
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 25 January 2023
Sec. Adrenal Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1070074
This article is part of the Research TopicAdvances in Diagnostics and Management of Adrenal TumorsView all 24 articles
 Sara Mellid1
Sara Mellid1 Eduardo Gil1Rocío Letón1Eduardo Caleiras2Emiliano Honrado3
Eduardo Gil1Rocío Letón1Eduardo Caleiras2Emiliano Honrado3 Susan Richter4
Susan Richter4 Nuria Palacios5Marcos Lahera6
Nuria Palacios5Marcos Lahera6 Juan C. Galofré7Adriá López-Fernández8
Juan C. Galofré7Adriá López-Fernández8 Maria Calatayud9
Maria Calatayud9 Aura D. Herrera-Martínez10
Aura D. Herrera-Martínez10 María A. Galvez10
María A. Galvez10 Xavier Matias-Guiu11Milagros Balbín12Esther Korpershoek13Eugénie S. Lim14
Xavier Matias-Guiu11Milagros Balbín12Esther Korpershoek13Eugénie S. Lim14 Francesca Maletta15
Francesca Maletta15 Sofia Lider16
Sofia Lider16 Stephanie M. J. Fliedner17
Stephanie M. J. Fliedner17 Nicole Bechmann4
Nicole Bechmann4 Graeme Eisenhofer4,18
Graeme Eisenhofer4,18 Letizia Canu19
Letizia Canu19 Elena Rapizzi19
Elena Rapizzi19 Irina Bancos20
Irina Bancos20 Mercedes Robledo1,21
Mercedes Robledo1,21 Alberto Cascón1,21*
Alberto Cascón1,21*Introduction: The percentage of patients diagnosed with pheochromocytoma and paraganglioma (altogether PPGL) carrying known germline mutations in one of the over fifteen susceptibility genes identified to date has dramatically increased during the last two decades, accounting for up to 35-40% of PPGL patients. Moreover, the application of NGS to the diagnosis of PPGL detects unexpected co-occurrences of pathogenic allelic variants in different susceptibility genes.
Methods: Herein we uncover several cases with dual mutations in NF1 and other PPGL genes by targeted sequencing. We studied the molecular characteristics of the tumours with co-occurrent mutations, using omic tools to gain insight into the role of these events in tumour development.
Results: Amongst 23 patients carrying germline NF1 mutations, targeted sequencing revealed additional pathogenic germline variants in DLST (n=1) and MDH2 (n=2), and two somatic mutations in H3-3A and PRKAR1A. Three additional patients, with somatic mutations in NF1 were found carrying germline pathogenic mutations in SDHB or DLST, and a somatic truncating mutation in ATRX. Two of the cases with dual germline mutations showed multiple pheochromocytomas or extra-adrenal paragangliomas - an extremely rare clinical finding in NF1 patients. Transcriptional and methylation profiling and metabolite assessment showed an “intermediate signature” to suggest that both variants had a pathological role in tumour development.
Discussion: In conclusion, mutations affecting genes involved in different pathways (pseudohypoxic and receptor tyrosine kinase signalling) co-occurring in the same patient could provide a selective advantage for the development of PPGL, and explain the variable expressivity and incomplete penetrance observed in some patients.
Over the last two decades, the number of genes identified as involved in the hereditary susceptibility to developing pheochromocytoma (PCC) or paraganglioma (PGL), collectively known as PPGLs, has grown to more than fifteen, increasing the percentage of patients carrying known germline predisposing mutations to 35-40% (1). Furthermore, the prevalence and penetrance of PPGL-associated mutations is highly variable and parent-of-origin effects have been identified for at least, three susceptibility genes (SDHD, SDHAF2 and MAX), making genetic diagnosis of the disease challenging. Comprehensive gene panels or exome sequencing, which allow the examination of all predisposing genes in one test, have replaced complicated algorithms employed for genetic testing of PPGL patients, and as a result of the shift from sequential gene testing to next-generation sequencing, unexpected genetic variants have been discovered. In this regard, two recent studies describe the presence of pathogenic germline variants in both NF1 and SDHD genes in patients with multiple PPGLs (2, 3), one of whom had no clinical sign of neurofibromatosis type 1 (NF1; MIM #162200). The presence of germline variants in more than one susceptibility gene co-occurring in the same patient adds a further layer of complexity to the genetic diagnosis of the disease, and suggests that a more intricate genetic condition accounts for the development of PPGL in some NF1 patients.
Development of PPGL in patients with NF1 is thought to be very rare (0.1-5.7%) (4–6). A recent study revealed that only 0.3% (1/342) of patients with NF1 developed PPGL (7), while an exhaustive study for the presence of PPGL amongst patients diagnosed with neurofibromatosis (n=156), found that up to 7.7% of patients developed PPGL (8). This percentage could be underestimated since the use of targeted NGS has led to the identification of NF1 germline mutations in PPGL patients without a clear clinical diagnosis of NF1 (3, 9–12). Nevertheless, the percentage of patients developing PPGL amongst NF1 patients contrasts with that observed for other PPGL syndromes, e.g. 20% in von Hippel-Lindau (VHL; MIM #193300) and 50% in multiple endocrine neoplasia type 2A (MEN2; MIM #171400) (13, 14). Moreover, PPGLs are multifocal or bilateral in 43-45% of VHL cases and in 50-80% of MEN2 cases, compared to 16% in NF1 patients. These differences point to still unknown mechanisms that could account for the development of PPGL in NF1 patients.
We recently identified three patients carrying NF1 germline mutations and additional variants in other PPGL susceptibility genes. Interestingly, two of the three cases showed quite uncommon clinical findings (bilateral PCC and extra-adrenal PGL). Therefore, herein we analyse, using a comprehensive gene panel, a series of NF1 patients with PPGL, enriched in cases with multiple PPGLs, to investigate the co-occurrence of NF1 mutations with causal variants in other PPGL susceptibility genes. Moreover, we study the molecular characteristics of the tumours with dual mutations, using various omic tools, to gain insight into the role of this genetic event in the development of PPGL.
Twenty-three patients diagnosed with PPGL and carrying a germline NF1 mutation were included in the study (Table 1). The series was enriched with patients developing bilateral PCC or extra-adrenal PGLs. In addition, three patients carrying known germline or somatic mutations in other PPGL susceptibility genes, and a somatic NF1 alteration were also included for discussion. A summary of the clinical data of the patients included in the study is provided in Table 2. Genomic DNA was extracted from available peripheral-blood leukocytes with the Maxwell Blood DNA-purification system (Promega). Tumour DNA was obtained from all patients with the DNeasy Blood and Tissue kit (QIAGEN) for frozen tissue and the Covaris S2 System (Covaris) for formalin-fixed paraffin-embedded (FFPE) tissue according to the manufacturers’ instructions. Informed written consent was obtained for all participants in the study. Ethical approval was granted by local ethical committees with the following reference numbers: 13-004137, 06/Q0104/133, 15/024, 88/11, 2011/0020149, 14/11.06.2020, PI54_2016-v2.
