 Adali Pecci
Adali Pecci María Florencia Ogara
María Florencia Ogara Rosario T. Sanz
Rosario T. Sanz Guillermo Pablo Vicent
Guillermo Pablo Vicent
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 04 November 2022
Sec. Molecular and Structural Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1037177
This article is part of the Research TopicSteroid Hormone Receptors and Cell Cycle in Breast CancerView all 7 articles
Steroid hormone receptors (SHRs) belong to a large family of ligand-activated nuclear receptors that share certain characteristics and possess others that make them unique. It was thought for many years that the specificity of hormone response lay in the ligand. Although this may be true for pure agonists, the natural ligands as progesterone, corticosterone and cortisol present a broader effect by simultaneous activation of several SHRs. Moreover, SHRs share structural and functional characteristics that range from similarities between ligand-binding pockets to recognition of specific DNA sequences. These properties are clearly evident in progesterone (PR) and glucocorticoid receptors (GR); however, the biological responses triggered by each receptor in the presence of its ligand are different, and in some cases, even opposite. Thus, what confers the specificity of response to a given receptor is a long-standing topic of discussion that has not yet been unveiled. The levels of expression of each receptor, the differential interaction with coregulators, the chromatin accessibility as well as the DNA sequence of the target regions in the genome, are reliable sources of variability in hormone action that could explain the results obtained so far. Yet, to add further complexity to this scenario, it has been described that receptors can form heterocomplexes which can either compromise or potentiate the respective hormone-activated pathways with its possible impact on the pathological condition. In the present review, we summarized the state of the art of the functional cross-talk between PR and GR in breast cancer cells and we also discussed new paradigms of specificity in hormone action.
Steroid hormones play diverse roles in the regulation of biological functions such as pregnancy, sex organ development, inflammation and immune responses, cholesterol distribution and brain function (1, 2). These effects are mediated by the members of the highly conserved Steroid Hormone receptor (SHR) sub-family that includes receptors for estrogens (ER), progestins (PR), androgens (AR), glucocorticoids (GR) and mineralocorticoids (MR) (3) (Figure 1A i). In fact, a general structural organization is common to all nuclear receptor family members, although the regulation of their quaternary structure may differ (5).
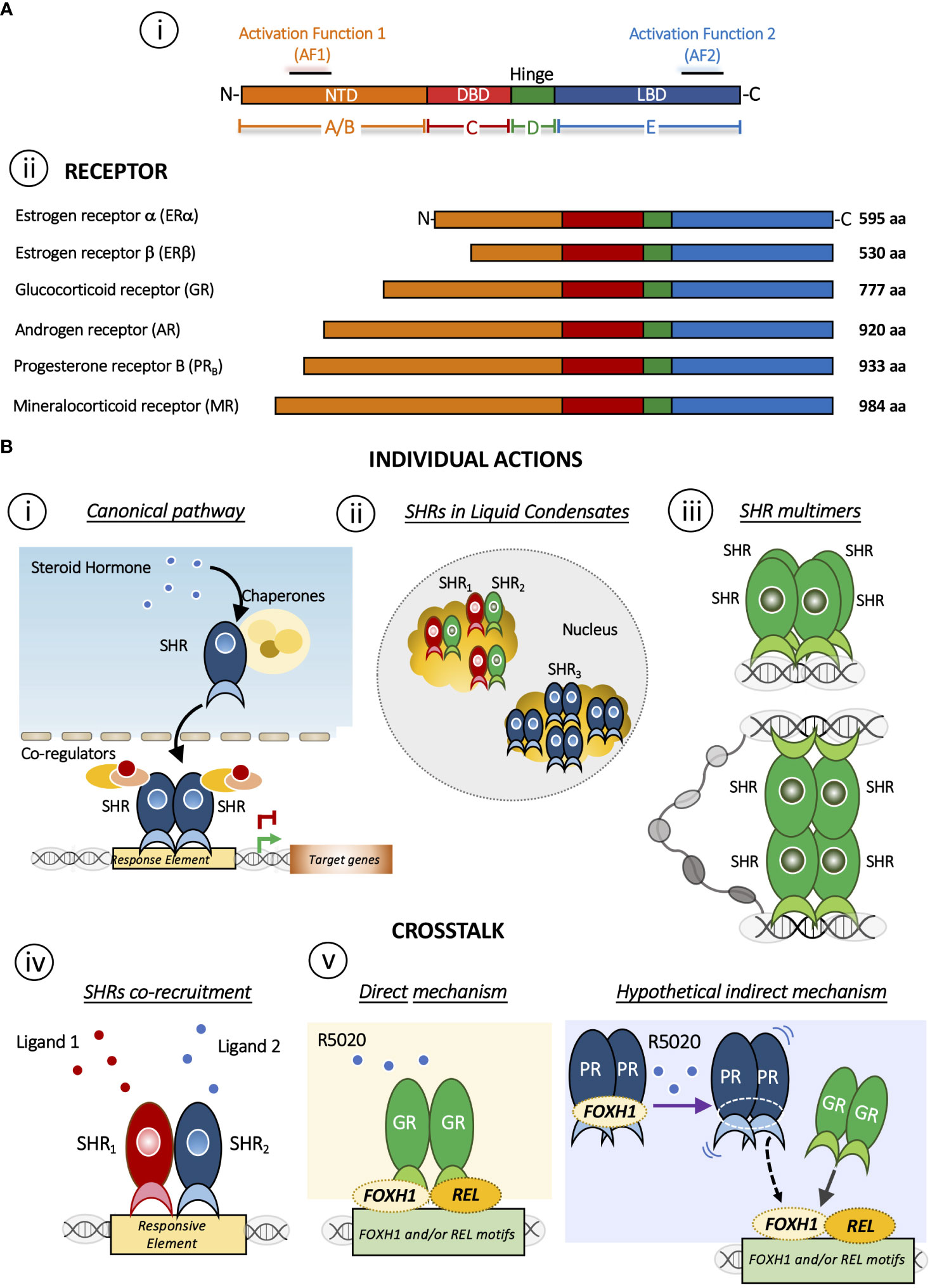
Figure 1 (A) Domain structure of SHRs. i) Basic domain structure of SHRs is composed of an unstructured N-terminal domain (NTD) that contains the Activation Function 1 (AF-1) surface, a zinc finger DNA-binding domain (DBD), a flexible hinge region, and a LBD that binds to ligands and interacts with co-regulator proteins through the Activation Function 2 (AF-2) surface. ii) Domain size and amino acid length of different members of the SHRs sub-family. The DBD and LBDs are the most conserved regions whereas the other domains are more variable in length and sequence composition. (B) Mechanisms of action of SHRs. Individual action i) The canonical pathway is shown. Steroid hormone binding to the steroid hormone receptor (SHR), often in the cytoplasm, causes the receptor to undergo a conformational change and translocate to the nucleus, where it interacts with specific DNA sequences to regulate transcription of target genes. ii) Distribution of SHRs in liquid condensates or foci in the nucleus [Reviewed in (4)]. iii) Steroid hormone receptor could form multimers or quaternary structure after DNA binding (5, 6). Interesting, this mechanism brings together regions that may be distant in the linear genome. Crosstalk between SHRs. iv) The SHRs co-recruitment to DNA response elements occurs in presence of both ligands. As a result, this crosstalk triggers a specific gene program. SHRs= ERα/PR (7); SHRs= GR/PR (8); SHRs= ERα/GR (9). v) Redirection mechanism in which the presence of R5050 in GC-free medium leads to the binding of GR to REL and FHOX1 motifs and repress the expression genes required for PR function (8). On one hand, it has been described that R5020 can directly activate and could drive GR binding (10) but R5020 activated PR could also participate indirectly stabilizing REL and/or FOXH1, increasing GR binding. This possibility, although feasible, requires further investigation.
