 Qiu Yang
Qiu Yang Bing Fu
Bing Fu Dan Luo4
Dan Luo4 Xiang Chen
Xiang Chen Li Tian
Li Tian Xijie Yu
Xijie Yu- 1Department of Endocrinology and Metabolism, Laboratory of Endocrinology and Metabolism, West China Hospital, Sichuan University, Chengdu, China
- 2Department of Endocrinology and Metabolism, Chengdu Fifth People’s Hospital, Chengdu, China
- 3Department of Medical Imaging, Chengdu Fifth People’s Hospital, Chengdu, China
- 4Department of General Surgery, Chengdu Fifth People’s Hospital, Chengdu, China
Dipeptidyl peptidase-4 (DPP4) is a ubiquitously occurring protease involved in various physiological and pathological processes ranging from glucose homeostasis, immunoregulation, inflammation to tumorigenesis. Recently, the benefits of DPP4 inhibitors as novel hypoglycemic agents on bone metabolism have attracted extensive attraction in many studies, indicating that DPP4 inhibitors may regulate bone homeostasis. The effects of DPP4 on bone metabolism are still unclear. This paper thoroughly reviews the potential mechanisms of DPP4 for interaction with adipokines, bone cells, bone immune cells, and cytokines in skeleton system. This literature review shows that the increased DPP4 activity may indirectly promote bone resorption and inhibit bone formation, increasing the risk of osteoporosis. Thus, bone metabolic balance can be improved by decreasing DPP4 activities. The substantial evidence collected and analyzed in this review supports this implication.
1 Introduction
The human dipeptidyl peptidase-4 (DPP4) is a cell surface glycoprotein widely expressed in various tissue compartments, including the lymph gland, the biliary tract, the kidneys, the liver, the intestinal system, and bone marrow (1, 2). It is a member of the serine peptidase/prolyl oligopeptidase family, which possesses enzyme functions and selectively cleaves off the penultimate alanine, proline, or serine in the N-terminus start site. Confirmed substrates for DPP4 in vivo included many incretins and cytokines (3, 4). In addition, DPP4 was previously known as the T-cell activation antigen cluster of differentiation-26 (CD-26). It has been known for playing a complex role in non-enzyme–dependent functions by acting as costimulatory proteins in immune cells such as T cells, monocytes, and dendritic cells. Functionally, it is involved in multiple physiological processes and pathologies ranging from glucose homeostasis, immunoregulation, inflammation to tumorigenesis.
Over the past decade, it has aroused public concern for its pleiotropic actions on bone metabolism. First, as a new oral hypoglycemic drug, DPP4 inhibitors might promote bone formation and inhibit bone absorption in addition to possessing practical hypoglycemic effects, reducing the risk of osteoporosis and fracture (5–9). Second, cells in the bone microenvironment, such as osteoclasts, bone marrow adipose tissue (BMAT), and immune cells, would secrete DPP4 (10–12). Third, the substrates of DPP4 are distributed on bone cells, bone immune cells, and cytokines in the skeleton system (3, 4). Therefore, it can be speculated that DPP4 may directly or indirectly regulate bone metabolism. This study reviews the regulation mechanisms of DPP4 on bone energy metabolism, bone immunity, and bone remodeling, aiming to provide a new target for the treatment of osteoporosis.
2 Structure and Functions of DPP4
2.1 Molecular Properties and Forms
DPP4/CD26 is a dimeric 240-kDa glycoprotein composed of two 120-kDa subunits and coded by a gene on chromosome 2q24.3 (13). DPP4 can be divided into two different forms: either as a soluble form (sDPP4) in circulation or a membrane-bound form (mDPP4) anchored to a cellular surface (14). mDPP4 consists of three domains with 766 residues, including a six–amino acid N-terminal cytoplasmic domain, a 22‐residue hydrophobic transmembrane domain, and an extracellular domain. The extracellular domain includes the cystein-rich region, glycosylated-rich region, adenosine deaminase (ADA)–binding region (340–343 a.a.), fibronectin-binding region (469– 479 a.a.), and c-terminal DPP4 catalytic sites (507–766 a.a. with active catalytic sites at 630, 708, and 740) (Figure 1). mDPP4 is widely expressed on the surface of epithelial, endothelial, stromal, preadipocytes, mature adipocytes, embryonic stem cells, hematopoietic stem cells, hematopoietic progenitor cells, and immune cells (11, 15–23) in different tissues and organs, such as the liver, gut, adipose tissue, and bone marrow (1, 2). sDPP4 contains 727 residues without a cytoplasmic domain, a transmembrane domain, and flexible region (24). sDPP4 is considered as the hydrolyzed form of mDPP4, which is widely present in serum, saliva, cerebrospinal, seminal fluid, and bile and has fully enzymatically active (25, 26). The protease responsible for generating sDPP4 is unclear. The different physiological function between sDPP4 and mDPP4 is not well known.
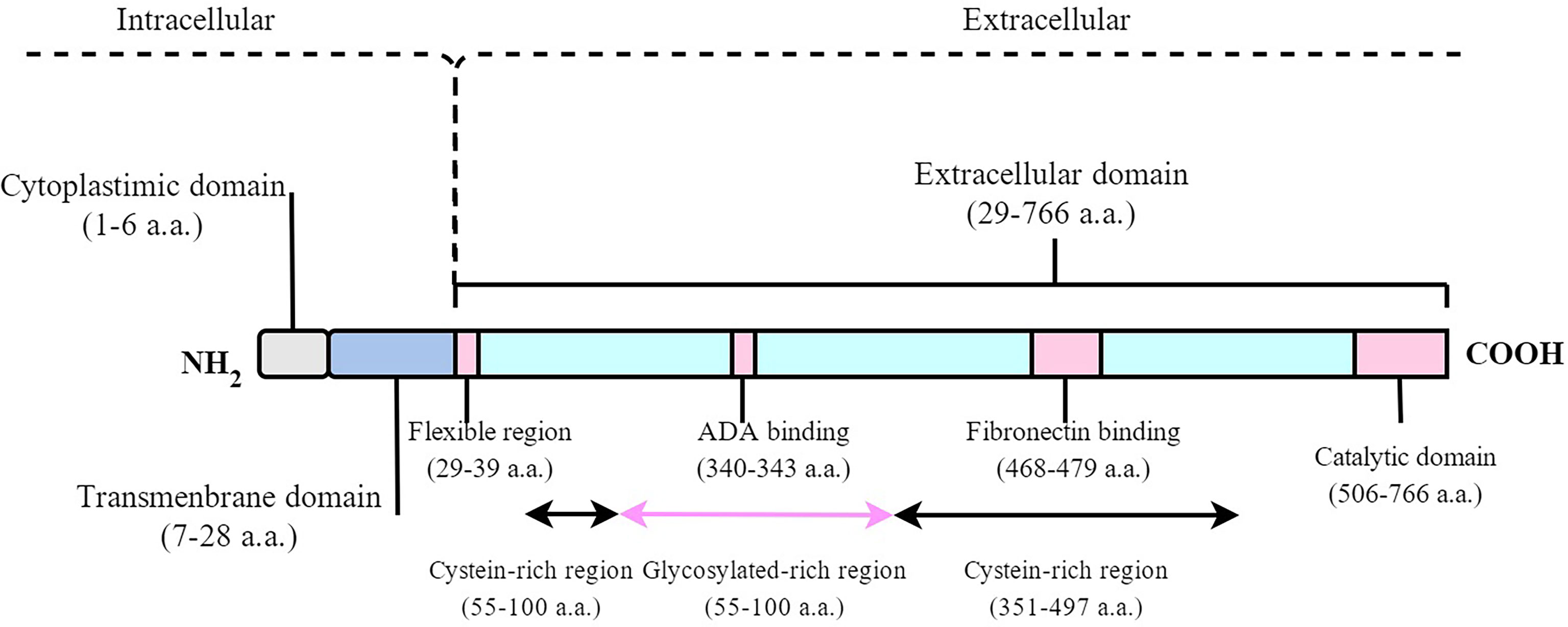
Figure 1 Schematic diagram of the human DPP4 molecule. mDPP4 consists of three domains with 766 residues, including a 6-amino acid N-terminal cytoplasmic domain, a 22-residue hydrophobic transmembrane domain, and an extracellular domain. sDPP4 contains 727 residues without cytoplasmic domain, transmembrane domain, and flexible region.
2.2 Multifunctional Properties
DPP4 is a member of the serine peptidase/prolyl oligopeptidase family, which selectively cleaves the penultimate alanine, proline, or serine in the N-terminus start site, including incretins and cytokines. Glucagon-like peptide-1 (GLP-1), glucagon-like peptide-2 (GLP-2), glucose-dependent insulinotropic peptide (GIP) (3), peptide YY (PYY), and neuropeptide Y (NPY) (4) are the most widely studied incretins. Besides the factors mentioned above, Ou et al. (2) identified many proteins as cleaved proteins for DPP4 from the National Center for Biotechnology Information database and Universal Protein Resource.
In addition, DPP4 plays a complex role in classical enzyme functions as well as various subcellular localization and non-enzyme-dependent functions. DPP4 performs its non-proteolytic functions as receptor or costimulatory protein in the fields of immunology. It is a receptor of ADA (27, 28), which plays an important role in immune regulation as the key enzyme for adenosine decomposition. The formation of DPP4-ADA complex reduces local concentration of adenosine, alleviating the inhibitory effect of high concentration adenosine on T lymphocyte proliferation and activation (29). The DPP4-ADA complex promotes the differentiation of primary T lymphocytes into helper T cells and memory T cells (30). Meanwhile, DPP4-ADA complex also plays a costimulatory role in immunological synapse between dendritic cells and CD4+ T cells, enhancing inflammatory response by inducing interleukin-6 (IL-6), interferon-γ (INF-γ), and tumor necrosis factor-α (TNF-α) secretion (31, 32). Furthermore, DPP4 also has costimulatory function by binding to CXC chemokine receptor-4 (CXCR4) (33), mannose 6 phosphate/insulin-like growth factor II receptor (M6P/IGF-IIR) (34), TCR/CD3 (30), CD45 (35), caveolin-1 (36), CARMA1 (37), and fibronectin III (38, 39). This evidence suggests the immunomodulation function of DPP4 in different signaling process.
3 Scientific Evidence of the Relationship Between DPP4 and Bone Metabolism
3.1 The Relationship Between Serum DPP4 Activity and BMD
A study of 744 postmenopausal Chinese women with normal glucose tolerance showed significant lower in lumbar and femoral neck bone marrow density (BMD) in the quartile with the highest DPP4 activity (40). However, a 13-year follow-up study (41) of 1,536 community-based elderly adults revealed that there was no significant association between basal DPP4 activity and hip BMD, lumbar spine BMD, or incidence of hip fractures. A cross-sectional study with 147 newly diagnosed T2D showed that elevated sDPP4 activity was positively associated with the risk of osteoporosis/osteopenia and fracture (42). However, a study (43) of 204 male Japanese diabetics showed no correlation between DPP4 levels and BMD in the lumbar spine or femoral neck. The relationship between DPP4 and BMD has been reported to depend on BMI. Durinx et al. (44) demonstrated that DPP4 activity was negatively correlated with spine BMD in obese postmenopausal women, which was not found in non-obese postmenopausal women. From the evidence mentioned above, it prompted that the correlation between DPP4 activity and bone metabolism might be influenced by aging and metabolic status. Durinx et al. (44) studied 481 healthy subjects aged between 19 and 61 years and revealed an age related decline in DPP4 activity. Indeed, previous studies have showed that DPP4 was a novel adipokine (45) released from immune cells, adipose cells, and bone marrow cells, which correspondingly indicated that sDPP4 concentrations and activity were linked to obesity, metabolic syndrome, T2D, and inflammatory diseases (10, 46, 47). When evaluating DPP4 activity and bone metabolism, stratified population analysis has to be taken into account.
3.2 The Impact of DPP4 Inhibitors on Bone Metabolism in the Pre-Clinical and Clinical Studies
A significant number of pre-clinical studies have shown a beneficial impact on osteogenesis for DPP4 inhibitors. Intraperitoneal injections of sitagliptin for 9 days significantly increased the osteogenic progenitors while decreased the adipogenic progenitors in non-fractured tibiae (5). Sitagliptin would increase the BMD of mice with a high-fat diet (6). In addition, the negligible BMD loss was observed in OVX non-diabetic adult rats treated with the higher sitagliptin doses, compared with thiazolidinediones or placebo for 12 weeks (7). In addition, DPP4 inhibitors showed the function of pro-osteoblastogenesis and anti-osteoclastogenesis. Anagliptin significantly promoted the osteoblastic differentiation of MC3T3-E1 cells and increased matrix deposition and mineralization (5). Linagliptin downregulated LPS-induced osteoclast formation and bone resorption (8). Nishida et al. (9) showed that the humanized anti-CD26 monoclonal antibody (hu-CD26mAb) inhibited osteoclast precursor differentiation.
