 Li Liu
Li Liu Lihong Shi1†
Lihong Shi1† Hongtao Xiao
Hongtao Xiao Yongxia Zhu
Yongxia Zhu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 18 March 2022
Sec. Cancer Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.839857
This article is part of the Research TopicEndocrine Effects of Emerging Cancer TherapiesView all 4 articles
Sex hormone-dependent cancers, including breast, ovary, and prostate cancer, contribute to the high number of cancer-related deaths worldwide. Steroid hormones promote tumor occurrence, development, and metastasis by acting on receptors, such as estrogen receptors (ERs), androgen receptors (ARs), and estrogen-related receptors (ERRs). Therefore, endocrine therapy targeting ERs, ARs, and ERRs represents the potential and pivotal therapeutic strategy in sex hormone-dependent cancers. Proteolysis-targeting chimeras (PROTACs) are a novel strategy that can harness the potential of the endogenous ubiquitin-proteasome system (UPS) to target and degrade specific proteins, rather than simply inhibiting the activity of target proteins. Small molecule PROTACs degrade a variety of proteins in cells, mice, and humans and are an emerging approach for novel drug development. PROTACs targeting ARs, ERs, ERRs, and other proteins in sex hormone-dependent cancers have been reported and may overcome the problem of resistance to existing endocrine therapy and receptor antagonist treatments. This review briefly introduces the PROTAC strategy and summarizes the progress on the development of small molecule PROTACs targeting oncoproteins in sex hormone-dependent cancers, focusing on breast and prostate cancers.
Great success has been achieved in drug discovery programs and human being health, particularly targeted therapy. There are two main types of targeted therapeutic drugs: monoclonal antibodies and small molecule drugs (1). Although targeted therapeutic drugs have fewer side effects than traditional chemotherapeutics, some limitations impede their widespread use (2). Monoclonal antibodies have potent specificity, however, their identification can be expensive, laborious, and time-consuming, meanwhile poor cell permeability limits their application (3). Whereas, treatment with small molecule drugs frequently results in the emergence of drug resistance (4, 5). New strategies to target protein degradation using small molecules have been developed (6, 7). One such attractive alternative technology involves PROteolysis TArgeting Chimeras (PROTACs), also known as protein degrader (7), which is an irreversible process of catalytic degradation of target proteins.
PROTACs can harness the endogenous ubiquitin-proteasome system (UPS) to target and degrade specific proteins (8, 9). PROTACs are heterobifunctional small molecules consisting of a target protein-binding ligand, an E3 recruiting ligand, and an interval linker. Rather than simply inhibiting the target protein activity, PROTACs can eliminate the entire target (9). The PROTAC strategy has been developed and validated for a range of targets, including kinases and protein targets that are “undruggable” via traditional inhibitors or non-enzymatic proteins (10, 11). Different studies have recently shown that embryonic ectoderm development (EED) -targeted or enhancer of zeste homolog 2-targeted (EZH2) -targeted PROTACs can potently degrade both target proteins and other core components of the polycomb repressive complex 2 (PRC2), suggesting that the PROTAC-mediated degradation mechanism can be a viable therapeutic modality (12–15). Due to this mechanism, the utilization of PROTACs can open new avenues for drug discovery efforts (11).
Over the last 20 years, PROTACs have evolved from peptides into small molecules that can degrade a variety of proteins in cells, mice, and humans (10, 11, 16). Small molecule PROTACs have gained the attention of both academic researchers and the pharmaceutical industry, including companies such as Arvinas, C4 Therapeutics, and Kymera Therapeutics (17). Some small molecule PROTACs have also undergone clinical trials (18, 19). In this review, we briefly introduce the PROTAC strategy and summarize the progress of small molecule PROTACs targeting oncoproteins in sex hormone-dependent cancers, with a particular focus on breast and prostate cancers. We also discuss the advantages and limitations of this emerging pharmacological modality.
PROTACs are bifunctional-hybrid molecules that comprise two ligands joined via a flexible chemical linker, with one of the ligands binding to the target protein and the other binding to the E3 ubiquitin ligase, facilitating poly-ubiquitination and subsequent proteasome-mediated degradation of the target protein (Figure 1). The concept of PROTACs was first proposed in cell lysates with peptide ligands by Deshaies and Crew in 2001 (9). Since then, PROTAC technology has evolved, and has been applied in cultured mammalian cells, in vivo, and even in clinical trials (10). There are more than 600 E3 ligase-encoding genes in the human genome. Numerous E3 ligases are employed in PROTAC technology, including cereblon (CRBN) (20), Von Hippel-Lindau (VHL) (21), a cellular inhibitor of apoptosis (cIAP) (22), and mouse double 2 homologue (MDM2) (23). CRBN and VHL are the most widely used because of the availability of drug-like small molecules that can recruit them (24–26). Other E3 ligases have also been studied and used to target protein degradation (27). In addition, diverse target proteins can be degraded efficiently by the utilization of PROTACs, including bromodomain and extra-terminal (BET) proteins (28), transcription factors (29), and tyrosine kinases [such as activin receptor-like kinases (ALK), Abelson tyrosine kinase (c-Abl), and Bruton’s tyrosine kinase (BTK)] (30).
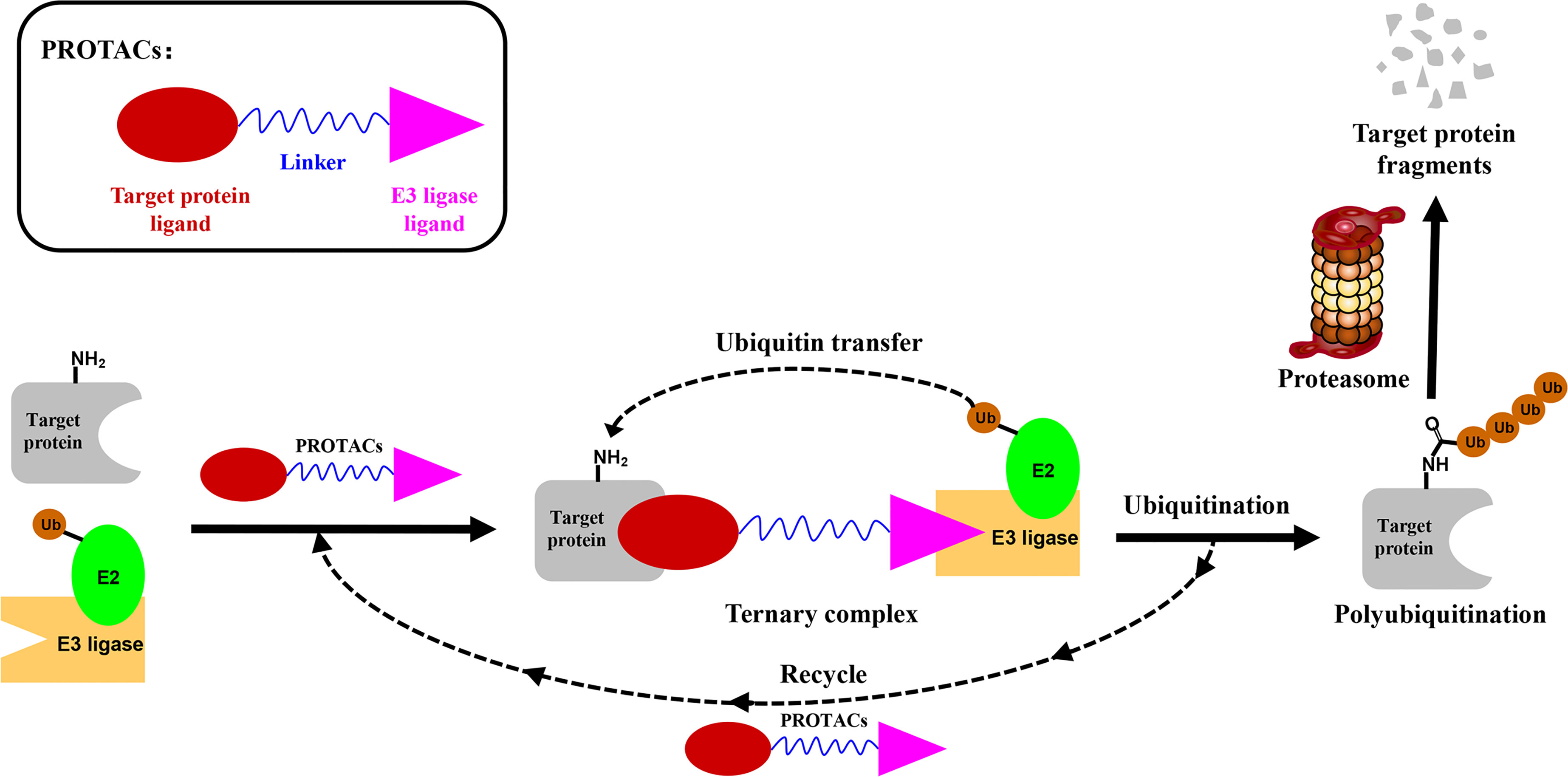
Figure 1 Schematic diagram of Proteolysis Targeting Chimeras (PROTACs) strategy. PROTACs comprise two ligands joined via a flexible chemical linker (blue font part), with one ligand (red font part) binding to the target protein and the other (violet font part) binding to the E3 ubiquitin ligase, followed by poly-ubiquitination and proteasome degradation of target protein.
Peptide-based PROTACs were the first-generation PROTACs, first introduced in 2001. They include Protac-1, which contains IκBα phosphopeptide as the E3 ubiquitin ligand recognized by the Skp1-Cullin-F (SCF) box complex (9). Protac-1 (Figure 2) recruits methionine aminopeptidase-2 (MetAP-2) to the SCF ubiquitin ligase, promoting MetAP-2 ubiquitination and inducing its degradation (9). In 2003, the PROTAC approach was used to target steroid hormone receptor proteins, including estrogen receptors (ERs) and androgen receptors (ARs) (31). Protac-2 and Protac-3 (Figure 2) consist of IκBα phosphopeptide and either estradiol or dihydroxytestosterone (DHT), which recruit ER or AR to the SCF ubiquitin ligase for ubiquitination and degradation, respectively (31). These three PROTAC molecules are all peptide-based and have cell permeability issues (9, 31).
