
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 16 November 2022
Sec. Cellular Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1048601
This article is part of the Research TopicInsights in Cellular Endocrinology: 2022View all 9 articles
 Pauline Raynaud1†
Pauline Raynaud1† Camille Gauthier1†
Camille Gauthier1† Vinesh Jugnarain1
Vinesh Jugnarain1 Frédéric Jean-Alphonse1,2Eric Reiter1,2Gilles Bruneau1
Frédéric Jean-Alphonse1,2Eric Reiter1,2Gilles Bruneau1 Pascale Crépieux1,2*
Pascale Crépieux1,2*Single-domain antibody fragments, also known as VHHs or nanobodies, have opened promising avenues in therapeutics and in exploration of intracellular processes. Because of their unique structural properties, they can reach cryptic regions in their cognate antigen. Intracellular VHHs/antibodies primarily directed against cytosolic proteins or transcription factors have been described. In contrast, few of them target membrane proteins and even less recognize G protein-coupled receptors. These receptors are major therapeutic targets, which reflects their involvement in a plethora of physiological responses. Hence, they elicit a tremendous interest in the scientific community and in the industry. Comprehension of their pharmacology has been obscured by their conformational complexity, that has precluded deciphering their structural properties until the early 2010’s. To that respect, intracellular VHHs have been instrumental in stabilizing G protein-coupled receptors in active conformations in order to solve their structure, possibly bound to their primary transducers, G proteins or β-arrestins. In contrast, the modulatory properties of VHHs recognizing the intracellular regions of G protein-coupled receptors on the induced signaling network have been poorly studied. In this review, we will present the advances that the intracellular VHHs have permitted in the field of GPCR signaling and trafficking. We will also discuss the methodological hurdles that linger the discovery of modulatory intracellular VHHs directed against GPCRs, as well as the opportunities they open in drug discovery.
Antibody fragments are currently revolutionizing the specific targeting of molecules inside the cell. Among those, paramount importance has been given to variable fragments from single-chain antibodies encountered in the blood of some Camelidae and cartilaginous fishes. These fragments represent the VHH (VH of heavy-chain only antibodies), and their recombinant analogues have been baptized as Nbs for nanobodies, or sdAbs for single domain antibodies. When compared to conventional immunoglobulins (Igs), they have smaller molecular weight (∼13 kDa) and size (diameter of 2.5 nm, length of 4 nm), and they consist in the minimum paratope able to recognize an epitope. They are composed of 4 framework regions (FR) and 3 complementarity-determining regions (CDR) that support complementarity to the epitope. A striking feature of these VHHs is the greater length of their CDR3 compared to that of a VH, that optimizes the contact surface with the epitope and compensates the lack of light chain. The length and flexibility of CDR3 are properties that are advantageous to reach poorly accessible, cryptic epitopes (1).
VHHs contain a canonical disulfide bond linking FR1 and FR3, and possibly additional bonds, for example between CDR2 and CDR3 in lama. These disulfide bonds are essential for thermal and thermodynamic stability (2), but apparently, reducing conditions do not compromise antigen binding affinity, nor mechanical stability (3). In addition, because of “hallmark” hydrophilic residues at solvent-exposed positions, they are less prone to protein aggregation than single-chain variable fragments (scFv) (4). These properties are of uttermost importance when intracellularly expressed VHHs (intra-VHHs) are being studied, because they are placed in a mildly reducing environment in the cytosol. Although we are aware that the terminology of “nanobody” given by Ablynx in 2001 is widely accepted, in the case of intracellular Nb we prefer the term “intra-VHH” over “intrabodies”, that does not explicitly indicate the format of the antibody (VHH, scFv, Ig). This is the term that will be used in this review.
Intra-VHHs are a new type of biological tools that target proteins present at different sub-cellular compartments [reviewed in (5)], such as the GTP-bound form of RhoA at the inner face of the plasma membrane (6), or lamin lining the nuclear envelope (7). They can be used to enhance, block or monitor (8) protein activation/activity, to track protein trafficking inside the cell when fused to fluorescent probes (7), to hinder protein/protein interactions, or to direct the antigen protein to degradation pathways (9). Some of them appear exquisitely sensitive to protein conformational dynamics, and this chaperone-like property has been particularly useful to decipher the conformational complexity of G protein-coupled receptors (GPCR) in their ligand-activated states.
GPCRs are involved in most physiological responses and as such, they bind ligands encompassing a vast array of biochemical classes, ranging from photons or ions to large glycoprotein hormones. Accordingly, they are major targets of the therapeutic arsenal, including treatments of hypertension, migraine, cancers, metabolic diseases, or disorders of the central nervous system, among other pathologies. But drug discovery of small chemicals with high potency, efficacy and selectivity is reaching a limit, and in 2017, the 481 referenced drugs against GPCRs only targeted 107 out of 850 receptors of this protein family (10). Conventional antibodies now open new avenues in the field, but still, GPCRs remain extremely difficult targets since i/they are highly hydrophobic in nature because of their seven transmembrane helices, ii/most of them expose very limited regions outside the cell membrane, iii/their wide conservation across the animal kingdom goes together with a low immunogenicity, which precludes a strong immune response in heterologous animals. Until now, only two therapeutic monoclonal antibodies that recognize extracellular regions of GPCRs are on the market: mogamulizumab, an anti-neoplasic Ig directed against C-C chemokine receptor type 4 (CCR4) in T cell leukemia (11) and erenumAb, an antagonist of the Calcitonin gene-related peptide type 1 receptor (CALCRL) used to treat migraine (12). The remarkable properties of VHHs now raise exciting prospects for future therapies, because they could stimulate GPCR activity in vivo by binding their orthosteric ligand-binding site, or they could modulate the activity of the endogenous ligand by binding to allosteric sites of the receptor. But this line of research is still in its infancy and so far, only the apelin receptor (APLNR) (13) and the metabotropic glutamate receptor 2 (GRM2) (14) have been targeted with agonistic/positive allosteric modulator VHHs, while others, such as CXCR4 (15), are recognized by antagonists. Interestingly, a VHH directed against the parathyroid hormone receptor (PTHR1) has recently been used as a tether of PTH(1–34), a bioactive N-terminal fragment of PTH, in order to improve its selectivity for the receptor (16).
In contrast, the use of intra-VHHs has been essentially limited to academic research. Remarkably, they have been instrumental in overcoming the major challenges associated to the conformational complexity of GPCR in their ligand-activated states. These receptors basally exist in transient conformational states due to rearrangements at the atomic level and at larger receptor portions (17). The corollary of GPCR intrinsic instability is that obtaining the necessary periodic organization required for resolving the 3D structure of ligand-bound GPCRs at high resolution has been a major stumbling block in the GPCR research field.
These transient conformational states are stabilized by ligand binding and by the subsequent coupling of their intracellular transducing partners, namely G proteins and β-arrestins (Figure 1). Hence, there is an allosteric communication whereby ligand binding induces conformational changes at the ligand-binding pocket that propagate to the inner side of the receptor. Allosteric communication goes also the other way round, since, reciprocally, transducer binding enhances the affinity of the receptor for the ligand (18, 19) (Figure 2). A decisive breakthrough has been achieved on the G protein and ligand binding pocket allosteric dialog (20–22), with the use of Nb80. Indeed, binding of this antibody to the β2-adrenergic receptor (ADRB2) induces a closed conformation of the orthosteric site that traps the agonist while decreasing its dissociation rate (20, 22). Reciprocal, allosteric coupling of the two regions is essential for full receptor activation. It explains why the crystal structure of ADRB2 bound to a potent agonist unexpectedly matched with the inactive crystal structure of the receptor (23), unless it was stabilized intracellularly (24).
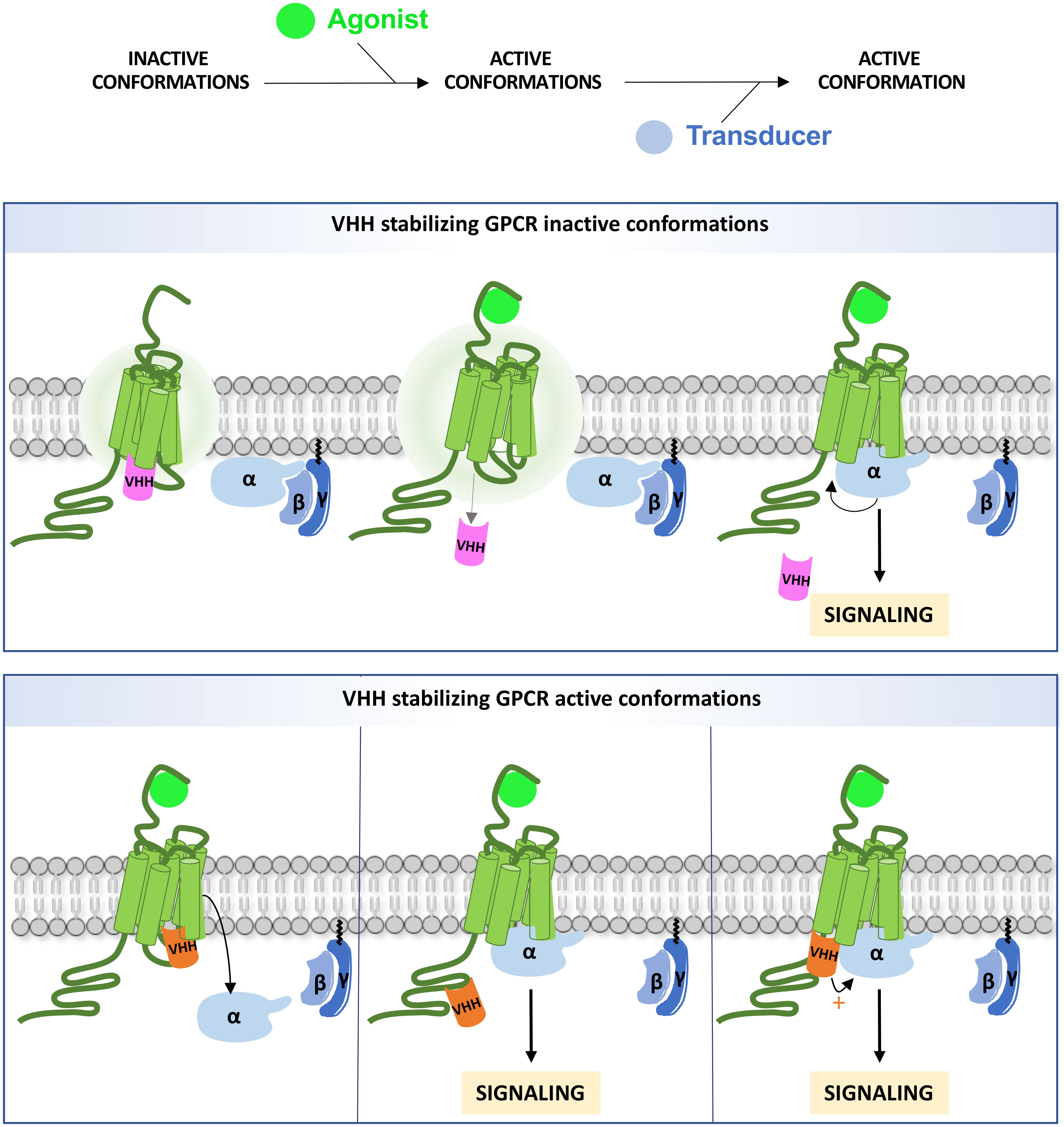
Figure 1 Intra-VHH interference on GPCR signaling. An intra-VHH may stabilize inactive conformations of the GPCR (upper panel) and dissociate upon stimulation. The size of the aura indicates the degree of intrinsic instability. Intra-VHHs may also stabilize active conformations (bottom panel), and compete with G proteins and/or β-arrestins (not shown on the figure) for binding to the receptor, depending on their affinity (left). Alternatively, intra-VHH and transducer binding may be compatible. In that case, GRK phosphorylation sites may be masked (middle) or the VHH may allosterically enhance Gα binding (right).