Data from animal studies suggested that DPP4 inhibitors (sitagliptin, saxagliptin, vildagliptin, and linagliptin) have an anabolic effect on bone. However, clinical studies of DPP4 inhibitors remained controversial (5). Dombrowski et al. (48) found that DPP4 inhibitors were associated with a decrease in the risk of osteoporotic fracture by matching 4,160 patients with DPP4 inhibitors to never users. A meta-analysis of 28 trials showed 40% reduction in fracture risk with the treatment of DPP4 inhibitors (11,880 T2D) compared to placebo or other treatments (9,175 T2D) in a duration of 24 weeks (49). A retrospective population–based (49) cohort study of T2D (N = 216,816) revealed that DPP4 inhibitor usage was not associated with change in fracture risk compared to controls and other non-insulin anti-diabetic drug usage. Since the average duration of 1.3 years might have been too short to show association with fracture risk.
In contrast, a post hoc pooled analysis of 20 randomized controlled studies in 9,156 patients with T2D revealed that the incidence rate for bone fracture was higher with saxagliptin versus control (50). Different study designs may explain the different findings because the studies included in the pooled analyses took higher doses than approved saxagliptin doses (2.5 mg/day) with almost up to 5 or 10 mg/day or even 100 mg/day. Meanwhile, another network meta-analysis (51) including 117 RCTs of 221,364 T2D, compared anti-diabetic drugs head-to-head. Specifically, trelagliptin, omarigliptin, sitagliptin, vildagliptin, and saxagliptin may increase the risk of bone fracture, whereas others (linagliptin and alogliptin) may show benefits. It indicated that fracture risk was independent of age, sex distribution, and the duration of exposure to anti-diabetic drugs.
With aging, diabetes, obesity, and menopause status were risk factors for osteoporotic and bone fractures, thus it is hard to conclude the association between DPP4 inhibitors and bone metabolism from the current clinical evidence. Further controlling confounding factors, long-term studies are needed to confirm the effect of DPP4 inhibitors on osteoporotic fracture in none-diabetes, non-menopausal, and different levels of obesity population.
4 Mechanisms of the Effects of DPP 4 on Bone Metabolism
Some previous studies have tried to prove whether the beneficial effects of DPP4 inhibitors on bone metabolism arise from a direct action on the bone cells. Ishida et al. (52) found that DPP4 inhibitor had no direct impact on the differentiation and proliferation of osteoclast precursors or on the ligand of receptor activator of nuclear factor κB (RANKL) expression in bone marrow stromal cells. Gallagher et al. (53) investigated the effect of a DPP4 inhibitor (MK-0626) on bone metabolism in an animal model of T2D. They revealed that MK-0626 had no adverse effects on bone in vivo or no direct effect on osteoblasts in vitro. The results also suggested that the effects of DPP4 on bone remodeling might mediate through indirect mechanisms instead of direct impacts.
4.1 DPP4 Takes Part in the Bone Metabolism Through a Network of Adipokines
There is an essential relationship between bone remodeling and energy metabolism since bone remodeling requires energy. A great number of observations have shown that adipose could regulate energy metabolism via adipokines based on the thesis of adipose-bone interactions (54). In the last decade, BMAT was found to be a kind of fat depot within the marrow niche with different origin, structure, and function.
It was shown that the BMAT is a metabolically active fat depot that plays a role in lipid storage and metabolic homeostasis (45). In addition, BMAT is involved in the bone–fat interaction through a complex network of adipokines such as adiponectin and leptin.
DPP4 is a newly discovered adipokine that originated from mature adipocytes (55–57) including bone marrow adipocytes (10, 11). Mulvihill et al. (11) revealed that bone marrow–derived cells contributed to ~40%–50% of sDPP4 activity in the fasting state by three gene knockout (KO) mice models. In addition, Weivoda et al. (12) utilized denosumab (DMAb) to pharmacologically ablate osteoclasts and RNA sequencing of bone biopsies in postmenopausal women, demonstrating that DPP4 is not only an osteoclast-derived protein but also links bone remodeling to energy metabolism. Thus, it was deducible DPP4 might play an essential role in the adipokines network of bone energy metabolism.
4.1.1 Adiponectin
Adiponectin is mainly from BMAT (58) and secreted from MSC-derived adipocytes (59). Its receptor is expressed on osteoblasts and osteoclasts. Adiponectin would promote the differentiation and activity of osteoblasts and inhibit the differentiation of preosteoclasts and bone resorption (60–62). Evidence had proved a negative correlation between the activity of DPP4 and the circulating level of adiponectin in lean and obese subjects (63). Meanwhile, a systematic review and meta-analysis of randomized controlled trials showed that treatment of DPP4 inhibitors would increase the plasma concentrations of adiponectin (64). Exendin-4, a GLP-1 receptor agonist, increased the expression and secretion of adiponectin via the Protein Kinase A (PKA) pathway in 3T3-L1 adipocytes (65). In addition, Piao et al. found a cross-talk between DPP4/GLP-1/GLP-1R and adiponectin/adiponectin receptor in ischemic vascular regeneration under chronic stress conditions (66). From the accumulated evidence, it was speculated that DPP4 might diminish the positive effect of adiponectin on bone mass.
4.1.2 Leptin
Leptin signaling is one of the critical pathways affecting human energy metabolism, and its biological functions are complicated. Leptin has a dual effect on bone tissue, which can either centrally inhibit bone formation by binding to leptin receptors in the hypothalamus or locally promoting bone formation and inhibiting bone resorption by binding to the expressed receptors on the surface of osteoblasts (67, 68). Leptin is not a substrate of DPP4 but has a putative DPP4 truncation site. Anagliptin (DPP4 inhibitor) ameliorated leptin resistance and attenuated food intake and body weight in diet-induced obesity mice (69). A group of Japanese T2D patients was treated with anagliptin and metformin or miglitol for 52 weeks, and reduced leptin concentration was observed (70). However, the exact function of truncated leptin on bone remains unknown.
4.2 DPP4 Regulates Bone Remodeling by Binding to Its Substrates Locating in the Skeletal System
DPP4 might indirectly regulate bone remodeling by binding to multiple peptides substrates including incretins, gastrointestinal peptides, and neuropeptides, whose receptors are widely expressed in skeletal system. Gastrointestinal peptides, such as GLP-1, GLP-2, and GIP have been shown to favor bone formation over resorption, whereas NPY and PYY have been shown to have catabolic effects on bone metabolism (Table 1).
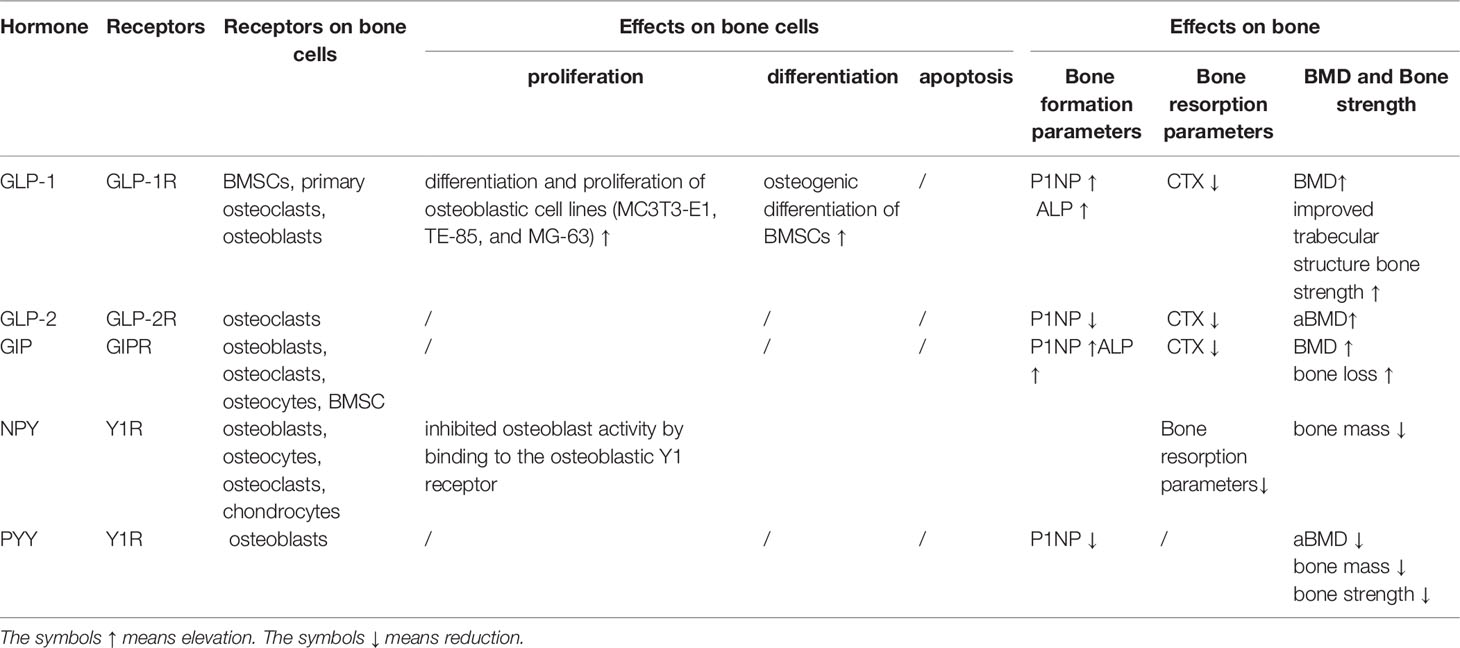
Table 1 Summary of the effects of the gut hormones and neuropeptide on bone metabolism.
4.2.1 Anabolic action
GLP-1 is an incretin hormone originating from the distal small intestine and would be degraded at the N-terminus by DPP4 (71–73). The GLP-1 receptor (GLP-1R) is expressed on human bone marrow stem cells (BMSCs) but not on mature osteoblasts (74) and MLO-Y4 osteocytic cell line (75). In human BMSCs, GLP-1 inhibited adipocyteogenesis but promotes bone formation through upregulating osteocalcin (OCN) and osteoprotegerin (OPG) (76–78). Furthermore, GLP-1 and GLP-1R agonists (exendin-4 and liraglutide) are proved to increase the proliferation and differentiation of osteoblasts (79–81). In animal studies, GLP-1R KO mice show decreased bone quality and strength and reduced cortical area (82). In contrast, GLP-1R agonists (liraglutide and exendin-4) increased aBMD and improved trabecular structure and bone strength (83, 84). The mechanisms of GLP-1R–mediated osteogenic action are exerted through a dual role: the cAMP/PKA/b-catenin/T cell factor (TCF) pathway to initiate osteoblast differentiation and the PKA/PI3K/Akt/GSK3b pathway to inhibit ß-catenin degradation and promote its nuclear accumulation in BMSCs, which resulted in the anabolic bone formation (74).
GLP-2 was co-secreted with GLP-1 in the intestine (85) and was deactivated by sDPP4 with a half-life of 5 to 7 min (86). The GLP-2 receptor (GLP-2R) was expressed on osteoclasts (87). Both short-term (14 days) (88) and long-term (4 months) (89) administrations of GLP-2 would reduce bone resorption (CTX) in a dose-dependent manner without influencing bone formation (OCN and P1NP). Four-months of treatment with GLP-2 enhanced hip BMD in postmenopausal women (89).
GIP mainly originates from the proximal small intestine (90) and is degraded by DPP4 with a half-life of about 4 min in human serum (91). The GIP receptor (GIPR) is expressed on BMSCs (92), osteoblasts (93), osteoclasts (94), and osteocytes (93). In vitro, stimulating osteoblasts with GIPR would reduce its apoptosis (91) and increase their viability, alkaline phosphatase activity, and type 1 collagen expression (93, 95). In vivo, GIPR KO mice exhibit low bone mass (91), altered bone microarchitecture, decreased markers of bone formation (96), and cortical osteopenia due to decreased bone formation and increased bone resorption. In clinical studies, reduced collagen type-1 (CTX-1) and increased procollagen type I N-terminal propeptide (P1NP) were observed in short-term GIP infusion in both healthy humans and patients with type 1 diabetes (97). These results indicate that GIP can uncouple formation and bone resorption and play an anabolic role in bone metabolism.