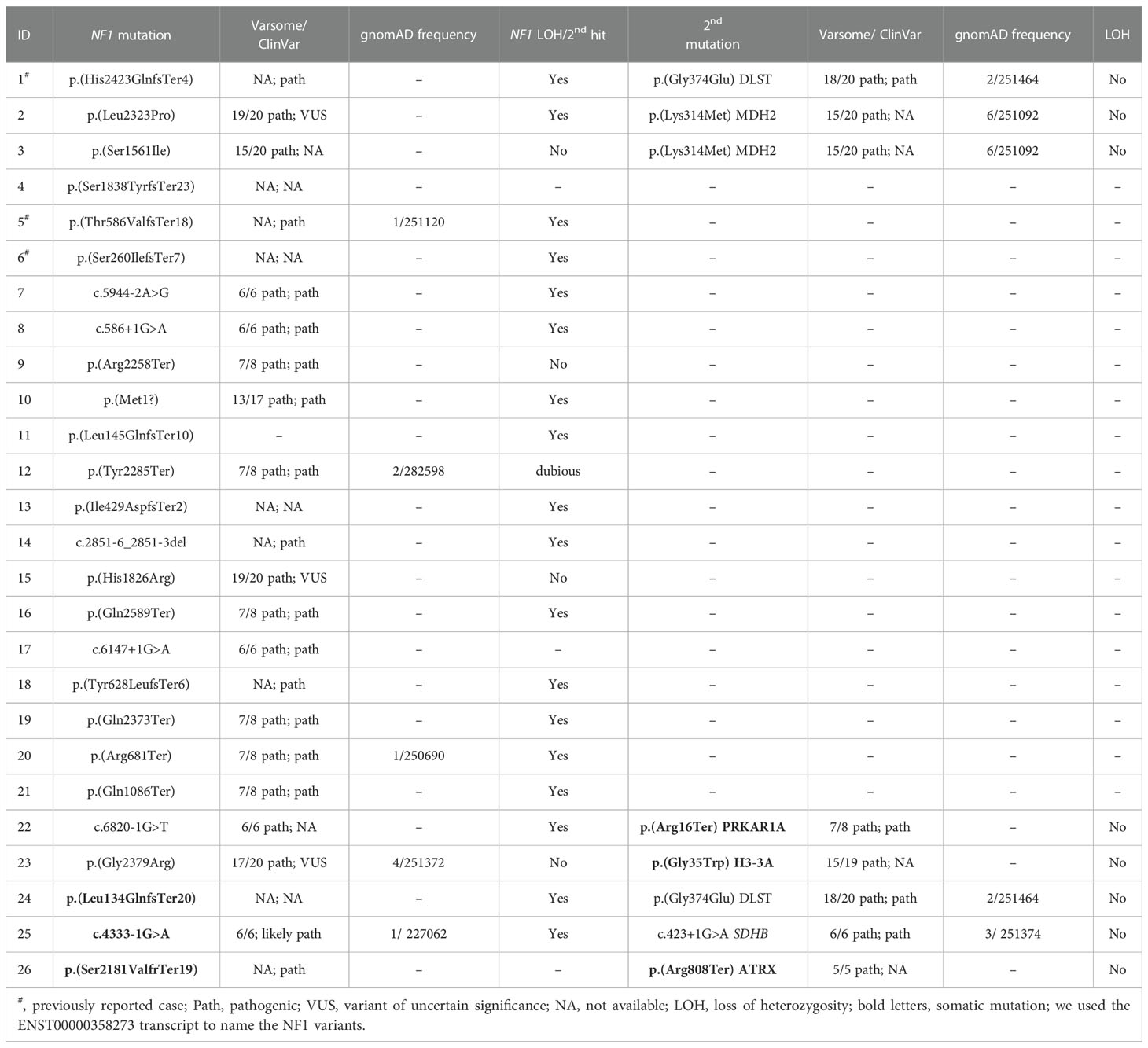
Table 1 Molecular findings identified in the patients included in the study.
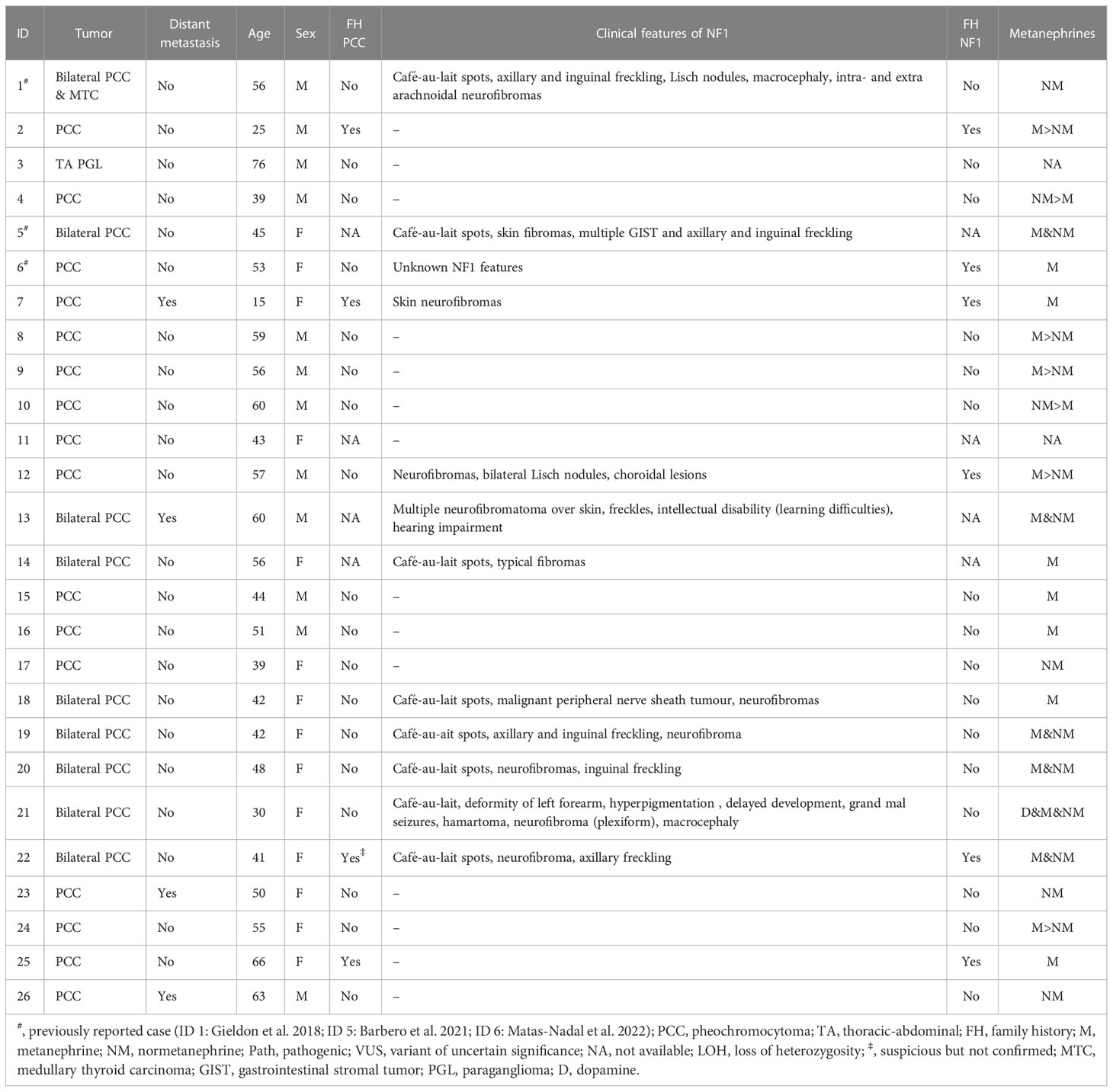
Table 2 Clinical data of the patients included in the study.
DNA obtained from blood or tumour samples was used for targeted sequencing. The diagnostic PPGL target panel included all susceptibility genes described so far, and some genes mutated somatically in PPGL, in total 33 genes (CSDE1, KIF1B, SDHA, SDHB, SDHC, SDHD, SDHAF1, SDHAF2, EGLN1, EGLN2, FH, MDH2, DLST, IDH2, IDH1, TMEM127, VHL, MET, RET, PTEN, HRAS, MEN1, KRAS, MAX, GOT2, NF1, PRKAR1A, ATRX, BRAF, EPAS1, SLC25A11, DNMT3A, H3F3A). TruSight (Illumina) oligo probes were designed for the genes of interest. DesignStudio software (Illumina) was used for the design of 891 amplicons. Once the library was prepared, sequencing was performed using a MiSeq sequencer (Illumina). Alignment of sequences was done by MiSeq Reporter and VariantStudio (Illumina), and variant calling was carried out by GATK software. Variants were filtered following standard quality and depth criteria and candidate regions without adequate coverage or quality were amplified by Sanger sequencing. The PredictSNP consensus classifier was used to predict the effect of the amino acid missense substitution identified (15). LOH analysis of the DLST locus was performed on tumour DNA from case 1 (see Figure 1) by high-density SNP-array analysis. Briefly, a genome-wide scan was conducted on 250 ng of tumour DNA with the Illumina Human610-Quad BeadChip (Illumina) and image data was analysed with the Chromosome Viewer tool contained in GenomeStudio 2010.2 (Illumina). The metric we used was the log-R ratio, which is the binary logarithm of the ratio of the observed to expected normalized R values for a given SNP, as previously reported (16).
