 Hui Xiang
Hui Xiang Dating Sun
Dating Sun Xin Liu
Xin Liu Zhi-Gang She
Zhi-Gang She Yonghong Chen
Yonghong Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 08 February 2022
Sec. Gut Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.812610
This article is part of the Research TopicPrebiotics in the Management of Obesity and Associated Metabolic DisordersView all 6 articles
Nonalcoholic steatohepatitis (NASH) is a serious disease threatening public health, and its pathogenesis remains largely unclear. Recent scientific research has shown that intestinal microbiota and its metabolites have an important impact on the development of NASH. A balanced intestinal microbiota contributes to the maintenance of liver homeostasis, but when the intestinal microbiota is disequilibrated, it serves as a source of pathogens and molecules that lead to NASH. In this review, we mainly emphasize the key mechanisms by which the intestinal microbiota and its metabolites affect NASH. In addition, recent clinical trials and animal studies on the treatment of NASH by regulating the intestinal microbiota through prebiotics, probiotics, synbiotics and FMT have also been briefly elaborated. With the increasing understanding of interactions between the intestinal microbiota and liver, accurate and personalized detection and treatment methods for NASH are expected to be established.
Non‐alcohol fatty liver disease (NAFLD) has become the most common chronic liver disease worldwide (1), with a global prevalence of more than 25% (2). Nonalcoholic steatohepatitis (NASH), the progressive stage of NAFLD, is characterized by hepatic steatosis, inflammation, ballooning and fibrosis (3). NASH is the leading cause of liver-related mortality worldwide because of its tendency to develop into cirrhosis and hepatocellular carcinoma (HCC), as well as its impact on extrahepatic diseases, such as cardiovascular disease (CVD) and chronic kidney disease (CKD) (4, 5). The pathogenesis of NASH, however, remains extensively elusive, which is one of the main factors hampering the development of pharmaceutical strategies of NASH. In recent years, accumulating studies have indicated that the intestinal microbiota mediates the progression of NASH by affecting gut barrier permeability, hepatic lipid metabolism, inflammation and fibrosis (6).
Approximately 40 trillion microbes inhabit the human gut (7), which mainly includes bacteria, archaea, viruses and fungi (8). Intestinal microbiota is usually associated with metabolic diseases of the human host, such as diabetes, obesity, CVD, NAFLD and NASH (5). To date, multiple clinical and preclinical studies have demonstrated that individuals with NASH usually have compositional changes in their intestinal microbiota (9). Intestinal microbiota dysbiosis has been shown to accelerate the development and progression of NASH (10). On account of this, more and more scientific research attempts to inhibit or treat NASH by intervening with the intestinal microbiota, such as probiotics, prebiotics and synbiotics supplementation and fecal microbiota transplantation (FMT) (10). The mechanisms by which intestinal dysbiosis promotes NASH will be highlighted in the review. Moreover, studies on the treatment of NASH by intervening intestinal microbiota have also been elaborated accordingly.
A clinical study by Zhu et al. showed significant increases of Bacteroidetes, Proteobacteria, Enterobacteriaceae and Escherichia and decrease of Firmicutes and Bifidobacterium in the NASH group compared with the healthy group (11). However, Mouzak et al. found that NASH was significantly associated with a lower proportion of Bacteroides (12). Del Chierico et al. demonstrated that compared with healthy controls, Ruminococcus, Blautia and Dorea increased in NASH patients, while Oscillospira reduced (13). In addition, the progression from nonalcoholic fatty liver (NAFL) to NASH is also accompanied by changes in intestinal microbiota, but the relevant studies are insufficient, currently. Del Chierico et al. found an increase in Firmicutes and a decrease in Bacteroidetes, Proteobacteria and Actinobacteria in NASH patients compared with NAFL (13). Similarly, Schwimmer et al. found that NAFLD patients had higher abundance of Oscillibacter, Lactonifactor, Akkermansia and Enterococcus, while NASH patients were accompanied by higher abundance of Lactobacillus and Oribacterium (14). Taken together, multiple clinical studies have shown that compositional changes of intestinal microbiota are common in patients with NAFLD/NASH, but such changes remain largely inconsistent and contradictory due to the heterogeneous of the relative distribution of intestinal microbiota (Table 1). Therefore, more studies are urgently needed to clarify the compositional changes of intestinal microbiota in each stage of NASH, even beginning from the healthy stage, and this has positive implications for future treatments of NASH by targeting individual intestinal microbiota.
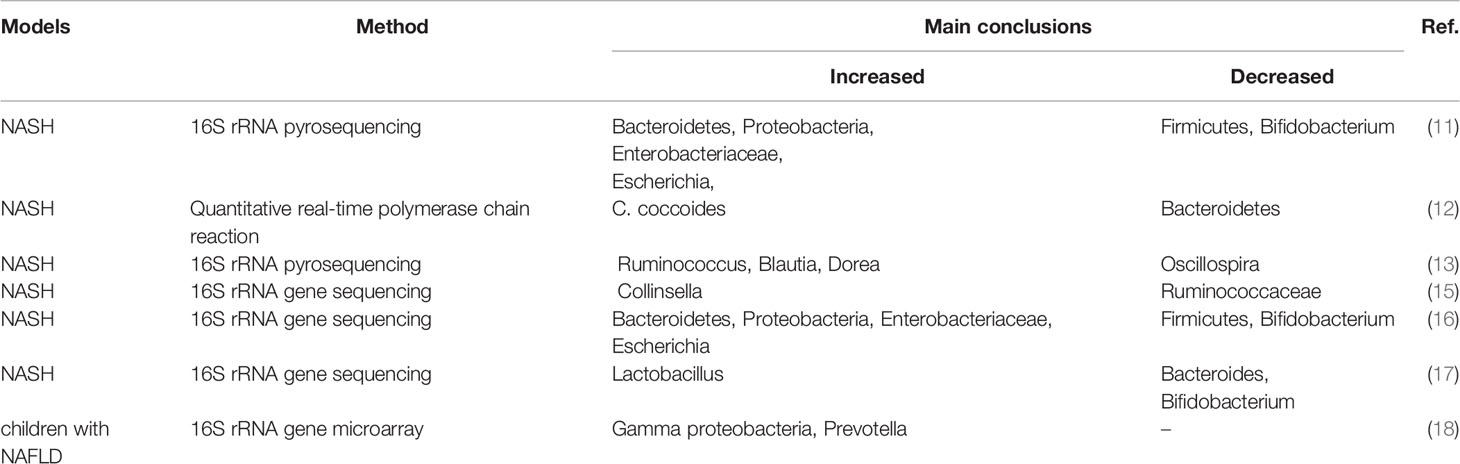
Table 1 Composition of intestinal microbiota in NASH patients.
Intestinal bacteria (such as Ruminococcus, Anaerostipes, Bacteroidetes, Akkermansia muciniphila and Lachnospiraceae) can degrade polysaccharides, dietary fiber and resistant starch into monosaccharides and SCFAs, and SCFAs of which mainly include acetate, propionate, butyrate (19, 20). Most SCFAs are utilized in the intestine (especially butyrate) to provide approximately 70% of the energy for intestinal epithelial cells (21). A small number of SCFAs enter the portal vein through monocarboxylate transporter 1 (MCT1) and sodium‐coupled monocarboxylate transporter 1 (SMCT1) receptors and subsequently infiltrate the liver (6). As a metabolic substrate and signaling molecule that regulates metabolism, SCFAs regulate hepatic metabolism through the gut -liver axis (Figure 1), which refers to the bilateral relationship between the gut and the liver through the portal system and biliary tract (22).
