 Väinö Lithovius
Väinö Lithovius Timo Otonkoski
Timo Otonkoski- 1Stem Cells and Metabolism Research Program, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 2Children’s Hospital, Helsinki University Hospital, Helsinki, Finland
Congenital hyperinsulinism (CHI) is a severe inherited neonatal disorder characterized by inappropriate insulin secretion caused by genetic defects of the pancreatic beta cells. Several open questions remain in CHI research, such as the optimal treatment for the most common type of CHI, caused by mutations in the genes encoding ATP-sensitive potassium channels, and the molecular mechanisms of newly identified CHI genes. Answering these questions requires robust preclinical models, particularly since primary patient material is extremely scarce and accurate animal models are not available. In this short review, we explain why pluripotent stem cell derived islets present an attractive solution to these issues and outline the current progress in stem-cell based modeling of CHI. Stem cell derived islets enable the study of molecular mechanisms of CHI and the discovery of novel antihypoglycemic drugs, while also providing a valuable model to study the biology of variable functional states of beta cells.
Introduction
Congenital hyperinsulinism (CHI), characterized by inappropriate insulin secretion from the pancreatic beta cells, is the most common cause of persistent childhood hypoglycemia. At least 15 causative genes have been identified (1), with 30-55% of patients remaining without a genetic diagnosis (2–5). Over 50% of all CHI patients (2, 5) carry a recessive loss-of-function mutation in the KATP-channel genes ABCC8 or KCNJ11 (KATPHI), which leads to abnormal membrane depolarization and constitutive insulin secretion. Clinically, this leads to severe hypoglycemia for which there is no optimal treatment. This represents an ongoing clinical challenge as the hypoglycemia is life-threatening in the first days of life and despite best contemporary treatment, many continue to suffer from learning difficulties in the long term (6–10). A robust preclinical model would be required for studies aiming to discover improved treatment options for KATPHI and to pinpoint molecular mechanisms of rare newly identified forms of CHI.
Pluripotent stem cells (PSCs) represent the epiblast cells of the early embryo, capable of differentiation to any cell type in the human body. Tissue differentiated from PSCs holds enormous promise in regenerative medicine to replace or repair a damaged or degenerated organ. PSCs can also serve as a powerful research tool by allowing limitless generation of difficult-to-procure tissue and would thus serve as an attractive solution for preclinical study of CHI. Pluripotent stem cell derived islets (SC-islets) could replace or complement rodent models and primary patient islet tissue, which both have important inherent weaknesses. Rodent islets are structurally and physiologically different from human islets (11–14) and these differences have manifested in KATP-channel knockout mouse models, which have presented with a much milder phenotype than the KATPHI patients (15–17). The availability of healthy primary islets is limited and faces issues of variable in vitro function (www.epicore.ualberta.ca/isletcore/). The availability of CHI patient islets presents an additional challenge due to the rarity of the disease. The limited tissue availability challenges any study that requires large amounts of tissue, such as screening for novel pharmacotherapeutics.
This review focuses on the use of PSC-derived pancreatic islets (SC-islets) for preclinical study of congenital hyperinsulinism. We outline the practical necessities in setting up SC-islet models and aim to identify relevant questions for CHI research where the SC-islets are particularly powerful.
SC-Islet Based Disease Modeling: Key Technologies and Current Progress
Genome Editing of Pluripotent Stem Cells
Pluripotent stem cells (PSCs) are derived from two main sources: preimplantation embryos (embryonic stem cells, ESCs) (18) and somatic cells that have been reprogrammed back to pluripotent state by overexpression of key genes (induced pluripotent stem cells, iPSCs) (19). iPSCs reprogrammed from a patient sample carry the disease-causing mutations of that individual and should thus phenocopy the disease, such as CHI, when differentiated. A similarly differentiated healthy iPSC line would serve as a non-isogenic control for this type of approach. Theoretically, this offers a disease model without the need for genome editing. In practice however, the differences in the donor genetic background exert a high degree of influence on the differentiation efficiency of stem cell lines, at least in islet differentiation, making it difficult to conclude whether the detected differences between patient and healthy cell lines are due to the disease gene or a differentiation-related artefact. Thus, it is often more practical to correct the disease-causing mutation with genome editing tools to yield an isogenic control cell line. Isogenic controls offer a clean look into the disease phenotype without differences in the genetic background. Generating isogenic controls with genome editing of the PSCs can be considered as the ideal approach for the effective modeling of a genetic disease such as CHI.
Another approach to create isogenic cell lines is to engineer the disease-causing mutation in a healthy hPSC line. This approach is often more straightforward than the patient cell line approach because the differentiation protocol used to derive the SC-islets can be optimized for one cell line and all the interesting mutations can be engineered to it. Furthermore, from the genome editing point of view, it is easier to generate a knock-out than a knock-in. In correcting a patient line, a knock-in is always required, but for generating a disease cell line a simple knock-out is often enough since many diseases are caused by loss-of-function mutations.
Multiple technologies are available for the genome editing itself, but recently CRISPR-based technologies have started to dominate, due to their relative ease and high efficiency. The basic CRISPR system consists of a guide RNA, able to target the editing to a specific locus in the genome; a protein exhibiting nuclease activity such as Cas9 or Cas12a creating a double stand break; and an RNA template containing the mutation-corrected sequence or a sequence knocking out the healthy gene which can be read during homology directed repair of the double strand break. This basic system has been expanded and optimized further in many ways, as reviewed here (20). Regardless of the specifics, genome editing technology has matured to a state where generating disease relevant stem cell lines for further differentiation is practical.