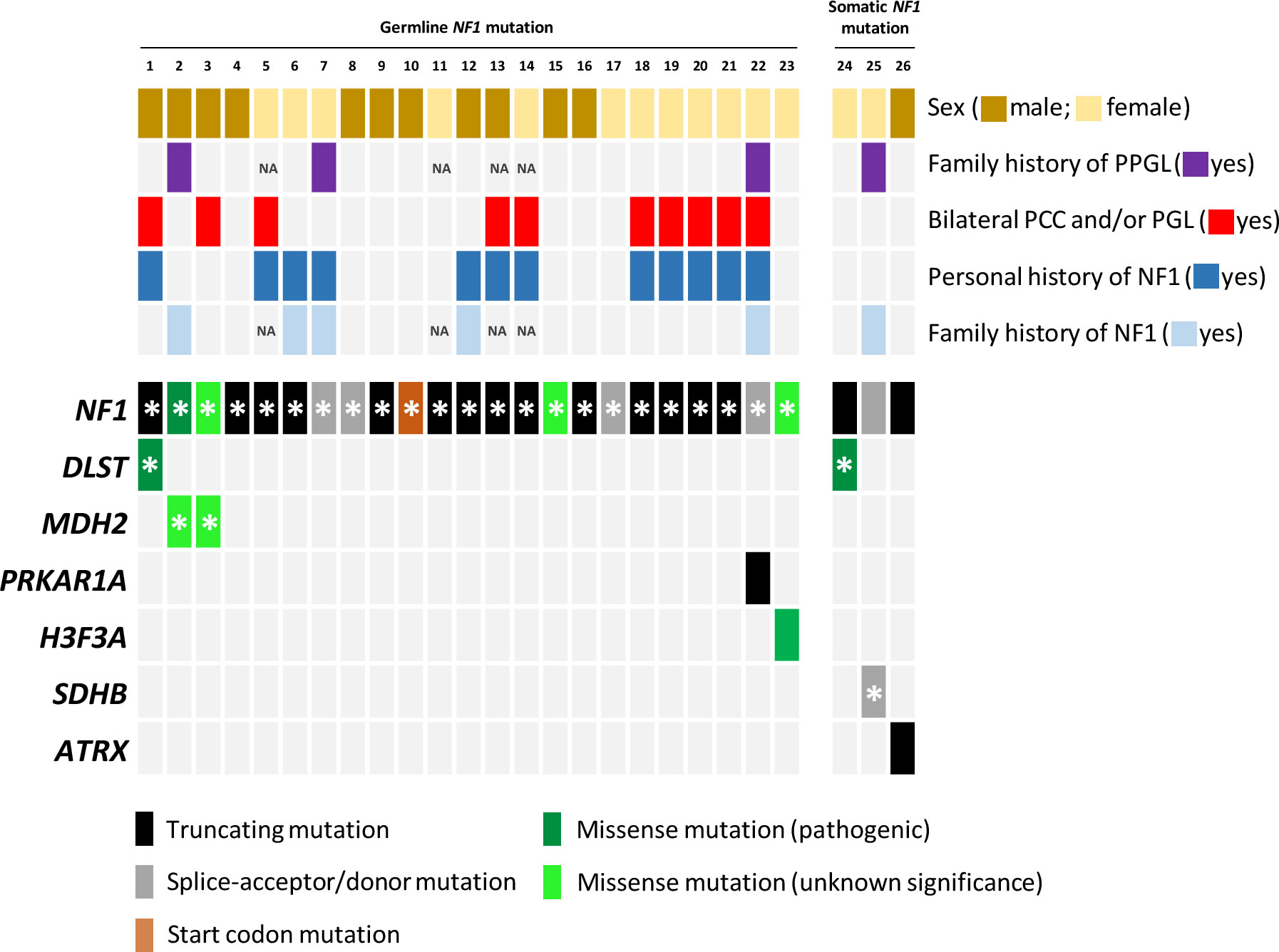
Figure 1 Mutational profile of the twenty-six samples included in the study. Each column represents a tumour, with coloured rectangles representing mutations that are germline (denoted with an asterisk) or somatic in each row. Missense variants that ClinVar does not classify as pathogenic mutations and/or that are absent in NF1 patients were categorized as variants of unknown significance. NA, not available.
Bisulfite conversion of tumour DNA from case 1 (carrying the germline DLST-p.Gly374Glu mutation) was performed using the EZ DNA Methylation Kit (Zymo Research) and genome-wide DNA methylation was assayed using the Infinium MethylationEPIC BeadChip (Illumina) at the Centro Nacional de Genotipado (CEGEN-ISCIII) (www.cegen.org), as previously described (17). This BeadChip interrogates over 850,000 methylation sites per sample at single-nucleotide resolution. M values were used for statistical analyses. Following, we profiled MethylationEPIC data obtained from the tumour with our series of 12 PPGLs carrying mutations in different susceptibility genes (including three cases with the DLST-p.Gly374Glu mutation). Hierarchical clustering of methylation data was performed using GeneCluster 2.0 (18). The methylome of the tumor carrying co-ocurrent mutations in DLST and NF1 (case 1) was deposited in the National Center for Biotechnology Information GEO database under the accession number GSE217207. Methylation data from the rest of the tumor samples used in this study were previously deposited under the accession numbers GSE111336 and GSE123185 (16).
FFPE tumour tissue (5–10 mg) from three available individuals (cases 1 and 24 with dual NF1/DLST variants, and case 3 with dual NF1/MDH2 variants) were immersed in 500 mL LC-MS/MS-grade methanol containing isotope-labelled internal standards and processed as previously described (19). An analysis of metabolites was carried out with an AB Sciex 5500 QTRAP mass spectrometer coupled to an Acquity ultra-high-performance liquid chromatographic system (Waters) as previously described (19). We compared the data with that from a series of tumours carrying either NF1 or DLST mutations previously analysed in our laboratory.
Immunohistochemical staining of DLST (11954; rabbit monoclonal 1:150, Cell Signaling Technology), and 5-hydroxymethylcytosine (5-hmC) (Active Motif; 39770) were performed with 3 mm FFPE sections from tumours 1 and 24, the two DLST/NF1-mutated tumour samples, as previously described (16, 20).
RNA libraries were built using QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina (Lexogen, 015), following the vendor’s protocol for FFPE RNA using 500 ng of total RNA from tumours 1 and 24, carriers of dual mutations in NF1 and DLST. We also used UMI Second Strands Synthesis Module for QuantSeq FWD (Lexogen, 081) and PCR Add-on Kit for Illumina (Lexogen, 020) to adjust the library amplification number of cycles. Libraries were applied to an Illumina flow cell for cluster generation and sequenced on NovaSeq6000. For the hierarchical clustering, performed with GeneCluster 2.0 (18), we used expression data from a list of genes differentially expressed in DLST mutated PPGLs (16) from the two NF1/DLST mutated tumours, three DLST- and seven NF1-mutated PPGLs. The transcriptomes of the tumors carrying co-ocurrent mutations in DLST and NF1 (cases 1 and 24) were deposited in GEO under the accession number GSE217206. RNA-Seq data for the rest of the tumors used in this study were previously deposited under the EGAS00001006044 accession number in the European Genome-phenome archive (Calsina et al., under review).