The SHR´s structure has been extensively discussed in excellent reviews (11–13). Briefly, SHRs are composed of three different domains: an N-terminal ligand-independent activation function 1 domain composed of an intrinsically disordered region (AF-1), a central DNA-binding domain (DBD), which links to the C-terminal ligand-binding and the activation function 2 domain (LBD/AF-2) through a hinge region. Particularly, the AF-2 domain is a primarily hydrophobic groove formed by residues from helices H3, H4 and H12 of the LBD/AF-2 domain, where the H12 position plays a critical role in the AF-2 spatial conformation and SHRs function. In fact, AF-2 interacts with specific residues present in particular coregulators’ amino acid motifs (LxxLL and I/LxxII for coactivators and corepressors families, respectively) arranged on one side of their amphipathic helix (7). Here we will focus on PR and GR, two receptors that shared several features ranging from several aspects of ligand-binding to the DNA sequences to which the receptor binds (Figure 1A ii). In the absence of ligands, these receptors are part of a protein complex associated with chaperones and co-chaperones, which increase the affinity of SHRs to their ligands in vivo (14). Early reconstitution experiments with GR (15) and with PR established that the central proteins in the activation pathway include Hsp40, Hsp70, Hsp90, HOP, and p23 (16, 17). Recently, has been shown that coordinated chaperone interactions enhance stability, function and regulation of GR (18).
Activation occurs when the ligand interacts with the receptor and initiates a signal transduction cascade which ultimately leads to changes in gene expression, whose canonical pathway is depicted in Figure 1B i. PR and GR present a heterogeneous distribution concentrated in liquid condensates or foci (Figure 1B ii). The formation of these discrete foci (19–29), containing ~40-80 receptor molecules have been reported (30). This nuclear compartmentalization would modulate the kinetics of biochemical reactions and thus would actively participate the transcription process (reviewed in (4)).
SHR´s mechanism of action involves genomic and nongenomic processes. Genomic actions result from the direct binding of ligand-activated receptor complexes to specific hormone responsive DNA elements located at the enhancers and promoters of target genes; and/or the ligand-receptor recruitment to other regions in the genome, relying on additional transcription factors such as FOXA1, GATA-3, STAT5, NFκ-B, and AP-1, among others (31, 32).
Several families of co-activators interact directly with ligand-receptor complexes. Some of them do so through the AF-1 domain (33, 34), while others by means of the LBD/AF-2 (35). In this way, the regulation of gene transcription by these receptors is closely associated with the reorganization of chromatin at target genes.
In addition to these direct genomic effects, steroid hormones induce rapid nongenomic responses similar to those initiated by peptide growth factors (36, 37). For example, progestins can activate Src/p21ras/Erk and the PI3K/Akt pathways, either via an interaction of the PR with ERα, which itself activates c-Src and PI3K, or by direct interaction of PR with the SH3 domain of c-Src (38–40). A growing body of evidence suggests that GR may also act via nongenomic mechanisms. Glucocorticoid activation of a membrane associated GR regulates gap junction intercellular communication and neural progenitor cell proliferation by a mechanism that requires c-Src activity and rapid MAPK-dependent phosphorylation of connexin-43 (41).
Traditionally, the genomic and nongenomic actions of steroid hormones have been considered as two independent pathways, but we found that both pathways converge in the modification of structural components of the target chromatin (42).
The breast develops predominantly after birth: a poorly developed ductal system initially begins to unfold during puberty and gains in complexity during adulthood (43). From pregnancy to lactation, lobuloalveolar growth is followed by the complete differentiation of the mammary epithelium and at weaning, a dramatic switch from survival to death signaling occurs, leading to mammary gland involution. During these periods experienced throughout a woman’s life, hormones promote first mammary gland development resulting in ductal elongation, then in adulthood, through recurrent estrous cycles trigger side branching and upon each pregnancy they control cyclical periods of cellular proliferation, differentiation and regression of the mammary epithelium (44). While ER is required at an earlier stage to induce ductal elongation, PR is needed later for side branching and alveologenesis (44–46). Progestins were described to be involved in driving cell proliferation, thus favoring breast cancer development but also to inhibit ER-dependent breast tumorigenesis (7, 47). Moreover, progestins also inhibit the production and secretion of milk and stimulate the proliferation of epithelial cells during late pregnancy (48). On the other hand, glucocorticoids (GCs) play a key role at puberty and pregnancy (49, 50); they promote lactation and the synthesis of milk proteins, maintaining the differentiation stage of the mammary epithelium (51–53). GR is expressed in all stages from normal to cancerous breast tissue (54, 55).