4.2.2 Catabolic Action
NPY is expressed in osteoblasts, osteoclasts, osteocytes, and chondrocytes (98). NPY inhibits the differentiation and activity of osteoblast by binding to Y1 receptors on osteoblasts. The reduced bone formation and bone mass (99) would be vanished by osteoblast-specific Y1 KO (100, 101). NPY was truncated by DPP4 with a half-life of 2 to 3 min and then lost the ability of binding to the Y1 receptor (102). Thus, it is valuable to evaluate the effect of DPP4 inhibition on NPY-related bone loss.
PYY shares the same receptors with NPY and is often co-secreted with GLP-1 and GLP-2R (103). The secretion form of PYY (PYY1−36) containing 36–amino acid molecular was degraded by DPP4 to form PYY3−36 (104). PYY1-36 might exert suppressive effects on osteoblast activity by binding to the Y1 receptor (101). PYY1-36 was negatively associated with aBMD and P1NP (105), indicating a catabolic impact on bone metabolism. Bone mass was reduced in transgenic mouse models with overproduction of PYY, whereas bone mass and strength was increased in PYY KO mice (106). Further studies are needed to clarify whether the DPP4 degraded form PYY3−36 would have an anti-osteogenic effects on BMD.
4.3 DPP4 Regulates Immune Responses in the Bone Microenvironment
It is well known that there is a dynamic cross-talk between bone remodeling and immune systems which is called “Osteoimmunology” (107). Several diversified immune cells, including CD4+ T cells and CD8+ T cells, were found in the bone marrow (108–110). Bone cells share the same microenvironment with immune cells in the bone marrow, including cytokines, chemokines, receptors, and transcription factors. Since DPP4 was expressed on the surface of osteoblasts (111) and osteoclasts (112) and a wide range of immune cells (i.e., macrophages, lymphocytes, natural killer cells, monocytes, and dendritic cells) (113–115), it is conceivable that DPP4 would exert its effect on the “osteoimmunology”.
4.3.1 T Cells
Naïve T cells played a potential protective role for bone in studies where T cell–deficient mice were presented with enhanced osteoclastogenesis and reduced BMD (116). Accordingly, activated T cells may disturb bone homeostasis and induce subsequent bone loss via the release of RANKL (117) and TNF-α (118) under pathological conditions such as estrogen deficiency (119, 120) and in inflammatory diseases (121). Activated T cells can be divided into cytotoxic CD8+ T cells and CD4+ T helpers cells, which is further subcategorized in Th1, Th2, Th17, and Treg cells. DPP4 is a marker of human activated T cells and an essential co-stimulatory molecule for T cells’ maturation, activation, and differentiation. It directly triggers T cell activation and proliferation by binding to ADA (30). Furthermore, DPP4 also has a costimulatory function by binding to CXCR4 (32), M6P/IGF-IIR (33), TCR/CD3 (30), CD45 (protein tyrosine phosphatase) (34), caveolin-1 (36), CARMA1 (37), and fibronectin III (38, 39). Synthetic inhibition or deficiency of DPP4 results in impaired development and maturation of naïve T cells (122–124). In addition, DPP4/CD26 plays an essential role in T cell differentiation.
Naïve CD4+ T cells differentiated into distinct effector T cell subsets of T helper l (Th1) and T helper 2 (Th2) (125). Both Th1 and Th2 cells are now known to inhibit osteoclast formation by secreting signature cytokines IFN-γ and IL-4, respectively (126). DPP4 is expressed at a higher level in Th1 cells but at a lower level in Th2 cells. Deleting of DPP4/CD26 in mice deregulated Th1 immune responses and reduced Th1 cytokines but upregulated Th2-type cytokines (127, 128). Accordingly, DPP4 was proposed to enhance the production of Th1 proimflamatory cytokines including IFN-γ, TNF-α, and IL-6 (115). As an osteoclastogenic subset of T cells, T helper 17 (Th17) cells secreted IFN-γ and IL-17, regulating pre-osteoclast proliferation, differentiation, and apoptosis, leading to pathological bone loss (129, 130). Treg cells (131–133) are known to have anti-osteoclastogenesis function by downregulating the expression in RANKL and macrophage colony-stimulating factor (M-CSF) (134, 135). DPP4/CD26 was highly expressed on Th17 cells (121), yet was used as a negative selection marker for Tregs. The absent/low DPP4 expression on the surface of Treg cells were important for the utilization and accumulation of adenosine to present as Treg/effector T cells (135). In addition, DPP4 inhibitors significantly increased the Treg expansion in non-obese diabetic mice (136). Hence, DPP4 may promote osteoclastogenesis by restraining proliferation and activization of Treg cells. Any imbalance within the two couple of lymphocytes (Th1, Th2, Th17, and Tregs) played a prominent role in immune regulation (137, 138). A study using solid-phase immobilized specific anti-CD3 mAb to stimulate T cells differentiation showed that the high expression of DPP4/CD26 played an indispensable role in the differentiation and functions of Th1 and Th17 lymphocytes. Thus, DPP4/CD26 could be involved in bone diseases such as osteoporosis by regulating naïve CD4+ T differentiation and the immune balance of its activated subsets.
CD8+ T cells were another kind of lymphocytes with both osteogenesis and osteoclastogenesis functions. A bi-directional regulatory loop between osteoclasts and CD25+FoxP3+CD8+ T cells was observed in previous researches (139). The naïve CD8+ T-cells primed by osteoclasts would express cytokines including FoxP3, CD25, CTLA-4, RANKL, and IFN-γ (140), which, in turn, potentially activated or suppressed osteoclast activity. The IFN-γ and CTLA-4 inhibited osteoclast activity (140, 141), whereas RANKL increased osteoclast activity. Healthy individuals taking DPP4 inhibitor sitagliptin 100 mg daily for 28 days displayed significant increase in the percentage of memory CD8+ T cells from days 0 to 3 compared to the placebo group (142). Therefore, these findings support further studies to verify regulation mechanisms of DPP4 on CD8+ T cells and clinical outcome of bi-directional regulatory loop.
4.3.2 B Cells
B cells have long been recognized as active RANK/RANKL/OPG axis regulators in osteoimmunology. B cells produced OPG (116) and RANKL (143) and played dual functions on bone homeostasis. Imbalanced production cytokines of B cells between RANKL and OPG might strongly link bone turnover and the immune response. DPP4 is expressed on B cells, a natural killer (144). Reduced DNA synthesis (144, 145) in B cells and impaired immunoglobulin isotype switching of B cells were observed in DPP4-deficient mice (146). However, an in vitro study showed no effect of DPP4 deficiency on B cells in rats (132), whereas B cell numbers were decreased markedly in later life by monitoring the long-term effect of DPP4 deficiency in vivo (147). DPP4 is expressed on B cells (144); in turn, it might be involved in the proliferation and function of B cells in physiological bone remodeling.
4.3.3 Macrophages
Osteomacs, a kind of bone macrophages, settles in the endosteal and periosteal surfaces of the bone (148). Loss of osteomacs leads to a complete loss of osteoblasts, which are presumed to play an essential role in maintaining mature osteoblasts (149). Macrophages are highly plastic in response to their microenvironment and could be typically divided into pro-inflammatory M1 and anti-inflammatory M2. Osteomacs cells polarization with alternatively types are important mediators in bone homeostasis (150). M1 enhances osteoclast differentiation and resorption by upregulation of RANKL, whereas M2 participated in the clearance of apoptotic cells (151). Treatment with DPP4 inhibitor Alogliptin was associated with reduced numbers of pro-inflammatory M1 macrophages and increased gene expression of M2 macrophage markers (152). Hiromura et al. (153) found that DPP4 inhibitors can induce the polarization and migration of M2 macrophages by CCL3 Caveolin-1 cell signal transcription factors in white fat. Other research (154) showed GLP-1 agonist (exenatide) would promote the polarization of M2 macrophages and the differentiation of bone marrow hematopoietic stem cells into osteoclasts through the PKA/STAT pathway. Therefore, it is speculated that DPP4, as the upper hydrolase signaling molecule of GLP-1, might regulate macrophage polarization through the GLP-1/GLP-1 receptor pathway. Accordingly, DPP4 was proposed to regulate M1/M2 polarization, resulting in a shift in macrophage phenotypes toward M1. Further studies concerning the direct role of DPP4 on osteomacs polarization would be helpful for understanding the mechanism of “osteoimmunology”.
4.4 DPP4 Cleaves Cytokines in Bone Microenvironment
There is increasing evidence that cytokines are critically responsible for bone resorption and formation changes. DPP4 can cleave X-Pro or X-Ala dipeptides from numerous cytokines in the bone microenvironment through its N-terminal dipeptidase activity. This cleavage can result in both inactive and active fragments of the targeted cytokines (Table 2). Chemokines promoted the migration of osteoclast precursor cells and facilitated the process of osteoclastogenesis and bone resorption. The NH2 terminal of many chemokines could be cleaved by DPP4, thus influencing their biologic activities. The widely studied CX3CL1/CX3CR1 (155), CXCL9 (156), CXCL10 (157–160), CXCL12/CXCR4 (157–160), and CCL5/CCR1 (161) signaling pathway were proved to play an important role in osteoclast formation and maturation. DPP4 removes N-terminal amino acids from their ligands or receptors and impaires its chemotactic activity (174–178). Other chemokines such as CCL2 (162, 163), CCL3 (164), CCL11 (165), and CCL20 (166) are known to have putative truncation sites for DPP4, which showed modifying effects on hematopoietic stem/progenitor cells with truncated forms (2).
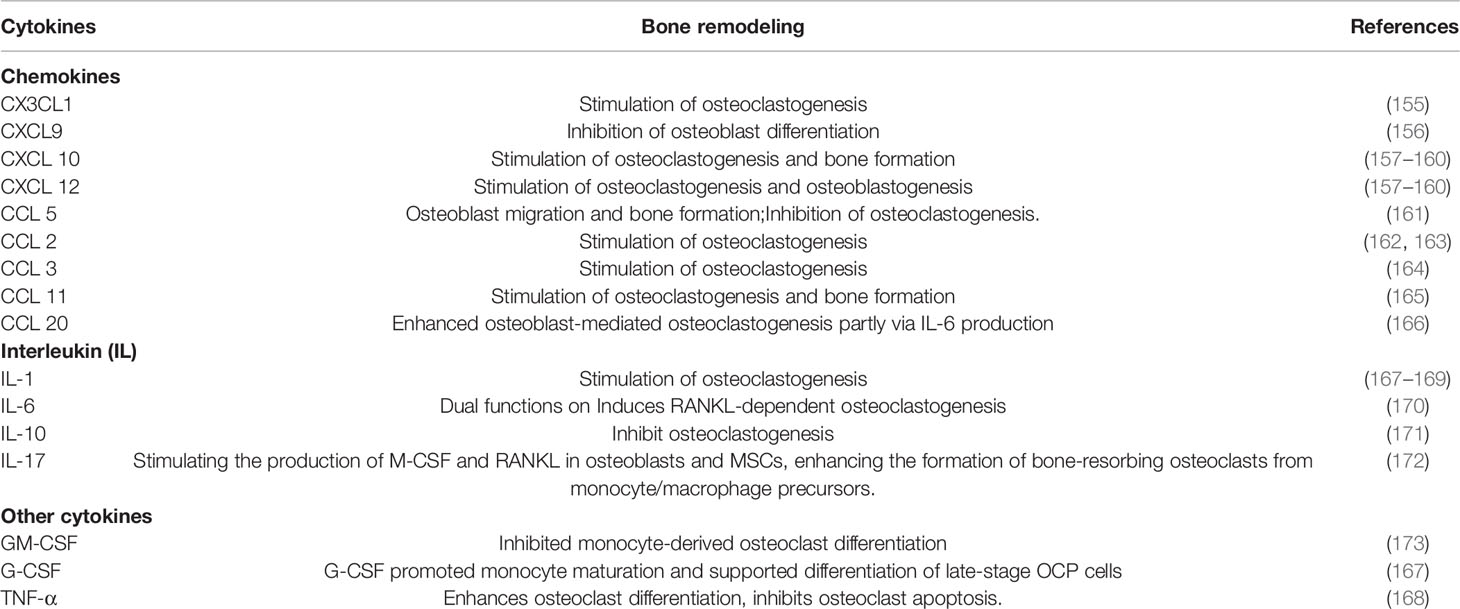
Table 2 The effects of cytokines on bone metabolism.
