 Nicole Aaron
Nicole Aaron Samantha Costa
Samantha Costa Clifford J. Rosen3,4*
Clifford J. Rosen3,4* Li Qiang
Li Qiang- 1Naomi Berrie Diabetes Center, Columbia University, New York, NY, United States
- 2Department of Pharmacology, Columbia University, New York, NY, United States
- 3Center for Clinical and Translational Research, Maine Medical Center Research Institute, Scarborough, ME, United States
- 4Graduate School of Biomedical Science and Engineering, University of Maine, Orono, ME, United States
- 5Department of Pathology, Columbia University, New York, NY, United States
Once considered an inert filler of the bone cavity, bone marrow adipose tissue (BMAT) is now regarded as a metabolically active organ that plays versatile roles in endocrine function, hematopoiesis, bone homeostasis and metabolism, and, potentially, energy conservation. While the regulation of BMAT is inadequately understood, it is recognized as a unique and dynamic fat depot that is distinct from peripheral fat. As we age, bone marrow adipocytes (BMAds) accumulate throughout the bone marrow (BM) milieu to influence the microenvironment. This process is conceivably signaled by the secretion of adipocyte-derived factors including pro-inflammatory cytokines and adipokines. Adipokines participate in the development of a chronic state of low-grade systemic inflammation (inflammaging), which trigger changes in the immune system that are characterized by declining fidelity and efficiency and cause an imbalance between pro-inflammatory and anti-inflammatory networks. In this review, we discuss the local effects of BMAT on bone homeostasis and the hematopoietic niche, age-related inflammatory changes associated with BMAT accrual, and the downstream effect on endocrine function, energy expenditure, and metabolism. Furthermore, we address therapeutic strategies to prevent BMAT accumulation and associated dysfunction during aging. In sum, BMAT is emerging as a critical player in aging and its explicit characterization still requires further research.
Introduction
All tissues are affected by aging, but diseases that weaken the skeleton constitute the most prevalent chronic impairments in the United States (1–3). Skeletal diseases and related conditions are of grave concern among the aging population as they have the potential to significantly compromise systemic and local functions and diminish quality of life. The increase in bone marrow adiposity (BMA) over a lifetime is thought to be a major contributor to age-associated chronic conditions such as osteoporosis, osteoarthritis, and cancer (4–7). Qualitative studies have reported changes in the bone marrow (BM) of humans since 1882 when Ernest Neumann recognized aging resulted in trabecular bone loss and most of the BM consisted of adipose tissue (8). Since then, studies in both rodents and humans have validated that aging is associated with a significant increase in bone marrow adipose tissue (BMAT) (9, 10) with a concurrent decline in bone mineral density (11). Over the years, considerable advancements have been made related to BM imaging and BMAT quantification in humans and rodents. In humans, quantitative magnetic resonance imaging (MRI) and spectroscopy (MRS) allows for noninvasive monitoring of BMAT development and expansion (12–15). This compares to osmium tetroxide and contrast enhanced computed tomography, considered the gold standard in rodents, which provides both volumetric and spatial quantification of BMAT (16). Notwithstanding the advances in methodologies, BMAT represents an understudied aspect of adipocyte biology. Distinct from peripheral adipose tissue, BMAT displays a unique response to physiological changes (i.e., aging, exercise, cold exposure, nutritional variations like high-fat diet and fasting) (17–20). Furthermore, given its unique location, BMAT directly influences mechanisms of bone remodeling, hematopoiesis, and inflammation within the BM microenvironment (21, 22).
In general, aging is associated with impaired tissue regeneration that is congruent with increased BMA and an inflammaging phenotype. Inflammaging is characterized by unresolved and uncontrolled inflammation and a dysfunctional immune response that exacerbate the aging process and age-related chronic diseases (23, 24). Furthermore, this process is believed to exacerbate the decline in the regenerative capacity of the skeleton (25) by affecting bone marrow stromal cell (BMSC) proliferation, frequency, and fate determination (25). With recent evidence supporting BMAT as an endocrine and paracrine organ capable of local regulation of the BM microenvironment, it is important to further understand the relationship between bone marrow adipocytes (BMAds) and the observed inflammaging phenotype in aging.
The Effects of BMA on Bone Marrow Stromal Cells and Hematopoietic Stem Cells
BMA and BMSC Potential
As we age, our capacity for tissue repair and regeneration in response to injury declines (Figure 1). Accordingly, bone repair is delayed and impaired as well. BMSCs are the foundation of bone regeneration by serving as the progenitor cells of osteoblasts as well as of adipocytes (26, 27). In addition, BMSCs support proliferation and differentiation of hematopoietic stem cells (HSCs), promote HSC engraftment in animal models, and can decrease inflammation under normal conditions (28). However, aging affects BMSCs through intrinsic and extrinsic factors. Intrinsically, BMSCs accumulate DNA damage, reactive oxygen species (ROS), and damaged proteins that may promote aging (29). Extrinsically, the composition of the BM niche and the growth factors and cytokines that are secreted into the local environment change with age (29) (Table 1). In particular, the increase in BMAds may disrupt the microenvironment structure and alter the fate of BMSCs. Age-related bone loss has thought to be driven in part by a decline in BMSC proliferation and function as well as increased commitment of BMSCs to adipogenic lineages (48). At the cellular level, the BMSC pool in the BM niche shows a biased differentiation towards adipogenesis at the cost of osteoblastogenesis in aging (48). Despite their regenerative capabilities, BMSCs were shown to have decreased differentiation potential when exposed to inflammatory environments (49). Josephson et al. revealed that skeletal stem/progenitor cell (SSPC) frequency significantly declined with increased age, and this directly correlated to a longer fracture healing time in a human cohort (25). Using in vivo and in vitro models, the authors recapitulated reduced bone healing commonly associated with advanced aging. SSPCs cultured with 52-week-old serum began to express pro-inflammatory cytokines (elevated IL-1a, TNF-a, RELA expression), illustrating the declined SSPC number and function were negatively affected by the cytokine milieu associated with age (25). The expansion of BMAT, which is known to actively produce pro-inflammatory factors, likely exacerbates this effect (45).
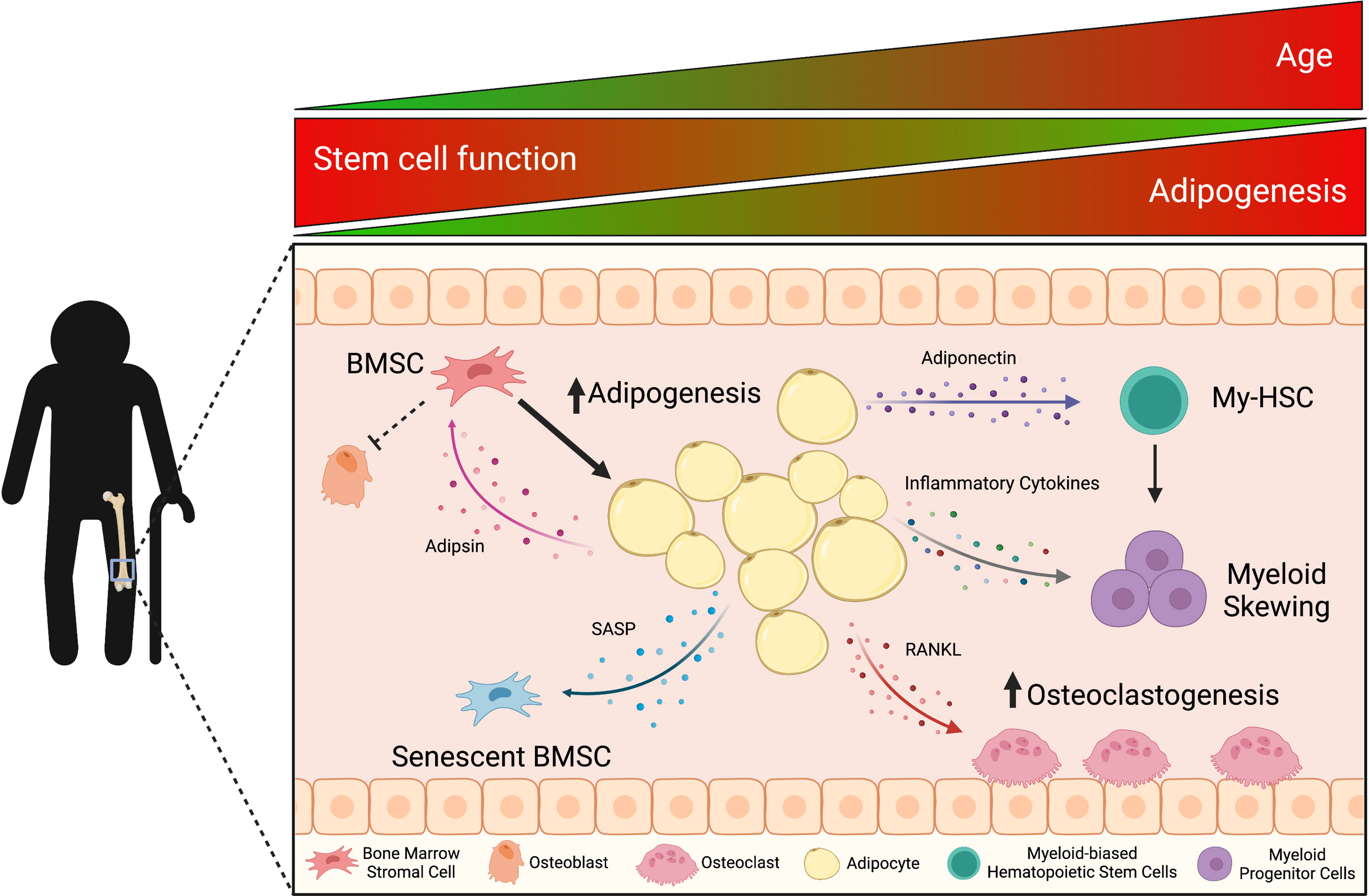
Figure 1 With age, the accumulation of bone marrow adipocyte (BMAd)-derived factors influences mechanisms of bone remodeling, hematopoiesis, and inflammation, which triggers a cascade effect within the bone marrow (BM) microenvironment. Aging is associated with increased bone marrow adiposity (BMA) and decreased bone mineral density. These classic characteristics of aging result from adipsin priming bone marrow stromal cells (BMSCs) towards adipogenesis and adipocytes (including pre-adipocytes in aged mice) secreting the pro-osteoclastic factor, RANKL. Adipocytes also secrete adiponectin and pro-inflammatory cytokines that skew hematopoietic stem cell differentiation towards the myeloid lineage, which is observed in the chronic inflammatory state of aging (inflammaging). In the BM, this pro-inflammatory microenvironment leads to senescence-associated secretory phenotype (SASP) factors decreasing BMSC potential and functionality. This figure was created using BioRender.com.