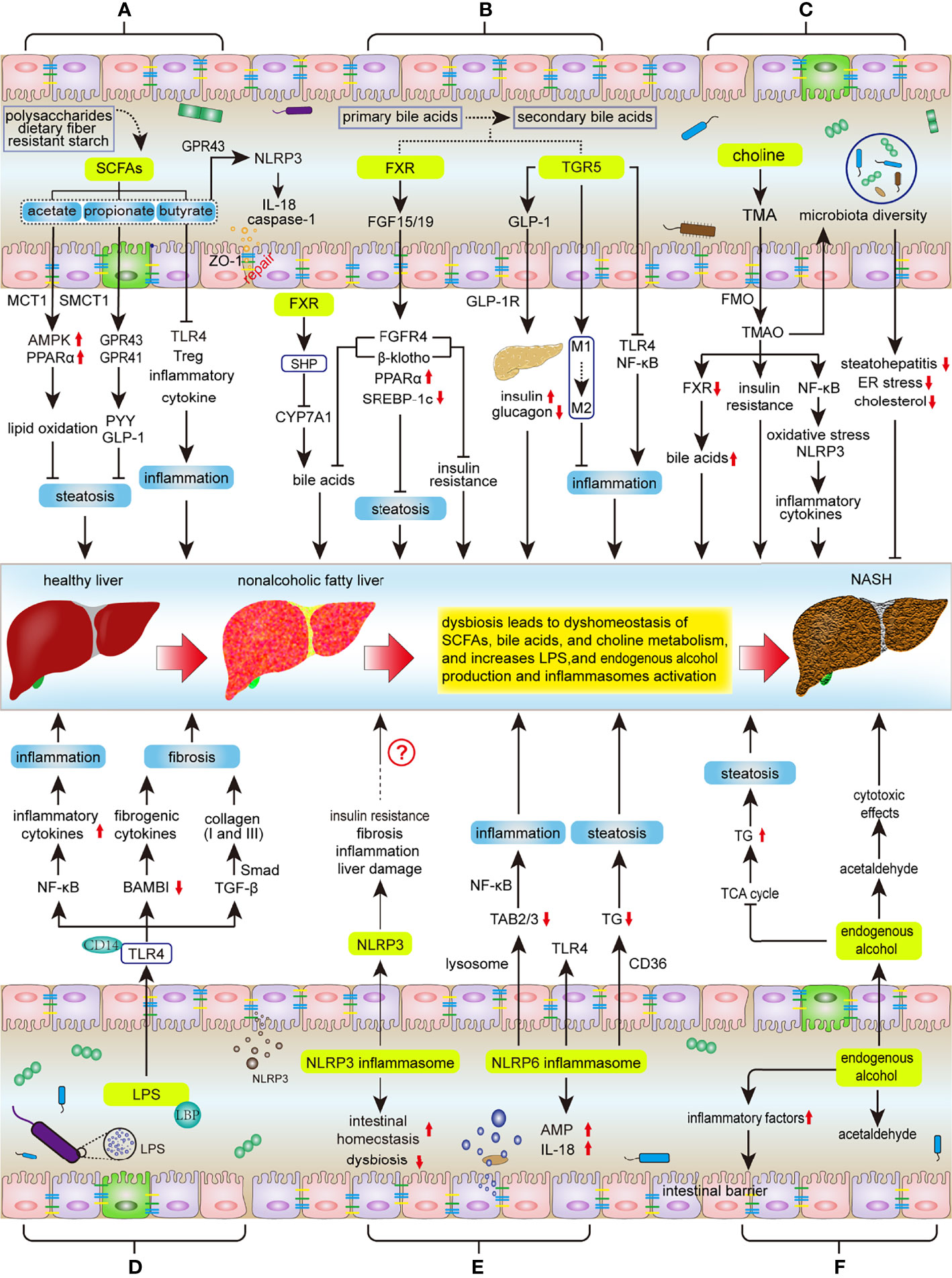
Figure 1 Key mechanisms involved in the regulation of intestinal microbiota during NASH progression. Intestinal dysbiosis results in disruption of intestinal SCFAs, bile acids, and choline metabolic homeostasis, as well as increases LPS and endogenous alcohol production and NLPR3/6 activation, subsequently affecting the progression of ANSH: (A) SCFAs inhibit hepatic steatosis, inflammation, and protect the integrity of the intestinal barrier. Dysbiosis decreases SCFA production, thereby promoting the NASH process. (B) The metabolism of bile acids is regulated by FXR and TGR5. FXR signaling suppresses hepatic steatosis and insulin resistance, as well as negative feedback inhibits bile acid synthesis; TGR5 can protect the liver from inflammation and insulin resistance. However, dysbiosis will reduce the activity of FXR and TGR5 signaling. (C) Intestinal microbiota metabolizes choline to TMAO, but the effect of TMAO on NASH is controversial. (D) LPS mainly affects the progress of NASH through LPS-TLR4 and NF-κB signaling pathways, including hepatic inflammation, fibrosis and liver injury. (E) Activation of NLRP3 in the liver promotes liver damage, but NLRP3 in the intestine maintains intestinal homeostasis and improves intestinal dysbiosis. NLRP6 inhibits NASH progression by inhibiting TLR4/NF-κB signaling and TG accumulation and promoting AMP and IL-18 secretion. (F) Intestinal microbiota increases the production of endogenous alcohol and promotes the progress of NASH. SCFAs, short chain fatty acids; MCT1, monocarboxylate transporter 1; SMCT1, sodium‐coupled monocarboxylate transporter 1; AMPK, AMP activated protein kinase; PPARα, Peroxisome proliferator-activated receptor α; GPR41/43, G protein-coupled receptor 41/43; IL-18, Interleukin 18; PYY, peptide YY; GLP1, Glucagon like peptide 1; TLR4, Toll-like Receptor 4; Treg, regulatory T; FXR, Farnesol X receptor; LRH-1, liver receptor homolog 1; CYP7A1, cholesterol 7a hydroxylated enzyme; FGF15/19, fibroblast growth factors 15/19; FGFR4, fibroblast growth factor receptor 4; SREBP-1c, sterol regulatory element-binding protein 1c; TGR5, Takeda G protein-coupled receptor 5; GLP-1R, GLP-1 receptor; NF-κB, nuclear factor-kappaB, TMA, trimethylamine; TMAO, trimethylamine-N-oxide; FMO, flavin monooxygenases; LPS, lipopolysaccharide; LBP, LPS binding protein; TGF-β, transforming growth factor-β; BAMBI, bone morphogenetic protein and active membrane-bound inhibitor; AMPs, antimicrobial peptides; NLRP3/6, nucleotide-binding domain, leucine-rich-repeat containing family, pyrin domain-containing 3/6; TAB2/3, TGF-β activated kinase 1 binding protein 2/3; TG, triglyceride; TCA, tricarboxylic acid.
Animal studies have shown that SCFAs can activate AMP-activated protein kinase (AMPK) to accelerate fatty acid oxidation and inhibit hepatic lipogenesis, which leads to a decrease of hepatic lipid accumulation (23). For instance, Araújo et al. found that acetate produced by E. coli is absorbed by intestinal epithelial cells and metabolized into acetyl-CoA and AMP, as well as upregulates the AMPK/PGC-1α/PPARα pathway, subsequently promoting lipid oxidation (24). Acetate and propionate act as ligands to activate G protein-coupled receptors 41 (GPR41) and GPR43, leading to an increase of peptide YY (PYY) and glucagon-like peptide-1 (GLP-1) secretion, and thereby inhibiting energy intake (23). Butyrate increases hepatic GLP-1 receptor (GLP-1R) expression by inhibiting histone deacetylase-2 (HDAC-2) and activating AMPK (25). In adipocytes, SCFAs promote leptin expression by binding to GPR41 to promote glycometabolism and lipid and energy metabolism and to inhibit fatty acid synthesis (6).
In addition, SCFAs are beneficial to reducing hepatic inflammatory responses. SCFAs beneficially maintain the integrity of the intestinal barrier, which prevents intestinal toxins (e.g. LPS) from invading the liver (20). In colon epithelial cells, SCFAs activate the NLRP3 inflammasome by binding to GPR43, leading to caspase-1 activation and IL-18 release, thereby promoting epithelial repair (26). Butyrate can reduce endotoxin levels and hepatic inflammation by decreasing the expression of Toll-like Receptor 4 (TLR4) and CD14 and ameliorating dysbiosis and intestinal barrier function (increased claudin-1 and ZO-1 expression) (27, 28). Moreover, SCFAs reduces the generation of regulatory T (Treg) cells and inflammatory cytokines by inhibiting the activity of histone acetyltransferases, thereby reducing hepatic inflammatory responses (20). In methionine/choline-deficient diet (MCD)-induced NASH mouse model, Deng et al. demonstrated that SCFAs (sodium acetate) alleviated hepatic steatosis and inflammation by activating AMPK and inhibiting macrophage proinflammatory activation, respectively (23). In line with this result, Olaniyi et al. found that acetate ameliorates hepatic lipid dysregulation by inhibiting HDAC and enhancing insulin sensitivity (29). Therefore, SCFAs can be regarded as a novel and viable therapeutic compound for preventing and alleviating NASH.