Some ILs, such as IL-1 (167–169), IL-6 (170), and IL-17 (172) known to enhance osteoclastogenesis or anti-osteoclastogenesis (IL-10) (171) have putative truncation site for DPP4 (173). Other cytokines, such as granulocyte macrophage (GM)–colony-stimulating factor (CSF) (173), G-CSF (167), and TNF-α (168), also take part in the regulation of immune and bone metabolism. DPP4 could induce truncated products with inactive function for GM-CSF, G-CSF (2), and TNF-α (27). The biochemical and biological functions of putative DPP4 truncation sites are poorly investigated. Previous research has shown that DPP4-cleaved substrates could get inactivated, modified receptor interactions, or modified bioactivities compared with the intact forms. For example, DPP4-cleaved GLP-1 can act through a different receptor from GLP-1R. As for PYY and NPY, the DPP4 cleavage form could enhance the selectivity of the Y2 receptor relative to the non-selective activity of the intact form. DPP4 cleavage of GIP induced an antagonistic feature for GIP-R signaling. The enzymatic activity of DPP4 has been implicated in certain disease states.
Nevertheless, the function of truncated forms of these cytokines has not been adequately appreciated. Further studies are needed to determine the functional difference between the truncated molecules and the full-length form of the protein.
5 Conclusion and Future Perspectives
This review introduces the role of DPP4 on bone metabolism and summarizes its potential mechanisms (Figure 2). Inhibition of DPP4 activity does not directly regulate bone remodeling, whereas DPP4 indirectly affects bone metabolism by regulating DPP4 substrates and immune cells in the bone microenvironment. We speculate that increased DPP4 activity might indirectly promote bone resorption and inhibit bone formation, thereby increasing the risk of osteoporosis, showing prospects of a potentially new understanding of the mechanism of DPP4 on osteoporosis. However, the study of DPP4 on bone metabolism is still in an early stage, and there are still various questions that need to be solved. Some suggestions are purposed for future research directions:
1. It is crucial for bone remodeling to keep the energy homeostasis in the bone microenvironment. Adipokines originating from BMAT would mediate bone remodeling and energy balance through paracrine because adiponectin and leptin may be regulated by adipocytokine DPP4 in the bone microenvironment. Future studies may focus on the relationship between DPP4 and other adipocytokines in the bone microenvironment or other regulatory signals of energy metabolism.
2. Because of the extensive cross-linking signals among bone cells cytokines of bone immune cells in the bone microenvironment, it is necessary to conduct in-depth studies to find the initiating or critical mechanisms of the dialogue between DPP4 and those factors. Thus, the mechanism of DPP4 promoting bone resorption and inhibiting bone formation in bone remodeling need to be was further verified.
3. It is valuable to modify DPP4 inhibitors from the distinct regulation mechanisms of bone metabolism between DPP4 inhibitors and DPP4. So as to achieve the dual effects of lowering glucose and reducing the risk of osteoporosis.
4. Substrates cleaved by DPP4 can inactivate, modify receptor interactions, or modify biological activity. Because DPP4 has a wide range of cytokine effects on the bone microenvironment, it is necessary to investigate cytokines’ biochemical and biological functions after the DPP4 truncation site to further clarify the role of DPP4 in proteolysis.
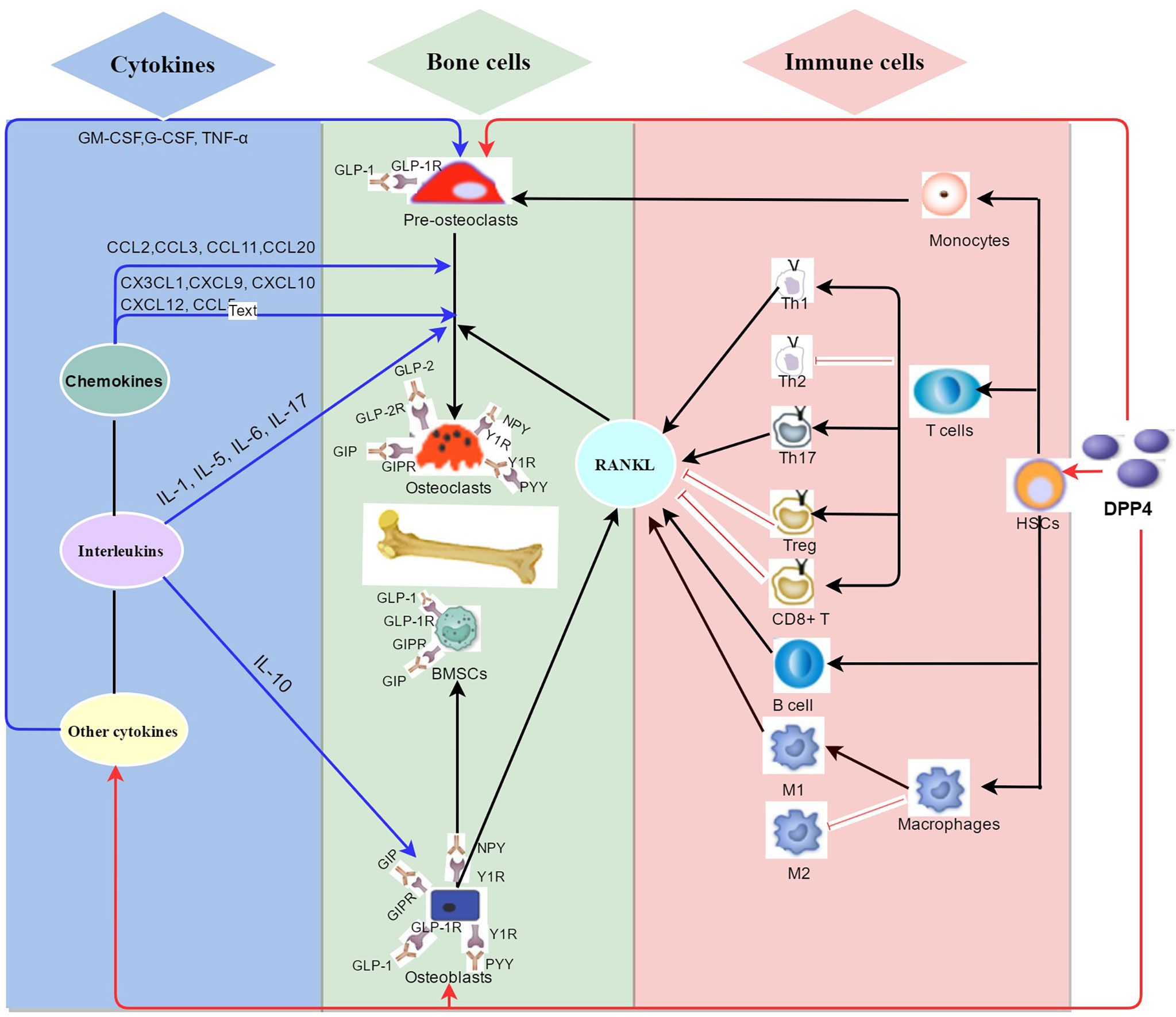
Figure 2 Summary of the potential mechanisms of DPP4 on bone metabolism in bone environment. As a newly discovered adipokine originated from mature adipocytes including bone marrow adipocytes, DPP4 plays a complex role in classical enzyme functions and non-enzyme functions in bone metabolism. (1) (red arrow) DPP4 might indirectly regulate bone remodeling by binding to multiple peptides substrates on bone cells such as glucagon-like peptide-1 (GLP-1), Glucagon-like peptide-2 (GLP-2), and glucose-dependent insulinotropic polypeptide (GIP), neuropeptide Y (NPY) and peptide YY (PYY). (2) (black arrow) DPP4 acts as receptor or costimulatory protein of different immunomodulation signaling process of diversified immune cells including CD4+ T cells, CD8+ T cells, B cells and macrophages. (3) (blue arrow) DPP4 hydrolyzes different sits on chemokines, interleukins, and other cytokines which take part in bone remodeling.
Author Contributions
QY,BF, and DL contributed equal to ideation and drafting and revising of the manuscript. HW and HC contributed to literature search. XC and LT revised the manuscript and contributed with intellectual ideas. XY contributed to the supervision of the study and revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Science and Technology Department of Sichuan Province (2022YFS0136), the Sichuan University (2018SCUH0093), the 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (Nos. 2020HXFH008, ZYJC18003), and the Scientific Research Project Of Sichuan Medical Association (S20036).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank AM, Bocian C, et al. Adipocyte Accumulation In the Bone Marrow During Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell (2017) 20(6):771–84.e6. doi: 10.1016/j.stem.2017.02.009
2. Ou X, O’Leary HA, Broxmeyer HE. Implications of DPP4 Modification of Proteins That Regulate Stem/Progenitor and More Mature Cell Types. Blood (2013) 122(2):161–9. doi: 10.1182/blood-2013-02-487470
3. Drucker DJ, Shi Q, Crivici A, Sumner-Smith M, Tavares W, Hill M, et al. Regulation of the Biological Activity of Glucagon-Like Peptide 2 In Vivo by Dipeptidyl Peptidase Iv. Nat Biotechnol (1997) 15(7):673–7. doi: 10.1038/nbt0797-673
4. Frerker N, Wagner L, Wolf R, Heiser U, Hoffmann T, Rahfeld JU, et al. Neuropeptide Y (NPY) Cleaving Enzymes: Structural and Functional Homologues of Dipeptidyl Peptidase 4. Peptides (2007) 28(2):257–68. doi: 10.1016/j.peptides.2006.09.027
5. Monami M, Dicembrini I, Antenore A, Mannucci E. Dipeptidyl Peptidase-4 Inhibitors and Bone Fractures: A Meta-Analysis of Randomized Clinical Trials. Diabetes Care (2011) 34(11):2474–6. doi: 10.2337/dc11-1099
6. Kyle KA, Willett TL, Baggio LL, Drucker DJ, Grynpas MD. Differential Effects of PPAR-{Gamma} Activation Versus Chemical or Genetic Reduction of DPP-4 Activity on Bone Quality in Mice. Endocrinology (2011) 152(2):457–67. doi: 10.1210/en.2010-1098
7. Cusick T, Mu J, Pennypacker BL, Li Z, Scott KR, Shen X, et al. Bone Loss in the Oestrogen-Depleted Rat Is Not Exacerbated by Sitagliptin, Either Alone or in Combination With a Thiazolidinedione. Diabetes Obes Metab (2013) 15(10):954–7. doi: 10.1111/dom.12109
8. Dong C, Yang H, Wang Y, Yan X, Li D, Cao Z, et al. Anagliptin Stimulates Osteoblastic Cell Differentiation and Mineralization. Biomed Pharmacother (2020) 129:109796. doi: 10.1016/j.biopha.2019.109796
9. Nishida H, Suzuki H, Madokoro H, Hayashi M, Morimoto C, Sakamoto M, et al. Blockade of CD26 Signaling Inhibits Human Osteoclast Development. J Bone Miner Res (2014) 29(11):2439–55. doi: 10.1002/jbmr.2277
10. Kim TY, Schafer AL. Diabetes and Bone Marrow Adiposity. Curr Osteoporos Rep (2016) 14(6):337–44. doi: 10.1007/s11914-016-0336-x
11. Mulvihill EE, Drucker DJ. Pharmacology, Physiology, and Mechanisms of Action of Dipeptidyl Peptidase-4 Inhibitors. Endocr Rev (2014) 35(6):992–1019. doi: 10.1210/er.2014-1035
12. Weivoda MM, Chew CK, Monroe DG, Farr JN, Atkinson EJ, Geske JR, et al. Identification of Osteoclast-Osteoblast Coupling Factors in Humans Reveals Links Between Bone and Energy Metabolism. Nat Commun (2020) 11(1):87. doi: 10.1038/s41467-019-14003-6