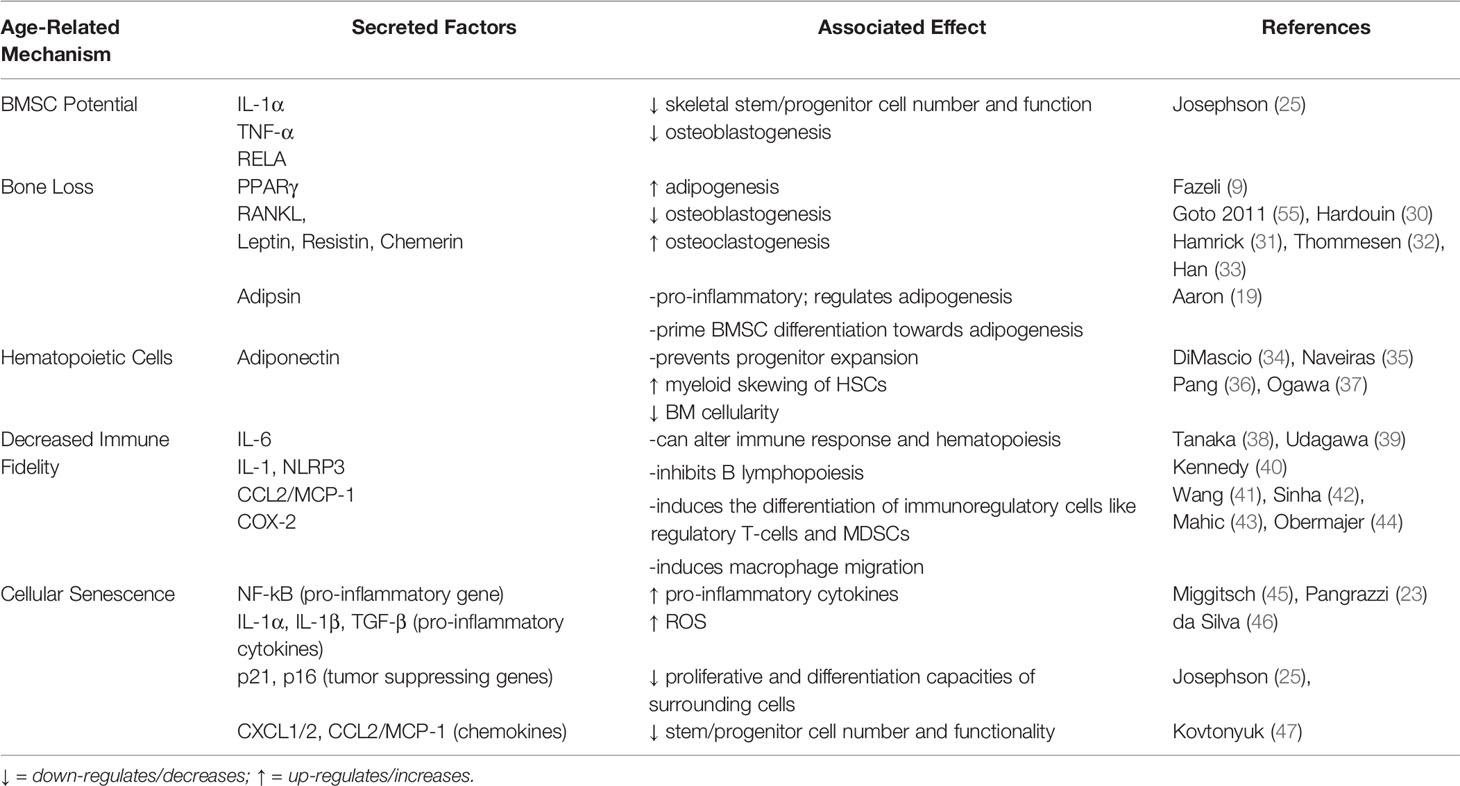
Table 1 BMAT-derived factors and the age-associated phenotype.
Adipogenesis and Bone Loss
Aging studies have shown increased BMAT coincides with decreased bone mass, suggestive of a link between bone formation and BMA. The general understanding is a common progenitor cell undergoes adipogenesis at the expense of osteogenesis (27, 48, 50, 51). For example, it has been shown that upregulation of PPARγ promotes the differentiation of BMSCs into adipocytes while repressing osteoblast differentiation. In aging, the increased expression of PPARγ in the BM leads to enhanced adipogenesis and reduced osteogenesis (9). In addition to expression, post-translational modification of PPARγ, particularly acetylation, is also critical to this lineage determination (19, 52) thus, PPARγ is appreciated as a critical lineage-switching regulator. However, this bifurcated differentiation path between adipocytes and osteoblasts has remained poorly understood, despite the elucidation of PPARγ expression in the BM. Recent studies have delineated mesenchymal progenitors to their bi-lineage differentiation stages and characterized non-proliferative, adiponectin-expressing BMAd precursors, termed MALPs (marrow adipogenic lineage precursor) (53). These are thought to secrete a number of factors that can drive bone loss such as RANKL. Upon maturation, BMAT is responsible for the release of adipokines and free fatty acids that potentially interfere with bone formation (19, 52). For example, adipsin is among the group of adipokines released by BMAT expansion that has been shown to retroactively affect BMSC differentiation by priming these cells toward adipogenesis (19).
Coinciding with the increase in BMA, often an age-related decline in trabecular bone volume, but not in cortical bone, is observed (9). The impaired skeletal health with aging is accounted for not only by defective bone formation capabilities but also by accelerated bone resorption through increased osteoclast number and/or activity (54). In contrast to the repressive function on osteoblasts, BMAds play a favorable role on osteoclasts. Primary human femoral BMAds were shown to express the pro-osteoclastogenic factor, RANKL, and through direct cell contact mediate the differentiation of osteoclast precursors (30, 55, 56). In murine studies, an age-dependent increase in osteoclastogenesis was observed (57). Additionally, RANKL expression was shown to be associated with BMAd differentiation and with pre-adipocytes in the BM of aged mice (30, 58). This creates a self-reinforcing cycle of osteoclastogenesis and adipogenesis which leads to increased deleterious effects on the bone architecture and increases the incidences of fractures within the elderly (54). Furthermore, osteoblasts in aged mice (16 months old) were found to exhibit markedly impaired adhesion to the bone surface and significantly reduced mineralization (59). Thus, the age-associated decline in bone mass is an integrative pathology of BMAds filling the BM cavity and their crosstalk to bone remodeling cells.