As the prominent bacteria in the intestine, intestinal microbiota such as Bifidobacterium, Bacteroides, Lactobacillus, Clostridium, Escherichia, Ruminococcus and Fusobacterium play a pivotal role in the metabolism of bile acids by metabolizing primary bile acids into secondary bile acids (10, 30). The metabolism of bile acid plays an important role in liver homeostasis and regulates the NASH process by activating the bile acid receptors, including farnesoid X receptor (FXR) and membrane Takeda G protein-coupled receptor 5(TGR5) (Figure 1) (31). In the liver, FXR activates small heterodimer partner (SHP), which forms a polymeride with liver receptor homolog 1 (LRH-1), thereby suppressing the activity of cholesterol 7a hydroxylated enzyme (CYP7A1, a rate-limiting enzyme of bile acid synthesis) (32). In the gut, FXR promotes the expression of fibroblast growth factors FGF15/19, and further activates fibroblast growth factor receptor 4 (FGFR4) and β-klotho in hepatocytes, thereby inhibiting bile acid synthesis and reducing hepatic steatosis and insulin resistance (33, 34). In addition, FXR can also inhibit hepatic lipogenesis by activating PPARα and repressing the expression of SREBP-1c in SHP-79- and FGF 15/19-dependent manners (35).
In a western diet-induced NASH model, TGR5 activation stimulates the production of GLP-1 in intestinal endocrine cells, thereby increasing insulin secretion and decreasing glucagon synthesis by binding to GLP-1R in β-cells (36). TGR5 also suppresses inflammation by promoting the transformation of macrophages from the M1 to M2 phenotype and suppressing the activity of TLR4-NF-κB pathway (37, 38). Furthermore, TGR5 increases energy expenses by increasing the activity of thyroid hormone activating enzyme deiodinase 2, which increases conversion of T4 to T3 (39).
As reported, the abundance of bacteria that converts primary bile acids to secondary bile acids was decreased under NASH condition (10). Intestinal dysbiosis contributes to the disorders of bile acid metabolism, and results in an insufficient activation of FXR and TGR5, ultimately leading to lipogenesis and inflammation (10). Therefore, restoring the intestinal microbiota involved in bile acid metabolism in NASH patients is a promising treatment option. Previous studies have showed that the administration of probiotics altered the bile acid composition (40), and bile acid-based therapies, including hepatic FXR agonists, FGF15/19 analog, are considered for NASH therapy (31). A recent clinical trial (NCT02443116) showed that aldafermin, a FGF19 analog, suppressed bile acid synthesis, led to an enrichment of Veillonella and corresponding changes in serum bile acids in patients with NASH (41). Obeticholic acid (OCA), a FXR agonist, has shown effectiveness in altering the intestinal microbiota, and effectively improves hepatic histological characteristics in patients with NASH (42, 43).
As an essential nutrient, choline was involved in liver lipid and cholesterol metabolism and signal transduction in bile acid enterohepatic circulation. Intestinal microbiota, such as Desulfovibrio desulfuricans, E. coli, Clostridium, Anaerococcus hydrogenalis and Klebsiella pneumoniae, can convert choline to trimethylamine (TMA) (44). TMA can be reabsorbed into the liver through the portal system, where it is metabolized by flavin monooxygenases (FMO) to generate trimethylamine-N-oxide (TMAO) (45). TMAO is correlated with the etiology and mortality of multiple diseases. Evidence indicates that higher serum level of TMAO was harmful to CVD and CKD (46), but controversial in NASH process (44, 47) (Figure 1).
Previous studies showed that TMAO aggravates liver steatosis by suppressing the activation of liver FXR signaling (48), upregulating glucose metabolism, and increasing insulin resistance (49). Furthermore, the increased TMAO levels induce the activation of the NF-κB pathway, promote oxidative stress and activate the NLRP3 inflammasome, thereby increasing the release of inflammatory cytokines such as IL-18 and IL-1β (46). However, recent evidence has demonstrated that TMAO modulated intestinal microbiota diversity, improved the histological alterations of steatohepatitis, alleviated hepatic endoplasmic reticulum (ER) stress, and inhibited the absorption of intestinal cholesterol in a high fat-high cholesterol (HFHC)-induced NASH mouse model (50). Despite this inconsistent observation, it is generally believed that decreasing choline levels and increasing toxic choline metabolites are crucial mechanisms by which the intestinal microbiota promotes NASH progression (51), which is the reason why MCD was considered a common dietary pattern for inducing NASH models.
LPS, a component of gram-negative bacteria, has been identified as a major factor in NASH (45, 52). The association between LPS and intestinal dysbiosis has been well reported (45), the increased Bilophila wadsworthia, Atopobium spp., Clostridium cocleatum and decreased Bifidobacterium, Bacteroides, and Eubacterium, all leading to an increase of serum LPS concentrations (53).
LPS deteriorates NASH progression mainly by inducing the hepatic inflammatory response and fibrosis via LPS/TLR4 and NF-κB signaling pathways(Figure 1) in hepatocytes, hepatic stellate cells (HSCs) and Kupfer cells (54). LPS binds to LPS-binding protein (LBP) and then activates CD14-TLR4 to form a complex, subsequently promoting the release of inflammatory cytokines such as NF-κB (54). Carpino et al. found that LPS induces the activation of macrophages and platelets through the TLR4 pathway, thereby eliciting liver damage (52). LPS also promotes the expression of TGF-β, which induces the transcription of type I and III collagen via Smad-dependent pathways, thereby promoting hepatic fibrosis (55). In HSC cell line, TLR4 directly downregulates the bone morphogenetic protein and active membrane-bound inhibitor (BAMBI) to produce fibrogenic cytokines and activate TGF-β-mediated HSCs (56). Moreover, LPS directly induces oxidative stress, which is one of the most important pathological events in the development of NASH (54), targeting intestinal microbiota offers a therapy potential to reduce LPS concentrations and ameliorate NASH (57, 58).
Inflammasomes are multiprotein complexes assembled from cytoplasmic pattern recognition receptor (PRR), which mainly include NLRP1, NLRP3, NLRC4, and AIM2 (59). To date, NLRP3 and NLRP6 are considered to be closely related to microbe-induced NASH (Figure 1). Intestinal dysbiosis promotes the entry of PAMPs, DAMPs and LPS into the portal circulation through the impaired intestinal barrier, which leads to the activation of NLRP3 and inflammation in the liver via TLR4 signaling and Kupffer cells (60). Mridha et al. found that MCC950, a NLRP3 selective inhibitor, reduces hepatic inflammation and fibrosis in MCD-fed mice by decreasing the expression of pro-IL-1β, and normalizing caspase 1, IL-6, IL-1β, and MCP-1 levels in the liver (61). HSCs engulfed NLRP3 particles increase IL-1β secretion and α-smooth muscle actin (α-SMA) expression, thereby inducing hepatic fibrosis (62). Moreover, Dong et al. found that NLRP3 activation in HSCs of mice exacerbated the progression of NASH to hepatic fibrosis through the TLR4-NF-κB signaling pathway (63). NLRP3 instigates insulin resistance (64), and participates in the transition from NAFLD to NASH (65). However, NLRP3 inflammasome deficiency exacerbates gut-liver axis derangement, dysbiosis, steatohepatitis and liver damage in HFHC-fed Nlrp3-/- mice (66). These studies suggest that NLRP3 may have different activities in different organs, thereby promoting liver damage and protecting intestinal permeability and preventing bacterial translocation (67).
NLRP6 is highly expressed in the small intestine and colon, especially in colonic goblet cells, myofibroblasts, and enterocytes (68). Intestinal microbiota induces NLRP6 signaling to produce IL-18 in mice, which is necessary for antimicrobial peptide (AMP) induction in the colonic mucosa (69). The deletion of NLRP6 alters the configuration of the intestinal microbiota, which leads to hepatic steatosis and inflammation via TLR4 signaling (70). In NASH mouse models, Huang et al. found that NLRP6 accelerates the degradation of TGF-β activated kinase 1 binding protein 2/3 (TAB2/3) through a lysosomal dependent pathway, thereby suppressing NF-κB mediated inflammatory responses (70). Also, NLRP6 inhibits hepatic TG accumulation by regulating CD36-mediated fatty acid uptake (70). Therefore, targeting the intestinal microbiota to regulate the activity of NLRP3 and NLRP6 is a promising therapeutic strategy to treat NASH.