PR is expressed from a single gene as two main isoforms, PRA (94 kDa) and PRB (116 kDa) (56, 57), both are transcribed from two distinct promoters and exhibit different transcriptional and biological activities as ligand-activated transcription factors. PRB is a full-length receptor; PRA is a truncated form of PRB lacking the N-terminal 164 amino acids (56, 58). Both isoforms are usually co-expressed at similar levels in normal breast while a significant increase in PRA or PRB was detected in breast cancer that correlates with lesion progression, from the normal state to malignancy. In this regard, the PRA/PRB ratio has been proposed as a prognostic and predictive factor for antiprogestin responsiveness in breast cancer (59). Some reports indicated that a high ratio of PRA/PRB is associated with worse prognosis, and recurrence after tamoxifen (60, 61), yet other studies concluded that higher PRA/PRB are related with biomarkers of better prognosis (59, 62). These contradictory results speak clearly that the mechanism of action of PR is more complex than we originally thought and requires further investigation. Moreover, PRA and PRB can form homodimers or heterodimers that exhibit distinct transcriptional regulatory functions by targeting different subsets of genes (63–66).
In the mammary gland, the luminal epithelium forms the inner layer of the ducts and the basal epithelium harbors myoepithelial cells that form the outer layer of the mature mammary ducts as well as stem and progenitor cells. The mammary ducts also comprise fibrous connective tissue, and variable amounts of adipose tissue (43, 67). In the adult mouse mammary gland, PR is expressed only in a fraction of the luminal epithelial cells, where progesterone promotes alveolar growth by a paracrine mechanism. In PR+ breast luminal cells progesterone upregulates RANKL expression. Then, RANKL binds to RANK expressed on the surface of neighboring PR- luminal cells or basal cells, activating downstream pathways of cell proliferation, expansion, and survival. Progesterone can also induce adjacent PR+ cell proliferation by a cell-autonomous, CCND1-dependent mechanism. Moreover, progesterone also elicits the proliferation of PR- luminal epithelial cells by a paracrine mechanism involving RANK and the NFκB signaling pathway (68).
Most of the research performed so far has been carried out in breast cancer cell lines, with very few studies conducted in normal human breast. Therefore, the role of PR in mammary physiology is underrepresented. This is justified by the loss of receptors detected in normal mammary cells once in culture. However, some steps have already been taken to overcome this issue. In 2009 Graham et al. reported the development and validation of a physiologically relevant model of matrix-embedded normal human breast cells, which was appropriate for studying hormone action in the normal breast (69). Moreover, Clarke et al. performed genome wide PR binding studies in breast cancer cells and in immortalized normal breast cells (70). Although PR binding was correlated with transcriptional outcome in both cell lines, there was a remarkably low overlap between the PR cistromes and in transcriptional targets. Moreover, distinct patterns of enrichment of known cofactor binding motifs were detected, with FOXA1 sites over-represented in breast cancer binding regions and NF1 and AP-1 motifs uniquely enriched in normal cells (70). What determines this difference? The expression levels and/or activation of the cofactors that participate in PR signaling could explain these variations. Hence, the pioneer factor FOXA1 would be a tumoral cofactor of PR. Conversely, NF1 and AP-1 would be the transcription factors chosen by PR in the normal context. An in-depth analysis of the differences in gene expression and PR binding in systems that adequately recapitulate both normal and breast cancer systems, may provide clinically valuable information on hormonal action. Thus, cofactor levels may modulate PR specificity (70).
Functional studies, performed in human breast, support that PR+/ER+ cells do not proliferate in direct response to hormone signals, but rather exert a paracrine effect on the surrounding PR-/ER- cells (71, 72). In contrast, almost 70% of breast cancers express ER and PR and require their ligands during breast cancer progression, suggesting that these cells switch from paracrine to autocrine mechanisms, as they acquire the ability to proliferate during the tumorigenic process. However, the mechanisms that drive this “switch” are not known. In this regard, the presence of tumoral cofactors such as FOXA1 or the availability of other receptors could help to redirect PR and/or ER binding towards an oncogenic program. Indeed, as it has been described for ER and PR, the formation of receptor heterocomplexes in the presence of both ligands could regulate the process towards malignancy in PR+/ER+ cells (7).
It is well known that GR mediates the effects of stress hormones, and of synthetic derivatives that are widely used in the clinic as anti-inflammatory and immunosuppressive agents (73). In the mammary gland, GR was found strongly localized in the nuclei of myoepithelial cells surrounding lobular and duct units and occasionally localized in the nuclei of stromal and endothelial cells (74). The GR nuclear localization indicates that the receptor is transcriptionally active, since the inactive GR resides mainly in the cytoplasm bound to heat shock proteins and immunophilins (14, 75).
In mammary development, GR was shown to be essential for cell proliferation during lobulo-alveolar development and to contribute to mammary lobular unit spatial formation (50). Of note, GCs also exert anti-proliferative and anti-apoptotic activity in mammary epithelial breast cancer cell lines (76, 77). These steroids are used in the treatment of metastatic breast cancer to reduce the side effects produced by the chemotherapy, and to treat symptoms related to advanced cancer. However, despite the fact that GR expression in mammary tissue declines from normal to precancerous lesions and to invasive breast carcinoma (54, 78), the increment in stress hormones during breast cancer progression results in GR activation even at distant metastatic sites. This, in turn, increases intra-tumor heterogeneity and colonization, therefore reducing cell survival. These observations suggest that caution is needed when including GCs in the treatment of breast cancer patients (79).