BMA and Hematopoietic Cells
While BMAds have a defined function as regulators of bone turnover, evidence also suggests BMAT impacts hematopoietic activity (45, 48). Human BMAds were reported to support differentiation of CD34+ HSCs into myeloid and lymphoid immune cells (60). Accordingly, myelopoiesis was shown to positively correlate with increased adipogenesis and reduced osteoblastogenesis in the senescence-accelerated mouse prone 6 (SAMP6) mouse model, representative of advanced aging (61). In diet-induced obese mice an enhancement in hematopoietic and lymphopoietic BM cell populations were correlated with increased marrow adiposity (62). In contrast, lipid-laden BMAds were linked to the suppression of growth and differentiation of HSCs (35, 63) and were considered negative regulators of the hematopoietic niche (64, 65). This suppressive activity was primarily attributed to reduced production of granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF) as well as increased secretion of neuropilin and lipocalin-2 (35, 66, 67). Of note, BMAds are a significant source of plasma adiponectin in mice during calorie restriction and in cancer patients receiving radiotherapy or chemotherapy (20). Moreover, increased BMA during aging has been negatively correlated to hematopoietic cell function during aging through the secretion of adiponectin (20). Adiponectin appears to positively affect multipotent stem cells proliferation, but not more committed progenitor cells (34), a phenomenon suspected in preserving the HSC pool while preventing progenitor expansion (35). This ultimately highlights the anti-inflammatory properties of adiponectin (68) and the dynamic relationship between BMAds and the hematopoietic niche. Overall, aging in humans and mice, a process associated with increased BMA (69–71), induces myeloid skewing in HSCs (36), while promoting an overall decrease in BM cellularity (37).
Age-Related BMAT Expansion Results in Decreased Immune Fidelity and Cellular Senescence
Decreased Immune Fidelity
With aging, inflammaging is thought to be a major contributor to the decline in fidelity and efficiency of the immune system. The immune system waxes and wanes in response to stimuli. A decline in immunocompetency or the capacity for a normal functioning immune system with aging can increase susceptibility to infections, decrease the number of T- and B-cells as myelopoiesis occurs (the process in which innate immune cells develop from myeloid progenitor cells), and increase the prevalence of autoimmune diseases (47). Gasparrini et al. analyzed cytokines produced by BMAT and found 53 proteins upregulated in aging (72), one of which they identified as IL-6, a well-known pro-inflammatory protein that can affect immune response, hematopoiesis, and suppress bone formation (38, 39). In vitro cultures of BMAds were shown to secrete adipocyte-derived soluble factors that inhibit B lymphopoiesis, particularly at the earliest progenitor stage in which differentiation into pre-pro B-cells occurs, while simultaneously promoting the differentiation and subsequent proliferation of HSCs towards the myeloid lineage (73). In humans and mice (74–76), B lymphopoiesis wanes in mid (77) and late stages of life (73, 78, 79). In mice, the decline in B lymphopoiesis has been attributed to BMAds altering the BM stroma and/or by direct action on hematopoietic progenitors (77–79). Kennedy et al. revealed that BMAds induce myeloid-derived suppressor cells (MDSCs), particularly in mononuclear cells (CD11b+Ly6C+Ly6G−), which inhibit B lymphopoiesis by producing IL-1 (80). Additionally, BMAds can also activate inflammasomes, such as the nod-like receptor 3 (NLRP3), which directly inhibit B lymphopoiesis (40). Activation of inflammasomes can stimulate thymic degeneration (81, 82) and exert a negative effect on T-cell proliferation (83), likely contributing to systemic inflammatory conditions associated with advanced age.
There is growing evidence to support the involvement of chemokines such as C-motif chemokine ligand 2/monocyte chemoattractant protein 1 (CCL2/MCP-1) and cyclooxygenase-2 (COX-2) in regulation of the BM microenvironment (84). During inflammatory events, high expression of COX-2 is often coupled with CCL2/MCP-1 upregulation (85–87). The major COX-2 metabolite, prostaglandin E2 (PGE2), is known to induce differentiation of immunoregulatory cells like regulatory T-cells and MDSCs (41–44). Cox-2 inhibitors prevent CCL2/MCP-1 production by activated macrophages (88, 89). Under normal physiological conditions, COX-2 expression in macrophages is low but is increased in response to pro-inflammatory stimuli (90). In fact, the COX-2 expression and PGE2 release by macrophages were shown to be stimulated by CCL2/MCP-1 and to be important for macrophage migration (91–93). In vitro studies using conditioned media from BMAds demonstrated that macrophages are highly stimulated by BMAd-derived factors and that invasiveness increases with age (94). Obesity phenocopies aging with increased BMA, which has been shown to induce CCL2/MCP-1 and COX-2 within the BM (94), emphasizing a close relationship between immune response and BMA.
Cellular Senescence
Aging studies have consistently shown a strong correlation between increased BMA and pro-inflammatory factors (18). It has been suggested that a sustained pro-inflammatory state may negatively impact the proliferative and differentiation capacities of surrounding cells. This effect is referred to as the “bystander effect” and most notably contributes to the accumulation of senescent cells in the BM, a process that naturally occurs with aging (46). Despite studies finding relatively low percentages (10–20%) of senescent cells in aged BMSCs, the bystander effect greatly impairs osteogenic capacities of non-senescent BMSCs, likely through senescence-associated secretory phenotype (SASP) factors (IL-1α, IL-1β, NF-κB, CXCL1/2, TGF-β, p21, p16, CCL2/MCP-1) and the resulting inflammation (95, 96).
BMAT expansion induces pro-inflammatory cytokines, which perpetuates the damaging effects on neighboring cells (46). In this pro-inflammatory microenvironment, BMSCs become senescent, resulting in decreased stem/progenitor cell number and decreased functionality (25, 47). In addition, the increased levels of pro-inflammatory cytokines promote ROS within the BM, further contributing to cellular senescence (23, 45). Flow cytometry analysis by Miggitsch et al. highlighted BMAds as a major contributor of ROS by determining higher ROS levels within femoral BMAds compared to subcutaneous white adipose tissue (WAT) from the thigh (45). Treatment of both tissues with ROS scavengers, N-acetylcysteine (NAC) and vitamin C, significantly reduced ROS levels within the BMAT compared to the WAT (45). The role of ROS in hematopoiesis has been well documented, thus these results demonstrate that BMAds limit the capacity of BMSCs to support the hematopoietic niche (97, 98).
Mimicking the potential effect of increased BMAT, Lo et al. showed that in conditions of elevated glucose in vitro, β-galactosidase activity and adipogenic differentiation markers (Pparγ and Fas) were notably increased while osteogeneic markers (Runx2 and Col1a1) were decreased in BMSCs, indicative of altered differentiation potential (99). This hyperglycemic condition induces inflammation and senescence through oxidant-mediated autophagy, ultimately contributing to dysfunction of bone development and hematopoiesis in the BM microenvironment (100). BMP-2, an established pro-osteoblastogenic protein, can stimulate bone production in healthy, non-senescent BMSCs. However, in senescent cells recombinant BMP-2 upregulates pathways of inflammation, adipogenesis, and cell apoptosis (101). In mouse models, FOXP1, a regulator of the pro-adipogenic CEBPβ/δ complex in BMAT, has been shown to attenuate senescence through repressing p16INK4A (encoded by CDKN2A), a cell cycle repressor that functions by inducing a G1 phase arrest (102). Collectively, BMAds play a critical role in inducing senescence of BMSCs, thereby determining the microenvironmental status in the BM compartment during aging.