Although NASH is defined as absence of alcohol intake, endogenous alcohol, produced by intestinal microbiota such as high-alcohol-producing Klebsiella pneumoniae (HiAlc-Kpn), Bacteroides, Bifidobacterium, and Escherichia, is a non-negligible pathogenic factor in NASH progression (11, 71). It has been reported that patients with NASH have more bacteria associated with elevated blood alcohol levels (72). Zhu et al. found that compared with healthy controls, the serum alcohol concentration was strikingly elevated, as well as the remarkably increased abundance of Escherichia in NASH patients (11). Similarly, Yuan et al. found that HiAlc-Kpn aggravates hepatic steatosis, inflammation, mitochondrial dysfunction and liver injury by producing excessive ethanol (71). On the one hand, alcohol increases the expression of intestinal inflammatory factors and destroys the intestinal barrier, which is associated with small intestinal bacterial overgrowth, and aggravates intestinal dysbiosis (73) (Figure 1). On the other hand, endogenous alcohol inhibits the tricarboxylic acid(TCA) cycle, and aggravates hepatic triglyceride accumulation and deposition (74). Furthermore, the toxic intermediates of alcohol metabolism (acetaldehyde) and alcohol disorders the function of intestinal tight junction proteins (75). Therefore, reducing the production of endogenous alcohol by targeting alcohol-producing bacteria in the intestine is a promising direction for NASH treatment.
To date, there are no Food and Drug Administration (FDA)-approved special drugs for NASH clinical treatment (76). Recently, an increasing number of clinical trials (Table 2) and animal experiments (Table 3) have attempted to prevent the development of NASH by targeting intestinal microbiota, including probiotics, prebiotics, synbiotics and FMT.

Table 2 Intestinal microbiota-targeted therapies of NASH-clinical trials.
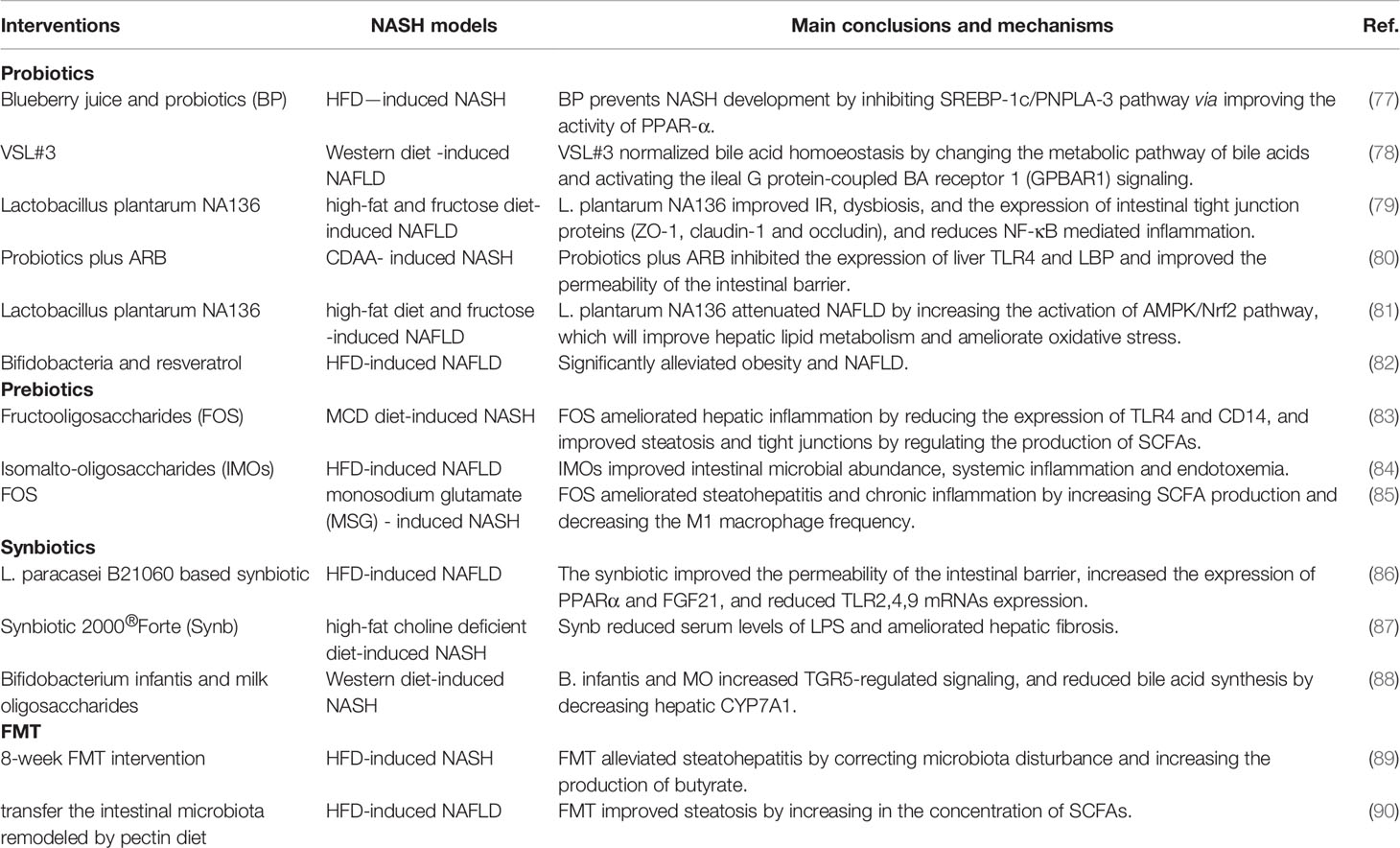
Table 3 Intestinal microbiota-targeted therapies of NASH-animal experiments.
Probiotics are living microorganisms that produce health benefits to the host, mainly including Lactobacillus, Bifidobacterium and Streptococcus (91). Both clinical and preclinical studies have congruously shown that probiotics dramatically ameliorate the histological spectrum of NASH (92, 93). For example, VSL#3 is a mixture of Streptococcus, Thermophilus, Bifidobacterium and Lactobacillus (94). The administration of VSL#3 significantly inhibited activation or production of JNK, NF-kB, α-SMA, metalloproteinases (MMP) and Cyclooxygenase 2 (COX-2), improved the intestinal permeability and alleviating oxidative stress, all of which were conductive to NASH therapy (94, 95).
Prebiotics, mainly including various oligofructoses (OFSs) or fructooligosaccharides (FOSs), refer to dietary supplements and indigestible food ingredients. Prebiotics improve host health by selectively stimulating the growth and activity of beneficial bacteria (96). Cani et al. found that prebiotics increased the production of proglucagon-derived peptide (GLP-2) in ob/ob mice, subsequently reducing plasma LPS levels and decreasing hepatic inflammatory and oxidative stress (97). Other studies have consistently shown that FOS attenuates hepatic lipid accumulation and steatohepatitis (84). In clinical trials, prebiotics have shown impressive efficacy in treating NASH, mainly by reducing serum alanine aminotransferase (ALT), aspartate aminotransferase (AST) levels and hepatic inflammation and increasing the abundance of Faecalibacterium prausnitzii and Bifidobacterium (98).
Synbiotics, the combination of probiotics and prebiotics, significantly inhibits NASH with liver histology improvement and inflammatory cytokines and endotoxin decrease (99). For instance, in a randomized clinical trial conducted by Ferolla et al. found that synbiotics supplementation effectively reduced hepatic steatosis and improved BMI and waist circumference (100). The mechanisms may be that synbiotics inhibited the subclinical pro-inflammatory signaling and reduced liver fat without altering the existing intestinal microbiota (100, 101).
FMT is an emerging and underexplored method to alternate the intestinal microbiota. FMT has been used in the treatment of a variety of gastrointestinal diseases, such as clostridioides difficile infections (102), diabetes (103) and irritable bowel syndrome (104). However, regrettably, there are no published clinical reports on the role of FMT in patients with NASH, but animal studies have shown that FMT treatment significantly improves the intestinal microbiota diversity and alleviates steatohepatitis (105–107). Zhou et al. found that FMT corrected the intestinal dysbiosis in HFD-fed mice and increased butyrate concentration and ZO-1 expression, thereby alleviating hepatic steatohepatitis (89). Vrieze et al. found that FMT from healthy donors to recipients with metabolic syndrome increased insulin sensitivity and butyric-producing microbiota of the recipient (108). Therefore, as a contemporary emerging therapeutic technology targeting the intestinal microbiota, FMT is undoubtedly regarded as a promising method for NASH treatment, but more clinical studies are urgently needed.