Activated GR undergoes phosphorylation, oligomerization and nuclear translocation (80). In the nucleus, the receptor is predominantly recruited to pre-accessible sites along with chromatin remodeling enzymes (81). Interestingly, GR is also able to initiate DNAse hypersensitive sites as a pioneer factor (82–84).
Like PR, chromatin remodeling factors regulate GR binding to DNA and thus, are involved in the overall function of GR. This points out the chromatin landscape as a major contributing factor to the GR-regulated cell-type specific gene expression (84–90). In fact, Johnson et al. reported that all GR binding events involving the SWI/SNF remodeling complex are either pre-recruited by other factors or recruited by the receptor itself (88).
In mouse mammary epithelial cells, 51% of pre-programmed GR binding sites are enriched in the pioneer transcription factor AP-1 (89), which along with GR triggers the recruitment of several remodelers such as Brg1, Chd4, and Snf2h (91). Nevertheless, activated GR induces de novo remodeling of chromatin at a minority (~15%) of GR binding sites in a highly tissue-specific manner (85, 89, 90, 92, 93). The current evidence suggests that in mammary epithelial cells this pioneering capacity of AP-1 would be relevant for regulating chromatin accessibility not only at GR target enhancers but also at other genomic regions (89).
The oligomeric status of the GR has also been considered to play a key role in the mechanism of action of the activated receptor. Moreover, in the late 1990s the oligomerization state was proposed as a parameter in the search for synthetic ligands with dissociate GC effects (6). In this sense, we have reported that hormone-activated GR adopts a dimer configuration in the nucleus of living murine mammary adenocarcinoma cells (94) and upon binding to a specific DNA binding site the GR dimer becomes a tetramer (Figure 1B iii) (5, 6). Of note, tetrameric configuration was also detected in activated PR complexes in the same cells (5). The influence of DNA binding on the quaternary structure of the GR proposes a similarity to an allosteric structural transition of the receptor once bound to its target DNA region (95–97).
Although at different concentrations, various hormones are present simultaneously in the bloodstream or locally at their target cells. For instance, estrogens that are governed by the menstrual cycle coincide at any time with high levels of GCs, which are regulated by stress and circadian cycles. These multiple signals converge to the same cell and, together, they participate in the cellular response. In this sense, the mechanisms of hormonal action need to be studied in an integrated manner, where different receptors could be activated simultaneously by their cognate ligands.
GCs exert an antagonistic effect on estrogen-dependent cell growth in ER+/GR+ breast carcinoma cells and reduce their cell proliferation through a functional crosstalk between both receptors (Figure 1B iv) (9, 32, 98–100). On the other hand, also in breast cancer models, it has been proposed that PR redirects ERα chromatin binding events. ERα and PR form a complex in the presence of both ligands, resulting in a unique gene expression program that is associated with good clinical outcome (Figure 1B iv) (7). In this case, it has been proposed that PR functions as a molecular rheostat to control ERα chromatin binding and transcriptional activity.
Furthermore, it was recently reported the generation of metastasis-competent circulating tumor cells (CTCs) in patients with breast cancer occur during sleep, in the rest phase; while CTCs generated during the active phase are devoid of metastatic ability. The authors found that key circadian rhythm hormones such as melatonin, testosterone and GCs dictate CTC generation dynamics (101). Treatment with the synthetic GC dexamethasone (DEX) or testosterone did not affect primary tumor size but resulted in a marked reduction in single CTCs and CTC clusters (101). These key effects of hormones that determine the metastatic capacity of cancer cells are likely to occur in the presence of both GCs and androgens, thus, a putative crosstalk between both activated receptors could be directly operating in this process.
Unlike the functional connections between ERα with PR and GR, or AR with ERα and GR, few studies have addressed the influence of GR on the transcriptional activity of PR and vice versa. Similarities in protein structure as well as in the DNA sequences to which the receptors bind are readily evident for PR and GR (>90% sequence identity between their DNA binding domains (DBD). However, the resulting biological response differs markedly. For example, the association of progestins with breast cancer incidence and progression contrasts with the growth suppressive action of GCs on ER+/PR+ breast tumor cells (102, 103). In fact, in those cell types, increased circulating levels of progestins and estrogens and/or overexpression of their receptors lead to uncontrolled cell division (47, 104). On the other hand, previous works suggested that GR would act as a suppressor of proliferation (52, 105) as well as a cell death inducer in tumoral mammary epithelium (106).
To address the existence of a potential crosstalk between PR and GR, we used PR+/GR+ breast cancer cell lines (107, 108) where we found that GC-free or DEX-activated GR inhibits PR-dependent cell proliferation and dedifferentiation through the modulation of certain PR-target genes, i.e. GREB1, STAT5A, ELF5 and SNAI1 (8). In the presence of DEX, the antagonistic effect increases and involves the formation of GR-PR protein complexes. By ChIP-seq and sequential ChIP analyses, we detected overlapped binding of GR and PR at key enhancer sites and confirmed co-recruitment of both receptors to shared sites (Figure 1B iv). Moreover, GC-free GR upon stimulation with the PR-agonist R5020, can bind to REL and FOXH1 motifs and repress the expression of nearby genes encoding for SWI/SNF and other chromatin remodeler complexes such as SMARCD2, ARID1A and INO80C (Figure 1B v). Thus, in the presence of the synthetic progestin R5020, relocated GR are bound to a subset of genes required for PR function, reinforcing the anti-progestational effect of GCs in ER+/PR+ breast cancer cells (8).
The mechanism behind the R5020-dependent GR binding to approximately 600 unique sites could be due at least by two non-mutually exclusive mechanisms: 1) a direct effect of R5020 on the GR or 2) an indirect mechanism whereby activated PR would stabilize GR binding to other transcription factors (i.e. REL or FOXH1) genomic regions (Figure 1B v).