13. Ohnuma K, Morimoto C. Gene Section (2013). Available at: http://AtlasGeneticsOncology.org.
14. Lambeir AM, Durinx C, Scharpé S, De Meester I. Dipeptidyl-Peptidase IV From Bench to Bedside: An Update on Structural Properties, Functions, and Clinical Aspects of the Enzyme DPP IV. Crit Rev Clin Lab Sci (2003) 40(3):209–94. doi: 10.1080/713609354
15. Mentlein R. Dipeptidyl-Peptidase IV (CD26)–Role in the Inactivation of Regulatory Peptides. Regul Pept (1999) 85(1):9–24. doi: 10.1016/s0167-0115(99)00089-0
16. De Meester I, Korom S, Van Damme J, Scharpé S. CD26, Let It Cut or Cut It Down. Immunol Today (1999) 20(8):367–75. doi: 10.1016/s0167-5699(99)01486-3
17. Morimoto C, Schlossman SF. The Structure and Function of CD26 in the T-Cell Immune Response. Immunol Rev (1998) 161:55–70. doi: 10.1111/j.1600-065x.1998.tb01571.x
18. von Bonin A, Hühn J, Fleischer B. Dipeptidyl-Peptidase IV/CD26 on T Cells: Analysis of an Alternative T-Cell Activation Pathway. Immunol Rev (1998) 161:43–53. doi: 10.1111/j.1600-065x.1998.tb01570.x
19. Yaron A, Naider F. Proline-Dependent Structural and Biological Properties of Peptides and Proteins. Crit Rev Biochem Mol Biol (1993) 28(1):31–81. doi: 10.3109/10409239309082572
20. Guo Y, Hangoc G, Bian H, Pelus LM, Broxmeyer HE. SDF-1/CXCL12 Enhances Survival Chemotaxis of Murine Embryonic Stem Cells and Production of Primitive and Definitive Hematopoietic Progenitor Cells. Stem Cells (2005) 23(9):1324–32. doi: 10.1634/stemcells.2005-0085
21. Christopherson KW 2nd, Hangoc G, Broxmeyer HE. Cell Surface Peptidase CD26/dipeptidylpeptidase IV Regulates CXCL12/Stromal Cell-Derived Factor-1 Alpha-Mediated Chemotaxis of Human Cord Blood CD34+ Progenitor Cells. J Immunol (2002) 169(12):7000–8. doi: 10.4049/jimmunol.169.12.7000
22. Christopherson KW 2nd, Hangoc G, Mantel CR, Broxmeyer HE. Modulation of Hematopoietic Stem Cell Homing and Engraftment by CD26. Science (2004) 305(5686):1000–3. doi: 10.1126/science.1097071
23. Zilleßen P, Celner J, Kretschmann A, Pfeifer A, Racké K, Mayer P. Metabolic Role of Dipeptidyl Peptidase 4 (DPP4) in Primary Human (Pre)Adipocytes. Sci Rep (2016) 6:23074. doi: 10.1038/srep23074
24. Chien CH, Tsai CH, Lin CH, Chou CY, Chen X. Identification of Hydrophobic Residues Critical for DPP-IV Dimerization. Biochemistry (2006) 45(23):7006–12. doi: 10.1021/bi060401c
25. Klemann C, Wagner L, Stephan M, von Hörsten S. Cut to the Chase: A Review of CD26/dipeptidyl Peptidase-4’s (DPP4) Entanglement in the Immune System. Clin Exp Immunol (2016) 185(1):1–21. doi: 10.1111/cei.12781
26. Mortier A, Gouwy M, Van Damme J, Proost P, Struyf S. CD26/Dipeptidylpeptidase IV-Chemokine Interactions: Double-Edged Regulation of Inflammation and Tumor Biology. J Leukoc Biol (2016) 99(6):955–69. doi: 10.1189/jlb.3MR0915-401R
27. Morrison ME, Vijayasaradhi S, Engelstein D, Albino AP, Houghton AN. A Marker for Neoplastic Progression of Human Melanocytes Is a Cell Surface Ectopeptidase. J Exp Med (1993) 177(4):1135–43. doi: 10.1084/jem.177.4.1135
28. De Meester I, Vanham G, Kestens L, Vanhoof G, Bosmans E, Gigase P, et al. Binding of Adenosine Deaminase to the Lymphocyte Surface via CD26. Eur J Immunol (1994) 24(3):566–70. doi: 10.1002/eji.1830240311
29. Dong RP, Kameoka J, Hegen M, Tanaka T, Xu Y, Schlossman SF, et al. Characterization of Adenosine Deaminase Binding to Human CD26 on T Cells and its Biologic Role in Immune Response. J Immunol (1996) 156(4):1349–55.
30. Martín M, Huguet J, Centelles JJ, Franco R. Expression of Ecto-Adenosine Deaminase and CD26 in Human T Cells Triggered by the TCR-CD3 Complex. Possible Role of Adenosine Deaminase as Costimulatory Molecule. J Immunol (1995) 155(10):4630–43.
31. Martinez-Navio JM, Casanova V, Pacheco R, Naval-Macabuhay I, Climent N, Garcia F, et al. Adenosine Deaminase Potentiates the Generation of Effector, Memory, and Regulatory CD4+ T Cells. J Leukoc Biol (2011) 89(1):127–36. doi: 10.1189/jlb.1009696
32. Yu DM, Slaitini L, Gysbers V, Riekhoff AG, Kähne T, Knott HM, et al. Soluble CD26 / Dipeptidyl Peptidase IV Enhances Human Lymphocyte Proliferation In Vitro Independent of Dipeptidyl Peptidase Enzyme Activity and Adenosine Deaminase Binding. Scand J Immunol (2011) 73(2):102–11. doi: 10.1111/j.1365-3083.2010.02488.x
33. Herrera C, Morimoto C, Blanco J, Mallol J, Arenzana F, Lluis C, et al. Comodulation of CXCR4 and CD26 in Human Lymphocytes. J Biol Chem (2001) 276(22):19532–9. doi: 10.1074/jbc.M004586200
34. Ikushima H, Munakata Y, Ishii T, Iwata S, Terashima M, Tanaka H, et al. Internalization of CD26 by Mannose 6-Phosphate/Insulin-Like Growth Factor II Receptor Contributes to T Cell Activation. Proc Natl Acad Sci USA (2000) 97(15):8439–44. doi: 10.1073/pnas.97.15.8439
35. Ishii T, Ohnuma K, Murakami A, Takasawa N, Kobayashi S, Dang NH, et al. CD26-Mediated Signaling for T Cell Activation Occurs in Lipid Rafts Through Its Association With CD45RO. Proc Natl Acad Sci USA (2001) 98(21):12138–43. doi: 10.1073/pnas.211439098
36. Ohnuma K, Yamochi T, Uchiyama M, Nishibashi K, Yoshikawa N, Shimizu N, et al. CD26 Up-Regulates Expression of CD86 on Antigen-Presenting Cells by Means of Caveolin-1. Proc Natl Acad Sci USA (2004) 101(39):14186–91. doi: 10.1073/pnas.0405266101
37. Ohnuma K, Uchiyama M, Yamochi T, Nishibashi K, Hosono O, Takahashi N, et al. Caveolin-1 Triggers T-Cell Activation via CD26 in Association With Carma1. J Biol Chem (2007) 282(13):10117–31. doi: 10.1074/jbc.M609157200
38. Piazza GA, Callanan HM, Mowery J, Hixson DC. Evidence for a Role of Dipeptidyl Peptidase IV in Fibronectin-Mediated Interactions of Hepatocytes With Extracellular Matrix. Biochem J (1989) 262(1):327–34. doi: 10.1042/bj2620327
39. Cheng HC, Abdel-Ghany M, Pauli BU. A Novel Consensus Motif in Fibronectin Mediates Dipeptidyl Peptidase IV Adhesion and Metastasis. J Biol Chem (2003) 278(27):24600–7. doi: 10.1074/jbc.M303424200
40. Zheng T, Yang L, Liu Y, Liu H, Yu J, Zhang X, et al. Plasma DPP4 Activities Are Associated With Osteoporosis in Postmenopausal Women With Normal Glucose Tolerance. J Clin Endocrinol Metab (2015) 100(10):3862–70. doi: 10.1210/jc.2015-2233
41. Carbone LD, Bužková P, Fink HA, Robbins JA, Bethel M, Isales CM, et al. DPP-4 Activity With BMD, Body Composition, and Incident Hip Fracture: The Cardiovascular Health Study. Osteoporos Int (2017) 28(5):1631–40. doi: 10.1007/s00198-017-3916-4
42. Qiu M, Zhai S, Liu D. DPP4 Activities Are Associated With Osteopenia/Osteoporosis and Fracture Risk in Newly Diagnosed Type 2 Diabetes. Int J Endocrinol (2020) 2020:8874272. doi: 10.1155/2020/8874272
43. Notsu M, Kanazawa I, Tanaka S, Yamaguchi T, Sugimoto T. Serum Dipeptidyl Peptidase-4 Is Associated With Multiple Vertebral Fractures in Type 2 Diabetes Mellitus. Clin Endocrinol (Oxf) (2016) 84(3):332–7. doi: 10.1111/cen.12971
44. Durinx C, Neels H, van der Auwera JC, Naelaerts K, Scharpe S, De Meester I. Reference Values for Plasma Dipeptidyl-Peptidase IV Activity and Their Association With Other Laboratory Parameters. Clin Chem Lab Med (2001) 39(2):155–9. doi: 10.1515/CCLM.2001.026
45. Griffith JF, Yeung DK, Ma HT, Leung JC, Kwok TC, Leung PC. Bone Marrow Fat Content in the Elderly: A Reversal of Sex Difference Seen in Younger Subjects. J Magn Reson Imaging (2012) 36(1):225–30. doi: 10.1002/jmri.23619
46. Drucker DJ. Dipeptidyl Peptidase-4 Inhibition and the Treatment of Type 2 Diabetes: Preclinical Biology and Mechanisms of Action. Diabetes Care (2007) 30(6):1335–43. doi: 10.2337/dc07-0228
47. Röhrborn D, Eckel J, Sell H. Shedding of Dipeptidyl Peptidase 4 is Mediated by Metalloproteases and Up-Regulated by Hypoxia in Human Adipocytes and Smooth Muscle Cells. FEBS Lett (2014) 588(21):3870–7. doi: 10.1016/j.febslet.2014.08.029
48. Dombrowski S, Kostev K, Jacob L. Use of Dipeptidyl Peptidase-4 Inhibitors and Risk of Bone Fracture in Patients With Type 2 Diabetes in Germany-A Retrospective Analysis of Real-World Data. Osteoporos Int (2017) 28(8):2421–8. doi: 10.1007/s00198-017-4051-y
49. Driessen JH, van Onzenoort HA, Henry RM, Lalmohamed A, van den Bergh JP, Neef C, et al. Use of Dipeptidyl Peptidase-4 Inhibitors for Type 2 Diabetes Mellitus and Risk of Fracture. Bone (2014) 68:124–30. doi: 10.1016/j.bone.2014.07.030
50. Hirshberg B, Parker A, Edelberg H, Donovan M, Iqbal N. Safety of Saxagliptin: Events of Special Interest in 9156 Patients With Type 2 Diabetes Mellitus. Diabetes Metab Res Rev (2014) 30(7):556–69. doi: 10.1002/dmrr.2502
51. Zhang YS, Zheng YD, Yuan Y, Chen SC, Xie BC. Effects of Anti-Diabetic Drugs on Fracture Risk: A Systematic Review and Network Meta-Analysis. Front Endocrinol (Lausanne) (2021) 12:735824. doi: 10.3389/fendo.2021.735824
52. Ishida M, Shen WR, Kimura K, Kishikawa A, Shima K, Ogawa S, et al. DPP-4 Inhibitor Impedes Lipopolysaccharide-Induced Osteoclast Formation and Bone Resorption In Vivo. BioMed Pharmacother (2019) 109:242–53. doi: 10.1016/j.biopha.2018.10.052
53. Gallagher EJ, Sun H, Kornhauser C, Tobin-Hess A, Epstein S, Yakar S, et al. The Effect of Dipeptidyl Peptidase-IV Inhibition on Bone in a Mouse Model of Type 2 Diabetes. Diabetes Metab Res Rev (2014) 30(3):191–200. doi: 10.1002/dmrr.2466
54. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine Regulation of Energy Metabolism by the Skeleton. Cell (2007) 130(3):456–69. doi: 10.1016/j.cell.2007.05.047
55. Gerich J. DPP-4 Inhibitors: What May Be the Clinical Differentiators? Diabetes Res Clin Pract (2010) 90(2):131–40. doi: 10.1016/j.diabres.2010.07.006
56. Sell H, Blüher M, Klöting N, Schlich R, Willems M, Ruppe F, et al. Adipose Dipeptidyl Peptidase-4 and Obesity: Correlation With Insulin Resistance and Depot-Specific Release From Adipose Tissue In Vivo and In Vitro. Diabetes Care (2013) 36(12):4083–90. doi: 10.2337/dc13-0496
