 Jinni Hong
Jinni Hong Tingting Fu1,2
Tingting Fu1,2 Cunyun Min
Cunyun Min Datao Lin
Datao Lin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Endocrinol. , 05 December 2022
Sec. Gut Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1053900
This article is part of the Research Topic The Cross-Talk Between Gut Microbiota and Endogenous Metabolites in Endocrine Diseases, volume II View all 7 articles
Background: The role of gut microbiota in diabetes mellitus (DM) and its complications has been widely accepted. However, the alternation of gut microbiota in diabetic microvascular complications (DC) remains to be determined.
Methods: Publications (till August 20th, 2022) on gut microbiota in patients with DC were retrieved from PubMed, Web of Science, Embase and Cochrane. Review Manager 5.3 was performed to estimate the standardized mean difference (SMD) and 95% confidence interval (CI) and calculate alpha diversity indices and the relative abundance of gut microbiota between patients in DC v.s. DM and DC v.s. healthy controls (HC).
Results: We included 13 studies assessing 329 patients with DC, 232 DM patients without DC, and 241 HC. Compared to DM, patients with DC shared a significantly lower Simpson index (SMD = -0.59, 95% CI [-0.82, -0.36], p < 0.00001), but a higher ACE index (SMD = 0.42, 95% CI[0.11, 0.74], p = 0.009). Compared to HC, DC patients held a lower ACE index (SMD = -0.61, 95% CI[-1.20, -0.02], p = 0.04). The relative abundances of phylum Proteobacteria (SMD = 0.03, 95% CI[0.01, 0.04], p = 0.003, v.s. HC) and genus Klebsiella (SMD = 0.00, 95% CI[0.00, 0.00], p < 0.00001, v.s. HC) were enriched, accompanying with depleted abundances of phylum Firmicutes (SMD = -0.06, 95% CI[-0.11, -0.01], p = 0.02, v.s. HC), genera Bifidobacterium (SMD = -0.01, 95% CI[-0.02,-0.01], p < 0.0001, v.s. DM), Faecalibacterium (SMD = -0.01, 95% CI[-0.02, -0.00], p = 0.009, v.s. DM; SMD = -0.02, 95% CI[-0.02, -0.01], p < 0.00001, v.s. HC) and Lactobacillus (SMD = 0.00, 95% CI[-0.00, -0.00], p < 0.00001, v.s. HC) in DC.
Conclusions: Gut microbiota perturbations with the depletion of alpha diversity and certain short-chain fatty acids (SCFAs)-producing bacteria were associated with the pathology of DC. Therefore, gut microbiota might serve as a promising approach for the diagnosis and treatment of DC. Further investigations are required to study the mechanisms by which gut dysbiosis acts on the onset and progression of DC.
Diabetes mellitus (DM) is an epidemic and accounts for 80% of premature deaths globally (1, 2). As of 2021, there are approximately 537 million DM patients in the world, with an extended increment to 700 million by 2045 (3). DM is hardly a disease with mere elevation in blood glucose, it brings along a plethora of microvascular complications including diabetic retinopathy (DR), diabetic kidney disease (DKD) and diabetic peripheral neuropathy (DPN) in most cases. These microvascular complications are responsible for the high mortality and morbidity rate in DM patients and account for marked social and economic burdens (4). Although numerous treatments for DM and resulting complications were available, cases are still on the rise. DR is the leading cause of blindness ophthalmic disorder in the population on working age, with a prevalence of 77.8% in 15 years of DM patients (5–7). DKD occurs in approximately 40% of DM, and 30%-90% of DM might suffer from DPN (8). These trends highlight the urgency for a better understanding of diabetic microvascular complications (DC) (9).
The rapid scientific interest in gut microbiota coincided with the global increase in DM (9). The advent of next-generation sequencing technology has greatly enhanced our understanding of gut microbiota and host health (10, 11). The gut microbiota was a complex microbial community, represented by 1500 different species (12, 13). DM and its complications have been linked with dysbiosis of the gut microbiota (14). Differences in gut microbiota composition have been observed in animal models as well as patients with DM and complications such as DKD, DR and DPN. In particular, perturbed Bacteroidetes/Firmicutes eubiosis was proven to be associated with increased intestinal permeability, with bacteria byproducts infiltrated through a leaky gut barrier, triggering inflammatory responses of diabetes. Species Lactobacillus fermentum, Akkermansia muciniphila, Bacteroides fragilis and Roseburia intestinalis were demonstrated to be linked to insulin sensitivity and glucose metabolism. Phyla Bacteroidetes, Actinobacteria and Mucoromycota were depleted, while genera Acidaminococcus, Escherichia and Enterobacter were enriched in patients with DR compared to HC (15). In DPN, the richness of Firmicutes and Actinobacteria was elevated, while Bacteroidetes was depleted. At the genus level, Bacteroides and Faecalibacterium were significantly decreased, whereas Lachnoclostridium and Ruminococcus were enriched (16). In DKD, there was a marked increase in genera Acidaminococcus, Selenomonadales, Bilophila and Shigella, as well as phylum Proteobacteria, and the richness of species Syntrophaceticus schinkii and Citrobacter farmeri was positively correlated with the urinary albumin creatinine ratio (17). Notably, Akkermansia in the gut may contribute to the effect of metformin, the most commonly prescribed drug for DM, on glucose metabolism (18, 19).
Although a plethora of studies had characterized the gut microbiota of DC with promising findings, the relationship between them was still controversial. A meta-analysis on more than 2000 studies on microbiota suggested the alternation of microbiota to differentiate healthy and diseased populations and serve as bio-markers for the diagnosis or treatment of DM (20). Therefore, we performed a meta-analysis of gut microbiota from patients with DC and explored the diversity and bacterial characteristics of the gut microbiota in DC.
The scheme of the review was registered in PROSPERO with registration number CRD42022353144. We followed the Preferred Reporting Items for the Systematic Reviews and Meta-analyses (PRISMA) reporting guidelines (21).
We searched PubMed, Web of Science, Embase and Cochrane databases for observational case-control studies or cross-sectional studies from inception to August 20th, 2022, with the search strategy: ((diabetic retinopathy) OR (diabetic microvascular complications) OR (diabetic peripheral neuropathy) OR (diabetic neuropathy) OR (diabetic kidney disease) OR (diabetic nephropathy)) AND (Microbiota OR Microbiome). This online strategy was augmented by a bibliographic search to identify other potentially eligible publications. Records were restricted to studies published in English and conducted on humans. All duplicated records were removed.
Two reviewers (JH and TF) independently screened titles, abstracts, and full-text articles for inclusion and resolved differences through consensus. The eligible criteria included studies reporting DM patients with DC, setting healthy controls (HC) or DM patients without DC as controls, and performing gut microbiota analysis and reporting alpha diversity or abundance measures as outcome results. Records in review form or without full-text available were excluded.
Using standardized forms, two reviewers (JH and TF) extracted information on the general study and participant characteristics, as well as the outcome results including alpha diversity indices and relative abundance of microbiota at the phylum and genus level. Alpha diversity referred to species richness and evenness within communities or habitats, including observed number of operational taxonomic units (OTUs), Chao1, ACE, Simpson and Shannon indices, which were usually plotted using the R package. Specially, OTUs and Chao1 reflected the number of species in the community, regardless of the abundance of each species in the community. ACE considered a wider range of rare species, and adjusted the coefficient of variation and sample coverage to make data more reasonable. The Simpson and Shannon indices reflected the diversity of the bacterial communities, which were affected by the species richness and evenness in the community. Mean and standard deviation (SD) were collected from text or tables in articles, and results presented in graphs were abstracted using GetData Graph Digitizer v.2.22 (Australia) software. Since a large part of data on microbiota was presented as a box-plot, when the mean and SD were not available, the median and quartile range were extracted and estimated by the calculator of Review Manager 5.3. A third reviewer (DL) confirmed the abstracted data.
Newcastle-Ottawa Quality Assessment Scale (NOS) was used to evaluate the literature quality (22), and a score ≥ 5 indicated adequate quality for inclusion in the present review (23). A funnel plot was constructed to assess publication bias.
Data were exported to Review Manager 5.3 (Nordic Cochrane Centre, Copenhagen, Denmark) software for statistical meta-analysis. We pooled the mean difference (MD) or standardized mean difference (SMD) for continuous outcomes, and a 95% confidence interval (CI) was utilized to estimate the prediction. The heterogeneity was evaluated by I2 statistic, and a fixed and random effect model was performed when I2 < 50% and I2 > 50%, respectively. Two-sided p values were statistically significant at less than 0.05.
A total of 566 potentially eligible articles were retrieved, including 16 records identified from other sources. By screening abstracts and titles, 518 articles were excluded for duplication or irrelevant topics, leaving 48 studies. After that, eight articles were abandoned for language restriction and 13 were due to no full-text. Then 27 full -text articles were screened for eligibility. The most common reason for exclusion was insufficient data (n = 6), five records were excluded because the control was not appropriate, one was an animal study, one was a review article, and one did not report data in applicable form, leaving 13 studies meet all the inclusion criteria (Figure 1).