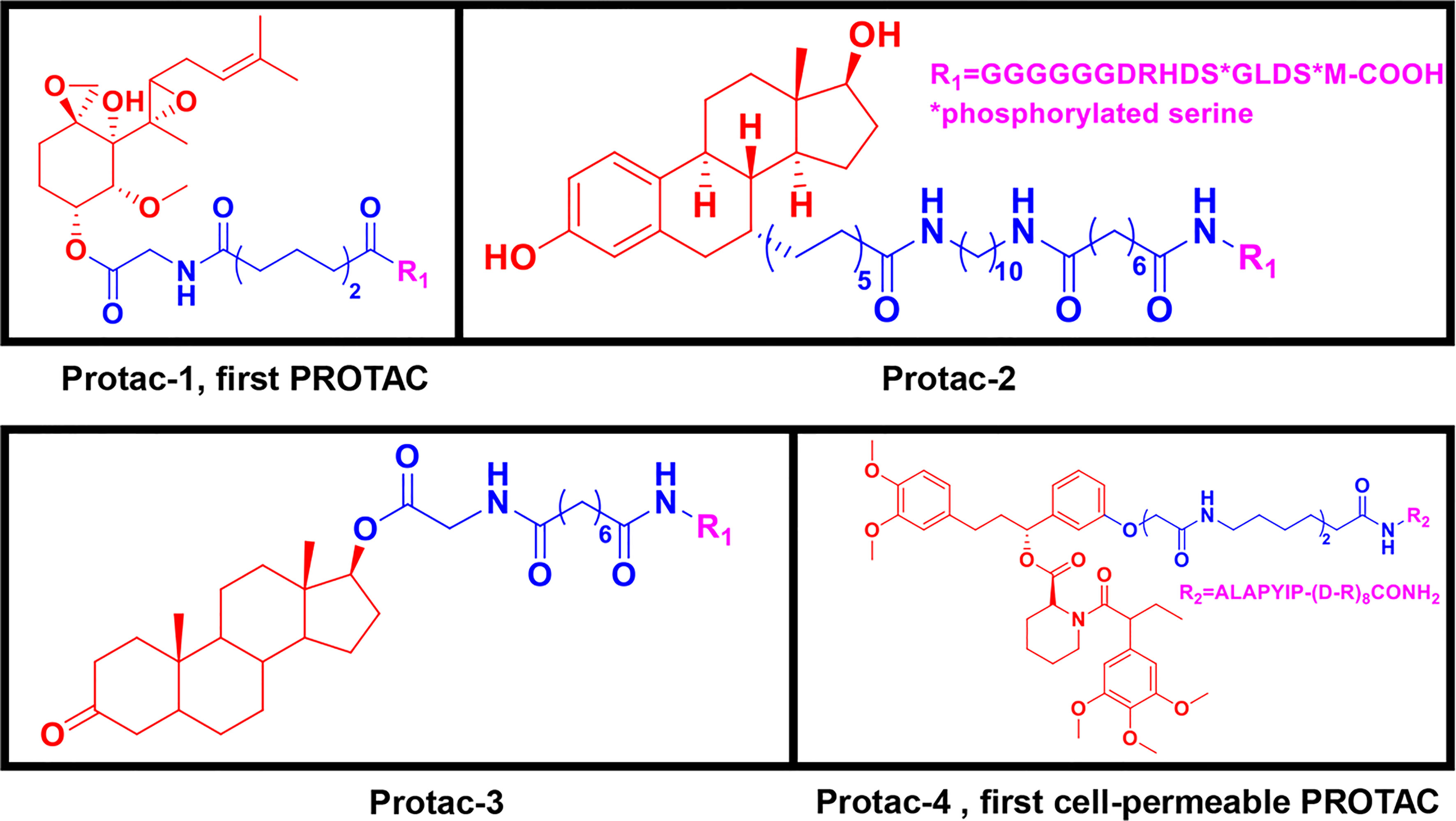
Figure 2 First-generation PROTACs, Peptide-based PROTACs. Chemical structures of Protac-1 (first PROTAC), Protc-2, Protac-3, and Protac-4 (first cell-permeable PROTAC).
To increase their cell permeability, Schneekloth et al. (32) designed the first cell-permeable peptide-based PROTAC (Protac-4) (Figure 2). Protac-4 contains ALAPYIP peptide with a ploy-D-arginine tag as the E3 ligase ligand, which is recognized by VHL. Protac-4 recruits the target protein, FK506-binding protein 12 (FKBP12), and induces its ubiquitination and subsequent degradation (32). Zhang et al. (33, 34) also developed a PROTAC molecule with a similar hydroxyl-proline peptide to hijack the VHL to target the ERα protein. This PROTAC can enter cells and inhibit breast cancer cell proliferation by degrading the ERα protein. Although additional strategies have been applied to peptide-based PROTAC molecules to improve cell permeability and new peptide-based PROTACs have been discovered, peptide-based PROTACs are still unattractive as drugs in clinical therapy owing to their vulnerable peptide bonds and poor delivery abilities (32, 35–37).
Following the early pioneering work, numerous small molecule-based PROTACs have been developed to overcome poor cell permeability and stability. In 2008, Schneekloth et al. (23) developed the first all small-molecule-based PROTAC targeting AR in HeLa cells. This PROTAC molecule (Figure 3) consists of a selective AR modulator (SARM) hydroxyflutamide, a non-steroidal AR ligand, and nutlin-3A, known as the MDM2 ligand, connected by a short soluble PEG linker. With an acceptable cell permeability, the SARM-nutlin PROTAC induced intracellular AR protein degradation in a UPS-dependent manner (23). All small-molecule-based PROTACs can efficiently induce intracellular protein degradation while bypass issues associated with peptide-based PROTACs.
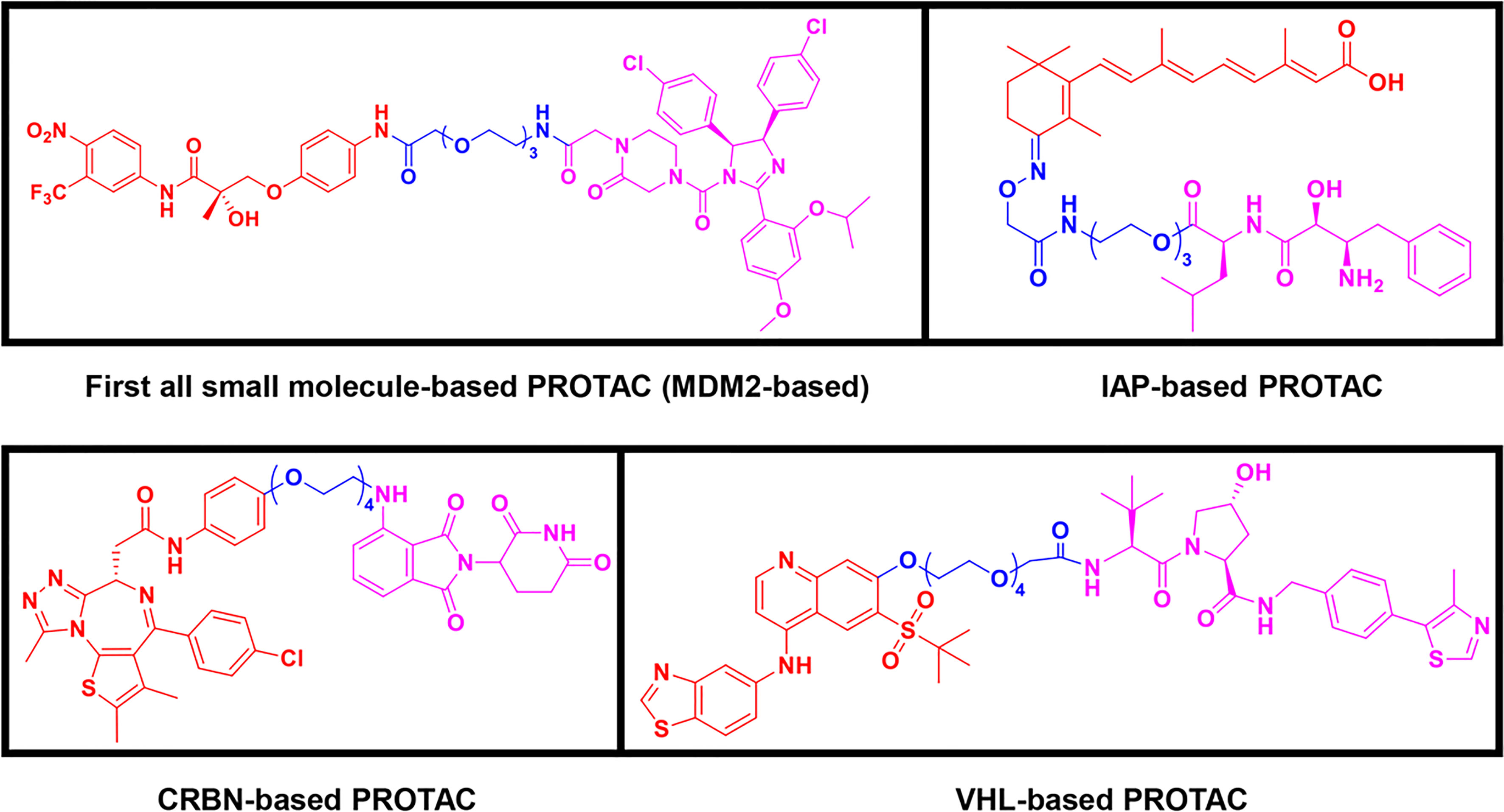
Figure 3 Second-generation PROTACs, Small molecule-based PROTACs. Chemical structures of first all small molecule-based PROTAC (MDM2-based), representative IAP-based PROTAC, representative CRBN-based PROTAC, and representative VHL-based PROTAC.
Since then, small molecules have been widely used as E3 ligase ligands in PROTACs, including nutlin-3A (23) and Idasanutlin (38) as MDM2 ligand, VH032 (39), VHL ligand 8 (40), VH298 (41, 42) and their derivatives (43, 44) as VHL ligands, thalidomide (45), lenalidomide (46, 47), pomalidomide (45), and TD-106 derivatives (48) as CRBN ligands, and bestatin-methyl ester (ME-BS) (49), MV1 (50), and LCL161 (51) as IAP ligands. Several small molecule-based PROTACs have been reported based on these afore-mentioned small molecule ligands for E3 ligases, including PROTACs against BET proteins (28, 38), kinases (52–55), nuclear hormone receptors (17, 40, 43, 56–58), as well as additional proteins (59, 60).
Homo-PROTACs are a special type of small molecule-based PTOTACs, which dimerize one particular E3 ubiquitin ligase and then induce auto-degradation. The first homo-PROTAC, the representative compound CM11, was reported in 2017 (25). CM11 composed two identical VHL ligands, and induced proteasome-dependent self-degradation of VHL in different cell lines. Pomalidomide-based homo-PROTACs exhibit highly potent degradation of CRBN, with only minimal effects on Ikaros (IKZF1) and Aiolos (IKZF3) (61, 62). Homo-PROTACs provide a useful chemical tool to investigate the biological functions of different E3 ligases.