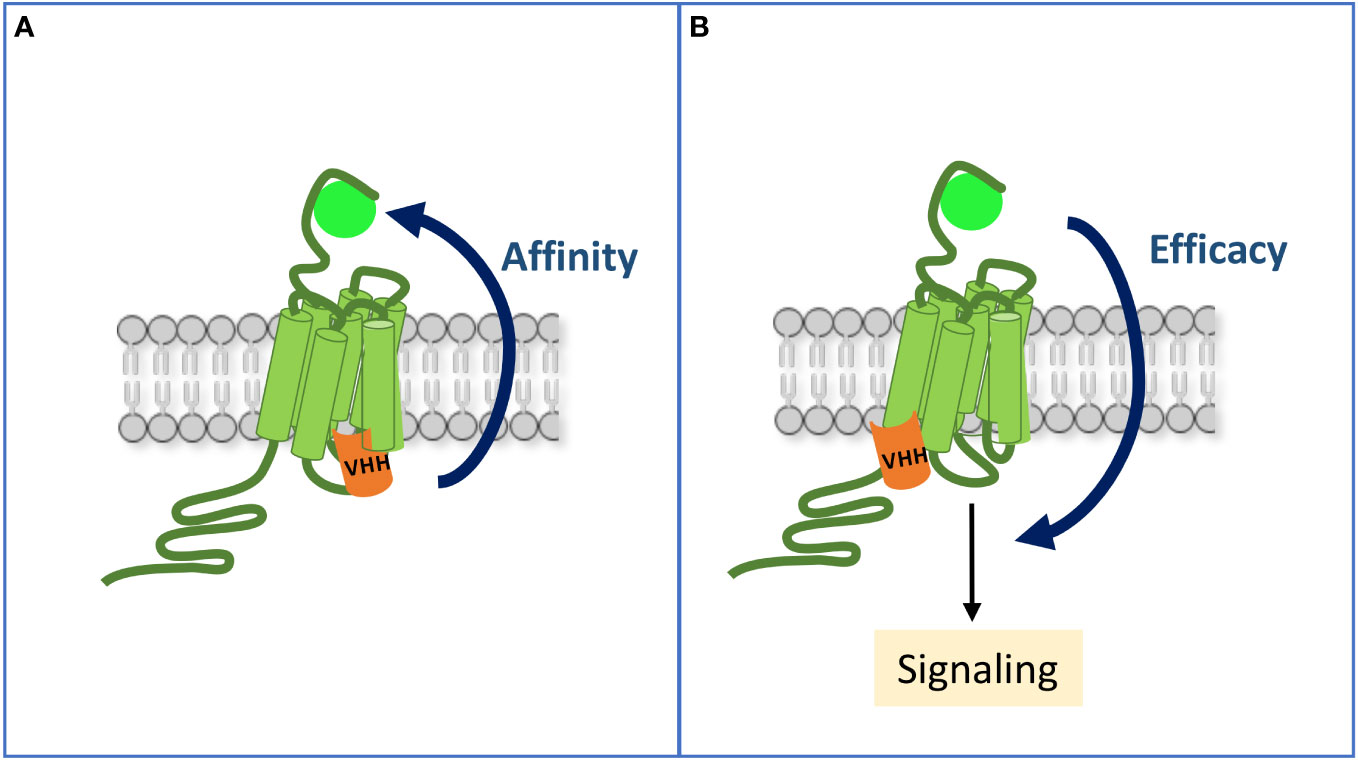
Figure 2 Allosteric communication between the ligand-binding pocket and the intracellular sites of an activated GPCR. (A) By mimicking occupancy of the receptor by a transducer, an intra-VHH increases its affinity for the ligand. (B) A VHH may also allosterically enhance the efficacy of the ligand to stimulate some signaling pathways, hence inducing a biased response when compared to the reference ligand alone.
Structural studies have provided detailed insights into the critical epitopes on GPCRs that could be targeted by intra-VHHs in order to be able to lock the receptors in the active, intermediate, or inactive conformations. For class A GPCRs, displacement of the transmembrane domains (TM), in particular TM6, results into a range of ‘open-active’ or ‘close-inactive’ states on the intracellular side of the GPCR, which in turn determines the degree of activity of the receptor, via G protein or β-arrestin coupling (25, 26). For instance, the structure of ADRB2 stabilized in the active conformation by Nb80 showed that this intra-VHH possesses G protein mimetic properties (24). Upon agonist binding, Nb80 was capable of maintaining the receptor-activated form by having its CDR3 inserted in the hydrophobic allosteric cytoplasmic pocket, formed by TM segments 3, 5, 6 and 7, otherwise occupied by the C-terminal α-helix of Gαs (27). Leading to major outward displacement of the TM6, Nb80 disrupts the critical ‘ionic lock’ interaction between R1313.50 of TM3 and E2686.30 of TM6, which would otherwise maintain the receptor in the inactive state (24). Similar G protein mimetic intra-VHHs targeting corresponding regions of muscarinic acetylcholine receptor M1 (CHRM1) (28) and mu-type opioid receptor (OPRM1) (29) have also been structurally described. Partial agonism was demonstrated with intra-VHH Nb71 bound to ADRB2, of which the TM6 had lesser outward displacement (30). Alternatively, acting as a negative allosteric modulator (NAM), intra-VHH Nb60 also had its CDR3 inserted in the allosteric cytoplasmic pocket of ADRB2 (31). However, the inactive state of the receptor was maintained since residues of the CDR3 of the intra-VHH bridged ‘the ionic lock’.
Since 2011, more than a hundred structures of active GPCR conformationally stabilized with intra-VHHs directly or via stabilization of G proteins, have been solved. By stabilizing discrete receptor conformational states, in the close future, these single domain antibodies are expected to provide significant insights onto the dynamics of the signaling network and integrated cell response induced by G protein and β-arrestin binding to a ligand- activated GPCR. However, very few VHHs directed against the intracellular parts of GPCRs have been extensively studied functionally. Here, we highlight the outcomes of intra-VHH binding to GPCR intra-cellular regions, or to their transducers, on the receptor signaling properties and trafficking. We will also discuss the methodological pitfalls that preclude extensive studies on GPCR modulation by intra-VHHs, and conclude with the therapeutical avenues that they open in drug discovery.
Upon agonist binding, GPCRs undergo an ensemble of conformational state transitions enabling the recruitment of G proteins and β-arrestins, that propagate the membrane signal to a complex intracellular signaling network. G protein-coupled receptor kinases (GRKs) also are direct binding partners that desensitize the receptor by phosphorylation and promote β-arrestin-dependent G protein uncoupling and internalization. In addition, GPCRs make direct interactions with less conventional partners, such as HOMER protein homologs (32), PDZ domain-containing protein GIPC (33), Na(+)/H(+) exchange regulatory cofactor NHERF1 (34), vesicle-fusing protein NSF (35) or Multiple PDZ domain protein (MPDZ) (36), that also deserve interest since they generally intervene in receptor targeting to specific subcellular compartments and cell signaling (37). Like G proteins, GRKs and β-arrestins, their interaction with GPCRs could also be modulated by intra-VHHs.
Since anti-GPCR intra-VHHs stabilize selective conformations of the receptor, they have been named “confobodies”, a registered trademark of Confo Therapeutics (38). They can recognize intracellular regions of the receptor devoted to signal transduction (Figure 1). Hence, their expected modulatory effect is a competition with direct transducers such as G proteins and β-arrestins by steric hindrance. Alternatively, they may also mask regulatory GRK phosphorylation sites on the receptor. In addition, some intra-VHHs may bind to an active conformation of the receptor without competing with the transducers, by positive allostery. So far, no intra-VHH enhancing the affinity of a transducer for a GPCR has been identified, nor, to our knowledge, intra-VHH forcing a new receptor conformation by induced fit.
The Nb80 intra-VHH and Gαs binding regions on ADRB2 overlap, which explains that the conformation of ADRB2 stabilized by Nb80 or by the Gαβγ trimer are structurally very close (24, 27). As such, this intra-VHH displaces Gαs and stabilizes active conformations of ADRB2 (27). Eighteen other anti- ADRB2 intra-VHHs have been selected from llama immunization with ADRB2 embedded in lipids and covalently bound to the BI-167107 high affinity agonist. These VHHs have been functionally characterized and most of them decreased or inhibited isoproterenol-induced cAMP production and β-arrestin recruitment (39). However, their inhibitory effect likely results from different causes, since 12 of them exhibited preference for agonist-bound ADRB2, which suggests a probable steric hindrance of transducer binding, while the 6 remaining ones, including Nb60, selectively bound to inactive conformations immobilized in the presence of antagonists that do not support transducer recruitment (39). The Nb71 intra-VHH preferentially binds to Isoproterenol-activated receptor, and decreased cAMP production. However, it impaired phosphorylation of ADRB2 by GRKs, hence precluding β-arrestin recruitment and proper receptor intracellular trafficking, which suggests that Nb71 recognizes the carboxy-terminal region of ADRB2 and not the Gs binding region as Nb80 for example, although both intra-VHHs functionally inhibit cAMP production. Four additional anti- ADRB2 intra-VHHs with even better affinity than Nb80 have been isolated from a yeast synthetic library by combining magnetic-bead and fluorescence-activated cell sorting (FACS) enrichments, and they all inhibited the activity of a cAMP-responsive reporter gene, in response to adrenaline, up to 45% of Emax (see Nb.c200 in Figure 3) (40).