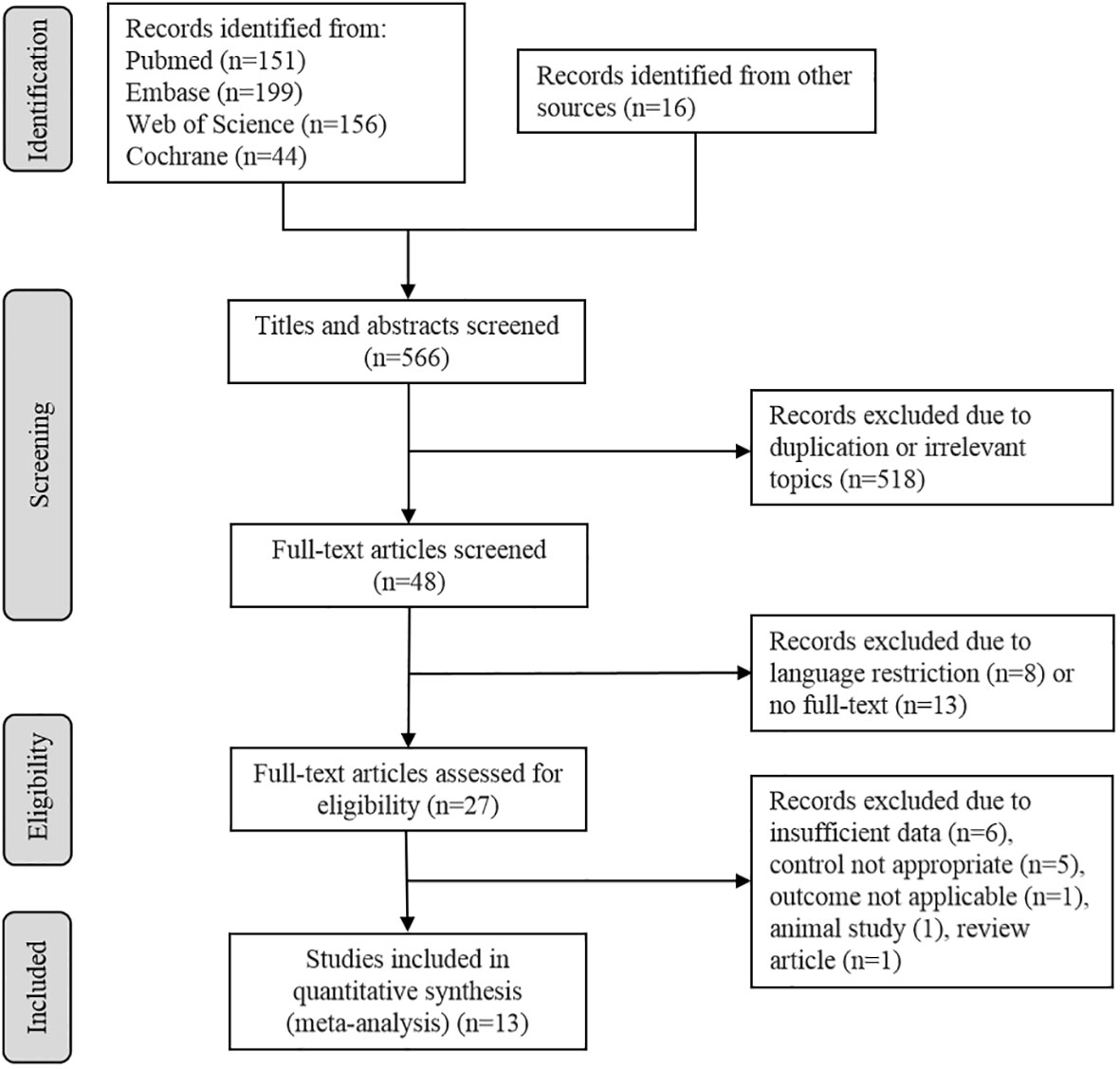
Figure 1 Flow diagram describes the selection process of the included studies.
The total 13 studies (15–17, 24–33) captured 329 patients with DC, 232 DM patients without DC, and 241 HC, with the territorial scope covering India (2/13), USA (2/13) and China (9/13). Seven (7/13) of the studies focused on DKD, five (5/13) focused on DR, and one (1/13) focused on DPN. The methodology of composition analysis also varied widely, with 16S rRNA gene sequencing being the most common (6/13), followed by 16S rDNA-based high-throughput sequencing (4/13) and three (3/13) with metagenomic sequencing. The basic characteristics of the articles included in the study were shown in Table 1.
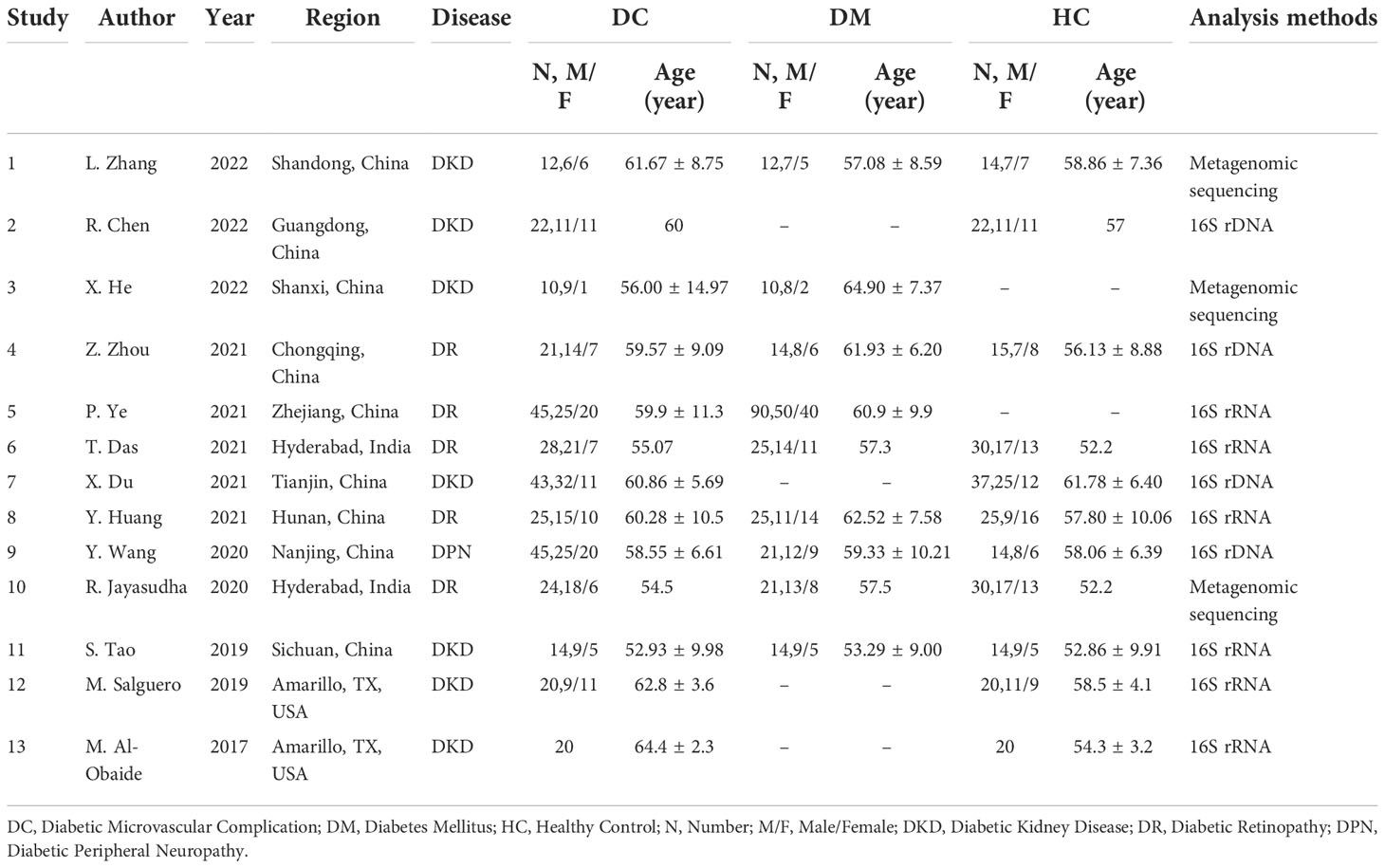
Table 1 Characteristics of the included studies.
The NOS scores showed one (1/13) study with a score of six, ten (10/13) with a score of seven, and two (2/13) with a score of eight, indicating a relatively high quality of the studies selected (Supplementary Table S1).
A total of ten trials were obtained to assess alpha diversity, including 279 DC patients, 222 DM patients and 201 HC. Five indices were obtained including estimates of richness (OTUs, ACE and Chao1), and diversity (Simpson and Shannon).