To overcome the off-tissue issues, which represent one of the major limitations of PROTACs, a new generation of controllable PROTACs was devised (63). Phospho-dependent PROTACs (phosphoPROTACs), the first controllable PROTACs, were developed by Hines et al. (64) in 2013 to specifically degrade targets by activating kinase-signaling (Figure 4). Since then, other research groups have independently reported light-inducible opto-PROTACs (Figure 4) and photo-switchable PROTACs (photoPROTACs, Figure 4), which use light, particularly ultraviolet A (UVA) or near-infrared, to achieve PROTACs control (65–68). These controllable PROTACs can potentially be used in clinical settings. However, their application is restricted to specific types of cancers because there is a lack of clear boundaries between tumor and normal tissues, and UV light can cause DNA damage and can penetrate tissues (8, 66).
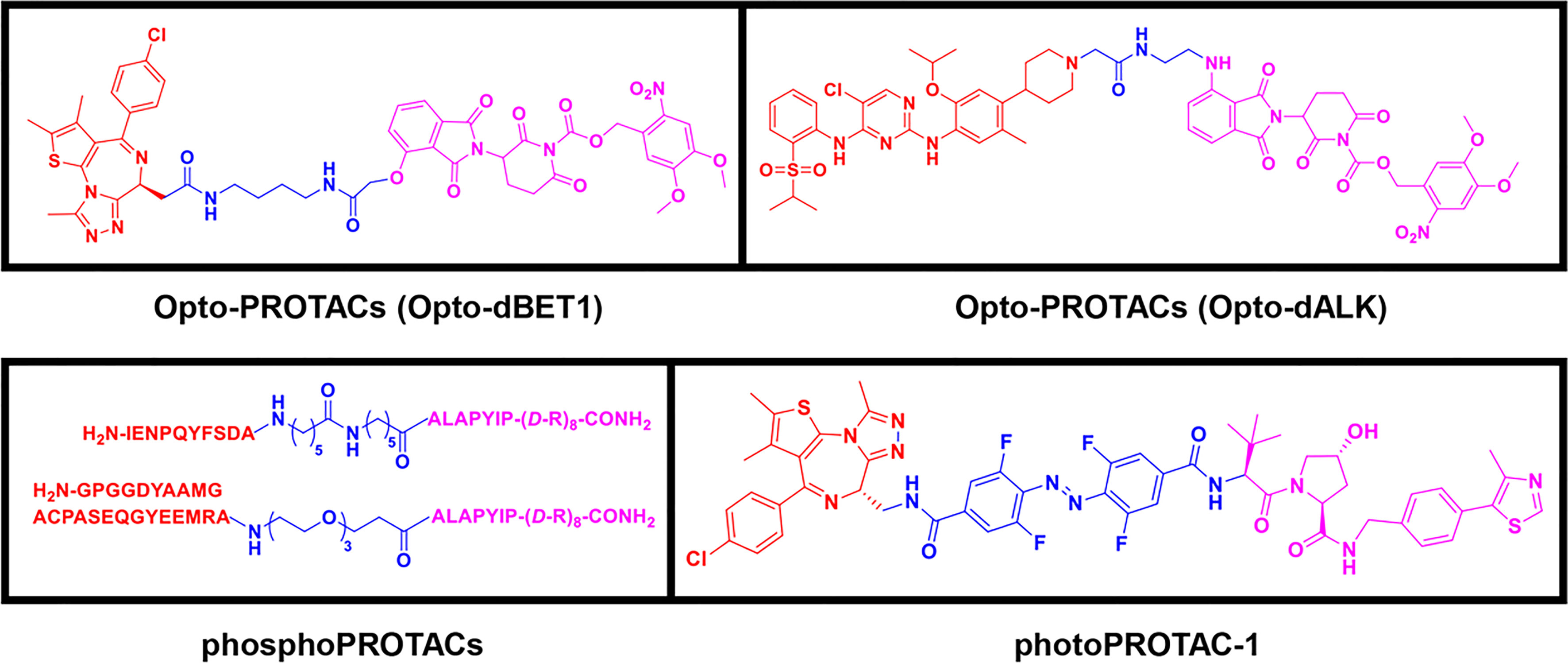
Figure 4 Third-generation PROTACs, Controllable PROTACs. Chemical structures of representative opto-PROTACs (opto-dBET1 and opto-dALK), representative phosphoPROTACs, representative photoPROTACs.
Sex hormone-dependent cancers, including breast, ovarian, and prostate cancers, currently contribute to a high number of cancer-related deaths worldwide (69). Sex hormone-dependent cancers affect both females and males since sex steroid hormones share common features (70). Some hormones can activate relevant pathways by binding to related receptors, including ER and AR, and then promote tumor occurrence, development, and metastasis (71). Therefore, ER and AR are potential therapeutic targets in sex hormone-dependent cancers (72).
Prostate cancer (PC) is the second leading cause of cancer-associated mortality in males in developed countries after lung cancer, and the incidence rate is increasing in developing countries (73, 74). Dihydrotestosterone (DHT) binds to inactivated AR, and cause the dissociation of heat shock protein (HSP) from AR-HSP complex. DHT-bound AR then translocate to the nucleus after suffer phosphorylation and dimerization. As a nuclear transcription factor, AR binds to the androgen response element (ARE) in DNA and starts the transcription of target genes after recruiting transcription regulators (Figure 5A). AR is the main driving force in PC development and has been identified as a pivotal therapeutic target. Endocrine therapy is one of the important strategies for PC treatment, including abiraterone by blocking androgen synthesis and AR antagonist enzalutamide by inhibiting AR function. However, most patients with PC will inevitably progress to castration-resistant PC (CRPC) (75). For metastatic CRPC, the main treatment plan involves using the small molecule chemotherapy drugs docetaxel and carbataxel (75), combined with the androgen small molecule inhibitors abiraterone and enzalutamide (76).
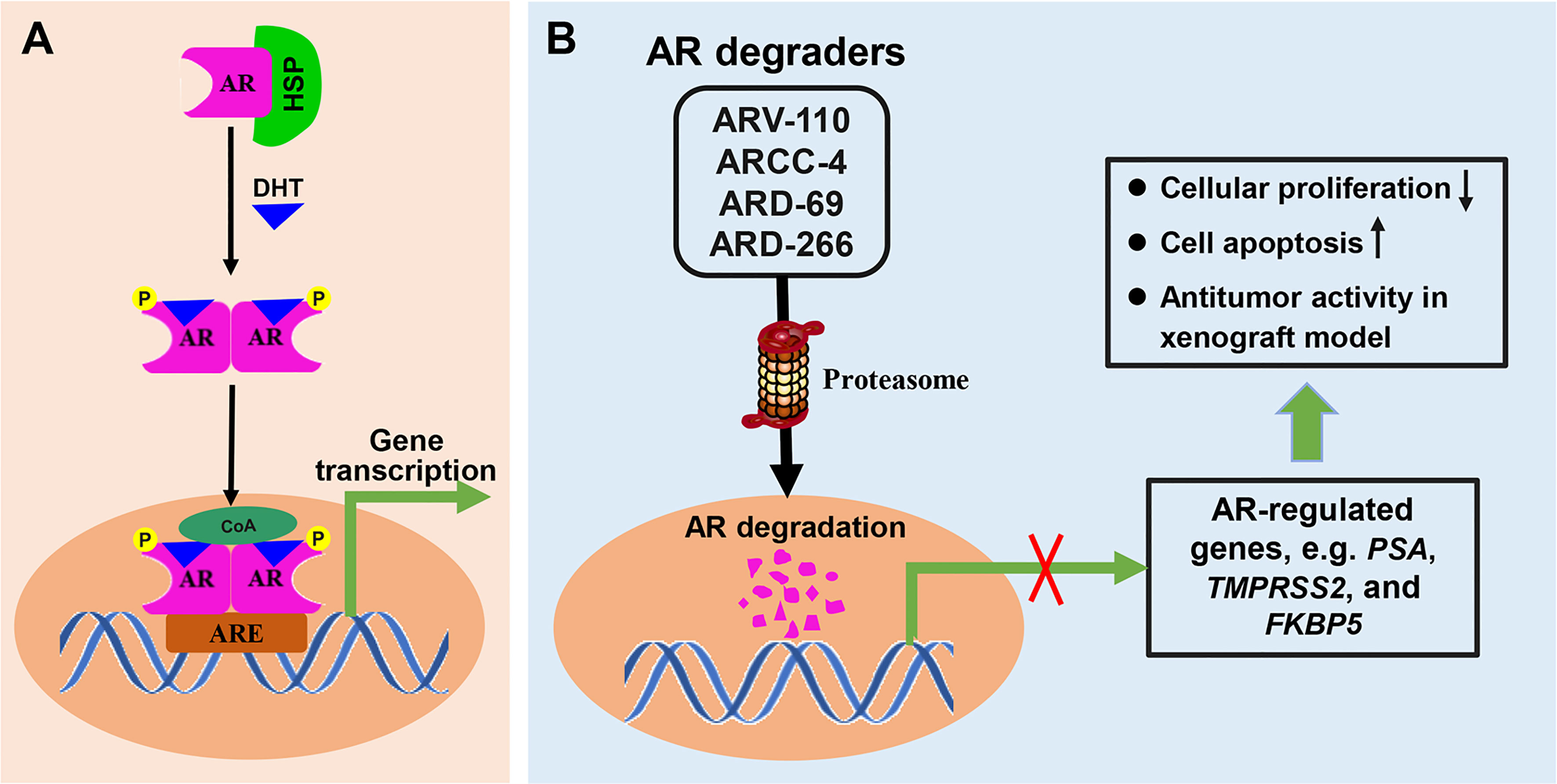
Figure 5 The mechanisms involved in AR signal pathway and PROTACs targeting AR. (A) Dihydrotestosterone (DHT) binds to inactivated AR, and cause the dissociation of heat shock protein (HSP) from AR-HSP complex. DHT-bound AR translocate to the nucleus after suffer phosphorylation and dimerization, then interacts with the androgen response element (ARE) and control target gene transcription. (B) PROTAC AR degraders can induce proteasome-mediated degradation of AR protein, and effectively suppress the mRNA levels of AR-regulated genes, which resulted in cellular proliferation inhibition, induction of cell apoptosis, and antitumor activity in xenograft model.
Unfortunately, approximately 25% of CRPC patients do not respond to these treatments, and the vast majority of responsive patients will ultimately develop resistance. AR signal pathway can be activated in CRPC because of AR mutation, AR amplification and AR alternate splicing variants formation, which cause the failure of traditional endocrine therapy. Nonetheless, small-molecule PROTACs targeting AR, which may overcome the problem of resistance to existing drug treatments, have been recently reported and provide an attractive direction (77). A number of other small-molecule PROTACs targeting AR have been discovered since the discovery of the first in 2008 (23). PROTAC AR degraders can induce proteasome-mediated degradation of AR protein in prostate cancer, block AR signaling and suppress the mRNA levels of AR-regulated genes, which results in cellular proliferation inhibition, induction of cell apoptosis, and antitumor activity in a xenograft model (Figure 5B). The subsequent sections of this review introduce these representative degraders.
