, Kovacs K, Goodman RS, Frohman LA. Acromegaly Due to Secretion of Growth Hormone by an Ectopic Pancreatic Islet-Cell Tumor. N Engl J Med (1985) 312:9–17. doi: 10.1056/NEJM198501033120103
24. Beuschlein F, Strasburger CJ, Siegerstetter V, Moradpour D, Lichter P, Bidlingmaier M, et al. Acromegaly Caused by Secretion of Growth Hormone by a non-Hodgkin's Lymphoma. N Engl J Med (2000) 342:1871–6. doi: 10.1056/NEJM200006223422504
25. Biswal S, Srinivasan B, Dutta P, Ranjan P, Vaiphei K, Singh RS, et al. Acromegaly Caused by Ectopic Growth Hormone: A Rare Manifestation of a Bronchial Carcinoid. Ann Thorac Surg (2008) 85:330–2. doi: 10.1016/j.athoracsur.2007.06.072
26. Ozkaya M, Sayiner ZA, Kiran G, Gul K, Erkutlu I, Elboga U. Ectopic Acromegaly Due to a Growth Hormone-Secreting Neuroendocrine-Differentiated Tumor Developed From Ovarian Mature Cystic Teratoma. Wien Klin Wochenschr (2015) 127:491–3. doi: 10.1007/s00508-015-0775-x
27. Krug S, Boch M, Rexin P, Pfestroff A, Gress T, Michl P, et al. Acromegaly in a Patient With a Pulmonary Neuroendocrine Tumor: Case Report and Review of Current Literature. BMC Res Notes (2016) 9:326. doi: 10.1186/s13104-016-2132-1
28. Guillemin R, Brazeau P, Böhlen P, Esch F, Ling N, Wehrenberg WB. Growth Hormone-Releasing Factor From a Human Pancreatic Tumor That Caused Acromegaly. Science (1982) 218:585–7. doi: 10.1126/science.6812220
29. Rivier J, Spiess J, Thorner M, Vale W. Characterization of a Growth Hormone-Releasing Factor From a Human Pancreatic Islet Tumour. Nature (1982) 300:276–8. doi: 10.1038/300276a0
30. Losa M, Schopohl J, von Werder K. Ectopic Secretion of Growth Hormone-Releasing Hormone in Man. J Endocrinol Invest (1993) 16:69–81. doi: 10.1007/BF03345835
31. Giustina A, Barkhoudarian G, Beckers A, Ben-Shlomo A, Biermasz N, Biller B, et al. Multidisciplinary Management of Acromegaly: A Consensus. Rev Endocr Metab Disord (2020) 21:667–78. doi: 10.1007/s11154-020-09588-z
32. Bolanowski M, Ruchała M, Zgliczyński W, Kos-Kudła B, Hubalewska-Dydejczyk A, Lewiński A. Diagnostics and Treatment of Acromegaly - Updated Recommendations of the Polish Society of Endocrinology. Endokrynol Pol (2019) 70:2–18. doi: 10.5603/EP.a2018.0093
33. Southgate HJ, Archbold GP, el-Sayed ME, Wright J, Marks V. Ectopic Release of GHRH and ACTH From an Adenoid Cystic Carcinoma Resulting in Acromegaly and Complicated by Pituitary Infarction. Postgrad Med J (1988) 64:145–8. doi: 10.1136/pgmj.64.748.145
34. Roth KA, Wilson DM, Eberwine J, Dorin RI, Kovacs K, Bensch KG, et al. Acromegaly and Pheochromocytoma: A Multiple Endocrine Syndrome Caused by a Plurihormonal Adrenal Medullary Tumor. J Clin Endocrinol Metab (1986) 63:1421–6. doi: 10.1210/jcem-63-6-1421
35. Vieira Neto L, Taboada GF, Corrêa LL, Polo J, Nascimento AF, Chimelli L, et al. Acromegaly Secondary to Growth Hormone-Releasing Hormone Secreted by an Incidentally Discovered Pheochromocytoma. Endocr Pathol (2007) 18:46–52. doi: 10.1007/s12022-007-0006-8