Regarding the direct effect, although R5020 is considered a PR specific agonist, it has been reported that R5020 can also activate GR (10). Thus, R5020 binding to GR would induce a unique conformational change to the receptor leading to its recruitment to REL and/or FOXH1-enriched regions. However, we can also speculate that R5020-activated PR could favor GR recruitment to REL and/or FOXH1 regions throughout an indirect mechanism. Accordingly, it has been reported that FOXH1 can act as a hormone-independent corepressor of AR in prostate cancer cells; indeed, a protein-protein interaction was identified between the AR AF-1 domain and FOXH1 independently of the presence of dihydrotestosterone (109). In this model, R5020-activated PR could release FOXH1/REL, which in turn would bind to their sites in the genome favoring GR loading (Figure 1B v, right panel). Whether GR and PR physically interact with FOXH1 and/or REL in mammary cells is unknown and more research is required to support this second hypothetical mechanism.
Given the concentration of different steroid hormones varies considerably over a wide range of time (from hours, days and even weeks), several receptors might be coexisting and simultaneously activated by their ligands. Thus, the mechanism of control of one receptor over the other/s could be very frequent and could be involved in relevant receptor-mediated functions. This brings up a complex scenario in which several activated hormone receptors could be interacting reciprocally, regulating the final transcriptional output and their function in the target cell. This crosstalk between receptors could be positive, implying synergism between hormone pathways, or on the contrary, negative, through competition for pathways, for co-regulators and for binding sites in the genome.
Thus, it will be key to elucidate the causes that establish this hierarchy between receptors. What determines which receptor leads and regulates the activity over the other/s? The circulating levels of ligands, receptor levels, and the cellular identity of the target cell (more adapted to respond to one stimulus than to another) are factors that could be involved in this complex scenario. Also, under these circumstances, heterologous complexes, composed of different activated receptors and with different stoichiometry, could be formed and reciprocally regulate the hormonal pathways involved.
We propose that the prevalence of one or the other mechanism is dependent upon which ligand and/or combination of ligands bind to each receptor. Under this hypothesis, new questions arise regarding GR and PR functional crosstalk. How does the ligand-dependent conformation acquired by each receptor influence the control of gene expression? Do the GR-PR heterocomplexes display the same activity as homo-PR or homo-GR complexes? Do the GR-PR heterocomplexes recruit the same set of co-regulators? Do they have similar intranuclear dynamics compared to homodimers? Recruitment of the GR to non-canonical sites requires the presence of the transcription factor that directly recognizes that site, or does it do so by a different mechanism?
To address these questions, state-of-the-art techniques that allow us to monitor simultaneously homo- and heterocomplexes populations of PR and GR in the same cell, are required. For this purpose, it is necessary to engineered cell lines through mutations targeting the interacting regions involved in the formation of homo- and heterodimers. Moreover, mass spectrometry will enable to identify the protein interactome of each receptor population; Next Generation Sequencing (NGS) including ChIP-seq and RNA-seq will help to decipher the defined cistromes for each receptor population; and high-resolution microscopy will allow to visualize the nuclear dynamics of homo- or hetero-complexes in response to hormone, including the formation of condensates. This will shed light on the complex mechanism by which SHRs act upon simultaneous activation. Therefore, and from a pharmacological point of view, understanding PR-GR crosstalk could contribute to the design of new endocrine combined therapies that minimize tumor resistance, colonization, and metastasis and thus, provide tumorigenesis vulnerability for therapeutic improvement in patients.
GV and AP wrote the manuscript. MO and RS searched for references and built the Figure. All authors contributed to the article and approved the submitted version.
Grants from the Agencia Estatal de Investigación AEI/10.13039/501100011033 [PID2019-105173RBI00]; Agencia Nacional de Programación Científica y Tecnológica, [Préstamo BID-PICT2019-0158] and University of Buenos Aires [20820180100745BA], Argentina. MO and AP are members of the CONICET-Argentina. GV is Researcher of the Spanish National Research Council (CSIC), Spain.
Authors thanks Dr. Diego M. Presman (IFIBYNE, University of Buenos Aires-CONICET) for advice on the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zhao L, Hu H, Gustafsson JA, Zhou S. Nuclear receptors in cancer inflammation and immunity. Trends Immunol (2020) 41:172–85. doi: 10.1124/pr.112.006833
2. Burris TP, Solt LA, Wang Y, Crumbley C, Banerjee S, Griffett K, et al. Nuclear receptors and their selective pharmacologic modulators. Pharmacol Rev (2013) 65:710–78. doi: 10.1038/nrd1551
3. Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov (2004) 3:950–64. doi: 10.1042/BCJ20200883
