 Kun Yu1‡
Kun Yu1‡ Zheng-Yuan Huang
Zheng-Yuan Huang Xue-Ling Xu
Xue-Ling Xu Jun Li
Jun Li Shou-Long Deng
Shou-Long Deng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 28 February 2022
Sec. Reproduction
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.827724
This article is part of the Research Topic Molecular and Cytogenetic Research Advances in Human Reproduction View all 10 articles
The physiological role of estrogen in the female endometrium is well established. On the basis of responses to steroid hormones (progesterone, androgen, and estrogen), the endometrium is considered to have proliferative and secretory phases. Estrogen can act in the endometrium by interacting with estrogen receptors (ERs) to induce mucosal proliferation during the proliferative phase and progesterone receptor (PR) synthesis, which prepare the endometrium for the secretory phase. Mouse knockout studies have shown that ER expression, including ERα, ERβ, and G-protein-coupled estrogen receptor (GPER) in the endometrium is critical for normal menstrual cycles and subsequent pregnancy. Incorrect expression of ERs can produce many diseases that can cause endometriosis, endometrial hyperplasia (EH), and endometrial cancer (EC), which affect numerous women of reproductive age. ERα promotes uterine cell proliferation and is strongly associated with an increased risk of EC, while ERβ has the opposite effects on ERα function. GPER is highly expressed in abnormal EH, but its expression in EC patients is paradoxical. Effective treatments for endometrium-related diseases depend on understanding the physiological function of ERs; however, much less is known about the signaling pathways through which ERs functions in the normal endometrium or in endometrial diseases. Given the important roles of ERs in the endometrium, we reviewed the published literature to elaborate the regulatory role of estrogen and its nuclear and membrane-associated receptors in maintaining the function of endometrium and to provide references for protecting female reproduction. Additionally, the role of drugs such as tamoxifen, raloxifene, fulvestrant and G-15 in the endometrium are also described. Future studies should focus on evaluating new therapeutic strategies that precisely target specific ERs and their related growth factor signaling pathways.
Graphical Abstract
The endometrium is the primary target tissue for estrogen. The main function of the endometrium is to prepare for implantation and to maintain the pregnancy after embryo implantation. Estrogen exerts a critical influence on female reproduction via the two main classical estrogen receptors (ERs), ERα and ERβ, and perhaps through G-protein-coupled estrogen receptor (GPER; formerly GPR30) (1). Recent descriptions of the phenotypes of ER-knockout mice have also revealed key roles of estrogen signaling in the endometrium. Estrogen acts through ERs that are associated with the onset and maintenance of disease and tumor events, particularly in uterine tissue. This review focuses on recent advances in our knowledge of the role of ERs in the endometrium, animal models of ER deficiency in the uterus, and how ERs are involved in endometrial diseases and pharmacological treatments.
ERα and ERβ are members of the nuclear receptor (NR) superfamily and consist of five domains (A/B, DNA-binding; D, ligand-binding; and F) (Figure 1). The structurally distinct amino-terminal A/B domains share 17% amino-acid identity between the ERs, acting as ligand-independent activation function 1 (AF-1); AF-1 is involved in both inter-molecular and intra-molecular interactions as well as in activating gene transcription (2). The near-identical central C region (97% amino-acid identity) is the DNA-binding domain that allows ERs to dimerize. Acting as a flexible hinge, the D domain (36% amino-acid identity) contains a nuclear localization signal, is important for receptor dimerization and binding to chaperone heat-shock proteins and links the C domain to the multifunctional carboxyl-terminal (E) domain. The E domain, also known as the ligand-binding domain (LBD), shares 56% amino-acid identity between the ERs and is a globular region that comprises an 17β-estradiol (E2)-binding site, a dimerization interface (homo- and heterodimerization), and ligand-dependent coregulator interaction activity (activation function 2, AF-2); the LBD works synergistically with the amino-terminal domain to regulate gene transcription (3–6). The F domain shares 18% amino-acid identity between the ERs and is located at the extreme carboxyl-terminus of the receptors. In ERα, the F domain appears to modulate transcriptional activity, co-activator interactions, dimerization, and stability; however, its role in ERβ is unclear (7–9).
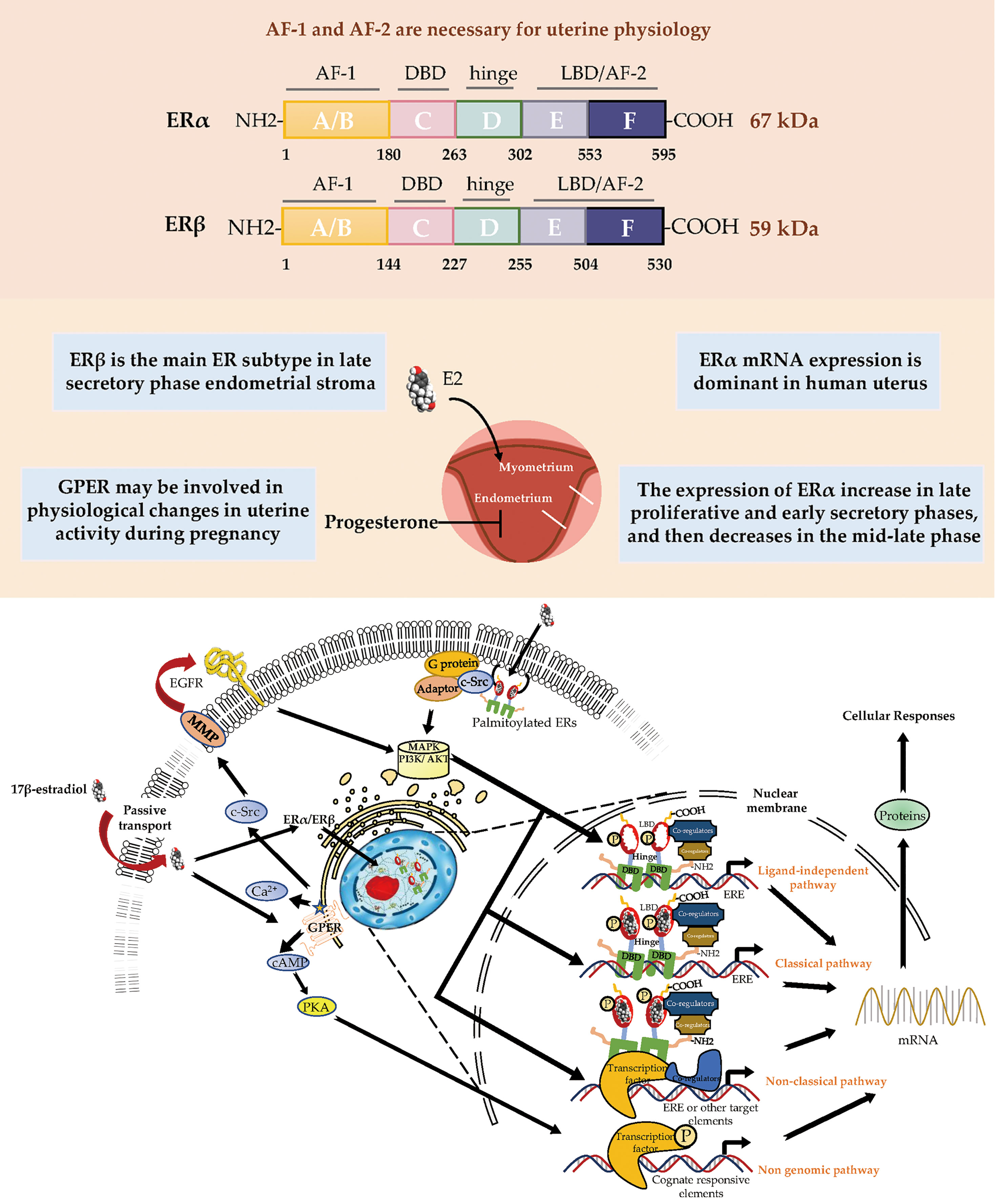
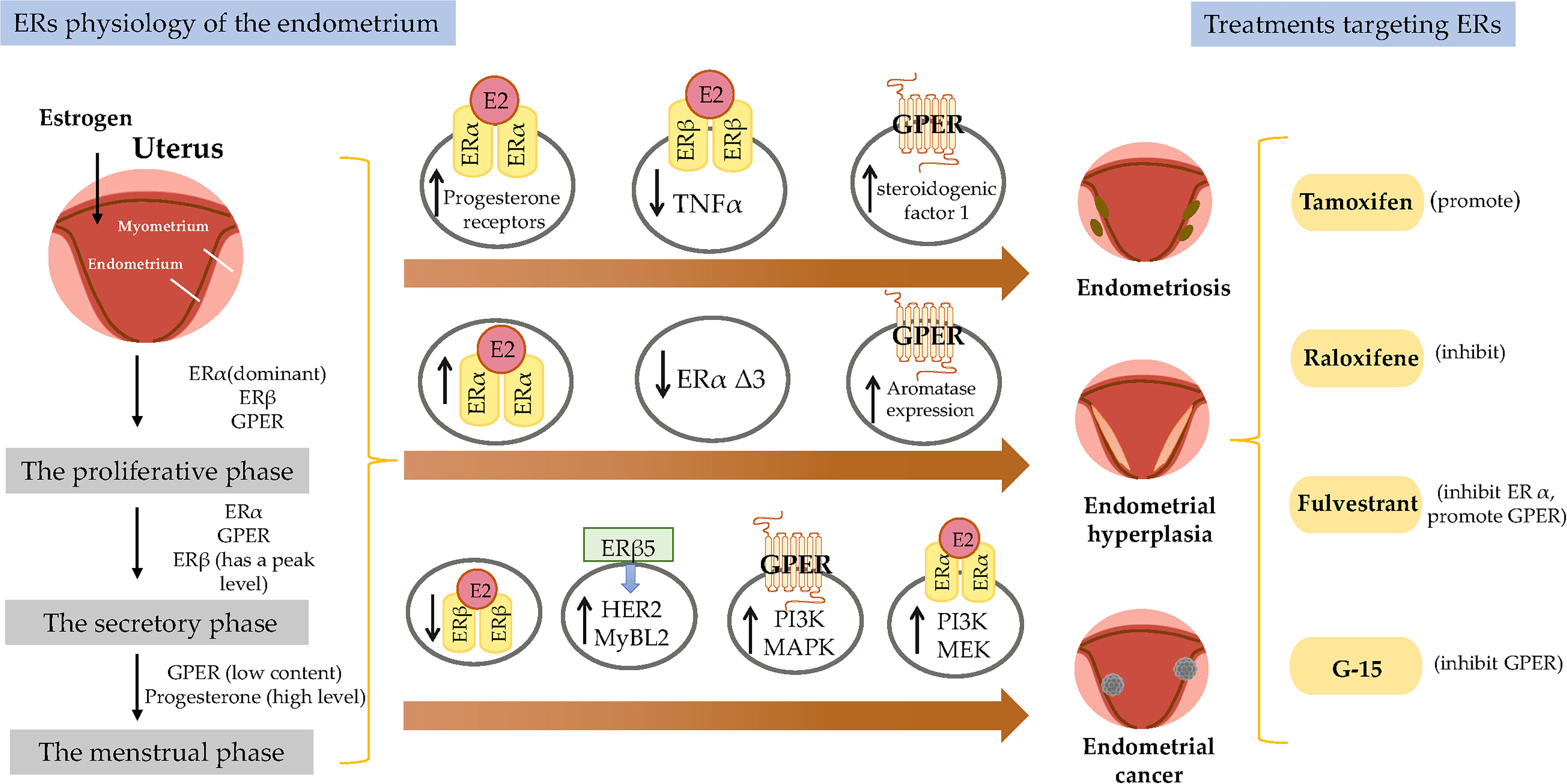
Figure 1 Estrogen receptor-mediated signaling pathways in the endometrial. E2 promotes endometrial growth, while progesterone and other progesterone hormones block endometrial growth and promote differentiation. E2 mediates its biological response by binding to ERs via genomic and non-genomic pathways. There are 3 main mechanisms of genomic regulation. Firstly, in genomic regulation, E2 ligands passively enter the cells by diffusion. ERα and ERβ are located in the cytosol. The binding of E2 to the ER promotes the formation of dimers, enters the nucleus and is directly binding to EREs, or to transcription factors which regulate transcription of its target genes. Secondly, the nonclassical pathway involves binding the E2-bound ER to TFs that are already bound to the DNA. The third mechanism is hormone-independent. The ER can regulate E2 responses by activating the signaling of growth factors via the phosphorylation of different serine (118/167) residues on the receptor. In non-genomic regulation, binding of E2 to ERs and GPR30 at the plasma membrane leads to various nongenomic responses, such as calcium signaling, PKC, and cAMP/PKA pathways. E2, 17β-estradiol; ER, estrogen receptor; GPER, G-protein-coupled estrogen receptor; EGFR, epidermal growth factor receptor; PKA, protein kinase A; SF-1, steroidogenic factor 1; TNFα, tumor necrosis factor α; PR, progesterone receptor; MAPK, mitogen-activated protein kinases; PI3K, phosphoinositide-3-kinase. ERE, estrogen response element.