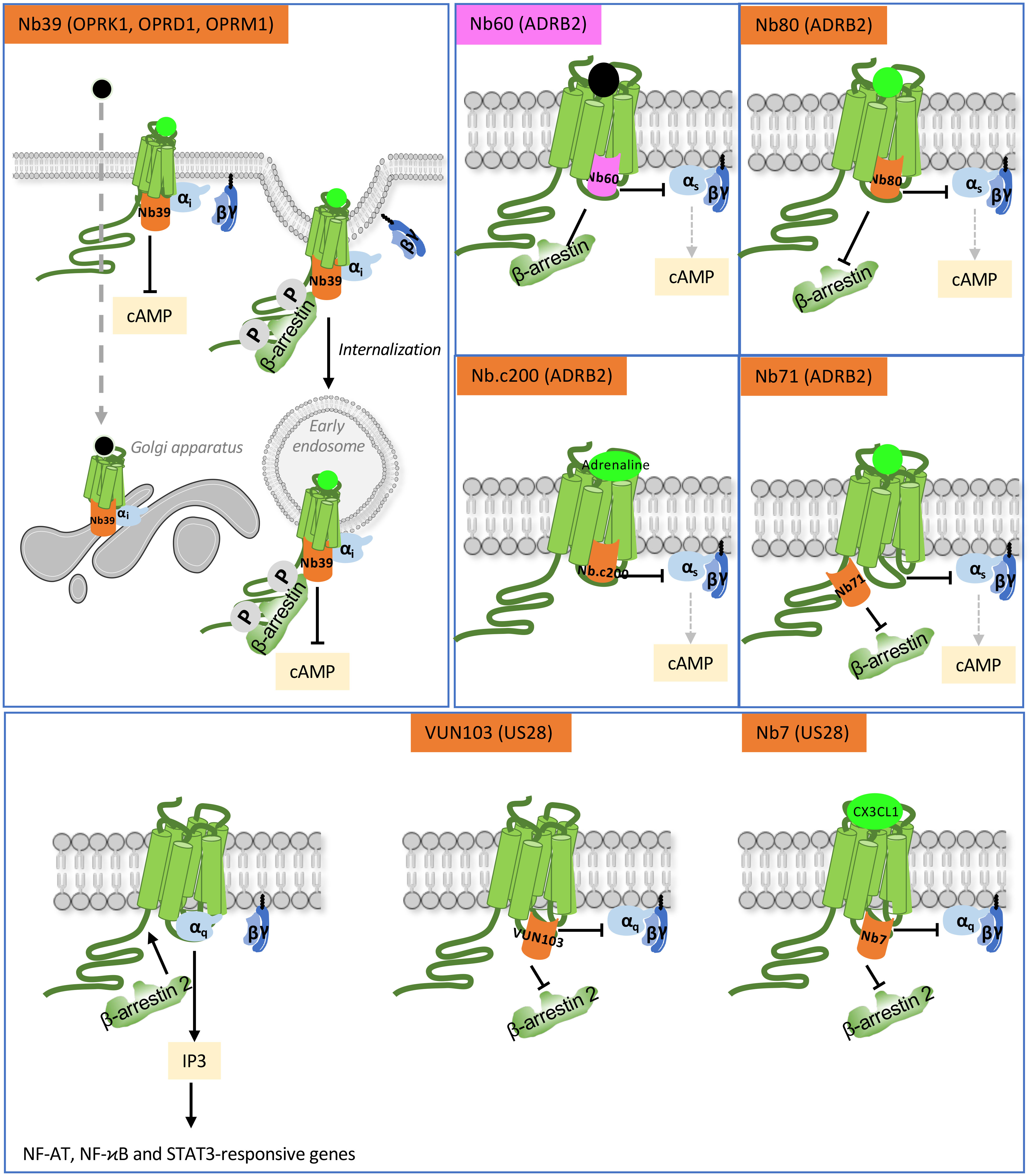
Figure 3 Functional action of intra-VHHs against OPRK1, OPRD1 and OPRK1 opioid receptors, ADRB2 and US28. Nb39 reveals the location of opioid receptors when stimulated by a peptide ligand (bright green) or a cell-permeant non-peptide ligand (black dot). Nb60 locks ADRB2 in an inactive conformation that precludes interaction with transducers. Nb80 stabilizes conformationally active ADRB2. Nb.c200 stabilizes ADRB2 even when stimulated by its natural, low-affinity agonist, adrenaline. Nb71 stabilizes active conformations of ADRB2 and prevents β-arrestin recruitment by limiting access to GRKs. US28 is a constitutively active GPCR. The VUN103 and Nb7 intra-VHH discriminate respectively between constitutively active and agonist-stimulated conformations of the receptor, because Nb7 has no affinity for the apo-conformation. Competition between VUN103 and Nb7 and the transducers hampers US28 signaling and expression of its target genes. Agonists are in bright green, antagonist are in light grey.
Biological characterization of intra-VHH is easier for GPCR with diversified ligands and well-described pharmacology. By using agonists and antagonists of a given receptor, it is possible to conclude whether a VHH preferentially stabilizes inactive forms of the receptor or if it binds constitutively the active receptor and is displaced by further recruitment of G proteins, or whether it only binds activated conformations and then prevents G protein binding, provided that it has a sufficient affinity for the receptor, as seems to be the case of Nb80 (estimated Kd ≃140 nM) (41). Several authors have developed a bioluminescence resonance energy transfer (BRET) assay to monitor the interaction of the intra-VHH and a GPCR (42–44). This is the case of the interaction of the kappa-type opioid receptor (OPRK1) with the Nb6 and Nb39 intra-VHHs (Figure 3) (42, 45). The profile of dose-response curves obtained in the presence of the salvinorin A (SalA) agonist indicates that Nb6 binds preferentially to inactive OPRK1, whereas Nb39 binds to the receptor only when fully active, and, as expected, enhances SalA binding affinity (42). Accordingly, increasing concentrations of the JDTIc antagonist prevent Nb39 binding to OPRK1 but has no effect on Nb6 binding that recognizes inactive conformations. Interestingly, domain swapping experiments grafting the OPRK1 intracellular loop (ICL3) 3 to seven heterologous receptors physiologically coupled to Gs, Gi or Gq, confer not only Gq recruitment ability but also antagonistic Nb6 binding (45), suggesting that this system can be used to monitor the activation state of Gq-coupled GPCRs. Thus, the Nb6 intra-VHH could be used similarly as the mini-Gq biosensor, and may possibly help de-orphanising orphan Gq-coupled GPCRs (45).
The human cytomegalovirus US28 receptor is an interesting example of a receptor exhibiting high constitutive activity. The latter is due to structural instability of the inactive state, and is only modestly enhanced by ligand binding (46). This observation suggests that constitutively active US28 and ligand-bound receptor have distinct conformations. In agreement, the conformation-specific VUN103 intra-VHH competes with Gq and β-arrestin 2 that constitutively interact with US28, and severely impairs inositol phosphate accumulation, nuclear factor of activated T cells (NFAT), signal transducer and activator of transcription 3 (STAT3) and NF-ϰB activation. In contrast, Nb7 does not affect constitutively active US28, but rather stabilizes US28 bound to its fractalkine CX3CL1 ligand, and inhibits the cell signaling it mediates (44) (Figure 3). Although both Nb7 and VUN103 bind ICL2 and ICL3 (46), they seem to recognize distinct active conformations of US28. Importantly, by interfering with US28 constitutive activity, VUN103 partially inhibited the growth and US28-dependent signaling of glioblastoma spheroids as well as glioblastoma cells.
Most G protein mimics described above have affinity for an agonist-bound GPCR, but none of them is able to allosterically enhance receptor-Gα interaction. By using viral evolution of genetically actuating sequences (VEGAS), intra-VHHs against D(2) dopamine receptor (DRD2), the pH-sensing GPR68 and the 5-hydroxytryptamine receptor 2A (HTR2A) receptor have been obtained (43). One of these, VGS-Nb2, was qualified as positive allosteric modulator (PAM). It directly associates to HTR2A and stimulates a serum-responsive element (SRE) responsive reporter gene in the absence of ligand and regardless of Gαq expression, hence inducing an active conformation of the receptor. This response was still enhanced after stimulation by 5HT via Gq, coupling, but not β-arrestin 2, whose recruitment to the receptor was decreased. Hence, intra-VHHs isolated by VEGAS are able of inducing receptor activity in the absence of ligand.
GPCR intrinsic instability is the major driver of biased signaling. The latter is determined by the selection of discrete conformational states, stabilized by a given ligand, that will ultimately lead to the recruitment of different transducers (e.g. G proteins and β-arrestins) with various efficacy or kinetics, according to the ligand (47–50). The high conformational dynamics and heterogeneity of GRKs and β-arrestins and their multiple modes of interactions with GPCR contribute to this diversity (51). The ability of a ligand to selectively stimulate a signaling pathway over the other, when compared to a reference ligand, has been mainly approached as an indirect measure of G protein vs β-arrestin-dependent downstream signaling, and estimated by calculation with the operational model and variants thereof (52–54). But it is not directly informative of receptor conformation. By providing important advance onto active receptor trafficking, intra-VHH conformational biosensors provide insights directly at the level of the receptor on the mechanisms underlying signaling bias.
GPCR signaling and trafficking are closely connected, since it has been unambiguously demonstrated that GPCR signaling does not exclusively take place at the plasma membrane, but also occurs in several intracellular compartments, ultimately leading to distinct biological outcomes (55–59). This conceptual advance has been enabled by repurposing intra-VHHs sensitive to GPCR conformational dynamics initially developed for structural biology. These antibodies can be converted to genetically-encoded conformational biosensors by fusion to a fluorescent protein derived from green-fluorescent protein (GFP). With these tools, it becomes possible to detect where activated receptors are located inside the cell. The first anti-GPCR intra-VHH biosensor was derived from Nb80, that contributed to the resolution of activated ADRB2 3D structure (24). By tracking isoproterenol-activated ADRB2 inside the cell, this biosensor has uncovered that receptor stimulation leads to cAMP production, not only from the plasma membrane but also from endosomes, once the receptor is internalized (60). This finding was confirmed with Nb37-GFP, that recognizes the alpha-5 helical domain of Gαs in the guanine nucleotide-free form that is an activation intermediate and is important for GPCR binding (61). At intra-cellular concentrations that do not inhibit Gαs activation, the Nb37-GFP biosensor is resident in the cytosol, and is recruited to the cell membrane upon ADRB2 activation. Even more importantly, this biosensor detects ligand-activated ADRB2 wherever it is located in the cell, hence providing precious informations on the sites of persistent ADRB2 cAMP signaling, such as the endosomes (Figure 4) (60). Similar observations were extended to the gastric inhibitory polypeptide receptor (GIPR), that also induces cAMP production once in the early endosomes (62). Single-molecule imaging with the Nb37-GFP biosensor has further uncovered that the alpha2A adrenergic receptor (ADRA2A) coupling to G protein is trapped to defined hot spots of interactions inside the plasma membrane, where signaling mainly occurs. These nanodomains are partially constituted by actin fibers, microtubules and clathrin-coated pits (63). Nanoclustering depends on the receptor conformational state, as demonstrated by single-molecule tracking of active and inactive conformers of ADRB2 bound to GFP-tagged Nb80, Nb37 or Nb60 biosensors in PC12 cells (64).