Differentiation of PSCs to Islets
Due to the curative potential of PSCs in cell replacement therapy for insulin deficient diabetes, enormous effort has been expended in developing protocols that can drive PSCs to differentiate in vitro to pancreatic islets (SC-islets). Starting with the breakthrough protocol published in 2006, showing for the first time that insulin positive cells can be differentiated from hPSCs through steps mimicking normal development (21), the progress in the field has been rapid. First evidence of glucose-regulated insulin release was provided in 2014 (22, 23), Since then, many further improvements have been made (24–26) and in the past two years the first protocols giving robust, dynamic glucose stimulated insulin secretion have been reported, achieving beta cell maturity at least in terms of insulin secretory function (27–35). This recent progress in the field has identified multiple conditions related to optimal late-stage maturation. These include keeping SC-islets appropriately sized by resizing or by culture format, keeping the proliferation rate low, normoglycemic culture conditions, lack of ALK5-inhibition, addition of WNT4, circadian entrainment, and reducing the number of unwanted cells that might compromise function on the islet level by sorting or by addition of aurora kinase inhibitor.
Given the recapitulation of the adult function in the state-of-the-art protocols and the fact that the protocols used to derive the mature SC-islet use the same signaling cues as the fetal islets during their in vivo development, SC-islets represent an excellent avenue for modeling CHI pathophysiology, including both developmental and insulin secretory defects. We have shown that all the main components of the stimulus-secretion coupling machinery of beta cells: metabolic processing of glucose, currents of the critical ion-channels, the insulin-secretion modulating amplifying mechanisms and the exocytosis machinery, are present in SC-islets (30). As most forms of CHI manifest in the neonatal period, achieving adult-like function might not even be necessary for some study questions. This is exemplified by existing SC-islet models for CHI, which have been successful even using less efficient differentiation protocols, as described in the following section.
Stem Cell-Based Models for CHI
Thus far, two studies have taken advantage of the SC-islet differentiation and genome editing technologies in modeling KATP-channel related CHI (KATPHI) (36, 37), as summarized in Table 1. Guo and colleagues (36), used healthy hESCs and introduced a knockout of the ABCC8 gene, encoding the KATP-channel subunit SUR1. The ABCC8 KO beta cells secreted around 2-fold more insulin in vitro and failed to respond to KATP-channel acting pharmaceuticals. Their beta-like cells could be inhibited with octreotide, and to a lesser degree with nicorandil and nifedipine. Thus, they replicated the KATPHI insulin secretion phenotype in vitro.

Table 1 Summary of published studies on stem cell based modeling of congenital hyperinsulinism.
We used iPSCs derived from a patient carrying the homozygous V187D-mutation (38) in the ABCC8 gene and compared them to mutation-corrected controls (37). The ABCC8 mutant beta-like cells secreted around 3-fold more insulin in low glucose compared to the corrected counterparts. They also failed to respond to KATP-channel acting pharmaceuticals but could be inhibited with clonidine and EGTA. Upon transplantation and in vivo maturation under the kidney capsule of immunocompromised mice, the mutant grafts secreted 7-fold higher levels of human C-peptide and caused 38% lower blood glucose upon fasting than the control grafts. We could thus replicate the cardinal phenotypic features of KATPHI both in vitro and in vivo. In addition to these features of the secretory function, we found that the KATP-inactivation directed the development of endocrine cells towards beta cells at the expense of alpha cells in the in vitro differentiation. This may have been due to the increased proliferation we detected in the ABCC8 mutant beta cells. The role of KATP-channel inactivation in proliferation was also found in a previous study using pancreas-derived mesenchymal stem cells derived from a KATPHI patient (39).
Potential Research Avenues for SC-Islet CHI Models
Development of Pharmacotherapy for Diazoxide-Resistant CHI
The first-line antihypoglycemic drug used in CHI, diazoxide, is an opener of the KATP-channel and as such, most of the recessive KATPHI patients are unresponsive to it (40, 41). Second-line treatments, such as somatostatin receptor agonists are widely used, but still the most severe patients must undergo pancreatectomy to control hypoglycemia. This is a suboptimal treatment which rarely results in euglycemia (42–44). There is an obvious need for more effective antihypoglycemic medication for diazoxide-resistant CHI.
SC-islets can be generated in limitless quantities and when derived with state-of-the-art differentiation protocols (27, 29, 30), they harbor the stimulus-secretion coupling machinery of adult primary islets: the KATP-channel related triggering pathway, as well as the neurohormonal and metabolic amplifying pathways (30). This covers the key pathways that modulate insulin secretion and could thus serve as pharmacological targets.
Potential antihypoglycemic targets on the beta cell include ion-channels, G-protein coupled receptors and transcription factors controlling beta cell function, as summarized in Figure 1. Depending on the genetic cause of CHI, some of these might be more advantageous than others. In the most common type of diazoxide resistant CHI, KATPHI, insulin hypersecretion occurs because the KATP-channel is inactive leading to constitutive depolarization and constantly elevated intracellular calcium (45). In this intracellular environment, a molecule acting upstream of the KATP-channel in the stimulus-secretion coupling machinery (ie. glucose uptake, glycolysis and the TCA cycle) is likely to be ineffective. Potential antihypoglycemic ion-channel acting drugs would act by decreasing the membrane potential independently of the KATP-channel (ie. by acting on K+, Na+ and Cl- channels) or by reducing intracellular Ca2+, the final trigger of exocytosis. Nifedipine and other Ca2+ channel blockers have been used in the treatment of KATPHI but have proven ineffective (46). Sikimic and colleagues identified DCEBIO, acting as an agonist of the repolarizing KCa3.1 channel, as a molecule abolishing the glucose induced Ca2+ oscillations in two KATPHI islet preparations (47). A potential downside with ion-channel acting molecules is that many of the target channels are also expressed in non-islet cells, such as cardiomyocytes and neurons, increasing the likelihood of serious side effects.