The uterus reacts to cyclical changes in estrogen and progesterone levels to prepare the embryo for implantation. Most known estrogen effects are mediated by ERα and ERβ, which regulate classical hormone signaling pathways (10). Both ERα and ERβ are expressed in the murine uterus (11). ER dimers bind to estrogen response elements (EREs) in the promoter regions of target genes, initiating the recruitment of co-activators, co-suppressors, and chromatin remodeling factors to activate or inhibit transcription of target genes. Despite the presence of similarities in initiation and termination, the ERE-dependent signaling pathway substantially differs between ERα and ERβ in terms of the mode and extent of transcription, presumably because their distinct amino-termini influence the magnitude of transcriptional responses (6, 12, 13). The amino-terminal A/B domains of ERα include the ligand-independent AF-1, while the LBD includes the ligand-dependent AF-2. Both AF-1 and AF-2 of ERα are necessary for uterine physiology because their functional integration is required to mediate transcription at full capacity in response to E2 in a tissue-specific manner (14, 15). In contrast, ERβ displays a lower affinity for ERE binding and considerably lower transcriptional activity in the E2-induced ERE-dependent genomic signaling pathway; moreover, it interacts with a different set of proteins, presumably because of the absence of AF-1 in its amino-terminus (6, 16). The N-terminal domain of ERβ is much shorter than the corresponding domain of ERα. Moreover, the amino-terminus of ERβ has been proven to impair receptor-ERE interactions and does not interact with the carboxyl-terminus (17, 18).
In the nonclassical pathway of estrogen action, ER can regulate genes that lack a canonical ERE. ER can activate or suppress gene expression by interacting with other transcription factors, such as activator protein-1 (AP-1), specificity protein-1 (SP-1), nuclear factor kappa-B (NF-κB) (19, 20). Jun and Fos are members of the AP-1 transcription factor family that can form Jun-Jun or Jun-Fos dimers; their basic regions can interact with a consensus sequence known as the TPA response element (21). Selective estrogen receptor modulators (SERMs) and selective estrogen receptor downregulators (SERDs) can activate the transcriptional responses mediated by ERα at TPA response element sites, while they cannot activate transcriptional responses mediated by ERβ (22–24). Another transcription factor, SP-1, binds to a consensus sequence known as the GC box element. ERα has been shown to tether to GC box response element-bound SP-1, for which the amino-terminus of ERα is critical (25). In addition, in ligand-independent genomic events, ER activates growth factor signaling by phosphorylating different serine (118/167) residues on the receptor, thereby regulating the E2 response.
Beginning in the 1990s, there was increasing evidence that rapid modulation (within seconds or minutes) of estrogen-mediated signaling pathways occurs via a subpopulation of ERs located in or adjacent to the plasma membrane; these non-genomic effects induced by E2 were determined by the observation that exposure of uterine or ovarian cells to E2 could rapidly induce ion influx, cyclic adenosine monophosphate (cAMP) production, and phosphoinositide-3-kinase (PI3K) activation (26–30). In the 2000s, membrane ERs were characterized; these primarily consisted of GPR30/GPER, ER-X, and the Gq membrane ER (31–33).
Among all membrane ERs, GPER has been recognized as the major mediator of the rapid cellular effects of E2 because stimulating GPER activates metalloproteinases and induces the release of heparin-binding epidermal growth factor, which binds and activates epidermal growth factor receptor (EGFR); this interaction leads to the activation of downstream signaling molecules such as extracellular signal-regulated kinase 1/2 (ERK1/2), followed by the production of cyclic cAMP, intracellular calcium mobilization, and PI3K activation (34–37). Additionally, GPER indirectly regulates transcriptional activity through signaling mechanisms that involve cAMP, ERK, and PI3K (38) (Figure 1).
Regarding targeted therapies for GPER, the first GPER-selective agonist G-1 was identified in 2006. Subsequently, a GPER-selective antagonist G-15 was identified in 2009, followed in 2011 by G-36, a more selective GPER antagonist than G-15 (39, 40). Fulvestrant is a SERD that causes degradation or downregulation of ER, but acts as a GPER agonist (41).
E2 activity is mediated by the two classical nuclear hormone receptors, ERα and ERβ. Transcriptional regulation arises from the direct interaction of the ER with components of cellular transcriptional machinery. Cells can express a set of coregulators that can enhance or decrease the transcriptional activity of steroid hormone receptors, called ER coactivators and corepressors, respectively. Coregulators are crucial for ER transcriptional activity and have led us to recognize that there is a great deal of sophistication in transcriptional regulation. ER is able to bind to cofactors through the AF-1 and AF-2 domains, acting synergistically in the recruitment of coregulators. Coregulators are both targets and propagators of posttranslational modification codes and are involved in many steps of the gene expression process, including chromatin modification and remodeling, transcription initiation, elongation of RNA chains, mRNA splicing, mRNA translation, miRNA processing, and degradation of the activated NR coregulator complexes (42).
Approximately 200 coactivators play a central role in NR-mediated promotion of gene expression and are associated with cancer and other diseases. Many additional ERα coregulators have been found, although little is known about Erβ (43). Investigation of coregulators of ERs began in the 1990s and the first identified ERα coregulator was called steroid receptor co-activator (SRC-1). Co-expression of SRC-1 reverses the ability of ER to inhibit human progesterone receptor (PR) activation (44). Coregulators of ERα include the SRC/p160 group, histone acetyltransferase cAMP response element binding protein (CREB)/p300, ATP-dependent chromatin remodeling complexes such as SWI/SNF, E3 ubiquitin protein ligases, and steroid RNA activator (45). Then transcriptional corepressors have the NR corepressor and thyroid hormone receptors (46, 47). Therefore, nuclear ERs play a variety of roles and functions in different cells and tissues that are mediated by various intermediaries and differential utilization of coregulators (48).
From studies of cancer cells, we have learned that a large number of coregulators have specific structural motifs that affect their contact with the ER ligand binding domain. The ER crystal structure suggests that helix 12 lays across the ligand binding pocket when binding agonists, resulting in a surface suitable for the binding of LXXLL motifs found on coregulators. Conversely, we know that co-inhibitory factors block ER-mediated gene transcription by directly interacting with unbound ERs, using their corepressor NR box or by competing with coactivators (49). It has also been reported that some post-translational modifications, such as phosphorylation, methylation, ubiquitination, SUMOylation and acetylation, can affect the action of coregulatory factors that target gene expression (50, 51).
The dynamic expression pattern of steroid hormones and their receptors in the endometrium during the menstrual cycle has been established and drive cyclical growth, shedding, and regeneration of the endometrium. During the follicular phase of the menstrual cycle, estrogen, working through ERs, induces growth of the endometrium, producing a measurable thickening of the mucosa (52). After ovulation, the corpus luteum continues to produce a large amount of estrogen ligand in addition to progesterone. The presence of progesterone inhibits estrogen-induced endometrial growth and transforms the endometrium into a receptive state for blastocyst implantation (1). Physiologically, progesterone plays an important role in preventing endometrial hyperplasia (EH) (53). In animal models, high levels of estrogen without progesterone interference can lead to EH or cancer, suggesting that the balance between estrogens and progestogens is often dominated by estrogens during cancer formation (54, 55) (Figure 1).
During postnatal development, the uterus undergoes a unique pattern of changes in ER expression. ERα is weak and the proliferative activity is high from postnatal days 1 to 7 in the uterine stroma. In contrast, the cell proliferation rate is decreased from day 7 to puberty, but ERα expression in the epithelial and stroma cells is increased (56). The low rate of proliferation in the prepubertal period is due to estrogen levels being too low to activate ERα (57, 58). However, in the proliferative phase, ERα immunoreactivity is significantly stronger than ERβ immunoreactivity in the nuclei of epithelial, stromal, and muscle cells, suggesting ERα mRNA expression is dominant in the human uterus. Immunohistochemical localization revealed increased expression of ERα and ERβ in the late proliferative and early secretory phases, and then a decrease in the mid-late phase (59). ERβ is the main ER subtype expressed in the endometrial stroma in the late secretory phase (60). ERα and ERβ are differentially expressed in endometrial vascular endothelial cells and perivascular cells surrounding endometrial blood vessels. Monoclonal antibodies and immunocytochemistry have shown that ERα is localized to muscle cells of uterine arteries (61). Analysis of immunostaining confirmed that endometrial endothelial cells only express ERβ, which may be the target of selective agonists or antagonists for the ERβ subtype (59) (Table 1). It is notable that nonradioactive in situ hybridization confirmed that ERα and ERβ were localized in both epithelial and stromal cells of endometriotic tissues (66) (Figure 1).
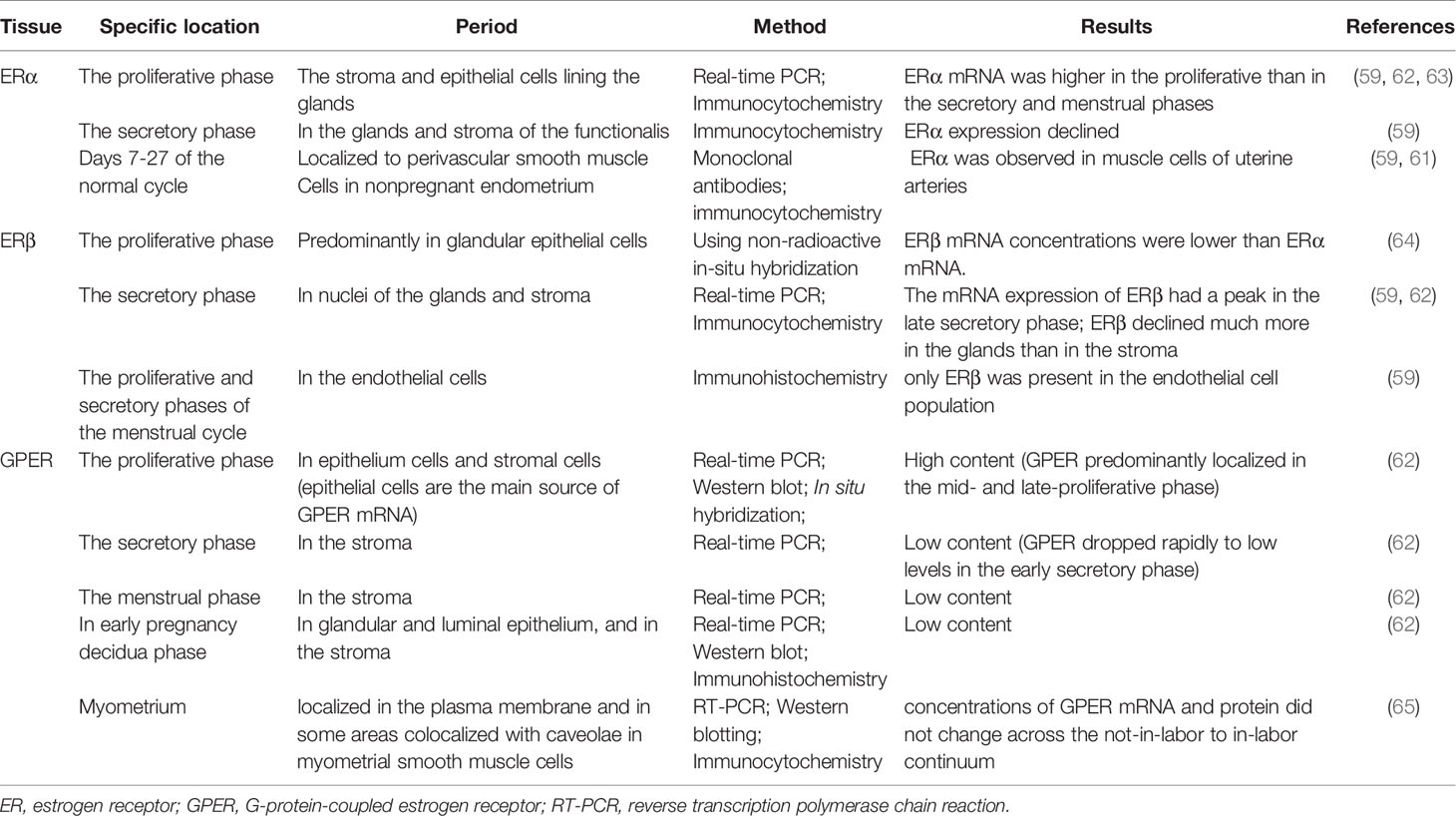
Table 1 ER expression by uterine phase.
The plasticity of the human endometrium is beyond other tissues, and it can still respond to steroid hormones and induce endometrial cycles after menopause. The post-menopausal endometrium has low circulating estrogen level and lacks progesterone, while androgen levels from adrenal glands are relatively unchanged. ERβ mRNA expression in the myometrium of postmenopausal women is significantly higher than in premenopausal women, while ERα expression is decreased (67). Thirty-one postmenopausal women underwent combined treatment with E2 and testosterone, which increased AR and ERβ expression in the endometrium (68). In contrast, levels of ERα and PRs in the endometrium were upregulated after estrogen-alone treatment (68). This study indicated that the antiproliferative effect of androgen treatment in the endometrium is associated with increased ERβ expression and that ERα may promote endometrial proliferation.
In humans, several studies have reported GPER mRNA and protein expression in the uterine epithelia (epithelial and stromal cells), endometrium, myometrium, and early pregnancy decidua (62, 65, 69). Real-time PCR and immunohistochemistry results showed that GPER mRNA was highly expressed in glandular epithelial cells in the mid- and late- proliferative phase, higher than during the secretory and menstrual phases. GPER expression levels were also found to decrease rapidly from the early secretory phase and remained low until the end of the cycle (62). In decidual tissue, GPER mRNA expression was localized in both the glandular and luminal epithelium and in the stroma (62). GPER activation enhanced contractile responses to oxytocin in the myometrium. These findings suggest that GPER may be involved in the physiological changes in human uterine activity during pregnancy (65). In general, several studies using cell and animal models have highlighted the importance of ER signaling in female reproduction. GPER is involved in cyclic alterations of endometrial estrogen action, and high GPER transcript levels were observed in the eutopic endometrium during the proliferative phase, whereas higher GPER mRNA expression has been shown during the secretory phase (70) (Table 1). GPER is also found in the endometrium of women with endometriosis (71).