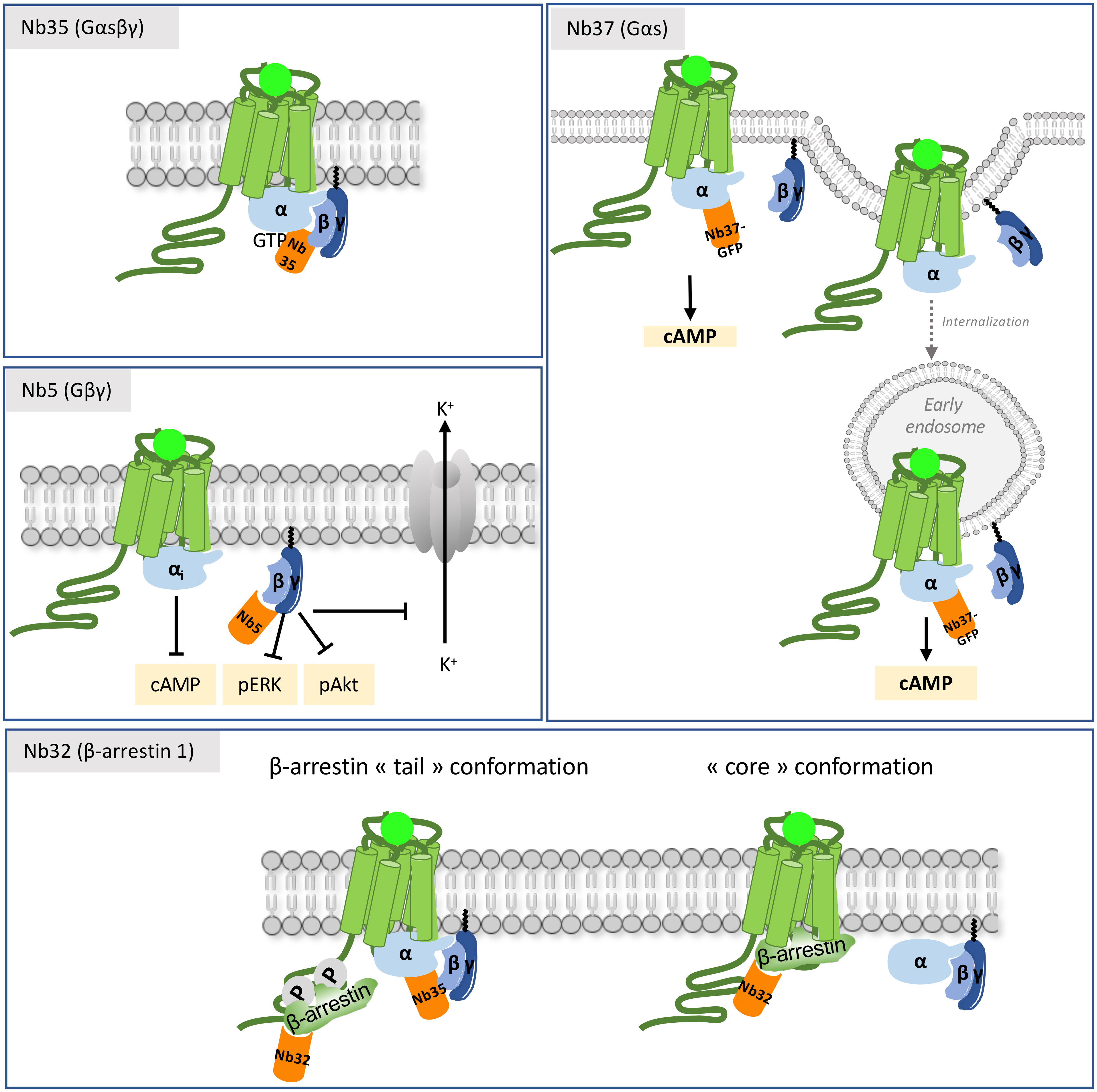
Figure 4 Mechanisms of action of intra-VHHs against GPCR transducers. Nb37 recognizes the nucleotide-free form of active Gαs, and has been used as a conformational biosensor to track receptor activation and signaling throughout the cell compartments. Nb35 stabilizes the interface between Gα and Gβγ, and prevents GTP release. Nb5 binds to Gβγ. It has a pronounced effect on Gβγ signaling (adenylate cyclase, pERK and pAkt) and on the K+ current of the GIRK channel in the striatum. Nb32 is an anti-β-arrestin 1 VHH. Nb35 and Nb32 have been used to reconstruct the structural arrangement of the megacomplex composed of an agonist-stimulated ADRB2-V2R chimera simultaneously bound to Gα and to β-arrestin 1.
The NbSmo8 biosensor was the first intra-VHH biosensor directed against a class F GPCR. Similarly to anti-class A biosensors, it has been shown to bind the Smoothened homolog (SMO) only when activated by agonist binding (65).
Diverse ligands stabilizing discrete active states of the receptor induce its interaction with the conformational intra-VHH biosensors in a concentration-dependent manner, without the signal amplification that happens when measuring downstream signaling. Therefore, these intra-VHH biosensors may potentially provide a more reliable access to efficacy and potency of each ligand-receptor pair. Biosensors to monitor OPRM1, OPRD1, OPRK1 opioid receptors activity have been developed, either to monitor receptor activation state by different ligands, by quantifying biosensor interaction (42, 66), or to track activated receptor inside the cell (45, 66, 67). One of the latter ones has been developed to discriminate the side-effects of non-peptide clinically-relevant drugs versus the beneficial therapeutical effects of natural peptide ligands of OPRM1 (67, 68). This biosensor, derived from Nb33, is almost similar to Nb39, the intra-VHH that binds selectively to activated OPRM1 and OPRK1 described above (29, 42, 69). By total internal reflection fluorescence (TIRF) time-lapse experiments in neurons, the Nb33 biosensor reveals that met-enkephalin- or β-endorphin-activated OPRM1 or OPRD1 signals transiently from the plasma membrane, and then from receptor internalized in the endosomes. In contrast, non-peptide opioid drugs, such as morphine or etorphine, induce an additional, very early stimulation wave in the Golgi, that is independent of receptor trafficking (Figure 3). Similar results have been gathered when probing OPRK1 with the Nb39 conformational biosensor in cells stimulated with the SalA permeant drug or with Dynorphin, the OPRK1 natural ligand (45). These observations, emphasizing the notion of spatio-temporal bias, have wide implications for the development of neuro-modulatory drugs that generally penetrate the cell, hence are prone to illegitimate signaling from the Golgi. The latter might be involved in the well-known side-effects of opioids such as toxicity and addiction. With the use of Nb6B9, a high-affinity derivative of Nb80, agonist-activated D1A dopamine receptor (D1DR) or β1-adrenergic receptor (ADRB1) has also been observed at the Golgi apparatus in addition to the plasma membrane (70).
Intra-VHHs directed against the C-C chemokine receptor type 7 (CCR7) have been recently derived by random mutagenesis of CDR1 and CDR3 of Nb80 (71). Interaction of Nb1, Nb5 and Nb38 with CCR7 was tracked by bimolecular fluorescence complementation (BiFC) using split-YFP, regardless of the receptor activation state. They co-localized with CCR7 at the plasma membrane in resting conditions, and also in membrane ruffles and endosomes upon CCL19 stimulation, confirming that they do not recognize selective conformations of the receptor. Accordingly, they also modestly interfered with Gi binding to the receptor and Ca2+ response.
Ligand binding induces conformational modifications of GPCRs that lead to the recruitment of intracellular transducers, among which G proteins and β-arrestins have been the most extensively studied. Intra-VHHs stabilizing G proteins or β-arrestins have served as tools to address the following questions, that are theoretically applicable to every GPCRs: i/the cryogenic electron microscopy (cryo-EM) structure of activated GPCR complexes with G proteins or β-arrestins, ii/the sub-cellular localization of signaling receptor, iii/the modulation of transducer activity and signaling outcomes, iv/understanding the relationships between β-arrestin conformational transitions and its activity, v/modulating the interactions of β-arrestins with their binding partners.
The structure of Gs-bound ADRB2 complexes has been resolved with the Nb35 as a structural chaperone (27). Because it recognizes the interface between Gαs Ras domain and Gβγ and prevents GTP dissociation when the trimer is bound to agonist-occupied receptor, Nb35 has helped solving the high resolution 3D structures of other active, Gs-coupled GPCRs, such as the thyrotropin receptor (TSHR) (72, 73), the lutropin-choriogonadotropic hormone receptor (LHCGR) (74) or the adenosine receptor A2a (ADORA2A) (75) among others, in active, transducer-bound conformation. In contrast to the paradigm establishing that β-arrestins binding precludes Gα binding on the receptor in order to desensitize it, it has been demonstrated by single-particle electron microscopy that Gαs and β-arrestin 1 can bind simultaneously the GPCRs that strongly interact with β-arrestins, such as the vasopressin V2 receptor (AVPR2) (76). In agreement, Nb35 has been used to stabilize the Gαβγ trimer within an activated ADRB2/AVPR2 chimeric receptor in complexation with both transducers (77) (Figure 4). The AVPR2 moiety represents the carboxyterminal region of the AVPR2 receptor that confers long-lasting interaction with β-arrestins, in contrast to ADRB2 that only transiently interacts with them (78). This study has profound biological impact because it revisits the role of β-arrestins as desensitizing agents presumably precluding Gs access to the receptor, by showing that β-arrestin and Gs binding is in fact not mutually exclusive. Gs occupies the core domain of the receptor, while β-arrestin 2 remains bound to the phosphorylated carboxy-terminal region of the receptor, in a so-called “tail” conformation. β-arrestins can also adopt a “core” conformation whereby they engage three structural elements, including the finger loop, into the core (transmembrane helices and intracellular loops) of the receptor for desensitization, their classically assigned role (77). Hence, by demonstrating that a GPCR coupled to β-arrestins can continue to signal through Gs, even if internalized, this “megaplex” hypothesis has provided a mechanistic clue on endosomal signaling, as presented above.