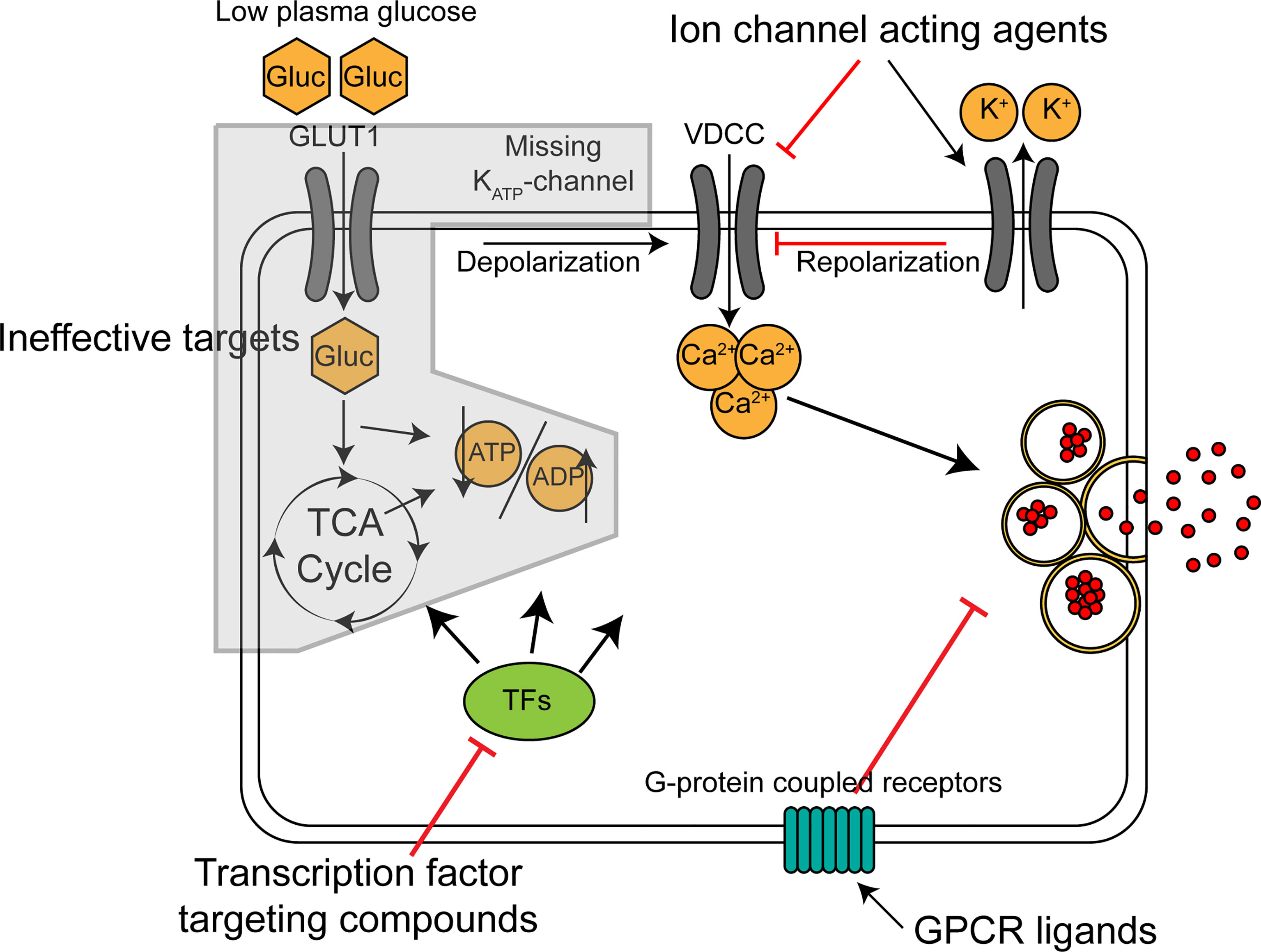
Figure 1 Mechanism of action for classes of potential antihypoglycemic agents for treatment of KATP-channel related CHI. Broad, schematic depiction of insulin secretion machinery in KATP-channel deficient beta cells in low glucose. GLUT1, glucose transporter; Gluc, glucose; VDCC, voltage-dependent calcium channel; TCA cycle, tricarboxylic acid cycle; TFs, transcription factors.
Several islet specific G-protein coupled receptors (GPCRs) have been identified (48). The role of these receptors in beta cells is to fine-tune the glucose-stimulated insulin secretion by affecting the amplifying pathways. They exert their action through cAMP and other second messengers, which sensitize or desensitize insulin granule exocytosis. Human islets express around 300 GPCRs (49). Highly expressed ones include receptors for the other islet hormones, glucagon and somatostatin, and the gut-derived incretins, GLP1 and GIP, but many beta cell GPCRs also have no known ligand (orphan receptors). This offers the potential for novel antihypoglycemic compounds to be found by screening. Many of the most potent GPCR-coupled amplifying pathways, such as the GLP1-receptor coupled pathway, are primarily stimulated only when the beta cells are already triggered by the KATP-channel closure and subsequent influx of calcium, restricting their activity mostly to high glucose conditions (50). The KATPHI cells are constantly in the state of high [Ca2+]i, which should allow the inappropriate activation of these pathways even in low glucose. Targeting GPCRs in KATPHI has been shown to be effective by the well-established use of somatostatin receptor agonists such as octreotide. More recently, studies by De León and associates have identified the GLP1-receptor inverse agonist exendin-(9-39) as an effective antihypoglycemic agent in KATP-KO mice (51) and in adult KATPHI patients (52).
