
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 16 February 2022
Sec. Pediatric Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.820001
This article is part of the Research Topic Controversies in Growth Hormone Treatment and Diagnosis View all 17 articles
 Meiping Chen
Meiping Chen Hui Miao
Hui Miao Hanting Liang
Hanting Liang Xiaoan Ke
Xiaoan Ke Hongbo Yang
Hongbo Yang Fengying Gong
Fengying Gong Linjie Wang
Linjie Wang Lian Duan
Lian Duan Shi Chen
Shi Chen Hui Pan
Hui Pan Huijuan Zhu*
Huijuan Zhu*Context: Clinical genetic evaluation has been demonstrated as an important tool to elucidate the causes of growth disorders. Genetic defects of collagen formation (the collagenopathies) have been reported to be associated with short stature and skeletal dysplasias. Etiological diagnosis of skeletal abnormality-related short stature is challenging, and less is known about recombinant human growth hormone (rhGH) therapy.
Objective: This is a single-center cohort study which aims at exploring the genetic architecture of short-stature children with skeletal abnormalities and evaluating the frequency of collagenopathies to determine their phenotype, including the rhGH treatment response.
Patients and Methods: One hundred and six children with short stature and skeletal abnormalities were enrolled who were evaluated by next-generation sequencing (NGS) to detect variants in the skeletal collagen genes including COL1A1, COL1A2, COL2A1, COL9A1, COL9A2, COL9A3, COL10A1, COL11A1, and COL11A2. The results were evaluated using American College of Medical Genetics and Genomics (ACMG) guidelines. Clinical characteristics and rhGH treatment response were summarized.
Results: Twenty-four pathogenic or likely pathogenic variants of collagen genes were found in 26 of 106 (24.5%) short-stature patients with skeletal abnormalities, of which COL2A1 mutations were the most common, accounting for about 57.7%. Other frequent mutations associated with skeletal development include FGFR3, ACAN, NPR2, COMP, and FBN1 in 12.2%, 0.9%, 0.8%, 0.4%, and 0.4%, respectively, resulting in significantly different degrees of short stature. An overview of clinical features of collagenopathies showed growth retardation, skeletal abnormalities, and heterogeneous syndromic abnormalities involving facial, eye, hearing, and cardiac abnormalities. The average height of 9 patients who received rhGH treatment improved from a median of -3.2 ± 0.9 SDS to -2.2 ± 1.3 SDS after 2.8 ± 2.1 years. The most significant height improvement of 2.3 SDS and 1.7 SDS was also seen in two patients who had been treated for more than 6 years.
Conclusions: A proband-based NGS revealed that distinct genetic architecture underlies short stature in varying degrees and clinical features. Skeletal abnormality-related short stature involving multiple systems should be tested for skeletal collagen gene mutation. Limited rhGH treatment data indicate an improved growth rate and height, and close monitoring of adverse reactions such as scoliosis is required.
Childhood linear growth is the result of chondrogenesis at the skeletal growth plate, the structure responsible for bone elongation and therefore overall body size (1). Recently, findings have uncovered a vast array of regulatory systems that implicate multiple aspects of the growth plate and long bone development and an accompanying vast array of genetic defects that can cause disorders of linear growth (2). Some sequence variations in genes affecting growth plate function can produce a phenotypic spectrum of short stature with skeletal dysplasia, ranging from severe skeletal deformity to disproportionately short stature, most of which show severe short stature (2). The etiological diagnosis for short stature with skeletal abnormalities is still a clinical challenge, and therapy for improving their severe short stature has been rarely attempted.
With the advances of broad sequencing approaches, clinical genetic evaluation has been demonstrated as an important tool to elucidate the causes of growth disorders from among the myriad possibilities, and an increasing number of short stature-associated genes have been discovered. These causative genes are involved in the physiological processes of the growth plate and long bone development, including normal production and action of multiple hormones, paracrine signaling, and extracellular matrix (ECM) molecules (e.g., cartilage oligomeric matrix protein, aggrecan, several different types of collagens produced by chondrocytes), as well as the normal function of multiple intracellular processes required for chondrocyte proliferation, hypertrophy, and extracellular matrix production (2, 3). Some typical genetic syndromes have been identified, such as Laron syndrome (MIM #262500) related to GHR, Leri–Weill dyschondrosteosis (MIM #127300) related to SHOX, Noonan syndrome (MIM #163950), and Silver–Russell syndrome (MIM #180860), which are associated with short stature and various multiorgan malformations (4–6). Genetic disorders associated with skeletal dysplasia include many genes involved in growth plate development, such as FGFR3, ACAN, NPR2, FBN1, and IHH, which can cause varying degrees of short stature with or without other minor abnormalities (7, 8). Recently, not only for ACAN (aggrecan) and COMP (cartilage oligomeric matrix protein) in ECM components but also for NPR2 (natriuretic peptide receptor 2) in paracrine signaling, we reported the phenotypic and genotypic spectra and efficacy of GH therapy for height gain. For collagen, the most abundant protein in the human body, however, the prevalence of collagen gene mutation in short-stature patients with skeletal abnormalities is yet unknown, the current clinical manifestations of the disease are heterogeneous, and the response data of growth hormone therapy are limited. Collagen types II, IX, X, and XI are present in a growth plate important to long bone development and joint health, and type I collagen is the primary collagen in bone for bone formation, growth, and remodeling, and subsequently mineralization to form bone tissue. Mutations in genes that encode skeletal collagen are not uncommon in the genetic causes for growth defects with skeletal abnormalities (9).
Subsequently, we analyzed 106 children with short stature by using a gene panel for short stature and whole-exome sequencing (WES) from our single-center cohort and searched for variants in the skeletal collagen genes. Variant interpretation, genotype–phenotype analyses, and the response to rhGH treatment of skeletal abnormality-related short stature were investigated.
One hundred and six children with short stature and skeletal abnormalities in our endocrinology department were included, 64 of whom received WES and 42 of whom received a short stature-targeted gene panel sequencing. Skeletal abnormalities were characterized by an intrinsic abnormality in growth and (re-)modeling of cartilage and bone, including the whole-body skeleton of axial bones, limbs, and craniofacial bones, which were assessed by a professional physician through physical examination and measurements. All probands fulfilled the following diagnostic criteria: height standard deviation (SD) ≤ -2 with skeletal abnormalities, absence of abnormal findings on clinical examination or in laboratory tests that could account for short statures, such as hypothyroidism and GHD, and known Noonan syndrome and Turner syndrome. Clinical materials of the first and follow-up visits of the probands, including history-taking, physical examination, and auxiliary examination, were collected. Information about rhGH therapy was also reviewed and recorded. Peripheral blood samples of patients and their available relatives were collected, and genomic DNA was obtained from peripheral blood leukocytes by using standard techniques. The patients or guardians signed informed consent forms regarding the research, and this study was performed with the approval of the Ethics Committee of Peking Union Medical College Hospital.
The 3-μg genomic DNA concentrations were sheared with a Covaris LE220 ultrasonic instrument (MA, USA) to a target of 100–500-bp average size. Then, the DNA fragments with a main fragment size of 150–200 bp were screened by magnetic beads to create a DNA library for each subject. The library was qualitatively controlled by Agilent 2100 Bioanalyzer (BGI, Shenzhen, China). All amplified libraries were subsequently sent to BGI for circularization and sequencing on the BGISEQ-500 platform, and the primary sequencing data were read out. To detect the potential variants in the family, bioinformatics processing and data analysis were performed after receiving the primary sequencing data. Sequencing data were aligned to the human genome reference (hg19) using the BWA (Burrows–Wheeler Aligner) Multi-Vision software package to analyze single-nucleotide variants (SNVs) and INDEL calling (10). All SNVs and indels were filtered and estimated via multiple databases, including NCBI dbSNP, HapMap, 1000 human genome dataset, and database of 100 Chinese healthy adults.
A capture panel (NimbleGen, Madison, USA) of short-stature genes was previously designed and assessed by our group. The capture panel covered all exons together with the flanking exon and intron boundaries (± 15 bp) of 466 genes. Sequencing was performed on an Illumina HiSeq 2500 or HiSeq 2000 platform in paired-end mode. In-house bioinformatic analysis was performed. The sequences were aligned to the reference human genome (HG19/HG20). The probe size was about 2.427 MB, and the theoretical capture efficiency of the probe was 98.83%.