Regarding richness, four studies provided data on ACE, and the pooled estimate showed a significant increase in ACE in DC (SMD = 0.42, 95% CI[0.11, 0.74], p = 0.009) in Figure 2A. Four and eight studies reported the OTUs and Chao1 index in DC v.s. DM respectively, with non-significant differences between groups (OTUs SMD = -0.15, 95% CI[-0.97, 0.67], p = 0.72, I2 = 89%; Chao1 SMD = 0. 21, 95% CI[-0.38, 0.81], p = 0.48, I2 = 88%) in Figures 2B, C. To explore the potential sources of the existed heterogeneity in OTUs, subgroup analysis on the nation and city was done in Supplementary Figures S1A, B. The heterogeneity decreased to 10% in nation except China, and further analysis revealed that city bias might be the source of heterogeneity. To explore the potential sources of the heterogeneity in Chao1, subgroup analysis on DC including DR, DPN and DKD was performed. Since the heterogeneity still existed, subgroup analysis on the nation was done, and results showed the heterogeneity vanished in the nation except China (Supplementary Figure S1C).
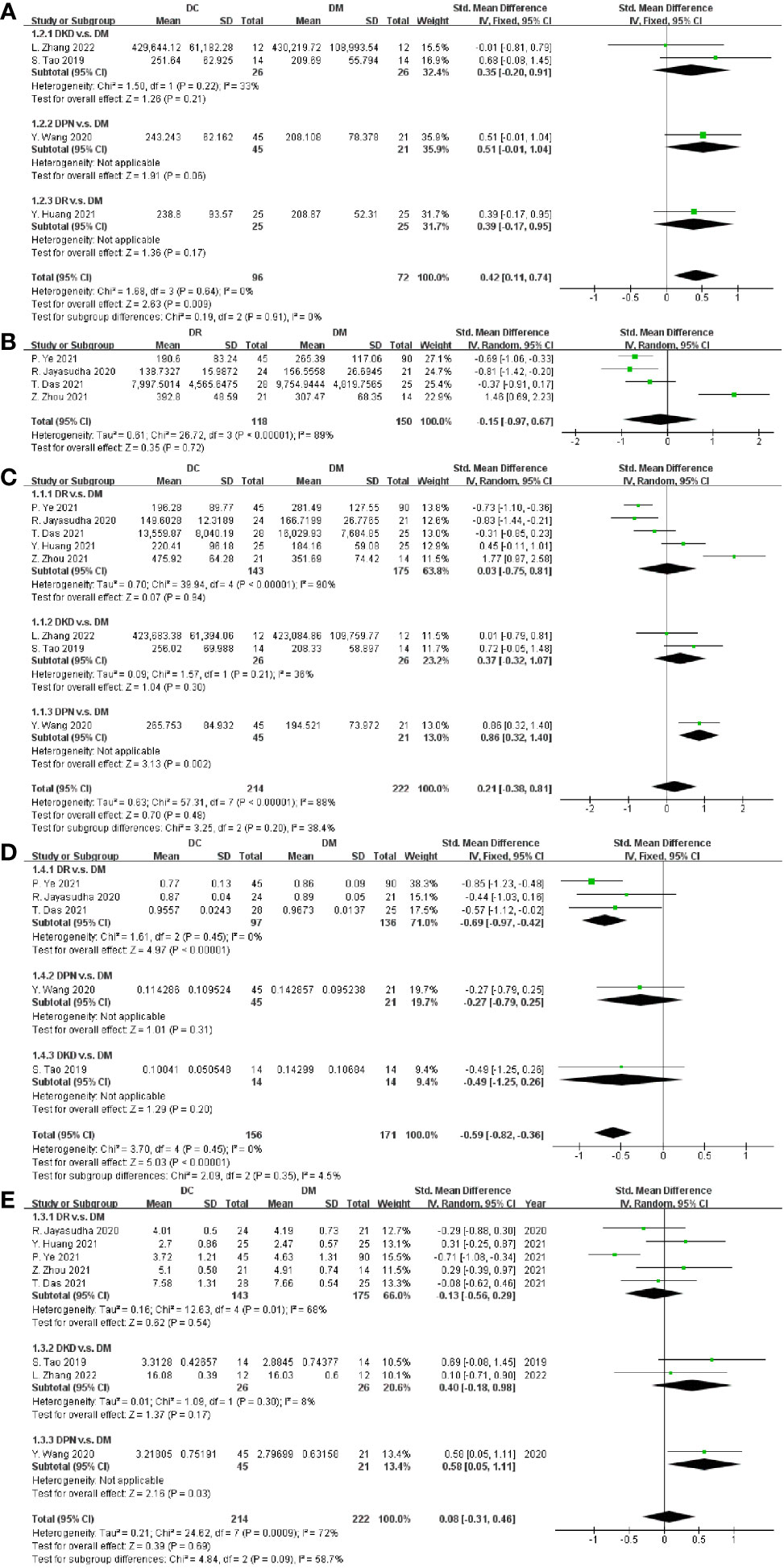
Figure 2 Forest plots of alpha diversity in DC v.s. DM. (A) ACE; (B) OTUs; (C) Chao1; (D) Simpson; (E) Shannon.
Regarding diversity, five studies provided data on DC, and the pooled estimate showed a significant decrease of the Simpson index in DC (SMD = -0.59, 95% CI [-0.82, -0.36], p < 0.00001), as shown in Figure 2D. Eight studies measured and reported the Shannon index in DC and DM, but our meta-analysis revealed no significant difference between them (SMD = 0.08, 95% CI [-0.31 0.46], p = 0.69), as depicted in Figure 2E. The type of DC and nation bias might be the sources of high heterogeneity (I2 = 72%), as depicted in Figure 2E and Supplementary Figure S1D.
Since only one study on DPN (16) was included in this study, to explore the source of heterogeneity caused by it, we performed a meta-analysis on studies excluding DPN and compared the results with meta-analysis on studies including DPN. As depicted in Supplementary Figure S2, a meta-analysis of studies excluding DPN was conducted. There was a significant increase in the Simpson index (SMD = -0.67, 95% CI [-0.93, -0.41], p < 0.00001), with no significant differences found in the Shannon index (SMD = -0.01, 95% CI [-0.39, 0.38], p = 0.97), ACE (SMD = 0.37, 95% CI[-0.02, 0.76], p = 0.06), and Chao1 (SMD = 0.11, 95% CI[-0.51, 0.74], p = 0.72) between DC and DM. Subgroup analysis on the type of DC and nation was performed in Supplementary Figure S3, and similar conclusions with studies including DPN were drawn that the type of DC and nation bias might be the sources of heterogeneity in the Shannon index and Chao1, respectively. Above all, the tendency of the results with or without DPN was consistent.
Furthermore, alpha diversity between DC and HC was compared in terms of OTUs, ACE, Chao1, Simpson and Shannon. Five studies reported a relative abundance of bacterial taxa in terms of OTUs, while a non-significant difference was found between DC and HC (SMD = -0.27, 95% CI[-1.15, 0.61], p = 0.55), as depicted in Figure 3A. Since there was a high heterogeneity (I2 = 92%), subgroup analysis on the type of DC and nation was performed. Nevertheless, the heterogeneity still existed in either subgroup of DC and nation, and the funnel plots were symmetric overall as shown in Supplementary Figure S4.

Figure 3 Forest plots of alpha diversity in DC v.s. HC. (A) OTUs; (B) ACE; (C) Chao1; (D) Simpson; (E) Shannon.
In Figure 3B, from the six trials included in this meta-analysis, we obtained that the SMD of ACE was -0.70 in DKD (95% CI: -1.64, 0.25), -0.43 in DPN (95% CI: -1.03, 0.18) and -0.46 in DR (95% CI: -1.02, 0.11), with no significant difference. Nevertheless, the overall SMD of DC was significantly decreased (SMD = -0.61, 95% CI[-1.20, -0.02], p = 0.04). Nine studies provided Chao1 data in DC v.s. HC, with a non-significant difference estimated after meta-analysis (SMD = -0.32, 95% CI[-0.80, 0.16], p = 0.19), as depicted in Figure 3C.
Regarding community diversity, Figures 3D, E showed that six and nine trails provided Simpson and Shannon data respectively. However, the differences in community diversity between DC and HC estimated by Simpson and Shannon were not significant, with Simpson SMD = -0.12, 95% CI[-0.35, 0.11], p = 0.29, and Shannon SMD = 0.16, 95% CI[-0.14, 0.45], p = 0.30. Nation bias assessments and funnel plots for the alpha diversity were shown in Supplementary Figure S5.
Since only one study on DPN was included in this meta-analysis, considering the representativeness and potential bias, a meta-analysis of studies excluding DPN was performed in Supplementary Figure S6. Compared with the meta-analysis of studies including DPN, similar results were found in ACE (SMD = -0.65, 95% CI[-1.36, 0.07], p = 0.08), Chao1 (SMD = -0.37, 95% CI[-0.90, 0.16], p = 0.17), the Simpson index (SMD = -0.07, 95% CI[-0.32, 0.17], p = 0.55), and the Shannon index (SMD = 0.12, 95% CI[-0.20, 0.44], p = 0.47) between DC and HC. Nation bias assessments and funnel plots of alpha diversity were shown in Supplementary Figure S7.
Five studies involving 187 participants described the distinct taxa at the phylum level. Examining taxonomic distribution at the phylum level did not reveal remarkable differences between DC and DM in Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, Fusobacteria, and Verrucomicrobia (Figure 4 and Supplementary Figure S8).