Due to the emergence of 16S ribosomal RNA sequencing and metagenomics technologies, great progress has been made in research on the intestinal microbiota in recent years. The intestinal microbiota is related to a variety of human diseases mainly through metabolic pathways, such as regulating lipometabolism and glycometabolism. Studies focusing on the intestinal microbiota provide satisfactory opportunities for the pathogenesis and treatment of NASH. However, great challenges have to be borne. Many pending issues and challenges in this field need to be elaborate urgently. For example, the changes in the composition of the intestinal microbiota in NASH patients and healthy individuals remain largely elusive and controversial. The specific mechanisms of dysbiosis affecting the progress of NASH need to be further understood. In addition, many studies have been conducted only on rodent models. More clinical evidence and/or results obtained from nonhuman primates are needed to validate the results from experiments conducted on rodent models. There have been many clinical trials using prebiotics, probiotics and synbiotics to treat NASH, but these trials are relatively new strategies that lack specific mechanisms and large clinical trials with comparative endpoints. Therefore, intestinal microbiota analysis based on metagenomics may become a promising direction for the diagnosis and treatment of NASH in the future. Ultimately, intestinal microbial targeted precision medicine in the treatment of NASH, as well as early, accurate and non-invasive diagnostic and prevention methods for NASH are expected to be established.
YC and Z-GS contributed to conception and design of the study. HX wrote the first draft of the manuscript. DS and XL reviewed and edited the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This work was supported by Chongqing University Three Gorges Hospital.
1. Loomba R, Lim JK, Patton H, El-Serag HB. AGA Clinical Practice Update on Screening and Surveillance for Hepatocellular Carcinoma in Patients With Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology (2020) 158:1822–30. doi: 10.1053/j.gastro.2019.12.053
2. Huang DQ, El-Serag HB, Loomba R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat Rev Gastroenterol Hepatol (2021) 18:223–38. doi: 10.1038/s41575-020-00381-6
3. Ajmera V, Loomba R. Imaging Biomarkers of NAFLD, NASH, and Fibrosis. Mol Metab (2021) 50:101167. doi: 10.1016/j.molmet.2021.101167
4. Kasper P, Martin A, Lang S, Kütting F, Goeser T, Demir M, et al. NAFLD and Cardiovascular Diseases: A Clinical Review. Clin Res Cardiol (2021) 110:921–37. doi: 10.1007/s00392-020-01709-7
5. Adams LA, Anstee QM, Tilg H, Targher G. Non-Alcoholic Fatty Liver Disease and Its Relationship With Cardiovascular Disease and Other Extrahepatic Diseases. Gut (2017) 66:1138–53. doi: 10.1136/gutjnl-2017-313884
6. Kolodziejczyk AA, Zheng D, Shibolet O, Elinav E. The Role of the Microbiome in NAFLD and NASH. EMBO Mol Med (2019) 11:e9302. doi: 10.15252/emmm.201809302
7. Sender R, Fuchs S, Milo R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell (2016) 164:337–40. doi: 10.1016/j.cell.2016.01.013
8. Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-Bacterial Mutualism in the Human Intestine. Science (2005) 307:1915–20. doi: 10.1126/science.1104816
9. Fan Y, Pedersen O. Gut Microbiota in Human Metabolic Health and Disease. Nat Rev Microbiol (2021) 19:55–71. doi: 10.1038/s41579-020-0433-9
10. Chen J, Vitetta L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. Int J Mol Sci (2020) 21:5214. doi: 10.3390/ijms21155214
11. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of Gut Microbiomes in Nonalcoholic Steatohepatitis (NASH) Patients: A Connection Between Endogenous Alcohol and NASH. Hepatology (2013) 57:601–9. doi: 10.1002/hep.26093
12. Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, et al. Intestinal Microbiota in Patients With Nonalcoholic Fatty Liver Disease. Hepatology (2013) 58:120–7. doi: 10.1002/hep.26319
13. Del Chierico F, Nobili V, Vernocchi P, Russo A, De Stefanis C, Gnani D, et al. Gut Microbiota Profiling of Pediatric Nonalcoholic Fatty Liver Disease and Obese Patients Unveiled by an Integrated Meta-Omics-Based Approach. Hepatology (2017) 65:451–64. doi: 10.1002/hep.28572
14. Schwimmer JB, Johnson JS, Angeles JE, Behling C, Belt PH, Borecki I, et al. Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease. Gastroenterology (2019) 157:1109–22. doi: 10.1053/j.gastro.2019.06.028
15. Astbury S, Atallah E, Vijay A, Aithal GP, Grove JI, Valdes AM. Lower Gut Microbiome Diversity and Higher Abundance of Proinflammatory Genus Collinsella Are Associated With Biopsy-Proven Nonalcoholic Steatohepatitis. Gut Microbes (2020) 11:569–80. doi: 10.1080/19490976.2019.1681861
16. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The Severity of Nonalcoholic Fatty Liver Disease Is Associated With Gut Dysbiosis and Shift in the Metabolic Function of the Gut Microbiota. Hepatology (2016) 63:764–75. doi: 10.1002/hep.28356
17. Sydor S, Best J, Messerschmidt I, Manka P, Vilchez-Vargas R, Brodesser S, et al. Altered Microbiota Diversity and Bile Acid Signaling in Cirrhotic and Noncirrhotic NASH-HCC. Clin Transl Gastroenterol (2020) 11:e00131. doi: 10.14309/ctg.0000000000000131
18. Michail S, Lin M, Frey MR, Fanter R, Paliy O, Hilbush B, et al. Altered Gut Microbial Energy and Metabolism in Children With Non-Alcoholic Fatty Liver Disease. FEMS Microbiol Ecol (2015) 91:1–9. doi: 10.1093/femsec/fiu002
19. Zhou D, Fan JG. Microbial Metabolites in Non-Alcoholic Fatty Liver Disease. World J Gastroenterol (2019) 25:2019–28. doi: 10.3748/wjg.v25.i17.2019
20. Zhang S, Zhao J, Xie F, He H, Johnston LJ, Dai X, et al. Dietary Fiber-Derived Short-Chain Fatty Acids: A Potential Therapeutic Target to Alleviate Obesity-Related Nonalcoholic Fatty Liver Disease. Obes Rev (2021) 22:e13316. doi: 10.1111/obr.13316
21. Ramakrishna BS. Role of the Gut Microbiota in Human Nutrition and Metabolism. J Gastroenterol Hepatol (2013) 28 Suppl 4:9–17. doi: 10.1111/jgh.12294
22. Albillos A, de Gottardi A, Rescigno M. The Gut-Liver Axis in Liver Disease: Pathophysiological Basis for Therapy. J Hepatol (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
23. Deng M, Qu F, Chen L, Liu C, Zhang M, Ren F, et al. SCFAs Alleviated Steatosis and Inflammation in Mice With NASH Induced by MCD. J Endocrinol (2020) 245:425–37. doi: 10.1530/joe-20-0018
24. Araújo JR, Tazi A, Burlen-Defranoux O, Vichier-Guerre S, Nigro G, Licandro H, et al. Fermentation Products of Commensal Bacteria Alter Enterocyte Lipid Metabolism. Cell Host Microbe (2020) 27:358–75.e7. doi: 10.1016/j.chom.2020.01.028