Potential Targets for Age-Related Bone Conditions
Senolytics
Senolytics are a class of drugs that selectively induce apoptosis in senescent cells. Studies have shown reductions in age-related chronic inflammation led to functional restoration of bone regeneration through decreased senescence, increased stem/progenitor cell number, and increased osteogenic gene expression (25). In a pharmacological rescue experiment, Zhou et al. showed that BMSCs from aged mice (27 months old) had lower proliferation rates (30%) than young, 3-month-old mice (45%) (95). Twenty-four-hour treatment with dasatinib (generic chemotherapy; tyrosine kinase inhibitor) and quercetin (flavonol; antioxidant and chelating abilities) increased proliferation rates of the old BMSCs to 40% but did not affect the proliferative rates of the young BMSCs (95, 103, 104). Furthermore, dasatinib and quercetin treatments have been shown to improve osteogenic capacity in the aged BMSCs and reduce their expression of several senescence-related and inflammation markers including p21, p16INK4A, IL-6, CXCL1 and MCP-1 (95) in multiple aged tissues (105, 106). Therefore, clearance of senescent cells by senolytics shows promise in improving osteogenesis of aged BMSCs and ameliorating BM inflammation.
miRNAs
In the past few decades, microRNAs (miRNAs) have emerged as key regulators of different aspects in development, homeostasis, and function. However, only a handful of miRNAs have been identified as capable of mediating adipocyte differentiation and function (107, 108). Multiple studies have implicated a potential role for miRNAs on post-transcriptional regulation of BMSC differentiation and aging (109, 110). For example, mice lacking miR-188, an age-associated miRNA found in the BM, showed substantial protection from bone loss and BMAT accumulation over time (109). In comparison, BMSCs transfected with miR-183-5p mimicked reduced cell proliferation and osteogenic differentiation and demonstrated increased cellular senescence (111). Therefore, miRNAs represent a unique class of therapeutic targets of bone inflammaging, given that their specific roles in the BM during aging become elucidated.
Antioxidants
Given the positive effect of low-glucose conditions on senescent BMSCs, methods for glucose reduction have the potential to improve BM health through increasing mitochondrial respiration (99). Studies have shown that restricted glucose conditions increase the presence of antioxidant enzymes and decrease superoxide production, highlighting a therapeutic role for antioxidant defenses (99). An antioxidant and free radical scavenger, apocynin, was used to establish potential inhibition of cellular senescence, even in a senescence-accelerated mouse model, while concurrently improving osteogenesis (112). Similarly, treatment of aged rats with the aforementioned ROS scavenger, NAC, displayed an improved bone phenotype (113). Natural antioxidants have the potential to ameliorate concerns of age-related BMAT expansion. For example, phloretin, a flavonoid commonly found in apples, activates osteogenic gene OPG while promoting BMAd apoptosis to promote osteoblast differentiation, even in aged BMSCs (114). Given what we know about the link between inflammation and aging, it is not surprising to note that in addition to antioxidants, nonsteroidal anti-inflammatory drugs such as aspirin have been shown to counteract the effects of BMSCs senescence by improving cell proliferation and osteogenic differentiation (115, 116).
Adipokines
Adipokines have the potential to regulate physiological functions including satiety, glucose homeostasis, energy expenditure (117), and inflammation (118). As a major regulator of the bone marrow niche with changes during inflammation and aging, adipokines are of great potential for future therapeutics. Numerous cytokines such as CCL2/MCP-1 (94), IL-6, and TNF-α (119) have elucidated roles in linking BMA with bone loss through inflammation. Of note, existing anti-TNF-α therapy infliximab and other TNF-α inhibitors have been shown to prevent age-related bone loss in various conditions (120, 121). In addition, pro-inflammatory adipokines shown to drive decline in bone health include leptin (31), resistin (32), chemerin (33), and adipsin (19). Among them, adipsin provides a straightforward relationship that might be of interest therapeutically by being produced abundantly in the BM and directly priming BMSCs. Furthermore, adipsin is involved in the alternative pathway (AP) of the complement system, a known activator of inflammation in the bone marrow further contributing to bone loss conditions (122–125). In human studies, patients with bone-related conditions such as post-menopausal bone loss and osteoarthritis displayed an increase in serum adipsin levels positively associated with other pro-inflammatory cytokines (126, 127). As such, current pharmacological advancements including the synthesis and pre-clinical characterization of adipsin inhibitors targeting the AP may be of interest in addressing inflammaging and bone loss (128).
Conclusion
The development of BMAT is a normal physiological process and is arguably of importance in regulating BM microenvironment, skeletal homeostasis, hematopoiesis, endocrine function, and energy expenditure, and metabolism. However, extensive BMAT accumulation that occurs with aging and in clinical conditions such as obesity, calorie restriction/anorexia (20, 129), and in response to chemotherapy and irradiation treatments (130–132), suggests that aberrant BMAT formation has pathological implications. The increased adiposity within the BM exacerbates age-related inflammation and contributes to reduced bone health through physical changes in the bone matrix and defects in the BM stroma and HSCs. Ultimately, the age-associated shift of BMSCs toward adipogenesis promotes increased ROS, reduced HSC potential, dysfunctional immune cell response through increased myelopoiesis, and cellular senescence. As such, therapeutic interventions to maintain BMAT in appropriate quantity and quality may improve overall bone health, inflammaging, and senescence, further contributing to increases in life expectancy and quality of life for the elderly population.
Author Contributions
NA and SC have contributed equally to this work and share first authorship. NA and SC wrote the manuscript. LQ and CR advised and edited the manuscript. All authors contributed to manuscript revision and approved the submitted version.
Funding
Funding was supplied by the NIH/NIDDK R24 DK092759-06 (CR), NIH U19AG060917 (CR), NIH/NIDDK R01 DK112943-05 (LQ), R01 DK128848 (LQ), and NIH/NIDDK F31 DK124926 (NA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to acknowledge Samantha Costa and Clifford Rosen (Maine Medical Center Research Institute) for the publication and licensing rights to use BioRender.com.
References
1. Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and Economic Burden of Osteoporosis-Related Fractures in the United States, 2005-2025. J Bone Miner Res (2007) 22:465–75. doi: 10.1359/jbmr.061113
2. Williams SA, Daigle SG, Weiss R, Wang Y, Arora T, Curtis JR. Economic Burden of Osteoporosis-Related Fractures in the US Medicare Population. Ann Pharmacother (2021) 55:821–9. doi: 10.1177/1060028020970518
3. Williams SA, Chastek B, Sundquist K, Barrera-Sierra S, Leader D, Weiss RJ, et al. Economic Burden of Osteoporotic Fractures in US Managed Care Enrollees. Am J Manage Care (2020) 26:E142–9. doi: 10.37765/ajmc.2020.43156
