
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 20 October 2022
Sec. Bone Research
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1042060
This article is part of the Research Topic Underlying Mechanisms and Treatment for Intervertebral Disc Disease View all 14 articles
 Lu-Ping Zhou†
Lu-Ping Zhou† Ren-Jie Zhang†Chong-Yu Jia†
Ren-Jie Zhang†Chong-Yu Jia† Liang KangZhi-Gang ZhangHua-Qing ZhangJia-Qi WangBo Zhang
Liang KangZhi-Gang ZhangHua-Qing ZhangJia-Qi WangBo Zhang Cai-Liang Shen*
Cai-Liang Shen*Ferroptosis, an iron-dependent form of programmed cell death marked by phospholipid peroxidation, is regulated by complex cellular metabolic pathways including lipid metabolism, iron balance, redox homeostasis, and mitochondrial activity. Initial research regarding the mechanism of ferroptosis mainly focused on the solute carrier family 7 member 11/glutathione/glutathione peroxidase 4 (GPX4) signal pathway. Recently, novel mechanisms of ferroptosis, independent of GPX4, have been discovered. Numerous pathologies associated with extensive lipid peroxidation, such as drug-resistant cancers, ischemic organ injuries, and neurodegenerative diseases, are driven by ferroptosis. Ferroptosis is a new therapeutic target for the intervention of IVDD. The role of ferroptosis in the modulation of intervertebral disc degeneration (IVDD) is a significant topic of interest. This is a novel research topic, and research on the mechanisms of IVDD and ferroptosis is ongoing. Herein, we aim to review and discuss the literature to explore the mechanisms of ferroptosis, the relationship between IVDD and ferroptosis, and the regulatory networks in the cells of the nucleus pulposus, annulus fibrosus, and cartilage endplate to provide references for future basic research and clinical translation for IVDD treatment.
Low back pain (LBP) is a common musculoskeletal disease in the world, and its prevention and treatment are the major challenges in public health programs, which contribute to severe socioeconomic and health burdens (1). Intervertebral disc (IVD) degeneration (IVDD) has been considered as the leading cause of LBP, thereby resulting in a series of structural changes, such as the decrease of intervertebral height, breakage of the existing nucleus pulposus (NP), fissure of annulus fibrosus (AF), calcification of cartilage endplate (CEP), and imbalance of extracellular matrix (ECM) metabolism (2). In recent years, many new ways of programmed cell death have been reported in studies on IVDD. In contrast to apoptosis, necroptosis, pyroptosis, autophagy, and other types of death procedures, ferroptosis is characterized by the iron-mediated accumulation of lipid peroxides, morphologically manifested as mitochondrial shrinkage, reduction of mitochondrial cristae, and rupture of the mitochondrial outer membrane, and it has been regarded as a new target for the treatment of IVDD (3).
The overload of cellular iron content, particularly ferrous iron, can induce lipid peroxidation of fatty acids (4). The abnormal mitochondrial oxidative phosphorylation pathway results from iron overload, which produces a large amount of reactive oxygen species (ROS) and ATP. When the ROS content exceeds the scavenging level of the antioxidant system, polyunsaturated fatty acids (PUFAs) on the cell membranes and organelle membranes are oxidized to form lipid peroxides, which directly or indirectly destroy cell structure and function, thereby resulting in cell damage or death. Initial research on the mechanism of ferroptosis primarily focuses on the solute carrier family 7 member 11 (SLC7A11)–glutathione (GSH)–glutathione peroxidase 4 (GPX4) signaling pathway. Recently, novel mechanisms of ferroptosis independent of GPX4 have been discovered, which are closely related to lipid metabolism, iron balance, and redox reactions.
Although ferroptosis has been extensively investigated in various physiological and pathological processes, such as tumors, injuries, viral infection, immune response, and metabolic disorders since the item was coined by Dixon et al. (5) in 2012, research regarding the relationship between ferroptosis and IVDD started relatively lately (6–9). To date, a growing number of studies have investigated the relationship between ferroptosis and IVDD. Herein, we aimed to review recent literature to explore the underlying mechanism of ferroptosis and its role in IVDD and to investigate new therapeutic targets for the treatment of IVDD.
Iron homeostasis is essential for various metabolic processes in mammalian organ systems (Figure 1). Iron absorption mainly has two sources (heme iron primarily from animal products, including beef, fish, chicken, and liver, and non-heme iron primarily from fruit, vegetables, eggs, and grains) in the intestine, depending on different receptors (10). The heme iron is transported through the intestinal epithelium by heme carrier protein 1 (11). For the non-heme iron, ferric iron is reduced to be ferrous iron by cytochrome b reductase 1, which is then transported by divalent metal transporter 1 (DMT1), a carrier protein, into the enterocytes (12). Ferrous iron is exported through the iron exporter, ferroportin 1 (FPN1) (12, 13). The ferrous iron is oxidized from +2 to +3 state by hephaestin, subsequently loading ferric iron onto transferrin (TF) for systematic transport in the bloodstream (14). Moreover, the systematic iron homeostasis is complemented by serum ferritin and non-TF bound iron and regulated by the hepcidin–FPN1–regulatory axis (15).
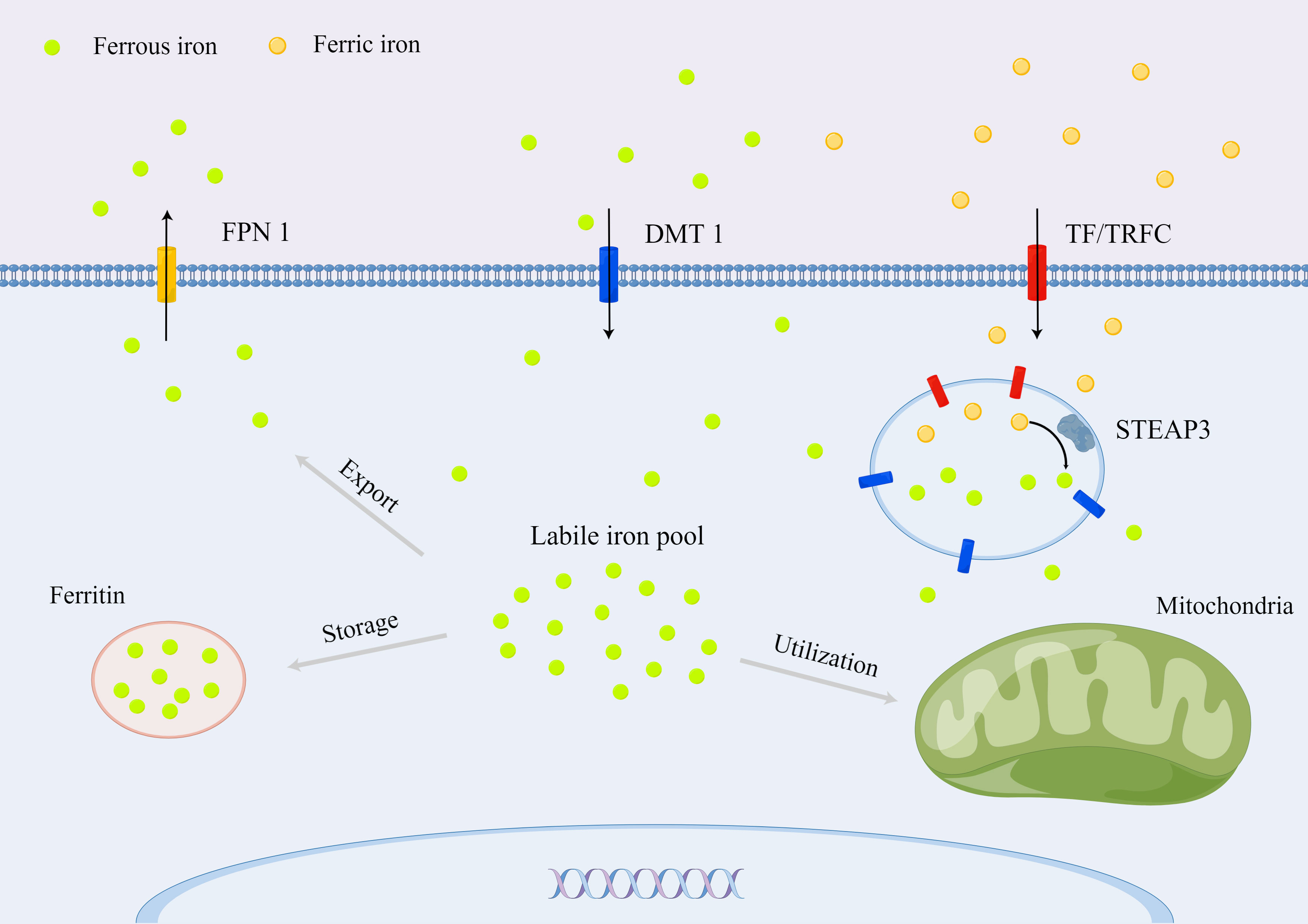
Figure 1 Cellular iron metabolism in mammals.
For intracellular iron homeostasis, ferric iron binding to the TF in the serum can be taken up by a transferrin receptor (TFRC) on the cell membrane (16). The ferric iron is released from the TF in the endosome because of the rapid drop of pH and then reduced by six transmembrane epithelial antigens of prostate 3 (STEAP3) to ferrous iron, which is subsequently transported into the cytoplasm through the solute carrier family 11 member 2 (SLC11A2)/DMT1 (17). The transported ferrous iron stored in ferritin or labile iron pool for further utilization is essential for metabolic and biochemical processes, such as the regulation of the iron-requiring enzymatic activity, iron–sulfur protein production, and oxygen transport (18). Excess iron can be extruded into the extracellular space via the iron-efflux protein metal transporter protein-1/FPN1/iron-regulated transporter-1, which is the product of the solute carrier family 40 member 1 (SLC40A1) gene (19). Moreover, the intracellular iron homeostasis is regulated by iron-responsive element binding protein 2, heme oxygenase 1 (HO-1), and iron regulatory proteins (20, 21).
Hematological disorders, such as hereditary hemochromatosis associated with gene mutations of HFE, hepcidin hormone, and TFRC, can contribute to a high serum ferritin level (22, 23). In addition, chronic renal failure receiving repeated hemodialysis and other chronic diseases receiving repeated blood transfusions, including myelodysplastic syndrome and thalassemias, can saturate the iron-binding capacity of TF in the cytoplasm, leading to chronic iron overload (24, 25).
Restrictive export and excessive import of iron result in intracellular iron overload. Genetic defects in SLC40A1 and STEAP3 mutations restrict iron export, but they have no effect on iron import (26, 27). Genetic mutations in SLC11A2 accelerate iron import, leading to intracellular iron overload (28).
Iron accumulation in IVD is commonly observed in aging patients suffering from diseases because of the lack of effective mechanisms to exert excess iron, including hereditary hemochromatosis and thalassemia (29, 30). Meanwhile, iron overload in IVD may result from neovascularization within the disc, which exposes tissues to high levels of heme, a major source of intracellular iron (31, 32). Neovascularization was initially reported in herniated NP using histological staining in 1993, and Shan et al. (31) found that the immature vessels during neovascularization in herniated IVD lead to the extravasation of red blood cells and the deposition of iron in this tissue.