Amongst the twenty-three germline NF1 mutations included in the study, three missense variants (identified in cases 3, 15 and 23; Table 1; Figure 1) were not classified or were classified as variants of uncertain significance by ClinVar, and have not been found in NF1 patients. However, most of the predictions included in Varsome suggest a pathological effect of the substitutions, so we took these variants into account throughout the current study. Overall, NF1 clinical features (Table 2) or a family history of NF1 were reported in ~57% of the individuals carrying NF1 germline mutations.
Targeted sequencing of a panel of 33 PPGL-related genes revealed three germline variants, one in DLST and two in MDH2, and two somatic mutations in H3-3A (previously known as H3F3A) and PRKAR1A amongst the twenty-three patients carrying NF1 germline mutations analysed (Table 1; Figure 1). The DLST-p.Gly374Glu variant, previously reported as pathogenic in PPGL patients (16, 21), was found in a patient with bilateral PCC. The MDH2-p.Lys314Met variant, identified in two unrelated patients, has not been previously described in PPGL patients and it is found in 6/251092 controls from gnomAD. Several software (Polyphen/SIFT, Mutation taster, MutationAssessor, CUPSAT, Mutant v3.0) predicted the variant as deleterious. The H3-3A variant, p.Gly35Trp, is a known hotspot mutation found in giant cell tumour of bone (22) and PPGL (23).
Three additional patients carrying NF1 somatic mutations were also included in the study: one patient with PCC, and family history of NF1, carrying a known SDHB germline pathogenic mutation (ClinVar ID:29896; LOVD-SDHB ID: 000047), one patient with a DLST pathogenic mutation p.Gly374Glu (ClinVar ID:635133) identified in the germline, and one case with an ATRX somatic truncating mutation.
All variants were validated by Sanger sequencing.
Targeted and Sanger sequencing revealed no LOH of the wild-type (WT) DLST allele in the two tumours carrying the p.Gly374Glu mutation. For one of the tumours, a genome-wide SNP array analysis confirmed the absence of LOH and discarded uniparental disomy (UPD) of chromosomal region 14q, on which DLST is located, a mechanism described in DLST-mutated PPGLs (Supplementary Figure 1) (16). This tumor showed LOH on the long arm of chromosome 17 including the NF1 locus.
It has been reported that the methylation profile observed in DLST-mutated tumours is intermediate between the CpG island methylator phenotype (CIMP) described for SDH-mutated PPGLs and the unmethylated profile exhibited by tumours belonging to the so called cluster 2 (i.e. tumours with mutations in NF1, RET, MAX or HRAS) (24). When we profiled one of the tumours carrying mutations in NF1 and DLST, it clustered together with DLST-mutated tumours and separated from cluster 2 samples (including one NF1-mutated PCC) (Figure 2A), suggesting a role of the DLST variant in the observed molecular phenotype.
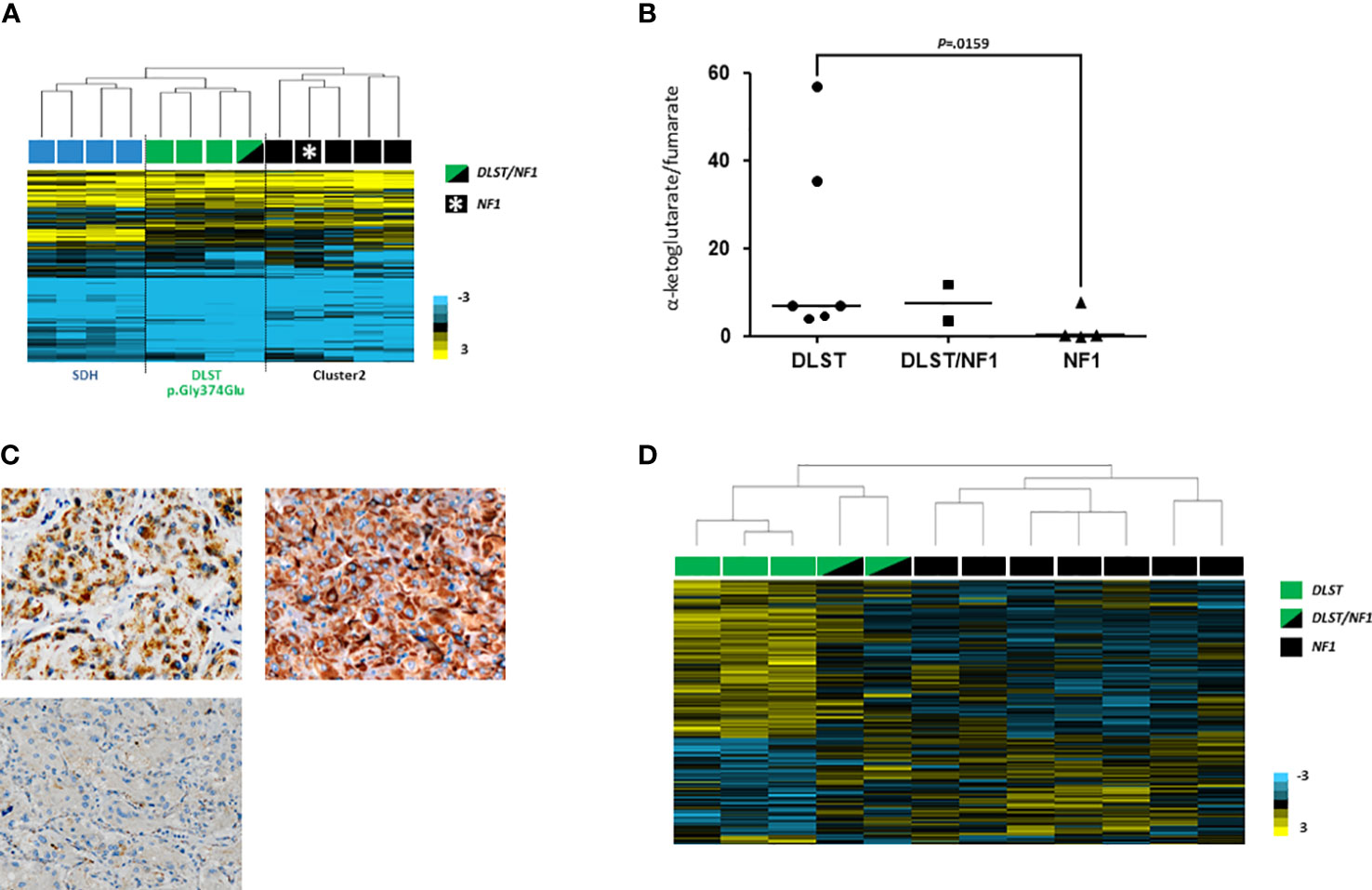
Figure 2 (A) Hierarchical clustering performed on the basis of methylation data from PPGLs as follows: one of the two DLST/NF1 (indicated with a green and black rectangle), four SDH-mutated (indicated with blue rectangles), three DLST-mutated (indicated with green rectangles) and five cluster 2-mutated (denoted in black rectangles) PPGLs. The DLST/NF1 PPGL was grouped with DLST-mutated tumours, and separated from cluster 2-mutated PPGLs. City-block and complete linkage characteristics were used for the analyses. (B) α-ketoglutarate/fumarate ratios assessed by LC-MS/MS in DLST-mutated tumours (n = 6), NF1-mutated PPGLs (n = 4), and the two tumours carrying dual NF1/DLST mutations. Black lines represent medians. A t test identified significant differences between DLST-mutated and NF1-mutated tumours. (C) Immunostaining for DLST was conducted in the two tumours carrying DLST/NF1 dual mutations (upper panel) revealing the characteristic cytoplasmic aggregates (x20). A VHL-mutated tumor was used as a negative control (lower panel) (D) Hierarchical clustering of the two NF1/DLST mutated tumours, three DLST- and seven NF1-mutated PPGLs based on gene expression data from a previously reported list of genes differentially expressed in DLST-mutated PPGLs. The two tumours carrying the NF1/DLST mutations were clustered between PPGLs carrying mutations in NF1 and DLST. Uncentered correlation and complete linkage characteristics were used for the analysis.