To predict the effect of variants, we used in silico prediction programs to assess (PolyPhen-2, Mutation Taster, Provean, and scale-invariant feature transform [SIFT]). Pathogenic variants were under the protocol issued by American College of Medical Genetics and Genomics (ACMG) guidelines (11). The Human Gene Mutation Database (HGMD) was used to screen mutations reported in published studies. All the potential pathogenic variants observed by whole-exome and targeted panel sequencing as well as segregation analysis within family members were validated and genotyped by Sanger sequencing. Statistical analysis was performed using SPSS.25 software. All charts were completed in GraphPad Prism 8.0.2 software. Wilcoxon signed-rank test was used to explore the difference of height SDS in patients before and after rhGH treatment. The Kruskal–Wallis rank test was used to compare the height SDS and the height SDS changes after rhGH treatment in patients with collagen genes, ACAN, and NPR2 mutations. p <0.05 was considered statistically significant.
Sixty-five patients were identified with genetic defects of cartilage extracellular matrix components in the 106 short-stature individuals with skeletal abnormalities, including 26 with collagen genes, 10 with ACAN, 4 with COMP, and 4 with fibrillin-1 (FBN1) mutation. Other frequent mutations in paracrine signaling of the growth plate development include fibroblast growth factor receptor 3 (FGFR3) covering 12.3% (13/106), NPR2 covering 7.5% (8/106), and fibroblast growth factor receptor 3 (PTH1R) (n = 2). The remaining causal genes were associated with a fundamental cellular process, including TRPV4 (n = 5), SHOX (n = 3), KIF22 (n = 2), TRAPPC2 (n = 1), ARSL (n = 1), RUNX2 (n = 1), CENPJ (n = 1), FAM111A (n = 1), and pathogenic copy number variant (CNV) (n = 1) (Figure 1 and Table S1).
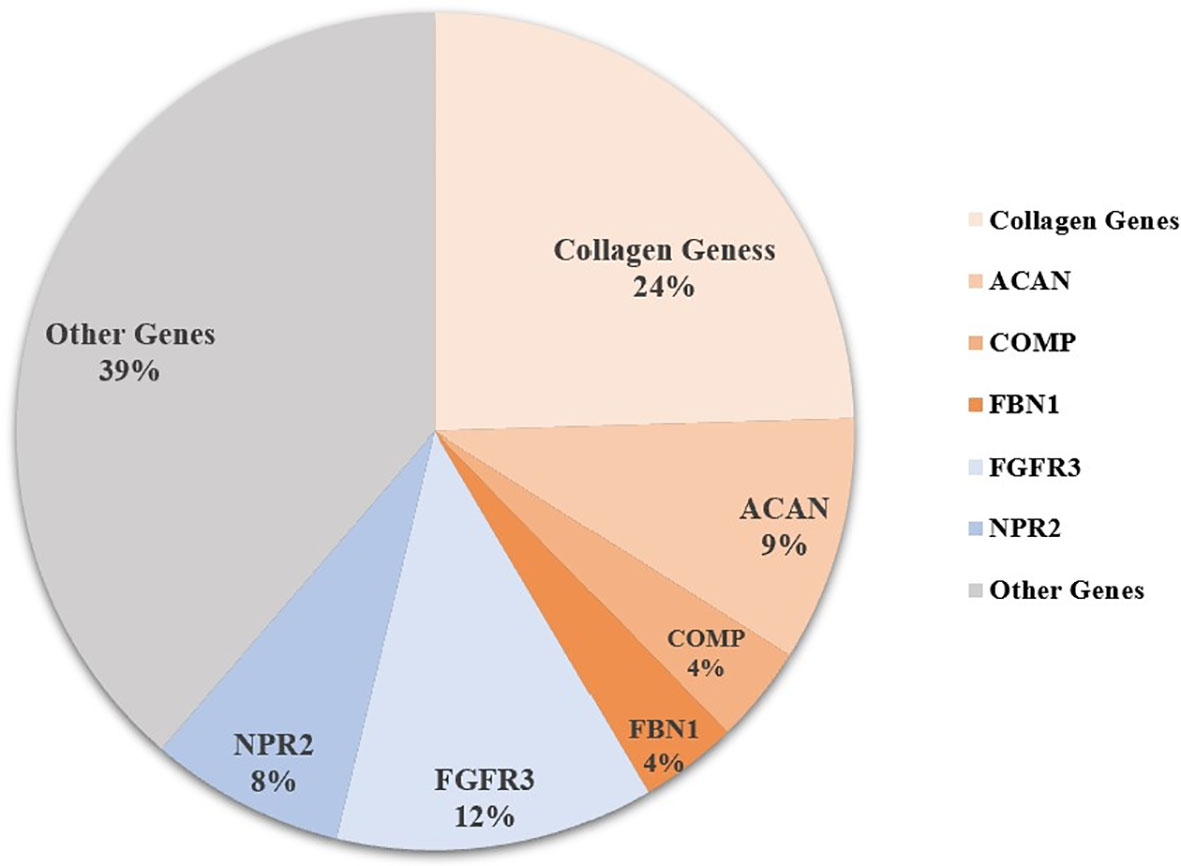
Figure 1 Genetic architecture of short statue with skeletal abnormalities from a single center. Other genes include PTH1R, TRPV4, SHOX, KIF22, TRAPPC2, ARSL, RUNX2, CENPJ, FAM111A, CNV (copy number variant), and some unknown causative genes.
Among this cohort, a total of 24 pathogenic or likely pathogenic rare variants in collagen genes were identified in 26 (24.5%) of the 106 short-stature individuals with skeletal abnormalities as per the ACMG guidelines, of which 5 variants were classified as pathogenic and 19 as likely pathogenic (Table 1). Type II collagenopathies were the most common. Fifteen patients, accounting for about 57.7%, had variants in the COL2A1, 3 had variants in the type IX collagen gene, 4 had variants in the type X collagen gene, and 2 had type XI collagen gene variants and 3 had variants in the type I collagen genes. Types of variant alleles include 18 (75.0%) missense mutations, 3 (12.5%) splicing mutations, 2 (8.2%) nonsense mutations, and 1 (4.2%) in-frame insertion mutation. Except for 4 biallelic heterozygous mutations, all the others were monoallelic heterozygous mutations. The results are summarized in Table 1. The mutations mainly occurred in the triple-helical region (17/24, 70.8%), followed by the C-terminal non-collagenous (NC1) domain (5/24; 20.8%) and the N-terminal non-collagenous (NC2) domain (2/24; 8.3%). Twenty-two of the variants are absent in public databases, whereas the c.1557+5C>T variant in COL11A2 has been identified in 1/7,442 of East Asian chromosomes by the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org), and the c.580G>A in COL2A1 is present at an extremely low allelic frequency (4/140282) in gnomAD.
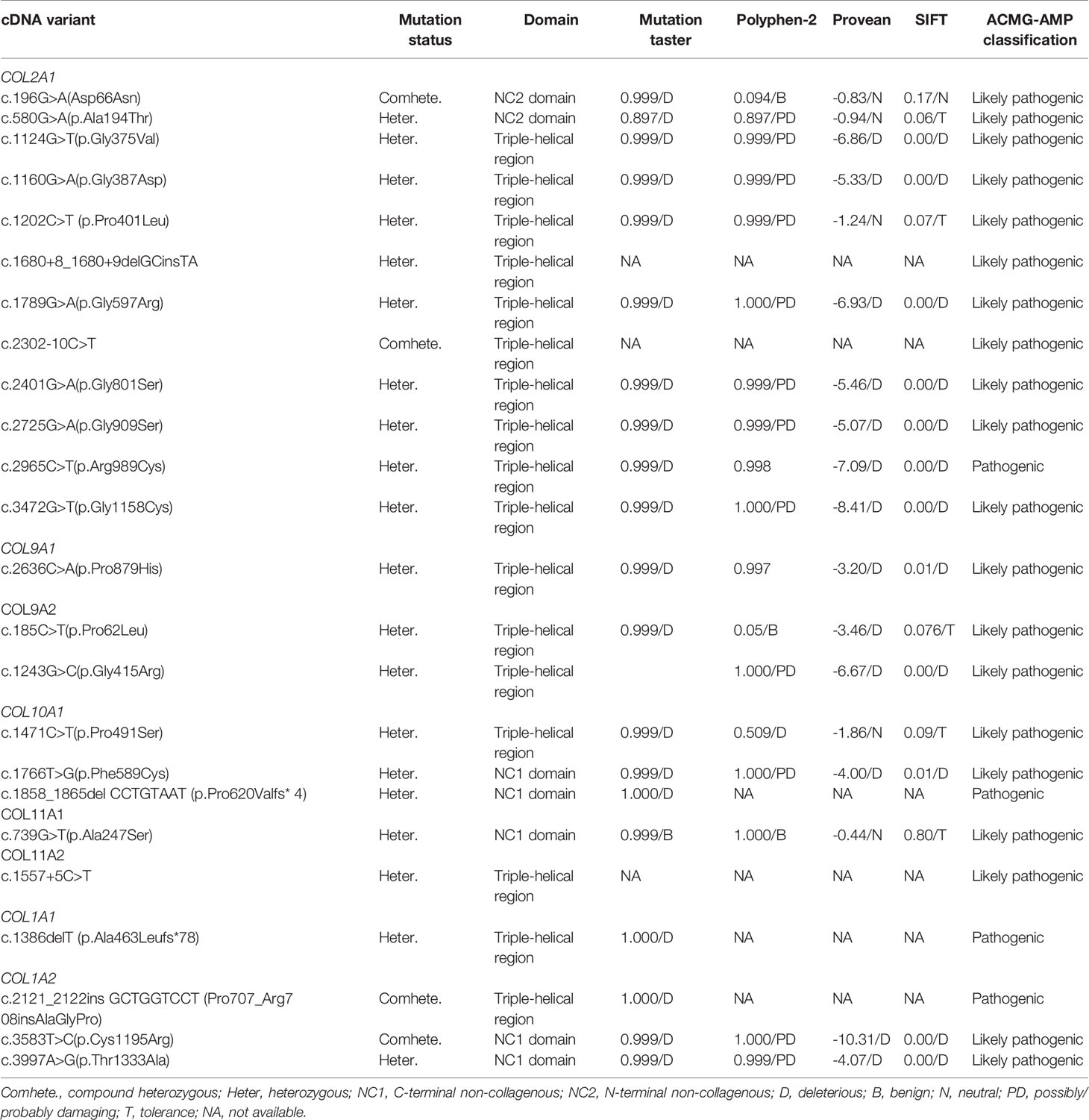
Table 1 In silico analysis and ACMG/AMP classification of collagen gene identified variants.