36. Boix E, Picó A, Pinedo R, Aranda I, Kovacs K. Ectopic Growth Hormone-Releasing Hormone Secretion by Thymic Carcinoid Tumour. Clin Endocrinol (Oxf) (2002) 57:131–4. doi: 10.1046/j.1365-2265.2002.01535.x
37. Jansson JO, Svensson J, Bengtsson BA, Frohman LA, Ahlman H, Wängberg B, et al. Acromegaly and Cushing's Syndrome Due to Ectopic Production of GHRH and ACTH by a Thymic Carcinoid Tumour: In Vitro Responses to GHRH and GHRP-6. Clin Endocrinol (Oxf) (1998) 48:243–50. doi: 10.1046/j.1365-2265.1998.3471213.x
38. Sugihara H, Shibasaki T, Tatsuguchi A, Okajima F, Wakita S, Nakajima Y, et al. A non-Acromegalic Case of Multiple Endocrine Neoplasia Type 1 Accompanied by a Growth Hormone-Releasing Hormone-Producing Pancreatic Tumor. J Endocrinol Invest (2007) 30:421–7. doi: 10.1007/BF03346321
39. Tadokoro R, Sato S, Otsuka F, Ueno M, Ohkawa S, Katakami H, et al. Metastatic Pancreatic Neuroendocrine Tumor That Progressed to Ectopic Adrenocorticotropic Hormone (ACTH) Syndrome With Growth Hormone-Releasing Hormone (GHRH) Production. Internal Med (Tokyo Japan) (2016) 55(20):2979–83. doi: 10.2169/internalmedicine.55.6827
40. Garby L, Caron P, Claustrat F, Chanson P, Tabarin A, Rohmer V, et al. Clinical Characteristics and Outcome of Acromegaly Induced by Ectopic Secretion of Growth Hormone-Releasing Hormone (GHRH): A French Nationwide Series of 21 Cases. J Clin Endocrinol Metab (2012) 97:2093–104. doi: 10.1210/jc.2011-2930
41. Kontogeorgos G, Mourouti G, Kyrodimou E, Liapi-Avgeri G, Parasi E. Ganglion Cell Containing Pituitary Adenomas: Signs of Neuronal Differentiation in Adenoma Cells. Acta Neuropathol (2006) 112:21–8. doi: 10.1007/s00401-006-0055-y
42. Bertherat J, Turpin G, Rauch C, Li JY, Epelbaum J, Sassolas G, et al. Presence of Somatostatin Receptors Negatively Coupled to Adenylate Cyclase in Ectopic Growth Hormone-Releasing Hormone- and Alpha-Subunit-Secreting Tumors From Acromegalic Patients Responsive to Octreotide. J Clin Endocrinol Metab (1994) 79:1457–64. doi: 10.1210/jcem.79.5.7962343
43. Liu SW, van de Velde CJ, Heslinga JM, Kievit J, Roelfsema F. Acromegaly Caused by Growth Hormone-Relating Hormone in a Patient With Multiple Endocrine Neoplasia Type I. Jpn J Clin Oncol (1996) 26:49–52. doi: 10.1093/oxfordjournals.jjco.a023178
44. Price DE, Absalom SR, Davidson K, Bolia A, Bell PR, Howlett TA. A Case of Multiple Endocrine Neoplasia: Hyperparathyroidism, Insulinoma, GRF-Oma, Hypercalcitoninaemia and Intractable Peptic Ulceration. Clin Endocrinol (Oxf) (1992) 37:187–8. doi: 10.1111/j.1365-2265.1992.tb02305.x
45. Moller DE, Moses AC, Jones K, Thorner MO, Vance ML. Octreotide Suppresses Both Growth Hormone (GH) and GH-Releasing Hormone (GHRH) in Acromegaly Due to Ectopic GHRH Secretion. J Clin Endocrinol Metab (1989) 68:499–504. doi: 10.1210/jcem-68-2-499
46. Harris PE, Bouloux PM, Wass JA, Besser GM. Successful Treatment by Chemotherapy for Acromegaly Associated With Ectopic Growth Hormone Releasing Hormone Secretion From a Carcinoid Tumour. Clin Endocrinol (Oxf) (1990) 32:315–21. doi: 10.1111/j.1365-2265.1990.tb00872.x