4. Stortz M, Presman DM, Pecci A, Levi V. Phasing the intranuclear organization of steroid hormone receptors. Biochem J (2021) 478:443–61. doi: 10.1073/pnas.1606774113
5. Presman DM, Ganguly S, Schiltz RL, Johnson TA, Karpova TS, Hager GL. DNA Binding triggers tetramerization of the glucocorticoid receptor in live cells. Proc Natl Acad Sci USA (2016) 113:8236–41. doi: 10.1080/21541264.2016.1249045
6. Presman DM, Hager GL. More than meets the dimer: What is the quaternary structure of the glucocorticoid receptor? Transcription (2017) 8:32–9. doi: 10.1038/nature14583
7. Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature (2015) 523:313–7. doi: 10.1093/nar/gkz857
8. Ogara MF, Rodriguez-Segui SA, Marini M, Nacht AS, Stortz M, Levi V, et al. The glucocorticoid receptor interferes with progesterone receptor-dependent genomic regulation in breast cancer cells. Nucleic Acids Res (2019) 47:10645–61. doi: 10.1074/jbc.M113.473819
9. Karmakar S, Jin Y, Nagaich AK. Interaction of glucocorticoid receptor (GR) with estrogen receptor (ER) alpha and activator protein 1 (AP1) in dexamethasone-mediated interference of ERalpha activity. J Biol Chem (2013) 288:24020–34. doi: 10.1210/endo-107-2-566
10. Svec F, Yeakley J, Harrison RW. 3rd, progesterone enhances glucocorticoid dissociation from the AtT-20 cell glucocorticoid receptor. Endocrinology (1980) 107:566–72. doi: 10.1042/BST20210419
11. Frank F, Ortlund EA, Liu X. Structural insights into glucocorticoid receptor function. Biochem Soc Trans (2021) 49:2333–43. doi: 10.1002/pro.3496
12. Weikum ER, Liu X, Ortlund EA. The nuclear receptor superfamily: A structural perspective. Protein Sci Publ Protein Soc (2018) 27:1876–92. doi: 10.1146/annurev-physiol-021909-135917
13. Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol (2010) 72:247–72. doi: 10.1016/j.celrep.2022.111039
14. Backe SJ, Sager RA, Regan BR, Sit J, Major LA, Bratslavsky G, et al. A specialized Hsp90 co-chaperone network regulates steroid hormone receptor response to ligand. Cell Rep (2022) 40:111039. doi: 10.1007/3-540-29717-0_5
15. Pratt WB, Morishima Y, Murphy M, Harrell M. Chaperoning of glucocorticoid receptors. Handb Exp Pharmacol (2006), 172:111–38. doi: 10.1074/jbc.271.22.12833
16. Dittmar KD, Hutchison KA, Owens-Grillo JK, Pratt WB. Reconstitution of the steroid receptor hsp90 heterocomplex assembly system of rabbit reticulocyte lysate. J Biol Chem (1996) 271:12833–9. doi: 10.1074/jbc.273.49.32973
17. Kosano H, Stensgard B, Charlesworth MC, McMahon N, Toft D. The assembly of progesterone receptor-hsp90 complexes using purified proteins. J Biol Chem (1998) 273:32973–9. doi: 10.1016/j.cell.2014.04.038
18. Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell (2014) 157:1685–97. doi: 10.1016/0014-4827(91)90057-2
19. Vazquez-Nin GH, Echeverria OM, Fakan S, Traish AM, Wotiz HH, Martin TE. Immunoelectron microscopic localization of estrogen receptor on pre-mRNA containing constituents of rat uterine cell nuclei. Exp Cell Res (1991) 192:396–404. doi: 10.1083/jcb.102.4.1191
20. Perrot-Applanat M, Groyer-Picard MT, Logeat F, Milgrom E. Ultrastructural localization of the progesterone receptor by an immunogold method: Effect of hormone administration. J Cell Biol (1986) 102:1191–9. doi: 10.1007/BF00215514
21. Isola JJ. The effect of progesterone on the localization of progesterone receptors in the nuclei of chick oviduct cells. Cell Tissue Res (1987) 249:317–23. doi: 10.1210/mend-5-2-217
22. Martins VR, Pratt WB, Terracio L, Hirst MA, Ringold GM, Housley PR. Demonstration by confocal microscopy that unliganded overexpressed glucocorticoid receptors are distributed in a nonrandom manner throughout all planes of the nucleus. Mol Endocrinol (1991) 5:217–25. doi: 10.1210/mend-5-2-217
23. Yang J, DeFranco DB. Differential roles of heat shock protein 70 in the in vitro nuclear import of glucocorticoid receptor and simian virus 40 large tumor antigen. Mol Cell Biol (1994) 14:5088–98. doi: 10.1073/pnas.95.6.2973
24. Fejes-Toth G, Pearce D, Naray-Fejes-Toth A. Subcellular localization of mineralocorticoid receptors in living cells: effects of receptor agonists and antagonists. Proc Natl Acad Sci United States America (1998) 95:2973–8. doi: 10.1074/jbc.M101755200
25. Tomura A, Goto K, Morinaga H, Nomura M, Okabe T, Yanase T, et al. The subnuclear three-dimensional image analysis of androgen receptor fused to green fluorescence protein. J Biol Chem (2001) 276:28395–401. doi: 10.1210/me.2002-0110
26. Matsuda K, Ochiai I, Nishi M, Kawata M. Colocalization and ligand-dependent discrete distribution of the estrogen receptor (ER)alpha and ERbeta. Mol Endocrinol (2002) 16:2215–30. doi: 10.1074/jbc.M105966200
27. Pearce D, Naray-Fejes-Toth A, Fejes-Toth G. Determinants of subnuclear organization of mineralocorticoid receptor characterized through analysis of wild type and mutant receptors. J Biol Chem (2002) 277:1451–6. doi: 10.1210/me.2005-0050
28. Schaaf MJ, Lewis-Tuffin LJ, Cidlowski JA. Ligand-selective targeting of the glucocorticoid receptor to nuclear subdomains is associated with decreased receptor mobility. Mol Endocrinol (2005) 19:1501–15. doi: 10.1210/me.2006-0041
29. Arnett-Mansfield RL, Graham JD, Hanson AR, Mote PA, Gompel A, Scurr LL, et al. Focal subnuclear distribution of progesterone receptor is ligand dependent and associated with transcriptional activity. Mol Endocrinol (2007) 21:14–29. doi: 10.1038/s41598-017-06676-0
30. Stortz M, Presman DM, Bruno L, Annibale P, Dansey MV, Burton G, et al. Mapping the dynamics of the glucocorticoid receptor within the nuclear landscape. Sci Rep (2017) 7:6219. doi: 10.1016/0039-128X(96)00030-X