The growth and phenotypic characteristics of individuals with collagen gene variations enrolled in the study are outlined in Table 2. The average age of patients with skeletal collagenopathies in this cohort was 7.4 ± 4.0 years for the 16 males and 10 females, and their bone age was consistent with chronological age with 6.9 ± 4.1 years, but we observed two type IX collagenopathy patients with a delayed bone age by about 3 years. The overall growth characteristics of patients with collagenopathies indicate growth retardation: four (33.3%) were born with small for gestational age (SGA); their average height Z-scores before rhGH treatment was -3.6 ± 1.4, and the calculated growth rate from 19 available individuals was 5.1 ± 1.7 cm/year. The average Z-score for weight and body mass index (BMI) was -1.3 ± 1.2 and 0.6 ± 1.4, respectively. Thirty-nine percent (9/23) was of familial short stature. The patients with proven collagenopathy and their affected parents had a median height Z score of -3.6 ± 1.3 and -4.1 ± 1.7, respectively.
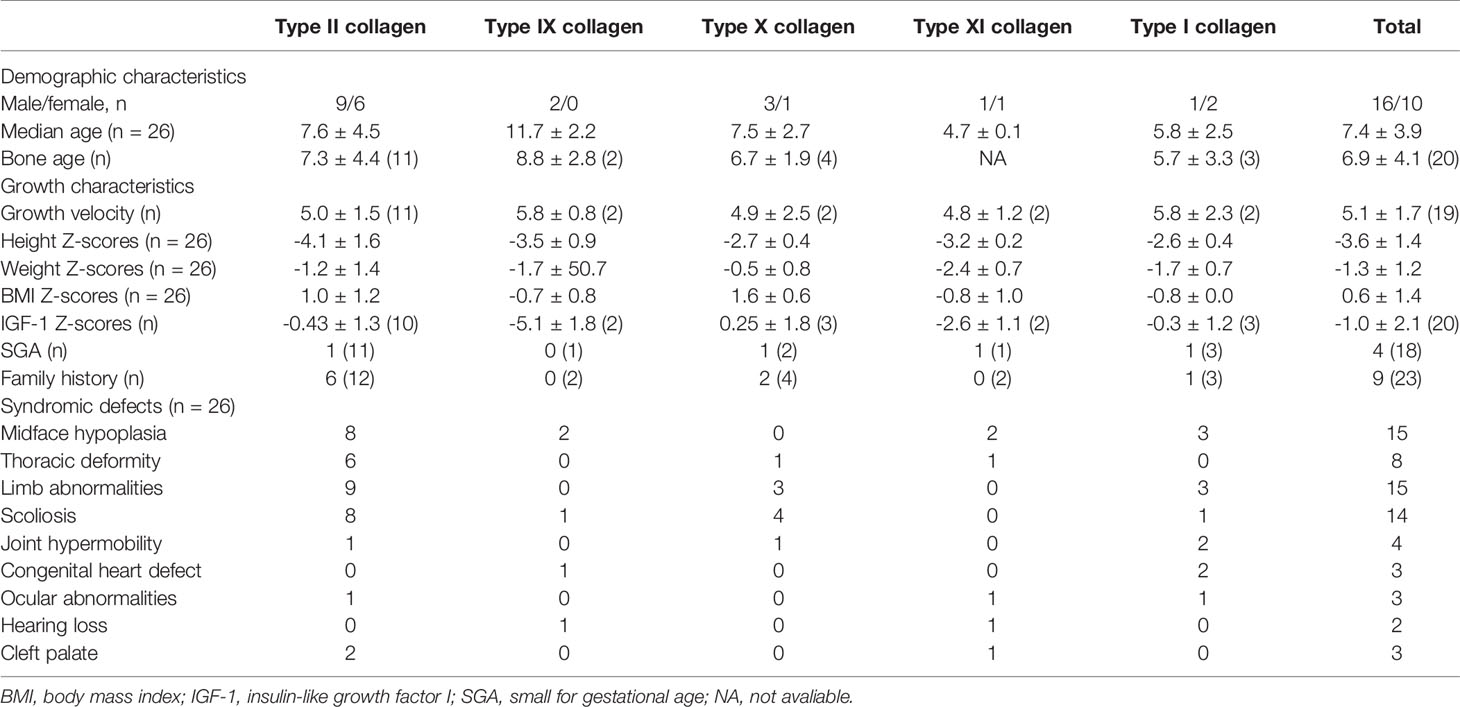
Table 2 Baseline characteristics of individuals with skeletal collagenopathies.
The main clinical manifestations are growth retardation, skeletal abnormalities, and heterogeneous syndromic abnormalities involving facial, eye, hearing, and cardiac abnormalities. All patients had skeletal abnormalities, among which limb abnormalities and spinal deformities are the most common, accounting for 57.7% and 53.8%, respectively. The main manifestations of bone involvement in skeletal collagenopathies include shorting and curving of long bone or phalanges of extremities, metaphyseal dysplasia of spine and limbs, arthrogryposis, joint laxity, scoliosis, or kyphosis. In addition, chest deformity, mainly presented as pectus carinatum, was also observed in 8 patients (30.8%). Fifty-eight percent of cases had facial abnormalities commonly observed in patients, mainly a low nasal bridge, high-arched palate, small jaw, or big or prominent ears. Cleft palate was observed in two patients with type II and one with type XI collagen gene mutations, and blue sclera was observed in two patients with type I and one with type IX collagen gene mutations. In addition, there were a small number of patients with other system defects, such as three with heart defects, three with congenital cataracts, strabismus, or amblyopia, and two with mixed deafness or ear deformity and congenital aural atresia.
Our data obtained from 15 probands with 12 kinds of COL2A1 mutations showed that the phenotypic spectrum of COL2A1 mutations included spinal deformity, abnormal cartilage development, midface hypoplasia, and ocular abnormalities. Almost all showed spine deformity including scoliosis, lordosis, dysplasia, and osteoporosis. The clinical manifestations of three boys (P.11, P.12, P.13 shown in Table S2) from three independent families with the same hot spot mutation (p.Arg989Cys) were consistent with spondyloepimetaphyseal dysplasia Strudwick type (SEDC) (MIM #184250), of which P.12 and P.13 had more severe skeletal manifestations and were diagnosed at 5.3 and 6.8 years of age, respectively, while P.11 was the milder type and diagnosed at a later age of 14.8 years. One novel missense variant (p.Ala194Thr) was identified and confirmed to segregate with the autosomal dominant short-stature phenotype in a family (P.3), which showed only short stature and flat round face with no obvious skeletal deformity in P.4. Besides, P.7 carries pathogenic variants in both COL2A1 and COL9A2 mutations resulting in a more severe skeletal deformity and short-trunk dwarfism, while the other two patients (P.16, P.17) in our study only carrying COL9A1/COL9A2 mutation had very mild symptoms, with only mild chondrodysplasia.