System XC¯, consisting of SLC7A11 and solute carrier family 3 member 2 (SLC3A2), is a Na+-dependent amino acid antiporter that is widely distributed in the plasma membrane and is responsible for the import of extracellular cystine and the export of intracellular glycine (33). Intracellular cystine is immediately reduced back to cysteine by depleting NADPH, which is a rate-limiting precursor amino acid for the synthesis of GSH, a tripeptide consisting of cysteine, glutamate, and glycine (34). GSH plays an important role in anti-oxidative stress, reduction of lipid peroxidation, and protection of tissue cells, which is a necessary cofactor of GPX4 for normal function (35). Compared with other members of the GPXs family, GPX4 can directly convert phospholipid hydroperoxides (PLOOHs), a form of lipid-based ROS, on cell membranes to nontoxic lipid alcohols (PLOHs) with sufficient cellular GSH, whereas the depletion of GSH results in the inactivation of GPX4 (36, 37) (Figure 2).
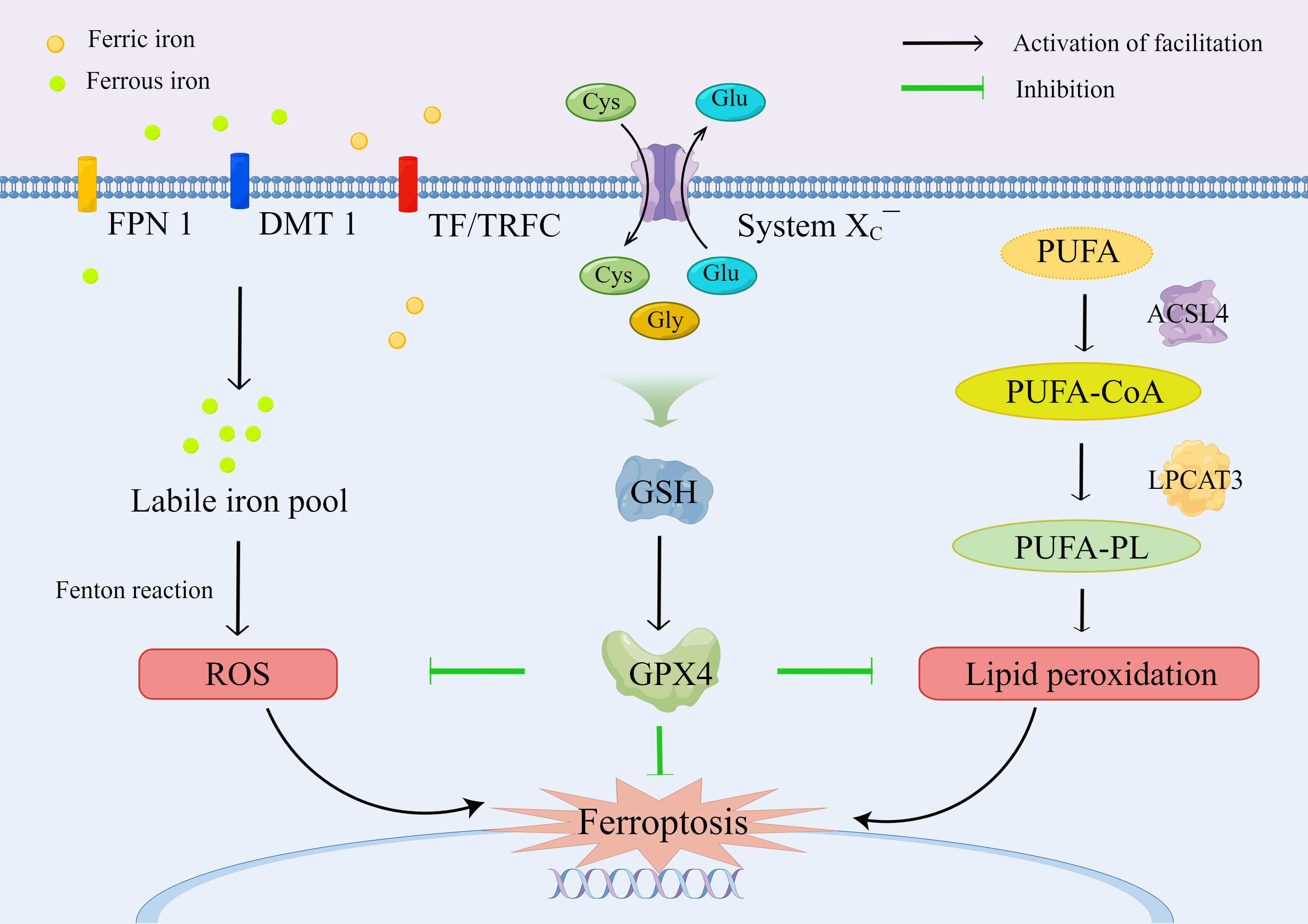
Figure 2 The molecular mechanism and regulation of ferroptosis.
GPX4 is the major neutralizing enzyme for PLOOHs, which protects the structure and function of cell membranes, and it has been regarded as a specific marker of ferroptosis, which plays an essential role in limiting lipid peroxidation (36, 38). Selenocysteine is the key group for the catalytic function of GPX4. PLOOH is reduced to PLOH, whereas the selenocysteine is oxidized to selenic acid intermediate (GPX4-SeOH). Subsequently, the selenium–glutathione adduct is produced after the reaction between GPX4-SeOH and GSH. Then, the selenium–glutathione adduct is converted back to selenocysteine by reacting with the equivalent of GSH. Similarly, the oxidized glutathione (GSSG) is produced from GSH, which is then reduced to GSH by glutathione reductase for recycling and utilization (35). Apart from ferroptosis, GPX4 plays a role in pyroptosis (39), apoptosis (40), necroptosis (41), and autophagy (42, 43), indicating that the regulation of PLOOHs may be a hallmark in the signaling pathway for the induction of regulated cell death.
Recently, the regulation of ferroptosis through the SLC7A11/GSH/GPX4 signaling pathway has been explored for the intervention of IVDD. Zhang et al. (44) demonstrated that the promotion of methylase expression upregulated GPX4 methylation in patients with hyperhomocysteinemia (HHcy), thereby inducing ferroptosis in NP cells (NPCs). In addition, the level of GPX4 protein was reduced after treatment with heme by simulating neovascularization in a heme-induced ferroptosis model (31). Moreover, ferroptosis in cartilage cells was modulated via the IL-6/miR-10a-5p/IL-6R axis in the inflammatory microenvironment (45), and the IL-6/STAT3/GPX4 signaling pathway might be implicated in this procedure (46).
GPX4 has been regarded as the primary enzyme that prevents ferroptosis through the conversion of lipid hydroperoxides into non-toxic lipid alcohols (36). However, the sensitivity of GPX4 inhibitors differs in cancer cell lines, indicating that additional independent pathways govern the regulation of ferroptosis (47). Therefore, current mechanisms of intracellular defense against ferroptosis can be divided into the SLC7A11-GSH-GPX4 signaling pathway and other signaling pathways independent of GPX4 (Figure 3).
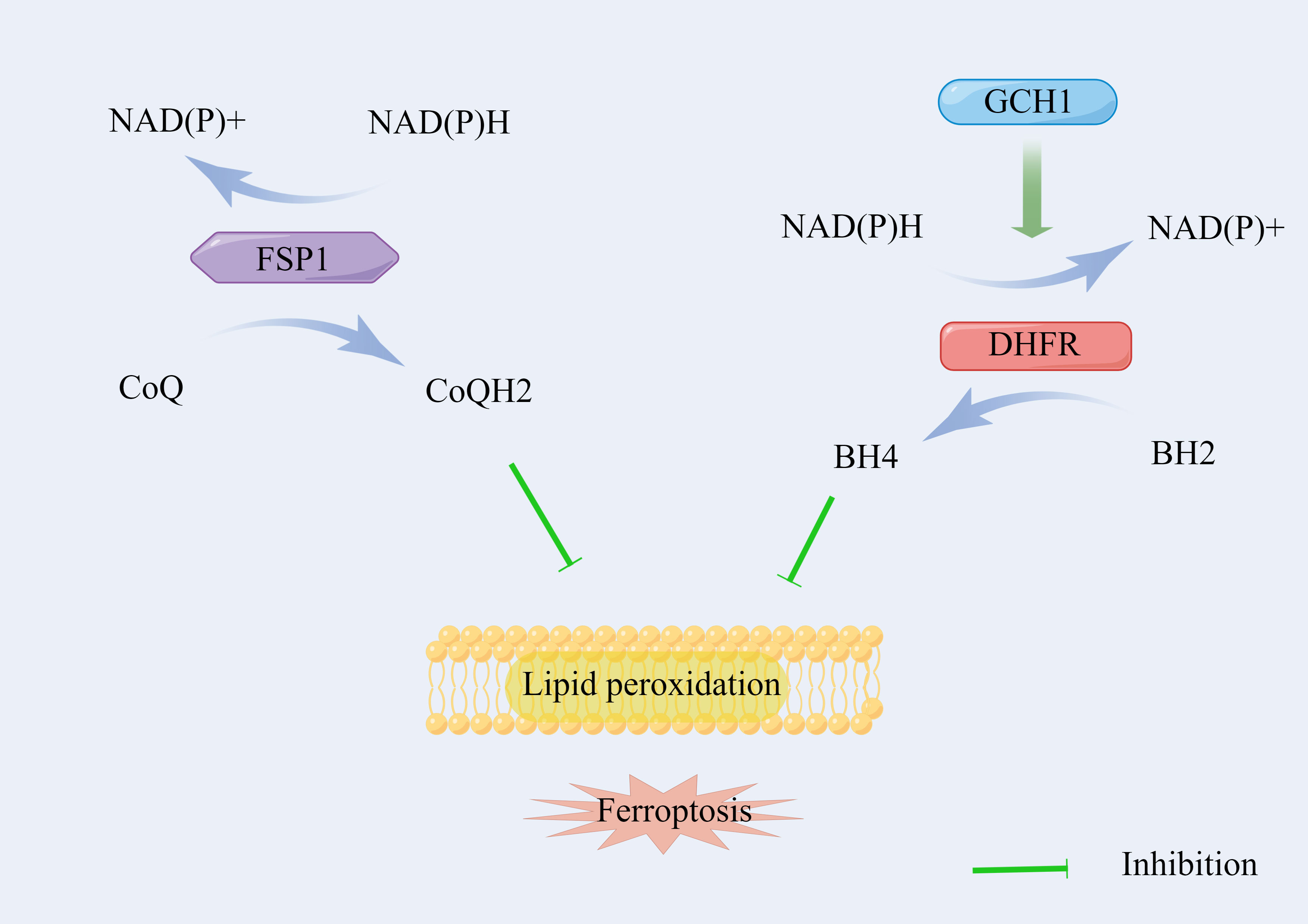
Figure 3 The signaling pathways of ferroptosis independent of SLC7A11/GSH/GPX4 axis.
Bersuker et al. (48) and Doll et al. (49) identified that ferroptosis suppressor protein 1 (FSP1), also known as apoptosis-inducing factor mitochondrial 2, acts parallel to the GSH-dependent GPX4 pathway with regard to the inhibition of phospholipid (PL) peroxidation and ferroptosis. FSP1 on the membrane reduces coenzyme Q (CoQ) by using NAD(P)H to ubiquinol (CoQH2), which serves as a lipophilic radical-trapping antioxidant (RTA), suppressing the propagation of lipid peroxides (50, 51). The loss of FPS1 improves PL peroxidation with the normal function of GPX4, indicating an independent mechanism of the FSP1/CoQH2/NAD(P)H pathway during ferroptosis.