LC-MS/MS analysis of the three tumours available did not reveal conclusive alterations in TCA-cycle metabolites. However, one of the tumours carrying the double mutation in NF1/DLST exhibited an accumulation of cis-aconitate and a slightly high ketoglutarate/fumarate ratio was observed in the two NF1/DLST-mutated tumours compared to tumours carrying only an NF1 mutation (Figure 2B). It is possible that a certain block in the TCA-cycle due to the DLST mutation causes this partial accumulation. Finally, a relatively high fumarate to succinate ratio was observed in the only available tumour carrying dual NF1/MDH2 variants (Supplementary Figure 2), something previously described in MDH2-mutated PPGL (19).
In order to investigate whether tumours carrying the NF1/DLST-p.Gly374Glu mutations showed other molecular features observed previously in DLST-mutated PPGLs, we carried out the IHC for DLST. The two tumours showed a highly positive staining (Figure 2C), something previously reported for PPGLs carrying mutations in DLST and other TCA-cycle genes (16, 21), supporting again a role of the DLST mutation in tumour development. Another molecular marker described for DLST-mutated tumours is the low level of 5-hmC nuclear staining (21), similar to the observed in SDH-mutated PPGLs (25). 5-hmC staining applied to the two DLST-mutated tumours were positive in one of the samples and heterogeneous in the second tumour, in which positive regions alternated with regions that had negative IHC (Figuress 3A, B). SDHB IHC carried out in the tumour carrying co-occurrent SDHB germline mutation and somatic NF1 mutation yielded a positive stain, which is atypical for pathogenic SDHB mutations (Figure 3C).
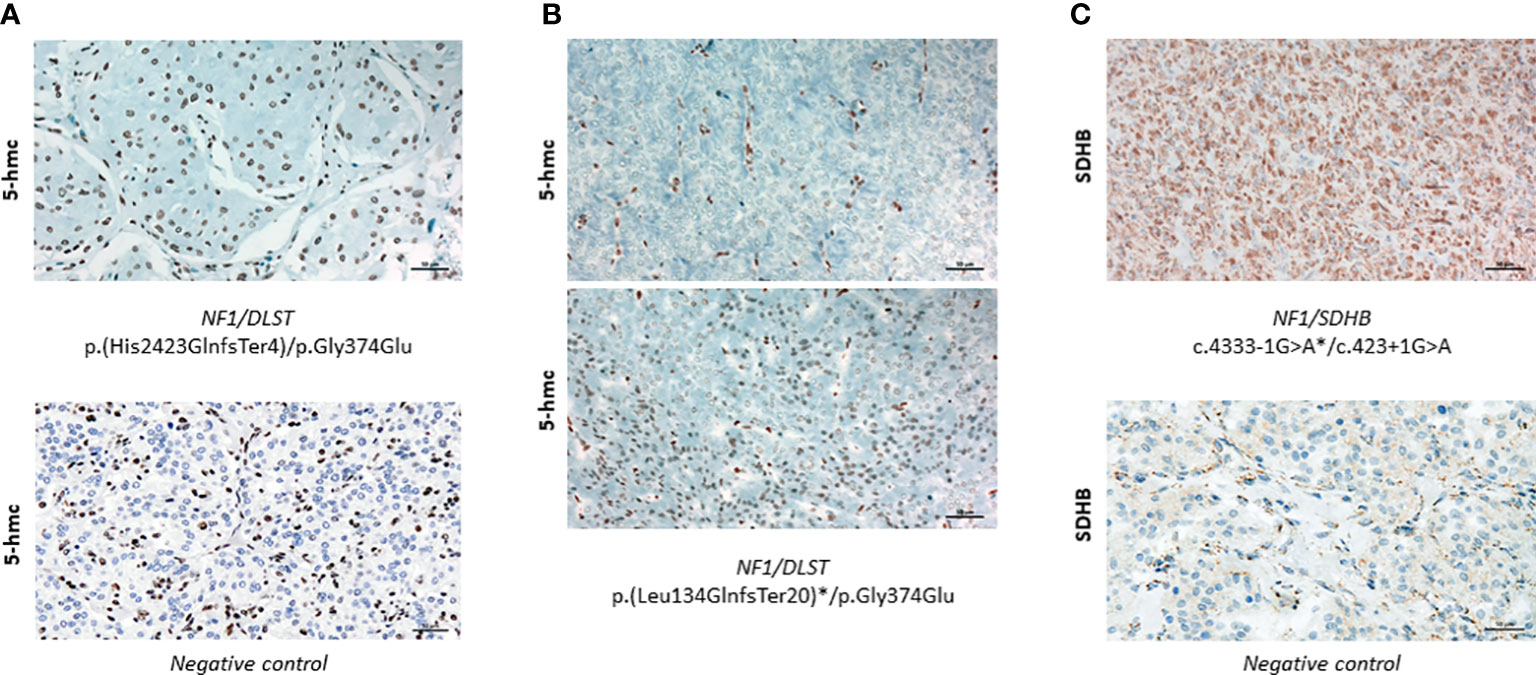
Figure 3 (A) Positive 5-hmC IHC performed in case 1 (upper panel) compared to a negative control (lower panel) (x20). (B) 5-hmC IHC performed in case 24, showing patches of negative (upper panel) and positive (lower panel) staining (x20)(C) Positive SDHB IHC performed in the tumour carrying SDHB/NF1 dual germline mutations (upper panel) compared to a negative control (lower panel) (x20). Asterisks denote a somatic mutation.
Hierarchical clustering of the two tumours carrying dual mutations in NF1 and DLST based on expression data for genes differentially expressed in DLST-mutated PPGLs grouped them in between DLST and NF1 mutated PPGLs, suggesting that an intermediate phenotype was caused by the coexistence of the two alterations (Figure 2D).
PPGL is the tumour with the highest degree of heritability attributable to the presence of germline mutations in known genes (up to 40% of cases) (26). Although mutations in some of the susceptibility genes are highly penetrant (e.g. germline mutations in VHL or RET), variants in other genes such as SDHB, SDHA (27) or TMEM127 (28) do not segregate with the disease in some pedigrees due to a low age-dependent penetrance. In this regard, the latest susceptibility genes identified in PPGL patients, SLC25A11 and DLST, have been found in patients with no relatives affected (16, 29), suggesting that these variants have a very low penetrance as well. Though penetrance of NF1 germline mutations is considered to be virtually complete after childhood, some studies have reported the presence of unexpected NF1 germline mutations in PPGL patients for whom a clinical diagnosis of the disease was not previously established (3, 9). The finding of 11 additional PPGL patients harbouring germline NF1 mutations without clinical manifestations of NF1 at diagnosis, underscores the importance of including NF1 testing in the genetic diagnosis of every PPGL patient, even in the absence of clinical features of the disease.
The penetrance of a given allele can be modified by the simultaneous presence of variants at other gene loci, which may also affect the clinical expressivity of the disease. In the case of NF1, modifier genes have been reported to contribute to the variable expressivity of the disease in a large series of patients (30, 31). The number of café-au-lait macules has been found to be influenced by common SNPs (32), polymorphisms in ADCY8 correlate with glioma risk in a sex-specific manner (33), and rare germline variants in various genes including ATM have been proposed as candidate modifiers of plexiform neurofibroma (34). NF1-associated PPGLs are mostly unilateral (84% of patients) and rarely extra-adrenal (6.1% of patients) (4, 5). Herein we describe two NF1 patients with multiple tumours harboring dual mutations, which, in addition to the previously reported patients carrying SDHD/NF1 mutations, suggest that the co-occurrence of NF1 mutations with variants affecting other PPGL susceptibility genes (i.e. DLST, MDH2, SDHD), or still unknown genes, could be responsible of this unusual clinical manifestation in some NF1 patients. In turn, no germline mutations affecting other PPGL susceptibility gene were found in the six exomes from patients with unilateral PCC and constitutive NF1 mutation included in The Cancer Genome Atlas program (24). Another possible explanation is that the development of PPGL in NF1 is an example of an oligogenic disease caused by the co-occurrence of mutations in two or more genes. In contrast to the thousands of reports in which mutations in single genes cause human diseases, there are only dozens of human disease phenotypes with evidence of digenic inheritance (35–37). The incidence of NF1, approximately 1:2600 to 3000 individuals, could indicate that the findings described in the present study are coincidental.