In the data obtained from three probands in two independent families with COL10A1 mutations, one missense variant 1766T>G (p.Phe589Cys) was de novo paternity and maternity confirmed in two identical twin brothers (P.18, P.19), and one truncating mutation c.1858_1865del CCTGTAAT (p.Pro620Valfs*4) was confirmed to segregate with the autosomal-dominant short-stature phenotype in five affected family members (P.21). Both types of mutations were located in the NC1 domain. All three probands and affected relatives exhibited typical metaphyseal chondrodysplasia type Schmid (SMCD) (MIN #156500) phenotypes with short bowed limbs, valgus knees, pronounced lumbar lordosis, posterior flexion of hips, enlarged large joints, and a faltering gait, which were consistent with radiographical findings. In addition, the proband with truncating mutations was born with flexion of the legs and had more severe forms of SMCD with additional manifestations such as short neck, pectus carinatum, beaded ribs, and widened epiphysis of the ribs. The two cases caused by missense variants exhibited relatively late-onset ages at around 2 years of age and mild or moderate manifestations.
Two novel variants in the type XI collagen gene were identified with one missense variant of COL11A1 [c.739G>T(p.Ala247Ser)] and one splice site alteration of COL11A2 (c.1557+5C>T). The girl (P.22) with the COL11A1 mutation had a phenotype consistent with mild Marshall syndrome (MIM #154780), with midfacial hypoplasia, cleft palate, a less severe ocular presentation, but striking ocular hypertelorism, and short stature with spondyloepiphyseal dysplasia (12). The boy (P.23) with a COL11A2 splice site alteration was characterized by Stickler syndrome (MIM #108300) with congenital cataract, sensorineural deafness, relatively short extremities with elbows valgus and joints pain, and typical midface hypoplasia.
Three patients with mutations in genes encoding type I collagen (COL1A1 and COL1A2) were identified, and their skeletal abnormalities were characterized primarily by osteoporosis, with less common abnormalities in the limb bones and skull. In addition to skeletal abnormalities, they also have abnormalities in many organs, such as cardiovascular, joints, ligaments, midface development, and ocular anomalies. One patient (P.26) with biallelic heterozygous mutations [c.2121_2122insGCTGGTCCT (p.Pro707_Arg708insAlaGlyPro) and c.3583T>C (p. Cys1195Arg)] and one (P.24) with heterozygous truncating mutation [c.1386delT (p. Ala463Leufs*78)] had more severe osteogenesis imperfecta, such as early-onset motor retardation, heart defects, reduced thoracolumbar bone density, and obvious joint relaxation, than did the heterozygous missense mutations [c.3997A>G (p. Thr1333Ala)] (P.25).
The clinical phenotypes of other skeletal abnormality-related short stature were also briefly summarized based on our recent report (13–15). Heterozygous mutations in ACAN can lead to spondyloepiphyseal dysplasia, Kimberley type (MIM #608361), or osteochondritis dissecans (MIM #165800), which was consistent with the clinical findings of the 10 patients in our cohort, presenting as mild midface hypoplasia, short neck, thoracic deformity, spine malformation, short fingers/toes, short metacarpal bones, internal rotation of the elbow (contrast to cubitus valgus), and café-au-lait spots, and none of them complained of bone or joint pain. Four patients with COMP mutations exhibited typical pseudoachondroplasia (PSACH) (MIM #177170) with severe short-limb dwarfism, joint pain and stiffness, and early-onset osteoarthritis, and 4 patients with mutations in FBN1 represented as acromelic dysplasia (MIM # 102370) shared severe short stature, short hands and feet, and joint limitations. Biallelic variations of NPR2 mutation can cause acromesomelic dysplasia, Maroteaux type (AMDM) (MIM #602875), while monoallelic variants result in short stature with non-specific skeletal deformities and Miura-type osteochondral dysplasia. Autosomal dominant mutations in FGFR3 causing achondroplasia (ACH) (MIM #100800) were found in 13 of our cohort. They appeared as short stature resulting from the shortening of the limbs with proximal segments affected disproportionally. In addition, the typical broad or protruding forehead, lumbar lordosis, and sacral kyphosis were seen in all these patients. Among the 8 cases of NPR2 mutation in our study, except for one case with compound heterozygous mutation characterized by disproportionate short stature, mesomelic limb shortening, and shortened and broadened fingers and toes, conforming to AMDM, the other 7 cases were heterozygous mutation with or without disproportionate short stature, facial anomalies, and non-specific skeletal deformities, including mesomelic limb shortening, cubitus valgus, brachydactyly, shortened metacarpals or metatarsals, clinodactyly, and cone-shaped epiphysis.
There were 9 patients with collagen gene mutation who had received rhGH treatment, two of whom had combined treatment with gonadotropin-releasing hormone agonists (GnRHa) (Table 3). The initial age of treatment was 7.9 ± 3.5 years. After 1 year of treatment, the growth rate increased from 6.0 ± 1.6 to 9.0 ± 1.3 cm/years, and the average height Z score significantly increased from -3.2 ± 0.9 to -2.5 ± 1.0 (p <.01). Their average height Z score at the last follow-up was significantly increased to -2.2 ± 1.2 (p <.001) (Figure 2). The average duration of treatment was 2.9 ± 1.9 years, and three of them were treated discontinuously. Two cases (P.5, P.10) of scoliosis occurred after initial treatment, both of which discontinued the therapy for orthopedic evaluation (P.10 underwent spinal orthopedic treatment) and continued treatment. One girl (P.20) received rhGH at age of 5.7 and discontinued for personal reasons 1.3 years later, followed by the diagnosis of central precocious puberty with advanced bone age (+2~3 years), breast development at 7 years of age, and menophania at 9.8 years of age, and rhGH treatment was restarted combined with GnRHa for 1 year at 10 years of age, with poor therapeutic response. After exclusion of these three patients from growth response, the six remaining patients had an average improvement in a height Z score of 1.3 ± 0.5 (range, 0.8 to 2.3). The height of the two patients who had been treated for more than 6 years significantly increased their height Z score by 2.26 and 1.71, respectively. No abnormalities or side effects were observed throughout the treatment.

Table 3 Overview of patients with skeletal collagenopathies with rhGH treatment.
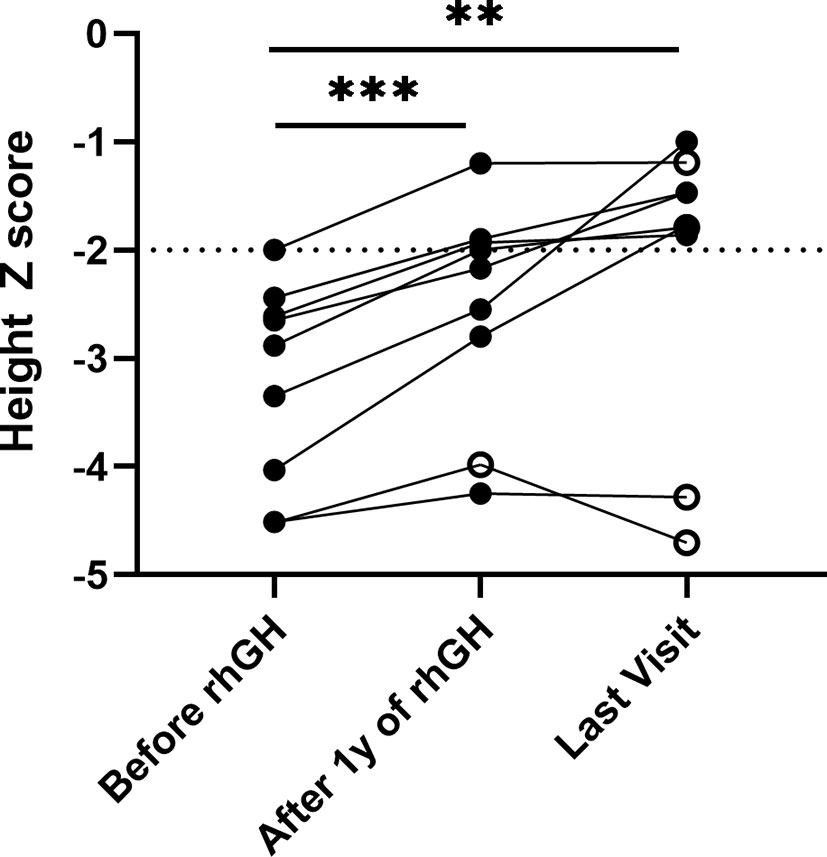
Figure 2 Height Z score in patients with collagenopathies with rhGH treatment. The median follow-up time was 2.25 years with a range of 1.12 to 6.50 years. Solid black dots indicate continued treatment up to the last follow-up, and hollow black dots indicate discontinuous treatment, **p <.01, ***p <.001.