The GTP cyclohydrolase-1 (GCH1)/6(R)-L-erythro5,6,7,8-tetrahydrobiopterin (BH4)/dihydrofolate reductase (DHFR) axis is another unique protective mechanism for ferroptosis, which is independent of the GSH-GPX4 system. Kraft et al. (52) identified GCH1 as a potent antagonist of ferroptosis using a whole-genome activation screen. The endogenous antioxidant BH4 on the membrane generated by the enzyme GCH1 also serves as a lipophilic RTA to selectively neutralize PUFA-PL-OOH, which alleviates sensitivity to ferroptosis. Moreover, BH4 also participates in the synthesis of CoQ to leverage oxidative damage under oxidative stress (52). Furthermore, DHFR serves as an essential regulator of ferroptosis by regenerating BH4 from dihydrobiopterin (BH2). The genetic or pharmacological loss of DHFR’s function can also induce ferroptosis (53).
Lipid peroxidation is an important process in ferroptosis. PUFA-containing PLs on the cell membrane are easily oxidized because of the highly active hydrogen atoms in the methylene bridge, destroying the structure and stability of the lipid bilayer, and disintegration of the cell membrane. Free PUFAs, such as adrenal acid and arachidonic acid, are catalyzed by acyl-CoA synthetase long-chain family member 4 (ACSL4) to generate PUFA-CoA, which is then transported to the cell membrane through lysophosphatidylcholine acyltransferase 3 (LPCAT3) by inserting acyl groups into lysophospholipids and synthesizing PUFA-PLs with PLs (54–56). PUFA oxidation mainly has two forms. First, PUFA can be oxidized through an enzymatic reaction. PUFA-PL is catalyzed by arachidonate lipoxygenase (ALOX) into PUFA-PL-OOH (57). Zou et al. (58) found that cytochrome P450 oxidoreductase (POR) promotes lipid peroxidation during ferroptosis in an ALOX-independent manner using systematic lipidomic profiling and suggested that POR is an essential mediator of ferroptosis. In addition, PUFAs are oxidized by other oxygenases, including NADPH oxidases (NOXs) and prostaglandin-endoperoxide synthase 2 (PTGS2/COX) (59, 60). Despite being upregulated during ferroptosis, PTGS2 might not be involved in the production of lipid peroxidation. Whether PTGS2 affects the procedure of ferroptosis still needs further investigation (61). Second, PUFA oxidation occurs through Fenton reaction in a non-enzymatic way. Ferric iron, hydroxyl radicals (HO ·), and OH- are generated during the reaction between ferrous iron and hydrogen peroxide (H2O2). Thus, free radical ions further cause oxidative damage to membrane lipids, particularly PUFAs. The inhibition of key molecules, such as ACSL4, LPCAT3, ALOX, POR, and NOXs, is of great significance to the reduction of lipid peroxidation, thereby counteracting ferroptosis.
The main mechanism of the biological toxicity of iron ions is the classical Fenton reaction, where ferrous iron reacts with hydrogen peroxide (H2O2). Among the products of the Fenton reaction, the hydroxyl radicals are largely destroyed, which can not only cause oxidative damage to cells by unspecifically attacking biomolecules, but also promote the peroxidation of lipid components to generate various oxidation products, the main products of which are lipid hydroperoxides (LOOHs) (57). LOOHs can be converted to oxygen radical intermediates, including lipid peroxyl radical (LOO ·) or alkoxyl (LO ·) (62). Given the high proportion of PUFAs on cell and plasma membranes, the oxygen radical intermediates cause cascade reactions, which further aggravate the destruction of the membranes, contributing to the disturbance of cellular homeostasis and activation of serious biochemical reactions. Furthermore, many different aldehydes that can be formed as secondary products, including 4-hydroxynonenal, malondialdehyde, hexanal, and propanal, can continuously react with PUFAs, destroy cells, and eventually lead to irreversible disruption of the structure and function of the cell membranes (63–65). Finally, the free radical ions and hydroxyl radicals generated from intracellular free ferrous ions through the Fenton reaction oxidize PUFA on the cell membrane and damage the protein in the cytoplasm and DNA in the nucleus (66). Moreover, iron ions are considered as the key components of various metabolic enzymes, including ALOX and POR. Therefore, homeostasis of iron ions is important for the normal functioning of organisms and cells.
The regulation of iron homeostasis can affect the sensitivity of cells to ferroptosis with regard to the uptake, storage, and efflux of iron. The knockdown of TF can suppress lapatinib-induced ferroptosis in SKBR3 cancer cell line, and the loss of TFRC can also decrease cystine starvation- or erastin-induced ferroptosis (67–69). Ferritin, composed of H and L subtypes, is the main iron storage protein primarily located in the cytoplasm, which stores around 70%–80% newly imported iron (35). The H subtype, ferritin heavy chain 1 (FTH1), can oxidize ferrous iron to ferric iron and combine with it to reduce free ferrous iron and subsequent Fenton reaction. SLC40A1, the only known iron exporter in mammalian cells, can influence ferroptosis by mediating iron output. Studies have shown that ferroptosis is promoted by the knockdown of SLC40A1, whereas this procedure is ameliorated by the overexpression of SLC40A1 (68, 70). In response to different types of ferroptosis inducers, the level of intracellular ferric ions will increase, and various protein transporters related to iron metabolism, such as TF, TFRC, ferritin, and SLC40A1, will be rearranged under the ferroptosis program.
In the oxidative stress microenvironment of IVDD simulated by the tert-butyl hydroperoxide (TBHP), Lu et al. (71) indicated that the intercellular iron overload resulted from FPN dysregulation that was regulated by metal-regulatory transcription factor 1 (MTF1), and the TBHP-indued ferroptosis was aggravated through the JNK/MTF1/FPN signal pathway. In addition, the levels of intercellular iron in AF cells (AFCs) and NPCs were increased by nuclear receptor coactivator 4 (NCOA4)-mediated ferritin selective autophagy during TBHP-indued ferroptosis (72).
Amino acid metabolism is an important part of the metabolic loop of organisms, and imbalances in intracellular and extracellular cysteine, cystine, glutamate, and GSH can induce ferroptosis. Intracellular cysteine is primarily used to synthesize antioxidant enzymes, including GSH and thioredoxin. When cysteine is deficient, cystathionine-β-synthase (CBS) and cystathionine gamma-lyase are activated under oxidative stress conditions, and cysteine is biosynthesized from methionine through the transsulfuration pathway, thereby reducing oxidative stress–induced ferroptosis (73). Liu et al. (74) demonstrated that the overexpression of CBS can confer ferroptosis resistance in ovarian cancer cells, and CBS has been identified as a new negative regulator of ferroptosis. By contrast, cysteinyl-tRNA synthetase (CARS) positively regulates ferroptosis by limiting the transsulfuration pathway. Hayano et al. (75) found that the loss of CARS contributed to the accumulation of cystathionine, induction of the transsulfuration pathway, and upregulation of genes associated with serine biosynthesis and transsulfuration.
The glutaminolysis pathway, in which glutamine is catabolized to glutamate, has also been implicated in the regulation of ferroptosis. Gao et al. (67) indicated that α-ketoglutarate converted from glutamine can cause cysteine deprivation and promote ferroptosis, and the limitation of glutaminolysis can reduce the heart triggered by ischemia–reperfusion injury through the inhibition of ferroptosis. Moreover, glutaminolysis is catalyzed by cytosolic glutaminase (GLS1) and mitochondrial glutaminase (GLS2); however, GLS2, instead of GLS1, is required for ferroptosis (67, 76, 77). Mitochondria play a crucial role in cysteine deprivation-induced ferroptosis, instead of GPX4 inhibition-induced ferroptosis, which is mediated by the potential hyperpolarization, mitochondrial tricarboxylic acid cycle, and electron transport chain of the mitochondrial membrane (78).
Glucose is the principal nutrient for biosynthesis and the main source of acetyl-CoA for the synthesis of fatty acids. Under nutritional deficiency, energy stress is induced by the depletion of intracellular ATP and subsequent improvement of intracellular AMP levels. In short-term and slight energy stress, the AMP-activated protein kinase (AMPK), a sensor of cellular energy status, participates in the adaptive response by promoting ATP-generating catabolism and maintaining cell survival (79). Lee et al. (80) found that the energy stress caused by glucose starvation can partly reduce ferroptosis by AMPK, and the activation of AMPK inhibits ferroptosis by the phosphorylation of acetyl-CoA carboxylase and restrains PUFA biosynthesis. In type 2 diabetic osteoporosis, high glucose levels can induce ferroptosis via increased ROS/lipid peroxidation/GSH depletion (81).
The transcription factors regulate ferroptosis-related target genes that serve as promoters or blockers, thereby affecting the sensitivity of ferroptosis through multiple roles in transcription-dependent or transcription-independent mechanisms (82). Many transcription factors, such as tumor protein 53, nuclear factor-erythroid 2 like 2 (NRF2), Yes 1-associated transcriptional regulator, MTF1, activating transcription factor 3 (ATF3), transcription factor AP-2 gamma, specificity protein 1, hypoxia-inducible factor 1 alpha, and egl-9 family hypoxia-inducible factor 2, have been found to be involved in a ferroptotic network, which has been a new potential treatment target (83–89).
DNA methylation and histone modification can regulate ferroptosis. Helicase can inhibit ferroptosis by DNA methylation through the induction of sterol-CoA desaturase 1 and fatty acid desaturase 2, epigenetic silencing of cytosolic long non-coding RNAs (IncRNA) LINC00472, and promotion of nuclear lncRNA 00336 (35, 90–92). Histone 2A ubiquitination (H2Aub) and histone 2 B ubiquitination (H2Bub) can induce SLC7A11 expression by histone modification to reduce sensitivity to ferroptosis. Bromodomain containing 4 (BRD4) epigenetically prevents ferroptosis by recognizing acetylated lysine residues on histones, and demethylase 3 B (KDM3B), a histone H3 lysine 9 demethylase, can activate the expression of SLC7A11 to reduce erastin-induced ferroptosis (93, 94).
The ferroptosis inducer (FINs) can be divided into four categories: The first category is the inhibiting activity of SCL7A11. The FINs inhibit the function of System XC¯ to reduce the uptake of cystine and the synthesis of GHS, including erastin, sulfasalazine, and sorafenib (5, 36). The second category refers to the FINs inhibiting GPX4. The FINs, including RSL3, ML162/DP17, and ML210/DP110, covalently react with selenocysteine to inhibit the activity of GPX4 (36, 95). The third category refers to organic peroxides that cause oxidative damage, including TBHP, artemisinin, and FINO2 (96, 97). The fourth category refers to the FINs resulting in iron overload, including exogenous hemin and hemoglobin (31, 98, 99). The inhibitors of ferroptosis include antioxidants (e.g., butylated hydroxytoluene, butylated hydroxyanisole, tetrahydronaphthyridinols, ferrostatin-1, liproxstain-1, vitamin E, and vitamin K), iron chelators (e.g., deferoxamine [DFO], deferasirox [DFP], deferiprone [DFX], and ciclopirox), ferroptosis-related enzyme inhibitors (e.g., ALOX inhibitors, including baicalein, zileuton, and cinnamyl-3,4-dihydroxya-cyanocinnamate; ACSL4 inhibitors, including thiazolidinediones and triacsin C; and NOX inhibitors, including diphenylene iodonium and 2-acetylphenothiazine), and protein degradation inhibitors (5, 35, 100–103) (Figure 4).