4. Ganguly P, El-Jawhari JJ, Giannoudis PV, Burska AN, Ponchel F, Jones EA. Age-Related Changes in Bone Marrow Mesenchymal Stromal Cells: A Potential Impact on Osteoporosis and Osteoarthritis Development. Cell Transplant (2017) 26:1520–9. doi: 10.1177/0963689717721201
5. Sanghani-Kerai A, Osagie-Clouard L, Blunn G, Coathup M. The Influence of Age and Osteoporosis on Bone Marrow Stem Cells From Rats. Bone Jt Res (2018) 7:289–97. doi: 10.1302/2046-3758.74.BJR-2017-0302.R1
6. Jaul E, Barron J. Age-Related Diseases and Clinical and Public Health Implications for the 85 Years Old and Over Population. Front Public Heal (2017) 5:335. doi: 10.3389/fpubh.2017.00335
7. Luo G, He Y, Yu X. Bone Marrow Adipocyte: An Intimate Partner With Tumor Cells in Bone Metastasis. Front Endocrinol (Lausanne) (2018) 9:339. doi: 10.3389/fendo.2018.00339
8. Nehlin JO, Jafari A, Tencerova M, Kassem M. Aging and Lineage Allocation Changes of Bone Marrow Skeletal (Stromal)Stem Cells. Bone (2019) 123:265–73. doi: 10.1016/j.bone.2019.03.041
9. Fazeli PK, Horowitz MC, MacDougald OA, Scheller EL, Rodeheffer MS, Rosen CJ, et al. Marrow Fat and Bone-New Perspectives. J Clin Endocrinol Metab (2013) 98:935–45. doi: 10.1210/jc.2012-3634
10. Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte Tissue Volume in Bone Marrow is Increased With Aging and in Patients With Osteoporosis. Biogerontology (2001) 2:165–71. doi: 10.1023/A:1011513223894
11. Shen W, Chen J, Punyanitya M, Shapses S, Heshka S, Heymsfield SB. MRI-Measured Bone Marrow Adipose Tissue is Inversely Related to DXA-Measured Bone Mineral in Caucasian Women. Osteoporos Int (2007) 18:641–7. doi: 10.1007/s00198-006-0285-9
12. Ruschke S, Pokorney A, Baum T, Eggers H, Miller JH, Hu HH, et al. Measurement of Vertebral Bone Marrow Proton Density Fat Fraction in Children Using Quantitative Water-Fat MRI. MAGMA (2017) 30:449–60. doi: 10.1007/S10334-017-0617-0
13. Karampinos DC, Ruschke S, Dieckmeyer M, Diefenbach M, Franz D, Gersing AS, et al. And Spectroscopy of Bone Marrow. J Magn Reson Imaging (2018) 47:332–53. doi: 10.1002/JMRI.25769
14. Baum T, Rohrmeier A, Syväri J, Diefenbach MN, Franz D, Dieckmeyer M, et al. Anatomical Variation of Age-Related Changes in Vertebral Bone Marrow Composition Using Chemical Shift Encoding-Based Water-Fat Magnetic Resonance Imaging. Front Endocrinol (Lausanne) (2018) 9:141. doi: 10.3389/FENDO.2018.00141
15. Singhal V, Bredella MA. Marrow Adipose Tissue Imaging in Humans. Bone (2019) 118:69–76. doi: 10.1016/j.bone.2018.01.009
16. Scheller EL, Troiano N, Vanhoutan JN, Bouxsein MA, Fretz JA, Xi Y, et al. Use of Osmium Tetroxide Staining With Microcomputerized Tomography to Visualize and Quantify Bone Marrow Adipose Tissue In Vivo, in: Methods Enzymol. Methods Enzymol (2014) 537:123–39. doi: 10.1016/B978-0-12-411619-1.00007-0
17. Suchacki KJ, Tavares AAS, Mattiucci D, Scheller EL, Papanastasiou G, Gray C, et al. Bone Marrow Adipose Tissue is a Unique Adipose Subtype With Distinct Roles in Glucose Homeostasis. Nat Commun (2020) 11:3097. doi: 10.1038/s41467-020-16878-2
18. Mancuso P, Bouchard B. The Impact of Aging on Adipose Function and Adipokine Synthesis. Front Endocrinol (Lausanne) (2019) 10:137. doi: 10.3389/fendo.2019.00137
19. Aaron N, Kraakman MJ, Zhou Q, Liu Q, Costa S, Yang J, et al. Adipsin Promotes Bone Marrow Adiposity by Priming Mesenchymal Stem Cells. Elife (2021) 10:e69209. doi: 10.7554/eLife.69209
20. Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, et al. Bone Marrow Adipose Tissue is an Endocrine Organ That Contributes to Increased Circulating Adiponectin During Caloric Restriction. Cell Metab (2014) 20:368–75. doi: 10.1016/j.cmet.2014.06.003
21. Ferland-McCollough D, Maselli D, Spinetti G, Sambataro M, Sullivan N, Blom A, et al. MCP-1 Feedback Loop Between Adipocytes and Mesenchymal Stromal Cells Causes Fat Accumulation and Contributes to Hematopoietic Stem Cell Rarefaction in the Bone Marrow of Patients With Diabetes. Diabetes (2018) 67:1380–94. doi: 10.2337/db18-0044
22. Veldhuis-Vlug AG, Rosen CJ. Clinical Implications of Bone Marrow Adiposity. J Intern Med (2018) 283:121–39. doi: 10.1111/joim.12718
23. Pangrazzi L, Meryk A, Naismith E, Koziel R, Lair J, Krismer M, et al. “Inflamm-Aging” Influences Immune Cell Survival Factors in Human Bone Marrow. Eur J Immunol (2017) 47:481–92. doi: 10.1002/eji.201646570
24. Chung HY, Kim DH, Lee EK, Chung KW, Chung S, Lee B, et al. Redefining Chronic Inflammation in Aging and Age-Related Diseases: Proposal of the Senoinflammation Concept. Aging Dis (2019) 10:367–82. doi: 10.14336/AD.2018.0324
25. Josephson AM, Bradaschia-Correa V, Lee S, Leclerc K, Patel KS, Lopez EM, et al. Age-Related Inflammation Triggers Skeletal Stem/Progenitor Cell Dysfunction. Proc Natl Acad Sci U S A (2019) 116:6995–7004. doi: 10.1073/pnas.1810692116
26. Owen M, Friedenstein AJ. Stromal Stem Cells: Marrow-Derived Osteogenic Precursors. Ciba Found Symp (1988) 136:42–60. doi: 10.1002/9780470513637.ch4
27. Tencerova M, Kassem M. The Bone Marrow-Derived Stromal Cells: Commitment and Regulation of Adipogenesis. Front Endocrinol (Lausanne) (2016) 7:127. doi: 10.3389/fendo.2016.00127
28. Noort WA, Kruisselbrink AB, In’t Anker PS, Kruger M, Van Bezooijen RL, De Paus RA, et al. Mesenchymal Stem Cells Promote Engraftment of Human Umbilical Cord Blood-Derived CD34+ Cells in NOD/SCID Mice. Exp Hematol (2002) 30:870–8. doi: 10.1016/S0301-472X(02)00820-2
29. Liu H, Xia X, Li B. Mesenchymal Stem Cell Aging: Mechanisms and Influences on Skeletal and non-Skeletal Tissues. Exp Biol Med (2015) 240:1099–106. doi: 10.1177/1535370215591828
30. Hardouin P, Rharass T, Lucas S. Bone Marrow Adipose Tissue: To Be or Not To Be a Typical Adipose Tissue? Front Endocrinol (Lausanne) (2016) 7:85. doi: 10.3389/fendo.2016.00085
31. Hamrick MW, Pennington C, Newton D, Xie D, Isales C. Leptin Deficiency Produces Contrasting Phenotypes in Bones of the Limb and Spine. Bone (2004) 34:376–83. doi: 10.1016/j.bone.2003.11.020
32. Thommesen L, Stunes AK, Monjo M, Grøsvik K, Tamburstuen MV, Kjøbli E, et al. Expression and Regulation of Resistin in Osteoblasts and Osteoclasts Indicate a Role in Bone Metabolism. J Cell Biochem (2006) 99:824–34. doi: 10.1002/JCB.20915
33. Han L, Zhang Y, Wan S, Wei Q, Shang W, Huang G, et al. Loss of Chemerin Triggers Bone Remodeling In Vivo and In Vitro. Mol Metab (2021) 53:101322. doi: 10.1016/j.molmet.2021.101322
34. DiMascio L, Voermans C, Uqoezwa M, Duncan A, Lu D, Wu J, et al. Identification of Adiponectin as a Novel Hemopoietic Stem Cell Growth Factor. J Immunol (2007) 178:3511–20. doi: 10.4049/jimmunol.178.6.3511
35. Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-Marrow Adipocytes as Negative Regulators of the Haematopoietic Microenvironment. Nature (2009) 460:259–63. doi: 10.1038/nature08099
36. Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, et al. Human Bone Marrow Hematopoietic Stem Cells are Increased in Frequency and Myeloid-Biased With Age. Proc Natl Acad Sci U S A (2011) 108:20012–7. doi: 10.1073/pnas.1116110108
37. Ogawa T, Kitagawa M, Hirokawa K. Age-Related Changes of Human Bone Marrow: A Histometric Estimation of Proliferative Cells, Apoptotic Cells, T Cells, B Cells and Macrophages. Mech Ageing Dev (2000) 117:57–68. doi: 10.1016/S0047-6374(00)00137-8
38. Tanaka T, Narazaki M, Kishimoto T. Il-6 in Inflammation, Immunity, And Disease. Cold Spring Harb Perspect Biol (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
39. Udagawa N, Takahashi N, Katagiri T, Tamura T, Wada S, Findlay DM, et al. Interleukin (IL)-6 Induction of Osteoclast Differentiation Depends on IL-6 Receptors Expressed on Osteoblastic Cells But Not on Osteoclast Progenitors. J Exp Med (1995) 182:1461–8. doi: 10.1084/jem.182.5.1461