Because of its negative regulatory action on Gα through re-association, the Gβγ complex has raised interest as intra-VHH target. Therefore Nb5, that recognizes Gβ subtypes 1-4, has been developed (79). By suppressing Gβγ signaling, Nb5 inhibits Gβγ-regulated G protein-gated inward rectifier potassium (GIRK) channels signaling in medium spiny neurons of the striatum excited with DRD2- or CHRM4-mediated inhibitory post-synaptic current (IPSC). In response to apelin, Nb5 indirectly limits the inhibition of the Gi-coupled apelin receptor on forskolin-stimulated cAMP, by suppressing the inhibitory action of Gβγ on adenylate cyclase. Nb5 also inhibits downstream kinases such as Akt and Erk. But despite its inhibitory action on Gβγ functioning, this intra-VHH has no effect on GTP-bound Gα (Figure 4) (79).
Intracellular antibodies against β-arrestins exhibiting interesting biological properties have also been characterized, although the first ones were not VHHs but scFv or Fabs. For example, scFv5 disrupted the interaction of β-arrestins with clathrin and inhibited the endocytosis of 8 different GPCRs, suggesting a generic value of this scFv as a tool to assess GPCR internalization (80). In addition, a Fab, Fab30, that recognized an active “tail” conformation of β-arrestin 1 when bound to a phosphorylated carboxyterminal region of V2R (81), was isolated. From Fab30, an intra-cellular scFv, named Ib30 by the authors, has been derived as a biosensor to monitor β-arrestin 1 recruitment to chimeric as well as GPCRs with a native carboxyterminal region, and then to track β-arrestin trafficking inside the cell (82). Importantly, Ib30 confers a gain-of-function in β-arrestin 1 conformation, since it restores its translocation to endosomes and its ability to stimulate ERK MAP kinases, otherwise impaired upon agonist-stimulation of the AVPR2 T360A phosphorylation mutant (83). Structural analysis and molecular dynamics indicate that only Lys11 of β-arrestin 1 phosphate sensor remains engaged by ionic interactions with the phosphorylated receptor and not Lys 294 in the polar core, nor Arg25, that are also involved in fully activated conformation of β-arrestin 1. This analysis explains that, when bound to the AVPR2 T360A mutant, β-arrestin 1 adopts a partially active conformation that can be allosterically modified to accommodate β2-adaptin interaction and subsequent internalization. The use of Ib30 as a biosensor to monitor β-arrestin 1 conformational changes has been extended to other GPCRs (82, 84, 85).
Finally, bona fide camelid anti-β-arrestin 1 intra-VHHs came out, and one of them, Nb32, has been very useful to investigate the functional outcomes of β-arrestin 1 binding to the GPCR core and/or to the tail (Figure 4) (86). This antibody binds β-arrestin 1 only when associated to a receptor. When comparing a β-arrestin 1 deleted of its finger loop, hence devoid of its ability to bind the core domain of the receptor, and the Nb32-stabilized β-arrestin 1, it appeared that β-arrestin 1 binding to the tail predominantly drives the receptor to internalization and promotes signaling, but is not able to promote G protein desensitization, as expected from the megaplex model (76, 77).
So far, the anti-β-arrestin antibody fragments that have been described allosterically modulate β-arrestin/GPCR interaction by stabilizing selective receptor conformations. In the future, it will be of great interest to stabilize selective, fully or partially engaged conformations of β-arrestins instead. In this way, it should be possible to control independently receptor-mediated signaling and trafficking. Beyond β-arrestin/GPCR interaction, only scFv5 has been shown to impair the interaction of β-arrestin 2 with one of its non-receptor interaction partner (80). This finding opens the promising possibility of interfering with β-arrestin signaling partners. This is of paramount interest because β-arrestins undergo interactions with hundreds of signaling proteins, including at least 17% of direct interactions, leading to diversified, yet essential biological responses (87–89). Hence, intra-VHHs directed against precise interfaces of β-arrestins might help to decipher the specificity of their signaling outputs.
Methodological hurdles that linger the development of VHHs against GPCR, such as the selection of the antigen (90), the advantages of synthetic libraries versus libraries from immunized animal (40, 91, 92), the comparison of display methods (5, 38, 40) have been previously reported. Here, we focus on the specificities that make anti-GPCR intra-VHH expression inside the cell and characterization particularly challenging (Figure 5).

Figure 5 Examples of methodological challenges associated to functional studies on intra-VHHs that modulate GPCR signaling, and the existing solutions.
One is that the anti-GPCR intra-VHHs isolated so far are generally conformational antibodies that recognize non-linear epitopes within the inner core domain of the receptor, as shown for Nb80. However, the intracellular loops, especially ICL3, and the carboxyterminal region of GPCR are intrinsically disordered, which may be a disadvantage to select specific, high-affinity VHHs.
The key parameter to be monitored when isolating an antibody against a selected target is direct antibody/antigen binding. But in vitro binding by enzyme-linked immunosorbent assay (ELISA), surface plasmon resonance (SPR) or interferometry can be a challenging task when the target is a GPCR that cannot be easily produced and purified in native, active conformation. Assaying intra-VHH-GPCR binding in cell can be an alternative by using BRET (42–44) or the developing Nanobit complementation technology (83, 93). Cytometry is also an efficient means (39), but low level of endogenous or exogenously expressed recombinant receptor and limited intracellular affinity of the VHH may lead to signal below the detection threshold. In addition, in vitro affinity may not be linearly correlated to efficacy in the reducing milieu of the cytoplasm, as shown for the anti-ADRB2 Nb.c203, which exhibits moderate affinity maturation in yeast when compared to the other intra-VHHs selected, but was the most efficient to decrease the Emax of adrenaline to produce cAMP. Similarly, the folding of a VHH produced in bacteria and expressed from inside the cell may be different, which adds another level of variability when assessing affinity. These observations underscore the necessity to test several intra-VHHs to obtain a maximum effect on receptor signaling.
Since the VHH is generally expressed inside the cell from a transfected plasmid, it can be difficult to accurately control its quantity from the beginning of the experiment. If it recognizes inactive conformations of the receptor, it would be bound to the GPCR prior to agonist stimulation. Therefore, if its affinity is high enough, it should prevent transducer binding and lead to functional inhibition of downstream signaling. But if it is not, then it should be displaced by the ligand-activated transducers, without any detectable functional outcome, which may lead to bewildering conclusions. One alternative is to express the VHH intracellularly from an inducible promoter, as previously done with anti-US28 VUN13 intra-VHH (44), which also minimizes off-target reactions. Quite interestingly, destabilizing mutations of an intra-VHH that are silent only in the presence of the specific antigen have been reported (94). These mutations constitutively drive the intra-VHH to the proteasome-degrading pathway, unless the antibody is bound to its cognate antigen. Hence, an intra-VHH may be permanently degraded until its cognate GPCR target adopts the suitable active conformation upon ligand binding. This work is of broad potential applications because the destabilizing mutations are present in the structurally conserved framework regions of the antibody. In addition, several optically-controlled intra-VHHs have been engineered [reviewed in (95)]. Additional endeavors have to be pursued, based on the introduction of the antibody with a cell-penetrating peptide (96), or the transfection of in vitro-transcribed RNA, to detect the intra-VHH as soon as 3 hours after introduction (97).
The fact that it is very difficult to date to control precisely the expression level of an intra-VHH severely compromises reproducibility between independent experiments, especially because the stoichiometric ratio between the receptor and the VHH, by altering ligand binding affinity, indirectly alters the efficacy to stimulate a given functional readout.
In several instance, the question remains as whether conformational intra-VHHs that have been reported are really specific of one receptor, or if they recognize active/inactive conformations that are common to several receptors of the same family (ex, the opioid receptors, adrenergic receptors, etc) or even receptors from distinct classes (ex, class A GPCR vs class F). As indicated above, Nb6B9 recognizes D1DR in addition to ADRB1 and ADRB2 (70). For class A GPCR, cross-reactivity could reflect the relative conformational conservation of their intracellular half, that accommodates to a limited number of G proteins and β-arrestins (98, 99).
The explosion of structures of GPCRs in active conformations since 2011 has paved the way for the rational, structure-based design of new drugs of potential therapeutic interest.
In drug discovery, one moonshot is to enhance binding affinity of biased agonists of therapeutical interest because, by stimulating various signaling pathways with distinct efficacy when compared to the natural agonist, they are expected to lead to less side-effects (Figure 6). If a GPCR is stabilized in a defined active conformation with an intra-VHH, then the affinity of not only its natural ligand will increase but also any other biased ligand with interesting signaling properties. This is the purpose of reverse pharmacology, where a receptor is locked in an active conformation with an intra-VHH, to screen for (allosteric/biased) ligands endowed with new pharmacological properties when compared to the reference agonist. A proof-of-concept of reverse pharmacology has been successfully applied to ADRB2 fused to the transducer-mimicking Nb80 and to the OPRM1-Nb33 fusion, in a fragment-based screening of 1000 small molecules (38, 100, 101). This comparative fragment screen has led to the pharmacological classification of multiple novel ligands as agonists, antagonists, inverse agonists, etc, on the basis of their efficacy to stimulate readouts of interest (100). Fragment-derived compounds binding the orthosteric site of ADRB2-Nb80 fusion with affinity in the nM range were selected, and they discriminate accurately the active and basal states of the receptor. Hence, this strategy raises hopes to the selection and characterization of ligands selective of a given receptor conformation of interest, in a cell-free system.