47. Ezzat S, Asa SL, Stefaneanu L, Whittom R, Smyth HS, Horvath E, et al. Somatotroph Hyperplasia Without Pituitary Adenoma Associated With a Long Standing Growth Hormone-Releasing Hormone-Producing Bronchial Carcinoid. J Clin Endocrinol Metab (1994) 78:555–60. doi: 10.1210/jcem.78.3.8126126
48. Lefebvre S, De Paepe L, Abs R, Rahier J, Selvais P, Maiter D. Subcutaneous Octreotide Treatment of a Growth Hormone-Releasing Hormone-Secreting Bronchial Carcinoid: Superiority of Continuous Versus Intermittent Administration to Control Hormonal Secretion. Eur J Endocrinol (1995) 133:320–4. doi: 10.1530/eje.0.1330320
49. Wilson DM, Ceda GP, Bostwick DG, Webber RJ, Minkoff JR, Pont A, et al. Acromegaly and Zollinger-Ellison Syndrome Secondary to an Islet Cell Tumor: Characterization and Quantification of Plasma and Tumor Human Growth Hormone-Releasing Factor. J Clin Endocrinol Metab (1984) 59:1002–5. doi: 10.1210/jcem-59-5-1002
50. Berger G, Trouillas J, Bloch B, Sassolas G, Berger F, Partensky C, et al. Multihormonal Carcinoid Tumor of the Pancreas. Secreting Growth Hormone-Releasing Factor as a Cause of Acromegaly. Cancer (1984) 54:2097–108. doi: 10.1002/1097-0142(19841115)54:10<2097::aid-cncr2820541009>3.0.co;2-x
51. Ch'ng JL, Christofides ND, Kraenzlin ME, Keshavarzian A, Burrin JM, Woolf IL, et al. Growth Hormone Secretion Dynamics in a Patient With Ectopic Growth Hormone-Releasing Factor Production. Am J Med (1985) 79:135–8. doi: 10.1016/0002-9343(85)90559-5
52. Ramsay JA, Kovacs K, Asa SL, Pike MJ, Thorner MO. Reversible Sellar Enlargement Due to Growth Hormone-Releasing Hormone Production by Pancreatic Endocrine Tumors in a Acromegalic Patient With Multiple Endocrine Neoplasia Type I Syndrome. Cancer (1988) 62:445–50. doi: 10.1002/1097-0142(19880715)62:2<445::aid-cncr2820620233>3.0.co;2-5
53. Giustina A, Barkan A, Beckers A, Biermasz N, Biller BMK, Boguszewski C, et al. A Consensus on the Diagnosis and Treatment of Acromegaly Comorbidities: An Update. J Clin Endocrinol Metab (2020) 105:e937–46. doi: 10.1210/clinem/dgz096
54. Rolla M, Jawiarczyk-Przybyłowska A, Halupczok-Żyła J, Kałużny M, Konopka BM, Błoniecka I, et al. Complications and Comorbidities of Acromegaly-Retrospective Study in Polish Center. Front Endocrinol (Lausanne) (2021) 12:642131. doi: 10.3389/fendo.2021.642131
55. Gomez-Pan A, Scanlon MF, Thorner MO, Rees LH, Schally AV, Hall R, et al. Effect of Somatostatin on Abnormal Growth Hormone and Prolactin Secretion in Patients With the Carcinoid Syndrome. Clin Endocrinol (Oxf) (1979) 10:575–81. doi: 10.1111/j.1365-2265.1979.tb02117.x
56. Thorner MO, Perryman RL, Cronin MJ, Rogol AD, Draznin M, Johanson A, et al. Somatotroph Hyperplasia. Successful Treatment of Acromegaly by Removal of a Pancreatic Islet Tumor Secreting a Growth Hormone-Releasing Factor. J Clin Invest (1982) 70:965–77. doi: 10.1172/jci110708