Transcription factors controlling beta cell insulin secretion could serve as a third group of targets for antihypoglycemic medication. Senniappan and colleagues demonstrated the validity of this approach using mTOR inhibitor rapamycin on diazoxide-resistant CHI patients (53). Since the initial report however, the use of rapamycin has been questioned due to low efficacy and high incidence of serious side effects (54, 55). These issues are likely due to mTOR being an important regulator of wide variety of cellular processes in most tissues, and as such, an agent acting on a transcription factor more specifically controlling beta cell insulin secretion related gene expression would be more ideal. Despite the existence of several beta cell-specific transcription factors that control the expression of insulin secretory machinery genes (e.g., MAFA or RFX6), small molecules specifically targeting them are missing, and thus the ion-channel and GPCR-related strategies are likely to provide more accessible targets.
The most clinically relevant parameter for identifying the potential drug would be its effectiveness in reducing the CHI SC-islets’ insulin secretion in low glucose. Measuring just intracellular calcium fluxes with a dye or a genetically encoded sensor could lead to failure in identifying a potential drug if the drug acts on the amplifying mechanisms of insulin secretion, whose activity does not result in further calcium fluxes. Any candidate identified in the in vitro screens should be validated in vivo. The first line strategy for this is to use CHI SC-islet grafts, which lead to hypoglycemia in the recipient mice (37). Conceivably measurements of mouse blood glucose and C-peptide secreted by the graft in drug-treated and non-treated mice engrafted with CHI SC-islets should reveal the effectiveness of a candidate molecule in vivo.
Discovery of Novel CHI Pathomechanisms
Around 50% of new CHI patients do not present a mutation in the genes previously identified as causative for CHI (2). Unraveling of the exact pathogenic mechanism should direct selection of the most appropriate treatment for each patient. Pinpointing the molecular mechanism could also aid discovery of antihypoglycemic pharmaceuticals and shed light to the role of the identified genes in the regulation of beta cell insulin secretion. Answering these questions requires a model system with sufficient fidelity to capture disease pathophysiology on many levels. SC-islets have been used to discover the molecular mechanism of multiple genes causing different types of monogenic diabetes, ranging from mechanisms related to beta cell development (56–58) to function (59, 60) and degeneration (61–63), as reviewed recently (64). In the case of novel CHI genes, several parameters can be used to unravel disease mechanisms. These include, at least, development of the endocrine populations during differentiation; insulin secretion under different stimuli in vitro; transcriptomic, epigenomic proteomic, and dynamic metabolomic studies. The stem cell based approach can provide even the large amounts of tissue required for these analyses.
The Use of CHI SC-Islets as a Model for Chronic Beta-Cell Hyperfunctionality and Glucotoxicity
The beta cell is highly specialized, focusing on the production and secretion of insulin. It is understandable that CHI mutations, which by definition accelerate this process, have profound consequences for the cellular biology of the beta cells. Huopio and colleagues established that, in addition to causing hypoglycemia in infancy (65), dominant KATPHI mutations predispose to T2DM later in life (66), providing clinical evidence for the detrimental consequences of long-term beta cell hyperactivity. Similar T2DM predisposing effect has been discovered in carriers of activating glucokinase mutations (GCK-HI) (67, 68).
Li and colleagues found that diverse cellular functions are disturbed or altered in CHI patient beta cells lacking KATP-channels, including glucose dependent metabolic pathways, expression of key transcription factors and receptors and the regulation of cell cycle (69). Additionally, the chronically elevated [Ca2+]i characteristic of KATPHI and GCK-HI compromises beta cell identity in (70) and causes double strand breaks and p53 activation (71). Related to these findings, the increased workload of CHI beta cells initially increases their proliferation and mass while later leading to beta cell dysfunction and apoptosis via glucotoxicity (71–74), thus paralleling the natural course of beta cells in type 2 diabetes. These examples highlight the possibility of using CHI SC-islets as a model to discover further consequences of the influence of chronically altered beta cell functional states on their biology. Indeed, we could capture the initially increased beta cell mass and proliferation in vitro, while the proliferation normalized in vivo (37). Again, the possibility for isogenic comparison between healthy and CHI SC-islets provides a specific means to link any alterations to specific mutations.
Conclusions
Stem cell derived islets represent a powerful tool for modeling diseases of the pancreatic beta cell, due to the potential to produce them in limitless quantities with high consistency and with high disease phenotype fidelity. In the case of CHI, the SC-islets can be harnessed to discover novel antihypoglycemic medications, to study molecular mechanisms of newly discovered CHI genes and to study the basic biology of a hyperactive beta cell. Thus, we believe that modeling of CHI with SC-islets will serve as a critical next step required for the development of specific and efficient antihypoglycemic drugs.
Author Contributions
VL wrote the initial draft and edited the manuscript. TO edited the manuscript and provided resources. All authors contributed to the article and approved the submitted version.
Funding
The studies in the Otonkoski laboratory were supported by the Academy of Finland (Center of Excellence MetaStem, grant no. 312437), the Novo Nordisk Foundation and the Sigrid Jusélius Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
VL acknowledges personal grant support by the Finnish Medical Foundation, Eemil Aaltosen Säätiö and the Orion research foundation.