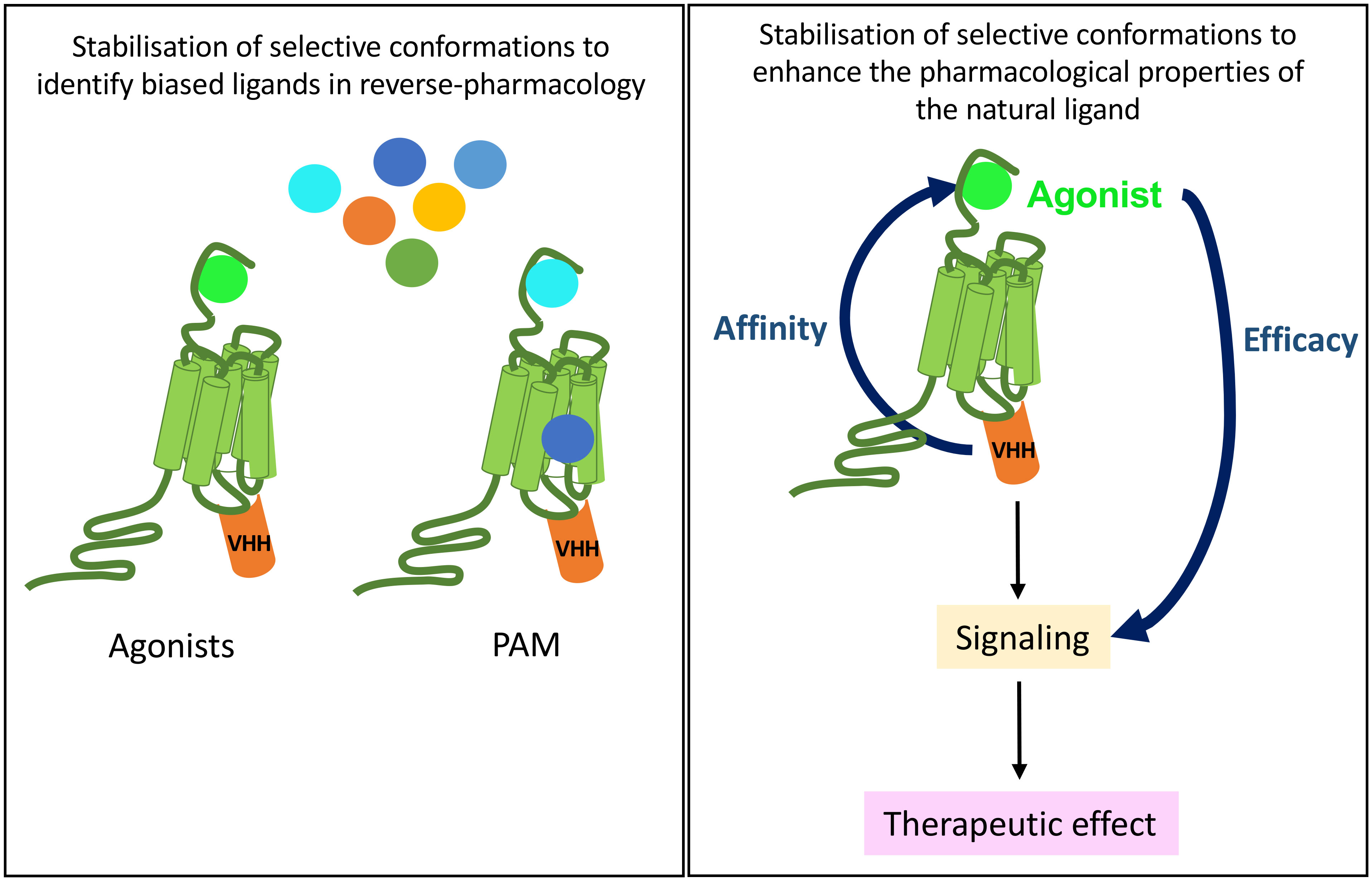
Figure 6 Potential therapeutical applications of anti-GPCR intra-VHHs. A GPCR immobilized by an intra-VHH in an active conformation of biological interest may serve as a target for drug discovery of new biased ligands, be they allosteric or orthosteric (left). In vivo, provided that intra-VHH may be vectorized into the appropriate target cell, they could allosterically modulate the pharmacological properties of the endogenous ligand, which, for example, would respect the endocrine physiological rhythms and control.
In the ADRB2-Nb80 complex, a β hairpin conformation of the VHH CDR3 engages interactions with the receptor overlapping the Gαs binding site (24). This prompted research groups to launch rational design programs using CDR3 β hairpin peptidomimetics to stabilize active conformations of ADRB2 for ligand screening (102). Two of these peptidomimetics inhibited cAMP production, as expected from competition with endogenous Gαs. Nevertheless, the effect was modest when compared to Nb80 inhibitory effect. These attempts still need more efforts for success, probably because peptidomimetics for ADRB2 fail to fully stabilize active conformations of the receptor (103, 104).
Importantly, intra-VHHs have the unique potential to isolate GPCR agonists, not only as small chemical compounds, but also in the VHH format, recognizing the extracellular regions of a receptor. As indicated above, VHHs with agonistic have only been described for APLNR and GRM2. Hence, intra-VHHs may increase the discovery of new agonistic VHHs to GPCR of clinical relevance.
Another application of intra-VHHs in drug discovery for GPCR can be foreseen through the example of atypical chemokine receptor 3 (ACKR3), that is naturally biased towards GRK and β-arrestin signaling, because of the absence of a kink of TM4 that leads to a clash of ICL2 and Gα (105, 106). Hence, it is conceivable that an intra-VHH stabilizing this compact conformation of the Gα-binding cleft would increase the probability to isolate new biased ligands of this receptor.
The exquisite properties of anti-GPCR VHHs, such as specificity, affinity, short serum half-life, open exciting opportunities for future therapeutics. Obviously, these properties are shared with intra-VHHs. However, in the latter case, even more critically than for in vitro applications, a major issue to be overcome is their functional delivery into target cells in vivo, although some progress are being made, as reviewed in (107). This is why, to date, no example of intracellularly delivered VHH has been reported.
Intra-VHHs have widely proven their utility as chaperones for structural studies of GPCR by X-ray crystallography or cryo-EM. In contrast, the characterization of anti-GPCR intra-VHHs with functional properties inside the cell is still poorly developed. So far, the functional impact of only very few intra-VHHs on GPCR signaling activity has been characterized, when considering that only ADRB2, OPRK1 and US28 and, to a lesser extent, HTR2A, have been targeted to date (Table 1).

Table 1 Summary of intra-VHHs modulating GPCR signaling and/or on trafficking cited in this paper.
One clear bottleneck is the difficulty to properly characterize their pharmacological properties in a complex cell system and to assess specificity despite likely intracellular off-targets. Screening for drugs that bind to conformationally stabilized binding pockets within GPCR might lead to more selective and efficient therapeutics. Intra-VHHs indirectly mimic the effect of biased agonists, from the intracellular side.
In the future, exploration of the functionality of intra-VHHs on a given GPCR will need to be systematically coupled to resolution of its structure. Only in this condition will it be possible to approach the issue of conformation/activity relationship, to correlate a (biased) ligand to a receptor conformation or ensemble of conformations, to a signaling pathway, or even network, and ultimately, integrated biological response. Still, one caveat is that the conformations that are co-crystallized do not necessarily predict accurately the conformations that are preferentially stabilized in a complex cell system.
All authors significantly contributed to conceptualization and writing of the manuscript, and to the design of the figures. All authors contributed to the article and approved the submitted version.
PR is funded by a joint fellowship from Région Centre Val de Loire and INRAE PHASE Department; CG and VJ are funded by the SELMAT grant of Région Centre Val de Loire ARD2020 Biomédicaments Program. CG is co-funded by the INTACT grant from Région Centre Val de Loire; GB and ER are funded by INRAE, FJ-A and PC are funded by the CNRS. This publication was funded with support from the French National Research Agency under the program “Investissements d’avenir” Grant Agreement LabEx MabImprove: ANR-10-LABX-53; “ARD2020 Biomédicaments” and “APR-IR INTACT” grants from Région Centre Val de Loire; Bill & Melinda Gates Foundation.
The authors would like to acknowledge Région Centre Val de Loire and the MAbImprove Labex for their constant support of our research on antibody fragments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be considered as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
GPCR, G protein-coupled receptor; VHH, VH of heavy-chain only antibodies; Nb, nanobody; sdAb single-domain antibody; CDR, complementarity-determining region.
1. Stijlemans B, Conrath K, Cortez-Retamozo V, Van Xong H, Wyns L, Senter P, et al. Efficient targeting of conserved cryptic epitopes of infectious agents by single domain antibodies. J Biol Chem (2004) 279(2):1256−61. doi: 10.1074/jbc.M307341200
2. Akazawa-Ogawa Y, Uegaki K, Hagihara Y. The role of intra-domain disulfide bonds in heat-induced irreversible denaturation of camelid single domain VHH antibodies. J Biochem (2016) 159(1):111−21. doi: 10.1093/jb/mvv082
3. Liu H, Schittny V, Nash MA. Removal of a conserved disulfide bond does not compromise mechanical stability of a VHH antibody complex. Nano Lett (2019) 19(8):5524−9. doi: 10.1021/acs.nanolett.9b02062
4. Cardinale A. Intracellular targeting and functional analysis of single-chain fv fragments in mammalian cells. Methods (2004) 34(2):171−8. doi: 10.1016/j.ymeth.2004.04.006
5. Cheloha RW, Harmand TJ, Wijne C, Schwartz TU, Ploegh HL. Exploring cellular biochemistry with nanobodies. J Biol Chem (2020) 295(45):15307−27. doi: 10.1074/jbc.REV120.012960
6. Keller L, Bery N, Tardy C, Ligat L, Favre G, Rabbitts TH, et al. Selection and characterization of a nanobody biosensor of GTP-bound RHO activities. Antibodies (2019) 8(1):8. doi: 10.3390/antib8010008
7. Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A, et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods (2006) 3(11):887−9. doi: 10.1038/nmeth953
8. Galli V, Sebastian R, Moutel S, Ecard J, Perez F, Roux A. Uncoupling of dynamin polymerization and GTPase activity revealed by the conformation-specific nanobody dynab. eLife (2017) 6:e25197. doi: 10.7554/eLife.25197
9. Fagbadebo FO, Kaiser PD, Zittlau K, Bartlick N, Wagner TR, Froehlich T, et al. A nanobody-based toolset to monitor and modify the mitochondrial GTPase Miro1. Front Mol Biosci (2022) 9:835302. doi: 10.3389/fmolb.2022.835302
10. Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: New agents, targets and indications. Nat Rev Drug Discovery (2017) 16(12):829−42. doi: 10.1038/nrd.2017.178
11. Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, et al. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res (2004) 64(6):2127−33. doi: 10.1158/0008-5472.CAN-03-2068
12. Shi L, Lehto SG, Zhu DXD, Sun H, Zhang J, Smith BP, et al. Pharmacologic characterization of AMG 334, a potent and selective human monoclonal antibody against the calcitonin gene-related peptide receptor. J Pharmacol Exp Ther (2015) 356(1):223−31. doi: 10.1124/jpet.115.227793
13. Ren H, Li J, Zhang N, Hu LA, Ma Y, Tagari P, et al. Function-based high-throughput screening for antibody antagonists and agonists against G protein-coupled receptors. Commun Biol (2020) 3(1):146. doi: 10.1038/s42003-020-0867-7
14. Scholler P, Nevoltris D, de Bundel D, Bossi S, Moreno-Delgado D, Rovira X, et al. Allosteric nanobodies uncover a role of hippocampal mGlu2 receptor homodimers in contextual fear consolidation. Nat Commun (2017) 8(1):1967. doi: 10.1038/s41467-017-01489-1
15. Jähnichen S, Blanchetot C, Maussang D, Gonzalez-Pajuelo M, Chow KY, Bosch L, et al. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc Natl Acad Sci U.S.A. (2010) 107(47):20565−70. doi: 10.1073/pnas.1012865107
16. Cheloha RW, Fischer FA, Woodham AW, Daley E, Suminski N, Gardella TJ, et al. Improved GPCR ligands from nanobody tethering. Nat Commun (2020) 11(1):2087. doi: 10.1038/s41467-020-15884-8
17. Latorraca NR, Venkatakrishnan AJ, Dror RO. GPCR dynamics: Structures in motion. Chem Rev (2017) 117(1):139−55. doi: 10.1021/acs.chemrev.6b00177