We pooled demographic and treatment information of all short-stature patients with skeletal abnormalities both in our cohort and partly from the literature review, including other extracellular matrix component genes (ACAN, COMP, and FBN1) and paracrine signaling genes (FGFR3 and NPR2), in which a subset of the 29 patients with ACAN mutation and 21 patients with NPR2 mutation were from the literature review, based on previous reports (Table 4) (13, 15). A total of 121 affected individuals diagnosed genetically are shown in Table 4. The gender proportion of males to females was 1.7 (73 males and 46 females). Their height Z score was −3.8 ± 2.0 from 114 affected individuals. The height Z score of males (70 cases) versus females (44 cases) was -3.9 ± 0.2 versus -3.7 ± 0.3 (p = .747), indicating that there is no difference in height between males and females. Six of the patients were adults with a height Z score of -6.8 ± 1.2, which was significantly lower than the 100 juvenile individuals with a height Z score of -3.7 ± 0.2 (p = .001), suggesting that the height impairment worsened with age and would be more severely affected in adults without any treatment. In addition, when the affected individuals were divided into extracellular matrix maintenance and paracrine signaling of the growth plate and long bone development according to physiological etiologies of short stature, there was no significant difference in their height impairment (p = .683). We compared the height Z score of collagenopathy patients with other different-causing gene mutations and found that the ACAN mutation resulted in milder short stature than the collagen gene mutation (p = .021), while the COMP mutation was the most severe (p = .009) (Table 4 and Figure 3).
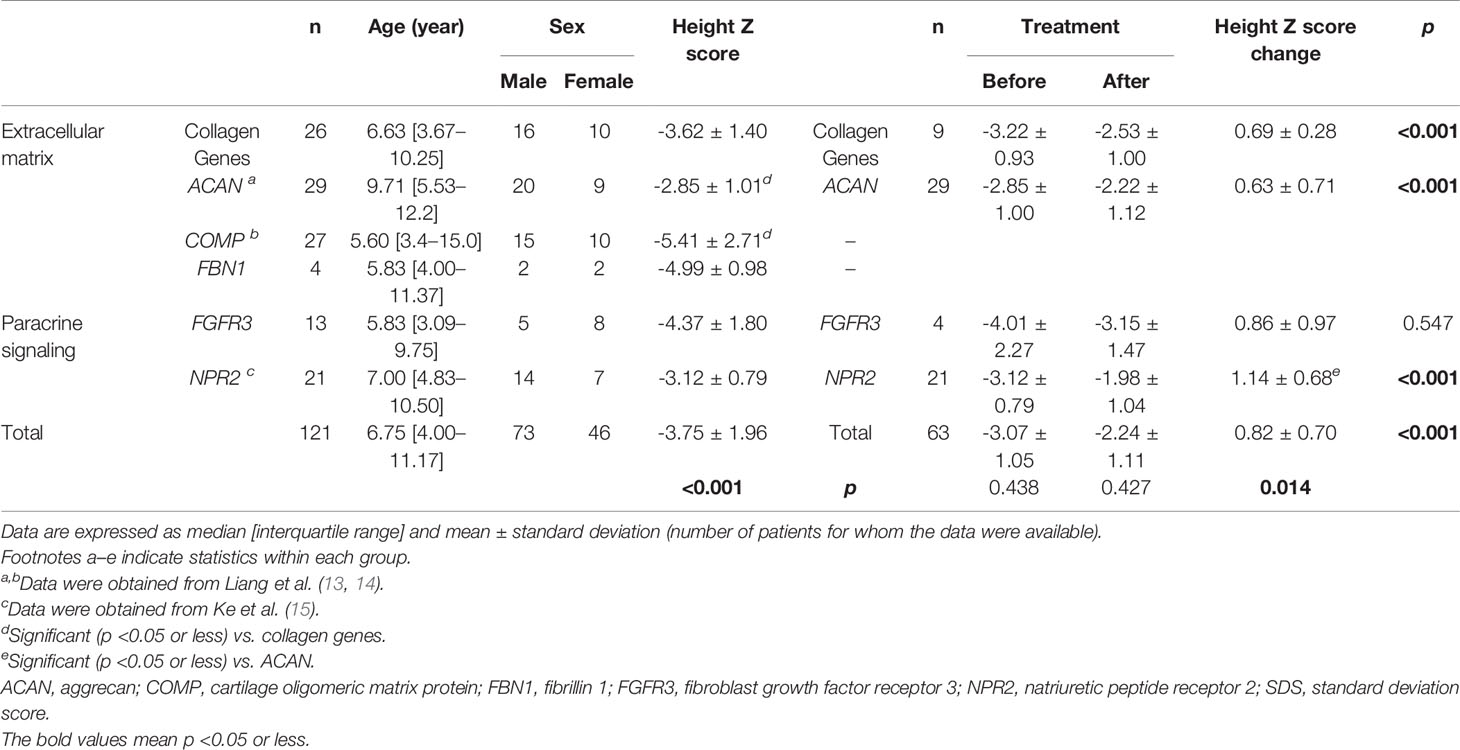
Table 4 Comparison of collagen gene-related short stature with other short stature genetic architecture.
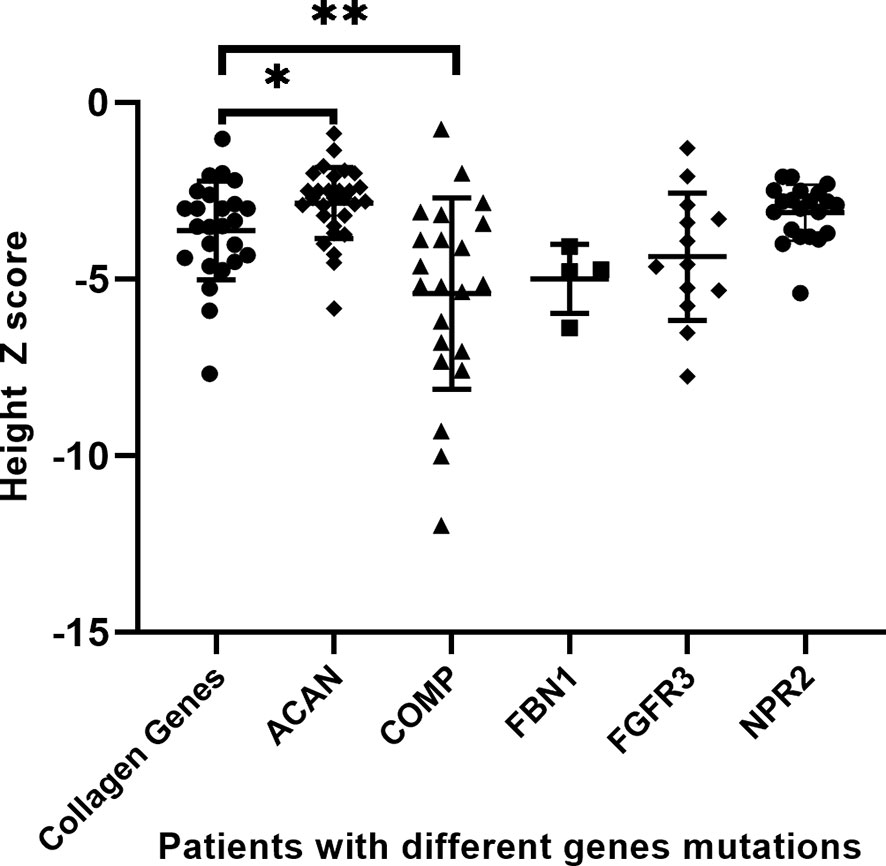
Figure 3 Comparison of different gene mutations leading to growth retardation. *p <.05, **p <.01.