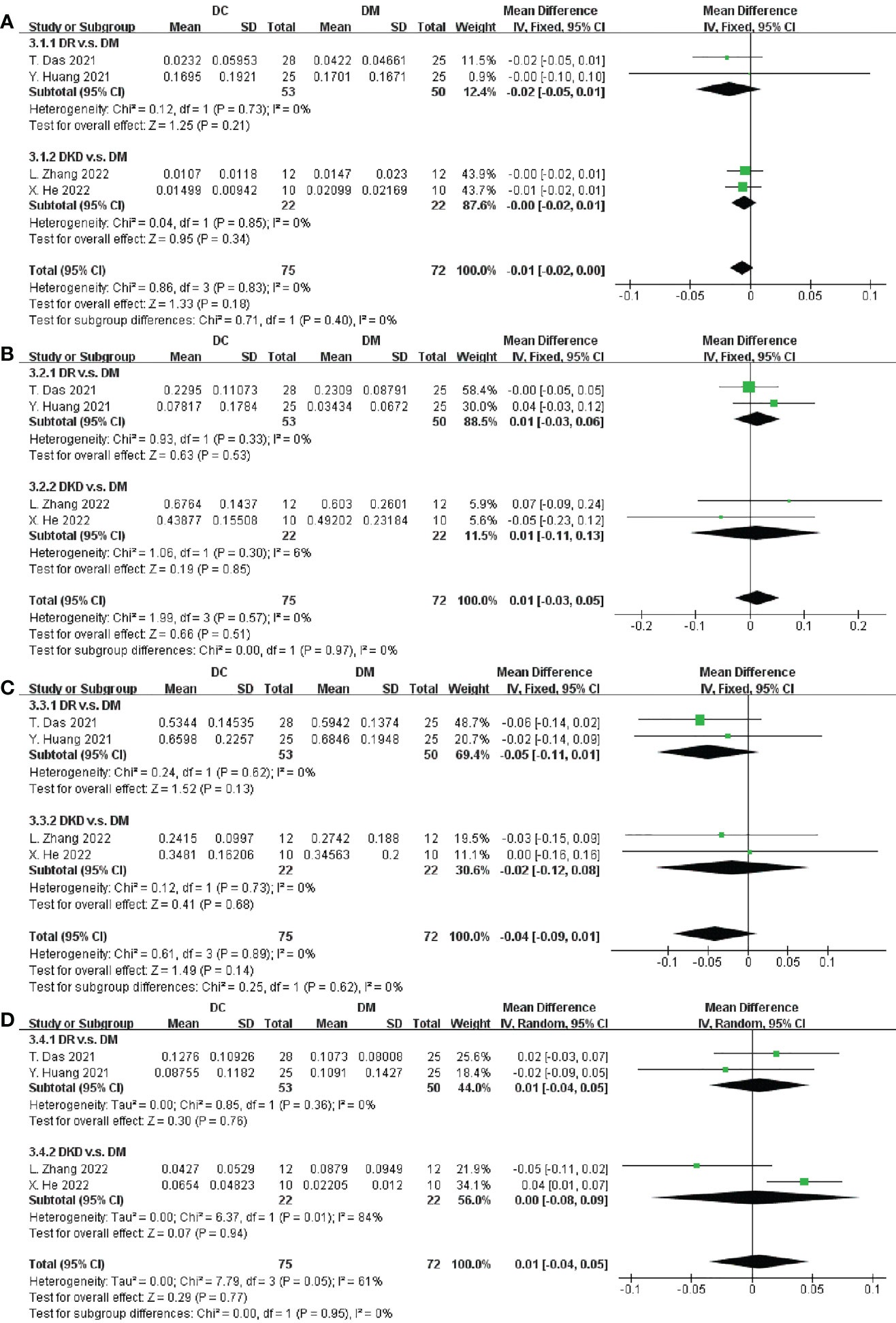
Figure 4 Forest plots of microbiota at the phylum level in DC v.s. DM. (A) Actinobacteria; (B) Bacteroidetes; (C) Firmicutes; (D) Proteobacteria.
We next performed an analysis of microbiota at the phylum level between DC and HC. Among the most abundant species, we observed that DC patients were enriched in Proteobacteria (SMD = 0.03, 95% CI[0.01, 0.04], p = 0.003), and depleted in Firmicutes (SMD = -0.06, 95% CI[-0.11, -0.01], p = 0.02), in Figures 5A, B. Specially, the abundance of Proteobacteria inDKD was significantly higher than that in HC (SMD = 0.03, 95% CI[0.01, 0.04], p = 0.008) and Firmicutes was significantly depleted in DR when compared to HC (SMD = -0.06, 95% CI[-0.12, -0.00], p = 0.03).
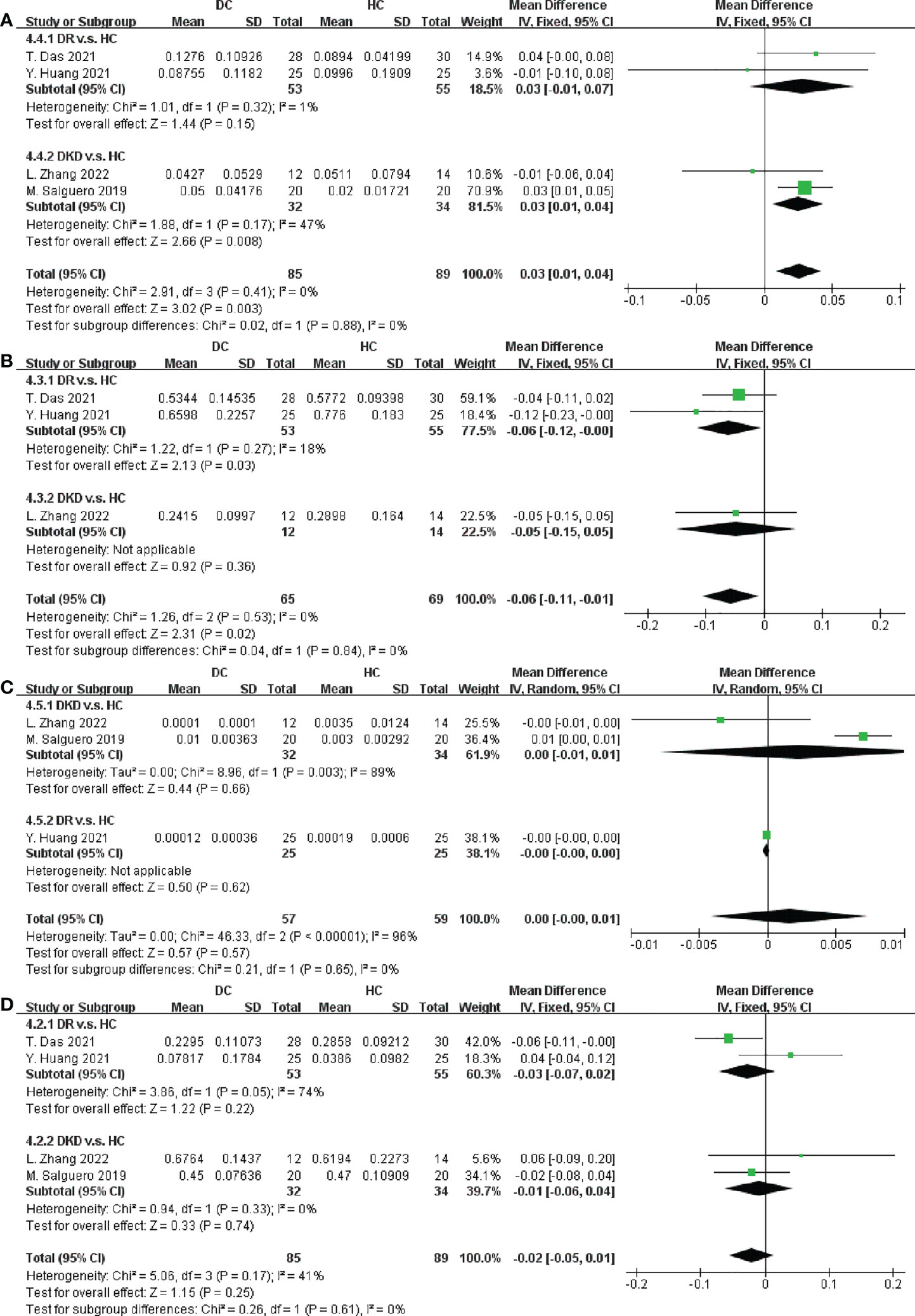
Figure 5 Forest plots of microbiota at the phylum level in DC v.s. HC. (A) Proteobacteria; (B) Firmicutes; (C) Fusobacteria; (D) Bacteroidetes.
Furthermore, data in Figures 5C, D and Supplementary Figure S9 demonstrated there were no significant differences in the relative abundance of Fusobacteria, Bacteroidetes, Actinobacteria and Verrucomicrobia between DC and HC.
From the 11 trials included in this meta-analysis, we obtained that there was a significantly deplete in richness of Bifidobacterium and Faecalibacterium in DC when compared to DM (SMD = -0.01, 95% CI[-0.02,-0.01], p < 0.0001; SMD = -0.01, 95% CI[-0.02, -0.00], p = 0.009, respectively), as depicted in Figures 6A, B. However, the abundances of Alistipes, Prevotella, Ruminococcus, Lachnospira, Roseburia, Clostridium, Blautia, Escherichia, Eubacterium, Parabacteroides, Mitsuokella and Lactobacillus varied, and we did not find any relatively consistent results after meta-analysis (Supplementary Figures S10 and 11).