57. Biermasz NR, Smit JW, Pereira AM, Frölich M, Romijn JA, Roelfsema F. Acromegaly Caused by Growth Hormone-Releasing Hormone-Producing Tumors: Long-Term Observational Studies in Three Patients. Pituitary (2007) 10:237–49. doi: 10.1007/s11102-007-0045-7
58. Spero M, White EA. Resolution of Acromegaly, Amenorrhea-Galactorrhea Syndrome, and Hypergastrinemia After Resection of Jejunal Carcinoid. J Clin Endocrinol Metab (1985) 60:392–5. doi: 10.1210/jcem-60-2-392
59. Li JY, Racadot O, Kujas M, Kouadri M, Peillon F, Racadot J. Immunocytochemistry of Four Mixed Pituitary Adenomas and Intrasellar Gangliocytomas Associated With Different Clinical Syndromes: Acromegaly, Amenorrhea-Galactorrhea, Cushing's Disease and Isolated Tumoral Syndrome. Acta Neuropathol (1989) 77:320–8. doi: 10.1007/BF00687585
60. Carroll DG, Delahunt JW, Teague CA, Cooke RR, Adams EF, Christofides ND, et al. Resolution of Acromegaly After Removal of a Bronchial Carcinoid Shown to Secrete Growth Hormone Releasing Factor. Aust N Z J Med (1987) 17:63–7. doi: 10.1111/j.1445-5994.1987.tb05054.x
61. Weiss DE, Vogel H, Lopes MB, Chang SD, Katznelson L. Ectopic Acromegaly Due to a Pancreatic Neuroendocrine Tumor Producing Growth Hormone-Releasing Hormone. Endocr Pract (2011) 17:79–84. doi: 10.4158/EP10165.CR
62. Puchner MJ, Lüdecke DK, Saeger W, Riedel M, Asa SL. Gangliocytomas of the Sellar Region–a Review. Exp Clin Endocrinol Diabetes (1995) 103:129–49. doi: 10.1016/j.clineuro.2016.08.002
63. Genka S, Soeda H, Takahashi M, Katakami H, Sanno N, Osamura Y, et al. Acromegaly, Diabetes Insipidus, and Visual Loss Caused by Metastatic Growth Hormone-Releasing Hormone-Producing Malignant Pancreatic Endocrine Tumor in the Pituitary Gland. Case Report. J Neurosurg (1995) 83:719–23. doi: 10.3171/jns.1995.83.4.0719
64. Saeger W, Puchner MJ, Lüdecke DK. Combined Sellar Gangliocytoma and Pituitary Adenoma in Acromegaly or Cushing's Disease. A Report of 3 Cases. Virchows Arch (1994) 425:93–9. doi: 10.1007/BF00193956
65. Shintani Y, Yoshimoto K, Horie H, Sano T, Kanesaki Y, Hosoi E, et al. Two Different Pituitary Adenomas in a Patient With Multiple Endocrine Neoplasia Type 1 Associated With Growth Hormone-Releasing Hormone-Producing Pancreatic Tumor: Clinical and Genetic Features. Endocr J (1995) 42:331–40. doi: 10.1507/endocrj.42.331
66. Suga K, Yamashita N, Chiba K, Ito T, Kaziwara Y, Yokoyama N. Multiple Endocrine Neoplasia Type 1 Producing Growth Hormone-Releasing Factor in an Endocrine Pancreatic Tumor. Acta Med Nagasaki (2002) 47:55–61. doi: 10.1097/MEG.0b013e32835f433f
67. Asa SL, Scheithauer BW, Bilbao JM, Horvath E, Ryan N, Kovacs K, et al. A Case for Hypothalamic Acromegaly: A Clinicopathological Study of Six Patients With Hypothalamic Gangliocytomas Producing Growth Hormone-Releasing Factor. J Clin Endocrinol Metab (1984) 58:796–803. doi: 10.1210/jcem-58-5-796
68. Scheithauer BW, Kovacs K, Randall RV, Horvath E, Okazaki H, Laws ER Jr. Hypothalamic Neuronal Hamartoma and Adenohypophyseal Neuronal Choristoma: Their Association With Growth Hormone Adenoma of the Pituitary Gland. J Neuropathol Exp Neurol (1983) 42:648–63. doi: 10.1097/00005072-198311000-00005