ARV-110 (Table 1) is a small-molecule PROTAC drug developed by Arvinas Inc. (New Haven, USA). ARV-110 uses PROTAC technology to degrade AR protein and has been developed as a potential treatment for metastatic CRPC, which is the second most prevalent cancer in men (78). AR activates the transcription of a variety of proteins and is closely associated with cell proliferation and apoptosis following binding to its ligand, thus fueling tumor progression. ARV-110 completely degraded AR in prostate vertebral body cancer (VCaP) and LNCaP cell lines, giving a half degradation concentration (DC50) of < 1 nM (77). ARV-110 inhibited the expression of prostate-specific antigen (PSA) and FK506 binding protein 5 (FKBP5), inhibited AR-dependent cell proliferation, and induced apoptosis in VCap cells. It also showed plasma PSA reduction potency similar to that of the traditional AR antagonist, enzalutamide, but at lower doses, and demonstrated efficacy in enzalutamide-resistant PC xenograft models (78). As an orally bioavailable AR PROTAC degrader, ARV-110 was evaluated for safety, tolerance, and pharmacokinetics in a phase I clinical trial (NCT03888612) in early 2019, and the results highlighted its acceptable safety and antineoplastic activity in a heavily pretreated cohort with metastatic CRPC (18, 79). A phase II clinical trial (NCT03888612) is currently underway to estimate the therapeutic potential of ARV-110 in men with metastatic CRPC who have failed to standard treatment.

Table 1 Components and properties of small-molecule PROTACs in sex hormone-dependent cancer.
In 2018, Salami et al. (80) synthesized a variety of enzalutamide-based VHL-recruiting PROTACs targeting AR, and ARCC-4 (Table 1) was screened out using cellular models of drug resistance. ARCC-4 was identified as a highly efficient degrader, with DC50 of about 5 nM and a maximum degradation (Dmax) of more than 98%, even in cells expressing high levels of AR protein. After head-to-head comparisons between the currently approved AR antagonist enzalutamide and ARCC-4 in different cell models of PC resistance, the antitumor efficacy of ARCC-4, evaluated by PSA expression reduction, induction of cell apoptosis, and AR-dependent cell proliferation inhibition, outperformed enzalutamide. Interestingly, ARCC-4 effectively degraded clinically relevant ARs with different point mutants, including F876L, T877A, L702H, H874Y, and M896V, and remained active despite elevated androgen levels. ARCC-4 had better anti-proliferative effects in AR mutant cancers, whereas enzalutamide failed.
Han et al. (56) designed, synthesized, and evaluated a series of PROTAC AR degraders using five different AR antagonists, ligands for high-affinity VHL E3 ligases, and an optimized linker, ARD-69 (Table 1) was identified as the most effective AR degrader. ARD-69 achieved DC50 values of 0.86, 0.76, and 10.4 nM in LNCaP, VCaP, and 22Rv1, respectively. After 24 h of treatment, ARD-69 almost completely degraded AR in LNCaP and VCaP cells, with the concentration reaching below 1 nM. ARD-69 also effectively and dose-dependently inhibited PSA, transmembrane protease serine 2 (TMPRSS2), and FKBP5 expression. Furthermore, ARD-69 inhibited LNCaP, VCaP, and 22Rv1 cell proliferation, with efficiencies 100 times higher relative to enzalutamide. Additionally, ARD-69 effectively reduced AR and PSA protein expression in VCaP xenograft tumor tissues. Altogether, these data demonstrate that ARD-69 is an extremely potent AR degrader in treating metastatic CRPC.
Wang et al. (40) also investigated how the binding affinity of the VHL ligand portion of the VHL protein influences the potency and efficiency of PROTAC degraders. Using the PROTAC degrader ARD-61 (Table 1) with a high-affinity VHL ligand (56, 81), they discovered another more potent PROTAC AR degrader, ARD-266 (Table 1), consisting of a low-affinity VHL ligand, a reoptimized shorter linker, and the same AR antagonist (40). ARD-266 effectively induced AR protein degradation in the AR-positive (AR+) PC cell lines LNCaP, VCaP, and 22Rv1, yielding DC50 values of 0.2–1 nM. In addition, AR protein levels in LNCaP and VCap cells decreased by more than 95% following treatment with 10 nM ARD-266 for 6 h. Notably, ARD-266 effectively inhibited AR-regulated gene expression in a dose-dependent manner. This was the first study to demonstrate that, even using a low-affinity ligand with nanomolar levels of E3 ligase complex, the PROTAC degrader can efficiently degrade the target protein. Development of additional AR degraders is currently underway (82).
Breast cancer is one of the most common malignant tumors in women and is associated with the highest cancer-related mortality. Approximately 70% of breast cancers are ER-positive (ER+) (92). The ERs located in the nucleus, including ERα and ERβ, can directly bind to DNA or indirectly bind to DNA through corresponding transcription factors to regulate the transcription of target genes and exert biological effects. Besides, ER can be phosphorylated and regulate gene transcription in a ligand independent pathway (Figure 6A). ER is, therefore, an important therapeutic target for ER+ breast cancer. Although approved treatments were relatively successful, resistance to previous anti-estrogen therapy is developing in ER+ breast cancer. Fulvestrant, a selective estrogen receptor degrader (SERD), is the only ER-degrading agent approved for the treatment of ER+ breast cancer following anti-estrogen therapy. Though fulvestrant is beneficial for treating ER+ breast cancer, clinical trials show that it has poor solubility and cannot be orally administered. In clinical practice, fulvestrant can only be administered intramuscularly, limiting the total amount of drug intake and resulting in incomplete receptor blockade. To improve drug delivery, new ER-degrading agents are under continuous development. The recent use of PROTAC technology to degrade ER in breast cancer cells has attracted increasing attention. PROTAC targeting ERα can degrade both ERα proteins no matter whether ERα-encoding genes is mutated, and reduce the expression of estrogen-regulated genes, which resulted in cell proliferation inhibition, induction of necrotic cell death, and significant antitumor activity in MCF7 and patient-derived xenograft models (Figure 6B).
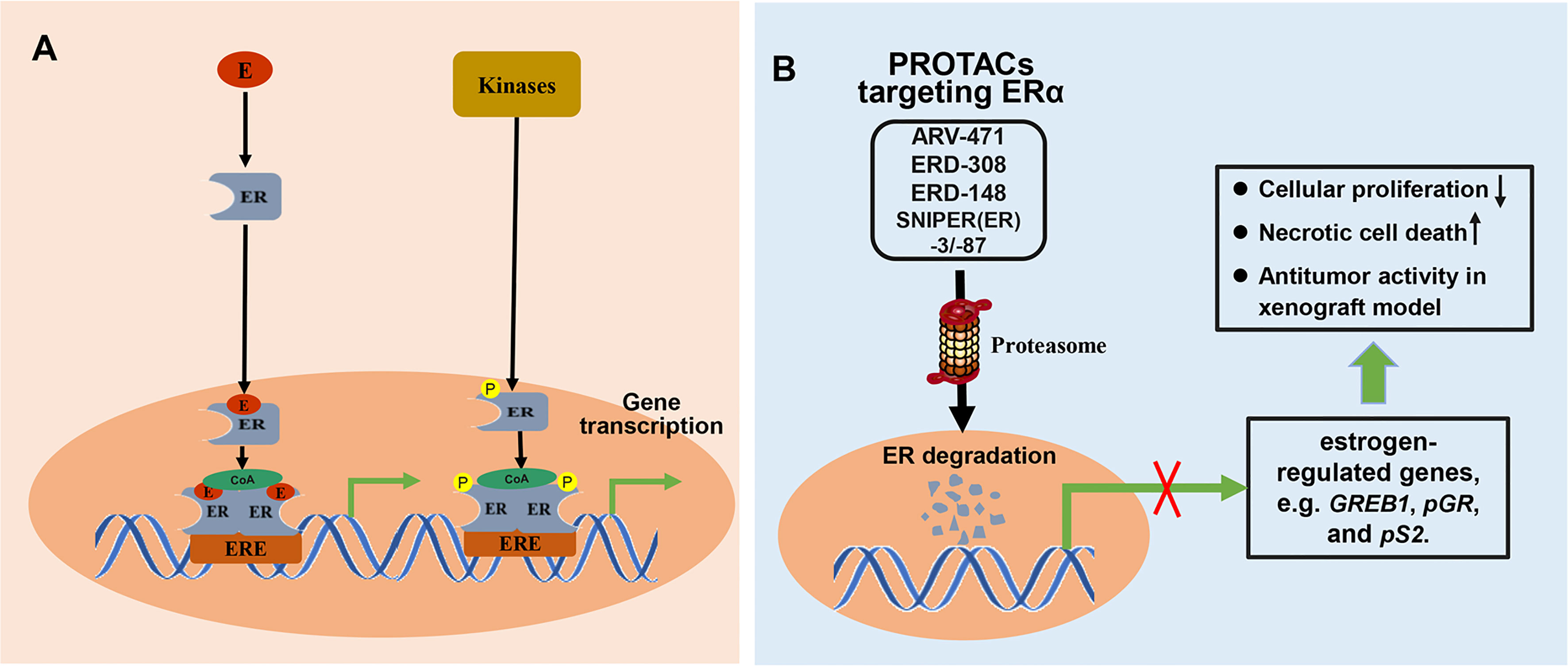
Figure 6 The mechanisms involved in ER signal pathway and PROTACs targeting ER. (A) Classically, ligands, like estrogen (E), bind to the ER to form ligand-bound ER. ligand-bound ER dimerizes and binds to estrogen response elements (ERE), and then activates gene transcription after recruiting coactivator (CoA). Besides, ER can be phosphorylated by related kinases and regulates gene transcription in a ligand independent pathway. (B) PROTAC targeting ER can induce proteasome-mediated degradation of ER protein, and effectively suppress the mRNA levels of estrogen-regulated genes, which resulted in cell proliferation inhibition, induction of necrotic cell death, and significant antitumor activity in xenograft model.