References
1. Gϋemes M, Rahman SA, Kapoor RR, Flanagan S, Houghton JAL, Misra S, et al. Hyperinsulinemic Hypoglycemia in Children and Adolescents: Recent Advances in Understanding of Pathophysiology and Management. Rev Endocr Metab Disord (2020) 21(4):577–97. doi: 10.1007/s11154-020-09548-7
2. Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and Molecular Characterisation of 300 Patients With Congenital Hyperinsulinism. Eur J Endocrinol (2013) 168(4):557–645. doi: 10.1530/EJE-12-0673
3. Männistö JME, Maria M, Raivo J, Kuulasmaa T, Otonkoski T, Huopio H, et al. Clinical and Genetic Characterization of 153 Patients With Persistent or Transient Congenital Hyperinsulinism. J Clin Endocrinol Metab (2020) 105(4):E1686–945. doi: 10.1210/clinem/dgz271
4. Rosenfeld E, Ganguly A, de Leon DD. Congenital Hyperinsulinism Disorders: Genetic and Clinical Characteristics. Am J Med Genet Part C: Semin Med Genet (2019) 181(4):682–925. doi: 10.1002/ajmg.c.31737
5. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and Phenotype Correlations in 417 Children With Congenital Hyperinsulinism. J Clin Endocrinol Metab (2013) 98(2):355–63. doi: 10.1210/jc.2012-2169
6. Muukkonen L, Männistö J, Jääskeläinen J, Hannonen R, Huopio H. The Effect of Hypoglycaemia on Neurocognitive Outcome in Children and Adolescents With Transient or Persistent Congenital Hyperinsulinism. Dev Med Child Neurol (2019) 61(4):451–575. doi: 10.1111/dmcn.14039
7. Roeper M, Dafsari RS, Hoermann H, Mayatepek E, Kummer S, Meissner T. Risk Factors for Adverse Neurodevelopment in Transient or Persistent Congenital Hyperinsulinism. Front Endocrinol (2020) 11:580642. doi: 10.3389/fendo.2020.580642
8. Shah R, Harding J, Brown J, Mckinlay C. Neonatal Glycaemia and Neurodevelopmental Outcomes: A Systematic Review and Meta-Analysis. Neonatology (2019) 115(2):116–265. doi: 10.1159/000492859
9. Lord K, Radcliffe J, Gallagher PR, Adzick NS, Stanley CA, de León DD. High Risk of Diabetes and Neurobehavioral Deficits in Individuals With Surgically Treated Hyperinsulinism. J Clin Endocrinol Metab (2015) 100(11):4133–395. doi: 10.1210/jc.2015-2539
10. Helleskov A, Melikyan M, Globa E, Shcherderkina I, Poertner F, Larsen AM, et al. Both Low Blood Glucose and Insufficient Treatment Confer Risk of Neurodevelopmental Impairment in Congenital Hyperinsulinism: A Multinational Cohort Study. Front Endocrinol (2017) 8:156. doi: 10.3389/fendo.2017.00156
11. Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren P-O, Caicedo A. The Unique Cytoarchitecture of Human Pancreatic Islets Has Implications for Islet Cell Function. Proc Natl Acad Sci USA (2006) 103(7):2334–95. doi: 10.1073/pnas.0510790103
12. Nair G, Hebrok M. Islet Formation in Mice and Men: Lessons for the Generation of Functional Insulin-Producing β-Cells From Human Pluripotent Stem Cells. Curr Opin Genet Dev (2015) 32:171–80. doi: 10.1016/j.gde.2015.03.004
13. Balboa D, Otonkoski T. Human Pluripotent Stem Cell Based Islet Models for Diabetes Research. Best Pract Res: Clin Endocrinol Metab (2015) 29(6):899–9095. doi: 10.1016/j.beem.2015.10.012
14. Rodriguez-Diaz R, Molano RD, Weitz JR, Abdulreda MH, Berman DM, Leibiger B, et al. Paracrine Interactions Within the Pancreatic Islet Determine the Glycemic Set Point. Cell Metab (2018) 27(3):549–58.e4. doi: 10.1016/j.cmet.2018.01.015
15. Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 Knockout Mice. A Model for K(ATP) Channel-Independent Regulation of Insulin Secretion. J Biol Chem (2000) 275(13):9270–7. doi: 10.1074/jbc.275.13.9270
16. Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, et al. Defective Insulin Secretion and Enhanced Insulin Action in K ATP Channel-Deficient Mice. Biochemistry (1998) 95:10402–6. doi: 10.1073/pnas.95.18.10402
17. Shiota C, Larsson O, Shelton K, Shiota M, Efanov A, Hoy M, et al. Sulfonylurea Receptor Type 1 Knock-Out Mice Have Intact Feeding-Stimulated Insulin Secretion Despite Marked Impairment in Their Response to Glucose. J Biol Chem (2002) 277:37176–83. doi: 10.1074/jbc.M206757200
18. Thomson JA. Embryonic Stem Cell Lines Derived From Human Blastocysts. Science (1998) 282(5391):1145–7. doi: 10.1126/science.282.5391.1145
19. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of Pluripotent Stem Cells From Adult Human Fibroblasts by Defined Factors. Cell (2007) 131(5):861–72. doi: 10.1016/j.cell.2007.11.019
20. Anzalone AV, Koblan LW, Liu DR. Genome Editing With CRISPR–Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat Biotechnol (2020) 38(7):824–44. doi: 10.1038/s41587-020-0561-9