Figure 4 The inducers (marked red) and inhibitors (marked blue) of ferroptosis.
The IVD is a special structure without blood vessels in an ischemic and hypoxic microenvironment under normal physiological conditions, the steady balance of which is the basis for the maintenance of normal function. IVDD is a chronic process that commonly causes LBP; however, the specific cause of IVDD remains unclear. Published studies have demonstrated that IVDD is a complex process with multifactorial interactions, which is primarily characterized by ECM destruction and cell phenotype changes, as well as apoptosis, autophagy, pyroptosis, necroptosis, and ferroptosis of IVD (31, 104–107). The term ferroptosis was first coined in 2012. Since then the molecular mechanism and regulatory network of ferroptosis have been exponentially investigated in many degenerative diseases, such as Parkinson’s disease, Alzheimer’s disease, kidney degeneration, atherosclerosis, osteoporosis, and osteoarthritis (OA) (108–113). Although studies on ferroptosis in IVDD have been conducted in recent years, increasing evidence has shown that ferroptosis is associated with IVDD and is involved in degenerative processes of NP, AF, CEP, and ECM (Table 1).
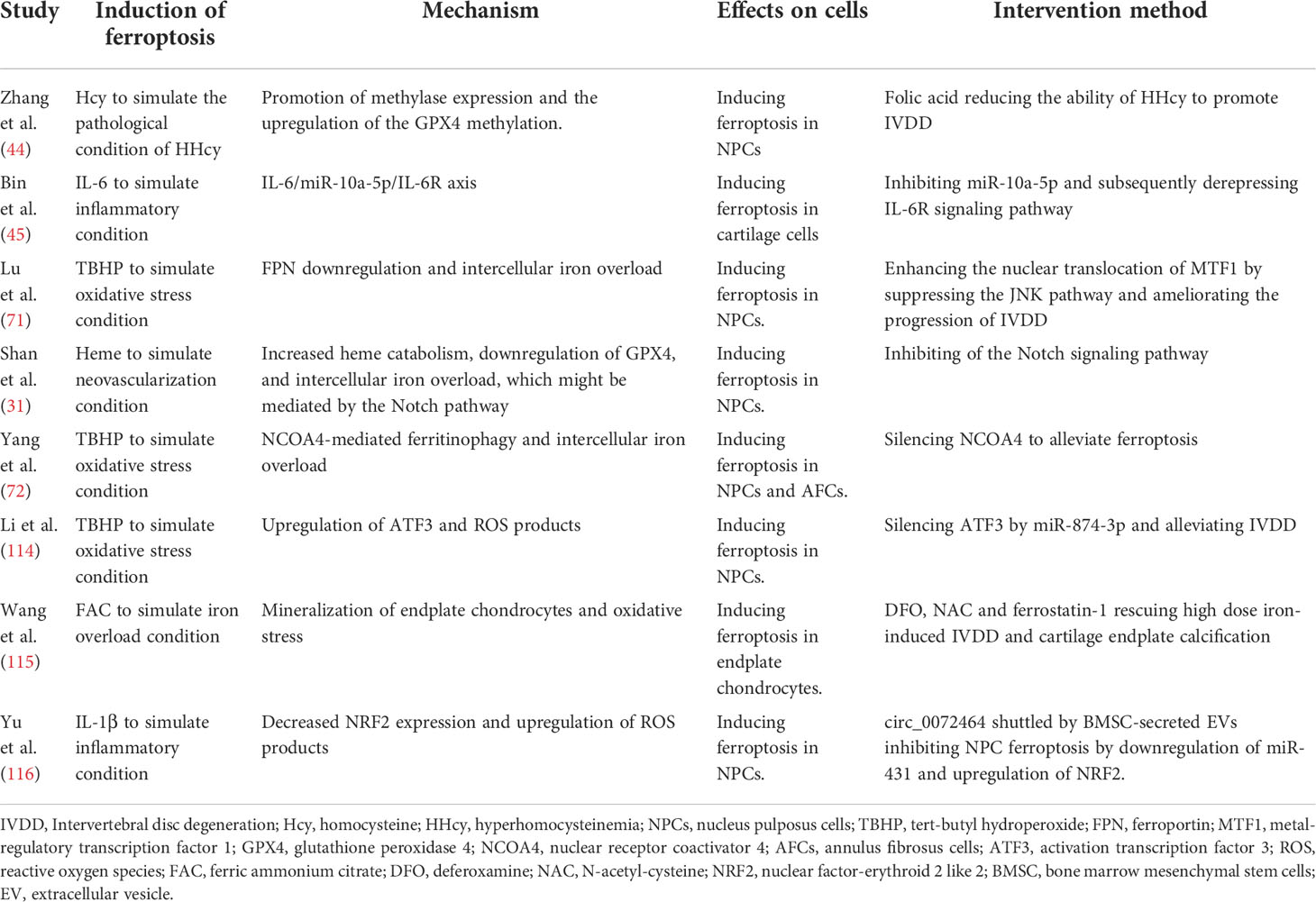
Table 1 Mechanism and intervention methods of ferroptosis in IVDD.
The property of resident progenitor cells in NP is altered by IVDD (117). The healthy NP primarily consists of chondrocyte-like cells, whereas the degenerative NP is primarily composed of chondrocyte-like cells, inflammatory cells, and fibroblast-like cells, which shrank extensively and became yellowish and fibrous (118). Zhang et al. (119) performed single-cell RNA sequencing analysis of NPCs isolated from normal controls and from patients with IVDD. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analyses revealed that ferroptosis pathways were enriched in mild IVDD. The pathways identified by scRNA-Seq were validated using a rat model of IVDD, and the levels of iron (a sign of ferroptosis), FTL, and HO-1 (two important regulators of ferroptosis) were assessed. They found that NP in the degenerative group was associated with remarkably higher iron levels and lower levels of ferritin light chain and HO-1 than in the control group, indicating that ferroptosis played a role in the progression of IVDD. Shan et al. (31) found that the increased level of iron primarily resulted from the high level of heme caused by neovascularization in degenerative NP, thereby inducing cytotoxicity and ferroptosis and accelerating the progression of IVDD. This result is also supported by scRNA-Seq analysis reporting endothelial cells only in IVDD samples, and the proportion of endothelial cells increased with the severity of IVDD (119).
Ferroptosis is regulated by multiple pathways during IVDD. Lu et al. (71) detected a decreased expression of FPN and the occurrence of ferroptosis under oxidative stress conditions simulated using TBHP in human NPCs in vitro and in vivo. They found that the downregulation of FPN, but not TFRC and DMT1, primarily accounted for the intercellular iron overload and ferroptosis in TBHP-induced human NPCs, whereas the overexpression of FPN inhibited ferroptosis through the JNK/MTF1/FPN signaling pathway. Therefore, the decreased nuclear translocation of MTF1 under TBHP treatment contributes to the reduced expression of FPN and ferroptosis in human NPCs. Meanwhile, hinokitiol, a natural tropolone derivative, can increase the nuclear translocation of MTF1, restore FPN, and attenuate TBHP-induced ferroptosis by suppressing the JNK pathway in human NPCs, as well as in the NP tissue of IVDD. Moreover, ferritinophagy is involved in TBHP-induced ferroptosis of NPCs through NCOA4-mediated ferritin-selective autophagy in an autophagy-dependent manner (72). NCOA4, a selective cargo receptor mutually combining with ferritin, transports ferritin to the autophagosomes with the occurrence of oxidative stress in IVD cells, thereby releasing free iron to induce ferroptosis (72, 120, 121). A newly published clinical study also proved that serum ferritin was negatively correlated with the degree of IVDD, which can be used as a clinical predictor of IVDD severity (122). Furthermore, Shan et al. (31) showed that heme-induced ferroptosis of human NPCs by the inhibition of the GPX4 protein can be rescued by DFO treatment. In addition, heme-induced ferroptosis might be mediated by the Notch signaling pathway, with substantial changes in the mRNA and protein levels of Notch1, Notch2, Jag1, Jag2, Hes1, Hes2, and Hey1.
MicroRNAs (miRNAs) and short non-coding RNAs (ncRNAs) primarily downregulate the expression of target genes and modulate the related downstream pathways by directly binding to the 3′-untranslated regions of the target genes, thereby regulating ferroptosis in human NPCs and IVDD. Li et al. (114) established a rat model of IVDD with TBHP and found that the overexpression of ATF3 induced ROS production and ferroptosis by suppressing SLC7A11. In addition, bioinformatics analysis and molecular experiments demonstrated that ATF3 is a direct target of miR-874-3p, indicating that the upregulation of ATF3 partially results from the downregulation of miR-874-3p in IVDD. Extracellular vesicles (EVs) derived from most cell types have been increasingly considered as important mediators of cell-to-cell communication and biomarkers of diseases, and they are involved in pathophysiological processes of IVDD (123, 124). Exosome-transported circular RNAs (circRNAs) have also been confirmed to exert effects on the regulation of IVDD (125). CircRNAs serve as miRNA sponges, contributing to the downregulation of miRNA and upregulation of miRNA downstream targets (126). Yu et al. (116) found that the uptake of EVs extracted from mouse bone marrow mesenchymal stem cells (BMSCs) by NPCs alleviated IVDD. In vitro and in vivo experiments showed that circ_0072464 shuttled by BMSC-derived EVs reduced ferroptosis in NPCs through the inhibition of miR-431 and upregulation of miR-431-mediated NRF2, indicating a potential biotherapeutic target for the treatment of IVDD.
Ferroptosis of NPCs and IVDD is also regulated by DNA methylation. HHcy, characterized by increased total homocysteine in plasma and its close relation to DNA methylation, results from the high concentration of homocysteine (Hcy) in serum caused by the deficiency of folic acid or the excessive intake of methionine (127–129). Zhang et al. (44) demonstrated that Hcy aggravates oxidative stress and induces ferroptosis in NPCs through the promotion of methylase expression and upregulation of GPX4 methylation. They also confirmed that HHcy is an independent risk factor for IVDD and that HHcy accelerates IVDD in vivo, which can be rescued by folic acid and the methylase inhibitor 5-AZA.
AF, which is divided into the inner (proteoglycan and collagen II rich) and outer regions (collagen I rich), has strong resistance to traction and compression, preventing the NP from protruding outwards (130). The AFCs in the outer region tend to be fibroblast-like and parallel to collagen fibers, whereas the AFCs in the inner region can be more oval (131). Yang et al. (72) investigated the expression of ferroptosis marker proteins in the AFCs of a rat model exposed to TBHP at different concentrations. They found decreased expression of FTH and GPX4 and increased expression of PTGS2 and ACSL4 with an increase in TBHP concentration in AFCs, indicating the existence of oxidative stress–induced ferroptosis in rat APCs. Moreover, ferroptosis in AFCs is upregulated by NCOA4-mediated ferritinophagy in response to TBHP treatment, indicating new insights into the treatment of IVDD. The composition and structure of AF are unique and critical for the maintenance of disc anisotropy, elastic mechanical loading, and homeostasis. However, most published research focuses on NPCs, and studies on the effects of ferroptosis on AFCs are rare. Therefore, further studies on the relationship between AFCs must be conducted in the future.