31. Beato M, Chavez S, Truss M. Transcriptional regulation by steroid hormones. Steroids (1996) 61:240–51. doi: 10.1016/j.mce.2013.03.002
32. Miranda TB, Morris SA, Hager GL. Complex genomic interactions in the dynamic regulation of transcription by the glucocorticoid receptor. Mol Cell Endocrinol (2013) 380:16–24. doi: 10.1126/science.1093686
33. Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, et al. RNAi-mediated targeting of heterochromatin by the RITS complex. Science (2004) 303:672–6. doi: 10.1038/nature02269
34. Bastow R, Mylne JS, Lister C, Lippman Z, Martienssen RA, Dean C. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature (2004) 427:164–7. doi: 10.1210/me.2003-0116
35. Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol (2003) 17:1681–92. doi: 10.1038/nrm1009
36. Losel R, Wehling M. Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol (2003) 4:46–56. doi: 10.1016/j.mce.2021.111453
37. Thiebaut C, Vlaeminck-Guillem V, Tredan O, Poulard C, Le Romancer M. Non-genomic signaling of steroid receptors in cancer. Mol Cell Endocrinol (2021) 538:111453. doi: 10.1128/MCB.23.6.1994-2008.2003
38. Ballare C, Uhrig M, Bechtold T, Sancho E, Di Domenico M, Migliaccio A, et al. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol Cell Biol (2003) 23:1994–2008. doi: 10.1016/S1097-2765(01)00304-5
39. Boonyaratanakornkit V, Scott MP, Ribon V, Sherman L, Anderson SM, Maller JL, et al. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-src family tyrosine kinases. Mol Cell (2001) 8:269–80. doi: 10.1093/emboj/17.7.2008
40. Migliaccio A, Piccolo D, Castoria G, Di Domenico M, Bilancio A, Lombardi M, et al. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J (1998) 17:2008–18. doi: 10.1073/pnas.1102821108
41. Samarasinghe RA, Di Maio R, Volonte D, Galbiati F, Lewis M, Romero G, et al. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci USA (2011) 108:16657–62. doi: 10.1016/j.molcel.2006.10.011
42. Vicent GP, Ballare C, Nacht AS, Clausell J, Subtil-Rodriguez A, Quiles I, et al. Induction of progesterone target genes requires activation of erk and msk kinases and phosphorylation of histone H3. Mol Cell (2006) 24:367–81. doi: 10.1152/physrev.00040.2018
43. Fu NY, Nolan E, Lindeman GJ, Visvader JE. Stem cells and the differentiation hierarchy in mammary gland development. Physiol Rev (2020) 100:489–523. doi: 10.1101/cshperspect.a003178
44. Brisken C, O'Malley B. Hormone action in the mammary gland. Cold Spring Harbor Perspect Biol 2 (2010), l12:a003178. doi: 10.1002/dvdy.1179
45. Hovey RC, Trott JF, Ginsburg E, Goldhar A, Sasaki MM, Fountain SJ, et al. Transcriptional and spatiotemporal regulation of prolactin receptor mRNA and cooperativity with progesterone receptor function during ductal branch growth in the mammary gland. Dev Dynamics an Off Publ Am Assoc Anat (2001) 222:192–205. doi: 10.1007/s10911-021-09496-1
46. Chen W, Wei W, Yu L, Ye Z, Huang F, Zhang L, et al. Mammary development and breast cancer: A notch perspective. J Mammary Gland Biol Neoplasia (2021) 26:309–20. doi: 10.1038/nrc.2016.116
47. Carroll JS, Hickey TE, Tarulli GA, Williams M, Tilley WD. Deciphering the divergent roles of progestogens in breast cancer. Nat Rev Cancer (2017) 17:54–64. doi: 10.1038/nrc.2016.116
48. Graham JD, Clarke CL. Physiological action of progesterone in target tissues. Endo Rev (1997) 18:502–19. doi: 10.1210/me.2004-0068
49. Wintermantel TM, Bock D, Fleig V, Greiner EF, Schutz G. The epithelial glucocorticoid receptor is required for the normal timing of cell proliferation during mammary lobuloalveolar development but is dispensable for milk production. Mol Endocrinol (2005) 19:340–9. doi: 10.1083/jcb.200403020
50. Murtagh J, McArdle E, Gilligan E, Thornton L, Furlong F, Martin F. Organization of mammary epithelial cells into 3D acinar structures requires glucocorticoid and JNK signaling. J Cell Biol (2004) 166:133–43. doi: 10.1016/0303-7207(94)90288-7
51. Groner B, Altiok S, Meier V. Hormonal regulation of transcription factor activity in mammary epithelial cells. Mol Cell Endocrinol (1994) 100:109–14. doi: 10.1002/jcp.22896
52. Hoijman E, Rocha-Viegas L, Kalko SG, Rubinstein N, Morales-Ruiz M, Joffe EB, et al. Glucocorticoid alternative effects on proliferating and differentiated mammary epithelium are associated to opposite regulation of cell-cycle inhibitor expression. J Cell Physiol (2012) 227:1721–30. doi: 10.1210/en.2010-0517
53. Bertucci PY, Quaglino A, Pozzi AG, Kordon EC, Pecci A. Glucocorticoid-induced impairment of mammary gland involution is associated with STAT5 and STAT3 signaling modulation. Endocrinology (2010) 151:5730–40. doi: 10.1093/nar/gkt327
54. Allegra JC, Lippman ME, Thompson EB, Simon R, Barlock A, Green L, et al. Distribution, frequency, and quantitative analysis of estrogen, progesterone, androgen, and glucocorticoid receptors in human breast cancer. Cancer Res (1979) 39:1447–54. doi: 10.1155/2017/1403054
55. Alyusuf R, Wazir JF, Brahmi UP, Fakhro AR, Bakhiet M. The immunoexpression of glucocorticoid receptors in breast carcinomas, lactational change, and normal breast epithelium and its possible role in mammary carcinogenesis. Int J Breast Cancer (2017) 2017:1403054. doi: 10.1002/j.1460-2075.1990.tb08280.x
56. Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, et al. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms a and b. EMBO J (1990) 9:1603–14. doi: 10.1002/j.1460-2075.1990.tb08280.x