21. D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, et al. Production of Pancreatic Hormone-Expressing Endocrine Cells From Human Embryonic Stem Cells. Nat Biotechnol (2006) 24(11):1392–401. doi: 10.1038/nbt1259
22. Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of Diabetes With Insulin-Producing Cells Derived in Vitro From Human Pluripotent Stem Cells. Nat Biotechnol (2014) 32(11):1121–33. doi: 10.1038/nbt.3033
23. Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of Functional Human Pancreatic β Cells. In Vitro Cell (2014) 159(2):428–39. doi: 10.1016/j.cell.2014.09.040
24. Nostro MC, Sarangi F, Yang C, Holland A, Elefanty AG, Stanley EG, et al. Efficient Generation of NKX6-1+ Pancreatic Progenitors From Multiple Human Pluripotent Stem Cell Lines. Stem Cell Rep (2015) 4(4):591–6045. doi: 10.1016/j.stemcr.2015.02.017
25. Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, et al. Controlled Induction of Human Pancreatic Progenitors Produces Functional Beta-Like Cells In Vitro. EMBO J (2015) 34(13):e201591058. doi: 10.15252/embj.201591058
26. Toyoda T, Kimura A, Tanaka H, Ameku T, Mima A, Hirose Y, et al. Rho-Associated Kinases and Non-Muscle Myosin IIs Inhibit the Differentiation of Human IPSCs to Pancreatic Endoderm. Stem Cell Rep (2017) 9(2):419–28. doi: 10.1016/j.stemcr.2017.07.005
27. Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, et al. Acquisition of Dynamic Function in Human Stem Cell-Derived Beta Cells. Stem Cell Rep (2019) 12(2):351–65. doi: 10.1016/j.stemcr.2018.12.012
28. Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L, Millman JR. Targeting the Cytoskeleton to Direct Pancreatic Differentiation of Human Pluripotent Stem Cells. Nat Biotechnol (2020) 38(4):460–70. doi: 10.1038/s41587-020-0430-6
29. Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, et al. Recapitulating Endocrine Cell Clustering in Culture Promotes Maturation of Human Stem-Cell-Derived β Cells. Nat Cell Biol (2019) 21(2):263–74. doi: 10.1038/s41556-018-0271-4
30. Balboa D, Barsby T, Lithovius V, Saarimäki-vire J, Omar-hmeadi M, Dyachok O, et al. Functional, Metabolic and Transcriptional Maturation of Human Pancreatic Islets Derived From Stem Cell. BioRxiv (2021) 447439:1–20. doi: 10.1038/s41587-022-01219-z
31. Yoshihara E, Connor CO, Gasser E, Wei Z, Oh TG, Tseng TW, et al. Immune-Evasive Human Islet-Like Organoids Ameliorate Diabetes. Nature (2020) 586:606–11. doi: 10.1038/s41586-020-2631-z
32. Helman A, Cangelosi AL, Davis JC, Straubhaar JR, Sabatini DM, Melton DA, et al. Article A Nutrient-Sensing Transition at Birth Triggers Glucose-Responsive Insulin Secretion Ll Article A Nutrient-Sensing Transition at Birth Triggers Glucose-Responsive Insulin Secretion. Cell Metab (2020) 31(5):1004–16.e5. doi: 10.1016/j.cmet.2020.04.004
33. Davis JC, Alves TC, Helman A, Liu DR, Kibbey RG, Melton DA, et al. Article Glucose Response by Stem Cell-Derived B Cells In Vitro Is Inhibited by a Bottleneck in Glycolysis Ll Ll Glucose Response by Stem Cell-Derived B Cells In Vitro Is Inhibited by a Bottleneck in Glycolysis. CellReports (2020) 31(6):107623. doi: 10.1016/j.celrep.2020.107623
34. Alvarez-Dominguez JR, Donaghey J, Rasouli N, Kenty JHR, Helman A, Charlton J, et al. Circadian Entrainment Triggers Maturation of Human. In Vitro Islets Cell Stem Cell (2020) 26(1):108–22.e10. doi: 10.1016/j.stem.2019.11.011
35. Mahaddalkar PU, Scheibner K, Pfluger S, Ansarullah MS, Beckenbauer J, Irmler M, et al. Generation of Pancreatic β Cells From CD177+ Anterior Definitive Endoderm. Nat Biotechnol (2020) 38(9):1061–72. doi: 10.1038/s41587-020-0492-5
36. Guo D, Liu H, Ru A, Gao GE, Nasir A. Modeling Congenital Hyperinsulinism With ABCC8 - Deficient Human Embryonic Stem Cells Generated by CRISPR / Cas9. Sci Rep (2017) 7(3156):1–8. doi: 10.1038/s41598-017-03349-w
37. Lithovius V, Saarimäki-Vire J, Balboa D, Ibrahim H, Montaser H, Barsby T, et al. SUR1-Mutant iPS Cell-Derived Islets Recapitulate the Pathophysiology of Congenital Hyperinsulinism. Diabetologia (2021) 64(3):630–40. doi: 10.1007/s00125-020-05346-7
38. Otonkoski T, Ämmälä C, Huopio H, Cote GJ, Chapman J, Cosgrove K, et al. A Point Mutation Inactivating the Sulfonylurea Receptor Causes the Severe Form of Persistent Hyperinsulinemic Hypoglycemia of Infancy in Finland. Diabetes (1999) 48:408–15. doi: 10.2337/diabetes.48.2.408
39. Kellaway SG, Mosinska K, Mohamed Z, Ryan A, Richardson S, Newbould M, et al. Increased Proliferation and Altered Cell Cycle Regulation in Pancreatic Stem Cells Derived From Patients With Congenital Hyperinsulinism. (2019) PLoS ONE 14(9):e0222350. doi: 10.1371/journal.pone.0222350