CEP interfaces the disc and vertebral body with a thin horizontal layer of semi-porous thickened cancellous bone and hyaline cartilage, serving as the predominant route for nutrition supply and waste product exchange within the IVD (131, 132). The degeneration of CEP has been regarded as the primary predictor of IVDD, which reduces tissue diffusivity and changes the biochemical microenvironment of IVD, leading to reduced glucose and oxygen concentrations, increased lactate levels, and decreased PH within the disc, thereby initiating IVDD (133–135). The overload of iron and ferroptosis plays a role in the degeneration and calcification of CEP. Wang et al. (115) explored the connection between iron overload and degeneration of CEP and found that oxidative stress mediated by iron overload induced endplate chondrocyte ferroptosis, which can be reversed by iron chelation, antioxidants, and ferroptosis inhibition, indicating that ferroptosis plays an important role in endplate chondrocyte degeneration. Bin et al. (45) demonstrated aberrant expression of interleukin (IL) 6 (IL-6) and its receptor in cartilage specimens obtained from patients with IVDD. Furthermore, they showed that cartilage cell ferroptosis is induced by IL-6 through oxidative stress and iron homeostasis. Furthermore, miR-10a-5p partially inhibited IL-6-induced ferroptosis by suppressing IL-6R expression, indicating that the IL-6/miR-10a-5p/IL-6R axis is a potential target for IVDD treatment.
Chondrocytes are the predominant cell type in ECP and articular cartilage. Thus, studies reporting the mechanisms of articular cartilage degeneration caused by ferroptosis in OA are inspiring and learnable for the intervention of IVDD. Jing et al. (136) indicated that iron overload in chondrocytes induced by pro-inflammatory cytokines contributes to oxidative stress and mitochondrial dysfunction through the upregulation of TRF1 and downregulation of FPN. In a study by Yao et al. (137), lipid ROS and ferroptosis in chondrocytes were induced by IL-1β and ferric ammonium citrate (FAC), but they were attenuated by Ferrostatin-1 that activated the NRF2 antioxidant system. The overexpression of NRF2 upregulated the level of GPX4 expression and ameliorated ferroptosis.
Under normal circumstances, the components of the ECM in the IVD are continuously updated through anabolism and catabolism, and the cells in the IVD associated with ECM form a coordinated functional system (138). However, the imbalance of anabolic and catabolic activities might result in ECM degradation, which is a pathological characteristic of IVDD (139). ECM metabolism is modulated by iron overload and ferroptosis.
The ECM of NP primarily consists of collagen II, proteoglycan, and chondroitin sulfate (140). In NPCs, the levels of collagen II, proteoglycan, matrix metalloproteinases (MMPs, particularly MMP13), disintegrin, and metalloproteinase with thrombospondin motifs (ADAMTSs, particularly ADAMTS4 and ADAMTS5) can reflect the degree of ECM degradation during the progression of IVDD (141, 142). The overexpression of circ_0072464 can promote the levels of collagen II and proteoglycan and reduce the levels of MMP13 and ADAMTS5 by sponging miR-431, upregulating NRF2, and suppressing ferroptosis (116). Meanwhile, the overexpression of ATF3 in NPCs not only aggravates TBHP-induced ferroptosis, apoptosis, and ROS production by suppressing SLC7A11 and superoxide dismutase 2, but also enhances ECM degradation by reducing the levels of proteoglycan and collagen II (114).
In endplate chondrocytes, the iron overload induced by FAC treatment enhanced the expression of MMP3 and MMP13 and reduced the expression of collagen II, thereby accelerating the degeneration of CEP and ECM (115). The study conducted by Camacho et al. (143) also demonstrated that iron overload was involved in chondrocyte-mediated ECM degradation. Meanwhile, erastin, an inducer of ferroptosis, reduces the expression of collagen II and increases the expression of MMP13 in chondrocytes (137). Furthermore, the inflammatory factor IL-1β accelerated iron uptake in chondrocytes, which was then promoted after co-treatment with FAC, because IL-1β can promote the expression of TFRC and DMT1 but downregulate the expression of FPN1, thereby aggravating iron accumulation in chondrocytes (136). Finally, co-treatment with IL-1β and FAC upregulated the expression of ECM-degrading enzymes, including MMPs and ADAMTS5.
Ferroptosis, characterized by iron-dependent lipid peroxidation and the accumulation of ROS within the IVD, is implicated in the pathogenesis of IVDD. Thus, ferroptosis opens a new therapeutic target for the intervention of IVDD with regard to the regulation of iron metabolism, chelation of iron, and antioxidants (113). Moreover, the key components in the signaling pathways are essential regulators of ferroptosis inhibition.
Classical iron chelators such as DFO, DFP, and DFX have been used for the clinical treatment of iron overload in thalassemia major (144). However, there is no clinical evidence of iron chelation therapy for the treatment of IVDD. Nevertheless, iron chelators have shown promising results in the inhibition of ferroptosis in vivo and in vitro in IVDD models. In addition, antioxidants, including Ferrostatin-1 and N-acetyl-cysteine (NAC), can exert protective effects against iron-induced abnormalities in IVDD. DFO and Fer‐1 reversed the decreased expression of FTH and GPX4 and the upward levels of autophagy and ferritinophagy induced by TBHP treatment in NPCs and AFCs (71, 72). In the tissue of CEP, the administration of DFO, NAC, and Ferrostatin-1 substantially inhibited iron overload-induced IVDD by alleviating endplate calcification and IVD collapse in a mouse model (115). Meanwhile, the FAC-induced ECM degradation and the decrease of mitochondrial membrane potential can be reversed by DFO or NAC (136). Furthermore, DFO partially reduced the inhibition of IL-6 on miR10a-5p in cartilage cell ferroptosis through the promotion of GPX4 and FPN1 and suppression of DMT1 expression (45). However, the long-term use of iron chelators may lead to iron deficiency in cells, which is detrimental to cellular metabolism. Thus, the safe application of iron chelators needs further investigation.
EVs, exosomes, and ncRNAs are involved in the regulation of gene expression related to ferroptosis, which has emerged as a potential therapeutic strategy for IVDD (116, 145, 146). The upregulation of miR-10a-5p inhibited IL-6R expression, thereby partially reducing IL-6-induced ferroptosis in chondrocytes (45). In addition, circ_0072464 shuttled by BMSC-secreted EVs suppresses ferroptosis in NPCs through the upregulation of miR-431-mediated NRF2 (116). At present, considerable research is needed to identify new ncRNAs and related mechanisms for the treatment of IVDD. Furthermore, the exact route of administration, safe dosing, and related dose toxicity of EVs, exosomes, and ncRNAs remain major problems in the application of regenerative medicine in clinical practice.
In addition, previous studies have demonstrated that hinokitiol ameliorates the activation of protein kinase B and mitogen-activated protein kinase to inhibit platelet activation and alleviate ferroptosis-related neurotoxicity through iron chelation and regulation of the NRF2 pathway (147, 148). Recently, Lu et al. (71) indicated that hinokitiol alleviates IVDD by upregulating MTF1, restoring FPN, and suppressing the JNK pathway, thereby attenuating TBHP-induced NPC ferroptosis. Furthermore, folic acid, a coenzyme in the methionine cycle, has been regarded as another ferroptosis-related therapeutic drug for IVDD caused by Hcy through the downregulation of GPX4 methylation and oxidative stress, thereby rescuing ferroptosis-induced NPC degeneration (44). In addition, other antioxidants and drugs, such as vitamin E, vitamin K, and curcumin, have exerted protective effects on ferroptosis (149–152). Moreover, the essential regulators of ferroptosis, such as ferritin, FPN1, NRF2, GPX4, GSH, HO-1, and TFRC, can be selected as the regulation targets for the treatment of IVDD. However, studies concerning the therapeutic effects of new bioactive compounds on ferroptosis-induced IVDD are rare and worthy of further studies.
The therapeutic efficacy of regulating iron homeostasis and ferroptosis to alleviate osteoporosis and OA was regarded as a potential option and reference for the treatment of IVDD. Icariin, the main active ingredient of Herba Epimedii, has antioxidant and antiosteoporosis functions by preventing iron overload-induced bone loss and regulating iron accumulation in vitro and in vivo (153). Icariin also attenuated IL-1β-induced degeneration of ECM and ROS in human OA cartilage via the activation of the Nrf2/ARE signaling pathway (154). Resveratrol, a member of the stilbene family of phenolic compounds, can reverse iron overload-induced bone loss by upregulating the levels of FOXO1 in osteoporotic mice (155). In addition, Tian et al. (156) demonstrated that NAC exerted protective effects against iron-mediated mitochondrial dysfunction and protected osteoblasts from iron overload-induced apoptosis. Moreover, melatonin (N-acetyl-5-methoxytryptamine), an effective endogenous antioxidant, can suppress high-glucose-induced ferroptosis by activating the Nrf2/HO-1 signaling pathway to improve bone microstructure in individuals with type 2 diabetes and osteoporosis (81). Furthermore, the upregulation of mitochondrial ferritin reduced osteoblastic ferroptosis under a high-glucose environment, whereas the deficiency of mitochondrial ferritin induced mitophagy via the ROS/PINK1/Parkin pathway in individuals with type 2 diabetes and osteoporosis (157). Therefore, mitochondrial ferritin might be another potential target for the treatment of type 2 diabetes and osteoporosis. Moreover, the endothelial cell‐secreted exosomes antagonized glucocorticoid‐induced osteoporosis in vitro and in vivo via the suppression of ferritinophagy‐dependent ferroptosis (158). Lu et al. (159) also indicated that EVs from endothelial progenitor cells suppressed the ferroptotic pathway of osteoblasts by restoring levels of GPX4 and System XC¯. Collectively, the pathophysiological progression of osteoporosis and OA is associated with iron metabolism disorder, ROS, and lipid peroxidation, which might provide potential therapeutic strategies for IVDD.
As a highly disabling disease, IVDD has attracted increasing attention worldwide. Growing evidence has shown that ferroptosis is involved in the pathophysiological processes of IVDD; thus, regulation of ferroptosis has become a new therapeutic target for IVDD. In this review, we summarized the pathogenesis and mechanisms of ferroptosis, the relationship between ferroptosis and IVDD, and the choice of IVDD treatment by inhibiting ferroptosis. Recent studies have demonstrated that ferroptosis is mainly regulated by SLC7A11/GSH/GPX4, FSP1/CoQH2/NAD(P)H, and GCH1/BH4/DHFR pathways. Ferroptosis is accompanied by metabolic imbalances of lipids, iron, amino acids, and glucose and is modulated by transcriptional and epigenetic regulation. The interaction and crosstalk between ferroptosis and IVD components in terms of NP, AF, CEP, and ECM provide remarkable insights into the prevention and treatment of IVDD. However, studies on ferroptosis in IVDD are still at a relatively early stage, and simulations of ferroptosis in IVDD are mainly induced by oxidative stress and inflammation. Other factors, such as hypoxia, acidic microenvironments, and compression, are needed to confirm the universality of ferroptosis in IVDD. Therefore, further research is required to investigate the specific mechanisms, molecular targets, and associated signaling pathways of ferroptosis to develop further understanding and effective options for intervention in IVDD.
L-PZ, R-JZ and C-LS conceptualized the review. L-PZ and R-JZ drafted the manuscript. L-PZ, R-JZ, C-YJ, LK, Z-GZ, H-QZ, J-QW, BZ, and C-LS revised the manuscript. All authors have read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (No. 81772408).