It is currently thought that E2-induced uterine epithelial cell proliferation is mediated by stromal ERα. ERα knockout mice showed no expression of estrogen responsive genes in the uterus, and the basic levels of PR mRNA in the αERKO uterus were equal to those in wild type mice (72). The levels of PR protein in the uterus of ERα knockout mice were at 60% of the level measured in a wild-type uterus, which is enough to induce the genomic responses mediated by progesterone but not enough to support embryo implantation (73). These observations suggest that ERα modulation of PR levels is not necessary for progesterone action in ERα knockout mouse uterus (Table 2).
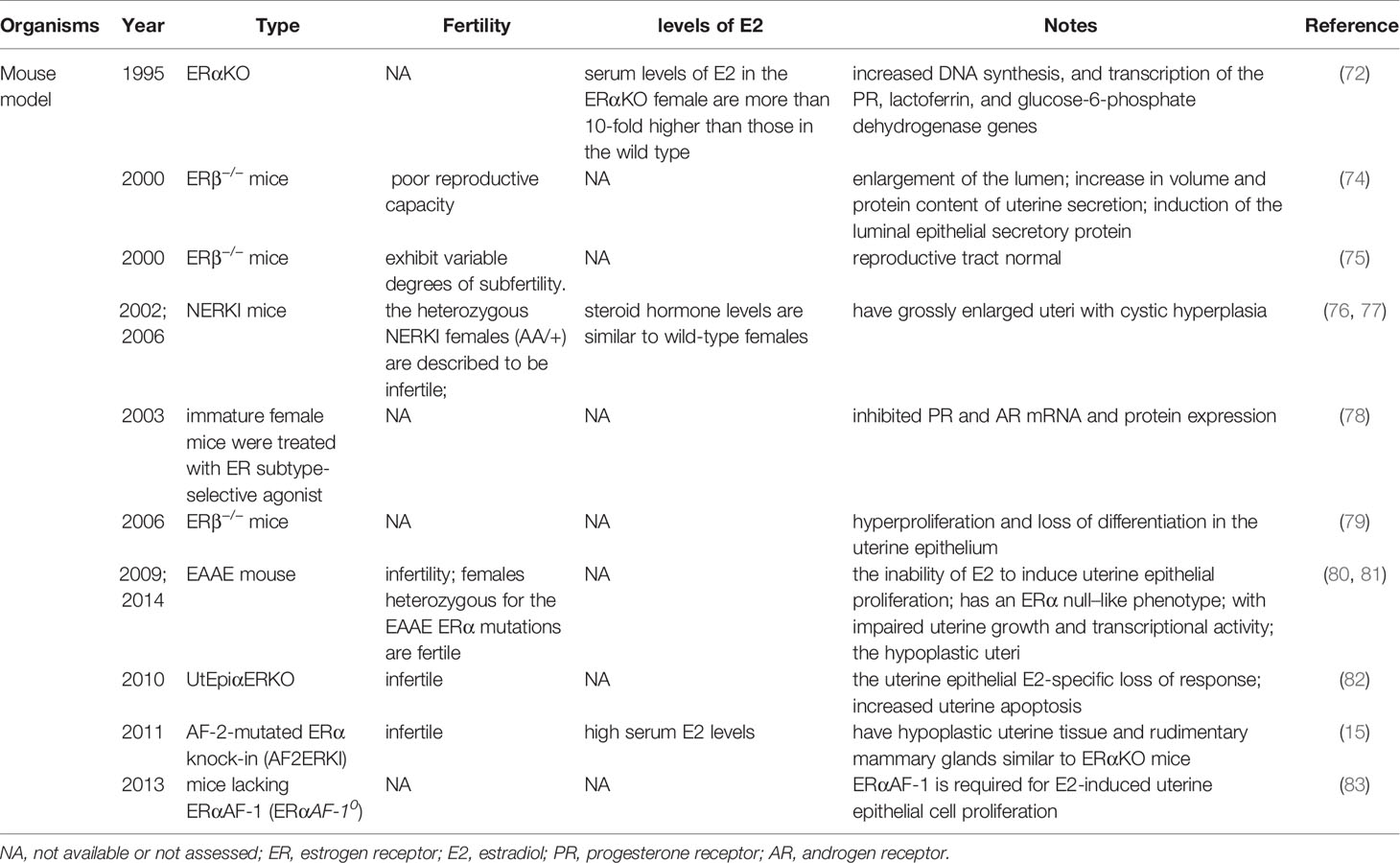
Table 2 Summary of mouse models involving ERα and ERβ.
ERs are dependent on AF-1 in the N-terminal domain and AF-2 in the C-terminal LBD to induce the specific conformational changes that are required for ER transcriptional activity (Figure 1). In the model of AF-2-mutated ERα knock-in (AF2ERKI), AF2ERKI homozygote female mice have hypoplastic uterine tissue and rudimentary mammary glands that are indistinguishable from ERα knockout mice (15). ERα AF-2 has been demonstrated to play a crucial role in the endometrial proliferative effect of E2. Furthermore, Abot et al. investigated the role of ERα AF-1 in the regulation of gene transcription and cell proliferation in the uterus. Targeted deletion of AF-1 in mice showed normal uterine development but delayed response to E2 (83). Nevertheless, in a study using the “EAAE” mouse, which has more severe DNA-binding domain mutations in ERα, uterine epithelial proliferation could not be induced through the estrogen signaling pathway, manifesting an ERα null-like phenotype with impaired uterine growth and transcriptional activity (80, 81) (Table 2). The ERα (EAAE/EAAE) mouse suggested that the DBD is necessary for estrogen action and the LBD is insufficient.
Nonclassical ERα knock-in (NERKI) mice with an ERα mutated at the DNA recognition helix that disrupts DNA binding but leaves nonclassical signaling intact have also been developed (Table 2). The uteri of NERKI mice are larger than those of ERα knockout mice but smaller than those of wild type mice. NERKI mice also have defective ovulation and underdeveloped mammary glands (76). The NERKI mice indicate that nonclassical ERα signaling plays a critical role in uterine growth and development, which is beneficial to restore the proliferation of luminal epithelial cells (77).
By establishing a uterine epithelial-specific ERα knockout (UtEpiαERKO) mouse line, it was found that while female UtEpiαERKO mice were infertile, they had regular estrous cycles and complete follicular development stages, indicating ovulation (82) (Table 2). Embryonic implantation was not observed in the uterus after natural mating or embryo transfer, suggesting that ERα in the uterine epithelium is necessary for embryo receptivity. E2 treatment to UtEpiαERKO mice stimulated epithelial cell growth, but apoptosis in the epithelial cells significantly increased compared with wild type mice. These studies might help to determine how the proliferation of uterine epithelial is mediated by ERβ in the stroma, while uterine epithelial-derived ERα is required subsequent to proliferation to prevent epithelial apoptosis ensuring the full uterine epithelial response (82).
Despite ERα being the predominant ER in the adult rodent uterus, transcripts encoding ERβ have also been detected in wild type and ERα−/− murine uteri (11). The uteri of untreated ERβ−/− mice exhibit exaggerated responsiveness to E2, as indicated by enlargement of the lumen, increased volume and protein content of uterine secretions, and induction of the luminal epithelial secretory protein (74). The increased cell proliferation and exaggerated response to E2 in ERβ-/- mice suggest ERβ inhibits ERα function, resulting in an anti-proliferative function in the immature uterus. ERβ can act as a regulator of ERα-mediated gene transcription in the uterus, alternatively, it is responsible for downregulating PR in the luminal epithelium. Paradoxically, there is evidence that ERβ can partially compensate for loss of ERα in the reproductive tract, as the uterine phenotype of ERαβ double knockout mice is similar to the aggravated uterine phenotype of ERα knockout mice, whereas the reproductive tract of ERβ knockout mice appears normal (75) (Table 2). Meanwhile, the uterine epithelium of ERβ−/− mice showed hyperproliferation and loss of differentiation (79). This suggests that the absence of ERβ predisposes the uterus to abnormal endometrial proliferation. Fully mapping ERβ expression in the endometrium may be useful in identifying women at higher risk of EH.
A study examining the potential synergistic regulation of gene expression and uterine growth by the two receptors for estradiol, ERα and ERβ, using ER subtype-selective agonist ligands showed that the ERβ agonist diarylpropionitrile did not increase uterine weight or luminal epithelial cell proliferation, but inhibited PR and AR mRNA and protein expression (78) (Table 2). Additionally, ERβ can modulate ERα activity in a response-specific and dose-dependent manner (78).
The structure of GPER is dramatically different from that of ERα and ERβ. No obvious developmental or functional defects have been observed in the reproductive organs of GPER knockout mice (84–86). Treating wild-type mice with G-1 stimulates uterine epithelial proliferation despite a lower potency relative to that of E2; conversely, blocking GPER with G-15 reduces the E2-mediated proliferative response by approximately 50% (40). Stable knockdown of GPER substantially eliminated the tumor growth induced by autocrine motility factor (AMF) in EC, with significantly longer survival times in tumor-bearing mice (87). Conversely, Gao et al. demonstrated that in an ovariectomized mouse uterus, GPER activation by high-concentration G-1 altered the expression of E2-dependent uterine genes and mediated inhibition of ERK1/2 and phosphorylation of ERα (Ser118) in the stromal compartment; thus, they concluded that GPER is a negative regulator of ERα-dependent uterine growth in response to E2, suggesting an interaction between non-genomic and genomic ERs (69).
Estrogen plays a different role in embryo implantation and angiogenesis. This suggests that although studies in mouse models provide evidence for the role of ovarian steroid hormones in regulating uterine function, extrapolation of mouse endometrial estrogen findings to human conditions needs to be considered carefully. Despite their compelling results, many of the reviewed studies were limited by a lack of replication, small sample sizes, retrospective designs, publication bias, and/or the use of non-standardized tools to diagnose conditions.
Endometriosis is the presence of endometrial cells outside the uterine cavity, which can invade local tissues and cause severe inflammation and adhesions. Approximately 15% of infertile women are reported to have endometriosis, although the true prevalence of endometriosis is unclear (88). Multiple cellular and molecular signaling pathways are likely to be involved in the pathogenesis of endometriosis.
Endometrial development and function are highly dependent on the cyclic secretion of sex steroid hormones and the expression of their cognate receptors (89). The endometrial cell types that are primarily targeted by steroid hormones include epithelial and stromal cells. Proliferation and differentiation of the endometrium are regulated by estrogens (90). Endometriosis typically involves higher levels of E2 than observed in a normal endometrium, which is due to higher gene expression levels of aromatase and 17β-hydroxysteroid dehydrogenase type 1 (91); the higher levels of E2 result in increased E2 binding and activation of ERs in endometriotic tissues, thereby stimulating estrogen-dependent growth. These higher levels of local E2 activity could contribute to the proliferation of endometriotic tissues (92). Estrogen-mediated changes in cell signaling presumably have important implications for the pathogenesis of endometriosis (93). For example, the invasion and migration of eutopic endometrial endometriosis stromal cells is regulated by the estrogen/H19/miR-216a-5p/ACTA2 axis (94). Alternatively, studies have implicated that tamoxifen and the phytoestrogen genistein can induce steroidogenic factor 1 (SF-1) target gene aromatase expression in a GPER-dependent manner to promote the proliferation of Ishikawa cells. Based on this finding, we hypothesized that GPRE/SF-1 may promote endometriosis by increasing local estrogen concentrations and mediating the proliferation of synthetic estrogens in combination with classical ER signaling (95) (Figure 2).
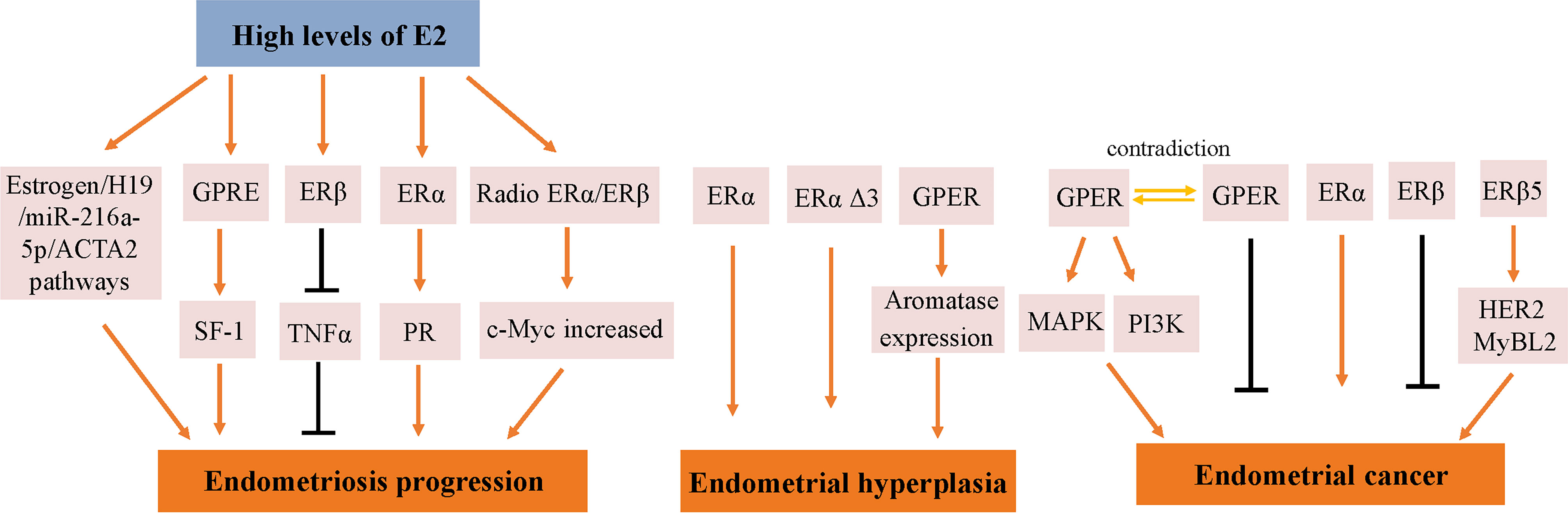
Figure 2 Molecular pathways regulated by ER in endometrial diseases. E2, estradiol; ER, estrogen receptor; GPER, G-protein-coupled estrogen receptor; SF-1, steroidogenic factor 1; TNFα, tumor necrosis factor α; PR, progesterone receptor; MAPK, mitogen-activated protein kinases; PI3K, phosphoinositide-3-kinase.