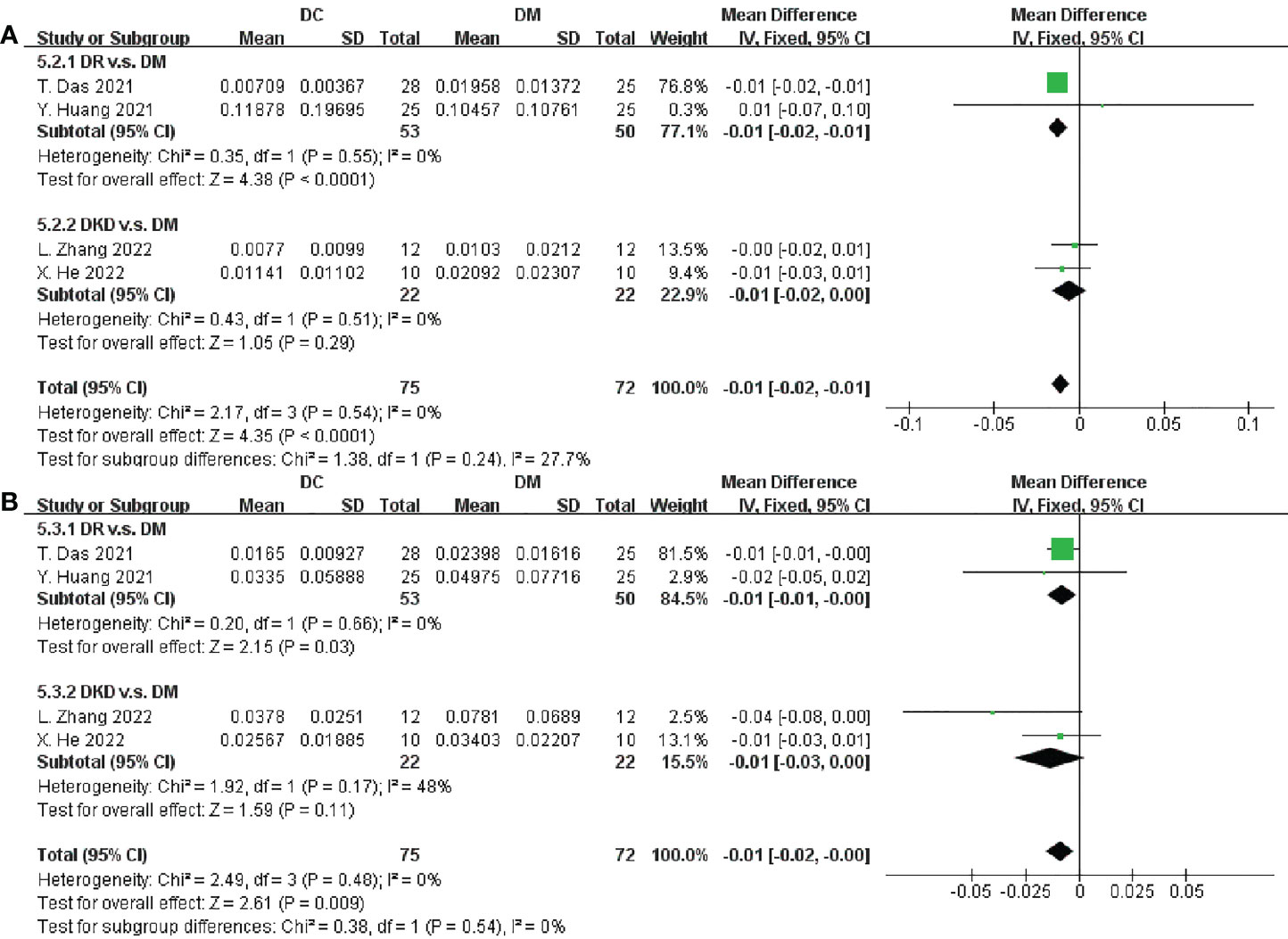
Figure 6 Forest plots of microbiota at the genus level in DC v.s. DM. (A) Bifidobacterium; (B) Faecalibacterium.
After that, the taxa between DC and HC were compared in Figure 7. Faecalibacterium and Lactobacillus were proven to be decreased in DC (SMD = -0.02, 95% CI[-0.02, -0.01], p < 0.00001; SMD = -0.00, 95% CI[-0.00, -0.00], p < 0.00001; respectively), as well as an increase of Klebsiella (SMD = 0.00, 95% CI[0.00, 0.00], p < 0.00001). Besides, no consistent results were found in the relative abundances of Streptococcus, Roseburia, Clostridium, Blautia, Escherichia, Eubacterium, Bifidobacterium and Lachnospira (Supplementary Figures S12 and S13). In this study, we summarized the alternation tendency of gut microbiota in DC compared to DM and HC respectively in Figure 8.
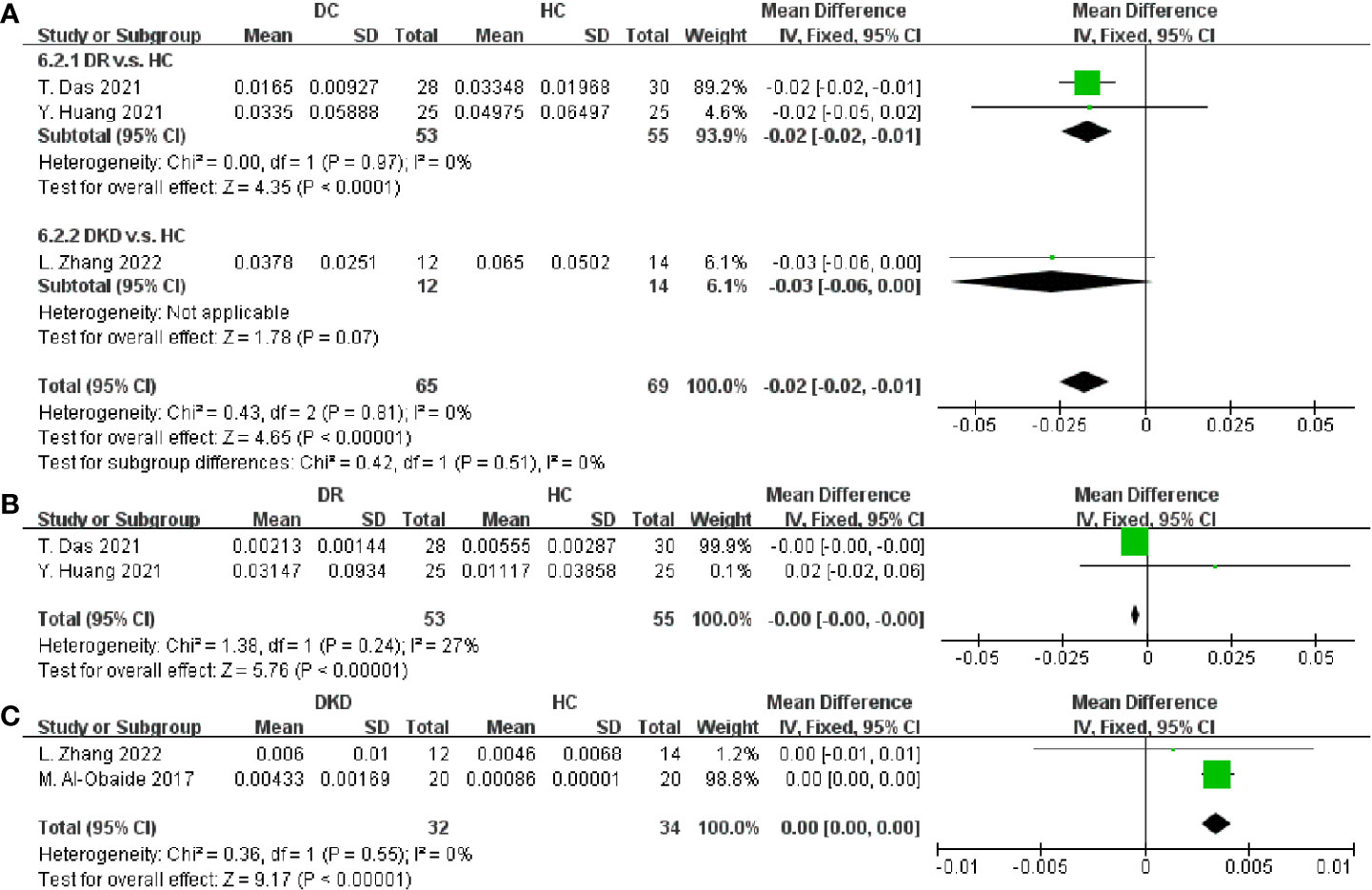
Figure 7 Forest plots of microbiota at the genus level in DC v.s. HC. (A) Faecalibacterium; (B) Lactobacillus; (C) Klebsiella.