ARV-471 (Table 1) is a small-molecule PROTAC degrader co-developed by Arvinas Inc. and Pfizer to target ER (19). It is used for locally advanced or metastatic ER+/human epidermal growth factor receptor 2 (HER2) negative breast cancer, and is currently in phase II clinical trial (NCT04072952). ARV-471 degraded ER in ER+ breast cancer cell lines, with a DC50 of 1.8 nM (83). ARV-471-mediated ER degradation reduced the expression of estrogen-regulated genes and inhibited the proliferation of estrogen-dependent cell lines, including MCF7 and T47D cells. In addition, ARV-471 degraded clinically related ESR1 mutants (Y537S and D538G) and inhibited the growth of cell lines with these mutations. It also showed significant antitumor activity in an estrogen-dependent MCF7 xenograft tumor model, with the ERα protein decreasing by more than 90% by the end of the experiment. Moreover, more significant tumor growth inhibition (131% TGI) was observed when ARV-471 was combined with a cyclin dependent kinase 4/6 (CDK4/6) inhibitor, significantly decreasing ERα protein levels. In a patient-derived xenograft model with an ESR1 Y537S mutation, ARV-471 completely inhibited growth at a dose of 10 mg/kg and significantly reduced the level of mutant ER protein, exhibiting superior inhibition compared with fulvestrant. In the future, ARV-471 will be studied as a monotherapy or in combination with other therapies, such as CDK 4/6 inhibitors.
In 2019, Hu et al. (57) designed, synthesized, and evaluated a series of PROTAC degraders targeting ERα and successfully discovered a highly potent degrader, ERD-308 (Table 1), consisting of an ER antagonist and VHL-1 as an E3 ligase. DC50 values of ERD-308 in MCF-7 and T47D ER+ breast cancer cells were 0.17 and 0.43 nM, respectively, and more than 95% ERα degradation was induced at concentrations as low as 5 nM in both cell lines. Compared with fulvestrant or RAD1901, two conventional SERD molecules, ERD-308 achieved more thorough ERα degradation and more effectively inhibited cell proliferation in MCF-7 cells. Moreover, ERD-308 inhibited the expression of ER-regulated genes and the proliferation inhibition ability was much higher than that of fulvestrant and the SERM molecule raloxifene. The discovery of ERD-308 may promote the development of a completely new class of therapeutics to treat ER+ breast cancer.
During the structure activity relationship (SAR) studies of the PROTAC degrader ERD-308, compound ERD-148 (Table 1) also displayed excellent degrading potency (57). ERD-148 has a hydrophobic linker, whereas ERD-308 has a polyethylene glycol unit (PEG) embedded in the linker. Further investigations were conducted to characterize the pharmacological activity of the PROTAC degrader ERD-148 in ER+, estrogen-dependent MCF-7 wild type, cY537S, and cD538G mutant cells (84). Results showed that ERD-148 inhibited the growth of ER-dependent cells, with IC50 values of 0.8 nM, 10.5 nM, and 6.1 nM, in MCF-7 wild type, cY537S, and cD538G mutant cells, respectively. ERD-148 significantly downregulated ERα expression at concentrations as low as 10 nM in MCF-7 wild type and Y537S mutant cells. Moreover, ERD-148 significantly downregulated the mRNA level of Growth regulation by estrogen in breast cancer 1 (GREB1), an ER-regulated gene. However, ERD-148 did not inhibit the growth of ER-negative and estrogen-independent MDA-MB-231 breast cancer cells.
In 2011, Itoh et al. (93) designed specific small molecular protein degradation inducers known as specific and non-genetic IAP-dependent protein erasers (SNIPERs). SNIPER is a class of PROTAC used for protein degradation via the UPS. SNIPER comprises bestatin (BS), a ligand that interacts with the IAPs and an appropriate ligand for the target protein (94). Using 4-hydroxytamoxifen (4-OHT) as an ERα ligand, they developed a series of SNIPER(ER)s targeting the ERα protein for degradation, including SNIPER(ER)-3 (Table 1) (85, 86). SNIPER(ER)-3 potently induced the degradation of ERα and inhibited the estrogen-dependent expression of presenili 2 (pS2) gene in ER+ estrogen-dependent MCF-7 cells. ER is degraded by proteasomes following cIAP1-mediated ubiquitylation. SNIPER(ER)-3 reduced the viability of MCF-7 cells expressing ERα, but not of U2OS and HeLa cells, which do not express ERα protein. SNIPER(ER)-3 also induced necrotic cell death, accompanied by the high mobility group box 1 (HMGB1) release, a necrosis marker, from the cells into the media. Unfortunately, BS is a nonspecific ligand that limits the bio-orthogonality and maximal potency of SNIPER(ER)s.
Several IAP antagonists were subsequently incorporated into SNIPER(ER)s, and a novel SNIPER against ER, called SNIPER(ER)-87 (Table 1), was developed (87). SNIPER(ER)-87 contains an LCL161 derivative as a ligand for IAP, reduces ERα protein levels at the nanomolar level, and preferentially recruits X-linked inhibitor of apoptosis protein (XIAP), but not cIAP, to ERα for degradation. SNIPER(ER)-87 induced more than 50% degradation of ERα at a concentration as low as 3 nM. Daily administration of SNIPER(ER)-87 suppressed tumor growth in the MCF-7 breast tumor xenograft model and induced ERα degradation in tumors. In addition, SNIPER(ER)-87 effectively inhibited β-estradiol-mediated ERα-dependent transcriptional activation.
Li et al. (88) designed and synthesized a series of novel PROTAC-like SERDs containing an oxabicycloheptene sulfonamide (OBHSA) core structure and different side chains (the basic side chains, long alkyl acid side chains, and glycerol ether side chains). The basic side chain was confirmed as the appropriate degron, exhibiting the best anti-proliferative activity and good ERα degradation efficacies. Compound 17e (cpd. 17e) (Table 1) was selected as the best compound. Notably, 1 μM of cpd. 17e completely degraded ERα and 500 nM of the cpd. 17e showed good degradation activity. Compared with the parent compound OBHSA-1, the basic side chain of cpd. 17e played a pivotal role in increasing the potency of ERα degradation. These results suggest new possibilities for the development of more effective PROTACs.
The estrogen-related receptor (ERR) is an orphan nuclear hormone receptor that can produce biological functions without binding to ligands. There are three ERR subtypes: ERRα (NR3B1), ERRβ (NR3B2), and ERRγ (NR3B3). Recent studies (95, 96) have found that ERRα is closely associated with estrogen-induced breast cancer, endometrial cancer, and other estrogen-dependent tumors. Both in vitro and in vivo, pharmacological inhibition and gene knockout of ERRα have been shown to slow down the progression of breast cancer (97, 98).
To design the effective small molecule PROTAC, Bondeson et al. (43) replaced the hypoxia-inducible factor 1α (HIF1α) peptide with a high-affinity small-molecule ligand of VHL, retaining the essential hydroxyproline part for VHL binding. A thiazolidinedione-based ligand, selectively binding to ERRα over other reported ERR isoforms, was incorporated to generate PROTAC cpd. 29 (Table 1). PROTAC cpd. 29 decreased the level of ERRα in MCF-7 breast cancer cells in a dose-dependent manner. The Dmax was 86%, and the DC50 was approximately 100 nM. In vivo experiments showed that PROTAC cpd. 29 possessed broad tissue distribution and knocked down ERRα in tumor xenografts. These results show that PROTACs offer a method for achieving in vivo protein knockdown with potential therapeutic applications.
Peng et al. (89) designed and synthesized a series of (E)-3-(4-((2,4-bis(trifluoromethyl)benzyl)oxy)-3-methoxyphenyl)-2-cyanoacrylamide derivatives as new ERRα degraders based on the PROTAC concept, and cpd. 6c (Table 1) was identified as one of the most potent and selective ERRα degraders. Cpd. 6c induced remarkable degradation of ERRα at a concentration of 3.0 nM. Additionally, 30 nM of cpd. 6c specifically degraded > 80% of ERRα and potently decreased the levels of proteins encoded by ERRα downstream target genes. Additional studies suggested that ternary complex and ubiquitin-proteasome were involved in cpd. 6c-mediated ERRα degradation.
Inhibitors of BET proteins have recently shown growth-inhibitory activity in preclinical models of CRPC, with BET being an attractive target in CRPC (99). In 2016, ARV-771 (Table 1), a VHL E3 ligase-based pan-BET PROTAC, was designed and synthesized (90). It rapidly degraded bromodomain protein 4 (BRD4) protein, with DC50 < 5 nM, and inhibited the expression of c-Myc, with IC50 < 1 nM in 22Rv1, LnCaP95, and VCaP CRPC cell lines. However, the ARV-771 downregulating activity of c-Myc expression was more than ten times higher than that of the BET inhibitor, JQ1. Moreover, ARV-771 induced significant poly (ADP-ribose) polymerase (PARP) cleavage, caspase activation, and apoptosis in 22Rv1 cells. Additionally, ARV-771 was efficacious in two different xenograft models of CRPC and resulted in tumor regression in enzalutamide-resistant 22Rv1 tumor xenografts.
Zhao et al. (91) designed and synthesized a small PROTAC molecule, the representative cpd. 3 (Table 1), based on niraparib as the PARP1 binding moiety and nutlin-3 derivatives as the E3 ligase-binding moiety. PROTAC cpd. 3 induced PARP1 cleavage and apoptosis in the MDA-MB-231 cell line. PROTAC cpd. 3 was also 5-fold more potent than niraparib, olaparib, and veliparib at degrading PARP1 when tested in MDA-MB-231 cells and exhibited no cytotoxicity to normal breast cells. This PARP1-targeting PROTAC-type compound represents a huge potential application value in the therapy of the MDA-MB-231 cell-like subtype of triple-negative breast cancers.
Steroid hormones play vital roles in the initiation and progression of sex hormone-dependent cancers, including breast and prostate cancers. Endocrine therapy targeting ERs, ARs, and ERRs represents a potential and pivotal therapeutic strategy for breast/prostate cancer therapy. The PROTAC approach is a novel therapeutic strategy that is particularly suited for abolishing the activity of target proteins, including ERs, ERRs, ARs, as well as other oncoproteins. Generally, PROTACs bind only to a small proportion of their target proteins and act like a catalyst, which is highly effective at degradation even if the target protein’s concentration varies considerably within cells. However, traditional small molecule regulators play their roles by occupying the active pocket sites and require high drug administration dosage to maintain activity, which increases the risk of off-target and adverse effects. Protein degradation using PROTACs also provides the opportunity to overcome resistance to endocrine therapy and receptor antagonists in sex hormone-dependent cancers. To improve cell permeability and stability, PROTACs have evolved from peptides to small molecules. Many successful cases of small molecule-based PROTACs have recently been reported in cultured cells, mice, and humans, demonstrating the feasibility of applying PROTACs in clinical settings.