The DLST-p.Gly374Glu variant has recently been demonstrated to be pathological in PPGL patients (PGL7; MIM # 618475) (16, 21). Despite the absence of LOH affecting the wild-type allele in the two patients in our series, the highly positive IHC for DLST as a molecular marker of TCA-cycle disruption (16), supports its role in tumour development. In addition, the intermediate expression profile of the two tumours between NF1 and DLST mutated PPGLs, the DLST-like methylation profile observed for one of them, and the accumulation of cis-aconitate and the heterogeneous 5-hmC staining observed in the other tumour, reinforces a role of the DLST mutation in tumour molecular characteristics. No additional cases with PPGL were identified in the two pedigrees carrying the DLST-p.Gly374Glu mutation, and the finding of the mutation in a healthy 58-year-old relative from one of the families (data not shown) supports the previously described low penetrance of the DLST-p.Gly374Glu variant (16). Interestingly, the patient carrying the NF1/DLST variants in the germline (case 1) also developed a medullary thyroid carcinoma (MTC) with LOH for the NF1 mutation (9). Neither NF1 nor DLST are clearly related to medullary thyroid malignancies, but the co-occurrence of both alterations could account for the presence of bilateral PCC and MTC in the same patient, resembling what happens in multiple endocrine neoplasia type 2. This case is especially intriguing, as clinical features of NF1 were not recognized until the diagnosis of bilateral PCC was made at age 56 years, and in addition the patient developed primary prostate and lung cancers during his lifetime.
The MDH2 variant, p.Lys314Met, identified in two of the NF1 patients involves one of the four lysines described as playing a role in the activation of MDH2 by acetylation (38, 39). Interestingly, the same lysine was found deleted in another patient with multiple noradrenergic PGLs (40), which supports the relevance of this residue for MDH2 function. The presence of a para-aortic PGL in one of the patients carrying the p.Lys314Met (case 3), reinforces the role of the variant in the disease since a thoracic-abdominal location of PGL, very rare in NF1 patients, is the most frequently found in MDH2 mutation carriers (19, 40).
The presence of NF1 somatic mutations is one of the main genetic findings occurring in sporadic PPGLs (accounts for 20-25% of all cases) (41), being mostly tumours with adrenal location and only 4% extra-adrenal (10, 42). It has been repeatedly suggested that there is a mutual exclusivity condition affecting driver events in PPGLs, something that points to the redundancy of the affected signals (43, 44). Finding a mutation in H3-3A in a tumour carrying a germline NF1 variant suggests that perhaps some mutations in NF1 do need another alteration that confers additional selective advantages to the tumour cells (44). In the majority of NF1 cases, the mutation in NF1 is sufficient for tumour development and there is no need for accumulation of mutations in other predisposing genes, although it has been suggested that a proportion of apparently NF1-sporadic PPGLs might actually have a germline susceptibility variant in other predisposing genes (44). This is the case of the patient described in the present study carrying a germline SDHB mutation and a somatic NF1 alteration, whose tumour showed no evidence of LOH and a positive SDHB IHC. There is another similar example of co-occurrence of a germline SDHB mutation and a somatic NF1 alteration in a patient who developed a para-aortic PGL (45). Recently, dual SDHB/NF1 loss has been reported as a successful mechanism to force tumour formation in a SDHB knockout mouse model (46). SDHB loss has been shown to be insufficient for tumour development in SDH-mouse models of PPGL (47), but the concomitant alteration of NF1 finally yielded SDH-like PPGLs, reinforcing the theory for a need of additional genetic events for tumour initiation and maintenance in SDHB PPGL, resulting in a lower penetrance of the mutations (48). A similar situation could explain the low penetrance of the p.Gly374Glu DLST germline variant observed in our pedigrees. It is noteworthy that all the NF1 cases described herein, as well as those reported in the literature, carry co-occurrent alterations in an apparently unrelated pathway (e.g. pseudohypoxic signalling), suggesting that co-occurrence of alterations in different signalling cascades could provide a selective advantage for the development of PPGL.
Somatic ATRX mutations in PPGLs carrying germline mutations in one of the major susceptibility genes (especially in SDHB-mutated cases) have been reported (24, 49), and this event has been described as an independent factor of poor prognosis (50). Moreover, a recent study described the co-existence of NF1 germline mutations and ATRX somatic alterations in different tumours from NF1 patients (51). Therefore, finding a PPGL carrying somatic mutations in NF1 and ATRX may not be unexpected, and warns of the malignant potential of the tumour carrying the ATRX mutation. Finally, PRKAR1A, mapped at 17q22-24, is the gene encoding the protein kinase A regulatory subunit type 1α which can interfere with the ERK1/2 cascade of the MAPK pathway and causes inhibition of cell proliferation (52). PRKAR1A is frequently mutated in patients with Carney complex syndrome (MIM #160980) (53), a rare multiple neoplasia syndrome characterized by both endocrine and non-endocrine manifestations. This gene can be rearranged with RET to form the thyroid tumour-specific chimeric oncogene PTC2 (54), and it is worth noting that dysregulated protein kinase A has been linked to cancer by activation of mechanisms that overlap but differ from those found in neurofibromatosis tumorigenesis (55). PRKAR1A haploinsufficiency is a general tumorigenic signal which may require inactivation of other tumour suppressors to function (56). Another inactivating variant in PRKAR1A has been found in a metastatic PCC carrying additional alterations in ATRX and MAML3 (24), but the significance of this somatic event in PPGL etiology is still unknown.
Because of the low penetrance of some mutations, variable expressivity, and parent-of-origin effects associated with some susceptibility genes, a large number of PPGL patients have no family history, masking the hereditary condition and making proper follow-up of individuals at risk for hereditary PPGL difficult. Herein, we propose a new layer of complexity in which mutations affecting genes involved in different pathways (e.g. pseudohypoxic and receptor tyrosine kinase signalling) (Supplementary Figure 3) co-occurring in the same patient, either in the germline or in the tumour, could provide a selective advantage for the development of PPGL. The widespread application of NGS to the diagnosis of PPGL will continue to identify unexpected co-occurrences of pathological mutations, which may provide new insights into the development of these tumors, as well as explain differences in their expressivity and incomplete penetrance.
The data presented in the study are deposited in the GEO database repository, accession numbers GSE217208, GSE111336 and GSE123185; and in the in the European Genome-phenome archive, accession number EGAS00001006044.
The studies involving human participants were reviewed and approved by Institutional Review Board of Mayo Clinic: IRB 13-004137; Cambridge East Medical Research Ethics Committee: Ref: 06/Q0104/133; Hospital Universitario 12 de Octubre: 15/024; ENS@T Ethics Committee: 88/11; Azienda Ospedaliera Universitaria Careggi: Prot. N. 2011/0020149; Ethics Committee Institutului National de Endocrinologie: 14/11.06.2020; The Instituto de Salud Carlos III (ISCIII) ethics committee: CEI PI 54_2016-v2. The patients/participants provided their written informed consent to participate in this study.
SM, EG, RL, EC, SR, GE, and EH were involved in data analysis, data interpretation, and final approval of the manuscript. NP, ML, JG, AL-F, MC, AH-M, MG, XM-G, MB, EK, EL, FM, SL, SF, NB, LC, ER, and IB were responsible for data collection and final approval of the manuscript. MR was responsible for study design, data interpretation, manuscript writing, and final approval of the manuscript. AC was responsible for study design, data analysis, data interpretation, manuscript writing, and final approval of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Instituto de Salud Carlos III (ISCIII), through the “Acción Estratégica en Salud” (AES) (projects PI18/00454 to AC and PI20/01169 to MR), cofounded by the European Regional Development Fund (ERDF). SM was supported by the Spanish Ministry of Science, Innovation and Universities “Formación del Profesorado Universitario— FPU” fellowship with ID number FPU19/04940.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.1070074/full#supplementary-material
Supplementary Figure 1 | SNP-array study of chromosomes 14 and 17 using tumour DNA from case 1 who carries the DLST-p.Gly374Glu/NF1-p.His2423GlnfsTer4 dual mutation showed no LOH. The lower panel shows the genomic plots of the log R ratio [log 2 (Rpatient/Rreference)], and the upper panel gives the allele frequency parameters along the chromosomes.