25. Zhou D, Chen YW, Zhao ZH, Yang RX, Xin FZ, Liu XL, et al. Sodium Butyrate Reduces High-Fat Diet-Induced Non-Alcoholic Steatohepatitis Through Upregulation of Hepatic GLP-1R Expression. Exp Mol Med (2018) 50:1–12. doi: 10.1038/s12276-018-0183-1
26. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-Sensing Receptors GPR43 and GPR109A Facilitate Dietary Fibre-Induced Gut Homeostasis Through Regulation of the Inflammasome. Nat Commun (2015) 6:6734. doi: 10.1038/ncomms7734
27. Ye J, Lv L, Wu W, Li Y, Shi D, Fang D, et al. Butyrate Protects Mice Against Methionine-Choline-Deficient Diet-Induced Non-Alcoholic Steatohepatitis by Improving Gut Barrier Function, Attenuating Inflammation and Reducing Endotoxin Levels. Front Microbiol (2018) 9:1967. doi: 10.3389/fmicb.2018.01967
28. Liu W, Luo X, Tang J, Mo Q, Zhong H, Zhang H, et al. A Bridge for Short-Chain Fatty Acids to Affect Inflammatory Bowel Disease, Type 1 Diabetes, and Non-Alcoholic Fatty Liver Disease Positively: By Changing Gut Barrier. Eur J Nutr (2021) 60:2317–30. doi: 10.1007/s00394-020-02431-w
29. Olaniyi KS, Amusa OA. Sodium Acetate-Mediated Inhibition of Histone Deacetylase Alleviates Hepatic Lipid Dysregulation and Its Accompanied Injury in Streptozotocin-Nicotinamide-Induced Diabetic Rats. BioMed Pharmacother (2020) 128:110226. doi: 10.1016/j.biopha.2020.110226
30. Jia W, Xie G, Jia W. Bile Acid-Microbiota Crosstalk in Gastrointestinal Inflammation and Carcinogenesis. Nat Rev Gastroenterol Hepatol (2018) 15:111–28. doi: 10.1038/nrgastro.2017.119
31. Li T, Chiang JYL. Bile Acid-Based Therapies for non-Alcoholic Steatohepatitis and Alcoholic Liver Disease. Hepatobiliary Surg Nutr (2020) 9:152–69. doi: 10.21037/hbsn.2019.09.03
32. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut Microbiota From Twins Discordant for Obesity Modulate Metabolism in Mice. Science (2013) 341:1241214. doi: 10.1126/science.1241214
33. Sun L, Pang Y, Wang X, Wu Q, Liu H, Liu B, et al. Ablation of Gut Microbiota Alleviates Obesity-Induced Hepatic Steatosis and Glucose Intolerance by Modulating Bile Acid Metabolism in Hamsters. Acta Pharm Sin B (2019) 9:702–10. doi: 10.1016/j.apsb.2019.02.004
34. Alvarez-Sola G, Uriarte I, Latasa MU, Fernandez-Barrena MG, Urtasun R, Elizalde M, et al. Fibroblast Growth Factor 15/19 (FGF15/19) Protects From Diet-Induced Hepatic Steatosis: Development of an FGF19-Based Chimeric Molecule to Promote Fatty Liver Regeneration. Gut (2017) 66:1818–28. doi: 10.1136/gutjnl-2016-312975
35. Chávez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology (2017) 152:1679–94.e3. doi: 10.1053/j.gastro.2017.01.055
36. Finn PD, Rodriguez D, Kohler J, Jiang Z, Wan S, Blanco E, et al. Intestinal TGR5 Agonism Improves Hepatic Steatosis and Insulin Sensitivity in Western Diet-Fed Mice. Am J Physiol Gastrointest Liver Physiol (2019) 316:G412–24. doi: 10.1152/ajpgi.00300.2018
37. Biagioli M, Carino A, Cipriani S, Francisci D, Marchianò S, Scarpelli P, et al. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice From Murine Colitis. J Immunol (2017) 199:718–33. doi: 10.4049/jimmunol.1700183
38. Hosseinkhani F, Heinken A, Thiele I, Lindenburg PW, Harms AC, Hankemeier T. The Contribution of Gut Bacterial Metabolites in the Human Immune Signaling Pathway of Non-Communicable Diseases. Gut Microbes (2021) 13:1–22. doi: 10.1080/19490976.2021.1882927
39. Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile Acids Induce Energy Expenditure by Promoting Intracellular Thyroid Hormone Activation. Nature (2006) 439:484–9. doi: 10.1038/nature04330
40. Winston JA, Theriot CM. Diversification of Host Bile Acids by Members of the Gut Microbiota. Gut Microbes (2020) 11:158–71. doi: 10.1080/19490976.2019.1674124
41. Loomba R, Ling L, Dinh DM, DePaoli AM, Lieu HD, Harrison SA, et al. The Commensal Microbe Veillonella as a Marker for Response to an FGF19 Analog in NASH. Hepatology (2021) 73:126–43. doi: 10.1002/hep.31523
42. Friedman ES, Li Y, Shen TD, Jiang J, Chau L, Adorini L, et al. FXR-Dependent Modulation of the Human Small Intestinal Microbiome by the Bile Acid Derivative Obeticholic Acid. Gastroenterology (2018) 155:1741–1752.e5. doi: 10.1053/j.gastro.2018.08.022
43. Siddiqui MS, Van Natta ML, Connelly MA, Vuppalanchi R, Neuschwander-Tetri BA, Tonascia J, et al. Impact of Obeticholic Acid on the Lipoprotein Profile in Patients With Non-Alcoholic Steatohepatitis. J Hepatol (2020) 72:25–33. doi: 10.1016/j.jhep.2019.10.006
44. Arias N, Arboleya S, Allison J, Kaliszewska A, Higarza SG, Gueimonde M, et al. The Relationship Between Choline Bioavailability From Diet, Intestinal Microbiota Composition, and Its Modulation of Human Diseases. Nutrients (2020) 12:2340. doi: 10.3390/nu12082340
45. Ji Y, Yin Y, Sun L, Zhang W. The Molecular and Mechanistic Insights Based on Gut-Liver Axis: Nutritional Target for Non-Alcoholic Fatty Liver Disease (NAFLD) Improvement. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21093066
46. Yang S, Li X, Yang F, Zhao R, Pan X, Liang J, et al. Gut Microbiota-Dependent Marker TMAO in Promoting Cardiovascular Disease: Inflammation Mechanism, Clinical Prognostic, and Potential as a Therapeutic Target. Front Pharmacol (2019) 10:1360. doi: 10.3389/fphar.2019.01360
47. León-Mimila P, Villamil-Ramírez H, Li XS, Shih DM, Hui ST, Ocampo-Medina E, et al. Trimethylamine N-Oxide Levels Are Associated With NASH in Obese Subjects With Type 2 Diabetes. Diabetes Metab (2020). doi: 10.1016/j.diabet.2020.07.010
48. Tan X, Liu Y, Long J, Chen S, Liao G, Wu S, et al. Trimethylamine N-Oxide Aggravates Liver Steatosis Through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol Nutr Food Res (2019) 63:e1900257. doi: 10.1002/mnfr.201900257
49. Janeiro MH, Ramírez MJ, Milagro FI, Martínez JA, Solas M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients (2018) 10:1398. doi: 10.3390/nu10101398
50. Zhao ZH, Xin FZ, Zhou D, Xue YQ, Liu XL, Yang RX, et al. Trimethylamine N-Oxide Attenuates High-Fat High-Cholesterol Diet-Induced Steatohepatitis by Reducing Hepatic Cholesterol Overload in Rats. World J Gastroenterol (2019) 25:2450–62. doi: 10.3748/wjg.v25.i20.2450
51. Hernandez GV, Smith VA, Melnyk M, Burd MA, Sprayberry KA, Edwards MS, et al. Dysregulated FXR-FGF19 Signaling and Choline Metabolism Are Associated With Gut Dysbiosis and Hyperplasia in a Novel Pig Model of Pediatric NASH. Am J Physiol Gastrointest Liver Physiol (2020) 318:G582–g609. doi: 10.1152/ajpgi.00344.2019
52. Carpino G, Del Ben M, Pastori D, Carnevale R, Baratta F, Overi D, et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology (2020) 72:470–85. doi: 10.1002/hep.31056
53. Natividad JM, Lamas B, Pham HP, Michel ML, Rainteau D, Bridonneau C, et al. Bilophila Wadsworthia Aggravates High Fat Diet Induced Metabolic Dysfunctions in Mice. Nat Commun (2018) 9:2802. doi: 10.1038/s41467-018-05249-7