Estrogen activity is mediated via genomic pathways including nuclear ERα/β, as well as by more rapid, non-genomic pathways, such as ERα36 and GPER (96). Elevated estrogen promotes the expression of ERα and ERβ, which reach their highest levels in the late proliferation phase (97); however, aberrant levels of ERs are observed in women with endometriosis. Compared with endometrial tissue, ERβ mRNA and protein levels were more than 100-fold increased, while the levels of ERα were several times lower (98); inhibiting the enhanced ERβ activity via an ERβ-selective antagonist also suppressed the growth of ectopic lesions in mice (99). Notably, ERβ activity stimulates endometriotic progression: ERβ inhibits tumor necrosis factor α (TNFα)-induced apoptosis through interactions with apoptotic mechanisms to avoid endogenous immune surveillance of surviving cells (99). ERβ directly induces Ras-like estrogen-regulated growth inhibitor gene expression in an estrogen-dependent manner to enhance the proliferative activity of endometriotic tissues (100). The role of ERβ is presumably more complicated, because greatly elevated levels of ERβ are present in both the nucleus and cytoplasm of endometriotic tissues (101).
It remains controversial whether ERα exhibits an endometriotic tissue-specific pattern (102). Studies of ERα knockout mice with endometriosis have shown that ERα causes cell adhesion and proliferation and that it regulates inflammatory signaling in ectopic lesions (103). E2 increases the expression of PRs mainly through ERα activation, thereby mediating the effects of progesterone on the endometrium and triggering the secretory phase of endometrial circulation. Estrogen responsiveness is considerably complex, as indicated by the results obtained from experiments in mice and the findings in human tissues contain splice-form variants of ERα and ERβ. Moreover, the differential effects of estrogen on endometrial cells may depend on the total amount of cellular ERs and/or the ratio of ERα to ERβ (104).
EH is a uterine pathology that involves a continuum of morphologic alterations that range from mild, reversible glandular hyperplasia to direct cancer precursors. Compared with the normal proliferative endometrium, the predominant characterization of EH is an increased endometrial gland-to-stroma ratio. During the reproductive period, the risk of EH is increased by conditions associated with intermittent or anovulation, such as Polycystic ovary syndrome. After menopause, when ovulation stops, EH is more common in women with increased circulating estrogen levels.
During the normal reproductive cycle, ERα expression in uterine epithelial cells is downregulated in the secretory phase. Hu et al. assessed the expression of ERα and ERβ in 114 patients using immunohistochemistry (105). The results indicated that from normal proliferation to simple and complex hyperplasia, the expression of ERα increased, while the expression of ERβ showed no significant change. Other studies have also revealed similar results. As early as 2003, Uchikawa et al. detected the expression levels of ERs in 20 normal endometrium samples, 36 hyperplastic endometrium samples, and 58 malignant endometrium samples, and found that ERα expression was increased in EHs compared with normal endometrium samples (106). In 2005, Bircan et al. described that ERα levels were significantly higher in EH than in the normal secretory endometrium (107).
However, different variants have different protein activities and different regulatory functions on ER signaling. ERα Δ3 lacks part of the DNA binding domain of exon 3 and inhibits estrogen-dependent transcription activation in a dominant negative way, which may potentially affect ERα signal transduction (108). The Δ3 variation is detected in prolactinoma, EH, and breast cancer, but not in the normal pituitary, normal endometrium, or endometrial cancer (EC) (109–111). For example, this variant was found in 19 of 21 EH cases, but not in any of 29 EC cases (110). Additionally, ERα Δ3 expression is reduced more than 30-fold in breast cancer compared with in the normal breast epithelium (112). It is speculated that the dominant negative activity of Δ3 may decrease normal estrogen signaling, and thus interfere with tumor progression and growth (Figure 2).
GPER is highly expressed in abnormal EH, and its expression trend follows that of ERα, which is gradually increased from the normal proliferative endometrium to simple EH, and then to a maximum in complex EH (Figure 2). This could imply that in normal and benign proliferation, GPER expression increases proportionally due to the induction of GPER by ERα (71, 113). Aromatase activity is not detected in the normal endometrium but is highly expressed in endometriosis and malignant endometrium cells (114, 115). Surprisingly, GPER activation increased aromatase expression in both endometriosis and malignant endometriosis cells (95). Thus, we speculate that the mechanism by which GPER regulates the growth of abnormal endometrial cells might be that it induces the expression of aromatase, increasing the synthesis of intracellular estrogen. In turn, estrogen activates intracellular GPER by the intracellular pathway, further increasing the abnormal proliferation rate of cells.
EC is an estrogen-dependent malignancy. The administration of estrogen alone for an extended period can increase the risk of EC in postmenopausal women. Some endometrial atypical hyperplasias can evolve into EC over a long period of time. EC is subdivided into two types on the basis of histopathology. Type I endometrial tumors, also known as low-grade endometrioid, which make up account for 75% of endometrial cases, are usually associated with high levels of ERα (116). Type II tumors include high-grade endometrioid tumors, serous tumors, clear cell tumors, carcinosarcomas, and mixed histology tumors. Carcinomas can typically be classified into two types according to their estrogenic state, type 1 (ER-positive EC) and type 2 (ER-negative EC).
In a previous study, the expression of GPER, estrogen, progesterone, EGFR, and Ki-67 in 47 EC patients treated between 1997 and 2001 based on early immunohistochemical methods showed that GPER was overexpressed in high-risk EC patients and was negatively correlated with PR expression (117). E2 and G-1 trigger the mitogen-activated protein kinase (MAPK) pathway in ER-negative KLE cells and ER-positive RL95-2 cells, which require GPER involvement (118). Li et al. investigated the relationship between AMF and GPER in EC (87). This mechanistic study demonstrated that the interaction between AMF and GPER activates PI3K signaling, which in turn accelerates the growth of EC cells (Figure 2). However, there have been inconsistent results regarding the expression of GPER in EC. In 2012, Krakstad et al. found decreased GPER mRNA and protein levels and increased ERα levels in high grade endometrial carcinoma, supporting the association between GPER loss and disease progression from primary to metastatic lesions (119). Furthermore, in another study, GPER expression in EC cells was found to be lower than in normal endometrial samples. Treating GPER-positive EC cells with the GPER agonist G-1 resulted in significant growth reduction, suggesting that GPER mRNA might be sufficient to mediate the antiproliferative effects of its ligand in EC (120) (Figure 2). Due to its high expression and mitogenic role in other tissues and cancers, among the three ERs encoded in the human genome, ERα and ERβ are considered to be the major mediators of pro-growth estrogen signaling in EC cells.
The results of studies investigating the expression and role of ERβ in EC are not yet fully known. ERβ may play a suppressive role in EC. Immunohistochemical results have shown decreased ERβ mRNA and protein levels in endometrioid EC compared with in normal proliferative endometrium or adjacent normal endometrium from post-menopausal control women (121, 122). In addition, Zhang et al. indicated that in ERβ knockout mice, there is an unusual proliferation of cells in the uterus (74). Nevertheless, ERβ also shows a potential tumorigenic effect. A C-terminally altered ERβ isoform, ERβ5, is upregulated in endometrial carcinoma and is associated with the expression of oncogenes such as HER2 and MyBL2 (123, 124) (Figure 2).
Analysis of The Cancer Genome Atlas data showed that the mean expression level of ERα in EC was 2.9-fold higher than that of ERβ (125). The reason for this may be that EC mainly affects postmenopausal women, and higher gonadotropin levels in postmenopausal women may downregulate ERβ (126). Because ERβ acts as a dominant negative regulator of ERα, the postmenopausal endometrium may promote uterine cell proliferation through unopposed ERα action (74). In terms of analyzing the relationship between ERα/ERβ and the clinical characteristics of EC patients, it was found that ERα expression is higher in the early stages of EC and decreased in advanced EC (127). Thus, ERα may promote the progression of EC by interacting with estrogen in endometrial atypical hyperplasia and the early stages of EC.
It is well known that obesity is one of the most common risk factors for EC because androgens are converted to estrogen in adipose tissue. In fact, ER mutations are present in 5.8% of primary ECs (128). In EC, ERα mutations are associated with worse outcomes and less obesity, so mutations in ERα might explain why women with normal body and without other risk factors also develop EC (129). Evaluation of an EC cell model that includes that D538G mutation revealed that mutant ER has estrogen-independent activity as well as an expanded set of genomic binding sites (130). Mutation confers estrogen-independent activity to ER, which causes gene expression changes. Understanding the molecular and pathological effects of ER mutations in EC will further our knowledge of ER mutant disease and may provide treatment options for patients with ER mutant tumors.
ER is used as a regulatory cofactor to regulate gene expression, and different transcription factors may be responsible for controlling the genomic interactions of ER in EC cells. Motif analysis of endometrial cancer-specific ER-bound sites, along with gene expression analysis, revealed that ETV4, a member of the ETS family, overlaps with 45% of ER binding sites in Ishikawa cells (131). In a recent study reported using CRISPR/Cas9 to knockout ETV4 in EC cells, genetic deletion of ETV4 resulted in a large reduction of ER binding signal at the majority of bound loci across the genome, leading to an expected decrease in the transcriptional response to E2 treatment and thus reduced cells growth (132). Qi et al. found that estrogen regulates the histone acetylation hMOF expression through activating the PI3K/Akt and Ras–Raf–MEK–ERK signaling pathway to promote cell cycle progression in EC cells (133). Unfortunately, although the role of ER in the development of EC has been demonstrated in numerous studies, there are still many gaps in our knowledge of ER in EC.
Hormone replacement therapy (HRT) has been used in menopausal women to relieve hot flashes, vaginal dryness, fluctuating emotions, irregular menses, chills, and sleeping problems. Pure antiestrogens represent an endocrine-targeted therapy for which the mechanism of action involves competition with ER ligands and ER downregulation. However, treating menopause symptoms with estrogen must be accompanied by a progestin component to avoid the effects of excess estrogen on the endometrium. Progesterone has been shown to downregulate ERs and stimulate direct PR-mediated effects that oppose estrogenic actions. In a postmenopausal estrogen/progestin interventions trial, women assigned to estrogen alone were more likely to develop simple, complex, or atypical hyperplasia. Combining conjugated equine estrogens with cyclic or continuous progestin protected the endometrium from hyperplastic changes associated with estrogen-only therapy (134). A small but not significant reduction in the risk of EC was observed in a sequential combination regimen with estrogen and progesterone (135). In the early 2000s, the Women’s Health Initiative raised numerous concerns regarding the use of hormone replacement therapy, as combined estrogen-progestin treatment was associated with a statistically significant increase in invasive breast cancer and in cardiovascular events after approximately five years of follow-up (136). Therefore, hormone replacement therapy use is now recommended to be relatively short-term (i.e., 3–5 years in post-menopausal women) and be administered at low doses; moreover, its use is very narrow and should be limited to women without a history of breast cancer and women who are not at increased risk of cardiovascular or thromboembolic disease (137). Consequently, novel ER modulators are necessary to maintain endocrine homeostasis.
Tamoxifen acts as an agonist in most estrogen target tissues, presumably in association with differences in the expression of co-activator and co-repressor proteins in different tissues, which result in the formation of distinct complexes with ERs (6, 138) (Figure 3). Tamoxifen stimulates ER dependent gene regulation in the uterus (139). In endometrial cells, tamoxifen-bound ERα is able to recruit coactivator proteins and to initiate gene transcription. This differential recruitment of coactivators contributes to the tissue specificity of tamoxifen ERα function (140). However, the estrogen agonist effects on the endometrium from partial agonists cannot be neglected and can manifest as increased endometrial thickness, endometrial polyps, leiomyomas, and EC (141–143). Tamoxifen may promote cancer development by upregulating ERα, PR, vascular endothelial growth factor, EGFR, mechanical target of rapamycin, IGF-1R, and C-MYC in EC cells (144, 145). In a randomized trial, Fornander et al. found that among 931 postmenopausal patients treated with tamoxifen, there was a 6.4-fold increase in the incidence of EC compared with controls (146). This could be related to the dose of tamoxifen used (40 mg per day), which is higher than what was used by other trials. Similarly, Fisher et al. demonstrated a significant increase in EC severity among women treated with tamoxifen, with a relative risk of 7.5 (147). It has been suggested that tamoxifen treatment has a cancer-promoting effect via GPER, which significantly stimulates the proliferation of endometrial cells (148). Because tamoxifen has a selective antiestrogenic effect in breast cancer but an agonistic estrogenic effect in the bones and the uterus/EC, it is not suitable for use in the general population due to the increased incidence of EC.