40. Kennedy DE, Knight KL. Inflammatory Changes in Bone Marrow Microenvironment Associated With Declining B Lymphopoiesis. J Immunol (2017) 198:3471–9. doi: 10.4049/jimmunol.1601643
41. Wang D, Dubois RN. Eicosanoids and Cancer. Nat Rev Cancer (2010) 10:181–93. doi: 10.1038/nrc2809
42. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 Promotes Tumor Progression by Inducing Myeloid-Derived Suppressor Cells. Cancer Res (2007) 67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174
43. Mahic M, Yaqub S, Johansson CC, Taskén K, Aandahl EM. FOXP3+CD4+CD25+ Adaptive Regulatory T Cells Express Cyclooxygenase-2 and Suppress Effector T Cells by a Prostaglandin E2-Dependent Mechanism. J Immunol (2006) 177:246–54. doi: 10.4049/jimmunol.177.1.246
44. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive Feedback Between PGE2 and COX2 Redirects the Differentiation of Human Dendritic Cells Toward Stable Myeloid-Derived Suppressor Cells. Blood (2011) 118:5498–505. doi: 10.1182/blood-2011-07-365825
45. Miggitsch C, Meryk A, Naismith E, Pangrazzi L, Ejaz A, Jenewein B, et al. Human Bone Marrow Adipocytes Display Distinct Immune Regulatory Properties. EBioMedicine (2019) 46:387–98. doi: 10.1016/j.ebiom.2019.07.023
46. da Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, et al. The Bystander Effect Contributes to the Accumulation of Senescent Cells In Vivo. Aging Cell (2019) 18:e12848. doi: 10.1111/acel.12848
47. Kovtonyuk LV, Fritsch K, Feng X, Manz MG, Takizawa H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front Immunol (2016) 7:502. doi: 10.3389/fimmu.2016.00502
48. Infante A, Rodríguez CI. Osteogenesis and Aging: Lessons From Mesenchymal Stem Cells. Stem Cell Res Ther (2018) 9:244. doi: 10.1186/s13287-018-0995-x
49. Planat-Benard V, Varin A, Casteilla L. MSCs and Inflammatory Cells Crosstalk in Regenerative Medicine: Concerted Actions for Optimized Resolution Driven by Energy Metabolism. Front Immunol (2021) 12:626755. doi: 10.3389/fimmu.2021.626755
50. Cawthorn WP, Scheller EL. Editorial: Bone Marrow Adipose Tissue: Formation, Function, and Impact on Health and Disease. Front Endocrinol (Lausanne) (2017) 8:112. doi: 10.3389/fendo.2017.00112
51. Jacobs FA, Sadie-Van Gijsen H, van de Vyver M, Ferris WF. Vanadate Impedes Adipogenesis in Mesenchymal Stem Cells Derived From Different Depots Within Bone. Front Endocrinol (Lausanne) (2016) 7:108. doi: 10.3389/fendo.2016.00108
52. Kraakman MJ, Liu Q, Postigo-Fernandez J, Ji R, Kon N, Larrea D, et al. PPAR? Deacetylation Dissociates Thiazolidinedione’s Metabolic Benefits From its Adverse Effects. J Clin Invest (2018) 128:2600–12. doi: 10.1172/JCI98709
53. Zhong L, Yao L, Tower RJ, Wei Y, Miao Z, Park J, et al. Single Cell Transcriptomics Identifies a Unique Adipose Lineage Cell Population That Regulates Bone Marrow Environment. Elife (2020) 9:e54695. doi: 10.7554/eLife.54695
54. Corrado A, Cici D, Rotondo C, Maruotti N, Cantatore FP. Molecular Basis of Bone Aging. Int J Mol Sci (2020) 21:3679. doi: 10.3390/ijms21103679
55. Goto H, Osaki M, Fukushima T, Sakamoto K, Hozumi A, Baba H, et al. Human Bone Marrow Adipocytes Support Dexamethasone-Induced Osteoclast Differentiation and Function Through RANKL Expression. Biomed Res (2011) 32:37–44. doi: 10.2220/biomedres.32.37
56. Goto H, Hozumi A, Osaki M, Fukushima T, Sakamoto K, Yonekura A, et al. Primary Human Bone Marrow Adipocytes Support TNF-α-Induced Osteoclast Differentiation and Function Through RANKL Expression. Cytokine (2011) 56:662–8. doi: 10.1016/j.cyto.2011.09.005
57. Cao JJ, Wronski TJ, Iwaniec U, Phleger L, Kurimoto P, Boudignon B, et al. Aging Increases Stromal/Osteoblastic Cell-Induced Osteoclastogenesis and Alters the Osteoclast Precursor Pool in the Mouse. J Bone Miner Res (2005) 20:1659–68. doi: 10.1359/JBMR.050503
58. Takeshita S, Fumoto T, Naoe Y, Ikeda K. Age-Related Marrow Adipogenesis is Linked to Increased Expression of RANKL. J Biol Chem (2014) 289:16699–710. doi: 10.1074/jbc.M114.547919
59. Lai P, Song Q, Yang C, Li Z, Liu S, Liu B, et al. Loss of Rictor With Aging in Osteoblasts Promotes Age-Related Bone Loss. Cell Death Dis (2016) 7:e2408–8. doi: 10.1038/cddis.2016.249
60. Corre J, Planat-Benard V, Corberand JX, Pénicaud L, Casteilla L, Laharrague P. Human Bone Marrow Adipocytes Support Complete Myeloid and Lymphoid Differentiation From Human CD34+ Cells. Br J Haematol (2004) 127:344–7. doi: 10.1111/j.1365-2141.2004.05198.x
61. Jilka RL. Osteoblast Progenitor Fate and Age-Related Bone Loss. J Musculoskelet Neuronal Interact (2002) 2:581–3.
62. Trottier MD, Naaz A, Li Y, Fraker PJ. Enhancement of Hematopoiesis and Lymphopoiesis in Diet-Induced Obese Mice. Proc Natl Acad Sci U S A (2012) 109:7622–9. doi: 10.1073/pnas.1205129109
63. Gimble JM, Nuttall ME. The Relationship Between Adipose Tissue and Bone Metabolism. Clin Biochem (2012) 45:874–9. doi: 10.1016/j.clinbiochem.2012.03.006
64. Lecka-Czernik B. Marrow Fat Metabolism is Linked to the Systemic Energy Metabolism. Bone (2012) 50:534–9. doi: 10.1016/j.bone.2011.06.032
65. Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The Essential Functions of Adipo-Osteogenic Progenitors as the Hematopoietic Stem and Progenitor Cell Niche. Immunity (2010) 33:387–99. doi: 10.1016/j.immuni.2010.08.017
66. Miharada K, Hiroyama T, Sudo K, Danjo I, Nagasawa T, Nakamura Y. Lipocalin 2-Mediated Growth Suppression is Evident in Human Erythroid and Monocyte/Macrophage Lineage Cells. J Cell Physiol (2008) 215:526–37. doi: 10.1002/jcp.21334
67. Belaid-Choucair Z, Lepelletier Y, Poncin G, Thiry A, Humblet C, Maachi M, et al. Human Bone Marrow Adipocytes Block Granulopoiesis Through Neuropilin-1-Induced Granulocyte Colony-Stimulating Factor Inhibition. Stem Cells (2008) 26:1556–64. doi: 10.1634/stemcells.2008-0068
68. Ouchi N, Walsh K. Adiponectin as an Anti-Inflammatory Factor. Clin Chim Acta (2007) 380:24–30. doi: 10.1016/j.cca.2007.01.026
69. Gimble JM, Nuttall ME. Bone and Fat: Old Questions, New Insights. Endocrine (2004) 23:183–8. doi: 10.1385/ENDO:23:2-3:183
70. Rosen CJ, Ackert-Bicknell C, Rodriguez JP, Pino AM. Marrow Fat and the Bone Microenvironment: Developmental, Functional, and Pathological Implications. Crit Rev Eukaryot Gene Expr (2009) 19:109–24. doi: 10.1615/CritRevEukarGeneExpr.v19.i2.20
71. Kawai M, de Paula FJA, Rosen CJ. New Insights Into Osteoporosis: The Bone-Fat Connection. J Intern Med (2012) 272:317–29. doi: 10.1111/j.1365-2796.2012.02564.x
72. Gasparrini M, Rivas D, Elbaz A, Duque G. Differential Expression of Cytokines in Subcutaneous and Marrow Fat of Aging C57BL/6J Mice. Exp Gerontol (2009) 44:613–8. doi: 10.1016/j.exger.2009.05.009
73. Bilwani FA, Knight KL. Adipocyte-Derived Soluble Factor(s) Inhibits Early Stages of B Lymphopoiesis. J Immunol (2012) 189:4379–86. doi: 10.4049/jimmunol.1201176
74. Rossi MID, Yokota T, Medina KL, Garrett KP, Comp PC, Schipul AH, et al. B Lymphopoiesis is Active Throughout Human Life, But There are Developmental Age-Related Changes. Blood (2003) 101:576–84. doi: 10.1182/blood-2002-03-0896
75. McKenna RW, Washington LBT, Aquino DB, Picker LJ, Kroft SH. Immunophenotypic Analysis of Hematogones (B-Lymphocyte Precursors) in 662 Consecutive Bone Marrow Specimens by 4-Color Flow Cytometry. Blood (2001) 98:2498–507. doi: 10.1182/blood.V98.8.2498
76. Nuñez C, Nishimoto N, Gartland GL, Billips LG, Burrows PD, Kubagawa H, et al. B Cells are Generated Throughout Life in Humans. J Immunol (1996) 156:866–72.