40. Banerjee I, Salomon-Estebanez M, Shah P, Nicholson J, Cosgrove KE, Dunne MJ. Therapies and Outcomes of Congenital Hyperinsulinism-Induced Hypoglycaemia. Diabetic Med (2019) 36(1):9–21. doi: 10.1111/dme.13823
41. Henquin JC, Nenquin M, Sempoux C, Guiot Y, Bellanné-Chantelot C, Otonkoski T, et al. In Vitro Insulin Secretion by Pancreatic Tissue From Infants With Diazoxide-Resistant Congenital Hyperinsulinism Deviates From Model Predictions. J Clin Invest (2011) 121(10):3932–42. doi: 10.1172/JCI58400
42. Adzick NS, de Leon DD, States LJ, Lord K, Bhatti TR, Becker SA, et al. Surgical Treatment of Congenital Hyperinsulinism: Results From 500 Pancreatectomies in Neonates and Children. J Pediatr Surg (2019) 54(1):27–32. doi: 10.1016/j.jpedsurg.2018.10.030
43. Beltrand J, Caquard M, Arnoux J-B, Laborde K, Velho G, Verkarre V, et al. Glucose Metabolism in 105 Children and Adolescents After Pancreatectomy for Congenital Hyperinsulinism. Diabetes Care (2012) 35(2):198–203. doi: 10.2337/dc11-1296
44. Arya VB, Senniappan S, Demirbilek H, Alam S, Flanagan SE, Ellard S, et al. Pancreatic Endocrine and Exocrine Function in Children Following Near-Total Pancreatectomy for Diffuse Congenital Hyperinsulinism. PloS One (2014) 9(5):4–9. doi: 10.1371/journal.pone.0098054
45. Thomas PM, Cote GJ, Wohilk N, Haddad B, Mathew PM, Rabl W, et al. Mutations in the Sulfonylurea Receptor Gene in Familial Persistent Hyperinsulinemic Hypoglycemia of Infancy. Science (1995) 268:426–29. doi: 10.1126/science.7716548
46. Güemes M, Shah P, Silvera S, Morgan K, Gilbert C, Hinchey L, et al. Assessment of Nifedipine Therapy in Hyperinsulinemic Hypoglycemia Due to Mutations in the Abcc8 Gene. J Clin Endocrinol Metab (2017) 102(3):822–30. doi: 10.1210/jc.2016-2916
47. Sikimic J, Hoffmeister T, Gresch A, Kaiser J, Barthlen W, Wolke C, et al. Possible New Strategies for the Treatment of Congenital Hyperinsulinism. Front Endocrinol (2020) 11:545638. doi: 10.3389/fendo.2020.545638
48. Regard JB, Sato IT, Coughlin SR. Anatomical Profiling of G Protein-Coupled Receptor Expression. Cell (2008) 135(3):561–71. doi: 10.1016/j.cell.2008.08.040
49. Amisten S, Salehi A, Rorsman P, Jones PM, Persaud SJ. An Atlas and Functional Analysis of G-Protein Coupled Receptors in Human Islets of Langerhans. Pharmacol Ther (2013) 139(3):359–91. doi: 10.1016/j.pharmthera.2013.05.004
50. Gromada J, Bokvist K, Ding WG, Holst JJ, Nielsen JH, Rorsman P. Glucagon-Like Peptide 1(7-36) Amide Stimulates Exocytosis in Human Pancreatic β-Cells by Both Proximal and Distal Regulatory Steps in Stimulus- Secretion Coupling. Diabetes (1998) 47(1):57–65. doi: 10.2337/diab.47.1.57
51. de León DD, Li C, Delson MI, Matschinsky FM, Stanley CA, Stoffers DA. Exendin-(9-39) Corrects Fasting Hypoglycemia in SUR-1-/- Mice by Lowering Camp in Pancreatic β-Cells and Inhibiting Insulin Secretion. J Biol Chem (2008) 283(38):25786–935. doi: 10.1074/jbc.M804372200
52. Calabria AC, Li C, Gallagher PR, Stanley CA, de León DD. GLP-1 Receptor Antagonist Exendin-(9-39) Elevates Fasting Blood Glucose Levels in Congenital Hyperinsulinism Owing to Inactivating Mutations in the ATP-Sensitive K+ Channel. Diabetes (2012) 61(10):2585–91. doi: 10.2337/db12-0166
53. Senniappan S, Alexandrescu S, Tatevian N, Shah P, Arya V, Flanagan S, et al. Sirolimus Therapy in Infants With Severe Hyperinsulinemic Hypoglycemia. N Engl J Med (2014) 370(12):1131–37. doi: 10.1056/NEJMoa1310967
54. Szymanowski M, Estebanez MS, Padidela R, Han B, Mosinska K, Stevens A, et al. MTOR Inhibitors for the Treatment of Severe Congenital Hyperinsulinism: Perspectives on Limited Therapeutic Success. J Clin Endocrinol Metab (2016) 101(12):4719–29. doi: 10.1210/jc.2016-2711
55. Güemes M, Dastamani A, Ashworth M, Morgan K, Ellard S, Flanagan SE, et al. Sirolimus: Efficacy and Complications in Children With Hyperinsulinemic Hypoglycemia: A 5-Year Follow-Up Study. J Endocr Soc (2019) 3(4):699–713. doi: 10.1210/js.2018-00417
56. Saarimäki-Vire J, Balboa D, Russell MA, Saarikettu J, Kinnunen M, Keskitalo S, et al. An Activating STAT3 Mutation Causes Neonatal Diabetes Through Premature Induction of Pancreatic Differentiation. Cell Rep (2017) 19(2):281–94. doi: 10.1016/j.celrep.2017.03.055
57. Teo AKK, Lau HH, Valdez IA, Dirice E, Tjora E, Raeder H, et al. Early Developmental Perturbations in a Human Stem Cell Model of MODY5/HNF1B Pancreatic Hypoplasia. Stem Cell Rep (2016) 6(3):357–67. doi: 10.1016/j.stemcr.2016.01.007
58. Vethe H, Ghila L, Berle M, Hoareau L, Haaland Ø, Scholz H, et al. The Effect of Wnt Pathway Modulators on Human IPSC-Derived Pancreatic Beta Cell Maturation. Front Endocrinol (2019) 10:293. doi: 10.3389/fendo.2019.00293