Supplementary Figure 2 | Fumarate/succinate ratios assessed by LC-MS/MS in MDH2-mutated PPGLs (n=5), NF1-mutated PPGLs (n=5), SDHx-mutated PPGLs (n=11), and one PPGL carrying dual NF1/MDH2 mutation. Black lines represent means. A t test identified differences between means.
Supplementary Figure 3 | Schematic representation of the three molecular processes involved in the development of PPGL in which we have found co-occurring mutations: pseudohypoxic signalling (shown in purple), receptor tyrosine kinase cascades (shown in green), and epigenetic modifications and chromatin remodelling (shown in blue). The proteins found mutated in PPGL are indicated with dark coloured ovals and white letters, and the proteins whose genes have been found mutated in the present study are indicated with a yellow asterisk.
1. Buffet A, Burnichon N, Favier J, Gimenez-Roqueplo AP. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab (2020) 34(2):101416. doi: 10.1016/j.beem.2020.101416
2. Chatzikyriakou P, Touska P, Moonim MT, Obholzer R, Afridi S, Sandison A, et al. Case report of a man with multiple paragangliomas and pathogenic germline variants in both NF1 and SDHD. Cancer Genet (2021) 256-257:110–4. doi: 10.1016/j.cancergen.2021.05.008
3. Ben Aim L, Pigny P, Castro-Vega LJ, Buffet A, Amar L, Bertherat J, et al. Targeted next-generation sequencing detects rare genetic events in pheochromocytoma and paraganglioma. J Med Genet (2019) 56(8):513–20. doi: 10.1136/jmedgenet-2018-105714
4. Jackson BS, De Villiers M, Montwedi D. Association between pheochromocytoma and neurofibromatosis type I: A rare entity in the African population. BMJ Case Rep (2021) 14(5):e238380. doi: 10.1136/bcr-2020-238380
5. Gruber LM, Erickson D, Babovic-Vuksanovic D, Thompson GB, Young WF Jr., Bancos I. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin Endocrinol (Oxf). (2017) 86(1):141–9. doi: 10.1111/cen.13163
6. Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. Von recklinghausen’s disease and pheochromocytomas. J Urol. (1999) 162(5):1582–6. doi: 10.1016/S0022-5347(05)68171-2
7. Darrigo Junior LG, Ferraz VEF, Cormedi MCV, Araujo LHH, Magalhaes MPS, Carneiro RC, et al. Epidemiological profile and clinical characteristics of 491 Brazilian patients with neurofibromatosis type 1. Brain Behav (2022) 12(6):e2599. doi: 10.1002/brb3.2599
8. Kepenekian L, Mognetti T, Lifante JC, Giraudet AL, Houzard C, Pinson S, et al. Interest of systematic screening of pheochromocytoma in patients with neurofibromatosis type 1. Eur J Endocrinol (2016) 175(4):335–44. doi: 10.1530/EJE-16-0233
9. Gieldon L, Masjkur JR, Richter S, Darr R, Lahera M, Aust D, et al. Next-generation panel sequencing identifies NF1 germline mutations in three patients with pheochromocytoma but no clinical diagnosis of neurofibromatosis type 1. Eur J Endocrinol (2018) 178(2):K1–9. doi: 10.1530/EJE-17-0714
10. Burnichon N, Buffet A, Parfait B, Letouze E, Laurendeau I, Loriot C, et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet (2012) 21(26):5397–405. doi: 10.1093/hmg/dds374
11. Albattal S, Alswailem M, Moria Y, Al-Hindi H, Dasouki M, Abouelhoda M, et al. Mutational profile and genotype/phenotype correlation of non-familial pheochromocytoma and paraganglioma. Oncotarget (2019) 10(57):5919–31. doi: 10.18632/oncotarget.27194
12. Parisien-La Salle S, Dumas N, Rondeau G, Latour M, Bourdeau I. Isolated pheochromocytoma in a 73-Year-Old man with no clinical manifestations of type 1 neurofibromatosis carrying an unsuspected deletion of the entire NF1 gene. Front Endocrinol (Lausanne). (2019) 10:546. doi: 10.3389/fendo.2019.00546
13. Glasker S, Neumann HPH, Koch CA, Vortmeyer A. Von Hippel-lindau disease. Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, editors. South Dartmouth (MA: Endotext (2000).
14. Amodru V, Taieb D, Guerin C, Romanet P, Paladino N, Brue T, et al. MEN2-related pheochromocytoma: current state of knowledge, specific characteristics in MEN2B, and perspectives. Endocrine (2020) 69(3):496–503. doi: 10.1007/s12020-020-02332-2
15. Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, et al. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PloS Comput Biol (2014) 10(1):e1003440. doi: 10.1371/journal.pcbi.1003440
16. Remacha L, Pirman D, Mahoney CE, Coloma J, Calsina B, Curras-Freixes M, et al. Recurrent germline DLST mutations in individuals with multiple pheochromocytomas and paragangliomas. Am J Hum Genet (2019) 104(4):651–64. doi: 10.1016/j.ajhg.2019.02.017
17. Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics (2009) 1(1):177–200. doi: 10.2217/epi.09.14
18. Reich M, Ohm K, Angelo M, Tamayo P, Mesirov JP. GeneCluster 2.0: An advanced toolset for bioarray analysis. Bioinformatics (2004) 20(11):1797–8. doi: 10.1093/bioinformatics/bth138
19. Cascón A, Comino-Méndez I, Currás-Freixes M, De Cubas AA, Contreras L, Richter S, et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Institute (2015) 107(5):djv053. doi: 10.1093/jnci/djv053
20. Remacha L, Comino-Mendez I, Richter S, Contreras L, Curras-Freixes M, Pita G, et al. Targeted exome sequencing of Krebs cycle genes reveals candidate cancer-predisposing mutations in pheochromocytomas and paragangliomas. Clin Cancer Res (2017) 23(20):6315–24. doi: 10.1158/1078-0432.CCR-16-2250
21. Buffet A, Zhang J, Rebel H, Corssmit EPM, Jansen JC, Hensen EF, et al. Germline DLST variants promote epigenetic modifications in pheochromocytoma-paraganglioma. J Clin Endocrinol Metab (2021) 106(2):459–71. doi: 10.1210/clinem/dgaa819
22. Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet (2013) 45(12):1479–82. doi: 10.1038/ng.2814
23. Toledo RA, Qin Y, Cheng ZM, Gao Q, Iwata S, Silva GM, et al. Recurrent mutations of chromatin-remodeling genes and kinase receptors in pheochromocytomas and paragangliomas. Clin Cancer Res (2016) 22(9):2301–10. doi: 10.1158/1078-0432.CCR-15-1841
24. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell (2017) 31(2):181–93. doi: 10.1016/j.ccell.2017.01.001
25. Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell (2013) 23(6):739–52. doi: 10.1016/j.ccr.2013.04.018
26. Wachtel H, Fishbein L. Genetics of pheochromocytoma and paraganglioma. Curr Opin Endocrinol Diabetes Obes (2021) 28(3):283–90. doi: 10.1097/MED.0000000000000634
27. Andrews KA, Ascher DB, Pires DEV, Barnes DR, Vialard L, Casey RT, et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J Med Genet (2018) 55(6):384–94. doi: 10.1136/jmedgenet-2017-105127
28. Toledo SP, Lourenco DM Jr., Sekiya T, Lucon AM, Baena ME, Castro CC, et al. Penetrance and clinical features of pheochromocytoma in a six-generation family carrying a germline TMEM127 mutation. J Clin Endocrinol Metab (2015) 100(2):E308–18. doi: 10.1210/jc.2014-2473