Although PROTACs offer considerable advantages and promising prospects in the clinical treatment of prostate and breast cancers, particularly hormone-resistant prostate and breast cancers, some issues still require consideration. First, PROTACs have a relatively high molecular weight (typically 700–1100 Da) and may not conform to Lipinski’s ‘rule of five’, which is a rule of thumb for evaluating whether a small molecule possesses pharmacological or biological properties of an orally active drug in humans (100). The high molecular weight of PROTACs reduces their cell permeability, tissue penetration, and metabolic attack, posing challenges for oral administration. To overcome the high molecular weight nature of typical PROTACs, heterobifunctional PROTACs can be formed intracellularly through the bio-orthogonal click combination of two tagged small molecule precursors in cells and can successfully induce target protein degradation, named in-cell click-formed proteolysis-targeting chimeras (CLIPTACs) (101).
In addition, it is cumbersome to predict the degradation efficiency of small-molecule PROTACs. For target protein ligands, high binary binding affinities do not always yield efficient degradation of target proteins (40), suggesting that it is difficult to identify the best ligand of the target protein for PROTAC construction. Linkerology plays the most pivotal role in PROTAC components, determining the biological and physicochemical properties of PROTACs. Optimization of length and rigidity of linker units are important for improving pharmaceutical performance. It is difficult to select an optimal linker tethering site on ligands of the target protein, even when the co-crystal structures of a specific ligand and the target protein are available.
Unintended on-target and off-target toxicities can also affect the clinical translation of PROTACs (102). E3 ligases contribute to the specificity in the degradation of target proteins. Complete degradation of certain proteins and degradation of untargeted proteins in a complex or close proximity to E3 ligase may be detrimental. The unintended on-target and off-target toxicities also affect PROTACs’ clinical translation (102). These toxicities can be avoided by incorporating a suitable E3 ligase, which is tissue-specific and tumor-selective, in the design of selective PROTACs. Light-controllable photo-PROTACs, whose action can be controlled under an external source of light to direct tumor-specific degradation of target proteins, have been reported by several research groups. Photo-PROTACs increase the potential for target degradation in the desired tissues.
The “hook effect” is another unavoidable problem. At high intracellular concentrations of small-molecule PROTACs, binary complexes are favored over ternary complexes, ultimately decreasing target degradation (16, 103). Since some target protein ligands have agonist/antagonist activities, PROTACs can function as traditional small molecule activators/inhibitors. This effect has been observed where PROTAC targets AR-containing mutations within the ligand-binding domain, as AR antagonists act as agonists for these mutations (104, 105). Consequently, generating tumor-specific/selective PROTACs is essential for reducing on- and off-target toxicities, while identifying the tumor-specific/selective E3 ligase is of primary importance. Thus, it is advisable to use suitable PK-PD models to predict the PROTAC dosage to avoid the hook effect.
Twenty years after the first PROTACs were synthesized in 2001, small-molecule PROTACs have not only drawn the attention of academic researchers, but also of pharmaceutical companies. Although there are concerns over their relatively high molecular weights and low oral availability, two orally active small-molecule PROTACs, ARV-110 (an AR degrader) and ARV-471 (an ER degrader), have been studied in phase I/II clinical trials. Small-molecule PROTACs are a potential strategy for drug discovery, providing a new way to treat sex hormone-dependent cancers. Since immune checkpoint inhibitors have achieved great success against some tumors by enhancing the antitumor immunity of immune cells, researchers inspired by this have attempted to design PROTACs that can potentially strengthen antitumor immunity (106, 107). Future development of small-molecule PROTACs shall focus on more “undruggable” proteins, and new E3 ligases with tissue-specific and tumor-specific expression patterns may be recruited. Overall, it is foreseeable that PROTACs will soon benefit patients.
YZ, HX, and YW designed the concept of the review and participated in manuscript writing, editing, revising, prepared the figures and supervised the entire process. LS, ZW, and LL collect literature data, participated in manuscript writing and formatting. LL and YZ prepared the structure of compounds. LL and JZ participated in manuscript writing, proof editing, and graphical design. All authors contributed to the article and approved the submitted version.
This work was supported by Excellent Youth Fund of Sichuan Cancer Hospital [grant number YB2021024], and Science and Technology Program of Sichuan Province [grant number 2021YFH0145], Scientific Research Project (Foundation) of the Health Planning Committee of Sichuan [grant number 20PJ108], and Scientific research project of Sichuan Medical Association [grant number S20023].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bashraheel SS, Domling A, Goda SK. Update on Targeted Cancer Therapies, Single or in Combination, and Their Fine Tuning for Precision Medicine. BioMed Pharmacother (2020) 125:110009. doi: 10.1016/j.biopha.2020.110009
2. Pisa R, Kapoor TM. Chemical Strategies to Overcome Resistance Against Targeted Anticancer Therapeutics. Nat Chem Biol (2020) 16(8):817–25. doi: 10.1038/s41589-020-0596-8
3. Kholodenko RV, Kalinovsky DV, Doronin II, Ponomarev ED, Kholodenko IV. Antibody Fragments as Potential Biopharmaceuticals for Cancer Therapy: Success and Limitations. Curr Med Chem (2019) 26(3):396–426. doi: 10.2174/0929867324666170817152554
4. Salami J, Crews CM. Waste Disposal—An Attractive Strategy for Cancer Therapy. Science (2017) 355(6330):1163–7. doi: 10.1126/science.aam7340
5. Boumahdi S, de Sauvage FJ. The Great Escape: Tumour Cell Plasticity in Resistance to Targeted Therapy. Nat Rev Drug Discovery (2020) 19(1):39–56. doi: 10.1038/s41573-019-0044-1
6. Groppe JC. Induced Degradation of Protein Kinases by Bifunctional Small Molecules: A Next-Generation Strategy. Expert Opin Drug Discovery (2019) 14(12):1237–53. doi: 10.1080/17460441.2019.1660641
7. Lebraud H, Heightman TD. Protein Degradation: A Validated Therapeutic Strategy With Exciting Prospects. Essays Biochem (2017) 61(5):517–27. doi: 10.1042/EBC20170030
8. Liu J, Ma J, Liu Y, Xia J, Li Y, Wang ZP, et al. PROTACs: A Novel Strategy for Cancer Therapy. Semin Cancer Biol (2020) 67:171–9. doi: 10.1016/j.semcancer.2020.02.006
9. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric Molecules That Target Proteins to the Skp1–Cullin–F Box Complex for Ubiquitination and Degradation. Proc Natl Acad Sci (2001) 98(15):8554–9. doi: 10.1073/pnas.141230798
10. Burslem GM, Crews CM. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell (2020) 181(1):102–14. doi: 10.1016/j.cell.2019.11.031
11. Bond MJ, Crews CM. Proteolysis Targeting Chimeras (PROTACs) Come of Age: Entering the Third Decade of Targeted Protein Degradation. RSC Chem Biol (2021) 2(3):725–42. doi: 10.1039/d1cb00011j
12. Hsu JH, Rasmusson T, Robinson J, Pachl F, Read J, Kawatkar S, et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem Biol (2020) 27(1):41–6.e17. doi: 10.1016/j.chembiol.2019.11.004
13. Liu Z, Hu X, Wang Q, Wu X, Zhang Q, Wei W, et al. Design and Synthesis of EZH2-Based PROTACs to Degrade the PRC2 Complex for Targeting the Noncatalytic Activity of EZH2. J Med Chem (2021) 64(5):2829–48. doi: 10.1021/acs.jmedchem.0c02234
14. Tu Y, Sun Y, Qiao S, Luo Y, Liu P, Jiang ZX, et al. Design, Synthesis, and Evaluation of VHL-Based EZH2 Degraders to Enhance Therapeutic Activity Against Lymphoma. J Med Chem (2021) 64(14):10167–84. doi: 10.1021/acs.jmedchem.1c00460
15. Potjewyd F, Turner AW, Beri J, Rectenwald JM, Norris-Drouin JL, Cholensky SH, et al. Degradation of Polycomb Repressive Complex 2 With an EED-Targeted Bivalent Chemical Degrader. Cell Chem Biol (2020) 27(1):47–56.e15. doi: 10.1016/j.chembiol.2019.11.006
16. Pettersson M, Crews CM. PROteolysis TArgeting Chimeras (PROTACs) - Past, Present and Future. Drug Discovery Today Technol (2019) 31:15–27. doi: 10.1016/j.ddtec.2019.01.002
17. Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, et al. PROTACs: Great Opportunities for Academia and Industry. Signal Transduction Targeted Ther (2019) 4(1):64. doi: 10.1038/s41392-019-0101-6
18. Petrylak DP, Gao X, Vogelzang NJ, Garfield MH, Taylor I, Dougan Moore M, et al. First-In-Human Phase I Study of ARV-110, an Androgen Receptor (AR) PROTAC Degrader in Patients (Pts) With Metastatic Castrate-Resistant Prostate Cancer (mCRPC) Following Enzalutamide (ENZ) and/or Abiraterone (ABI). Am Soc Clin Oncol (2020) 38(15-suppl):3500. doi: 10.1200/JCO.2020.38.15_suppl.3500
19. Snyder LB, Flanagan JJ, Qian Y, Gough SM, Andreoli M, Bookbinder M, et al. The Discovery of ARV-471, an Orally Bioavailable Estrogen Receptor Degrading PROTAC® for the Treatment of Patients With Breast Cancer. Am Soc Clin Oncol (2021) 81(13 Supplement):44. doi: 10.1158/1538-7445.AM2021-44
20. Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol (2015) 22(6):755–63. doi: 10.1016/j.chembiol.2015.05.009
21. Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, et al. Targeting the Von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules to Disrupt the VHL/HIF-1α Interaction. J Am Chem Soc (2012) 134(10):4465–8. doi: 10.1021/ja209924v
22. Itoh Y, Ishikawa M, Naito M, Hashimoto YJ. Protein Knockdown Using Methyl Bestatin– Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J Am Chem Soc (2010) 132(16):5820–6. doi: 10.1021/ja100691p
23. Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg Med Chem Lett (2008) 18(22):5904–8. doi: 10.1016/j.bmcl.2008.07.114
24. Asatsuma-Okumura T, Ito T, Handa H. Molecular Mechanisms of Cereblon-Based Drugs. Pharmacol Ther (2019) 202:132–9. doi: 10.1016/j.pharmthera.2019.06.004
25. Maniaci C, Hughes SJ, Testa A, Chen W, Lamont DJ, Rocha S, et al. Homo-PROTACs: Bivalent Small-Molecule Dimerizers of the VHL E3 Ubiquitin Ligase to Induce Self-Degradation. Nat Commun (2017) 8(1):830. doi: 10.1038/s41467-017-00954-1
26. Bricelj A, Steinebach C, Kuchta R, Gütschow M, Sosič I. E3 Ligase Ligands in Successful PROTACs: An Overview of Syntheses and Linker Attachment Points. Front Chem (2021) 9:707317(463). doi: 10.3389/fchem.2021.707317
27. Chen Y, Jin J. The Application of Ubiquitin Ligases in the PROTAC Drug Design. Acta Biochim Biophys Sin (Shanghai) (2020) 52(7):776–90. doi: 10.1093/abbs/gmaa053
28. Yang CY, Qin C, Bai L, Wang S. Small-Molecule PROTAC Degraders of the Bromodomain and Extra Terminal (BET) Proteins - A Review. Drug Discovery Today Technol (2019) 31:43–51. doi: 10.1016/j.ddtec.2019.04.001
29. Liu J, Chen H, Kaniskan H, Xie L, Chen X, Jin J, et al. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J Am Chem Soc (2021) 143(23):8902–10. doi: 10.1021/jacs.1c03852
30. Tan L, Gray NS. When Kinases Meet PROTACs. Chin J Chem (2018) 36(010):971–7. doi: 10.1002/cjoc.201800293
31. Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, et al. Development of Protacs to Target Cancer-Promoting Proteins for Ubiquitination and Degradation. Mol Cell Proteomics (2003) 2(12):1350–8. doi: 10.1074/mcp.T300009-MCP200
32. Schneekloth JS Jr, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, et al. Chemical Genetic Control of Protein Levels: Selective In Vivo Targeted Degradation. J Am Chem Soc (2004) 126(12):3748–54. doi: 10.1021/ja039025z
33. Zhang D, Baek S-H, Ho A, Lee H, Jeong Y, Kim K. Targeted Degradation of Proteins by Small Molecules: A Novel Tool for Functional Proteomics. Combinatorial Chem High Throughput Screening (2004) 7(7):689–97. doi: 10.2174/1386207043328364
34. Cyrus K, Wehenkel M, Choi EY, Lee H, Swanson H, Kim KB. Jostling for Position: Optimizing Linker Location in the Design of Estrogen Receptor-Targeting PROTACs. ChemMedChem (2010) 5(7):979–85. doi: 10.1002/cmdc.201000146
35. Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ, et al. Targeting Steroid Hormone Receptors for Ubiquitination and Degradation in Breast and Prostate Cancer. Oncogene (2008) 27(57):7201–11. doi: 10.1038/onc.2008.320
36. Ma D, Zou Y, Chu Y, Liu Z, Liu G, Chu J, et al. A Cell-Permeable Peptide-Based PROTAC Against the Oncoprotein CREPT Proficiently Inhibits Pancreatic Cancer. Theranostics (2020) 10(8):3708–21. doi: 10.7150/thno.41677
37. Jiang Y, Deng Q, Zhao H, Xie M, Chen L, Yin F, et al. Development of Stabilized Peptide-Based PROTACs Against Estrogen Receptor α. ACS Chem Biol (2017) 13(3):628–35. doi: 10.1021/acschembio.7b00985
38. Hines J, Lartigue S, Dong H, Qian Y, Crews CM. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of P53. Cancer Res (2019) 79(1):251–62. doi: 10.1158/0008-5472.CAN-18-2918
39. Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, et al. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat Chem Biol (2017) 13(5):514–21. doi: 10.1038/nchembio.2329
40. Han X, Zhao L, Xiang W, Qin C, Miao B, Xu T, et al. Discovery of Highly Potent and Efficient PROTAC Degraders of Androgen Receptor (AR) by Employing Weak Binding Affinity VHL E3 Ligase Ligands. J Med Chem (2019) 62(24):11218–31. doi: 10.1021/acs.jmedchem.9b01393
41. Qiu S, Jia Y, Sun Y, Han P, Xu J, Wen G, et al. Von Hippel-Lindau (VHL) Protein Antagonist VH298 Improves Wound Healing in Streptozotocin-Induced Hyperglycaemic Rats by Activating Hypoxia-Inducible Factor- (HIF-) 1 Signalling. J Diabetes Res (2019) 2019:1897174. doi: 10.1155/2019/1897174
42. Frost J, Galdeano C, Soares P, Gadd MS, Grzes KM, Ellis L, et al. Potent and Selective Chemical Probe of Hypoxic Signalling Downstream of HIF-α Hydroxylation via VHL Inhibition. Nat Commun (2016) 7:13312. doi: 10.1038/ncomms13312
43. Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, et al. Catalytic In Vivo Protein Knockdown by Small-Molecule PROTACs. Nat Chem Biol (2015) 11(8):611–7. doi: 10.1038/nchembio.1858
44. Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, et al. Small-Molecule Inhibitors of the Interaction Between the E3 Ligase VHL and HIF1alpha. Angew Chem Int Ed Engl (2012) 51(46):11463–7. doi: 10.1002/anie.201206231
45. Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, et al. Structure of the DDB1–CRBN E3 Ubiquitin Ligase in Complex With Thalidomide. Nature (2014) 512(7512):49–53. doi: 10.1038/nature13527
46. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science (2014) 343(6168):301–5. doi: 10.1126/science.1244851
47. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science (2014) 343(6168):305–9. doi: 10.1126/science.1244917
48. Kim SA, Go A, Jo SH, Park SJ, Jeon YU, Kim JE, et al. A Novel Cereblon Modulator for Targeted Protein Degradation. Eur J Med Chem (2019) 166:65–74. doi: 10.1016/j.ejmech.2019.01.023
49. Sekine K, Takubo K, Kikuchi R, Nishimoto M, Kitagawa M, Abe F, et al. Small Molecules Destabilize Ciap1 by Activating Auto-Ubiquitylation. J Biol Chem (2008) 283(14):8961–8. doi: 10.1074/jbc.M709525200
50. Tomoshige S, Nomura S, Ohgane K, Hashimoto Y, Ishikawa M. Degradation of Huntingtin Mediated by a Hybrid Molecule Composed of IAP Antagonist Linked to Phenyldiazenyl Benzothiazole Derivative. Bioorg Med Chem Lett (2018) 28(4):707–10. doi: 10.1016/j.bmcl.2018.01.012
51. Ohoka N, Morita Y, Nagai K, Shimokawa K, Ujikawa O, Fujimori I, et al. Derivatization of Inhibitor of Apoptosis Protein (IAP) Ligands Yields Improved Inducers of Estrogen Receptor α Degradation. J Biol Chem (2018) 293(18):6776–90. doi: 10.1074/jbc.RA117.001091
52. Kang CH, Lee DH, Lee CO, Du Ha J, Park CH, Hwang JY. Induced Protein Degradation of Anaplastic Lymphoma Kinase (ALK) by Proteolysis Targeting Chimera (PROTAC). Biochem Biophys Res Commun (2018) 505(2):542–7. doi: 10.1016/j.bbrc.2018.09.169
53. Tovell H, Testa A, Zhou H, Shpiro N, Crafter C, Ciulli A, et al. Design and Characterization of SGK3-PROTAC1, an Isoform Specific SGK3 Kinase PROTAC Degrader. ACS Chem Biol (2019) 14(9):2024–34. doi: 10.1021/acschembio.9b00505
54. Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem Biol (2018) 25(1):67–77.e3. doi: 10.1016/j.chembiol.2017.09.009
55. Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD, et al. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat Commun (2019) 10(1):131. doi: 10.1038/s41467-018-08027-7
56. Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang CY, et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J Med Chem (2019) 62(2):941–64. doi: 10.1021/acs.jmedchem.8b01631
57. Hu J, Hu B, Wang M, Xu F, Miao B, Yang CY, et al. Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (Er). J Med Chem (2019) 62(3):1420–42. doi: 10.1021/acs.jmedchem.8b01572
58. Shibata N, Nagai K, Morita Y, Ujikawa O, Ohoka N, Hattori T, et al. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J Med Chem (2018) 61(2):543–75. doi: 10.1021/acs.jmedchem.7b00168
59. Schiedel M, Herp D, Hammelmann, Swyter, Lehotzky A, Robaa D, et al. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J Med Chem (2018) 61(2):482–91. doi: 10.1021/acs.jmedchem.6b01872
60. Yang K, Song Y, Xie H, Wu H, Wu Y-T, Leisten ED, et al. Development of the First Small Molecule Histone Deacetylase 6 (HDAC6) Degraders. Bioorg Med Chem Lett (2018) 28(14):2493–7. doi: 10.1016/j.bmcl.2018.05.057
61. Steinebach C, Lindner S, Udeshi ND, Mani DC, Kehm H, Köpff S, et al. Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem Biol (2018) 13(9):2771–82. doi: 10.1021/acschembio.8b00693
62. Lindner S, Steinebach C, Udeshi N, Mani D, Kehm H, Köpff S, et al. Pomalidomide-Based Homo-Protacs for the Chemical Knockdown of Cereblon. ACS Chem Biol (2018) 132:260. doi: 10.1021/acschembio.8b00693
63. Liu J, Peng Y, Wei WJ. Light-Controllable PROTACs for Temporospatial Control of Protein Degradation. Front Cell Dev Biol (2021) 9:678077. doi: 10.3389/fcell.2021.678077
64. Hines J, Gough JD, Corson TW, Crews CM. Posttranslational Protein Knockdown Coupled to Receptor Tyrosine Kinase Activation With phosphoPROTACs. Proc Natl Acad Sci USA (2013) 110(22):8942–7. doi: 10.1073/pnas.1217206110
65. Xue G, Wang K, Zhou D, Zhong H, Pan Z. Light-Induced Protein Degradation With Photocaged PROTACs. J Am Chem Soc (2019) 141(46):18370–4. doi: 10.1021/jacs.9b06422
66. Liu J, Chen H, Ma L, He Z, Wang D, Liu Y, et al. Light-Induced Control of Protein Destruction by Opto-PROTAC. Sci Adv (2020) 6(8):eaay5154. doi: 10.1126/sciadv.aay5154