54. Ferro D, Baratta F, Pastori D, Cocomello N, Colantoni A, Angelico F, et al. New Insights Into the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Gut-Derived Lipopolysaccharides and Oxidative Stress. Nutrients (2020) 12:2762. doi: 10.3390/nu12092762
55. Fabregat I, Moreno-Càceres J, Sánchez A, Dooley S, Dewidar B, Giannelli G, et al. TGF-β Signalling and Liver Disease. FEBS J (2016) 283:2219–32. doi: 10.1111/febs.13665
56. Liu C, Chen X, Yang L, Kisseleva T, Brenner DA, Seki E. Transcriptional Repression of the Transforming Growth Factor β (TGF-β) Pseudoreceptor BMP and Activin Membrane-Bound Inhibitor (BAMBI) by Nuclear Factor κB (NF-κB) P50 Enhances TGF-β Signaling in Hepatic Stellate Cells. J Biol Chem (2014) 289:7082–91. doi: 10.1074/jbc.M113.543769
57. Chen Y, Feng R, Yang X, Dai J, Huang M, Ji X, et al. Yogurt Improves Insulin Resistance and Liver Fat in Obese Women With Nonalcoholic Fatty Liver Disease and Metabolic Syndrome: A Randomized Controlled Trial. Am J Clin Nutr (2019) 109:1611–9. doi: 10.1093/ajcn/nqy358
58. Mu J, Tan F, Zhou X, Zhao X. Lactobacillus Fermentum CQPC06 in Naturally Fermented Pickles Prevents Non-Alcoholic Fatty Liver Disease by Stabilizing the Gut-Liver Axis in Mice. Food Funct (2020) 11:8707–23. doi: 10.1039/d0fo01823f
59. Yan Z, Qi Z, Yang X, Ji N, Wang Y, Shi Q, et al. The NLRP3 Inflammasome: Multiple Activation Pathways and Its Role in Primary Cells During Ventricular Remodeling. J Cell Physiol (2021) 236:5547–63. doi: 10.1002/jcp.30285
60. Knorr J, Wree A, Tacke F, Feldstein AE. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin Liver Dis (2020) 40:298–306. doi: 10.1055/s-0040-1708540
61. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental NASH in Mice. J Hepatol (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
62. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, et al. Hepatocyte Pyroptosis and Release of Inflammasome Particles Induce Stellate Cell Activation and Liver Fibrosis. J Hepatol (2021) 74:156–67. doi: 10.1016/j.jhep.2020.07.041
63. Dong Z, Zhuang Q, Ning M, Wu S, Lu L, Wan X. Palmitic Acid Stimulates NLRP3 Inflammasome Activation Through TLR4-NF-κB Signal Pathway in Hepatic Stellate Cells. Ann Transl Med (2020) 8:168. doi: 10.21037/atm.2020.02.21
64. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat Med (2011) 17:179–88. doi: 10.1038/nm.2279
65. Zhang NP, Liu XJ, Xie L, Shen XZ, Wu J. Impaired Mitophagy Triggers NLRP3 Inflammasome Activation During the Progression From Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis. Lab Invest (2019) 99:749–63. doi: 10.1038/s41374-018-0177-6
66. Pierantonelli I, Rychlicki C, Agostinelli L, Giordano DM, Gaggini M, Fraumene C, et al. Lack of NLRP3-Inflammasome Leads to Gut-Liver Axis Derangement, Gut Dysbiosis and a Worsened Phenotype in a Mouse Model of NAFLD. Sci Rep (2017) 7:12200. doi: 10.1038/s41598-017-11744-6
67. De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, et al. Dysbiosis Contributes to Fibrogenesis in the Course of Chronic Liver Injury in Mice. Hepatology (2014) 59:1738–49. doi: 10.1002/hep.26695
68. Ghimire L, Paudel S, Jin L, Jeyaseelan S. The NLRP6 Inflammasome in Health and Disease. Mucosal Immunol (2020) 13:388–98. doi: 10.1038/s41385-020-0256-z
69. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell (2015) 163:1428–43. doi: 10.1016/j.cell.2015.10.048
70. Huang C, Liu Q, Tang Q, Jing X, Wu T, Zhang J, et al. Hepatocyte-Specific Deletion of Nlrp6 in Mice Exacerbates the Development of Non-Alcoholic Steatohepatitis. Free Radic Biol Med (2021) 169:110–21. doi: 10.1016/j.freeradbiomed.2021.04.008
71. Yuan J, Chen C, Cui J, Lu J, Yan C, Wei X, et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella Pneumoniae. Cell Metab (2019) 30:675–688.e7. doi: 10.1016/j.cmet.2019.08.018
72. Zhu L, Baker RD, Zhu R, Baker SS. Gut Microbiota Produce Alcohol and Contribute to NAFLD. Gut (2016) 65:1232. doi: 10.1136/gutjnl-2016-311571
73. Meroni M, Longo M, Dongiovanni P. Alcohol or Gut Microbiota: Who Is the Guilty? Int J Mol Sci (2019) 20. doi: 10.3390/ijms20184568
74. Alves-Bezerra M, Cohen DE. Triglyceride Metabolism in the Liver. Compr Physiol (2017) 8:1–8. doi: 10.1002/cphy.c170012
75. Elamin E, Masclee A, Troost F, Dekker J, Jonkers D. Cytotoxicity and Metabolic Stress Induced by Acetaldehyde in Human Intestinal LS174T Goblet-Like Cells. Am J Physiol Gastrointest Liver Physiol (2014) 307:G286–94. doi: 10.1152/ajpgi.00103.2014
76. Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic Steatohepatitis: A Review. Jama (2020) 323:1175–83. doi: 10.1001/jama.2020.2298
77. Ren T, Zhu J, Zhu L, Cheng M. The Combination of Blueberry Juice and Probiotics Ameliorate Non-Alcoholic Steatohepatitis (NASH) by Affecting SREBP-1c/PNPLA-3 Pathway via PPAR-α. Nutrients (2017) 9:198. doi: 10.3390/nu9030198
78. Jena PK, Sheng L, Li Y, Wan YY. Probiotics VSL#3 Are Effective in Reversing Non-Alcoholic Steatohepatitis in a Mouse Model. Hepatobiliary Surg Nutr (2020) 9:170–82. doi: 10.21037/hbsn.2019.09.07
79. Zhao Z, Chen L, Zhao Y, Wang C, Duan C, Yang G, et al. Lactobacillus Plantarum NA136 Ameliorates Nonalcoholic Fatty Liver Disease by Modulating Gut Microbiota, Improving Intestinal Barrier Integrity, and Attenuating Inflammation. Appl Microbiol Biotechnol (2020) 104:5273–82. doi: 10.1007/s00253-020-10633-9
80. Sawada Y, Kawaratani H, Kubo T, Fujinaga Y, Furukawa M, Saikawa S, et al. Combining Probiotics and an Angiotensin-II Type 1 Receptor Blocker has Beneficial Effects on Hepatic Fibrogenesis in a Rat Model of Non-Alcoholic Steatohepatitis. Hepatol Res (2019) 49:284–95. doi: 10.1111/hepr.13281
81. Zhao Z, Wang C, Zhang L, Zhao Y, Duan C, Zhang X, et al. Lactobacillus Plantarum NA136 Improves the Non-Alcoholic Fatty Liver Disease by Modulating the AMPK/Nrf2 Pathway. Appl Microbiol Biotechnol (2019) 103:5843–50. doi: 10.1007/s00253-019-09703-4
82. Hu D, Yang W, Mao P, Cheng M. Combined Amelioration of Prebiotic Resveratrol and Probiotic Bifidobacteria on Obesity and Nonalcoholic Fatty Liver Disease. Nutr Cancer (2020) 73:1–10. doi: 10.1080/01635581.2020.1767166
83. Matsumoto K, Ichimura M, Tsuneyama K, Moritoki Y, Tsunashima H, Omagari K, et al. Fructo-Oligosaccharides and Intestinal Barrier Function in a Methionine-Choline-Deficient Mouse Model of Nonalcoholic Steatohepatitis. PloS One (2017) 12:e0175406. doi: 10.1371/journal.pone.0175406
84. Singh DP, Khare P, Zhu J, Kondepudi KK, Singh J, Baboota RK, et al. A Novel Cobiotic-Based Preventive Approach Against High-Fat Diet-Induced Adiposity, Nonalcoholic Fatty Liver and Gut Derangement in Mice. Int J Obes (Lond) (2016) 40:487–96. doi: 10.1038/ijo.2015.197
85. Takai A, Kikuchi K, Ichimura M, Tsuneyama K, Moritoki Y, Matsumoto K, et al. Fructo-Oligosaccharides Ameliorate Steatohepatitis, Visceral Adiposity, and Associated Chronic Inflammation via Increased Production of Short-Chain Fatty Acids in a Mouse Model of Non-Alcoholic Steatohepatitis. BMC Gastroenterol (2020) 20:46. doi: 10.1186/s12876-020-01194-2