18. De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem (1980) 255(15):7108−17.
19. Murphree LJ, Marshall MA, Rieger JM, MacDonald TL, Linden J. Human a 2A adenosine receptors: High-affinity agonist binding to receptor-G protein complexes containing gβ 4. Mol Pharmacol (2002) 61(2):455−62. doi: 10.1124/mol.61.2.455
20. DeVree BT, Mahoney JP, Vélez-Ruiz GA, Rasmussen SGF, Kuszak AJ, Edwald E, et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature (2016) 535(7610):182−6. doi: 10.1038/nature18324
21. Warne T, Edwards PC, Doré AS, Leslie AGW, Tate CG. Molecular basis for high-affinity agonist binding in GPCRs. Science (2019) 364(6442):775−8. doi: 10.1126/science.aau5595
22. Chen Y, Fleetwood O, Pérez-Conesa S, Delemotte L. Allosteric effect of nanobody binding on ligand-specific active states of the β2 adrenergic receptor. J Chem Inf Model (2021) 61(12):6024−37. doi: 10.1021/acs.jcim.1c00826
23. Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature (2011) 469(7329):236−40. doi: 10.1038/nature09665
24. Rasmussen SGF, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature (2011) 469(7329):175−80. doi: 10.1038/nature09648
25. Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, et al. Structural insights into the dynamic process of β 2 -adrenergic receptor signaling. Cell (2015) 161(5):1101−11. doi: 10.1016/j.cell.2015.04.043
26. Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, et al. Single-molecule analysis of ligand efficacy in β2AR–g-protein activation. Nature (2017) 547(7661):68−73. doi: 10.1038/nature22354
27. Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the β2 adrenergic receptor–gs protein complex. Nature (2011) 477(7366):549−55. doi: 10.1038/nature10361
28. Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature (2013) 504(7478):101−6. doi: 10.1038/nature12735
29. Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, et al. Structural insights into µ-opioid receptor activation. Nature (2015) 524(7565):315−21. doi: 10.1038/nature14886
30. Masureel M, Zou Y, Picard LP, van der Westhuizen E, Mahoney JP, Rodrigues JPGLM, et al. Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist. Nat Chem Biol (2018) 14(11):1059−66. doi: 10.1038/s41589-018-0145-x
31. Staus DP, Strachan RT, Manglik A, Pani B, Kahsai AW, Kim TH, et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature (2016) 535(7612):448−52. doi: 10.1038/nature18636
32. Ango F, Prézeau L, Muller T, Tu JC, Xiao B, Worley PF, et al. Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein homer. Nature (2001) 411(6840):962−5. doi: 10.1038/35082096
33. Hu LA, Chen W, Martin NP, Whalen EJ, Premont RT, Lefkowitz RJ. GIPC interacts with the β1-adrenergic receptor and regulates β1-adrenergic receptor-mediated ERK activation. J Biol Chem (2003) 278(28):26295−301. doi: 10.1074/jbc.M212352200
34. Hall RA, Premont RT, Chow CW, Blitzer JT, Pitcher JA, Claing A, et al. The beta2-adrenergic receptor interacts with the Na+/H+-exchanger regulatory factor to control Na+/H+ exchange. Nature (1998) 392(6676):626−30. doi: 10.1038/33458
35. Cong M, Perry SJ, Hu LA, Hanson PI, Claing A, Lefkowitz RJ. Binding of the β2 adrenergic receptor toN-ethylmaleimide-sensitive factor regulates receptor recycling. J Biol Chem (2001) 276(48):45145−52. doi: 10.1074/jbc.M106087200
36. Parker LL, Backstrom JR, Sanders-Bush E, Shieh BH. Agonist-induced phosphorylation of the serotonin 5-HT2C receptor regulates its interaction with multiple PDZ protein 1. J Biol Chem (2003) 278(24):21576−83. doi: 10.1074/jbc.M210973200
37. Bockaert J, Fagni L, Dumuis A, Marin P. GPCR interacting proteins (GIP). Pharmacol Ther (2004) 103(3):203−21. doi: 10.1016/j.pharmthera.2004.06.004
38. Laeremans T, Sands ZA, Claes P, De Blieck A, De Cesco S, Triest S, et al. Accelerating GPCR drug discovery with conformation-stabilizing VHHs. Front Mol Biosci (2022) 9:863099. doi: 10.3389/fmolb.2022.863099
39. Staus DP, Wingler LM, Strachan RT, Rasmussen SGF, Pardon E, Ahn S, et al. Regulation of β2 -adrenergic receptor function by conformationally selective single-domain intrabodies. Mol Pharmacol (2014) 85(3):472−81. doi: 10.1124/mol.113.089516
40. McMahon C, Baier AS, Pascolutti R, Wegrecki M, Zheng S, Ong JX, et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat Struct Mol Biol (2018) 25(3):289−96. doi: 10.1038/s41594-018-0028-6
41. Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, et al. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature (2013) 502(7472):575−9. doi: 10.1038/nature12572
42. Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, et al. Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell (2018) 172(1−2):55–67.e15. doi: 10.1016/j.cell.2017.12.011
43. English JG, Olsen RHJ, Lansu K, Patel M, White K, Cockrell AS, et al. VEGAS as a platform for facile directed evolution in mammalian cells. Cell (2019) 178(3):748–761.e17. doi: 10.1016/j.cell.2019.05.051
44. De Groof TWM, Bergkamp ND, Heukers R, Giap T, Bebelman MP, Goeij-de Haas R, et al. Selective targeting of ligand-dependent and -independent signaling by GPCR conformation-specific anti-US28 intrabodies. Nat Commun (2021) 12(1):4357. doi: 10.1038/s41467-021-24574-y
45. Che T, English J, Krumm BE, Kim K, Pardon E, Olsen RHJ, et al. Nanobody-enabled monitoring of kappa opioid receptor states. Nat Commun (2020) 11(1):1145. doi: 10.1038/s41467-020-14889-7
46. Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, et al. Structural biology. structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science (2015) 347(6226):1113−7. doi: 10.1126/science.aaa5026
47. Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ. Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci U.S.A. (2008) 105(29):9988−93. doi: 10.1073/pnas.0804246105
48. Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discovery (2013) 12(3):205−16. doi: 10.1038/nrd3954
49. Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, et al. Structural features for functional selectivity at serotonin receptors. Science (2013) 340(6132):615−9. doi: 10.1126/science.1232808
50. Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, et al. Structural basis for molecular recognition at serotonin receptors. Science (2013) 340(6132):610−4. doi: 10.1126/science.1232807
51. Chen Q, Tesmer JJG. G Protein–coupled receptor interactions with arrestins and GPCR kinases: The unresolved issue of signal bias. J Biol Chem (2022) 298(9):102279. doi: 10.1016/j.jbc.2022.102279
52. Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci (1983) 220:1219141–62. doi: 10.1098/rspb.1983.0093
53. Kenakin T. Biased receptor signaling in drug discovery. barker EL, éditeur. Pharmacol Rev (2019) 71(2):267−315. doi: 10.1124/pr.118.016790
54. De Pascali F, Ayoub MA, Benevelli R, Sposini S, Lehoux J, Gallay N, et al. Pharmacological characterization of low molecular weight biased agonists at the follicle stimulating hormone receptor. IJMS (2021) 22(18):9850. doi: 10.3390/ijms22189850
55. Jong YJI, Kumar V, O’Malley KL. Intracellular metabotropic glutamate receptor 5 (mGluR5) activates signaling cascades distinct from cell surface counterparts. J Biol Chem (2009) 284(51):35827−38. doi: 10.1074/jbc.M109.046276
56. Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, et al. Persistent cAMP-signals triggered by internalized G-Protein–coupled receptors. PloS Biol (2009) 7(8):e1000172. doi: 10.1371/journal.pbio.1000172
57. Jean-Alphonse F, Bowersox S, Chen S, Beard G, Puthenveedu MA, Hanyaloglu AC. Spatially restricted G protein-coupled receptor activity via divergent endocytic compartments. J Biol Chem (2014) 289(7):3960−77. doi: 10.1074/jbc.M113.526350
58. Sposini S, Jean-Alphonse FG, Ayoub MA, Oqua A, West C, Lavery S, et al. Integration of GPCR signaling and sorting from very early endosomes via opposing APPL1 mechanisms. Cell Rep (2017) 21(10):2855−67. doi: 10.1016/j.celrep.2017.11.023
59. White AD, Peña KA, Clark LJ, Maria CS, Liu S, Jean-Alphonse FG, et al. Spatial bias in cAMP generation determines biological responses to PTH type 1 receptor activation. Sci Signal (2021) 14(703):eabc5944. doi: 10.1126/scisignal.abc5944
60. Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature (2013) 495(7442):534−8. doi: 10.1038/nature12000
61. Westfield GH, Rasmussen SGF, Su M, Dutta S, DeVree BT, Chung KY, et al. Structural flexibility of the gαs α-helical domain in the β 2 -adrenoceptor gs complex. Proc Natl Acad Sci USA (2011) 108(38):16086−91. doi: 10.1073/pnas.1113645108
62. Ismail S, Gherardi MJ, Froese A, Zanoun M, Gigoux V, Clerc P, et al. Internalized receptor for glucose-dependent insulinotropic peptide stimulates adenylyl cyclase on early endosomes. Biochem Pharmacol (2016) 120:33−45. doi: 10.1016/j.bcp.2016.09.009
63. Sungkaworn T, Jobin ML, Burnecki K, Weron A, Lohse MJ, Calebiro D. Single-molecule imaging reveals receptor–G protein interactions at cell surface hot spots. Nature (2017) 550(7677):543−7. doi: 10.1038/nature24264
64. Gormal RS, Padmanabhan P, Kasula R, Bademosi AT, Coakley S, Giacomotto J, et al. Modular transient nanoclustering of activated β2-adrenergic receptors revealed by single-molecule tracking of conformation-specific nanobodies. Proc Natl Acad Sci U.S.A. (2020) 117(48):30476−87. doi: 10.1073/pnas.2007443117
65. Deshpande I, Liang J, Hedeen D, Roberts KJ, Zhang Y, Ha B, et al. Smoothened stimulation by membrane sterols drives hedgehog pathway activity. Nature (2019) 571(7764):284−8. doi: 10.1038/s41586-019-1355-4
66. Stoeber M, Jullié D, Li J, Chakraborty S, Majumdar S, Lambert NA, et al. Agonist-selective recruitment of engineered protein probes and of GRK2 by opioid receptors in living cells. eLife (2020) 9:e54208. doi: 10.7554/eLife.54208.sa2
67. Stoeber M, Jullié D, Lobingier BT, Laeremans T, Steyaert J, Schiller PW, et al. A genetically encoded biosensor reveals location bias of opioid drug action. Neuron (2018) 98(5):963–976.e5. doi: 10.1016/j.neuron.2018.04.021
68. Oberhauser L, Stoeber M. Biosensors monitor ligand-selective effects at kappa opioid receptors. In: Liu-Chen LY, Inan S, editors. The kappa opioid receptor. handbook of experimental pharmacology (Cham: Springer), vol. 271 (2021). p. 65−82.