57. Romacho T, Vallejo S, Villalobos LA, Wronkowitz N, Indrakusuma I, Sell H, et al. Soluble Dipeptidyl Peptidase-4 Induces Microvascular Endothelial Dysfunction Through Proteinase-Activated Receptor-2 and Thromboxane A2 Release. J Hypertens (2016) 34(5):869–76. doi: 10.1097/HJH.0000000000000886
58. Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, et al. Bone Marrow Adipose Tissue Is an Endocrine Organ That Contributes to Increased Circulating Adiponectin During Caloric Restriction. Cell Metab (2014) 20(2):368–75. doi: 10.1016/j.cmet.2014.06.003
59. Xiao L, Sobue T, Esliger A, Kronenberg MS, Coffin JD, Doetschman T, et al. Disruption of the Fgf2 Gene Activates the Adipogenic and Suppresses the Osteogenic Program in Mesenchymal Marrow Stromal Stem Cells. Bone (2010) 47(2):360–70. doi: 10.1016/j.bone.2010.05.021
60. Oshima K, Nampei A, Matsuda M, Iwaki M, Fukuhara A, Hashimoto J, et al. Adiponectin Increases Bone Mass by Suppressing Osteoclast and Activating Osteoblast. Biochem Biophys Res Commun (2005) 331(2):520–6. doi: 10.1016/j.bbrc.2005.03.210
61. Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, et al. Adiponectin, an Adipocyte-Derived Plasma Protein, Inhibits Endothelial NF-kappaB Signaling Through a cAMP-Dependent Pathway. Circulation (2000) 102(11):1296–301. doi: 10.1161/01.cir.102.11.1296
62. Wu Y, Tu Q, Valverde P, Zhang J, Murray D, Dong LQ, et al. Central Adiponectin Administration Reveals New Regulatory Mechanisms of Bone Metabolism in Mice. Am J Physiol Endocrinol Metab (2014) 306(12):E1418–1430. doi: 10.1152/ajpendo.00048.2014
63. Kirino Y, Sei M, Kawazoe K, Minakuchi K, Sato Y. Plasma Dipeptidyl Peptidase 4 Activity Correlates With Body Mass Index and the Plasma Adiponectin Concentration in Healthy Young People. Endocr J (2012) 59(10):949–53. doi: 10.1507/endocrj.ej12-0158
64. Sahebkar A, Ponzo V, Bo S. Effect of Dipeptidyl Peptidase-4 Inhibitors on Plasma Adiponectin: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Curr Med Chem (2016) 23(13):1356–69. doi: 10.2174/0929867323666160405111354
65. Kim Chung le T, Hosaka T, Yoshida M, Harada N, Sakaue H, Sakai T, et al. Exendin-4, a GLP-1 Receptor Agonist, Directly Induces Adiponectin Expression Through Protein Kinase A Pathway and Prevents Inflammatory Adipokine Expression. Biochem Biophys Res Commun (2009) 390(3):613–8. doi: 10.1016/j.bbrc.2009.10.015
66. Piao L, Zhao G, Zhu E, Inoue A, Shibata R, Lei Y, et al. Chronic Psychological Stress Accelerates Vascular Senescence and Impairs Ischemia-Induced Neovascularization: The Role of Dipeptidyl Peptidase-4/Glucagon-Like Peptide-1-Adiponectin Axis. J Am Heart Assoc (2017) 6(10):e006421. doi: 10.1161/JAHA.117.006421
67. Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, et al. Leptin Inhibits Bone Formation Through a Hypothalamic Relay: A Central Control of Bone Mass. Cell (2000) 100(2):197–207. doi: 10.1016/s0092-8674(00)81558-5
68. Cornish J, Callon KE, Bava U, Lin C, Naot D, Hill BL, et al. Leptin Directly Regulates Bone Cell Function In Vitro and Reduces Bone Fragility In Vivo. J Endocrinol (2002) 175(2):405–15. doi: 10.1677/joe.0.1750405
69. Kohno D, Furusawa K, Kitamura T. Anagliptin Suppresses Diet-Induced Obesity Through Enhancing Leptin Sensitivity and Ameliorating Hyperphagia in High-Fat High-Sucrose Diet Fed Mice. Endocr J (2020) 67(5):523–9. doi: 10.1507/endocrj.EJ19-0389
70. Osonoi T, Saito M, Hariya N, Goto M, Mochizuki K. Add-On Therapy With Anagliptin in Japanese Patients With Type-2 Diabetes Mellitus Treated With Metformin and Miglitol Can Maintain Higher Concentrations of Biologically Active GLP-1/Total GIP and a Lower Concentration of Leptin. Peptides (2016) 86:118–25. doi: 10.1016/j.peptides.2016.10.011
71. Kieffer TJ, McIntosh CH, Pederson RA. Degradation of Glucose-Dependent Insulinotropic Polypeptide and Truncated Glucagon-Like Peptide 1 In Vitro and In Vivo by Dipeptidyl Peptidase IV. Endocrinology (1995) 136(8):3585–96. doi: 10.1210/endo.136.8.7628397
72. Deacon CF, Johnsen AH, Holst JJ. Degradation of Glucagon-Like Peptide-1 by Human Plasma In Vitro Yields an N-Terminally Truncated Peptide That Is a Major Endogenous Metabolite In Vivo. J Clin Endocrinol Metab (1995) 80(3):952–7. doi: 10.1210/jcem.80.3.7883856
73. Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-Like Peptide-1-(7-36)Amide Is Transformed to Glucagon-Like Peptide-1-(9-36)Amide by Dipeptidyl Peptidase IV in the Capillaries Supplying the L Cells of the Porcine Intestine. Endocrinology (1999) 140(11):5356–63. doi: 10.1210/endo.140.11.7143
74. Meng J, Ma X, Wang N, Jia M, Bi L, Wang Y, et al. Activation of GLP-1 Receptor Promotes Bone Marrow Stromal Cell Osteogenic Differentiation Through β-Catenin. Stem Cell Rep (2016) 6(4):579–91. doi: 10.1016/j.stemcr.2016.02.002
75. Pereira M, Jeyabalan J, Jørgensen CS, Hopkinson M, Al-Jazzar A, Roux JP, et al. Chronic Administration of Glucagon-Like Peptide-1 Receptor Agonists Improves Trabecular Bone Mass and Architecture in Ovariectomised Mice. Bone (2015) 81:459–67. doi: 10.1016/j.bone.2015.08.006
76. Nuche-Berenguer B, Portal-Núñez S, Moreno P, González N, Acitores A, López-Herradón A, et al. Presence of a Functional Receptor for GLP-1 in Osteoblastic Cells, Independent of the cAMP-Linked GLP-1 Receptor. J Cell Physiol (2010) 225(2):585–892. doi: 10.1002/jcp.22243
77. Nuche-Berenguer B, Moreno P, Esbrit P, Dapía S, Caeiro JR, Cancelas J, et al. Effect of GLP-1 Treatment on Bone Turnover in Normal, Type 2 Diabetic, and Insulin-Resistant States. Calcif Tissue Int (2009) 84(6):453–61. doi: 10.1007/s00223-009-9220-3
78. Sun HX, Lu N, Luo X, Zhao L, Liu JM. Liraglutide, the Glucagon-Like Peptide-1 Receptor Agonist, Has Anabolic Bone Effects in Diabetic Goto-Kakizaki Rats. J Diabetes (2015) 7(4):584–8. doi: 10.1111/1753-0407.12282
79. Pacheco-Pantoja EL, Dillon JP, Wilson PJ, Fraser WD, Gallagher JA. C-Fos Induction by Gut Hormones and Extracellular ATP in Osteoblastic-Like Cell Lines. Purinergic Signal (2016) 1(4):647–51. doi: 10.1007/s11302-016-9526-3
80. Feng Y, Su L, Zhong X, Guohong W, Xiao H, Li Y, et al. Exendin-4 Promotes Proliferation and Differentiation of MC3T3-E1 Osteoblasts by MAPKs Activation. J Mol Endocrinol (2016) 56(3):189–99. doi: 10.1530/JME-15-0264
81. Wu X, Li S, Xue P, Li Y. Liraglutide, a Glucagon-Like Peptide-1 Receptor Agonist, Facilitates Osteogenic Proliferation and Differentiation in MC3T3-E1 Cells Through Phosphoinositide 3-Kinase (PI3K)/protein Kinase B (AKT), Extracellular Signal-Related Kinase (ERK)1/2, and cAMP/Protein Kinase A (PKA) Signaling Pathways Involving β-Catenin. Exp Cell Res (2017) 360(2):281–91. doi: 10.1016/j.yexcr.2017.09.018
82. Mieczkowska A, Mansur S, Bouvard B, Flatt PR, Thorens B, Irwin N, et al. Double Incretin Receptor Knock-Out (DIRKO) Mice Present With Alterations of Trabecular and Cortical Micromorphology and Bone Strength. Osteoporos Int (2015) 26(1):209–18. doi: 10.1007/s00198-014-2845-8
83. Ma X, Meng J, Jia M, Bi L, Zhou Y, Wang Y, et al. Exendin-4, a Glucagon-Like Peptide-1 Receptor Agonist, Prevents Osteopenia by Promoting Bone Formation and Suppressing Bone Resorption in Aged Ovariectomized Rats. J Bone Miner Res (2013) 28(7):1641–52. doi: 10.1002/jbmr.1898
84. Iepsen EW, Lundgren JR, Hartmann B, Pedersen O, Hansen T, Jørgensen NR, et al. GLP-1 Receptor Agonist Treatment Increases Bone Formation and Prevents Bone Loss in Weight-Reduced Obese Women. J Clin Endocrinol Metab (2015) 100(8):2909–17. doi: 10.1210/jc.2015-1176
85. Hartmann B, Johnsen AH, Orskov C, Adelhorst K, Thim L, Holst JJ. Structure, Measurement, and Secretion of Human Glucagon-Like Peptide-2. Peptides (2000) 21(1):73–80. doi: 10.1016/s0196-9781(99)00176-x
86. Holst JJ. Gut Hormones as Pharmaceuticals. From Enteroglucagon to GLP-1 and GLP-2. Regul Pept (2000) 93(1-3):45–51. doi: 10.1016/s0167-0115(00)00185-3
87. Janssen P, Rotondo A, Mulé F, Tack J. Review Article: A Comparison of Glucagon-Like Peptides 1 and 2. Aliment Pharmacol Ther (2013) 37(1):18–36. doi: 10.1111/apt.12092
88. Henriksen DB, Alexandersen P, Hartmann B, Adrian CL, Byrjalsen I, Bone HG, et al. Disassociation of Bone Resorption and Formation by GLP-2: A 14-Day Study in Healthy Postmenopausal Women. Bone (2007) 40(3):723–9. doi: 10.1016/j.bone.2006.09.025
89. Henriksen DB, Alexandersen P, Hartmann B, Adrian CL, Byrjalsen I, Bone HG, et al. Four-Month Treatment With GLP-2 Significantly Increases Hip BMD: A Randomized, Placebo-Controlled, Dose-Ranging Study in Postmenopausal Women With Low BMD. Bone (2009) 45(5):833–42. doi: 10.1016/j.bone.2009.07.008
90. Berlier JL, Kharroubi I, Zhang J, Dalla Valle A, Rigutto S, Mathieu M, et al. Glucose-Dependent Insulinotropic Peptide Prevents Serum Deprivation-Induced Apoptosis in Human Bone Marrow-Derived Mesenchymal Stem Cells and Osteoblastic Cells. Stem Cell Rev Rep (2015) 11(6):841–51. doi: 10.1007/s12015-015-9616-6
91. Tsukiyama K, Yamada Y, Yamada C, Harada N, Kawasaki Y, Ogura M, et al. Gastric Inhibitory Polypeptide as an Endogenous Factor Promoting New Bone Formation After Food Ingestion. Mol Endocrinol (2006) 20(7):1644–51. doi: 10.1210/me.2005-0187
92. Baggio LL, Drucker DJ. Biology of Incretins: GLP-1 and GIP. Gastroenterology (2007) 132(6):2131–57. doi: 10.1053/j.gastro.2007.03.054
93. Bollag RJ, Zhong Q, Phillips P, Min L, Zhong L, Cameron R, et al. Osteoblast-Derived Cells Express Functional Glucose-Dependent Insulinotropic Peptide Receptors. Endocrinology (2000) 141(3):1228–35. doi: 10.1210/endo.141.3.7366
94. Zhong Q, Itokawa T, Sridhar S, Ding KH, Xie D, Kang B, et al. Effects of Glucose-Dependent Insulinotropic Peptide on Osteoclast Function. Am J Physiol Endocrinol Metab (2007) 292(2):E543–8. doi: 10.1152/ajpendo.00364.2006