Figure 8 Microbiota at the phylum and genus level. (A) DC v.s. DM; (B) DC v.s. HC.
To our knowledge, this meta-analysis was the first to assess alpha diversity and microbiota perturbations in DC compared with DM and HC. This study yielded three major insights into the gut microbial changes in DC. Firstly, patients with DC exhibited a significantly higher richness but lower diversity in gut microbiota when compared to DM. And when compared to HC, DC held a lower richness in gut microbiota. Secondly, the abundance of phylum Proteobacteria was enriched and Firmicutes was depleted in DC when compared to HC. Last, the abundances of genera Bifidobacterium and Faecalibacterium were depleted in DC when compared to DM. And when compared to HC, DC exhibited lower abundances of Faecalibacterium and Lactobacillus but a higher abundance of Klebsiella. All these changes may be associated with the pathology of DC, and serve as promising targets for the management of DC.
One common indicator of dysbiosis is a modified overall microbial alpha diversity, which denotes the relative abundance of microbial species in space and time within a specific community. Alpha diversity indices include OTUs, ACE, Chao1, Simpson and Shannon. ACE and Chao1 focus on species richness. Simpson and Shannon strengthen diversity. Generally, lower alpha diversity was observed in obesity and diabetes, and was considered detrimental to the host (34). Alpha diversity in richness and diversity declined in DR and DKD when compared to HC (28, 29). In contrast, no difference was found in gut microbiota between DM patients with or without cognitive impairment (35). DPN resulted in a more severe disruption of microbiota community richness than DM (16). These findings indicated that the profile of gut microbiota was altered in patients with DC, but further exploration was needed due to the extremely inconsistent findings.
Phylum Proteobacteria was gram-negative bacteria and proved to be significantly different in gut microbiota between DM patients with or without gastrointestinal autonomic neuropathy (36). Proteobacteria was proven to be negatively related to human health and found with a higher abundance in the obese population than the non-obese population (34, 37). The mechanism might be related to the regulation of bile acids, a metabolic controller (38). In line with these results, Proteobacteria was found significantly enriched in DC when compared to HC in this study, concluding that with the risk of DC increased, the abundance of Proteobacteria increased. These findings may further reveal that the alternation of gut microbiota at the phylum level is closely related to DC.
The phyla Firmicutes and Bacteroidetes accounted for more than 90% of the total community of human gut microbiota. Alterations affecting Firmicutes and Bacteroidetes were first described in obese subjects who exhibited increased abundances of Firmicutes at the expense of Bacteroidetes (39). Firmicutes was positively linked with inflammation and the modulation of metabolism, and was supposed to be more efficient at calorie absorption and weight gain (40–42). Bacteroidota was significantly reduced in DR patients, and Firmicutes to Bacteroidetes ratio was proven elevated in gestational diabetes patients (15). However, in opposition to these results, no change or even decreased Firmicutes to Bacteroidetes ratio in obese subjects was reported (43–45). In our study, Firmicutes were significantly depleted in DC compared to HC. The reason might be linked to less bacterial diversity in obese patients than the non-obese subjects (46). Therefore, further research involving this parameter should be high on the list.
In line with the results presented in this study concerning the significant decrease of genera Bifidobacterium, Lactobacillus and Faecalibacterium in DC (p < 0.05), similar results were observed in DR in Zhou’s study (31). Bifidobacteria and Lactobacillus were gram-positive microorganisms, which participated in the restoration of the intestinal mucosal barrier (47, 48). Both of them were inversely associated with inflammation, hyperglycemia, and insulin resistance (49–51). Bifidobacteria was demonstrated to produce bacteriocin that blocks adherence to the mucosa and preserves gut barrier function (52). DM patients presented a significantly lower richness of Bifidobacterium compared to healthy subjects (53, 54). Probiotics, as well as yogurt or milk containing Bifidobacterium and Lactobacillus, decreased fasting blood glucose and glycosylated hemoglobin type A1c (HbA1c) in DM patients (55, 56). Drawing a conclusion that Bifidobacteria and Lactobacillus decreased as the risk of DC increased. The mechanism might be linked to the depletion of potential intestinal pathogens, enhancement of intestinal antioxidant ability and digestive enzyme activity (57). Faecalibacterium was gram-negative, butyrate-producing bacteria, which was demonstrated to be negatively related to HbA1c (26). Faecalibacterium was proven to increase the intestinal synthesis of glucagon-like peptide-1, peptide YY, acetate and butyrate help to maintain glucose homeostasis (58). In this study, Faecalibacterium was depleted in DC, corroborating previous reports that linked Faecalibacterium to positive metabolic outcomes (29, 33, 59–61). These findings revealed that the alternation of gut microbiota at the genus level was closely related to DC, and the increase of Bifidobacterium, Lactobacillus and Faecalibacterium might be responsible for the pathology of DC. Klebsiella is a natural inhabitant of the gastrointestinal tract microbiome of healthy humans and animals, but it often causes extraintestinal infections, including urinary tract infections, pneumoniae and septicemia (62). Recently, Klebsiella was demonstrated to be increased in DKD (63), which was proven to be increased in DC when compared to HC in this study.
Microbiota symbiosis helped regulate metabolism and reduce the risk of DM. Gut microbiota dysbiosis was proven to lead imbalance of intestinal microbial bi-products, and cause insulin resistance, with mechanism unclear. Recently, the mechanism of how microbiota affect DM and its complications has raised academic interests. Firstly, microbiota dysbiosis led to the production of short-chain fatty acids (SCFAs) including butyrate, propionate, and acetate, which then impaired the intestinal barrier integrity, activated the inflammation signaling cascades and thus promoted the multi-organs damage (64). Secondly, microbiota-derived trimethylamine nitrogen oxide (TMAO) increased the accumulation of cholesterol, led to insulin resistance (65), and increased the risk of DM (66, 67). Third, changes in gut microbiota composition were proven to affect gut permeability and inflammation in DM (68, 69). Microbiota-derived lipopolysaccharide (LPS) triggered downstream inflammatory pathways and pro-inflammatory cytokine expression cascades, leading to inflammatory reactions and aggravating insulin resistance (70, 71). Last but not the least, microbiota-derived aromatic amino acids including indoxyl sulfate and p-cresyl sulfate, also known as uremic toxins, might induce mitochondrial dysfunction, podocytes injuries, thicken the glomerular basement membrane, and ultimately lead to renal micro-inflammation, and perivascular fibrosis in DKD (72, 73).
In this meta-analysis, genus Bifidobacterium belonged to the phylum Actinobacteria, genera Faecalibacterium and Lactobacillus belonged to the phylum Firmicutes, genus Klebsiella belonged to the phylum Proteobacteria. Phyla Actinobacteria, Firmicutes and Proteobacteria were both SCFAs-producing and LPS-producing bacteria. Therefore, we hypothesized that SCFAs-induced intestinal barrier integrity impairment and LPS-induced inflammation might be important parts of mechanism of DC. Nevertheless, direct targets of gut microbiota and the potential mechanisms needed to be further elucidated.
In spite of these interesting findings, our study was not without limitations. First, the language of included literature was limited to English, which may increase the possibility of language bias or publication bias. Secondly, since the gut microbiota was closely related to race and living environment, and the included studies originated mostly from Asia (11/13), in which nine studies were conducted in China and two in India. To explore the potential source of heterogeneity caused by it, the subgroup analysis on the nation has been performed in some comparison. The generalization of these results can only represent the characteristics of Asian populations to some extent. Third, in the 13 studies included, there were seven (7/13) studies focused on DKD, five (5/13) focused on DR, and only one (1/13) focused on DPN. To explore the potential source of heterogeneity caused by different microvascular complications of DM, the subgroup analysis on the type of microvascular complications has been performed. Considering the representativeness and potential bias of DPN, a meta-analysis of studies excluding DPN was performed to compare. Although DR and DKD were more representative microvascular complications in some ways, it would be better if a larger number of studies on DPN could be included in this meta-analysis. Fourth, the heterogeneity was high, although subgroup analysis on the type of DC, nation and city was performed, it was far from enough to explain the source of heterogeneity. More studies should be included and more factors related to microbiota such as ethnicity, eating habits, living environment, obesity condition, and even drugs on the changes of gut microbiota. Last, a different method of gene sequencing was also a potential bias on the results.
In conclusion, we demonstrated that the alternation of alpha diversity, the abundances of phyla Proteobacteria and Firmicutes as well as the abundances of genera Bifidobacterium, Faecalibacterium Lactobacillus and Klebsiella may be associated with DC. The changes of microbial features may be noninvasive biomarkers for monitoring the management of DC. However, our findings still need to be verified by further large-scale, multi-center and high-quality studies. Nevertheless, conflicting results have also been reported, and therefore further studies would be necessary to fully understand the relation between gut microbiota and DC.
All data may be obtained within the article, the supplementary files, or from the original articles.
JH, DL, and CM designed the study. JH and TF performed the literature search, screened for eligible studies, and extracted data, which were then analyzed by WL and YD. JH wrote the first draft of the manuscript and all authors contributed to subsequent drafts and approved the final version for submission.