67. Pfaff P, Samarasinghe KTG, Crews CM, Carreira EM. Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent Ence (2019) 5(10):1682–90. doi: 10.1021/acscentsci.9b00713
68. Trauner D, Arp CJ. PHOTACs Enable Optical Control of Protein Degradation. Synfacts (2020) 16(05):0591–. doi: 10.1126/sciadv.aay5064
69. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
70. McKenna NJ, Lanz RB, O’Malley BW. Nuclear Receptor Coregulators: Cellular and Molecular Biology. Endocr Rev (1999) 20(3):321–44. doi: 10.1210/edrv.20.3.0366
71. Folkerd EJ, Dowsett MJ. Influence of Sex Hormones on Cancer Progression. J Clin Oncol (2010) 28(26):4038–44. doi: 10.1200/JCO.2009.27.4290
72. Ulm M, Ramesh AV, McNamara KM, Ponnusamy S, Sasano H, Narayanan R. Therapeutic Advances in Hormone-Dependent Cancers: Focus on Prostate, Breast and Ovarian Cancers. Endocr Connect (2019) 8(2):R10–26. doi: 10.1530/EC-18-0425
73. Litwin MS, Tan HJ. The Diagnosis and Treatment of Prostate Cancer: A Review. Jama (2017) 317(24):2532–42. doi: 10.1001/jama.2017.7248
74. Hamdy FC, Donovan JL, Lane JA, Mason M, Metcalfe C, Holding P, et al. 10-Year Outcomes After Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. New Engl J Med (2016) 375(15):1415–24. doi: 10.1056/NEJMoa1606220
75. Karantanos T, Corn PG, Thompson TC. Prostate Cancer Progression After Androgen Deprivation Therapy: Mechanisms of Castrate Resistance and Novel Therapeutic Approaches. Oncogene (2013) 32(49):5501–11. doi: 10.1038/onc.2013.206
76. Cindolo L, Natoli C, De Nunzio C, De Tursi M, Valeriani M, Giacinti S, et al. Abiraterone Acetate for Treatment of Metastatic Castration-Resistant Prostate Cancer in Chemotherapy-Naive Patients: An Italian Analysis of Patients’ Satisfaction. Clin Genitourinary Cancer (2017) 15(5):520–5. doi: 10.1016/j.clgc.2017.04.003
77. Neklesa T, Snyder L, Willard R, Vitale N, Pizzano J, Gordon D, et al. ARV-110: An Oral Androgen Receptor PROTAC Degrader for Prostate Cancer. J Clin Oncol (2019) 37:259–. doi: 10.1200/JCO.2019.37.7_suppl.259
78. Snyder LB, Neklesa TK, Chen X, Dong H, Ferraro C, Gordon DA, et al. Discovery of ARV-110, a First in Class Androgen Receptor Degrading PROTAC® for the Treatment of Men With Metastatic Castration Resistant Prostate Cancer. Am Assoc Cancer Res (2021) 81(4_Suppl):43. doi: 10.1158/1538-7445.AM2021-43
79. Petrylak DP. Potential of Targeted Protein Degradation: Early Results for ARV-110. Oncol Times (2020) 42(21):27–8. doi: 10.1097/01.COT.0000722628.60363.59
80. Salami J, Alabi S, Willard RR, Vitale NJ, Wang J, Dong H, et al. Androgen Receptor Degradation by the Proteolysis-Targeting Chimera ARCC-4 Outperforms Enzalutamide in Cellular Models of Prostate Cancer Drug Resistance. Commun Biol (2018) 1:100. doi: 10.1038/s42003-018-0105-8
81. Kregel S, Wang C, Han X, Xiao L, Fernandez-Salas E, Bawa P, et al. Androgen Receptor Degraders Overcome Common Resistance Mechanisms Developed During Prostate Cancer Treatment. Neoplasia (2020) 22(2):111–9. doi: 10.1016/j.neo.2019.12.003
82. Kargbo RB. Androgen Receptor Degradation for Therapeutic Intervention of Prostate Cancer Drug Resistance. ACS Med Chem Lett (2021) 12(4):518–20. doi: 10.1021/acsmedchemlett.1c00120
83. Flanagan JJ, Qian Y, Gough SM, Andreoli M, Bookbinder M, Cadelina G, et al. Abstract P5-04-18: ARV-471, an Oral Estrogen Receptor PROTAC Degrader for Breast Cancer. Cancer Res (2019) 79:P5–04. doi: 10.1158/1538-7445.SABCS18-P5-04-18
84. Gonzalez TL, Hancock M, Sun S, Gersch CL, Larios JM, David W, et al. Targeted Degradation of Activating Estrogen Receptor Alpha Ligand-Binding Domain Mutations in Human Breast Cancer. Breast Cancer Res Treat (2020) 180(3):611–22. doi: 10.1007/s10549-020-05564-y
85. Okuhira K, Demizu Y, Hattori T, Ohoka N, Shibata N, Nishimaki-Mogami T, et al. Development of Hybrid Small Molecules That Induce Degradation of Estrogen Receptor-Alpha and Necrotic Cell Death in Breast Cancer Cells. Cancer Sci (2013) 104(11):1492–8. doi: 10.1111/cas.12272
86. Okuhira K, Demizu Y, Hattori T, Ohoka N, Shibata N, Kurihara M, et al. Molecular Design, Synthesis, and Evaluation of SNIPER(ER) That Induces Proteasomal Degradation of ERalpha. Methods Mol Biol (2016) 1366:549–60. doi: 10.1007/978-1-4939-3127-9_42
87. Ohoka N, Okuhira K, Ito M, Nagai K, Shibata N, Hattori T, et al. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-Dependent Protein Erasers (SNIPERs). J Biol Chem (2017) 292(11):4556–70. doi: 10.1074/jbc.M116.768853
88. Li Y, Zhang S, Zhang J, Hu Z, Xiao Y, Huang J, et al. Exploring the PROTAC Degron Candidates: OBHSA With Different Side Chains as Novel Selective Estrogen Receptor Degraders (SERDs). Eur J Med Chem (2019) 172:48–61. doi: 10.1016/j.ejmech.2019.03.058
89. Peng L, Zhang Z, Lei C, Li S, Zhang Z, Ren X, et al. Identification of New Small-Molecule Inducers of Estrogen-Related Receptor Alpha (ERRalpha) Degradation. ACS Med Chem Lett (2019) 10(5):767–72. doi: 10.1021/acsmedchemlett.9b00025
90. Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, et al. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc Natl Acad Sci USA (2016) 113(26):7124–9. doi: 10.1073/pnas.1521738113
91. Zhao Q, Lan T, Su S, Rao Y. Induction of Apoptosis in MDA-MB-231 Breast Cancer Cells by a PARP1-Targeting PROTAC Small Molecule. Chem Commun (Camb) (2019) 55(3):369–72. doi: 10.1039/c8cc07813k
92. Burstein HJ, Temin S, Anderson H, Buchholz TA, Davidson NE, Gelmon KE, et al. Adjuvant Endocrine Therapy for Women With Hormone Receptor-Positive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline Focused Update. J Clin Oncol (2014) 32(21):2255–69. doi: 10.1200/JCO.2013.54.2258
93. Itoh Y, Kitaguchi R, Ishikawa M, Naito M, Hashimoto Y. Design, Synthesis and Biological Evaluation of Nuclear Receptor-Degradation Inducers. Bioorg Med Chem (2011) 19(22):6768–78. doi: 10.1016/j.bmc.2011.09.041
94. Naito M, Ohoka N, Shibata N. SNIPERs—Hijacking IAP Activity to Induce Protein Degradation. Drug Discov Today Technol (2019) 31:35–42. doi: 10.1016/j.ddtec.2018.12.002
95. Fujimoto J, Sato E. Clinical Implication of Estrogen-Related Receptor (ERR) Expression in Uterine Endometrial Cancers. J Steroid Biochem Mol Biol (2009) 116(1-2):71–5. doi: 10.1016/j.jsbmb.2009.04.012
96. Kraus RJ, Ariazi EA, Farrell ML, Mertz JE. Estrogen-Related Receptor Alpha 1 Actively Antagonizes Estrogen Receptor-Regulated Transcription in MCF-7 Mammary Cells. J Biol Chem (2002) 277(27):24826–34. doi: 10.1074/jbc.M202952200
97. Dwyer MA, Joseph JD, Wade HE, Eaton ML, Kunder RS, Kazmin D, et al. WNT11 Expression is Induced by Estrogen-Related Receptor Alpha and Beta-Catenin and Acts in an Autocrine Manner to Increase Cancer Cell Migration. Cancer Res (2010) 70(22):9298–308. doi: 10.1158/0008-5472.Can-10-0226
98. Stein RA, Chang CY, Kazmin DA, Way J, Schroeder T, Wergin M, et al. Estrogen-Related Receptor Alpha is Critical for the Growth of Estrogen Receptor-Negative Breast Cancer. Cancer Res (2008) 68(21):8805–12. doi: 10.1158/0008-5472.Can-08-1594
99. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic Targeting of BET Bromodomain Proteins in Castration-Resistant Prostate Cancer. Nature (2014) 510(7504):278–82. doi: 10.1038/nature13229
100. Lipinski CA. Rule of Five in 2015 and Beyond: Target and Ligand Structural Limitations, Ligand Chemistry Structure and Drug Discovery Project Decisions. Advanced Drug Deliv Rev (2016) 101:34–41. doi: 10.1016/j.addr.2016.04.029
101. Lebraud H, Wright DJ, Johnson CN, Heightman TDJ. Protein Degradation by in-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent Sci (2016) 2(12):927–34. doi: 10.1021/acscentsci.6b00280
102. Moreau K, Coen M, Zhang AX, Pachl F, Castaldi MP, Dahl G, et al. Proteolysis-Targeting Chimeras in Drug Development: A Safety Perspective. Br J Pharmacol (2020) 177(8):1709–18. doi: 10.1111/bph.15014
103. Roy RD, Rosenmund C, Stefan MIJ. Cooperative Binding Mitigates the High-Dose Hook Effect. BMC Syst Biol (2017) 11(1):1–10. doi: 10.1186/s12918-017-0447-8
104. Prekovic S, van Royen ME, Voet AR, Geverts B, Houtman R, Melchers D, et al. The Effect of F877L and T878A Mutations on Androgen Receptor Response to Enzalutamide. Mol Cancer Ther (2016) 15(7):1702–12. doi: 10.1158/1535-7163.MCT-15-0892
105. Paiva SL, Crews CM. Targeted Protein Degradation: Elements of PROTAC Design. Curr Opin Chem Biol (2019) 50:111–9. doi: 10.1016/j.cbpa.2019.02.022
106. Moser SC, Voerman JSA, Buckley DL, Winter GE, Schliehe C. Acute Pharmacologic Degradation of a Stable Antigen Enhances Its Direct Presentation on MHC Class I Molecules. Front Immunol (2017) 8:1920. doi: 10.3389/fimmu.2017.01920
Keywords: sex hormone-dependent cancers, PROTACs, small molecule inhibitors, estrogen receptors, androgen receptors
Citation: Liu L, Shi L, Wang Z, Zeng J, Wang Y, Xiao H and Zhu Y (2022) Targeting Oncoproteins for Degradation by Small Molecule-Based Proteolysis-Targeting Chimeras (PROTACs) in Sex Hormone-Dependent Cancers. Front. Endocrinol. 13:839857. doi: 10.3389/fendo.2022.839857
Received: 20 December 2021; Accepted: 22 February 2022;
Published: 18 March 2022.
Edited by:
Claire Perks, University of Bristol, United KingdomReviewed by:
Fraydoon Rastinejad, University of Oxford, United KingdomCopyright © 2022 Liu, Shi, Wang, Zeng, Wang, Xiao and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongxia Zhu, emh1eW9uZ3hpYTA0MDlAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.