69. Livingston KE, Mahoney JP, Manglik A, Sunahara RK, Traynor JR. Measuring ligand efficacy at the mu-opioid receptor using a conformational biosensor. eLife (2018) 7:e32499. doi: 10.7554/eLife.32499
70. Puri NM, Romano GR, Lin TY, Mai QN, Irannejad R. The organic cation transporter 2 regulates dopamine D1 receptor signaling at the golgi apparatus. eLife (2022) 11:e75468. doi: 10.7554/eLife.75468
71. Jakobs BD, Spannagel L, Purvanov V, Uetz-von Allmen E, Matti C, Legler DF. Engineering of nanobodies recognizing the human chemokine receptor CCR7. IJMS (2019) 20(10):2597. doi: 10.3390/ijms20102597
72. Duan J, Xu P, Luan X, Ji Y, He X, Song N, et al. Hormone- and antibody-mediated activation of the thyrotropin receptor. Nature (2022) 609(7928):854–9. doi: 10.1038/s41586-022-05173-3
73. Faust B, Billesbølle CB, Suomivuori CM, Singh I, Zhang K, Hoppe N, et al. Autoantibody mimicry of hormone action at the thyrotropin receptor. Nature (2022) 609(7928):846−53. doi: 10.1038/s41586-022-05159-1
74. Duan J, Xu P, Cheng X, Mao C, Croll T, He X, et al. Structures of full-length glycoprotein hormone receptor signalling complexes. Nature (2021) 598(7882):688−92. doi: 10.1038/s41586-021-03924-2
75. García-Nafría J, Lee Y, Bai X, Carpenter B, Tate CG. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. Elife (2018) 7:e35946. doi: 10.7554/eLife.35946
76. Thomsen ARB, Plouffe B, Cahill TJ, Shukla AK, Tarrasch JT, Dosey AM, et al. GPCR-G protein-β-Arrestin super-complex mediates sustained G protein signaling. Cell (2016) 166(4):907−19. doi: 10.1016/j.cell.2016.07.004
77. Nguyen AH, Thomsen ARB, Cahill TJ, Huang R, Huang LY, Marcink T, et al. Structure of an endosomal signaling GPCR–G protein–β-arrestin megacomplex. Nat Struct Mol Biol (2019) 26(12):1123−31. doi: 10.1038/s41594-019-0330-y
78. Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem (2000) 275(22):17201−10. doi: 10.1074/jbc.M910348199
79. Gulati S, Jin H, Masuho I, Orban T, Cai Y, Pardon E, et al. Targeting G protein-coupled receptor signaling at the G protein level with a selective nanobody inhibitor. Nat Commun (2018) 9(1):1996. doi: 10.1038/s41467-018-04432-0
80. Ghosh E, Srivastava A, Baidya M, Kumari P, Dwivedi H, Nidhi K, et al. A synthetic intrabody-based selective and generic inhibitor of GPCR endocytosis. Nat Nanotech (2017) 12(12):1190−8. doi: 10.1038/nnano.2017.188
81. Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, et al. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature (2013) 497(7447):137−41. doi: 10.1038/nature12120
82. Baidya M, Kumari P, Dwivedi-Agnihotri H, Pandey S, Sokrat B, Sposini S, et al. Genetically encoded intrabody sensors report the interaction and trafficking of β-arrestin 1 upon activation of g-protein–coupled receptors. J Biol Chem (2020) 295(30):10153−67. doi: 10.1074/jbc.RA120.013470
83. Baidya M, Chaturvedi M, Dwivedi-Agnihotri H, Ranjan A, Devost D, Namkung Y, et al. Allosteric modulation of GPCR-induced β-arrestin trafficking and signaling by a synthetic intrabody. Nat Commun (2022) 13(1):4634. doi: 10.1038/s41467-022-32386-x
84. Pandey S, Kumari P, Baidya M, Kise R, Cao Y, Dwivedi-Agnihotri H, et al. Intrinsic bias at non-canonical, β-arrestin-coupled seven transmembrane receptors. Mol Cell (2021) 81(22):4605–4621.e11. doi: 10.1016/j.molcel.2021.09.007
85. Dwivedi-Agnihotri H, Sarma P, Deeksha S, Kawakami K, Inoue A, Shukla AK. An intrabody sensor to monitor conformational activation of β-arrestins. Methods Cell Biol (2022) 169:267−78. doi: 10.1016/bs.mcb.2021.12.023
86. Cahill TJ, Thomsen ARB, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, et al. Distinct conformations of GPCR–β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci USA (2017) 114(10):2562−7. doi: 10.1073/pnas.1701529114
87. Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab (2006) 17(4):159−65. doi: 10.1016/j.tem.2006.03.008
88. Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, et al. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U.S.A. (2007) 104(29):12011−6. doi: 10.1073/pnas.0704849104
89. Crépieux P, Poupon A, Langonné-Gallay N, Reiter E, Delgado J, Schaefer MH, et al. A comprehensive view of the β-arrestinome. Front Endocrinol (2017) 8:32. doi: 10.3389/fendo.2017.00032
90. Manglik A, Kobilka BK, Steyaert J. Nanobodies to study G protein–coupled receptor structure and function. Annu Rev Pharmacol Toxicol (2017) 57(1):19−37. doi: 10.1146/annurev-pharmtox-010716-104710
91. Moutel S, Bery N, Bernard V, Keller L, Lemesre E, de Marco A, et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. eLife (2016) 5:e16228. doi: 10.7554/eLife.16228.025
92. Zimmermann I, Egloff P, Hutter CA, Arnold FM, Stohler P, Bocquet N, et al. Synthetic single domain antibodies for the conformational trapping of membrane proteins. Elife (2018) 7:e34317. doi: 10.7554/eLife.34317
93. Vasudevan L, Stove CP. A novel nanobody-based bio-assay using functional complementation of a split nanoluciferase to monitor mu- opioid receptor activation. Anal Bioanal Chem (2020) 412(29):8015−22. doi: 10.1007/s00216-020-02945-6
94. Tang JC, Drokhlyansky E, Etemad B, Rudolph S, Guo B, Wang S, et al. Detection and manipulation of live antigen-expressing cells using conditionally stable nanobodies. Elife (2016) 5:e15312. doi: 10.7554/eLife.15312
95. Wagner TR, Rothbauer U. Nanobodies – little helpers unravelling intracellular signaling. Free Radical Biol Med (2021) 176:46−61. doi: 10.1016/j.freeradbiomed.2021.09.005
96. Herce HD, Schumacher D, Schneider AFL, Ludwig AK, Mann FA, Fillies M, et al. Cell-permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells. Nat Chem (2017) 9(8):762−71. doi: 10.1038/nchem.2811
97. Zhou X, Hao R, Chen C, Su Z, Zhao L, Luo Z, et al. Rapid delivery of Nanobodies/VHHs into living cells via expressing In vitro-transcribed mRNA. Mol Ther - Methods Clin Dev (2020) 17:401−8. doi: 10.1016/j.omtm.2020.01.008
98. Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci (2012) 33(1):17−27. doi: 10.1016/j.tips.2011.09.003
99. Venkatakrishnan AJ, Deupi X, Lebon G, Heydenreich FM, Flock T, Miljus T, et al. Diverse activation pathways in class a GPCRs converge near the G-protein-coupling region. Nature (2016) 536(7617):484−7. doi: 10.1038/nature19107
100. Pardon E, Betti C, Laeremans T, Chevillard F, Guillemyn K, Kolb P, et al. Nanobody-enabled reverse pharmacology on G-Protein-Coupled receptors. Angew Chem Int Ed (2018) 57(19):5292−5. doi: 10.1002/anie.201712581
101. Chevillard F, Rimmer H, Betti C, Pardon E, Ballet S, van Hilten N, et al. Binding-site compatible fragment growing applied to the design of β 2 -adrenergic receptor ligands. J Med Chem (2018) 61(3):1118−29. doi: 10.1021/acs.jmedchem.7b01558
102. Martin C, Moors SLC, Danielsen M, Betti C, Fabris C, Sejer Pedersen D, et al. Rational design of Nanobody80 loop peptidomimetics: Towards biased β 2 adrenergic receptor ligands. Chem Eur J (2017) 23(40):9632−40. doi: 10.1002/chem.201701321
103. Sencanski M, Glisic S, Šnajder M, Veljkovic N, Poklar Ulrih N, Mavri J, et al. Computational design and characterization of nanobody-derived peptides that stabilize the active conformation of the β2-adrenergic receptor (β2-AR). Sci Rep (2019) 9(1):16555. doi: 10.1038/s41598-019-52934-8
104. Van holsbeeck K, Martins JC, Ballet S. Downsizing antibodies: Towards complementarity-determining region (CDR)-based peptide mimetics. Bioorganic Chem (2022) 119:105563. doi: 10.1016/j.bioorg.2021.105563
105. Yen YC, Schafer CT, Gustavsson M, Eberle SA, Dominik PK, Deneka D, et al. Structures of atypical chemokine receptor 3 reveal the basis for its promiscuity and signaling bias. Sci Adv (2022) 8(28):eabn8063. doi: 10.1126/sciadv.abn8063
106. Kleist AB, Jenjak S, Sente A, Laskowski LJ, Szpakowska M, Calkins MM, et al. Conformational selection guides β-arrestin recruitment at a biased G protein–coupled receptor. Science (2022) 377(6602):222−8. doi: 10.1126/science.abj4922
Keywords: G protein-coupled receptor, conformations, intracellular VHHs, Cell signaling, G proteins, β-arrestins, biosensor
Citation: Raynaud P, Gauthier C, Jugnarain V, Jean-Alphonse F, Reiter E, Bruneau G and Crépieux P (2022) Intracellular VHHs to monitor and modulate GPCR signaling. Front. Endocrinol. 13:1048601. doi: 10.3389/fendo.2022.1048601
Received: 19 September 2022; Accepted: 24 October 2022;
Published: 16 November 2022.
Edited by:
Ralf Jockers, Université de Paris, FranceCopyright © 2022 Raynaud, Gauthier, Jugnarain, Jean-Alphonse, Reiter, Bruneau and Crépieux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pascale Crépieux, cGFzY2FsZS5jcmVwaWV1eEBpbnJhZS5mcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.