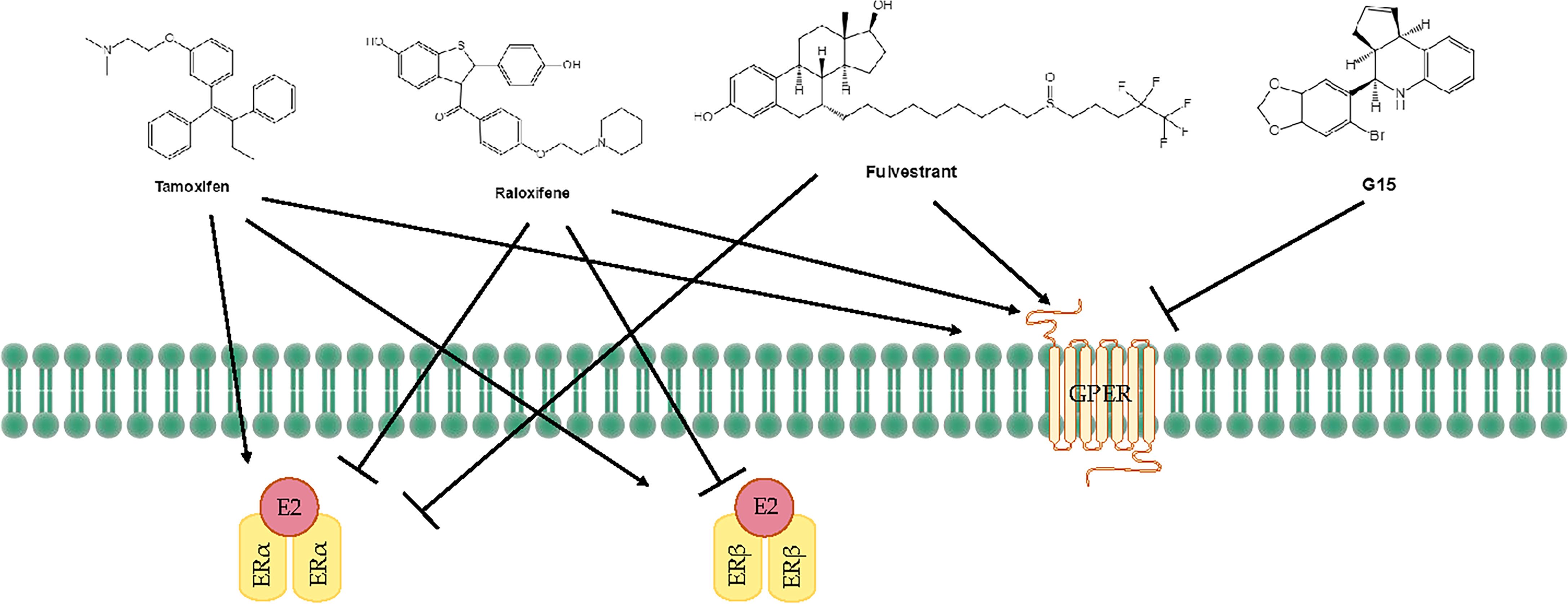
Figure 3 Treatments for endometrial diseases. ER, estrogen receptor; GPER, G-protein-coupled estrogen receptor; EGFR, epidermal growth factor receptor; ERE, estrogen response element; MMP, matrix metallopeptidase; LBD, ligand-binding domain; P, phosphorylation.
The mechanism of action of raloxifene occurs through binding to ERα and ERβ. This binding results in activation and blockade of estrogenic pathways in tissues that express ERs. The crystal structures of the LBD of ER in complex with the endogenous estrogen, E2, and the selective antagonist raloxifene, have shown that they are all bind in the same site within the core of the domain, but raloxifene induces a transcriptionally inactive LBD conformation (149). Raloxifene has estrogenic antagonistic effects in the uterus and breast. ERα blockade using raloxifene indicated that E2 alters endometrial cell proliferation via ERα. The antagonistic effect of raloxifene on estradiol-treated endometrial epithelial Ishikawa cells has been demonstrated by the altered expression of genes such as HOXA 10, leukemia inhibitory factor, PR, and EMX2 (150). Notably, raloxifene does not exhibit the endometrial side effects observed with tamoxifen (151, 152). In estrogen-stimulated ovariectomized rat surgical models of endometriosis, treatment with 10 mg/kg raloxifene for 7 to 14 days resulted in reduced uterine volumes (153). Adult female rhesus monkeys with spontaneous endometriosis treated with 10 mg/kg/d raloxifene for 90 days showed degeneration of endometrial tissue and attenuated uterine volume (154). Although raloxifene does not induce breast tenderness, EH, or EC, it may augment the risk of thromboembolic disease (1/1000 cases per year) as well as hot flashes (in 4%–6% of cases). Furthermore, raloxifene reduces proliferative markers in the epithelium of lesions in rodent models but not in the stroma, while the stromal component is the major contributor to endometriotic lesions (155). Petrie et al. have demonstrated that GPER plays an important role in the estrogen-mediated signaling of the ERα−/ERβ− EC cell line Hec50 and the SERM raloxifene is an agonist for GPER (156). In contrast to tamoxifen or raloxifene, antagonists such as fulvestrant (also known as SERDs) show complete antagonism. It completely inhibits estrogen-mediated changes in gene transcription and therefore has no agonist activity. The large side chain that originates from the B ring prevents H12 of ERα from docking in the agonist conformation, thereby preventing co-activator binding and transcriptional activation (149, 157) (Figure 3). Other effects of fulvestrant include inhibition of receptor dimerization and nucleocytoplasmic shuttling of the ER (158). Tamoxifen, raloxifene, or fulvestrant have been shown to be agonists of GPER. However, pharmacological inhibition of GPER activity in vivo prevents estrogen-mediated tumor growth, and the selective GPER antagonist G-15 retarded the growth of endometrial carcinoma (40) (Figure 3).
This review provides a summary of the body of published systematic reviews and meta-analyses of the effects of different ERs on the endometrium. The endometrium is a dynamic tissue that undergoes proliferation, secretion, and menstrual during the menstrual cycle of the female reproductive age. The current findings show that unopposed endometrial exposure to estrogen increases the risk of EH and cancer and emphasize the importance of modulating ERs to control the development of endometrial diseases. Multiple variants of ER are involved in endometrial pathophysiology and signaling pathways. ERα promotes uterine cell proliferation and is strongly associated with an increased risk of EC, because it plays an important role in tumor development and metastasis by activating signaling pathways involved in promoting proliferation, resisting apoptosis, stimulating migration and invasion, and inducing angiogenesis. In contrast, the responses mediated by ERβ has a key opposite effect in the endometrium. GPER is normally expressed in the endometrial but is highly expressed in abnormal EH, whereas paradoxically expressed in patients with EC. More research is needed to elucidate the disease mechanisms that involve ERs.
It is well known that the identification of risk factors that are strongly associated with endometrial risk can help the identification of high-risk groups of women, which will benefit from targeted prevention strategies. The mechanisms of action of hormone therapy involves competition with the ER ligands and ER downregulation. Unfortunately, the role of HRT has been debated. Estrogen receptor modulators provide potential treatment for high-risk women. Among SERMs, tamoxifen therapy improves survival in Erα-positive primary and advanced breast cancer. However, in endometrial, many authors have confirmed that tamoxifen use may cause endometrial thickness and polyps (54, 159). Raloxifene, a second SERM, has a high affinity to ERα, with a relative binding affinity of 46% for human ERα compared with E2 (160). It has anti-estrogen effects on the uterus and protects the endometrium from hyperplasia and irregular bleeding caused by estrogen hyperstimulation. Fulvestrant is a non-agonistic ER antagonist that blocks the ER and inhibits the proliferative effects of estrogen on tumor cells. To better treat or cure endometrial disease, a deeper knowledge of the roles of ERα, ERβ or GPER and their interactions is required.
Although there are still many diseases for which estrogens have been implicated but the role of their receptors has not been elucidated. Endometrium-associated diseases may require simultaneous attacks on multiple targets or a systems approach for effective treatment. With the increased understanding of the molecular basis and the pathways related to specific disease progression, the era of molecularly targeted therapies has emerged as a most promising direction of research. To develop personalized therapies, the molecular regulation of endometriosis, EH and EC needs to be carefully studied. Because endometrial disease is essentially a hormone-dependent manifestation of high ER and PR expression, targeting ER may be a viable approach to develop novel treatment strategies for this disease. Screening of various compounds by molecular simulation can help to identify promising selective agonists or antagonists for the prevention and treatment of estrogen-affected endometrial diseases.
Further work is needed to develop new, more bioavailable SERMs/SERDs with better pharmacological properties, and therapies that inhibit all types of ERs. Furthermore, due to low bioavailability, it is expected to improve existing formulations to address the barriers to optimal SERM or SERD use and efficacy profiles.
KY, X-LX, and Z-YH reviewed the literature, wrote the manuscript, and designed the figures and tables. JL revised the draft. X-WF and S-LD made substantial contributions to the conception and design of the work and provided input into manuscript content and composition. All authors contributed to the article and approved the submitted version.
This work was supported by National Nature Science Foundation Project of China (No. 32072722,31101714, and 31372307).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Wei Yan, Ph.D. and Ryan Chastain-Gross, Ph.D. (from Liwen Bianji, Edanz Group China www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript. We thank James P. Mahaffey, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the English text of a draft of this manuscript.
1. Critchley HOD, Saunders PTK. Hormone Receptor Dynamics in a Receptive Human Endometrium. Reprod Sci (2009) 16:191–9. doi: 10.1177/1933719108331121
2. Marino M, Galluzzo P, Ascenzi P. Estrogen Signaling Multiple Pathways to Impact Gene Transcription. Curr Genomics (2006) 7:497–508. doi: 10.2174/138920206779315737
3. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of Estrogen Action. Physiol Rev (2001) 81:1535–65. doi: 10.1152/physrev.2001.81.4.1535
4. Mosselman S, Polman J, Dijkema R. ER Beta: Identification and Characterization of a Novel Human Estrogen Receptor. FEBS Lett (1996) 392:49–53. doi: 10.1016/0014-5793(96)00782-x
5. Kumar R, Johnson BH, Thompson EB. Overview of the Structural Basis for Transcription Regulation by Nuclear Hormone Receptors. Essays Biochem (2004) 40:27–39. doi: 10.1042/bse0400027
6. Yasar P, Ayaz G, User SD, Gupur G, Muyan M. Molecular Mechanism of Estrogen-Estrogen Receptor Signaling. Reprod Med Biol (2017) 16:4–20. doi: 10.1002/rmb2.12006
7. Arao Y, Hamilton KJ, Coons LA, Korach KS. Estrogen Receptor Alpha L543A,L544A Mutation Changes Antagonists to Agonists, Correlating With the Ligand Binding Domain Dimerization Associated With DNA Binding Activity. J Biol Chem (2013) 288:21105–16. doi: 10.1074/jbc.M113.463455
8. Koide A, Zhao C, Naganuma M, Abrams J, Deighton-Collins S, Skafar DF, et al. Identification of Regions Within the F Domain of the Human Estrogen Receptor Alpha That are Important for Modulating Transactivation and Protein-Protein Interactions. Mol Endocrinol (2007) 21:829–42. doi: 10.1210/me.2006-0203
9. Montano MM, Muller V, Trobaugh A, Katzenellenbogen BS. The Carboxy-Terminal F Domain of the Human Estrogen Receptor: Role in the Transcriptional Activity of the Receptor and the Effectiveness of Antiestrogens as Estrogen Antagonists. Mol Endocrinol (1995) 9:814–25. doi: 10.1210/mend.9.7.7476965
10. Paech K, Webb P, Kuiper GGJM, Nilsson S, Gustafsson JA, Kushner PJ, et al. Differential Ligand Activation of Estrogen Receptors ER Alpha and ER Beta at AP1 Sites. Science (1997) 277:1508–10. doi: 10.1126/science.277.5331.1508
11. Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue Distribution and Quantitative Analysis of Estrogen Receptor-Alpha (ER Alpha) and Estrogen Receptor-Beta (ER Beta) Messenger Ribonucleic Acid in the Wild-Type and ER Alpha-Knockout Mouse. Endocrinology (1997) 138:4613–21. doi: 10.1210/en.138.11.4613
12. Li X, Huang J, Fluharty BR, Huang Y, Nott SL, Muyan M. What are Comparative Studies Telling Us About the Mechanism of ERbeta Action in the ERE-Dependent E2 Signaling Pathway? J Steroid Biochem Mol Biol (2008) 109:266–72. doi: 10.1016/j.jsbmb.2008.03.001
13. Huang J, Li X, Qiao T, Bambara RA, Hilf R, Muyan MA. Tale of Two Estrogen Receptors (ERs): How Differential ER-Estrogen Responsive Element Interactions Contribute to Subtype-Specific Transcriptional Responses. Nucl Recept Signal (2006) 4:e015. doi: 10.1621/nrs.04015
14. Billon-Gales A, Krust A, Fontaine C, Abot A, Flouriot G, Toutain C, et al. Activation Function 2 (AF2) of Estrogen Receptor-Alpha is Required for the Atheroprotective Action of Estradiol But Not to Accelerate Endothelial Healing. Proc Natl Acad Sci U.S.A. (2011) 108:13311–6. doi: 10.1073/pnas.1105632108