In short-stature patients with ACAN and NPR2 mutation, rhGH had a significant effect on height gain as reported previously (Figure 4) (13, 15). In ACAN-related short stature, rhGH treatment significantly increased height and the height Z score (from -2.9 ± 1.0 to -2.2 ± 1.1) after 2.8 ± 0.4 years of administration. For NPR2-related short stature, height Z scores were significantly improved from -3.1 ± 0.8 to -2.0 ± 1.0 after 3.8 ± 0.6 years of treatment. Moreover, the growth response for rhGH treatment in ACAN-related short stature was better than NPR2 (p = .014) (Figure 4). For FGFR3-related short, height Z scores were improved from -4.0 ± 2.3 to -3.2 ± 1.5, but this was not significant. However, given the severe PSACH phenotype caused by COMP mutations, limited benefit, and possibly serious complications, growth-promoting therapies were not recommended.
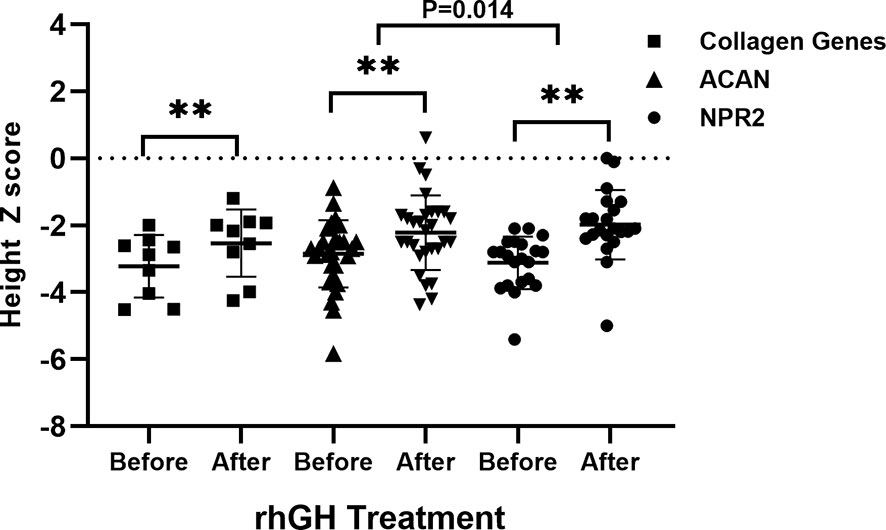
Figure 4 Comparison of growth response to rhGH between patients with collagen gene, ACAN, and NPR2 mutations. **p <.01.
In this study, we performed NGS in short-stature patients with skeletal abnormalities and identified causal variants of skeletal collagen genes in 26 (24.5%) of the 106 individuals, among which 15 patients (14%) carried COL2A1 mutations. Our molecular diagnosis rate of patients with skeletal abnormalities was 76.4% (81/106) and skeletal collagen genes accounted for 24.5% (26/106). COL2A1 mutations were detected in 15 patients (14%), which was slightly higher than that of a recent NGS study on 82 short Chinese patients with clear signs of bone dysplasia (positive rate = 11%) (16). A recent study evaluating oligosymptomatic collagenopathies yielded a lower molecular diagnostic rate of 11.5% in 87 FSS patients treated with rhGH (17). Meanwhile, our study revealed the genetic architecture of short stature with skeletal abnormalities and proposed that mutations of collagen genes (especially COL2A1), FGFR3, ACAN, NPR2, COMP, and FBN1 are common for short stature due to skeletal abnormalities in outpatient clinics in pediatric endocrinology. More importantly, we provided initial information about the phenotypical spectrum of collagenopathies by proving that short stature with skeletal abnormalities and heterogeneous syndromic abnormalities may be caused by mutations in the collagen genes.
Longitudinal growth of the skeleton is a result of endochondral ossification taking place in the epiphyseal growth plates of the long bones. The cartilaginous growth plate consists of extracellular matrix (ECM) and linear columns of differentiated chondrocytes that are organized into resting, proliferating, mature, and hypertrophic zones, which are continuously replaced by trabecular bone with the increase in length (1, 18, 19). Thus, multiple processes involved in the growth plate and long bone development, including basic cellular processes, extracellular matrix maintenance, paracrine signaling, and hormonal signaling, may underlie the distinct genetic architectures and physiological etiology of short stature with skeletal abnormalities. Collagens are a family of structurally related proteins that play a wide variety of roles in the ECM, are characterized by a basic structural coiled-coil right-handed triple helix, and are composed of three polypeptide chains (α chains). Both type II and X collagens are homotrimers composed of 3 identical chains encoded by the COL2A1 and COL10A1 genes, respectively. Both type IX and XI collagens are heterotrimers encoded by 3 different chains of COL9A1, COL9A2, COL9A3 genes, and COL11A1, COL11A2, and COL11A3, respectively. Type I collagen is also a heterotrimeric molecule encoded by COL1A1 and COL1A2. These genes encode procollagens, which are synthesized in the endoplasmic reticulum and contain a short N-telopeptide non-triple-helical (NC2) domain, a long triple-helical domain, and a short C-telopeptide non-triple-helical (NC1) domain, and then posttranslational modification generates mature collagens, which are secreted into the extracellular matrix (20, 21). In our study, of the 23 variants, 16 occurred in the triple-helical domain, highlighting the importance of this domain in the collagen genes. Consistent with previously reported results, all pathogenic variants of COL10A1 in our study were in the NC1 domain, which contains motifs required for normal assembly of the collagen trimer (22). In addition, type II collagen fibrils have covalently linked type IX fibrils on their surface and at their core is a fibril template of type XI collagen. This association of types II, IX, and XI collagens can explain some of the phenotypic overlaps among the resulting conditions (23).
Since collagen is one of the components of many tissues and organs, genetic defects of collagen formation can affect almost every organ system and tissue in the body, and the clinical features often overlap, showing a variable syndrome in addition to bone phenotype. Patients with skeletal collagenopathies described in our study typically manifest growth retardation, skeletal abnormalities, and heterogeneous syndromic abnormalities involving facial, eye, hearing, and cardiac abnormalities, including osteogenesis imperfecta, a variety of chondrodysplasias, rarely, some forms of osteoporosis, osteoarthritis, joint hypermobility, and extra-skeletal features, for example, myopia, astigmatism, cataracts, sensorineural and conductive hearing loss, and mitral or tricuspid regurgitation. Other frequent gene mutations related to skeletal development, including FGFR3, ACAN, NPR2, COMP, and, FBN1, usually cause varying degrees of short stature, with or without other mild abnormalities, such as slight growth ratio imbalance and skeletal non-specific abnormalities (such as short finger/toe, short thumb, or midfacial dysplasia) (7, 8). In addition, one patient (P.6) with COL2A1 mutation showed unexpected obesity and severe skeletal deformities and arthrogryposis, suggesting that weight gain and obesity may also be major concerns of epiphyseal dysplasia and contribute to the morbidity associated with joint problems. The underlying collagen mutation disrupts normal cartilage architecture, resulting in premature cartilage degeneration, and patients with these disorders often require joint replacement in the third to fourth decades of life (21). Both of our two patients with type IX collagen gene mutation showed a mild type of multiple epiphyseal dysplasias, which may be related to its high clinical heterogeneity and complex genetic background, and the late onset of the phenotype may also be one of the reasons as there was also a case that reported that symptoms did not appear until the age of 45 (24). Type X collagen is synthesized exclusively by hypertrophic chondrocytes in the cartilage growth plates of growing bones undergoing endochondral ossification, and its role of an extracellular scaffold or in the mineralization of hypertrophic growth plate has been proposed. Pathogenic variants in COL10A1 cause reduced levels of functional type X collagen in the growth plate and contribute to the development of SMCD phenotypes, a disorder characterized by dwarfism and an expanded growth plate hypertrophic zone, which was seen in three of our patients with two novel missense and truncating variants (21). In the 2 patients with type XI collagen gene mutation, besides the typical skeletal and orofacial manifestations, P.22 with COL11A1 missense mutation showed a less severe ocular presentation, while the boy with a COL11A2 splice site alteration presented with obvious ocular anomalies of congenital cataracts. These findings are different from previous variants associated with Stickler syndrome caused by mutations in genes encoding type XI collagens, in which ocular anomalies are predominantly present in COL11A1 mutation and the COL11A2 heterozygous mutation usually causes non-ocular Stickler syndrome (25).