This study was funded by the Traditional Chinese Medicine Bureau of Guangdong Province (Nos. 20231003, 20211003 and 20223001), the National Natural Science Foundation of China (No. 82202560), the Natural Science Foundation of Guangdong Province (No. 2021A1515220050), the Fundamental Research Funds for the Central University (No. 22qntd4813) and the second batch of central subsidy funds for traditional Chinese medicine departments in 2022 (construction of national traditional Chinese medicine specialty) (No. 2022CZ0047).
The authors acknowledged Guangdong Provincial People’s Hospital and Sun Yat-sen University for the academic supports.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.1053900/full#supplementary-material
1. Khalili L, Alipour B, Asghari JM, Hassanalilou T, Mesgari AM, Faraji I. Probiotic assisted weight management as a main factor for glycemic control in patients with type 2 diabetes: a randomized controlled trial. Diabetol Metab Syndr (2019) 11:5. doi: 10.1186/s13098-019-0400-7
2. Sivakumar PM, Prabhawathi V, Zarrabi A, Akthar S, Prabhakar PK. Current trends in the therapeutic strategies for diabetes management. Curr Med Chem (2021) 28:4616–37. doi: 10.2174/0929867328666210218183914
3. Saeedi P, Salpea P, Karuranga S, Petersohn I, Malanda B, Gregg EW, et al. Mortality attributable to diabetes in 20-79 years old adults 2019 estimates: Results from the international diabetes federation diabetes atlas, 9(th) edition. Diabetes Res Clin Pract (2020) 162:108086. doi: 10.1016/j.diabres.2020.108086
4. Cho NH, Shaw JE, Karuranga S, Huang Y, Da RFJ, Ohlrogge AW, et al. IDF diabetes atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract (2018) 138:271–81. doi: 10.1016/j.diabres.2018.02.023
5. Ahsan H. Diabetic retinopathy–biomolecules and multiple pathophysiology. Diabetes Metab Syndr (2015) 9:51–4. doi: 10.1016/j.dsx.2014.09.011
6. Leasher JL, Bourne RR, Flaxman SR, Jonas JB, Keeffe J, Naidoo K, et al. Global estimates on the number of people blind or visually impaired by diabetic retinopathy: A meta-analysis from 1990 to 2010. Diabetes Care (2016) 39:1643–9. doi: 10.2337/dc15-2171
7. Cui Y, Zhang M, Zhang L, Zhang L, Kuang J, Zhang G, et al. Prevalence and risk factors for diabetic retinopathy in a cross-sectional population-based study from rural southern China: Dongguan eye study. BMJ Open (2019) 9:e023586. doi: 10.1136/bmjopen-2018-023586
8. Javed S, Hayat T, Menon L, Alam U, Malik RA. Diabetic peripheral neuropathy in people with type 2 diabetes: too little too late. Diabetes Med (2020) 37:573–9. doi: 10.1111/dme.14194
9. Iatcu CO, Steen A, Covasa M. Gut microbiota and complications of type-2 diabetes. Nutrients (2021) 14:166. doi: 10.3390/nu14010166
10. Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol (2021) 19:55–71. doi: 10.1038/s41579-020-0433-9
11. Lin D, Song Q, Zhang Y, Liu J, Chen F, Du S, et al. Bacillus subtilis attenuates hepatic and intestinal injuries and modulates gut microbiota and gene expression profiles in mice infected with schistosoma japonicum. Front Cell Dev Biol (2021) 9:766205. doi: 10.3389/fcell.2021.766205
12. Dominguez-Bello MG, Godoy-Vitorino F, Knight R, Blaser MJ. Role of the microbiome in human development. Gut (2019) 68:1108–14. doi: 10.1136/gutjnl-2018-317503
13. Lin D, Song Q, Liu J, Chen F, Zhang Y, Wu Z, et al. Potential gut microbiota features for non-invasive detection of schistosomiasis. Front Immunol (2022) 13:941530. doi: 10.3389/fimmu.2022.941530
14. Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J, et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol (2020) 17:279–97. doi: 10.1038/s41575-020-0269-9
15. Das T, Jayasudha R, Chakravarthy S, Prashanthi GS, Bhargava A, Tyagi M, et al. Alterations in the gut bacterial microbiome in people with type 2 diabetes mellitus and diabetic retinopathy. Sci Rep (2021) 11:2738. doi: 10.1038/s41598-021-82538-0
16. Wang Y, Ye X, Ding D, Lu Y. Characteristics of the intestinal flora in patients with peripheral neuropathy associated with type 2 diabetes. J Int Med Res (2020) 48:300060520936806. doi: 10.1177/0300060520936806
17. He X, Sun J, Liu C, Yu X, Li H, Zhang W, et al. Compositional alterations of gut microbiota in patients with diabetic kidney disease and type 2 diabetes mellitus. Diabetes Metab Syndr Obes (2022) 15:755–65. doi: 10.2147/DMSO.S347805
18. Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Manneras-Holm L, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med (2017) 23:850–8. doi: 10.1038/nm.4345
19. Lee CB, Chae SU, Jo SJ, Jerng UM, Bae SK. The relationship between the gut microbiome and metformin as a key for treating type 2 diabetes mellitus. Int J Mol Sci (2021) 22:3566. doi: 10.3390/ijms22073566
20. Armour CR, Nayfach S, Pollard KS, Sharpton TJ. A metagenomic meta-analysis reveals functional signatures of health and disease in the human gut microbiome. mSystems (2019) 4:e00332-18. doi: 10.1128/mSystems.00332-18
21. Hutton B, Salanti G, Caldwell DM, Chaimani A, Schmid CH, Cameron C, et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: checklist and explanations. Ann Intern Med (2015) 162:777–84. doi: 10.7326/M14-2385
22. Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol (2010) 25:603–5. doi: 10.1007/s10654-010-9491-z
23. Nano J, Muka T, Cepeda M, Voortman T, Dhana K, Brahimaj A, et al. Association of circulating total bilirubin with the metabolic syndrome and type 2 diabetes: A systematic review and meta-analysis of observational evidence. Diabetes Metab (2016) 42:389–97. doi: 10.1016/j.diabet.2016.06.002
24. Al-Obaide M, Singh R, Datta P, Rewers-Felkins KA, Salguero MV, Al-Obaidi I, et al. Gut microbiota-dependent trimethylamine-n-oxide and serum biomarkers in patients with T2DM and advanced CKD. J Clin Med (2017) 6:86. doi: 10.3390/jcm6090086
25. Salguero MV, Al-Obaide M, Singh R, Siepmann T, Vasylyeva TL. Dysbiosis of gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp Ther Med (2019) 18:3461–9. doi: 10.3892/etm.2019.7943
26. Tao S, Li L, Li L, Liu Y, Ren Q, Shi M, et al. Understanding the gut-kidney axis among biopsy-proven diabetic nephropathy, type 2 diabetes mellitus and healthy controls: an analysis of the gut microbiota composition. Acta Diabetol (2019) 56:581–92. doi: 10.1007/s00592-019-01316-7
27. Jayasudha R, Das T, Kalyana CS, Sai PG, Bhargava A, Tyagi M, et al. Gut mycobiomes are altered in people with type 2 diabetes mellitus and diabetic retinopathy. PloS One (2020) 15:e0243077. doi: 10.1371/journal.pone.0243077
28. Du X, Liu J, Xue Y, Kong X, Lv C, Li Z, et al. Alteration of gut microbial profile in patients with diabetic nephropathy. Endocrine (2021) 73:71–84. doi: 10.1007/s12020-021-02721-1
29. Huang Y, Wang Z, Ma H, Ji S, Chen Z, Cui Z, et al. Dysbiosis and implication of the gut microbiota in diabetic retinopathy. Front Cell Infect Microbiol (2021) 11:646348. doi: 10.3389/fcimb.2021.646348
30. Ye P, Zhang X, Xu Y, Xu J, Song X, Yao K. Alterations of the gut microbiome and metabolome in patients with proliferative diabetic retinopathy. Front Microbiol (2021) 12:667632. doi: 10.3389/fmicb.2021.667632
31. Zhou Z, Zheng Z, Xiong X, Chen X, Peng J, Yao H, et al. Gut microbiota composition and fecal metabolic profiling in patients with diabetic retinopathy. Front Cell Dev Biol (2021) 9:732204. doi: 10.3389/fcell.2021.732204
32. Chen R, Zhu D, Yang R, Wu Z, Xu N, Chen F, et al. Gut microbiota diversity in middle-aged and elderly patients with end-stage diabetic kidney disease. Ann Transl Med (2022) 10:750. doi: 10.21037/atm-22-2926
33. Zhang L, Wang Z, Zhang X, Zhao L, Chu J, Li H, et al. Alterations of the gut microbiota in patients with diabetic nephropathy. Microbiol Spectr (2022) 10:e0032422. doi: 10.1128/spectrum.00324-22
34. Caudet J, Trelis M, Cifre S, Soriano JM, Rico H, Merino-Torres JF. Interplay between intestinal bacterial communities and unicellular parasites in a morbidly obese population: A neglected trinomial. Nutrients (2022) 14:3211. doi: 10.3390/nu14153211