15. Arao Y, Hamilton KJ, Ray MK, Scott G, Mishina Y, Korach KS. Estrogen Receptor Alpha AF-2 Mutation Results in Antagonist Reversal and Reveals Tissue Selective Function of Estrogen Receptor Modulators. Proc Natl Acad Sci U.S.A. (2011) 108:14986–91. doi: 10.1073/pnas.1109180108
16. Li X, Huang J, Yi P, Bambara RA, Hilf R, Muyan M. Single-Chain Estrogen Receptors (ERs) Reveal That the ERalpha/beta Heterodimer Emulates Functions of the ERalpha Dimer in Genomic Estrogen Signaling Pathways. Mol Cell Biol (2004) 24:7681–94. doi: 10.1128/MCB.24.17.7681-7694.2004
17. Yi P, Bhagat S, Hilf R, Bambara RA, Muyan M. Differences in the Abilities of Estrogen Receptors to Integrate Activation Functions Are Critical for Subtype-Specific Transcriptional Responses. Mol Endocrinol (2002) 16:1810–27. doi: 10.1210/me.2001-0323
18. Huang J, Li X, Maguire CA, Hilf R, Bambara RA, Muyan M. Binding of Estrogen Receptor β to Estrogen Response Element in Situ Is Independent of Estradiol and Impaired by Its Amino Terminus. Mol Endocrinol (2005) 19:2696–712. doi: 10.1210/me.2005-0120
19. McDonnell DP, Norris JD. Connections and Regulation of the Human Estrogen Receptor. Science (2002) 296:1642–4. doi: 10.1126/science.1071884
20. Ray A, Prefontaine KE, Ray P. Down-Modulation of Interleukin-6 Gene-Expression by 17-Beta-Estradiol in the Absence of High-Affinity DNA-Binding by the Estrogen-Receptor. J Biol Chem (1994) 269:12940–6. doi: 10.1016/S0021-9258(18)99966-7
21. Karin M, Liu Z, Zandi E. AP-1 Function and Regulation. Curr Opin Cell Biol (1997) 9:240–6. doi: 10.1016/s0955-0674(97)80068-3
22. Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, et al. The Estrogen Receptor Enhances AP-1 Activity by Two Distinct Mechanisms With Different Requirements for Receptor Transactivation Functions. Mol Endocrinol (1999) 13:1672–85. doi: 10.1210/mend.13.10.0357
23. Teyssier C, Belguise K, Galtier F, Chalbos D. Characterization of the Physical Interaction Between Estrogen Receptor α and JUN Proteins. J Biol Chem (2001) 276:36361–9. doi: 10.1074/jbc.M101806200
24. Cheung E, Acevedo ML, Cole PA, Kraus WL. Altered Pharmacology and Distinct Coactivator Usage for Estrogen Receptor-Dependent Transcription Through Activating Protein-1. Proc Natl Acad Sci U.S.A. (2005) 102:559–64. doi: 10.1073/pnas.0407113102
25. Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, et al. Ligand-, Cell-, and Estrogen Receptor Subtype (α/β)-Dependent Activation at GC-Rich (Sp1) Promoter Elements. J Biol Chem (2000) 275:5379–87. doi: 10.1074/jbc.275.8.5379
26. Aronica SM, Kraus WL, Katzenellenbogen BS. Estrogen Action via the cAMP Signaling Pathway: Stimulation of Adenylate Cyclase and cAMP-Regulated Gene Transcription. Proc Natl Acad Sci U.S.A. (1994) 91:8517–21. doi: 10.1073/pnas.91.18.8517
27. Tesarik J, Mendoza C. Direct non-Genomic Effects of Follicular Steroids on Maturing Human Oocytes: Oestrogen Versus Androgen Antagonism. Hum Reprod Update (1997) 3:95–100. doi: 10.1093/humupd/3.2.95
28. Morley P, Whitfield JF, Vanderhyden BC, Tsang BK, Schwartz J.-L.J.E. A. New, Nongenomic Estrogen Action: The Rapid Release of Intracellular Calcium. Endocrinology (1992) 131:1305–12. doi: 10.1210/endo.131.3.1505465
29. Moriarty K, Kim K, Bender J. Estrogen Receptor-Mediated Rapid Signaling. Endocrinology (2006) 147:5557–63. doi: 10.1210/en.2006-0729
30. Micevych P, Kuo J, Christensen A. Physiology of Membrane Oestrogen Receptor Signalling in Reproduction. J Neuroendocrinol (2009) 21:249–56. doi: 10.1111/j.1365-2826.2009.01833.x
31. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science (2005) 307:1625–30. doi: 10.1126/science.1106943
32. Rainville J, Pollard K, Vasudevan N. Membrane-Initiated non-Genomic Signaling by Estrogens in the Hypothalamus: Cross-Talk With Glucocorticoids With Implications for Behavior. Front Endocrinol (Lausanne) (2015) 6:18. doi: 10.3389/fendo.2015.00018
33. Smith A, Rønnekleiv O, Kelly MJS. Gq-mER Signaling has Opposite Effects on Hypothalamic Orexigenic and Anorexigenic Neurons. Steroids (2014) 81:31–5. doi: 10.1016/j.steroids.2013.11.007
34. Kelly MJ, Levin ER. Rapid Actions of Plasma Membrane Estrogen Receptors. Trends Endocrinol Metab (2001) 12:152–6. doi: 10.1016/s1043-2760(01)00377-0
35. Filardo EJ, Quinn JA, Frackelton AR Jr., Bland KI. Estrogen Action via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and cAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-To-MAPK Signaling Axis. Mol Endocrinol (2002) 16:70–84. doi: 10.1210/mend.16.1.0758
36. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol (2000) 14:1649–60. doi: 10.1210/mend.14.10.0532
37. Prossnitz ER, Barton M. Estrogen Biology: New Insights Into GPER Function and Clinical Opportunities. Mol Cell Endocrinol (2014) 389:71–83. doi: 10.1016/j.mce.2014.02.002
38. Meyer MR, Haas E, Prossnitz ER, Barton M. Non-Genomic Regulation of Vascular Cell Function and Growth by Estrogen. Mol Cell Endocrinol (2009) 308:9–16. doi: 10.1016/j.mce.2009.03.009
39. Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, et al. Identification of a GPER/GPR30 Antagonist With Improved Estrogen Receptor Counterselectivity. J Steroid Biochem Mol Biol (2011) 127:358–66. doi: 10.1016/j.jsbmb.2011.07.002
40. Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. In Vivo Effects of a GPR30 Antagonist. Nat Chem Biol (2009) 5:421–7. doi: 10.1038/nchembio.168
41. Jacenik D, Cygankiewicz AI, Krajewska WM. The G Protein-Coupled Estrogen Receptor as a Modulator of Neoplastic Transformation. Mol Cell Endocrinol (2016) 429:10–8. doi: 10.1016/j.mce.2016.04.011
42. Lonard DM, O’Malley B W. Nuclear Receptor Coregulators: Judges, Juries, and Executioners of Cellular Regulation. Mol Cell (2007) 27:691–700. doi: 10.1016/j.molcel.2007.08.012
43. Fuentes N, Silveyra P. Estrogen Receptor Signaling Mechanisms. Adv Protein Chem Struct Biol (2019) 116:135–70. doi: 10.1016/bs.apcsb.2019.01.001
44. Oñate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and Characterization of a Coactivator for the Steroid Hormone Receptor Superfamily. Science (1995) 270:1354–7. doi: 10.1126/science.270.5240.1354
45. Lonard DM, O’Malley BW. The Expanding Cosmos of Nuclear Receptor Coactivators. Cell (2006) 125:411–4. doi: 10.1016/j.cell.2006.04.021
46. Chen JD, Evans RM. A Transcriptional Co-Repressor That Interacts With Nuclear Hormone Receptors. Nature (1995) 377:454–7. doi: 10.1038/377454a0
47. Wong MM, Guo C, Zhang J. Nuclear Receptor Corepressor Complexes in Cancer: Mechanism, Function and Regulation. Am J Clin Exp Urol (2014) 2:169–87.
48. Manavathi B, Samanthapudi VS, Gajulapalli VN. Estrogen Receptor Coregulators and Pioneer Factors: The Orchestrators of Mammary Gland Cell Fate and Development. Front Cell Dev Biol (2014) 2:34. doi: 10.3389/fcell.2014.00034
49. Hu X, Lazar MA. The CoRNR Motif Controls the Recruitment of Corepressors by Nuclear Hormone Receptors. Nature (1999) 402:93–6. doi: 10.1038/47069
50. Han SJ, Lonard DM, O’Malley BW. Multi-Modulation of Nuclear Receptor Coactivators Through Posttranslational Modifications. Trends Endocrinol Metab (2009) 20:8–15. doi: 10.1016/j.tem.2008.10.001
51. O’Malley BW, McKenna NJ. Coactivators and Corepressors: What’s in a Name? Mol Endocrinol (2008) 22:2213–4. doi: 10.1210/me.2008-0201
52. Groothuis PG, Dassen HHNM, Romano A, Punyadeera C. Estrogen and the Endometrium: Lessons Learned From Gene Expression Profiling in Rodents and Human. Hum Reprod Update (2007) 13:405–17. doi: 10.1093/humupd/dmm009
53. Persson I. Cancer Risk in Women Receiving Estrogen-Progestin Replacement Therapy. Maturitas (1996) 23:S37–45. doi: 10.1016/0378-5122(96)01010-9
54. Vollmer G. Endometrial Cancer: Experimental Models Useful for Studies on Molecular Aspects of Endometrial Cancer and Carcinogenesis. Endocr-Relat Cancer (2003) 10:23–42. doi: 10.1677/erc.0.0100023
55. Newbold RR, Bullock BC, Mclachlan JA. Uterine Adenocarcinoma in Mice Following Developmental Treatment With Estrogens - A Model for Hormonal Carcinogenesis. Cancer Res (1990) 50:7677–81.
56. Li SF. Relationship Between Cellular DNA-Synthesis, Pcna Expression and Sex Steroid-Hormone Receptor Status in the Developing Mouse Ovary, Uterus and Oviduct. Histochemistry (1994) 102:405–13. doi: 10.1007/Bf00268912
57. Andrews WW, Mizejewski GJ, Ojeda SR. Development of Estradiol-Positive Feedback on Luteinizing-Hormone Release in the Female Rat - a Quantitative Study. Endocrinology (1981) 109:1404–13. doi: 10.1210/endo-109-5-1404
58. Meijsroelofs HMA, Uilenbroek JTJ, Degreef WJ, Dejong FH, Kramer P. Gonadotropin and Steroid Levels Around Time of 1st Ovulation in Rat. J Endocrinol (1975) 67:275–82. doi: 10.1677/joe.0.0670275
59. Critchley HOD, Brenner RM, Henderson TA, Williams K, Nayak NR, Slayden OD, et al. Estrogen Receptor Beta, But Not Estrogen Receptor Alpha, is Present in the Vascular Endothelium of the Human and Nonhuman Primate Endometrium. J Clin Endocr Metab (2001) 86:1370–8. doi: 10.1210/jc.86.3.1370
60. Lecce G, Meduri G, Ancelin M, Bergeron C, Perrot-Applanat M. Presence of Estrogen Receptor Beta in the Human Endometrium Through the Cycle: Expression in Glandular, Stromal, and Vascular Cells. J Clin Endocr Metab (2001) 86:1379–86. doi: 10.1210/jc.86.3.1379
61. Perrotapplanat M, Groyerpicard MT, Garcia E, Lorenzo F, Milgrom E. Immunocytochemical Demonstration of Estrogen and Progesterone Receptors in Muscle-Cells of Uterine Arteries in Rabbits and Humans. Endocrinology (1988) 123:1511–9. doi: 10.1210/endo-123-3-1511
62. Kolkova Z, Noskova V, Ehinger A, Hansson S, Casslen BG. Protein-Coupled Estrogen Receptor 1 (GPER, GPR 30) in Normal Human Endometrium and Early Pregnancy Decidua(Dagger). Mol Hum Reprod (2010) 16:743–51. doi: 10.1093/molehr/gaq043
63. Snijders MPML, Degoeij AFPM, Debetstebaerts MJC, Rousch MJM, Koudstaal J, Bosman FT. Immunocytochemical Analysis of Estrogen-Receptors and Progesterone Receptors in the Human Uterus Throughout the Menstrual-Cycle and After the Menopause. J Reprod Fertil (1992) 94:363–71. doi: 10.1530/jrf.0.0940363
64. Matsuzaki S, Fukaya T, Suzuki T, Murakami T, Sasano H, Yajima A. Oestrogen Receptor Alpha and Beta mRNA Expression in Human Endometrium Throughout the Menstrual Cycle. Mol Hum Reprod (1999) 5:559–64. doi: 10.1093/molehr/5.6.559
65. Maiti K, Paul JW, Read M, Chan EC, Riley SC, Nahar P. Smith, R. G-1-Activated Membrane Estrogen Receptors Mediate Increased Contractility of the Human Myometrium. Endocrinology (2011) 152:2448–55. doi: 10.1210/en.2010-0979
66. Matsuzaki S, Murakami T, Uehara S, Canis M, Sasano H, Okamura K. Expression of Estrogen Receptor Alpha and Beta in Peritoneal and Ovarian Endometriosis. Fertil Steril (2001) 75:1198–205. doi: 10.1016/S0015-0282(01)01783-6