Although precise genotype–phenotypic correlations in collagen genes have not yet been established, we have made some interesting findings. Phenotypic severity might vary among patients with the same mutation, and in patients with COL2A1 mutations, age at diagnosis might also be associated with disease severity, which was consistent with a previous study of COL2A1 based on a large database (20). The genotype–phenotype correlation of COL10A1 cases in this study was also consistent with previous reports (26, 27). Most of the identified mutations were present in the NC1 domain, which had motifs that control the formation of stable collagen X molecules by promoting the formation of the triple helix. The three cases of COL10A1 mutation we reported all showed SMCD with short-limbed short stature, bowed legs, and a waddling gait, while two cases caused by missense variants exhibited relatively late-onset ages and moderate manifestations than the truncating one. More importantly, we compared the demographic and growth characteristics of patients with different disease-causing genes and found that children with skeletal dysplasia were usually severely short, and the heights of adults who did not receive treatment were significantly more impaired. In addition, among the short-stature individuals with causing genes, compared with individuals with collagen gene mutations, those with ACAN mutations showed a milder short stature, while PSACH with COMP mutations was at the severe end of the dwarfism spectrum and was associated with significant limb shortening (28). Overall, we still suggest that a larger phenotypic spectrum of collagen gene mutations would help construct a solid basis for further research of the genotype–phenotype correlation.
GH therapy has been introduced in several syndromic disorders with short stature, i.e., Noonan syndrome, Prader–Willi syndrome (PWS), and Silver–Russell syndrome (SRS), while there are limited data on the effect of rhGH treatment on children with short stature and skeletal dysplasia (29–31). Recently, we demonstrated a good effect of rhGH treatment in patients with NPR2 heterozygous mutation, and the efficacy was negatively correlated with the initial age of treatment and was associated with gender and the gene positions of mutation (15). A study evaluating the efficiency of rhGH treatment with collagenopathy in a cohort also demonstrated a height Z score improvement from a median of –3.1 to –2.6 and to –2.2 after 1 and 3 years of therapy, respectively (17). Our study provides new evidence for the evaluation of rhGH therapy for skeletal collagenopathies. Consistent with previous results, the individual height Z score benefit after rhGH replacement varied considerably in this study (range: -0.18 to 2.26). During an average duration of 2.8 ± 2.1 years, the height Z score of 9 patients who received rhGH treatment improved from a median of -3.2 ± 0.9 to -2.2 ± 1.3 at the last follow-up, respectively. The most significant height Z score improvement of 2.3 and 1.7 was also seen in two patients who had been treated for more than 6 years. Limb and spinal deformities were a problem frequently seen in children with skeletal collagenopathies (57.7% and 53.8%). There was a concern about whether rhGH treatment would increase the frequency or severity of this finding, especially scoliosis. Two cases (P.5, P.10) of scoliosis occurred after initial treatment, both of which discontinued the therapy for orthopedic evaluation (P.10 underwent spinal orthopedic treatment) and continued treatment. This was also the main reason why rhGH is not recommended for patients with COMP mutations leading to PSACH, usually accompanied by severe osteoarthropathy. A previous clinical trial also demonstrated that in cases of PSACH, the height Z score was worse after rhGH therapy (32). Except for those two patients with the COL2A1 mutation who developed scoliosis after initiating treatment, no exacerbation of scoliosis or other skeletal deformities was observed in the remaining patients. However, we do not think that the occurrence of this scoliosis was completely caused by rhGH treatment, because both patients carry hotspot mutation of COL2A1 leading to SEDC, and the incidence of this disease is relatively high, which has been reported as 48% of 93 patients with molecularly confirmed SEDC or a related disorder in the previous literature (33). In addition, for the general short-stature population with skeletal dysplasia, ACAN-related short stature was more responsive to rhGH treatment than NPR2-related short stature, and significant height improvement was not seen in FGFR3-related short-stature patients in this cohort. While the effectiveness of rhGH for ACH caused by FGFR3 mutation had been proposed in previous cohort studies (34), the very small sample size in our study may be the reason for the difference. A similar difference in rhGH efficacy was also observed in two patients with osteogenesis imperfecta patients who received bisphosphonates with or without rhGH. During the 2-year follow-up, the height Z score of P.24 combined with rhGH therapy was improved from -3.0 to -1.8, while the height Z score of patients treated with bisphosphonates alone was decreased from -2.0 to -3.3, which reveals the effectiveness of bisphosphonate combined with rhGH in patients with osteogenesis imperfecta. Moreover, there are quite a few studies that have confirmed the effectiveness of the combination therapy, not only in terms of growth velocity but also in bone mineral density and bone turnover (35–37). There were no complications such as fractures in both patients. Overall, this evidence suggests that rhGH treatment tolerability and efficacy in improving growth in patients with skeletal abnormalities vary greatly and careful consideration of indications for therapy and cautious observation during therapy are crucial for each patient.
Potential limitations of this work warrant consideration. First of all, in terms of molecular genetic testing approaches, WES may not detect large CNVs. Therefore, multiple ligation-dependent probe amplification (MLPA) or chromosomal microarray (CMA) analysis should be performed to screen for large deletions and insertions in genes belonging to the short-stature gene panel. In addition, some of the newly discovered variants still lack further validation, so we only evaluated their pathogenicity according to the ACMG guidelines, combined with their clinical phenotypes, pedigree verification, and in silico prediction programs. Furthermore, there are signs or clinical manifestations that have not been identified in some of our patients due to young age or too short follow-up. It is also worth mentioning that, unfortunately, we did not routinely assess bone mass in some children without a clear history of fractures. Moreover, some evidence for segregation of the variants with short stature within the families is lacking in some suspect pedigrees because of some unavailable relatives, which can strongly support the mutations’ pathogenicity. Lastly, more accurate genotype–phenotype correlations and evidence for the treatment of recombinant growth hormone deserve further study in a larger cohort of skeletal collagenopathies children with short stature.
In conclusion, skeletal collagenopathies are relatively frequent in syndromic-related short stature, and screening for collagen mutations should be considered in short-stature children with skeletal abnormalities. Although long-term studies evaluating rhGH treatment are insufficient and large cohort studies regarding rhGH dose, the optimal age to start treatment, and adverse events are lacking, initial information provided by our study about the efficacy of rhGH treatment for skeletal collagenopathies indicates an improved growth rate and height. Before starting rhGH treatment, patients with collagenopathy-related short stature should be extensively evaluated, and close monitoring of adverse reactions such as scoliosis is required.
The original contributions presented in the study are publicly available. This data can be found here: National Genomics Data Center (NGDC), PRJCA008063.