95. Pacheco-Pantoja EL, Ranganath LR, Gallagher JA, Wilson PJ, Fraser WD. Receptors and Effects of Gut Hormones in Three Osteoblastic Cell Lines. BMC Physiol (2011) 11:12. doi: 10.1186/1472-6793-11-12
96. Xie D, Cheng H, Hamrick M, Zhong Q, Ding KH, Correa D, et al. Glucose-Dependent Insulinotropic Polypeptide Receptor Knockout Mice Have Altered Bone Turnover. Bone (2005) 37(6):759–69. doi: 10.1016/j.bone.2005.06.021
97. Christensen MB, Lund A, Calanna S, Jørgensen NR, Holst JJ, Vilsbøll T, et al. Glucose-Dependent Insulinotropic Polypeptide (GIP) Inhibits Bone Resorption Independently of Insulin and Glycemia. J Clin Endocrinol Metab (2018) 103(1):288–94. doi: 10.1210/jc.2017-01949
98. Lee NJ, Herzog H. NPY Regulation of Bone Remodelling. Neuropeptides (2009) 43(6):457–63. doi: 10.1016/j.npep.2009.08.006
99. Igwe JC, Jiang X, Paic F, Ma L, Adams DJ, Baldock PA, et al. Neuropeptide Y Is Expressed by Osteocytes and Can Inhibit Osteoblastic Activity. J Cell Biochem (2009) 108(3):621–30. doi: 10.1002/jcb.22294
100. Khor EC, Wee NK, Baldock PA. Influence of Hormonal Appetite and Energy Regulators on Bone. Curr Osteoporos Rep (2013) 11(3):194–202. doi: 10.1007/s11914-013-0157-0
101. Lee NJ, Nguyen AD, Enriquez RF, Doyle KL, Sainsbury A, Baldock PA, et al. Osteoblast Specific Y1 Receptor Deletion Enhances Bone Mass. Bone (2011) 48(3):461–7. doi: 10.1016/j.bone.2010.10.174
102. Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Rose W, Rone J, Movafagh S, Ji H, et al. Neuropeptide Y: A Novel Angiogenic Factor From the Sympathetic Nerves and Endothelium. Circ Res (1998) 83(2):187–95. doi: 10.1161/01.res.83.2.187
103. Adrian TE, Ferri GL, Bacarese-Hamilton AJ, Fuessl HS, Polak JM, Bloom SR. Human Distribution and Release of a Putative New Gut Hormone, Peptide YY. Gastroenterology (1985) 89(5):1070–7. doi: 10.1016/0016-5085(85)90211-2
104. Medeiros MD, Turner AJ. Processing and Metabolism of Peptide-YY: Pivotal Roles of Dipeptidylpeptidase-IV, Aminopeptidase-P, and Endopeptidase-24.11. Endocrinology (1994) 134(5):2088–94. doi: 10.1210/endo.134.5.7908871
105. Russell M, Stark J, Nayak S, Miller KK, Herzog DB, Klibanski A, et al. Peptide YY in Adolescent Athletes With Amenorrhea, Eumenorrheic Athletes and non-Athletic Controls. Bone (2009) 45(1):104–9. doi: 10.1016/j.bone.2009.03.668
106. Haderslev KV, Jeppesen PB, Hartmann B, Thulesen J, Sorensen HA, Graff J, et al. Short-Term Administration of Glucagon-Like Peptide-2. Effects on Bone Mineral Density and Markers of Bone Turnover in Short-Bowel Patients With No Colon. Scand J Gastroenterol (2002) 37(4):392–8. doi: 10.1080/003655202317316006
107. Weitzmann MN, Ofotokun I. Physiological and Pathophysiological Bone Turnover - Role of the Immune System. Nat Rev Endocrinol (2016) 12(9):518–32. doi: 10.1038/nrendo.2016.91
108. Fu Y, Wang T, Xiu L, Shi X, Bian Z, Zhang Y, et al. Levamisole Promotes Murine Bone Marrow Derived Dendritic Cell Activation and Drives Th1 Immune Response In Vitro and In Vivo. Int Immunopharmacol (2016) 31:57–65. doi: 10.1016/j.intimp.2015.12.015
109. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In Vivo Imaging of Treg Cells Providing Immune Privilege to the Haematopoietic Stem-Cell Niche. Nature (2011) 474(7350):216–9. doi: 10.1038/nature10160
110. Glatman Zaretsky A, Konradt C, Dépis F, Wing JB, Goenka R, Atria DG, et al. T Regulatory Cells Support Plasma Cell Populations in the Bone Marrow. Cell Rep (2017) 18(8):1906–16. doi: 10.1016/j.celrep.2017.01.067
111. Stanley KT, VanDort C, Motyl C, Endres J, Fox DA. Immunocompetent Properties of Human Osteoblasts: Interactions With T Lymphocytes. J Bone Miner Res (2006) 21(1):29–36. doi: 10.1359/JBMR.051004
112. Nishida H, Suzuki H, Madokoro H, Hayashi M, Morimoto C, Sakamoto M, et al. Blockade of CD26 Signaling Inhibits Human Osteoclast Development. J Bone Miner Res (2014) 29(11):2439–55. doi: 10.1002/jbmr.2277
113. Ghorpade DS, Ozcan L, Zheng Z, Nicoloro SM, Shen Y, Chen E, et al. Hepatocyte-Secreted DPP4 in Obesity Promotes Adipose Inflammation and Insulin Resistance. Nature (2018) 555(7698):673–7. doi: 10.1038/nature26138
114. Wronkowitz N, Görgens SW, Romacho T, Villalobos LA, Sánchez-Ferrer CF, Peiró C, et al. Soluble DPP4 Induces Inflammation and Proliferation of Human Smooth Muscle Cells via Protease-Activated Receptor 2. Biochim Biophys Acta (2014) 1842(9):1613–21. doi: 10.1016/j.bbadis.2014.06.004
115. Zhong J, Rao X, Deiuliis J, Braunstein Z, Narula V, Hazey J, et al. A Potential Role for Dendritic Cell/Macrophage-Expressing DPP4 in Obesity-Induced Visceral Inflammation. Diabetes (2013) 62(1):149–57. doi: 10.2337/db12-0230
116. Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. B Cells and T Cells are Critical for the Preservation of Bone Homeostasis and Attainment of Peak Bone Mass In Vivo. Blood (2007) 109(9):3839–48. doi: 10.1182/blood-2006-07-037994
117. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, Cytokine Profile and Function of Human Interleukin 17-Producing Helper T Cells. Nat Immunol (2007) 8(9):950–7. doi: 10.1038/ni1497
118. Yasuda K, Takeuchi Y, Hirota K. The Pathogenicity of Th17 Cells in Autoimmune Diseases. Semin Immunopathol (2019) 41(3):283–97. doi: 10.1007/s00281-019-00733-8
119. Hirahara K, Nakayama T. CD4+ T-Cell Subsets in Inflammatory Diseases: Beyond the Th1/Th2 Paradigm. Int Immunol (2016) 28(4):163–71. doi: 10.1093/intimm/dxw006
120. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface Phenotype and Antigenic Specificity of Human Interleukin 17-Producing T Helper Memory Cells. Nat Immunol (2007) 8(6):639–46. doi: 10.1038/ni1467
121. Bengsch B, Seigel B, Flecken T, Wolanski J, Blum HE, Thimme R. Human Th17 Cells Express High Levels of Enzymatically Active Dipeptidylpeptidase IV (Cd26). J Immunol (2012) 188(11):5438–47. doi: 10.4049/jimmunol.1103801
122. Zhuge F, Ni Y, Nagashimada M, Nagata N, Xu L, Mukaida N, et al. DPP-4 Inhibition by Linagliptin Attenuates Obesity-Related Inflammation and Insulin Resistance by Regulating M1/M2 Macrophage Polarization. Diabetes (2016) 65(10):2966–79. doi: 10.2337/db16-0317
123. Kitagawa N, Hamaguchi M, Majima S, Fukuda T, Kimura T, Hashimoto Y, et al. Dipeptidyl Peptidase-4 Inhibitors Have Adverse Effects for the Proliferation of Human T Cells. J Clin Biochem Nutr (2018) 63(2):106–12. doi: 10.3164/jcbn.17-64
124. Augustyns K, Bal G, Thonus G, Belyaev A, Zhang XM, Bollaert W, et al. The Unique Properties of Dipeptidyl-Peptidase IV (DPP IV/CD26) and the Therapeutic Potential of DPP IV Inhibitors. Curr Med Chem (1999) 6(4):311–27.
125. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two Types of Murine Helper T Cell Clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins. J Immunol (1986) 136(7):2348–57.
126. Fox SW, Chambers TJ. Interferon-Gamma Directly Inhibits TRANCE-Induced Osteoclastogenesis. Biochem Biophys Res Commun (2000) 276(3):868–72. doi: 10.1006/bbrc.2000.3577
127. Kubota T, Flentke GR, Bachovchin WW, Stollar BD. Involvement of Dipeptidyl Peptidase IV in an In Vivo Immune Response. Clin Exp Immunol (1992) 89(2):192–7. doi: 10.1111/j.1365-2249.1992.tb06931.x
128. Gliddon DR, Howard CJ. CD26 is Expressed on a Restricted Subpopulation of Dendritic Cells In Vivo. Eur J Immunol (2002) 32(5):1472–81. doi: 10.1002/1521-4141(200205)32:5<1472::AID-IMMU1472>3.0.CO;2-Q
129. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T Cell Subsets and Their Signature Cytokines in Autoimmune and Inflammatory Diseases. Cytokine (2015) 74(1):5–17. doi: 10.1016/j.cyto.2014.09.011
130. Adamopoulos IE, Chao CC, Geissler R, Laface D, Blumenschein W, Iwakura Y, et al. Interleukin-17A Upregulates Receptor Activator of NF-kappaB on Osteoclast Precursors. Arthritis Res Ther (2010) 12(1):R29. doi: 10.1186/ar2936
131. Vora KA, Porter G, Peng R, Cui Y, Pryor K, Eiermann G, et al. Genetic Ablation or Pharmacological Blockade of Dipeptidyl Peptidase IV Does Not Impact T Cell-Dependent Immune Responses. BMC Immunol (2009) 10:19. doi: 10.1186/1471-2172-10-19
132. Coburn MC, Hixson DC, Reichner JS. In Vitro Immune Responsiveness of Rats Lacking Active Dipeptidylpeptidase IV. Cell Immunol (1994) 158(2):269–80. doi: 10.1006/cimm.1994.1275
133. Shashkova EV, Trivedi J, Cline-Smith AB, Ferris C, Buchwald ZS, Gibbs J, et al. Osteoclast-Primed Foxp3+ CD8 T Cells Induce T-Bet, Eomesodermin, and IFN-γ To Regulate Bone Resorption. J Immunol (2016) 197(3):726–35. doi: 10.4049/jimmunol.1600253
134. Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, et al. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol Rev (2017) 97(4):1295–349. doi: 10.1152/physrev.00036.2016
135. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and Accumulation of Immunosuppressive Adenosine by Human CD4+CD25highFOXP3+ Regulatory T Cells. J Biol Chem (2010) 285(10):7176–86. doi: 10.1074/jbc.M109.047423
136. Tian L, Gao J, Hao J, Zhang Y, Yi H, O’Brien TD, et al. Reversal of New-Onset Diabetes Through Modulating Inflammation and Stimulating Beta-Cell Replication in Nonobese Diabetic Mice by a Dipeptidyl Peptidase IV Inhibitor. Endocrinology (2010) 151(7):3049–60. doi: 10.1210/en.2010-0068
137. Gong P, Shi B, Wang J, Cao P, Diao Z, Wang Y, et al. Association Between Th1/Th2 Immune Imbalance and Obesity in Women With or Without Polycystic Ovary Syndrome. Gynecol Endocrinol (2018) 34(8):709–14. doi: 10.1080/09513590.2018.1428301
138. Lee GR. The Balance of Th17 Versus Treg Cells in Autoimmunity. Int J Mol Sci (2018) 19(3):730. doi: 10.3390/ijms19030730
139. Buchwald ZS, Kiesel JR, DiPaolo R, Pagadala MS, Aurora R. Osteoclast Activated FoxP3+ CD8+ T-Cells Suppress Bone Resorption In Vitro. PLoS One (2012) 7(6):e38199. doi: 10.1371/journal.pone.0038199