Figures were created by Figdraw (www.figdraw.com). We would like to thank Figdraw for its help in creating the figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Yang S, Zhang F, Ma J, Ding W. Intervertebral disc ageing and degeneration: The antiapoptotic effect of oestrogen. Ageing Res Rev (2020) 57:100978. doi: 10.1016/j.arr.2019.100978
2. Millecamps M, Stone LS. Delayed onset of persistent discogenic axial and radiating pain after a single-level lumbar intervertebral disc injury in mice. Pain (2018) 159(9):1843–55. doi: 10.1097/j.pain.0000000000001284
3. Xiong Y, Chen L, Lin Z, Hu Y, Panayi AC, Zhou W, et al. The regulatory role of ferroptosis in bone homeostasis. Stem Cells Int (2022) 2022:3568597. doi: 10.1155/2022/3568597
4. Hassan W, Noreen H, Khalil S, Hussain A, Rehman S, Sajjad S, et al. Ethanolic extract of nigella sativa protects Fe(II) induced lipid peroxidation in rat’s brain, kidney and liver homogenates. Pak J Pharm Sci (2016) 29(1):231–7.
5. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
6. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol (2021) 18(5):280–96. doi: 10.1038/s41571-020-00462-0
7. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
8. Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, et al. PKCbetaII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol (2022) 24(1):88–98. doi: 10.1038/s41556-021-00818-3
9. Liu P, Wang W, Li Z, Li Y, Yu X, Tu J, et al. Ferroptosis: A new regulatory mechanism in osteoporosis. Oxid Med Cell Longev (2022) 2022:2634431. doi: 10.1155/2022/2634431
10. Saboor M, Zehra A, Hamali HA, Mobarki AA. Revisiting iron metabolism, iron homeostasis and iron deficiency anemia. Clin Lab (2021) 67(3). doi: 10.7754/Clin.Lab.2020.200742
11. Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, et al. Identification of an intestinal heme transporter. Cell (2005) 122(5):789–801. doi: 10.1016/j.cell.2005.06.025
12. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell (2017) 168(3):344–61. doi: 10.1016/j.cell.2016.12.034
13. Canonne-Hergaux F, Donovan A, Delaby C, Wang HJ, Gros P. Comparative studies of duodenal and macrophage ferroportin proteins. Am J Physiol Gastrointest Liver Physiol (2006) 290(1):G156–63. doi: 10.1152/ajpgi.00227.2005
14. Chaston T, Chung B, Mascarenhas M, Marks J, Patel B, Srai SK, et al. Evidence for differential effects of hepcidin in macrophages and intestinal epithelial cells. Gut (2008) 57(3):374–82. doi: 10.1136/gut.2007.131722
15. Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta (2012) 1823(9):1434–43. doi: 10.1016/j.bbamcr.2012.01.014
16. Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutr (2017) 106(Suppl 6):1559s–66s. doi: 10.3945/ajcn.117.155804
17. Montalbetti N, Simonin A, Simonin C, Awale M, Reymond JL, Hediger MA. Discovery and characterization of a novel non-competitive inhibitor of the divalent metal transporter DMT1/SLC11A2. Biochem Pharmacol (2015) 96(3):216–24. doi: 10.1016/j.bcp.2015.05.002
18. Lin L, Wang S, Deng H, Yang W, Rao L, Tian R, et al. Endogenous labile iron pool-mediated free radical generation for cancer chemodynamic therapy. J Am Chem Soc (2020) 142(36):15320–30. doi: 10.1021/jacs.0c05604
19. Lymboussaki A, Pignatti E, Montosi G, Garuti C, Haile DJ, Pietrangelo A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J Hepatol (2003) 39(5):710–5. doi: 10.1016/s0168-8278(03)00408-2
20. Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol (2006) 2(8):406–14. doi: 10.1038/nchembio807
21. Menon AV, Liu J, Tsai HP, Zeng L, Yang S, Asnani A, et al. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood (2022) 139(6):936–41. doi: 10.1182/blood.2020008455
22. Adams PC, Barton JC. Haemochromatosis. Lancet (2007) 370(9602):1855–60. doi: 10.1016/s0140-6736(07)61782-6
23. Rostoker G. The changing landscape of iron overload disorders at the beginning of the 21st century. Presse Med (2017) 46(12 Pt 2):e269–71. doi: 10.1016/j.lpm.2017.10.011
24. Taher AT, Saliba AN. Iron overload in thalassemia: different organs at different rates. Hematol Am Soc Hematol Educ Program (2017) 2017(1):265–71. doi: 10.1182/asheducation-2017.1.265
25. Gattermann N, Muckenthaler MU, Kulozik AE, Metzgeroth G, Hastka J. The evaluation of iron deficiency and iron overload. Dtsch Arztebl Int (2021) 118(49):847–56. doi: 10.3238/arztebl.m2021.0290
26. Grandchamp B, Hetet G, Kannengiesser C, Oudin C, Beaumont C, Rodrigues-Ferreira S, et al. A novel type of congenital hypochromic anemia associated with a nonsense mutation in the STEAP3/TSAP6 gene. Blood (2011) 118(25):6660–6. doi: 10.1182/blood-2011-01-329011
27. Le Lan C, Mosser A, Ropert M, Detivaud L, Loustaud-Ratti V, Vital-Durand D, et al. Sex and acquired cofactors determine phenotypes of ferroportin disease. Gastroenterology (2011) 140(4):1199–1207.e1-2. doi: 10.1053/j.gastro.2010.12.049
28. Bardou-Jacquet E, Island ML, Jouanolle AM, Détivaud L, Fatih N, Ropert M, et al. A novel N491S mutation in the human SLC11A2 gene impairs protein trafficking and in association with the G212V mutation leads to microcytic anemia and liver iron overload. Blood Cells Mol Dis (2011) 47(4):243–8. doi: 10.1016/j.bcmd.2011.07.004
29. Li J, Wang S, Duan J, Le P, Li C, Ding Y, et al. The protective mechanism of resveratrol against hepatic injury induced by iron overload in mice. Toxicol Appl Pharmacol (2021) 424:115596. doi: 10.1016/j.taap.2021.115596
30. Haidar R, Mhaidli H, Musallam KM, Taher AT. The spine in β-thalassemia syndromes. Spine (Phila Pa 1976) (2012) 37(4):334–9. doi: 10.1097/BRS.0b013e31821bd095
31. Shan L, Xu X, Zhang J, Cai P, Gao H, Lu Y, et al. Increased hemoglobin and heme in MALDI-TOF MS analysis induce ferroptosis and promote degeneration of herniated human nucleus pulposus. Mol Med (2021) 27(1):103. doi: 10.1186/s10020-021-00368-2
32. Chifman J, Laubenbacher R, Torti SV. A systems biology approach to iron metabolism. Adv Exp Med Biol (2014) 844:201–25. doi: 10.1007/978-1-4939-2095-2_10
33. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife (2014) 3:e02523. doi: 10.7554/eLife.02523
34. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: Discoveries, rediscoveries, and open issues. Antioxid Redox Signal (2018) 29(1):61–74. doi: 10.1089/ars.2017.7115
35. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy (2021) 17(9):2054–81. doi: 10.1080/15548627.2020.1810918
36. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
37. Ursini F, Maiorino M, Gregolin C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta (1985) 839(1):62–70. doi: 10.1016/0304-4165(85)90182-5
38. Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med (2014) 76:208–26. doi: 10.1016/j.freeradbiomed.2014.07.046
39. Fan R, Sui J, Dong X, Jing B, Gao Z. Wedelolactone alleviates acute pancreatitis and associated lung injury via GPX4 mediated suppression of pyroptosis and ferroptosis. Free Radic Biol Med (2021) 173:29–40. doi: 10.1016/j.freeradbiomed.2021.07.009
40. Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao X, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol (2021) 14(1):19. doi: 10.1186/s13045-020-01016-8
41. Basit F, van Oppen LM, Schöckel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis (2017) 8(3):e2716. doi: 10.1038/cddis.2017.133
42. Liu Y, Wang Y, Liu J, Kang R, Tang D. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther (2021) 28(1-2):55–63. doi: 10.1038/s41417-020-0182-y
43. Han D, Jiang L, Gu X, Huang S, Pang J, Wu Y, et al. SIRT3 deficiency is resistant to autophagy-dependent ferroptosis by inhibiting the AMPK/mTOR pathway and promoting GPX4 levels. J Cell Physiol (2020) 235(11):8839–51. doi: 10.1002/jcp.29727
44. Zhang X, Huang Z, Xie Z, Chen Y, Zheng Z, Wei X, et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic Biol Med (2020) 160:552–65. doi: 10.1016/j.freeradbiomed.2020.08.029
45. Bin S, Xin L, Lin Z, Jinhua Z, Rui G, Xiang Z. Targeting miR-10a-5p/IL-6R axis for reducing IL-6-induced cartilage cell ferroptosis. Exp Mol Pathol (2021) 118:104570. doi: 10.1016/j.yexmp.2020.104570
46. Zhang Z, Tang J, Song J, Xie M, Liu Y, Dong Z, et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic Biol Med (2022) 181:130–42. doi: 10.1016/j.freeradbiomed.2022.01.020
47. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun (2019) 10(1):1617. doi: 10.1038/s41467-019-09277-9
48. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
49. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
50. Marshall KR, Gong M, Wodke L, Lamb JH, Jones DJ, Farmer PB, et al. The human apoptosis-inducing protein AMID is an oxidoreductase with a modified flavin cofactor and DNA binding activity. J Biol Chem (2005) 280(35):30735–40. doi: 10.1074/jbc.M414018200
51. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol (2016) 12(7):497–503. doi: 10.1038/nchembio.2079
52. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, et al. GTP cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci (2020) 6(1):41–53. doi: 10.1021/acscentsci.9b01063
53. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol (2020) 16(12):1351–60. doi: 10.1038/s41589-020-0613-y
54. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
55. Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun (2016) 478(3):1338–43. doi: 10.1016/j.bbrc.2016.08.124
56. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol (2015) 10(7):1604–9. doi: 10.1021/acschembio.5b00245
57. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
58. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol (2020) 16(3):302–9. doi: 10.1038/s41589-020-0472-6
59. Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res (2020) 18(1):79–90. doi: 10.1158/1541-7786.Mcr-19-0691
60. Li Y, Wang J, Chen S, Wu P, Xu S, Wang C, et al. miR-137 boosts the neuroprotective effect of endothelial progenitor cell-derived exosomes in oxyhemoglobin-treated SH-SY5Y cells partially via COX2/PGE2 pathway. Stem Cell Res Ther (2020) 11(1):330. doi: 10.1186/s13287-020-01836-y
61. Xu Y, Liu Y, Li K, Yuan D, Yang S, Zhou L, et al. COX-2/PGE2 pathway inhibits the ferroptosis induced by cerebral ischemia reperfusion. Mol Neurobiol (2022) 59(3):1619–31. doi: 10.1007/s12035-021-02706-1
62. Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem (2005) 12(10):1161–208. doi: 10.2174/0929867053764635
63. Benedetti A, Comporti M, Esterbauer H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim Biophys Acta (1980) 620(2):281–96. doi: 10.1016/0005-2760(80)90209-x
64. Cheeseman KH, Beavis A, Esterbauer H. Hydroxyl-radical-induced iron-catalysed degradation of 2-deoxyribose. Quantit deter malondialdehyde. Biochem J (1988) 252(3):649–53. doi: 10.1042/bj2520649
65. Esterbauer H, Zollner H. Methods for determination of aldehydic lipid peroxidation products. Free Radic Biol Med (1989) 7(2):197–203. doi: 10.1016/0891-5849(89)90015-4
66. Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev (2014) 2014:360438. doi: 10.1155/2014/360438
67. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell (2015) 59(2):298–308. doi: 10.1016/j.molcel.2015.06.011
68. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis (2016) 7(7):e2307. doi: 10.1038/cddis.2016.208
69. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol (2008) 15(3):234–45. doi: 10.1016/j.chembiol.2008.02.010
70. Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci (2018) 22(12):3826–36. doi: 10.26355/eurrev_201806_15267
71. Lu S, Song Y, Luo R, Li S, Li G, Wang K, et al. Ferroportin-dependent iron homeostasis protects against oxidative stress-induced nucleus pulposus cell ferroptosis and ameliorates intervertebral disc degeneration in vivo. Oxid Med Cell Longev (2021) 2021:6670497. doi: 10.1155/2021/6670497
72. Yang RZ, Xu WN, Zheng HL, Zheng XF, Li B, Jiang LS, et al. Involvement of oxidative stress-induced annulus fibrosus cell and nucleus pulposus cell ferroptosis in intervertebral disc degeneration pathogenesis. J Cell Physiol (2021) 236(4):2725–39. doi: 10.1002/jcp.30039
73. McBean GJ. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids (2012) 42(1):199–205. doi: 10.1007/s00726-011-0864-8
74. Liu N, Lin X, Huang C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br J Cancer (2020) 122(2):279–92. doi: 10.1038/s41416-019-0660-x
75. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ (2016) 23(2):270–8. doi: 10.1038/cdd.2015.93
76. Cassago A, Ferreira AP, Ferreira IM, Fornezari C, Gomes ER, Greene KS, et al. Mitochondrial localization and structure-based phosphate activation mechanism of glutaminase c with implications for cancer metabolism. Proc Natl Acad Sci USA (2012) 109(4):1092–7. doi: 10.1073/pnas.1112495109
77. Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev (2016) 30(8):918–30. doi: 10.1101/gad.275891.115
78. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell (2019) 73(2):354–63.e3. doi: 10.1016/j.molcel.2018.10.042
79. Neumann D, Viollet B. AMP-activated protein kinase signalling. Int J Mol Sci (2019) 20(3):766. doi: 10.3390/ijms20030766
80. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol (2020) 22(2):225–34. doi: 10.1038/s41556-020-0461-8
81. Ma H, Wang X, Zhang W, Li H, Zhao W, Sun J, et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the Nrf2/HO-1 signaling pathway in type 2 diabetic osteoporosis. Oxid Med Cell Longev (2020) 2020:9067610. doi: 10.1155/2020/9067610
82. Dai C, Chen X, Li J, Comish P, Kang R, Tang D. Transcription factors in ferroptotic cell death. Cancer Gene Ther (2020) 27(9):645–56. doi: 10.1038/s41417-020-0170-2
83. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer (2014) 14(5):359–70. doi: 10.1038/nrc3711
84. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol (2013) 53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320
85. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system xc(). Cell Death Differ (2020) 27(2):662–75. doi: 10.1038/s41418-019-0380-z
86. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature (2019) 572(7769):402–6. doi: 10.1038/s41586-019-1426-6
87. Wang X, Sun D, Tai J, Chen S, Yu M, Ren D, et al. TFAP2C promotes stemness and chemotherapeutic resistance in colorectal cancer via inactivating hippo signaling pathway. J Exp Clin Cancer Res (2018) 37(1):27. doi: 10.1186/s13046-018-0683-9
88. Liu J, Yang M, Kang R, Klionsky DJ, Tang D. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy (2019) 15(11):2033–5. doi: 10.1080/15548627.2019.1659623
89. Yang M, Chen P, Liu J, Zhu S, Kroemer G, Klionsky DJ, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv (2019) 5(7):eaaw2238. doi: 10.1126/sciadv.aaw2238
90. Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ (2019) 26(11):2329–43. doi: 10.1038/s41418-019-0304-y
91. Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu X, et al. EGLN1/c-myc induced lymphoid-specific helicase inhibits ferroptosis through lipid metabolic gene expression changes. Theranostics (2017) 7(13):3293–305. doi: 10.7150/thno.19988
92. Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res (2018) 78(13):3484–96. doi: 10.1158/0008-5472.Can-17-3454
93. Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis (2019) 10(5):331. doi: 10.1038/s41419-019-1564-7
94. Wang Y, Zhao Y, Wang H, Zhang C, Wang M, Yang Y, et al. Histone demethylase KDM3B protects against ferroptosis by upregulating SLC7A11. FEBS Open Bio (2020) 10(4):637–43. doi: 10.1002/2211-5463.12823
95. Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol (2020) 16(5):497–506. doi: 10.1038/s41589-020-0501-5
96. Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol (2018) 14(5):507–15. doi: 10.1038/s41589-018-0031-6
97. Wenz C, Faust D, Linz B, Turmann C, Nikolova T, Bertin J, et al. T-BuOOH induces ferroptosis in human and murine cell lines. Arch Toxicol (2018) 92(2):759–75. doi: 10.1007/s00204-017-2066-y
98. Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol (2018) 314(3):H659–h668. doi: 10.1152/ajpheart.00452.2017
99. Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight (2017) 2(7):e90777. doi: 10.1172/jci.insight.90777
100. Li L, Wang K, Jia R, Xie J, Ma L, Hao Z, et al. Ferroportin-dependent ferroptosis induced by ellagic acid retards liver fibrosis by impairing the SNARE complexes formation. Redox Biol (2022) 56:102435. doi: 10.1016/j.redox.2022.102435
101. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
102. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. Tang: The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep (2017) 20(7):1692–704. doi: 10.1016/j.celrep.2017.07.055
103. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA (2019) 116(8):2996–3005. doi: 10.1073/pnas.1819728116
104. Zhou W, Shi Y, Wang H, Chen L, Yu C, Zhang X, et al. Exercise-induced FNDC5/irisin protects nucleus pulposus cells against senescence and apoptosis by activating autophagy. Exp Mol Med (2022) 54(7):1038–48. doi: 10.1038/s12276-022-00811-2
105. Bahar ME, Hwang JS, Ahmed M, Lai TH, Pham TM, Elashkar O, et al. Targeting autophagy for developing new therapeutic strategy in intervertebral disc degeneration. Antioxid (Basel) (2022) 11(8):1571. doi: 10.3390/antiox11081571
106. Li F, Xie W, Chen Z, Zhou Z, Wang Z, Xiao J, et al. Neuropeptide y and receptors are associated with the pyroptosis of nucleus pulposus in aging and degenerative intervertebral discs of rats. Neuropeptides (2022) 96:102284. doi: 10.1016/j.npep.2022.102284
107. Cao C, Chen S, Song Z, Liu Z, Zhang M, Ma Z, et al. Inflammatory stimulation mediates nucleus pulposus cell necroptosis through mitochondrial function disfunction and oxidative stress pathway. Front Biosci (Landmark Ed) (2022) 27(4):111. doi: 10.31083/j.fbl2704111
108. Ma J, Li X, Fan Y, Yang D, Gu Q, Li D, et al. miR-494-3p promotes erastin-induced ferroptosis by targeting REST to activate the interplay between SP1 and ACSL4 in parkinson’s disease. Oxid Med Cell Longev (2022) 2022:7671324. doi: 10.1155/2022/7671324
109. He DL, Fan YG, Wang ZY. Energy crisis links to autophagy and ferroptosis in alzheimer’s disease: current evidence and future avenues. Curr Neuropharmacol (2022). doi: 10.2174/1570159x20666220817140737
110. Fan X, Zhang X, Liu LC, Zhang S, Pelger CB, Lughmani HY, et al. Hemopexin accumulates in kidneys and worsens acute kidney injury by causing hemoglobin deposition and exacerbation of iron toxicity in proximal tubules. Kidney Int (2022). doi: 10.1016/j.kint.2022.07.024
111. Li M, Xin S, Gu R, Zheng L, Hu J, Zhang R, et al. Novel diagnostic biomarkers related to oxidative stress and macrophage ferroptosis in atherosclerosis. Oxid Med Cell Longev (2022) 2022:8917947. doi: 10.1155/2022/8917947
112. Li M, Yang N, Hao L, Zhou W, Li L, Liu L, et al. Melatonin inhibits the ferroptosis pathway in rat bone marrow mesenchymal stem cells by activating the PI3K/AKT/mTOR signaling axis to attenuate steroid-induced osteoporosis. Oxid Med Cell Longev (2022) 2022:8223737. doi: 10.1155/2022/8223737
113. Sun K, Guo Z, Hou L, Xu J, Du T, Xu T, et al. Iron homeostasis in arthropathies: From pathogenesis to therapeutic potential. Ageing Res Rev (2021) 72:101481. doi: 10.1016/j.arr.2021.101481
114. Li Y, Pan D, Wang X, Huo Z, Wu X, Li J, et al. Silencing ATF3 might delay TBHP-induced intervertebral disc degeneration by repressing NPC ferroptosis, apoptosis, and ECM degradation. Oxid Med Cell Longev (2022) 2022:4235126. doi: 10.1155/2022/4235126
115. Wang W, Jing X, Du T, Ren J, Liu X, Chen F, et al. Iron overload promotes intervertebral disc degeneration via inducing oxidative stress and ferroptosis in endplate chondrocytes. Free Radic Biol Med (2022) 190:234–46. doi: 10.1016/j.freeradbiomed.2022.08.018
116. Yu X, Xu H, Liu Q, Wang Y, Wang S, Lu R, et al. circ_0072464 shuttled by bone mesenchymal stem cell-secreted extracellular vesicles inhibits nucleus pulposus cell ferroptosis to relieve intervertebral disc degeneration. Oxid Med Cell Longev (2022) 2022:2948090. doi: 10.1155/2022/2948090
117. Mizrahi O, Sheyn D, Tawackoli W, Ben-David S, Su S, Li N, et al. Nucleus pulposus degeneration alters properties of resident progenitor cells. Spine J (2013) 13(7):803–14. doi: 10.1016/j.spinee.2013.02.065