69. Asada H, Otani M, Furuhata S, Inoue H, Toya S, Ogawa Y. Mixed Pituitary Adenoma and Gangliocytoma Associated With Acromegaly–Case Report. Neurol Med Chir (Tokyo) (1990) 30:628–32. doi: 10.1530/EDM-19-0099
70. Slowik F, Fazekas I, Bálint K, Gazsó L, Pásztor E, Czirják S, et al. Intrasellar Hamartoma Associated With Pituitary Adenoma. Acta Neuropathol (1990) 80:328–33. doi: 10.1007/BF00294652
71. Osella G, Orlandi F, Caraci P, Ventura M, Deandreis D, Papotti M, et al. Acromegaly Due to Ectopic Secretion of GHRH by Bronchial Carcinoid in a Patient With Empty Sella. J Endocrinol Invest (2003) 26:163–9. doi: 10.1007/BF03345146
72. Altstadt TJ, Azzarelli B, Bevering C, Edmondson J, Nelson PB. Acromegaly Caused by a Growth Hormone-Releasing Hormone-Secreting Carcinoid Tumor: Case Report. Neurosurgery (2002) 50:1356–60. doi: 10.1097/00006123-200206000-00029
73. Melmed S, Ziel FH, Braunstein GD, Downs T, Frohman LA. Medical Management of Acromegaly Due to Ectopic Production of Growth Hormone-Releasing Hormone by a Carcinoid Tumor. J Clin Endocrinol Metab (1988) 67:395–9. doi: 10.1210/jcem-67-2-395
74. Drange MR, Melmed S. Long-Acting Lanreotide Induces Clinical and Biochemical Remission of Acromegaly Caused by Disseminated Growth Hormone-Releasing Hormone-Secreting Carcinoid. J Clin Endocrinol Metab (1998) 83:3104–9. doi: 10.1210/jcem.83.9.5088
75. Fainstein Day P, Frohman L, Garcia Rivello H, Reubi JC, Sevlever G, Glerean M, et al. Ectopic Growth Hormone-Releasing Hormone Secretion by a Metastatic Bronchial Carcinoid Tumor: A Case With a non Hypophysial Intracranial Tumor That Shrank During Long Acting Octreotide Treatment. Pituitary (2007) 10:311–9. doi: 10.1007/s11102-007-0019-9
76. Schulte HM, Benker G, Windeck R, Olbricht T, Reinwein D. Failure to Respond to Growth Hormone Releasing Hormone (GHRH) in Acromegaly Due to a GHRH Secreting Pancreatic Tumor: Dynamics of Multiple Endocrine Testing. J Clin Endocrinol Metab (1985) 61:585–7. doi: 10.1210/jcem-61-3-585
77. Athanassiadi K, Exarchos D, Tsagarakis S, Johannesson A. Acromegaly Caused by Ectopic Growth Hormone-Releasing Hormone Secretion by a Carcinoid Bronchial Tumor: A Rare Entity. J Thorac Cardiovasc Surg (2004) 128:631–2. doi: 10.1016/j.jtcvs.2004.02.033
78. Barkan AL, Shenker Y, Grekin RJ, Vale WW. Acromegaly From Ectopic Growth Hormone-Releasing Hormone Secretion by a Malignant Carcinoid Tumor. Successful Treatment With Long-Acting Somatostatin Analogue SMS 201-995. Cancer (1988) 61:221–6. doi: 10.1002/1097-0142(19880115)61:2<221::aid-cncr2820610203>3.0.co;2-3
79. Othman NH, Ezzat S, Kovacs K, Horvath E, Poulin E, Smyth HS, et al. Growth Hormone-Releasing Hormone (GHRH) and GHRH Receptor (GHRH-R) Isoform Expression in Ectopic Acromegaly. Clin Endocrinol (Oxf) (2001) 55:135–40. doi: 10.1046/j.1365-2265.2001.01268.x