59. Balboa D, Saarimäki-Vire J, Borshagovski D, Survila M, Lindholm P, Galli E, et al. Insulin Mutations Impair Beta-Cell Development in a Patient-Derived iPSC Model of Neonatal Diabetes. ELife (2018) 7:e38519. doi: 10.7554/eLife.38519
60. Zeng H, Guo M, Zhou T, Tan L, Chong CN, Zhang T, et al. An Isogenic Human ESC Platform for Functional Evaluation of Genome-Wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell Stem Cell (2016) 19(3):326–40. doi: 10.1016/j.stem.2016.07.002
61. de Franco E, Lytrivi M, Ibrahim H, Montaser H, Wakeling MN, Fantuzzi F, et al. YIPF5 Mutations Cause Neonatal Diabetes and Microcephaly Through Endoplasmic Reticulum Stress. J Clin Invest (2020) 130(12):6338–53. doi: 10.1172/JCI141455
62. Montaser H, Patel KA, Balboa D, Ibrahim H, Lithovius V, Näätänen A, et al. Loss of MANF Causes Childhood-Onset Syndromic Diabetes Due to Increased Endoplasmic Reticulum Stress. Diabetes (2021) 70(4):1006–18. doi: 10.2337/db20-1174
63. Maxwell KG, Augsornworawat P, Velazco-Cruz L, Kim MH, Asada R, Hogrebe NJ, et al. Gene-Edited Human Stem Cell–Derived β Cells From a Patient With Monogenic Diabetes Reverse Preexisting Diabetes in Mice. Sci Trans Med (2020) 12(540):eaax9106. doi: 10.1126/scitranslmed.aax9106
64. Balboa D, Iworima DG, Kieffer TJ. Human Pluripotent Stem Cells to Model Islet Defects in Diabetes. Front Endocrinol (Lausanne) (2021) 12:1–19. doi: 10.3389/fendo.2021.642152
65. Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko H-l, Rahier J, et al. Dominantly Inherited Hyperinsulinism Caused by a Mutation in the Sulfonylurea Receptor Type 1. J Clin Invest (2000) 106: (7):897–906. doi: 10.1172/JCI9804
66. Huopio H, Otonkoski T, Vauhkonen I, Reimann F, Ashcroft FM, Laakso M. A New Subtype of Autosomal Dominant Diabetes Attributable to a Mutation in the Gene for Sulfonylurea Receptor 1. Lancet (2003) 361(9354):301–7. doi: 10.1016/S0140-6736(03)12325-2
67. Langer S, Waterstradt R, Hillebrand G, Santer R, Baltrusch S. The Novel GCK Variant P.Val455Leu Associated With Hyperinsulinism Is Susceptible to Allosteric Activation and Is Conducive to Weight Gain and the Development of Diabetes. Diabetologia (2021) 64(12):2687–700. doi: 10.1007/s00125-021-05553-w
68. Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, et al. Familial Hyperinsulinism Caused by an Activating Glucokinase Mutation. N Engl J Med (1998) 338(4):226–30. doi: 10.1056/NEJM199801223380404
69. Li C, Ackermann AM, Boodhansingh KE, Bhatti TR, Liu C, Schug J, et al. Functional and Metabolomic Consequences of ATP-Dependent Potassium Channel Inactivation in Human Islets. Diabetes (2017) 66:db170029. doi: 10.2337/db17-0029
70. Stancill JS, Cartailler JP, Clayton HW, O’Connor JT, Dickerson MT, Dadi PK, et al. Chronic β-Cell Depolarization Impairs β-Cell Identity by Disrupting a Network of Ca2+-Regulated Genes. Diabetes (2017) 66(8):2175–87. doi: 10.2337/db16-1355
71. Tornovsky-Babeay S, Dadon D, Ziv O, Tzipilevich E, Kadosh T, Haroush RS-B, et al. Type 2 Diabetes and Congenital Hyperinsulinism Cause DNA Double-Strand Breaks and P53 Activity in β Cells. Cell Metab (2014) 19(1):109–21. doi: 10.1016/j.cmet.2013.11.007
72. Tornovsky-Babeay S, Weinberg-Corem N, Schyr RB-H, Avrahami D, Lavi J, Feleke E, et al. Biphasic Dynamics of Beta Cell Mass in a Mouse Model of Congenital Hyperinsulinism: Implications for Type 2 Diabetes. Diabetologia (2021) 64(5):1133–43. doi: 10.1007/s00125-021-05390-x
73. Kassem S, Bhandari S, Rodríguez-Bada P, Motaghedi R, Heyman M, García-Gimeno MA, et al. Large Islets, Beta-Cell Proliferation, and a Glucokinase Mutation. N Engl J Med (2010) 362(14):1348–50. doi: 10.1056/nejmc0909845
Keywords: congenital hyperinsulinism, stem cell derived islets, disease modeling, drug screening and discovery, insulin secretion, hypoglycemia, genetic defects
Citation: Lithovius V and Otonkoski T (2022) Stem Cell Based Models in Congenital Hyperinsulinism – Perspective on Practicalities and Possibilities. Front. Endocrinol. 13:837450. doi: 10.3389/fendo.2022.837450
Received: 16 December 2021; Accepted: 27 January 2022;
Published: 18 February 2022.
Edited by:
Indraneel (Indi) Banerjee, The University of Manchester, United KingdomReviewed by:
Hail Kim, Korea Advanced Institute of Science and Technology, South KoreaCopyright © 2022 Lithovius and Otonkoski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Väinö Lithovius, dmFpbm8ubGl0aG92aXVzQGhlbHNpbmtpLmZp; Timo Otonkoski, dGltby5vdG9ua29za2lAaGVsc2lua2kuZmk=