77. Johnson KM, Owen K, Witte PL. Aging and Developmental Transitions in the B Cell Lineage. Int Immunol (2002) 14:1313–23. doi: 10.1093/intimm/dxf092
78. Stephan RP, Reilly CR, Witte PL. Impaired Ability of Bone Marrow Stromal Cells to Support B-Lymphopoiesis With Age. Blood (1998) 91:75–88. doi: 10.1182/blood.v91.1.75
79. Frasca D, Blomberg BB. Effects of Aging on B Cell Function. Curr Opin Immunol (2009) 21:425–30. doi: 10.1016/j.coi.2009.06.001
80. Kennedy DE, Knight KL. Inhibition of B Lymphopoiesis by Adipocytes and IL-1–Producing Myeloid-Derived Suppressor Cells. J Immunol (2015) 195:2666–74. doi: 10.4049/jimmunol.1500957
81. Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: Disturbed Interplay Between Autophagy and Inflammasomes. Aging (Albany NY) (2012) 4:166–75. doi: 10.18632/aging.100444
82. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat Med (2011) 17:179–89. doi: 10.1038/nm.2279
83. Wang H, Leng Y, Gong Y. Bone Marrow Fat and Hematopoiesis. Front Endocrinol (Lausanne) (2018) 9:694. doi: 10.3389/fendo.2018.00694
84. Craig MJ, Loberg RD. CCL2 (Monocyte Chemoattractant Protein-1) in Cancer Bone Metastases. Cancer Metastasis Rev (2006) 25:611–9. doi: 10.1007/s10555-006-9027-x
85. Chen YC, Sosnoski DM, Gandhi UH, Novinger LJ, Prabhu KS, Mastro AM. Selenium Modifies the Osteoblast Inflammatory Stress Response to Bone Metastatic Breast Cancer. Carcinogenesis (2009) 30:1941–8. doi: 10.1093/carcin/bgp227
86. Hsieh PS, Lu KC, Chiang CF, Chen CH. Suppressive Effect of COX2 Inhibitor on the Progression of Adipose Inflammation in High-Fat-Induced Obese Rats. Eur J Clin Invest (2010) 40:164–71. doi: 10.1111/j.1365-2362.2009.02239.x
87. Anderson GD, Hauser SD, McGarity KL, Bremer ME, Isakson PC, Gregory SA. Selective Inhibition of Cyclooxygenase (COX)-2 Reverses Inflammation and Expression of COX-2 and Interleukin 6 in Rat Adjuvant Arthritis. J Clin Invest (1996) 97:2672–9. doi: 10.1172/JCI118717
88. Muta M, Matsumoto G, Nakashima E, Toi M. Mechanical Analysis of Tumor Growth Regression by the Cyclooxygenase-2 Inhibitor, DFU, in a Walker256 Rat Tumor Model: Importance of Monocyte Chemoattractant Protein-1 Modulation. Clin Cancer Res (2006) 12:264–72. doi: 10.1158/1078-0432.CCR-05-1052
89. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 Blockade Suppresses Gliomagenesis by Inhibiting Myeloid-Derived Suppressor Cells. Cancer Res (2011) 71:2664–74. doi: 10.1158/0008-5472.CAN-10-3055
90. Eliopoulos AG, Dumitru CD, Wang CC, Cho J, Tsichlis PN. Induction of COX-2 by LPS in Macrophages is Regulated by Tpl2-Dependent CREB Activation Signals. EMBO J (2002) 21:4831–40. doi: 10.1093/emboj/cdf478
91. Nakao S, Kuwano T, Tsutsumi-Miyahara C, Ueda SI, Kimura YN, Hamano S, et al. Infiltration of COX-2-Expressing Macrophages is a Prerequisite for IL-1β-Induced Neovascularization and Tumor Growth. J Clin Invest (2005) 115:2979–91. doi: 10.1172/JCI23298
92. Tanaka S, Tatsuguchi A, Futagami S, Gudis K, Wada K, Seo T, et al. Monocyte Chemoattractant Protein 1 and Macrophage Cyclooxygenase 2 Expression in Colonic Adenoma. Gut (2006) 55:54–61. doi: 10.1136/gut.2004.059824
93. Tajima T, Murata T, Aritake K, Urade Y, Hirai H, Nakamura M, et al. Lipopolysaccharide Induces Macrophage Migration via Prostaglandin D 2 and Prostaglandin E2. J Pharmacol Exp Ther (2008) 326:493–501. doi: 10.1124/jpet.108.137992
94. Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I. Bone Marrow Fat: Linking Adipocyte-Induced Inflammation With Skeletal Metastases. Cancer Metastasis Rev (2014) 33:527–43. doi: 10.1007/s10555-013-9484-y
95. Zhou Y, Xin X, Wang L, Wang B, Chen L, Liu O, et al. Senolytics Improve Bone Forming Potential of Bone Marrow Mesenchymal Stem Cells From Aged Mice. NPJ Regen Med (2021) 6:34. doi: 10.1038/s41536-021-00145-z
96. Khosla S, Farr JN, Kirkland JL. Inhibiting Cellular Senescence: A New Therapeutic Paradigm for Age-Related Osteoporosis. J Clin Endocrinol Metab (2018) 103:1282–90. doi: 10.1210/jc.2017-02694
97. Zhu H, Kwak H-J, Liu P, Bajrami B, Xu Y, Park S-Y, et al. Reactive Oxygen Species–Producing Myeloid Cells Act as a Bone Marrow Niche for Sterile Inflammation–Induced Reactive Granulopoiesis. J Immunol (2017) 198:2854–64. doi: 10.4049/jimmunol.1602006
98. Khatri R, Krishnan S, Roy S, Chattopadhyay S, Kumar V, Mukhopadhyay A. Reactive Oxygen Species Limit the Ability of Bone Marrow Stromal Cells to Support Hematopoietic Reconstitution in Aging Mice. Stem Cells Dev (2016) 25:948–58. doi: 10.1089/scd.2015.0391
99. Lo T, Ho JH, Yang MH, Lee OK. Glucose Reduction Prevents Replicative Senescence and Increases Mitochondrial Respiration in Human Mesenchymal Stem Cells. Cell Transplant (2011) 20:813–25. doi: 10.3727/096368910X539100
100. Chang TC, Hsu MF, Wu KK. High Glucose Induces Bone Marrow-Derived Mesenchymal Stem Cell Senescence by Upregulating Autophagy. PloS One (2015) 10:e0126537. doi: 10.1371/journal.pone.0126537
101. Cho JH, Lee JH, Lee KM, Lee CK, Shin DM. BMP-2 Induced Signaling Pathways and Phenotypes: Comparisons Between Senescent and Non-Senescent Bone Marrow Mesenchymal Stem Cells. Calcif Tissue Int (2021) 1–15. doi: 10.1007/s00223-021-00923-3
102. Li H, Liu P, Xu S, Li Y, Dekker JD, Li B, et al. FOXP1 Controls Mesenchymal Stem Cell Commitment and Senescence During Skeletal Aging. J Clin Invest (2017) 127:1241–53. doi: 10.1172/JCI89511
103. Heim KE, Tagliaferro AR, Bobilya DJ. Flavonoid Antioxidants: Chemistry, Metabolism and Structure-Activity Relationships. J Nutr Biochem (2002) 13:572–84. doi: 10.1016/S0955-2863(02)00208-5
104. Lindauer M, Hochhaus A. Dasatinib, in: Recent Results Cancer Res. Recent Results Cancer Res (2018) 212:29–68. doi: 10.1007/978-3-319-91439-8_2
105. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat Med (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
106. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting Cellular Senescence Prevents Age-Related Bone Loss in Mice. Nat Med (2017) 23:1072–9. doi: 10.1038/nm.4385
107. Arner P, Kulyté A. MicroRNA Regulatory Networks in Human Adipose Tissue and Obesity. Nat Rev Endocrinol (2015) 11:276–88. doi: 10.1038/nrendo.2015.25
108. Brandão BB, Guerra BA, Mori MA. Shortcuts to a Functional Adipose Tissue: The Role of Small non-Coding RNAs. Redox Biol (2017) 12:82–102. doi: 10.1016/j.redox.2017.01.020