35. Zhang Y, Lu S, Yang Y, Wang Z, Wang B, Zhang B, et al. The diversity of gut microbiota in type 2 diabetes with or without cognitive impairment. Aging Clin Exp Res (2021) 33:589–601. doi: 10.1007/s40520-020-01553-9
36. Du Y, Neng Q, Li Y, Kang Y, Guo L, Huang X, et al. Gastrointestinal autonomic neuropathy exacerbates gut microbiota dysbiosis in adult patients with type 2 diabetes mellitus. Front Cell Infect Microbiol (2021) 11:804733. doi: 10.3389/fcimb.2021.804733
37. Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care (2011) 34:392–7. doi: 10.2337/dc10-1676
38. Prawitt J, Caron S, Staels B. Bile acid metabolism and the pathogenesis of type 2 diabetes. Curr Diabetes Rep (2011) 11:160–6. doi: 10.1007/s11892-011-0187-x
39. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature (2006) 444:1022–3. doi: 10.1038/4441022a
40. Krajmalnik-Brown R, Ilhan ZE, Kang DW, DiBaise JK. Effects of gut microbes on nutrient absorption and energy regulation. Nutr Clin Pract (2012) 27:201–14. doi: 10.1177/0884533611436116
41. Kumar H, Lund R, Laiho A, Lundelin K, Ley RE, Isolauri E, et al. Gut microbiota as an epigenetic regulator: pilot study based on whole-genome methylation analysis. mBio (2014) 5:e02113-14. doi: 10.1128/mBio.02113-14
42. Welcome MO. Gut microbiota disorder, gut epithelial and blood-brain barrier dysfunctions in etiopathogenesis of dementia: Molecular mechanisms and signaling pathways. Neuromol Med (2019) 21:205–26. doi: 10.1007/s12017-019-08547-5
43. Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr (2011) 94:58–65. doi: 10.3945/ajcn.110.010132
44. Patil DP, Dhotre DP, Chavan SG, Sultan A, Jain DS, Lanjekar VB, et al. Molecular analysis of gut microbiota in obesity among Indian individuals. J Biosci (2012) 37:647–57. doi: 10.1007/s12038-012-9244-0
45. Tims S, Derom C, Jonkers DM, Vlietinck R, Saris WH, Kleerebezem M, et al. Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J (2013) 7:707–17. doi: 10.1038/ismej.2012.146
46. Aguirre M, Venema K. Does the gut microbiota contribute to obesity? going beyond the gut feeling. Microorganisms (2015) 3:213–35. doi: 10.3390/microorganisms3020213
47. Robles-Vera I, Toral M, de la Visitacion N, Sanchez M, Romero M, Olivares M, et al. The probiotic lactobacillus fermentum prevents dysbiosis and vascular oxidative stress in rats with hypertension induced by chronic nitric oxide blockade. Mol Nutr Food Res (2018) 62:e1800298. doi: 10.1002/mnfr.201800298
48. Alessandri G, van Sinderen D, Ventura M. The genus bifidobacterium: From genomics to functionality of an important component of the mammalian gut microbiota running title: Bifidobacterial adaptation to and interaction with the host. Comput Struct Biotechnol J (2021) 19:1472–87. doi: 10.1016/j.csbj.2021.03.006
49. Moroti C, Souza ML, de Rezende CM, Cavallini DC, Sivieri K. Effect of the consumption of a new symbiotic shake on glycemia and cholesterol levels in elderly people with type 2 diabetes mellitus. Lipids Health Dis (2012) 11:29. doi: 10.1186/1476-511X-11-29
50. Cani PD. Microbiota and metabolites in metabolic diseases. Nat Rev Endocrinol (2019) 15:69–70. doi: 10.1038/s41574-018-0143-9
51. Zou Y, Chen T. Engineered akkermansia muciniphila: A promising agent against diseases (Review). Exp Ther Med (2020) 20:285. doi: 10.3892/etm.2020.9415
52. Riviere A, Selak M, Lantin D, Leroy F, De Vuyst L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front Microbiol (2016) 7:979. doi: 10.3389/fmicb.2016.00979
53. Wu X, Ma C, Han L, Nawaz M, Gao F, Zhang X, et al. Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr Microbiol (2010) 61:69–78. doi: 10.1007/s00284-010-9582-9
54. Sroka-Oleksiak A, Mlodzinska A, Bulanda M, Salamon D, Major P, Stanek M, et al. Metagenomic analysis of duodenal microbiota reveals a potential biomarker of dysbiosis in the course of obesity and type 2 diabetes: A pilot study. J Clin Med (2020) 9:369. doi: 10.3390/jcm9020369
55. Ejtahed HS, Mohtadi-Nia J, Homayouni-Rad A, Niafar M, Asghari-Jafarabadi M, Mofid V. Probiotic yogurt improves antioxidant status in type 2 diabetic patients. Nutrition (2012) 28:539–43. doi: 10.1016/j.nut.2011.08.013
56. Ostadrahimi A, Taghizadeh A, Mobasseri M, Farrin N, Payahoo L, Beyramalipoor GZ, et al. Effect of probiotic fermented milk (kefir) on glycemic control and lipid profile in type 2 diabetic patients: a randomized double-blind placebo-controlled clinical trial. Iran J Public Health (2015) 44:228–37.
57. Dowarah R, Verma AK, Agarwal N. The use of lactobacillus as an alternative of antibiotic growth promoters in pigs: A review. Anim Nutr (2017) 3:1–6. doi: 10.1016/j.aninu.2016.11.002
58. Huda MN, Kim M, Bennett BJ. Modulating the microbiota as a therapeutic intervention for type 2 diabetes. Front Endocrinol (Lausanne) (2021) 12:632335. doi: 10.3389/fendo.2021.632335
59. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of crohn disease patients. Proc Natl Acad Sci U.S.A. (2008) 105:16731–6. doi: 10.1073/pnas.0804812105
60. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature (2013) 500:541–6. doi: 10.1038/nature12506
61. Thingholm LB, Ruhlemann MC, Koch M, Fuqua B, Laucke G, Boehm R, et al. Obese individuals with and without type 2 diabetes show different gut microbial functional capacity and composition. Cell Host Microbe (2019) 26:252–264.e10. doi: 10.1016/j.chom.2019.07.004
62. Navon-Venezia S, Kondratyeva K, Carattoli A. Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol Rev (2017) 41:252–75. doi: 10.1093/femsre/fux013
63. Wang Y, Zhao J, Qin Y, Yu Z, Zhang Y, Ning X, et al. The specific alteration of gut microbiota in diabetic kidney diseases-a systematic review and meta-analysis. Front Immunol (2022) 13:908219. doi: 10.3389/fimmu.2022.908219
64. Schippa S, Conte MP. Dysbiotic events in gut microbiota: impact on human health. Nutrients (2014) 6:5786–805. doi: 10.3390/nu6125786
65. Oellgaard J, Winther SA, Hansen TS, Rossing P, von Scholten BJ. Trimethylamine n-oxide (TMAO) as a new potential therapeutic target for insulin resistance and cancer. Curr Pharm Des (2017) 23:3699–712. doi: 10.2174/1381612823666170622095324
66. Fernandes J, Su W, Rahat-Rozenbloom S, Wolever TM, Comelli EM. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes (2014) 4:e121. doi: 10.1038/nutd.2014.23
67. Shan Z, Sun T, Huang H, Chen S, Chen L, Luo C, et al. Association between microbiota-dependent metabolite trimethylamine-n-oxide and type 2 diabetes. Am J Clin Nutr (2017) 106:888–94. doi: 10.3945/ajcn.117.157107
68. Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut (2009) 58:1091–103. doi: 10.1136/gut.2008.165886
69. Boutagy NE, McMillan RP, Frisard MI, Hulver MW. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie (2016) 124:11–20. doi: 10.1016/j.biochi.2015.06.020
70. Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, et al. CD14 controls the LPS-induced endocytosis of toll-like receptor 4. Cell (2011) 147:868–80. doi: 10.1016/j.cell.2011.09.051
71. Wen L, Duffy A. Factors influencing the gut microbiota, inflammation, and type 2 diabetes. J Nutr (2017) 147:1468S–75S. doi: 10.3945/jn.116.240754
72. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell (2013) 155:1451–63. doi: 10.1016/j.cell.2013.11.024
Keywords: diabetic kidney disease (DKD), diabetic peripheral neuropathy (DPN), diabetic retinopathy (DR), gut microbiota (GM), diabetic microvascular complication
Citation: Hong J, Fu T, Liu W, Du Y, Min C and Lin D (2022) Specific alterations of gut microbiota in diabetic microvascular complications: A systematic review and meta-analysis. Front. Endocrinol. 13:1053900. doi: 10.3389/fendo.2022.1053900
Received: 26 September 2022; Accepted: 11 November 2022;
Published: 05 December 2022.
Edited by:
Van B. Lu, Western University, CanadaReviewed by:
Tianshu Zeng, Huazhong University of Science and Technology, ChinaCopyright © 2022 Hong, Fu, Liu, Du, Min and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinni Hong, aG9uZ2ppbm5pQGdkcGgub3JnLmNu; Cunyun Min, bWluY3VueXVuQGdkcGgub3JnLmNu; Datao Lin, bGluZHQ1QG1haWwuc3lzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.