80. Morel O, Giraud P, Bernier MO, Le Jeune JJ, Rohmer V, Jallet P. Ectopic Acromegaly: Localization of the Pituitary Growth Hormone-Releasing Hormone Producing Tumor by In-111 Pentetreotide Scintigraphy and Report of Two Cases. Clin Nucl Med (2004) 29:841–3. doi: 10.1097/00003072-200412000-00025
81. Agha, Farrell L, Downey P, Keeling P, Leen E, Sreenan S. Acromegaly Secondary to Growth Hormone Releasing Hormone Secretion. Ir J Med Sci (2004) 173:215–6. doi: 10.1007/BF02914554
82. von Werder K, Losa M, Müller OA, Schweiberer L, Fahlbusch R, Del Pozo E. Treatment of Metastasising GRF-Producing Tumour With a Long-Acting Somatostatin Analogue. Lancet (1984) 2:282–3. doi: 10.1016/S0140-6736(84)90320-9
83. Losa M, Wolfram G, Mojto J, Schopohl J, Spiess Y, Huber R, et al. Presence of Growth Hormone-Releasing Hormone-Like Immunoreactivity in Human Tumors: Characterization of Immunological and Biological Properties. J Clin Endocrinol Metab (1990) 70:62–8. doi: 10.1210/jcem-70-1-62
84. Platts JK, Child DF, Meadows P, Harvey JN. Ectopic Acromegaly. Postgrad Med J (1997) 73(860):349–51. doi: 10.1136/pgmj.73.860.349
85. Sasaki A, Sato S, Yumita S, Hanew K, Miura Y, Yoshinaga K. Multiple Forms of Immunoreactive Growth Hormone-Releasing Hormone in Human Plasma, Hypothalamus, and Tumor Tissues. J Clin Endocrinol Metab (1989) 68:180–5. doi: 10.1007/s11102-006-0267-0
86. Zatelli MC, Maffei P, Piccin D, Martini C, Rea F, Rubello D, et al Somatostatin Analogs In Vitro Effects in a Growth Hormone-Releasing Hormone-Secreting Bronchial Carcinoid. J Clin Endocrinol Metab (2005) 90:2104–9. doi: 10.1210/jc.2004-2156
87. Kawa S, Ueno T, Iijima A, Midorikawa T, Fujimori Y, Tokoo M, et al. Growth Hormone-Releasing Hormone (GRH)-Producing Pancreatic Tumor With No Evidence of Multiple Endocrine Neoplasia Type 1. Dig Dis Sci (1997) 42:1480–5. doi: 10.1023/a:1018818811199
88. Shalet SM, Beardwell CG, MacFarlane IA, Ellison ML, Norman CM, Rees LH, et al. Acromegaly Due to Production of a Growth Hormone Releasing Factor by a Bronchial Carcinoid Tumor. Clin Endocrinol (Oxf) (1979) 10:61–7. doi: 10.1111/j.1365-2265.1979.tb03034.x
89. Christofides ND, Stephanou A, Suzuki H, Yiangou Y, Bloom SR. Distribution of Immunoreactive Growth Hormone-Releasing Hormone in the Human Brain and Intestine and its Production by Tumors. J Clin Endocrinol Metab (1984) 59:747–51. doi: 10.1210/jcem-59-4-747
90. Furrer J, Hättenschwiler A, Komminoth P, Pfammatter T, Wiesli P. Carcinoid Syndrome, Acromegaly, and Hypoglycemia Due to an Insulin-Secreting Neuroendocrine Tumor of the Liver. J Clin Endocrinol Metab (2001) 86:2227–30. doi: 10.1210/jcem.86.5.7461
91. Scheithauer BW, Carpenter PC, Bloch B, Brazeau P. Ectopic Secretion of a Growth Hormone-Releasing Factor. Report of a Case of Acromegaly With Bronchial Carcinoid Tumor. Am J Med (1984) 76:605–16. doi: 10.1016/0002-9343(84)90284-5
92. Doga M, Bonadonna S, Burattin A, Giustina A. Ectopic Secretion of Growth Hormone-Releasing Hormone (GHRH) in Neuroendocrine Tumors: Relevant Clinical Aspects. Ann Oncol (2001) 12 Suppl 2:S89–94. doi: 10.1093/annonc/12.suppl_2.s89