118. Peng B, Chen J, Kuang Z, Li D, Pang X, Zhang X. Expression and role of connective tissue growth factor in painful disc fibrosis and degeneration. Spine (Phila Pa 1976) (2009) 34(5):E178–82. doi: 10.1097/BRS.0b013e3181908ab3
119. Zhang Y, Han S, Kong M, Tu Q, Zhang L, Ma X. Single-cell RNA-seq analysis identifies unique chondrocyte subsets and reveals involvement of ferroptosis in human intervertebral disc degeneration. Osteoarthritis Cartilage (2021) 29(9):1324–34. doi: 10.1016/j.joca.2021.06.010
120. Masaldan S, Clatworthy SAS, Gamell C, Meggyesy PM, Rigopoulos AT, Haupt S, et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol (2018) 14:100–15. doi: 10.1016/j.redox.2017.08.015
121. Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med (2019) 133:162–8. doi: 10.1016/j.freeradbiomed.2018.05.074
122. Guo Y, Li C, Shen B, Chen X, Hu T, Wu D. Is intervertebral disc degeneration associated with reduction in serum ferritin? Eur Spine J (2022). doi: 10.1007/s00586-022-07361-1
123. Li Y, Zhao J, Yu S, Wang Z, He X, Su Y, et al. Extracellular vesicles long RNA sequencing reveals abundant mRNA, circRNA, and lncRNA in human blood as potential biomarkers for cancer diagnosis. Clin Chem (2019) 65(6):798–808. doi: 10.1373/clinchem.2018.301291
124. DiStefano TJ, Vaso K, Danias G, Chionuma HN, Weiser JR, Iatridis JC. Extracellular vesicles as an emerging treatment option for intervertebral disc degeneration: Therapeutic potential, translational pathways, and regulatory considerations. Adv Healthc Mater (2022) 11(5):e2100596. doi: 10.1002/adhm.202100596
125. Song J, Chen ZH, Zheng CJ, Song KH, Xu GY, Xu S, et al. Exosome-transported circRNA_0000253 competitively adsorbs MicroRNA-141-5p and increases IDD. Mol Ther Nucleic Acids (2020) 21:1087–99. doi: 10.1016/j.omtn.2020.07.039
126. Du WW, Zhang C, Yang W, Yong T, Awan FM, Yang BB. Identifying and characterizing circRNA-protein interaction. Theranostics (2017) 7(17):4183–91. doi: 10.7150/thno.21299
127. Shen W, Gao C, Cueto R, Liu L, Fu H, Shao Y, et al. Homocysteine-methionine cycle is a metabolic sensor system controlling methylation-regulated pathological signaling. Redox Biol (2020) 28:101322. doi: 10.1016/j.redox.2019.101322
128. Sonkar SK, Kumar S, Singh NK, Tandon R. Hyperhomocysteinemia induced locked-in syndrome in a young adult due to folic acid deficiency. Nutr Neurosci (2021) 24(10):781–3. doi: 10.1080/1028415x.2019.1681064
129. Komorniak N, Szczuko M, Kowalewski B, Stachowska E. Nutritional deficiencies, bariatric surgery, and serum homocysteine level: Review of current literature. Obes Surg (2019) 29(11):3735–42. doi: 10.1007/s11695-019-04100-2
130. van den Akker GGH, Koenders MI, van de Loo FAJ, van Lent P, Blaney Davidson E, van der Kraan PM. Transcriptional profiling distinguishes inner and outer annulus fibrosus from nucleus pulposus in the bovine intervertebral disc. Eur Spine J (2017) 26(8):2053–62. doi: 10.1007/s00586-017-5150-3
131. Raj PP. Intervertebral disc: anatomy-physiology-pathophysiology-treatment. Pain Pract (2008) 8(1):18–44. doi: 10.1111/j.1533-2500.2007.00171.x
132. Ashinsky BG, Bonnevie ED, Mandalapu SA, Pickup S, Wang C, Han L, et al. Intervertebral disc degeneration is associated with aberrant endplate remodeling and reduced small molecule transport. J Bone Miner Res (2020) 35(8):1572–81. doi: 10.1002/jbmr.4009
133. van der Werf M, Lezuo P, Maissen O, van Donkelaar CC, Ito K. Inhibition of vertebral endplate perfusion results in decreased intervertebral disc intranuclear diffusive transport. J Anat (2007) 211(6):769–74. doi: 10.1111/j.1469-7580.2007.00816.x
134. Grunhagen T, Wilde G, Soukane DM, Shirazi-Adl SA, Urban JP. Nutrient supply and intervertebral disc metabolism. J Bone Joint Surg Am (2006) 88 Suppl:2, 30–5. doi: 10.2106/jbjs.E.01290
135. Wong J, Sampson SL, Bell-Briones H, Ouyang A, Lazar AA, Lotz JC, et al. Nutrient supply and nucleus pulposus cell function: effects of the transport properties of the cartilage endplate and potential implications for intradiscal biologic therapy. Osteoarthritis Cartilage (2019) 27(6):956–64. doi: 10.1016/j.joca.2019.01.013
136. Jing X, Du T, Li T, Yang X, Wang G, Liu X, et al. The detrimental effect of iron on OA chondrocytes: Importance of pro-inflammatory cytokines induced iron influx and oxidative stress. J Cell Mol Med (2021) 25(12):5671–80. doi: 10.1111/jcmm.16581
137. Yao X, Sun K, Yu S, Luo J, Guo J, Lin J, et al. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J Orthop Translat (2021) 27:33–43. doi: 10.1016/j.jot.2020.09.006
138. Zhang S, Liu W, Chen S, Wang B, Wang P, Hu B, et al. Extracellular matrix in intervertebral disc: basic and translational implications. Cell Tissue Res (2022), 390(1):1–22. doi: 10.1007/s00441-022-03662-5
139. Hu B, Lv X, Wei L, Wang Y, Zheng G, Yang C, et al. Sensory nerve maintains intervertebral disc extracellular matrix homeostasis Via CGRP/CHSY1 axis. Adv Sci (Weinh) (2022) e2202620. doi: 10.1002/advs.202202620
140. Francisco V, Pino J, González-Gay M, Lago F, Karppinen J, Tervonen O, et al. A new immunometabolic perspective of intervertebral disc degeneration. Nat Rev Rheumatol (2022) 18(1):47–60. doi: 10.1038/s41584-021-00713-z
141. Choi H, Tessier S, Silagi ES, Kyada R, Yousefi F, Pleshko N, et al. A novel mouse model of intervertebral disc degeneration shows altered cell fate and matrix homeostasis. Matrix Biol (2018) 70:102–22. doi: 10.1016/j.matbio.2018.03.019
142. Liu W, Xia P, Feng J, Kang L, Huang M, Wang K, et al. MicroRNA-132 upregulation promotes matrix degradation in intervertebral disc degeneration. Exp Cell Res (2017) 359(1):39–49. doi: 10.1016/j.yexcr.2017.08.011
143. Camacho A, Simao M, Ea HK, Cohen-Solal M, Richette P, Branco J, et al. Iron overload in a murine model of hereditary hemochromatosis is associated with accelerated progression of osteoarthritis under mechanical stress. Osteoarthritis Cartilage (2016) 24(3):494–502. doi: 10.1016/j.joca.2015.09.007
144. Di Maggio R, Maggio A. The new era of chelation treatments: effectiveness and safety of 10 different regimens for controlling iron overloading in thalassaemia major. Br J Haematol (2017) 178(5):676–88. doi: 10.1111/bjh.14712
145. Ran R, Liao HY, Wang ZQ, Gong CY, Zhou KS, Zhang HH. Mechanisms and functions of long noncoding RNAs in intervertebral disc degeneration. Pathol Res Pract (2022) 235:153959. doi: 10.1016/j.prp.2022.153959
146. Bhujel B, Shin HE, Choi DJ, Han I. Mesenchymal stem cell-derived exosomes and intervertebral disc regeneration: Review. Int J Mol Sci (2022) 23(13):7306. doi: 10.3390/ijms23137306
147. Lin KH, Kuo JR, Lu WJ, Chung CL, Chou DS, Huang SY, et al. Hinokitiol inhibits platelet activation ex vivo and thrombus formation in vivo. Biochem Pharmacol (2013) 85(10):1478–85. doi: 10.1016/j.bcp.2013.02.027
148. Xi J, Zhang Z, Wang Z, Wu Q, He Y, Xu Y, et al. Hinokitiol functions as a ferroptosis inhibitor to confer neuroprotection. Free Radic Biol Med (2022) 190:202–15. doi: 10.1016/j.freeradbiomed.2022.08.011
149. Wang YM, Gong FC, Qi X, Zheng YJ, Zheng XT, Chen Y, et al. Mucin 1 inhibits ferroptosis and sensitizes vitamin e to alleviate sepsis-induced acute lung injury through GSK3β/Keap1-Nrf2-GPX4 pathway. Oxid Med Cell Longev (2022) 2022:2405943. doi: 10.1155/2022/2405943
150. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature (2022) 608(7924):778–83. doi: 10.1038/s41586-022-05022-3
151. Wei Z, Shaohuan Q, Pinfang K, Chao S. Curcumin attenuates ferroptosis-induced myocardial injury in diabetic cardiomyopathy through the Nrf2 pathway. Cardiovasc Ther (2022) 2022:3159717. doi: 10.1155/2022/3159717
152. Hirata Y, Okazaki R, Sato M, Oh-Hashi K, Takemori H, Furuta K. Effect of ferroptosis inhibitors oxindole-curcumin hybrid compound and N,N-dimethylaniline derivatives on rotenone-induced oxidative stress. Eur J Pharmacol (2022) 928:175119. doi: 10.1016/j.ejphar.2022.175119
153. Jing X, Du T, Chen K, Guo J, Xiang W, Yao X, et al. Icariin protects against iron overload-induced bone loss via suppressing oxidative stress. J Cell Physiol (2019) 234(7):10123–37. doi: 10.1002/jcp.27678
154. Zuo S, Zou W, Wu RM, Yang J, Fan JN, Zhao XK, et al. Icariin alleviates IL-1β-Induced matrix degradation by activating the Nrf2/ARE pathway in human chondrocytes. Drug Des Devel Ther (2019) 13:3949–61. doi: 10.2147/dddt.S203094
155. Zhao L, Wang Y, Wang Z, Xu Z, Zhang Q, Yin M. Effects of dietary resveratrol on excess-iron-induced bone loss via antioxidative character. J Nutr Biochem (2015) 26(11):1174–82. doi: 10.1016/j.jnutbio.2015.05.009
156. Tian Q, Wu S, Dai Z, Yang J, Zheng J, Zheng Q, et al. Iron overload induced death of osteoblasts in vitro: involvement of the mitochondrial apoptotic pathway. PeerJ (2016) 4:e2611. doi: 10.7717/peerj.2611
157. Wang X, Ma H, Sun J, Zheng T, Zhao P, Li H, et al. Mitochondrial ferritin deficiency promotes osteoblastic ferroptosis Via mitophagy in type 2 diabetic osteoporosis. Biol Trace Elem Res (2022) 200(1):298–307. doi: 10.1007/s12011-021-02627-z
158. Yang RZ, Xu WN, Zheng HL, Zheng XF, Li B, Jiang LS, et al. Exosomes derived from vascular endothelial cells antagonize glucocorticoid-induced osteoporosis by inhibiting ferritinophagy with resultant limited ferroptosis of osteoblasts. J Cell Physiol (2021) 236(9):6691–705. doi: 10.1002/jcp.30331
Keywords: ferroptosis, intervertebral disc degeneration, iron, lipid peroxidation, therapeutic implication
Citation: Zhou L-P, Zhang R-J, Jia C-Y, Kang L, Zhang Z-G, Zhang H-Q, Wang J-Q, Zhang B and Shen C-L (2022) Ferroptosis: A potential target for the intervention of intervertebral disc degeneration. Front. Endocrinol. 13:1042060. doi: 10.3389/fendo.2022.1042060
Received: 12 September 2022; Accepted: 04 October 2022;
Published: 20 October 2022.
Edited by:
Baoshan Xu, Tianjin Hospital, ChinaReviewed by:
Changwing Li, Xinqiao Hospital, ChinaCopyright © 2022 Zhou, Zhang, Jia, Kang, Zhang, Zhang, Wang, Zhang and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cai-Liang Shen, c2hlbmNhaWxpYW5nQGFobXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.