The studies involving human participants were reviewed and approved by the Peking Union Medical College Hospital Ethics Committee. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
MC and HZ designed the study. MC, HM, and HL collected the data. MC and XK conducted the data collection and analysis. FG guided the experimental study. MC and HZ drafted the manuscript. HY, LW, LD, SC, and HP interpreted the data and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS 2021-I2M-1-003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.820001/full#supplementary-material
1. Krakow D, Rimoin DL. The Skeletal Dysplasias. Genet Med (2010) 12(6):327–41. doi: 10.1097/GIM.0b013e3181daae9b
2. Baron J, Sävendahl L, De Luca F, Dauber A, Phillip M, Wit JM, et al. Short and Tall Stature: A New Paradigm Emerges. Nat Rev Endocrinol (2015) 11(12):735–46. doi: 10.1038/nrendo.2015.165
3. Grunauer M, Jorge AAL. Genetic Short Stature. Growth Horm IGF Res (2018) 38:29–33. doi: 10.1016/j.ghir.2017.12.003
4. Kruszka P, Porras AR, Addissie YA, Moresco A, Medrano S, Mok GTK, et al. Noonan Syndrome in Diverse Populations. Am J Med Genet A (2017) 173(9):2323–34. doi: 10.1002/ajmg.a.38362
5. Laron Z, Werner H. Laron Syndrome - A Historical Perspective. Rev Endocr Metab Disord (2021) 22(1):31–41. doi: 10.1007/s11154-020-09595-0
6. Jee YH, Baron J, Nilsson O. New Developments in the Genetic Diagnosis of Short Stature. Curr Opin Pediatr (2018) 30(4):541–7. doi: 10.1097/mop.0000000000000653
7. Plachy L, Strakova V, Elblova L, Obermannova B, Kolouskova S, Snajderova M, et al. High Prevalence of Growth Plate Gene Variants in Children With Familial Short Stature Treated With Gh. J Clin Endocrinol Metab (2019) 104(10):4273–81. doi: 10.1210/jc.2018-02288
8. Gkourogianni A, Andrew M, Tyzinski L, Crocker M, Douglas J, Dunbar N, et al. Clinical Characterization of Patients With Autosomal Dominant Short Stature Due to Aggrecan Mutations. J Clin Endocrinol Metab (2017) 102(2):460–9. doi: 10.1210/jc.2016-3313
9. Jobling R, D’Souza R, Baker N, Lara-Corrales I, Mendoza-Londono R, Dupuis L, et al. The Collagenopathies: Review of Clinical Phenotypes and Molecular Correlations. Curr Rheumatol Rep (2014) 16(1):394. doi: 10.1007/s11926-013-0394-3
10. Li H, Durbin R. Fast and Accurate Short Read Alignment With Burrows-Wheeler Transform. Bioinformatics (2009) 25(14):1754–60. doi: 10.1093/bioinformatics/btp324
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
12. Shanske AL, Bogdanow A, Shprintzen RJ, Marion RW. The Marshall Syndrome: Report of a New Family and Review of the Literature. Am J Med Genet (1997) 70(1):52–7. doi: 10.1002/(sici)1096-8628(19970502)70:1<52::aid-ajmg11>3.0.co;2-w
13. Liang H, Miao H, Pan H, Yang H, Gong F, Duan L, et al. Growth-Promoting Therapies May Be Useful In Short Stature Patients With Nonspecific Skeletal Abnormalities Caused By Acan Heterozygous Mutations: Six Chinese Cases And Literature Review. Endocr Pract (2020) 26(11):1255–68. doi: 10.4158/ep-2019-0518
14. Liang H, Miao H, Pan H, Yang H, Chen S, Gong F, et al. Clinical Characteristics of Pseudoachondroplasia and Analysis of COMP Gene Mutation. Chin J Endocrinol Metab (2019) 35(12):1006–13. doi: 10.3760/cma.j.issn.1000-6699.2019.12.003
15. Ke X, Liang H, Miao H, Yang H, Wang L, Gong F, et al. Clinical Characteristics of Short-Stature Patients With an NPR2 Mutation and the Therapeutic Response to rhGH. J Clin Endocrinol Metab (2021) 106(2):431–41. doi: 10.1210/clinem/dgaa842
16. Zhang H, Yang R, Wang Y, Ye J, Han L, Qiu W, et al. A Pilot Study of Gene Testing of Genetic Bone Dysplasia Using Targeted Next-Generation Sequencing. J Hum Genet (2015) 60(12):769–76. doi: 10.1038/jhg.2015.112
17. Plachy L, Dusatkova P, Maratova K, Petruzelkova L, Elblova L, Kolouskova S, et al. Familial Short Stature-A Novel Phenotype of Growth Plate Collagenopathies. J Clin Endocrinol Metab (2021) 106(6):1742–9. doi: 10.1210/clinem/dgab084
18. Long F, Ornitz DM. Development of the Endochondral Skeleton. Cold Spring Harb Perspect Biol (2013) 5(1):a008334. doi: 10.1101/cshperspect.a008334
19. Ballock RT, O’Keefe RJ. Physiology and Pathophysiology of the Growth Plate. Birth Defects Res C Embryo Today (2003) 69(2):123–43. doi: 10.1002/bdrc.10014
20. Zhang B, Zhang Y, Wu N, Li J, Liu H, Wang J. Integrated Analysis of COL2A1 Variant Data and Classification of Type II Collagenopathies. Clin Genet Mar (2020) 97(3):383–95. doi: 10.1111/cge.13680
21. Carter EM, Raggio CL. Genetic and Orthopedic Aspects of Collagen Disorders. Curr Opin Pediatrics (2009) 21(1):46–54. doi: 10.1097/MOP.0b013e32832185c5
22. Rajpar MH, McDermott B, Kung L, Eardley R, Knowles L, Heeran M, et al. Targeted Induction of Endoplasmic Reticulum Stress Induces Cartilage Pathology. PloS Genet (2009) 5(10):e1000691. doi: 10.1371/journal.pgen.1000691
23. Blaschke UK, Eikenberry EF, Hulmes DJ, Galla HJ, Bruckner P. Collagen XI Nucleates Self-Assembly and Limits Lateral Growth of Cartilage Fibrils. J Biol Chem (2000) 275(14):10370–8. doi: 10.1074/jbc.275.14.10370
24. Czarny-Ratajczak M, Lohiniva J, Rogala P, Kozlowski K, Perälä M, Carter L, et al. A Mutation in COL9A1 Causes Multiple Epiphyseal Dysplasia: Further Evidence for Locus Heterogeneity. Am J Hum Genet (2001) 69(5):969–80. doi: 10.1086/324023
25. Vuoristo MM, Pappas JG, Jansen V, Ala-Kokko L. A Stop Codon Mutation in COL11A2 Induces Exon Skipping and Leads to non-Ocular Stickler Syndrome. Am J Med Genet A (2004) 130a(2):160–4. doi: 10.1002/ajmg.a.30111
26. Bateman JF, Freddi S, McNeil R, Thompson E, Hermanns P, Savarirayan R, et al. Identification of Four Novel COL10A1 Missense Mutations in Schmid Metaphyseal Chondrodysplasia: Further Evidence That Collagen X NC1 Mutations Impair Trimer Assembly. Hum Mutat (2004) 23(4):396. doi: 10.1002/humu.9222
27. Kong L, Shi L, Wang W, Zuo R, Wang M, Kang Q. Identification of Two Novel COL10A1 Heterozygous Mutations in Two Chinese Pedigrees With Schmid-Type Metaphyseal Chondrodysplasia. BMC Med Genet (2019) 20(1):200. doi: 10.1186/s12881-019-0937-1
28. Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, et al. Nosology and Classification of Genetic Skeletal Disorders: 2015 Revision. Am J Med Genet A (2015) 167a(12):2869–92. doi: 10.1002/ajmg.a.37365
29. Lindgren AC, Lindberg A. Growth Hormone Treatment Completely Normalizes Adult Height and Improves Body Composition in Prader-Willi Syndrome: Experience From KIGS (Pfizer International Growth Database). Horm Res (2008) 70(3):182–7. doi: 10.1159/000145019
30. Dahlgren J. GH Therapy in Noonan Syndrome: Review of Final Height Data. Horm Res Dec (2009) 72:46–8. doi: 10.1159/000243779
31. Toumba M, Albanese A, Azcona C, Stanhope R. Effect of Long-Term Growth Hormone Treatment on Final Height of Children With Russell-Silver Syndrome. Horm Res Paediatr (2010) 74(3):212–7. doi: 10.1159/000295924
32. Kanazawa H, Tanaka H, Inoue M, Yamanaka Y, Namba N, Seino Y. Efficacy of Growth Hormone Therapy for Patients With Skeletal Dysplasia. J Bone Miner Metab (2003) 21(5):307–10. doi: 10.1007/s00774-003-0425-7
33. Terhal PA, Nievelstein RJ, Verver EJ, Topsakal V, van Dommelen P, Hoornaert K, et al. A Study of the Clinical and Radiological Features in a Cohort of 93 Patients With a COL2A1 Mutation Causing Spondyloepiphyseal Dysplasia Congenita or a Related Phenotype. Am J Med Genet A (2015) 167a(3):461–75. doi: 10.1002/ajmg.a.36922
34. Pinto G, Cormier-Daire V, Le Merrer M, Samara-Boustani D, Baujat G, Fresneau L, et al. Efficacy and Safety of Growth Hormone Treatment in Children With Hypochondroplasia: Comparison With an Historical Cohort. Horm Res Paediatr (2014) 82(6):355–63. doi: 10.1159/000364807
35. Vieira NE, Marini JC, Hopkins E, Abrams SA, Yergey AL. Effect of Growth Hormone Treatment on Calcium Kinetics in Patients With Osteogenesis Imperfecta Type III and IV. Bone (1999) 25(4):501–5. doi: 10.1016/s8756-3282(99)00186-6
36. Antoniazzi F, Mottes M, Fraschini P, Brunelli PC, Tatò L. Osteogenesis Imperfecta: Practical Treatment Guidelines. Paediatr Drugs (2000) 2(6):465–88. doi: 10.2165/00128072-200002060-00005
Keywords: short stature, skeletal abnormalities, collagenopathies, next-generation sequencing, growth hormone treatment
Citation: Chen M, Miao H, Liang H, Ke X, Yang H, Gong F, Wang L, Duan L, Chen S, Pan H and Zhu H (2022) Clinical Characteristics of Short-Stature Patients With Collagen Gene Mutation and the Therapeutic Response to rhGH. Front. Endocrinol. 13:820001. doi: 10.3389/fendo.2022.820001
Received: 22 November 2021; Accepted: 06 January 2022;
Published: 16 February 2022.
Edited by:
Robert Rapaport, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Dimitra Micha, Amsterdam University Medical Center, NetherlandsCopyright © 2022 Chen, Miao, Liang, Ke, Yang, Gong, Wang, Duan, Chen, Pan and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huijuan Zhu, c2hlbmd4aW4yMDA0QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.