140. Kiesel JR, Buchwald ZS, Aurora R. Cross-Presentation by Osteoclasts Induces FoxP3 in CD8+ T Cells. J Immunol (2009) 182(9):5477–87. doi: 10.4049/jimmunol.0803897
141. Axmann R, Herman S, Zaiss M, Franz S, Polzer K, Zwerina J, et al. CTLA-4 Directly Inhibits Osteoclast Formation. Ann Rheum Dis (2008) 67(11):1603–9. doi: 10.1136/ard.2007.080713
142. Price JD, Linder G, Li WP, Zimmermann B, Rother KI, Malek R, et al. Effects of Short-Term Sitagliptin Treatment on Immune Parameters in Healthy Individuals, a Randomized Placebo-Controlled Study. Clin Exp Immunol (2013) 174(1):120–8. doi: 10.1111/cei.12144
143. Manabe N, Kawaguchi H, Chikuda H, Miyaura C, Inada M, Nagai R, et al. Connection Between B Lymphocyte and Osteoclast Differentiation Pathways. J Immunol (2001) 167(5):2625–31. doi: 10.4049/jimmunol.167.5.2625
144. Hatano R, Ohnuma K, Yamamoto J, Dang NH, Morimoto C. CD26-Mediated Co-Stimulation in Human CD8(+) T Cells Provokes Effector Function via Pro-Inflammatory Cytokine Production. Immunology (2013) 138(2):165–72. doi: 10.1111/imm.12028
145. Bühling F, Junker U, Reinhold D, Neubert K, Jäger L, Ansorge S. Functional Role of CD26 on Human B Lymphocytes. Immunol Lett (1995) 45(1-2):47–51. doi: 10.1016/0165-2478(94)00230-o
146. Yan S, Marguet D, Dobers J, Reutter W, Fan H. Deficiency of CD26 Results in a Change of Cytokine and Immunoglobulin Secretion After Stimulation by Pokeweed Mitogen. Eur J Immunol (2003) 33(6):1519–27. doi: 10.1002/eji.200323469
147. Klemann C, Schade J, Pabst R, Leitner S, Stiller J, von Hörsten S, et al. CD26/dipeptidyl Peptidase 4-Deficiency Alters Thymic Emigration Patterns and Leukcocyte Subsets in F344-Rats Age-Dependently. Clin Exp Immunol (2009) 155(2):357–65. doi: 10.1111/j.1365-2249.2008.03839.x
148. Hume DA. Differentiation and Heterogeneity in the Mononuclear Phagocyte System. Mucosal Immunol (2008) 1(6):432–41. doi: 10.1038/mi.2008.36
149. Zhu LL, Blair H, Cao J, Yuen T, Latif R, Guo L, et al. Blocking Antibody to the β-Subunit of FSH Prevents Bone Loss by Inhibiting Bone Resorption and Stimulating Bone Synthesis. Proc Natl Acad Sci USA (2012) 109(36):14574–9. doi: 10.1073/pnas.1212806109
150. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic Cell Clearance: Basic Biology and Therapeutic Potential. Nat Rev Immunol (2014) 14(3):166–80. doi: 10.1038/nri3607
151. Horwood NJ. Macrophage Polarization and Bone Formation: A Review. Clin Rev Allergy Immunol (2016) 51(1):79–86. doi: 10.1007/s12016-015-8519-2
152. Shah Z, Kampfrath T, Deiuliis JA, Zhong J, Pineda C, Ying Z, et al. Long-Term Dipeptidyl-Peptidase 4 Inhibition Reduces Atherosclerosis and Inflammation via Effects on Monocyte Recruitment and Chemotaxis. Circulation (2011) 124(21):2338–49. doi: 10.1161/CIRCULATIONAHA.111.041418
153. Hiromura M, Nohtomi K, Mori Y, Kataoka H, Sugano M, Ohnuma K, et al. Caveolin-1, a Binding Protein of CD26, is Essential for the Anti-Inflammatory Effects of Dipeptidyl Peptidase-4 Inhibitors on Human and Mouse Macrophages. Biochem Biophys Res Commun (2018) 495(1):223–9. doi: 10.1016/j.bbrc.2017.11.016
154. shi D, Fujiwara Y, Komohara Y, Mizuta H, Takeya M. Glucagon-Like Peptide-1 (GLP-1) Induces M2 Polarization of Human Macrophages via STAT3 Activation. Biochem Biophys Res Commun (2012) 425(2):304–8. doi: 10.1016/j.bbrc.2012.07.086
155. Koizumi K, Saitoh Y, Minami T, Takeno N, Tsuneyama K, Miyahara T, et al. Role of CX3CL1/fractalkine in Osteoclast Differentiation and Bone Resorption. J Immunol (2009) 183(12):7825–31. doi: 10.4049/jimmunol.0803627
156. Liu Z, Liang W, Kang D, Chen Q, Ouyang Z, Yan H, et al. Increased Osteoblastic Cxcl9 Contributes to the Uncoupled Bone Formation and Resorption in Postmenopausal Osteoporosis. Clin Interv Aging (2020) 15:1201–12. doi: 10.2147/CIA.S254885
157. Grassi F, Cristino S, Toneguzzi S, Piacentini A, Facchini A, Lisignoli G. CXCL12 Chemokine Up-Regulates Bone Resorption and MMP-9 Release by Human Osteoclasts: CXCL12 Levels Are Increased in Synovial and Bone Tissue of Rheumatoid Arthritis Patients. J Cell Physiol (2004) 199(2):244–51. doi: 10.1002/jcp.10445
158. Zannettino AC, Farrugia AN, Kortesidis A, Manavis J, To LB, Martin SK, et al. Elevated Serum Levels of Stromal-Derived Factor-1alpha Are Associated With Increased Osteoclast Activity and Osteolytic Bone Disease in Multiple Myeloma Patients. Cancer Res (2005) 65(5):1700–9. doi: 10.1158/0008-5472.CAN-04-1687
159. Zhu W, Liang G, Huang Z, Doty SB, Boskey AL. Conditional Inactivation of the CXCR4 Receptor in Osteoprecursors Reduces Postnatal Bone Formation Due to Impaired Osteoblast Development. J Biol Chem (2011) 286(30):26794–805. doi: 10.1074/jbc.M111.250985
160. Chung NC, Chen YR, Huang HY, Chuang WP, Lai DM. Imbalanced Osteogenesis and Adipogenesis in Mice Deficient in the Chemokine Cxcl12/Sdf1 in the Bone Mesenchymal Stem/Progenitor Cells. J Bone Miner Res (2018) 33(4):679–90. doi: 10.1002/jbmr.3340
161. Wintges K, Beil FT, Albers J, Jeschke A, Schweizer M, Claass B, et al. Impaired Bone Formation and Increased Osteoclastogenesis in Mice Lacking Chemokine (C-C Motif) Ligand 5 (Ccl5). J Bone Miner Res (2013) 28(10):2070–80. doi: 10.1002/jbmr.1937
162. Stashenko P, Dewhirst FE, Rooney ML, Desjardins LA, Heeley JD. Interleukin-1 Beta Is a Potent Inhibitor of Bone Formation In Vitro. J Bone Miner Res (1987) 2(6):559–65. doi: 10.1002/jbmr.5650020612
163. Nguyen L, Dewhirst FE, Hauschka PV, Stashenko P. Interleukin-1 Beta Stimulates Bone Resorption and Inhibits Bone Formation In Vivo. Lymphokine Cytokine Res (1991) 10(1-2):15–21.
164. Sims NA. Cell-Specific Paracrine Actions of IL-6 Family Cytokines From Bone, Marrow and Muscle That Control Bone Formation and Resorption. Int J Biochem Cell Biol (2016) 79:14–23. doi: 10.1016/j.biocel.2016.08.003
165. Dhodapkar KM, Barbuto S, Matthews P, Kukreja A, Mazumder A, Vesole D, et al. Dendritic Cells Mediate the Induction of Polyfunctional Human IL17-Producing Cells (Th17-1 Cells) Enriched in the Bone Marrow of Patients With Myeloma. Blood (2008) 112(7):2878–85. doi: 10.1182/blood-2008-03-143222
166. Evans KE, Fox SW. Interleukin-10 Inhibits Osteoclastogenesis by Reducing NFATc1 Expression and Preventing its Translocation to the Nucleus. BMC Cell Biol (2007) 8:4. doi: 10.1186/1471-2121-8-4
167. Yang X, Pande S, Scott C, Friesel R. Macrophage Colony-Stimulating Factor Pretreatment of Bone Marrow Progenitor Cells Regulates Osteoclast Differentiation Based Upon the Stage of Myeloid Development. J Cell Biochem (2019) 120(8):12450–60. doi: 10.1002/jcb.28512
168. Lu X, Gilbert L, He X, Rubin J, Nanes MS. Transcriptional Regulation of the Osterix (Osx, Sp7) Promoter by Tumor Necrosis Factor Identifies Disparate Effects of Mitogen-Activated Protein Kinase and NF Kappa B Pathways. J Biol Chem (2006) 28(10):6297–306. doi: 10.1074/jbc.M507804200
169. Broxmeyer HE, Hoggatt J, O’Leary HA, Mantel C, Chitteti BR, Cooper S, et al. Dipeptidylpeptidase 4 Negatively Regulates Colony-Stimulating Factor Activity and Stress Hematopoiesis. Nat Med (2012) 18(12):1786–96. doi: 10.1038/nm.2991
170. Yokota K, Sato K, Miyazaki T, Kitaura H, Kayama H, Miyoshi F, et al. Combination of Tumor Necrosis Factor α and Interleukin-6 Induces Mouse Osteoclast-Like Cells With Bone Resorption Activity Both In Vitro and In Vivo. Arthritis Rheumatol (2014) 66(1):121–9. doi: 10.1002/art.38218
171. Zhang Q, Chen B, Yan F, Guo J, Zhu X, Ma S, et al. Interleukin-10 Inhibits Bone Resorption: A Potential Therapeutic Strategy in Periodontitis and Other Bone Loss Diseases. BioMed Res Int (2014) 2014:284836. doi: 10.1155/2014/284836
172. Lubberts E, Koenders MI, van den Berg WB. The Role of T-Cell Interleukin-17 in Conducting Destructive Arthritis: Lessons From Animal Models. Arthritis Res Ther (2005) 7(1):29–37. doi: 10.1186/ar1478
173. Nomura K, Kuroda S, Yoshikawa H, Tomita T. Inflammatory Osteoclastogenesis Can Be Induced by GM-CSF and Activated Under TNF Immunity. Biochem Biophys Res Commun (2008) 367(4):881–7. doi: 10.1016/j.bbrc.2008.01.023
174. Broxmeyer HE, Capitano M, Campbell TB, Hangoc G, Cooper S. Modulation of Hematopoietic Chemokine Effects In Vitro and In Vivo by DPP-4/Cd26. Stem Cells Dev (2016) 25(8):575–85. doi: 10.1089/scd.2016.0026
175. Binder NB, Niederreiter B, Hoffmann O, Stange R, Pap T, Stulnig TM, et al. Estrogen-Dependent and C-C Chemokine Receptor-2-Dependent Pathways Determine Osteoclast Behavior in Osteoporosis. Nat Med (2009) 15(4):417–24. doi: 10.1038/nm.1945
176. Hopwood B, Tsykin A, Findlay DM, Fazzalari NL. Gene Expression Profile of the Bone Microenvironment in Human Fragility Fracture Bone. Bone (2009) 44(1):87–101. doi: 10.1016/j.bone.2008.08.120
177. Wan H, Qian TY, Hu XJ, Huang CY, Yao WF. Correlation of Serum CCL3/MIP-1α Levels With Disease Severity in Postmenopausal Osteoporotic Females. Balkan Med J (2018) 35(4):320–5. doi: 10.4274/balkanmedj.2017.1165
Keywords: dipeptidyl peptidase-4, bone metabolism, osteoimmunology, adipokines, bone microenvironment, cytokines
Citation: Yang Q, Fu B, Luo D, Wang HB, Cao HY, Chen X, Tian L and Yu X (2022) The Multiple Biological Functions of Dipeptidyl Peptidase-4 in Bone Metabolism. Front. Endocrinol. 13:856954. doi: 10.3389/fendo.2022.856954
Received: 17 January 2022; Accepted: 24 March 2022;
Published: 02 May 2022.
Edited by:
Daniela Merlotti, University of Siena, ItalyReviewed by:
Francesco Grassi, Rizzoli Orthopedic Institute (IRCCS), ItalySubhashis Pal, Emory University, United States
Copyright © 2022 Yang, Fu, Luo, Wang, Cao, Chen, Tian and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xijie Yu, eGlqaWV5dUBob3RtYWlsLmNvbQ==