29. Buffet A, Morin A, Castro-Vega LJ, Habarou F, Lussey-Lepoutre C, Letouze E, et al. Germline mutations in the mitochondrial 2-Oxoglutarate/Malate carrier SLC25A11 gene confer a predisposition to metastatic paragangliomas. Cancer Res (2018) 78(8):1914–22. doi: 10.1158/0008-5472.CAN-17-2463
30. Sabbagh A, Pasmant E, Laurendeau I, Parfait B, Barbarot S, Guillot B, et al. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum Mol Genet (2009) 18(15):2768–78. doi: 10.1093/hmg/ddp212
31. Pasmant E, Vidaud M, Vidaud D, Wolkenstein P. Neurofibromatosis type 1: from genotype to phenotype. J Med Genet (2012) 49(8):483–9. doi: 10.1136/jmedgenet-2012-100978
32. Pemov A, Sung H, Hyland PL, Sloan JL, Ruppert SL, Baldwin AM, et al. Genetic modifiers of neurofibromatosis type 1-associated cafe-au-lait macule count identified using multi-platform analysis. PloS Genet (2014) 10(10):e1004575. doi: 10.1371/journal.pgen.1004575
33. Warrington NM, Sun T, Luo J, McKinstry RC, Parkin PC, Ganzhorn S, et al. The cyclic AMP pathway is a sex-specific modifier of glioma risk in type I neurofibromatosis patients. Cancer Res (2015) 75(1):16–21. doi: 10.1158/0008-5472.CAN-14-1891
34. Yu Y, Choi K, Wu J, Andreassen PR, Dexheimer PJ, Keddache M, et al. NF1 patient missense variants predict a role for ATM in modifying neurofibroma initiation. Acta Neuropathol. (2020) 139(1):157–74. doi: 10.1007/s00401-019-02086-w
35. Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science (1994) 264(5165):1604–8. doi: 10.1126/science.8202715
36. Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, et al. Triallelic inheritance in bardet-biedl syndrome, a mendelian recessive disorder. Science (2001) 293(5538):2256–9. doi: 10.1126/science.1063525
37. Mukherjee S, Cogan JD, Newman JH, Phillips JA 3rd, Hamid R, Undiagnosed Diseases N, et al. Identifying digenic disease genes via machine learning in the undiagnosed diseases network. Am J Hum Genet (2021) 108(10):1946–63. doi: 10.1016/j.ajhg.2021.08.010
38. Xu W, Li Y, Liu C, Zhao S. Protein lysine acetylation guards metabolic homeostasis to fight against cancer. Oncogene (2014) 33(18):2279–85. doi: 10.1038/onc.2013.163
39. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science (2010) 327(5968):1000–4. doi: 10.1126/science.1179689
40. Calsina B, Curras-Freixes M, Buffet A, Pons T, Contreras L, Leton R, et al. Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients. Genet Med (2018) 20(12):1652–62. doi: 10.1038/s41436-018-0068-7
41. Group NGSiPS, Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol (2017) 13(4):233–47. doi: 10.1038/nrendo.2016.185
42. Welander J, Larsson C, Backdahl M, Hareni N, Sivler T, Brauckhoff M, et al. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum Mol Genet (2012) 21(26):5406–16. doi: 10.1093/hmg/dds402
43. Dahia PL. The genetic landscape of pheochromocytomas and paragangliomas: Somatic mutations take center stage. J Clin Endocrinol Metab (2013) 98(7):2679–81. doi: 10.1210/jc.2013-2191
44. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat Rev Cancer. (2014) 14(2):108–19. doi: 10.1038/nrc3648
45. Choi YM, Lim J, Jeon MJ, Lee YM, Sung TY, Hong EG, et al. Mutation profile of aggressive pheochromocytoma and paraganglioma with comparison of TCGA data. Cancers (Basel) (2021) 13(10):2389. doi: 10.3390/cancers13102389
46. Armstrong N, Storey CM, Noll SE, Margulis K, Soe MH, Xu H, et al. SDHB knockout and succinate accumulation are insufficient for tumorigenesis but dual SDHB/NF1 loss yields SDHx-like pheochromocytomas. Cell Rep (2022) 38(9):110453. doi: 10.1016/j.celrep.2022.110453
47. Lussey-Lepoutre C, Buffet A, Morin A, Goncalves J, Favier J. Rodent models of pheochromocytoma, parallels in rodent and human tumorigenesis. Cell Tissue Res (2018) 372(2):379–92. doi: 10.1007/s00441-018-2797-y
48. Barbolosi D, Crona J, Serre R, Pacak K, Taieb D. Mathematical modeling of disease dynamics in SDHB- and SDHD-related paraganglioma: Further step in understanding hereditary tumor differences and future therapeutic strategies. PloS One (2018) 13(8):e0201303. doi: 10.1371/journal.pone.0201303
49. Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D’Andrea K, Merrill S, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun (2015) 6:6140. doi: 10.1038/ncomms7140
50. Job S, Draskovic I, Burnichon N, Buffet A, Cros J, Lepine C, et al. Telomerase activation and ATRX mutations are independent risk factors for metastatic pheochromocytoma and paraganglioma. Clin Cancer Res (2019) 25(2):760–70. doi: 10.1158/1078-0432.CCR-18-0139
51. Tong S, Devine WP, Shieh JT. Tumor and constitutional sequencing for neurofibromatosis type 1. JCO Precis Oncol (2022) 6:e2100540. doi: 10.1200/PO.21.00540
52. Robinson-White A, Hundley TR, Shiferaw M, Bertherat J, Sandrini F, Stratakis CA. Protein kinase-a activity in PRKAR1A-mutant cells, and regulation of mitogen-activated protein kinases ERK1/2. Hum Mol Genet (2003) 12(13):1475–84. doi: 10.1093/hmg/ddg160
53. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase a type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet (2000) 26(1):89–92. doi: 10.1038/79238
54. Romei C, Elisei R. RET/PTC translocations and clinico-pathological features in human papillary thyroid carcinoma. Front Endocrinol (Lausanne). (2012) 3:54. doi: 10.3389/fendo.2012.00054
55. Jones GN, Tep C, Towns WH 2nd, Mihai G, Tonks ID, Kay GF, et al. Tissue-specific ablation of Prkar1a causes schwannomas by suppressing neurofibromatosis protein production. Neoplasia (2008) 10(11):1213–21. doi: 10.1593/neo.08652
Keywords: pheochromocytoma, NF1, germline mutation, DLST, MDH2, co-occurrent mutations
Citation: Mellid S, Gil E, Letón R, Caleiras E, Honrado E, Richter S, Palacios N, Lahera M, Galofré JC, López-Fernández A, Calatayud M, Herrera-Martínez AD, Galvez MA, Matias-Guiu X, Balbín M, Korpershoek E, Lim ES, Maletta F, Lider S, Fliedner SMJ, Bechmann N, Eisenhofer G, Canu L, Rapizzi E, Bancos I, Robledo M and Cascón A (2023) Co-occurrence of mutations in NF1 and other susceptibility genes in pheochromocytoma and paraganglioma. Front. Endocrinol. 13:1070074. doi: 10.3389/fendo.2022.1070074
Received: 14 October 2022; Accepted: 09 November 2022;
Published: 25 January 2023.
Edited by:
Marta Araujo-Castro, Ramón y Cajal University Hospital, SpainReviewed by:
Delmar Muniz Lourenco Jr., Clinical Hospital, University of São Paulo, BrazilCopyright © 2023 Mellid, Gil, Letón, Caleiras, Honrado, Richter, Palacios, Lahera, Galofré, López-Fernández, Calatayud, Herrera-Martínez, Galvez, Matias-Guiu, Balbín, Korpershoek, Lim, Maletta, Lider, Fliedner, Bechmann, Eisenhofer, Canu, Rapizzi, Bancos, Robledo and Cascón. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alberto Cascón, YWNhc2NvbkBjbmlvLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.