86. Raso GM, Simeoli R, Iacono A, Santoro A, Amero P, Paciello O, et al. Effects of a Lactobacillus Paracasei B21060 Based Synbiotic on Steatosis, Insulin Signaling and Toll-Like Receptor Expression in Rats Fed a High-Fat Diet. J Nutr Biochem (2014) 25:81–90. doi: 10.1016/j.jnutbio.2013.09.006
87. Cortez-Pinto H, Borralho P, Machado J, Lopes MT, Gato IV, Santos AM, et al. Microbiota Modulation With Synbiotic Decreases Liver Fibrosis in a High Fat Choline Deficient Diet Mice Model of Non-Alcoholic Steatohepatitis (NASH). GE Port J Gastroenterol (2016) 23:132–41. doi: 10.1016/j.jpge.2016.01.004
88. Jena PK, Sheng L, Nagar N, Wu C, Barile D, Mills DA, et al. Synbiotics Bifidobacterium Infantis and Milk Oligosaccharides Are Effective in Reversing Cancer-Prone Nonalcoholic Steatohepatitis Using Western Diet-Fed FXR Knockout Mouse Models. J Nutr Biochem (2018) 57:246–54. doi: 10.1016/j.jnutbio.2018.04.007
89. Zhou D, Pan Q, Shen F, Cao HX, Ding WJ, Chen YW, et al. Total Fecal Microbiota Transplantation Alleviates High-Fat Diet-Induced Steatohepatitis in Mice via Beneficial Regulation of Gut Microbiota. Sci Rep (2017) 7:1529. doi: 10.1038/s41598-017-01751-y
90. Houron C, Ciocan D, Trainel N, Mercier-Nomé F, Hugot C, Spatz M, et al. Gut Microbiota Reshaped by Pectin Treatment Improves Liver Steatosis in Obese Mice. Nutrients (2021) 13:3725. doi: 10.3390/nu13113725
91. Ebrahimzadeh Leylabadlo H, Ghotaslou R, Samadi Kafil H, Feizabadi MM, Moaddab SY, Farajnia S, et al. Non-Alcoholic Fatty Liver Diseases: From Role of Gut Microbiota to Microbial-Based Therapies. Eur J Clin Microbiol Infect Dis (2020) 39:613–27. doi: 10.1007/s10096-019-03746-1
92. Mohammed SK, Magdy YM, El-Waseef DA, Nabih ES, Hamouda MA, El-Kharashi OA. Modulation of Hippocampal TLR4/BDNF Signal Pathway Using Probiotics Is a Step Closer Towards Treating Cognitive Impairment in NASH Model. Physiol Behav (2020) 214:112762. doi: 10.1016/j.physbeh.2019.112762
93. Sharpton SR, Maraj B, Harding-Theobald E, Vittinghoff E, Terrault NA. Gut Microbiome-Targeted Therapies in Nonalcoholic Fatty Liver Disease: A Systematic Review, Meta-Analysis, and Meta-Regression. Am J Clin Nutr (2019) 110:139–49. doi: 10.1093/ajcn/nqz042
94. Meroni M, Longo M, Dongiovanni P. The Role of Probiotics in Nonalcoholic Fatty Liver Disease: A New Insight Into Therapeutic Strategies. Nutrients (2019) 11:2642. doi: 10.3390/nu11112642
95. Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and Antibodies to TNF Inhibit Inflammatory Activity and Improve Nonalcoholic Fatty Liver Disease. Hepatology (2003) 37:343–50. doi: 10.1053/jhep.2003.50048
96. Holscher HD. Dietary Fiber and Prebiotics and the Gastrointestinal Microbiota. Gut Microbes (2017) 8:172–84. doi: 10.1080/19490976.2017.1290756
97. Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, et al. Changes in Gut Microbiota Control Inflammation in Obese Mice Through a Mechanism Involving GLP-2-Driven Improvement of Gut Permeability. Gut (2009) 58:1091–103. doi: 10.1136/gut.2008.165886
98. Bomhof MR, Parnell JA, Ramay HR, Crotty P, Rioux KP, Probert CS, et al. Histological Improvement of Non-Alcoholic Steatohepatitis With a Prebiotic: A Pilot Clinical Trial. Eur J Nutr (2019) 58:1735–45. doi: 10.1007/s00394-018-1721-2
99. Kobyliak N, Abenavoli L, Mykhalchyshyn G, Kononenko L, Boccuto L, Kyriienko D, et al. A Multi-Strain Probiotic Reduces the Fatty Liver Index, Cytokines and Aminotransferase Levels in NAFLD Patients: Evidence From a Randomized Clinical Trial. J Gastrointestin Liver Dis (2018) 27:41–9. doi: 10.15403/jgld.2014.1121.271.kby
100. Ferolla SM, Couto CA, Costa-Silva L, Armiliato GN, Pereira CA, Martins FS, et al. Beneficial Effect of Synbiotic Supplementation on Hepatic Steatosis and Anthropometric Parameters, But Not on Gut Permeability in a Population With Nonalcoholic Steatohepatitis. Nutrients (2016) 8:397. doi: 10.3390/nu8070397
101. Shoelson SE, Herrero L, Naaz A. Obesity, Inflammation, and Insulin Resistance. Gastroenterology (2007) 132:2169–80. doi: 10.1053/j.gastro.2007.03.059
102. Kelly CR, Yen EF, Grinspan AM, Kahn SA, Atreja A, Lewis JD, et al. Fecal Microbiota Transplantation Is Highly Effective in Real-World Practice: Initial Results From the FMT National Registry. Gastroenterology (2021) 160:183–192.e3. doi: 10.1053/j.gastro.2020.09.038
103. de Groot P, Nikolic T, Pellegrini S, Sordi V, Imangaliyev S, Rampanelli E, et al. Faecal Microbiota Transplantation Halts Progression of Human New-Onset Type 1 Diabetes in a Randomised Controlled Trial. Gut (2021) 70:92–105. doi: 10.1136/gutjnl-2020-322630
104. Holvoet T, Joossens M, Vázquez-Castellanos JF, Christiaens E, Heyerick L, Boelens J, et al. Fecal Microbiota Transplantation Reduces Symptoms in Some Patients With Irritable Bowel Syndrome With Predominant Abdominal Bloating: Short- and Long-Term Results From a Placebo-Controlled Randomized Trial. Gastroenterology (2021) 160:145–157.e8. doi: 10.1053/j.gastro.2020.07.013
105. Witjes JJ, Smits LP, Pekmez CT, Prodan A, Meijnikman AS, Troelstra MA, et al. Donor Fecal Microbiota Transplantation Alters Gut Microbiota and Metabolites in Obese Individuals With Steatohepatitis. Hepatol Commun (2020) 4:1578–90. doi: 10.1002/hep4.1601
106. Delaune V, Orci LA, Lacotte S, Peloso A, Schrenzel J, Lazarevic V, et al. Fecal Microbiota Transplantation: A Promising Strategy in Preventing the Progression of Non-Alcoholic Steatohepatitis and Improving the Anti-Cancer Immune Response. Expert Opin Biol Ther (2018) 18:1061–71. doi: 10.1080/14712598.2018.1518424
107. Craven L, Rahman A, Nair Parvathy S, Beaton M, Silverman J, Qumosani K, et al. Allogenic Fecal Microbiota Transplantation in Patients With Nonalcoholic Fatty Liver Disease Improves Abnormal Small Intestinal Permeability: A Randomized Control Trial. Am J Gastroenterol (2020) 115:1055–65. doi: 10.14309/ajg.0000000000000661
Keywords: intestinal microbiota, nonalcoholic steatohepatitis, SCFAs, choline, inflammation, gut-liver axis
Citation: Xiang H, Sun D, Liu X, She Z-G and Chen Y (2022) The Role of the Intestinal Microbiota in Nonalcoholic Steatohepatitis. Front. Endocrinol. 13:812610. doi: 10.3389/fendo.2022.812610
Received: 10 November 2021; Accepted: 04 January 2022;
Published: 08 February 2022.
Edited by:
Jo Edward Lewis, University of Cambridge, United KingdomReviewed by:
Yan Chen, University of Chinese Academy of Sciences, ChinaCopyright © 2022 Xiang, Sun, Liu, She and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Xiang, eGlhbmdodWk5MzEyQDE2My5jb20=; Zhi-Gang She, emdzaGVAd2h1LmVkdS5jbg==; Yonghong Chen, d3pjaHktMTIzQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.