93. Penny ES, Penman E, Price J, Rees LH, Sopwith AM, Wass J. Circulating Growth Hormone Releasing Factor Concentrations in Normal Subjects and Patients With Acromegaly. Br Med J (Clin Res ed) (1984) 289(6443):453–5. doi: 10.1136/bmj.289.6443.453
94. Asa SL, Kovacs K, Stefaneanu L, Horvath E, Billestrup N, Gonzalez-Manchon C, et al. Pituitary Adenomas in Mice Transgenic for Growth Hormone-Releasing Hormone. Endocrinology (1992) 131:2083–9. doi: 10.1210/en.131.5.2083
95. Matsuno A, Katakami H, Sanno N, Ogino Y, Osamura RY, Matsukura S, et al. Pituitary Somatotroph Adenoma Producing Growth Hormone (GH)-Releasing Hormone (GHRH) With an Elevated Plasma GHRH Concentration: A Model Case for Autocrine and Paracrine Regulation of GH Secretion by GHRH. J Clin Endocrinol Metab (1999) 84:3241–7. doi: 10.1210/jcem.84.9.6008
96. Nasr C, Mason A, Mayberg M, Staugaitis SM, Asa SL. Acromegaly and Somatotroph Hyperplasia With Adenomatous Transformation Due to Pituitary Metastasis of a Growth Hormone-Releasing Hormone-Secreting Pulmonary Endocrine Carcinoma. J Clin Endocrinol Metab (2006) 91:4776–80. doi: 10.1210/jc.2006-0610
97. Horvath E, Kovacs K, Scheithauer BW. Pituitary Hyperplasia. Pituitary (1999) 1(3-4):169–79. doi: 10.1023/a:1009952930425
98. Bolanowski M, Schopohl J, Marciniak M, Rzeszutko M, Zatonska K, Daroszewski J, et al. Acromegaly Due to GHRH-Secreting Large Bronchial Carcinoid. Complete Recovery Following Tumor Surgery. Exp Clin Endocrinol Diabetes (2002) 110:188–92. doi: 10.1055/s-2002-32151
99. Krassowski J, Zgliczyński W, Jeske W, Zgliczyński S. Comment of Long-Acting Lanreotide Inducing Clinical and Biochemical Remission of Acromegaly Caused by Disseminated GHRH Secreting Carcinoid. J Clin Endocrinol Metab (1999) 84:1761–2. doi: 10.1210/jcem.84.5.5677-5
100. de Jager CM, de Heide LJ, van den Berg G, Wolthuis A, van Schelven WD. Acromegaly Caused by a Growth Hormone-Releasing Hormone Secreting Carcinoid Tumour of the Lung: The Effect of Octreotide Treatment. Neth J Med (2007) 65:263–6. doi: 10.1097/00006123-200206000-00029
101. Panzuto F, Nasoni S, Falconi M, Corleto VD, Capurso G, Cassetta S, et al. Prognostic Factors and Survival in Endocrine Tumor Patients: Comparison Between Gastrointestinal and Pancreatic Localization. Endocr Relat Cancer (2005) 12:1083–92. doi: 10.1677/erc.1.01017
Keywords: acromegaly, ectopic, GHRH, neuroendocrine tumors, pituitary hyperplasia
Citation: Zendran I, Gut G, Kałużny M, Zawadzka K and Bolanowski M (2022) Acromegaly Caused by Ectopic Growth Hormone Releasing Hormone Secretion: A Review. Front. Endocrinol. 13:867965. doi: 10.3389/fendo.2022.867965
Received: 01 February 2022; Accepted: 14 April 2022;
Published: 09 June 2022.
Edited by:
Gianluca Tamagno, Hermitage Medical Clinic, IrelandReviewed by:
Roberto Salvatori, Johns Hopkins University, United StatesCopyright © 2022 Zendran, Gut, Kałużny, Zawadzka and Bolanowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcin Kałużny, bWFyY2luLmthbHV6bnlAdW13LmVkdS5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.