67. Sakaguchi H, Fujimoto J, Aoki I, Tamaya T. Expression of Estrogen Receptor Alpha and Beta in Myometrium of Premenopausal and Postmenopausal Women. Steroids (2003) 68:11–9. doi: 10.1016/S0039-128x(02)00111-3
68. Zang H, Sahlin L, Masironi B, Hirschberg AL. Effects of Testosterone and Estrogen Treatment on the Distribution of Sex Hormone Receptors in the Endometrium of Postmenopausal Women. Menopause (2008) 15:233–9. doi: 10.1097/gme.0b013e318148bb99
69. Gao F, Ma XH, Ostmann AB, Das SK. GPR30 Activation Opposes Estrogen-Dependent Uterine Growth via Inhibition of Stromal ERK1/2 and Estrogen Receptor Alpha (ER Alpha) Phosphorylation Signals. Endocrinology (2011) 152:1434–47. doi: 10.1210/en.2010-1368
70. Yuguchi H, Tanabe A, Hayashi A, Tanaka Y, Okuda K, Yamashita Y, et al. The Expression Status of G Protein-Coupled Receptor GPR30 Is Associated With the Clinical Characteristics of Endometriosis. Endocr Res (2013) 38:223–31. doi: 10.3109/07435800.2013.774011
71. Plante BJ, Lessey BA, Taylor RN, Wang W, Bagchi MK, Yuan LW, et al. Protein-Coupled Estrogen Receptor (GPER) Expression in Normal and Abnormal Endometrium. Reprod Sci (2012) 19:684–93. doi: 10.1177/1933719111431000
72. Couse JF, Curtis SW, Washburn TF, Lindzey J, Lindzey J Fau - Golding TS, Golding Ts Fau - Lubahn DB, et al. Analysis of Transcription and Estrogen Insensitivity in the Female Mouse After Targeted Disruption of the Estrogen Receptor Gene. Mol Endocrinol (1995) 9:1441–54. doi: 10.1210/mend.9.11.8584021
73. Curtis SW, Clark J, Myers P, Korach KS. Disruption of Estrogen Signaling Does Not Prevent Progesterone Action in the Estrogen Receptor or Knockout Mouse Uterus. P Natl Acad Sci USA (1999) 96:3646–51. doi: 10.1073/pnas.96.7.3646
74. Zhang WH, Saji S, Makinen S, Cheng GJ, Jensen EV, Warner M, et al. Estrogen Receptor (ER) Beta, a Modulator of ER Alpha in the Uterus. P Natl Acad Sci USA (2000) 97:5936–41. doi: 10.1073/pnas.97.11.5936
75. Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of Single and Compound Knockouts of Estrogen Receptors Alpha (ER Alpha) and Beta (ER Beta) on Mouse Reproductive Phenotypes. Development (2000) 127:4277–91. doi: 10.1242/dev.127.19.4277
76. Jakacka M, Ito M, Martinson F, Ishikawa T, Lee EJ, Jameson JL. An Estrogen Receptor (ER)alpha Deoxyribonucleic Acid-Binding Domain Knock-In Mutation Provides Evidence for Nonclassical ER Pathway Signaling In Vivo. Mol Endocrinol (2002) 16:2188–201. doi: 10.1210/me.2001-0174
77. O’Brien JE, Peterson TJ, Tong MH, Lee EJ, Pfaff LE, Hewitt SC, et al. Estrogen-Induced Proliferation of Uterine Epithelial Cells is Independent of Estrogen Receptor Alpha Binding to Classical Estrogen Response Elements. J Biol Chem (2006) 281:26683–92. doi: 10.1074/jbc.M601522200
78. Frasor J, Barnett DH, Danes JM, Hess R, Parlow AF, Katzenellenbogen BS. Response-Specific and Ligand Dose-Dependent Modulation of Estrogen Receptor (ER) Alpha Activity by ER Beta in the Uterus. Endocrinology (2003) 144:3159–66. doi: 10.1210/en.2002-0143
79. Wada-Hiraike O, Hiraike H, Okinaga H, Imamov O, Barros RPA, Morani A, et al. Role of Estrogen Receptor Beta in Uterine Stroma and Epithelium: Insights From Estrogen Receptor Beta(-/-) Mice. P Natl Acad Sci USA (2006) 103:18350–5. doi: 10.1073/pnas.0608861103
80. Hewitt SC, Li L, Grimm SA, Winuthayanon W, Hamilton KJ, Pockette B, et al. Novel DNA Motif Binding Activity Observed In Vivo With an Estrogen Receptor Alpha Mutant Mouse. Mol Endocrinol (2014) 28:899–911. doi: 10.1210/me.2014-1051
81. Ahlbory-Dieker DL, Stride BD, Leder G, Schkoldow J, Trolenberg S, Seidel H, et al. DNA Binding by Estrogen Receptor-Alpha Is Essential for the Transcriptional Response to Estrogen in the Liver and the Uterus. Mol Endocrinol (2009) 23:1544–55. doi: 10.1210/me.2009-0045
82. Winuthayanon W, Hewitt SC, Orvis GD, Behringer RR, Korach KS. Uterine Epithelial Estrogen Receptor Alpha is Dispensable for Proliferation But Essential for Complete Biological and Biochemical Responses. P Natl Acad Sci USA (2010) 107:19272–7. doi: 10.1073/pnas.1013226107
83. Abot A, Fontaine C, Raymond-Letron I, Flouriot G, Adlanmerini M, Buscato M, et al. The AF-1 Activation Function of Estrogen Receptor Alpha Is Necessary and Sufficient for Uterine Epithelial Cell Proliferation In Vivo. Endocrinology (2013) 154:2222–33. doi: 10.1210/en.2012-2059
84. Isensee J, Meoli L, Zazzu V, Nabzdyk C, Witt H, Soewarto D, et al. Expression Pattern of G Protein-Coupled Receptor 30 in LacZ Reporter Mice. Endocrinology (2009) 150:1722–30. doi: 10.1210/en.2008-1488
85. Hewitt SC, Harrell JC, Korach KS. Lessons in Estrogen Biology From Knockout and Transgenic Animals. Annu Rev Physiol (2005) 67:285–308. doi: 10.1146/annurev.physiol.67.040403.115914
86. Otto C, Fuchs I, Kauselmann G, Kern H, Zevnik B, Andreasen P, et al. GPR30 Does Not Mediate Estrogenic Responses in Reproductive Organs in Mice. Biol Reprod (2009) 80:34–41. doi: 10.1095/biolreprod.108.071175
87. Li YR, Jia YH, Bian YD, Tong H, Qu JJ, Wang K, et al. Autocrine Motility Factor Promotes Endometrial Cancer Progression by Targeting GPER-1. Cell Commun Signal (2019) 17:22. doi: 10.1186/s12964-019-0336-4
88. Bulletti C, Coccia ME, Battistoni S, Borini A. Endometriosis and Infertility. J Assist Reprod Genet (2010) 27:441–7. doi: 10.1007/s10815-010-9436-1
89. Giudice LC. Endometrium in PCOS: Implantation and Predisposition to Endocrine CA. Best Pract Res Clin Endocrinol Metab (2006) 20:235–44. doi: 10.1016/j.beem.2006.03.005
90. Shang K, Jia X, Qiao J, Kang J, Guan Y. Endometrial Abnormality in Women With Polycystic Ovary Syndrome. Reprod Sci (2012) 19:674–83. doi: 10.1177/1933719111430993
91. Delvoux B, Groothuis P, D’Hooghe T, Kyama C, Dunselman G, Romano A. Increased Production of 17β-Estradiol in Endometriosis Lesions is the Result of Impaired Metabolism. J Clin Endocrinol Metab (2009) 94:876–83. doi: 10.1210/jc.2008-2218
92. Zhang H, Zhao X, Liu S, Li J, Wen Z, Li M. 17βe2 Promotes Cell Proliferation in Endometriosis by Decreasing PTEN via Nfκb-Dependent Pathway. Mol Cell Endocrinol (2010) 317:31–43. doi: 10.1016/j.mce.2009.11.009
93. Tang ZR, Zhang R, Lian ZX, Deng SL, Yu K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells (2019) 8:1123. doi: 10.3390/cells8101123
94. Xu Z, Zhang L, Yu Q, Zhang Y, Yan L, Chen Z-J. The Estrogen-Regulated lncRNA H19/miR-216a-5p Axis Alters Stromal Cell Invasion and Migration via ACTA2 in Endometriosis. Mol Hum Reprod (2019) 25:550–61. doi: 10.1093/molehr/gaz040
95. Lin BC, Suzawa M, Blind RD, Tobias SC, Bulun SE, Scanlan TS, et al. Stimulating the GPR30 Estrogen Receptor With a Novel Tamoxifen Analogue Activates SF-1 and Promotes Endometrial Cell Proliferation. Cancer Res (2009) 69:5415–23. doi: 10.1158/0008-5472.Can-08-1622
96. Lin SL, Tu BB, Du XG, Yan LY, Qiao J. Lower Expression of ER-Alpha36 is Associated With the Development of Endometrial Hyperplasia in PCOS Patients. Histol Histopathol (2013) 28:1491–8. doi: 10.14670/HH-28.1491
97. Artimani T, Saidijam M, Aflatoonian R, Amiri I, Ashrafi M, Shabab N, et al. Estrogen and Progesterone Receptor Subtype Expression in Granulosa Cells From Women With Polycystic Ovary Syndrome. Gynecol Endocrinol (2015) 31:379–83. doi: 10.3109/09513590.2014.1001733
98. Bulun SE, Monsavais D, Pavone ME, Dyson M, Xue Q, Attar E, et al. Role of Estrogen Receptor-Beta in Endometriosis. Semin Reprod Med (2012) 30:39–45. doi: 10.1055/s-0031-1299596
99. Han SJ, Jung SY, Wu S-P, Hawkins SM, Park MJ, Kyo S, et al. Estrogen Receptor β Modulates Apoptosis Complexes and the Inflammasome to Drive the Pathogenesis of Endometriosis. Cell (2015) 163:960–74. doi: 10.1016/j.cell.2015.10.034
100. Monsivais D, Dyson MT, Yin P, Coon JS, Navarro A, Feng G, et al. ERbeta- and Prostaglandin E2-Regulated Pathways Integrate Cell Proliferation via Ras-Like and Estrogen-Regulated Growth Inhibitor in Endometriosis. Mol Endocrinol (2014) 28:1304–15. doi: 10.1210/me.2013-1421
101. Cheng CW, Licence D, Cook E, Luo F, Arends MJ, Smith SK, et al. Activation of Mutated K-Ras in Donor Endometrial Epithelium and Stroma Promotes Lesion Growth in an Intact Immunocompetent Murine Model of Endometriosis. J Pathol (2011) 224:261–9. doi: 10.1002/path.2852
102. Han SJ, O’Malley BW, DeMayo FJ. An Estrogen Receptor Alpha Activity Indicator Model in Mice. Genesis (2009) 47:815–24. doi: 10.1002/dvg.20572
103. Burns KA, Rodriguez KF, Hewitt SC, Janardhan KS, Young SL, Korach KS. Role of Estrogen Receptor Signaling Required for Endometriosis-Like Lesion Establishment in a Mouse Model. Endocrinology (2012) 153:3960–71. doi: 10.1210/en.2012-1294
104. Hapangama D, Kamal A, Bulmer J. Estrogen Receptor β: The Guardian of the Endometrium. Hum Reprod Update (2014) 21:174–93. doi: 10.1093/humupd/dmu053
105. Hu K, Zhong G, He F. Expression of Estrogen Receptors ER Alpha and ER Beta in Endometrial Hyperplasia and Adenocarcinoma. Int J Gynecol Cancer (2005) 15:537–41. doi: 10.1111/j.1525-1438.2005.15321.x
106. Uchikawa J, Shiozawa T, Shih HC, Miyamoto T, Feng YZ, Kashima H, et al. Expression of Steroid Receptor Coactivators and Corepressors in Human Endometrial Hyperplasia and Carcinoma With Relevance to Steroid Receptors and Ki-67 Expression. Cancer (2003) 98:2207–13. doi: 10.1002/cncr.11760
107. Bircan S, Ensari A, Ozturk S, Erdogan N, Dundar I, Ortac F. Immunohistochemical Analysis of C-Myc, C-Jun and Estrogen Receptor in Normal, Hyperplastic and Neoplastic Endometrium. Pathol Oncol Res (2005) 11:32–9. doi: 10.1007/Bf03032403
108. Wang YL, Miksicek RJ. Identification of a Dominant Negative Form of the Human Estrogen-Receptor. Mol Endocrinol (1991) 5:1707–15. doi: 10.1210/mend-5-11-1707
109. Zhang QX, Hilsenbeck SG, Fuqua SAW, Borg A. Multiple Splicing Variants of the Estrogen Receptor are Present in Individual Human Breast Turners. J Steroid Biochem (1996) 59:251–60. doi: 10.1016/S0960-0760(96)00120-3
110. Horvath G, Leser G, Hahlin M, Henriksson M. Exon Deletions and Variants of Human Estrogen Receptor mRNA in Endometrial Hyperplasia and Adenocarcinoma. Int J Gynecol Cancer (2000) 10:128–36. doi: 10.1046/j.1525-1438.2000.00009.x
111. Chaidarun SS, Klibanski A, Alexander JM. Tumor-Specific Expression of Alternatively Spliced Estrogen Receptor Messenger Ribonucleic Acid Variants in Human Pituitary Adenomas. J Clin Endocr Metab (1997) 82:1058–65. doi: 10.1210/jc.82.4.1058
112. Herynk MH, Fuqua SAW. Estrogen Receptor Mutations in Human Disease. Endocr Rev (2004) 25:869–98. doi: 10.1210/er.2003-0010
113. Levin ER. Plasma Membrane Estrogen Receptors. Trends Endocrin Met (2009) 20:477–82. doi: 10.1016/j.tem.2009.06.009
114. Brosens J, Verhoeven H, Campo R, Gianaroli L, Gordts S, Hazekamp J, et al. High Endometrial Aromatase P450 mRNA Expression is Associated With Poor IVF Outcome. Hum Reprod (2004) 19:352–6. doi: 10.1093/humrep/deh075