109. Li CJ, Cheng P, Liang MK, Chen YS, Lu Q, Wang JY, et al. MicroRNA-188 Regulates Age-Related Switch Between Osteoblast and Adipocyte Differentiation. J Clin Invest (2015) 125:1509–22. doi: 10.1172/JCI77716
110. Xu J, Huang Z, Lin L, Fu M, Song Y, Shen Y, et al. MiRNA-130b is Required for the ERK/FOXM1 Pathway Activation-Mediated Protective Effects of Isosorbide Dinitrate Against Mesenchymal Stem Cell Senescence Induced by High Glucose. Int J Mol Med (2015) 35:59–71. doi: 10.3892/ijmm.2014.1985
111. Davis C, Dukes A, Drewry M, Helwa I, Johnson MH, Isales CM, et al. MicroRNA-183-5p Increases With Age in Bone-Derived Extracellular Vesicles, Suppresses Bone Marrow Stromal (Stem) Cell Proliferation, and Induces Stem Cell Senescence. Tissue Eng - Part A (2017) 23:1231–40. doi: 10.1089/ten.tea.2016.0525
112. Sun J, Ming L, Shang F, Shen L, Chen J, Jin Y. Apocynin Suppression of NADPH Oxidase Reverses the Aging Process in Mesenchymal Stem Cells to Promote Osteogenesis and Increase Bone Mass. Sci Rep (2015) 5:1–11. doi: 10.1038/srep18572
113. Geißler S, Textor M, Schmidt-Bleek K, Klein O, Thiele M, Ellinghaus A, et al. In Serum Veritas-in Serum Sanitas? Cell non-Autonomous Aging Compromises Differentiation and Survival of Mesenchymal Stromal Cells via the Oxidative Stress Pathway. Cell Death Dis (2013) 4:e970–e970. doi: 10.1038/cddis.2013.501
114. Casado-díaz A, Rodríguez-ramos Á, Torrecillas-baena B, Dorado G, Quesada-gómez JM, Gálvez-moreno MÁ. Flavonoid Phloretin Inhibits Adipogenesis and Increases Opg Expression in Adipocytes Derived From Human Bone-Marrow Mesenchymal Stromal-Cells. Nutrients (2021) 13:4185. doi: 10.3390/nu13114185
115. Yamaza T, Miura Y, Bi Y, Liu Y, Akiyama K, Sonoyama W, et al. Pharmacologic Stem Cell Based Intervention as a New Approach to Osteoporosis Treatment in Rodents. PloS One (2008) 3:e2615. doi: 10.1371/journal.pone.0002615
116. Wang L, Zhao Y, Liu Y, Akiyama K, Chen C, Qu C, et al. IFN-γ and TNF-α Synergistically Induce Mesenchymal Stem Cell Impairment and Tumorigenesis via Nfκb Signaling. Stem Cells (2013) 31:1383–95. doi: 10.1002/stem.1388
117. Fasshauer M, Blüher M. Adipokines in Health and Disease. Trends Pharmacol Sci (2015) 36:461–70. doi: 10.1016/j.tips.2015.04.014
118. Blüher M. Clinical Relevance of Adipokines. Diabetes Metab J (2012) 36:317–27. doi: 10.4093/dmj.2012.36.5.317
119. Walsh NC, Gravallese EM. Bone Loss in Inflammatory Arthritis: Mechanisms and Treatment Strategies. Curr Opin Rheumatol (2004) 16:419–27. doi: 10.1097/01.bor.0000127824.42507.68
120. Poutoglidou F, Pourzitaki C, Manthou ME, Samoladas E, Saitis A, Malliou F, et al. Infliximab Prevents Systemic Bone Loss and Suppresses Tendon Inflammation in a Collagen-Induced Arthritis Rat Model. Inflammopharmacology (2021) 29:661–72. doi: 10.1007/s10787-021-00815-w
121. Nigil Haroon N, Sriganthan J, Al Ghanim N, Inman RD, Cheung AM. Effect of TNF-Alpha Inhibitor Treatment on Bone Mineral Density in Patients With Ankylosing Spondylitis: A Systematic Review and Meta-Analysis. Semin Arthritis Rheum (2014) 44:155–61. doi: 10.1016/j.semarthrit.2014.05.008
122. Roche-Molina M, Sanz-Rosa D, Cruz FM, García-Prieto J, López S, Abia R, et al. Induction of Sustained Hypercholesterolemia by Single Adeno-Associated Virus-Mediated Gene Transfer of Mutant Hpcsk9. Arterioscler Thromb Vasc Biol (2015) 35:50–9. doi: 10.1161/ATVBAHA.114.303617
123. Szulc P, Naylor K, Hoyle NR, Eastell R, Leary ET. Use of CTX-I and PINP as Bone Turnover Markers: National Bone Health Alliance Recommendations to Standardize Sample Handling and Patient Preparation to Reduce Pre-Analytical Variability. Osteoporos Int (2017) 28:2541–56. doi: 10.1007/s00198-017-4082-4
124. Ehrnthaller C, Huber-Lang M, Nilsson P, Bindl R, Redeker S, Recknagel S, et al. Complement C3 and C5 Deficiency Affects Fracture Healing. PloS One (2013) 8:e81341. doi: 10.1371/journal.pone.0081341
125. Bloom AC, Collins FL, van’t Hof RJ, Ryan ES, Jones E, Hughes TR, et al. Deletion of the Membrane Complement Inhibitor CD59a Drives Age and Gender-Dependent Alterations to Bone Phenotype in Mice. Bone (2016) 84:253–61. doi: 10.1016/j.bone.2015.12.014
126. Azizieh FY, Shehab D, Al Jarallah K, Mojiminiyi O, Gupta R, Raghupathy R. Circulatory Pattern of Cytokines, Adipokines and Bone Markers in Postmenopausal Women With Low BMD. J Inflamm Res (2019) 12:99–108. doi: 10.2147/JIR.S203590
127. Zhu J, Ruan G, Cen H, Meng T, Zheng S, Wang Y, et al. Association of Serum Levels of Inflammatory Markers and Adipokines With Joint Symptoms and Structures in Participants With Knee Osteoarthritis. Rheumatology (2021) 0:1–9. doi: 10.1093/rheumatology/keab479
128. Karki RG, Powers J, Mainolfi N, Anderson K, Belanger DB, Liu D, et al. Design, Synthesis, and Preclinical Characterization of Selective Factor D Inhibitors Targeting the Alternative Complement Pathway. J Med Chem (2019) 62:4656–68. doi: 10.1021/acs.jmedchem.9b00271
129. Devlin MJ, Cloutier AM, Thomas NA, Panus DA, Lotinun S, Pinz I, et al. Caloric Restriction Leads to High Marrow Adiposity and Low Bone Mass in Growing Mice. J Bone Miner Res (2010) 25:2078–88. doi: 10.1002/JBMR.82
130. Bolan PJ, Arentsen L, Sueblinvong T, Zhang Y, Moeller S, Carter JS, et al. Water-Fat MRI for Assessing Changes in Bone Marrow Composition Due to Radiation and Chemotherapy in Gynecologic Cancer Patients. J Magn Reson Imaging (2013) 38:1578–84. doi: 10.1002/JMRI.24071
131. Carmona R, Pritz J, Bydder M, Gulaya S, Zhu H, Williamson CW, et al. Fat Composition Changes in Bone Marrow During Chemotherapy and Radiation Therapy. Int J Radiat Oncol Biol Phys (2014) 90:155–63. doi: 10.1016/J.IJROBP.2014.05.041
Keywords: inflammation, bone marrow adipocytes, inflammaging, aging, bone marrow adipose tissue (BMAT)
Citation: Aaron N, Costa S, Rosen CJ and Qiang L (2022) The Implications of Bone Marrow Adipose Tissue on Inflammaging. Front. Endocrinol. 13:853765. doi: 10.3389/fendo.2022.853765
Received: 13 January 2022; Accepted: 16 February 2022;
Published: 11 March 2022.
Edited by:
Jason Horton, Upstate Medical University, United StatesReviewed by:
Michael Dieckmeyer, Technical University of Munich, GermanyCopyright © 2022 Aaron, Costa, Rosen and Qiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clifford J. Rosen, cm9zZW5jQG1tYy5vcmc=; Li Qiang, bHEyMTIzQGN1bWMuY29sdW1iaWEuZWR1
†These authors have contributed equally to this work and share first authorship