115. Bulun SE, Fang ZJ, Imir G, Gurates B, Tamura M, Yilmaz B, et al. Aromatase and Endometriosis. Semin Reprod Med (2004) 22:45–50. doi: 10.1055/s-2004-823026
116. Sanderson PA, Critchley HOD, Williams ARW, Arends MJ, Saunders PTK. New Concepts for an Old Problem: The Diagnosis of Endometrial Hyperplasia. Hum Reprod Update (2017) 23:232–54. doi: 10.1093/humupd/dmw042
117. Smith HO, Leslie KK, Singh M, Qualls CR, Revankar CM, Joste NE, et al. GPR30: A Novel Indicator of Poor Survival for Endometrial Carcinoma. Am J Obstet Gynecol (2007) 196:386.e1–11. doi: 10.1016/j.ajog.2007.01.004
118. He YY, Cai B, Yang YX, Liu XL, Wan XP. Estrogenic G Protein-Coupled Receptor 30 Signaling is Involved in Regulation of Endometrial Carcinoma by Promoting Proliferation, Invasion Potential, and Interleukin-6 Secretion via the MEK/ERK Mitogen-Activated Protein Kinase Pathway. Cancer Sci (2009) 100:1051–61. doi: 10.1111/j.1349-7006.2009.01148.x
119. Krakstad C, Trovik J, Wik E, Engelsen IB, Werner HMJ, Birkeland E, et al. Loss of GPER Identifies New Targets for Therapy Among a Subgroup of ER Alpha-Positive Endometrial Cancer Patients With Poor Outcome. Brit J Cancer (2012) 106:1682–8. doi: 10.1038/bjc.2012.91
120. Skrzypczak M, Schuller S, Lattrich C, Ignatov A, Ortmann O, Treeck OG. Protein-Coupled Estrogen Receptor (GPER) Expression in Endometrial Adenocarcinoma and Effect of Agonist G-1 on Growth of Endometrial Adenocarcinoma Cell Lines. Steroids (2013) 78:1087–91. doi: 10.1016/j.steroids.2013.07.007
121. Paul M, Cholewa K, Mazurek U, Witek A, Wilczok T. Estrogen Receptor Beta Delta 6 (ER Beta Delta 6) Isoform in Human Endometrial Hyperplasia and Adenocarcinoma. Cancer Invest (2004) 22:211–8. doi: 10.1081/Cnv-120030209
122. Smuc T, Rizner TL. Aberrant Pre-Receptor Regulation of Estrogen and Progesterone Action in Endometrial Cancer. Mol Cell Endocrinol (2009) 301:74–82. doi: 10.1016/j.mce.2008.09.019
123. Skrzypczak M, Bieche I, Szymczak S, Tozlu S, Lewandowski S, Girault I, et al. Evaluation of mRNA Expression of Estrogen Receptor Beta and its Isoforms in Human Normal and Neoplastic Endometrium. Int J Cancer (2004) 110:783–7. doi: 10.1002/ijc.20224
124. Haring J, Skrzypczak M, Stegerer A, Lattrich C, Weber F, Gorse R, et al. Estrogen Receptor Beta Transcript Variants Associate With Oncogene Expression in Endometrial Cancer. Int J Mol Med (2012) 29:1127–36. doi: 10.3892/ijmm.2012.929
125. Rodriguez AC, Blanchard Z, Maurer KA, Gertz J. Estrogen Signaling in Endometrial Cancer: A Key Oncogenic Pathway With Several Open Questions. Horm Cancer-Us (2019) 10:51–63. doi: 10.1007/s12672-019-0358-9
126. Byers M, Kuiper GGJM, Gustafsson JA, ParkSarge OK. Estrogen Receptor-Beta mRNA Expression in Rat Ovary: Down-Regulation by Gonadotropins. Mol Endocrinol (1997) 11:172–82. doi: 10.1210/mend.11.2.9887
127. Hu GL, Zhang JB, Zhou XY, Liu JW, Wang Q, Zhang B. Roles of Estrogen Receptor Alpha and Beta in the Regulation of Proliferation in Endometrial Carcinoma. Pathol Res Pract (2020) 216:153149. doi: 10.1016/j.prp.2020.153149
128. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-Of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors From 33 Types of Cancer. Cell (2018) 173:291–304. doi: 10.1016/j.cell.2018.03.022
129. Backes FJ, Walker CJ, Goodfellow PJ, Hade EM, Agarwal G, Mutch D, et al. Estrogen Receptor-Alpha as a Predictive Biomarker in Endometrioid Endometrial Cancer. Gynecol Oncol (2016) 141:312–7. doi: 10.1016/j.ygyno.2016.03.006
130. Blanchard Z, Vahrenkamp JM, Berrett KC, Arnesen S, Gertz J. Estrogen-Independent Molecular Actions of Mutant Estrogen Receptor 1 in Endometrial Cancer. Genome Res (2019) 29:1429–41. doi: 10.1101/gr.244780.118
131. Gertz J, Savic D, Varley KE, Partridge EC, Safi A, Jain P, et al. Distinct Properties of Cell-Type-Specific and Shared Transcription Factor Binding Sites. Mol Cell (2013) 52:25–36. doi: 10.1016/j.molcel.2013.08.037
132. Rodriguez AC, Vahrenkamp JM, Berrett KC, Clark KA, Guillen KP, Scherer SD, et al. ETV4 Is Necessary for Estrogen Signaling and Growth in Endometrial Cancer Cells. Cancer Res (2020) 80:1234–45. doi: 10.1158/0008-5472.can-19-1382
133. Qi Y, Tan M, Zheng M, Jin S, Wang H, Liu J, et al. Estrogen/estrogen Receptor Promotes the Proliferation of Endometrial Carcinoma Cells by Enhancing hMOF Expression. Jpn J Clin Oncol (2020) 50:241–53. doi: 10.1093/jjco/hyz174
134. Judd HL, Mebane-Sims I, Legault C, Wasilauskas C, Johnson S, Merino M. Effects of Hormone Replacement Therapy on Endometrial Histology in Postmenopausal Women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. The Writing Group for the PEPI Trial. Jama (1996) 275:370–5. doi: 10.1001/jama.1996.03530290040035
135. D’Alonzo M, Bounous VE, Villa M, Biglia N. Current Evidence of the Oncological Benefit-Risk Profile of Hormone Replacement Therapy. Med (Kaunas Lithuania) (2019) 55:573. doi: 10.3390/medicina55090573
136. Utian WH, Speroff L, Ellman H, Dart C. Comparative Controlled Trial of a Novel Oral Estrogen Therapy, Estradiol Acetate, for Relief of Menopause Symptoms. Menopause (2005) 12:708–15. doi: 10.1097/01.gme.0000184220.63459.a8
137. Marjoribanks J, Farquhar C, Roberts H, Lethaby A, Lee J. Long-Term Hormone Therapy for Perimenopausal and Postmenopausal Women. Cochrane Database Syst Rev (2017) 1:CD004143. doi: 10.1002/14651858.CD004143.pub5
138. Vajdos FF, Hoth LR, Geoghegan KF, Simons SP, LeMotte PK, Danley DE, et al. The 2.0 A Crystal Structure of the ERalpha Ligand-Binding Domain Complexed With Lasofoxifene. Protein Sci (2007) 16:897–905. doi: 10.1110/ps.062729207
139. Shang Y. Molecular Mechanisms of Oestrogen and SERMs in Endometrial Carcinogenesis. Nat Rev Cancer (2006) 6:360–8. doi: 10.1038/nrc1879
141. Deligdisch L. Hormonal Pathology of the Endometrium. Mod Pathol (2000) 13:285–94. doi: 10.1038/modpathol.3880050
142. Nijkang NP, Anderson L, Markham R, Manconi F. Endometrial Polyps: Pathogenesis, Sequelae and Treatment. SAGE Open Med (2019) 7:2050312119848247. doi: 10.1177/2050312119848247
143. Polin SA, Ascher SM. The Effect of Tamoxifen on the Genital Tract. Cancer Imaging (2008) 8:135. doi: 10.1102/1470-7330.2008.0020
144. Bai JX, Yan B, Zhao ZN, Xiao X, Qin WW, Zhang R, et al. Tamoxifen Represses miR-200 microRNAs and Promotes Epithelial-To-Mesenchymal Transition by Up-Regulating C-Myc in Endometrial Carcinoma Cell Lines. Endocrinology (2013) 154:635–45. doi: 10.1210/en.2012-1607
145. Tergas AI, Buell-Gutbrod R, Gwin K, Kocherginsky M, Temkin SM, Fefferman A, et al. Clinico-Pathologic Comparison of Type II Endometrial Cancers Based on Tamoxifen Exposure. Gynecol Oncol (2012) 127:316–20. doi: 10.1016/j.ygyno.2012.07.105
146. Fornander T, Cedermark B, Mattsson A, Skoog L, Theve T, Askergren J, et al. Adjuvant Tamoxifen in Early Breast-Cancer - Occurrence of New Primary Cancers. Lancet (1989) 1:117–20. doi: 10.1016/s0140-6736(89)91141-0
147. Fisher B, Costantino JP, Redmond CK, Fisher ER, Wickerham DL, Cronin WM, et al. Endometrial Cancer in Tamoxifen-Treated Breast-Cancer Patients - Findings From the National Surgical Adjuvant Breast and Bowel Project (Nsabp) B-14. J Natl Cancer I (1994) 86:527–37. doi: 10.1093/jnci/86.7.527
148. Ignatov T, Eggemann H, Semczuk A, Smith B, Bischoff J, Roessner A, et al. Role of GPR30 in Endometrial Pathology After Tamoxifen for Breast Cancer. Am J Obstet Gynecol (2010) 203:595.e9–16. doi: 10.1016/j.ajog.2010.07.034
149. Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular Basis of Agonism and Antagonism in the Oestrogen Receptor. Nature (1997) 389:753–8. doi: 10.1038/39645
150. Kulak J Jr., Ferriani RA, Komm BS, Taylor HS. Tissue Selective Estrogen Complexes (TSECs) Differentially Modulate Markers of Proliferation and Differentiation in Endometrial Cells. Reprod Sci (2013) 20:129–37. doi: 10.1177/1933719112463251
151. DeMichele A, Troxel AB, Berlin JA, Weber AL, Bunin GR, Turzo E, et al. Impact of Raloxifene or Tamoxifen Use on Endometrial Cancer Risk: A Population-Based Case-Control Study. J Clin Oncol (2008) 26:4151–9. doi: 10.1200/JCO.2007.14.0921
152. Carneiro ALB, Spadella APC, Souza FA, Alves KBF, Araujo-Neto JT, Haidar MA, et al. Effects of Raloxifene Combined With Low-Dose Conjugated Estrogen on the Endometrium in Menopausal Women at High Risk for Breast Cancer. Clinics (Sao Paulo Brazil) (2021) 76:e2380. doi: 10.6061/clinics/2021/e2380
153. Swisher DK, Tague RM, Seyler DE. Effect of the Selective Estrogen-Receptor Modulator Raloxifene on Explanted Uterine Growth in Rats. Drug Dev Res (1995) 36:43–5. doi: 10.1002/ddr.430360107
154. Buelke-Sam J, Bryant HU, Francis PC. The Selective Estrogen Receptor Modulator, Raloxifene: An Overview of Nonclinical Pharmacology and Reproductive and Developmental Testing. Reprod Toxicol (1998) 12:217–21. doi: 10.1016/S0890-6238(98)00003-3
155. Altintas D, Kokcu A, Kandemir B, Tosun M, Cetinkaya MB. Comparison of the Effects of Raloxifene and Anastrozole on Experimental Endometriosis. Eur J Obstet Gyn R B (2010) 150:84–7. doi: 10.1016/j.ejogrb.2010.02.004
156. Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, et al. Protein-Coupled Estrogen Receptor-Selective Ligands Modulate Endometrial Tumor Growth. Obstet Gynecol Int (2013) 2013:472720. doi: 10.1155/2013/472720
157. Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell (1998) 95:927–37. doi: 10.1016/s0092-8674(00)81717-1
158. Dauvois S, White R, Parker MG. The Antiestrogen ICI 182780 Disrupts Estrogen Receptor Nucleocytoplasmic Shuttling. J Cell Sci (1993) 106(Pt 4):1377–88. doi: 10.1242/jcs.106.4.1377
159. Berlière M, Charles A, Galant C, Donnez J. Uterine Side Effects of Tamoxifen: A Need for Systematic Pretreatment Screening. Obstet Gynecol (1998) 91:40–4. doi: 10.1016/s0029-7844(97)00591-7
Keywords: endometrium, estrogen receptor α, estrogen receptor β, G-protein-coupled estrogen receptor, human
Citation: Yu K, Huang Z-Y, Xu X-L, Li J, Fu X-W and Deng S-L (2022) Estrogen Receptor Function: Impact on the Human Endometrium. Front. Endocrinol. 13:827724. doi: 10.3389/fendo.2022.827724
Received: 02 December 2021; Accepted: 01 February 2022;
Published: 28 February 2022.
Edited by:
Shi Qinghua, University of Science and Technology of China, ChinaReviewed by:
Xin Du, The 901th Hospital of the Joint Logistics Support Force of PLA, ChinaCopyright © 2022 Yu, Huang, Xu, Li, Fu and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang-Wei Fu, ZnV4aWFuZ3dlaUBjYXUuZWR1LmNu; Shou-Long Deng, ZGVuZ3Nob3Vsb25nQGNuaWxhcy5vcmc=; Jun Li, bGlqdW44MjAzQDE2My5jb20=
†ORCID: Xiang-Wei Fu, orcid.org/0000-0003-1638-6852
Shou-Long Deng, orcid.org/0000-0003-1187-3037
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.