57. Giulianelli S, Riggio M, Guillardoy T, Perez Pinero C, Gorostiaga MA, Sequeira G, et al. FGF2 induces breast cancer growth through ligand-independent activation and recruitment of ERalpha and PRBDelta4 isoform to MYC regulatory sequences. Int J Cancer (2019) 145:1874–88. doi: 10.1186/bcr2097
58. Cork DM, Lennard TW, Tyson-Capper AJ. Alternative splicing and the progesterone receptor in breast cancer. Breast Cancer Res BCR (2008) 10:207. doi: 10.1093/jnci/djw317
59. Rojas PA, May M, Sequeira GR, Elia A, Alvarez M, Martinez P, et al. Progesterone receptor isoform ratio: A breast cancer prognostic and predictive factor for antiprogestin responsiveness. J Natl Cancer Institute 109 (2017) 7:1–9. doi: 10.1016/j.jsbmb.2019.105560
60. Pateetin P, Pisitkun T, McGowan E, Boonyaratanakornkit V. Differential quantitative proteomics reveals key proteins related to phenotypic changes of breast cancer cells expressing progesterone receptor a. J Steroid Biochem Mol Biol (2020) 198:105560. doi: 10.1158/1078-0432.CCR-03-0141
61. Hopp TA, Weiss HL, Hilsenbeck SG, Cui Y, Allred DC, Horwitz KB, et al. Breast cancer patients with progesterone receptor PR-a-rich tumors have poorer disease-free survival rates. Clin Cancer Res an Off J Am Assoc Cancer Res (2004) 10:2751–60. doi: 10.1158/1078-0432.CCR-10-2950
62. Pathiraja TN, Shetty PB, Jelinek J, He R, Hartmaier R, Margossian AL, et al. Progesterone receptor isoform-specific promoter methylation: association of PRA promoter methylation with worse outcome in breast cancer patients. Clin Cancer Res an Off J Am Assoc Cancer Res (2011) 17:4177–86. doi: 10.1210/me.2005-0126
63. Graham JD, Yager ML, Hill HD, Byth K, O'Neill GM, Clarke CL. Altered progesterone receptor isoform expression remodels progestin responsiveness of breast cancer cells. Mol Endocrinol (2005) 19:2713–35. doi: 10.1210/me.2004-0287
64. Jacobsen BM, Schittone SA, Richer JK, Horwitz KB. Progesterone-independent effects of human progesterone receptors (PRs) in estrogen receptor-positive breast cancer: PR isoform-specific gene regulation and tumor biology. Mol Endocrinol (2005) 19:574–87. doi: 10.1002/ijc.21186
65. Leo JC, Wang SM, Guo CH, Aw SE, Zhao Y, Li JM, et al. Gene regulation profile reveals consistent anticancer properties of progesterone in hormone-independent breast cancer cells transfected with progesterone receptor. Int J Cancer (2005) 117:561–8. doi: 10.1371/journal.pone.0045993
66. Khan JA, Bellance C, Guiochon-Mantel A, Lombes M, Loosfelt H. Differential regulation of breast cancer-associated genes by progesterone receptor isoforms PRA and PRB in a new bi-inducible breast cancer cell line. PloS One (2012) 7:e45993. doi: 10.1002/wdev.35
67. Macias H, Hinck L. Mammary gland development. Wiley Interdiscip Rev Dev Biol (2012) 1:533–57. doi: 10.1186/bcr3166
68. Tanos T, Rojo L, Echeverria P, Brisken C. ER and PR signaling nodes during mammary gland development. Breast Cancer Res BCR (2012) 14:210. doi: 10.1007/s10911-009-9160-6
69. Graham JD, Mote PA, Salagame U, Balleine RL, Huschtscha LI, Clarke CL. Hormone-responsive model of primary human breast epithelium. J Mammary Gland Biol Neoplasia (2009) 14:367–79. doi: 10.1371/journal.pone.0035859
70. Clarke CL, Graham JD. Non-overlapping progesterone receptor cistromes contribute to cell-specific transcriptional outcomes. PloS One (2012) 7:e35859. doi: 10.1073/pnas.95.9.5076
71. Brisken C, Park S, Vass T, Lydon JP, O'Malley BW, Weinberg RA. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci USA (1998) 95:5076–81. doi: 10.1023/A:1005805831460
72. Clarke RB, Howell A, Anderson E. Estrogen sensitivity of normal human breast tissue in vivo and implanted into athymic nude mice: analysis of the relationship between estrogen-induced proliferation and progesterone receptor expression. Breast Cancer Res Treat (1997) 45:121–33. doi: 10.1016/j.jaci.2013.09.007
73. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol (2013) 132:1033–44. doi: 10.1002/path.1982
74. Lien HC, Lu YS, Cheng AL, Chang WC, Jeng YM, Kuo YH, et al. Differential expression of glucocorticoid receptor in human breast tissues and related neoplasms. J Pathol (2006) 209:317–27. doi: 10.1016/j.tem.2021.07.005
75. Mazaira GI, Piwien Pilipuk G, Galigniana MD. Corticosteroid receptors as a model for the Hsp90*immunophilin-based transport machinery. Trends Endocrinol Metab: TEM (2021) 32:827–38. doi: 10.1002/j.1460-2075.1990.tb08280.x
76. Moran TJ, Gray S, Mikosz CA, Conzen SD. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res (2000) 60:867–72. doi: 10.1038/bjc.1995.343
77. Harris RA, Hiles ID, Page MJ, O'Hare MJ. The induction of apoptosis in human mammary luminal epithelial cells by expression of activated c-neu and its abrogation by glucocorticoids. Br J Cancer (1995) 72:386–92. doi: 10.1007/s10549-011-1689-6
78. Vilasco M, Communal L, Mourra N, Courtin A, Forgez P, Gompel A. Glucocorticoid receptor and breast cancer. Breast Cancer Res Treat (2011) 130:1–10. doi: 10.1038/s41586-019-1019-4
79. Obradovic MMS, Hamelin B, Manevski N, Couto JP, Sethi A, Coissieux MM, et al. Glucocorticoids promote breast cancer metastasis. Nature (2019) 567:540–4. doi: 10.3389/fimmu.2019.01545
80. Timmermans S, Souffriau J, Libert C. A general introduction to glucocorticoid biology. Front Immunol (2019) 10:1545. doi: 10.1080/10253890802506409
81. Biddie SC, Hager GL. Glucocorticoid receptor dynamics and gene regulation. Stress (2009) 12:193–205. doi: 10.1016/0092-8674(84)90523-3
82. Zaret KS, Yamamoto KR. Reversible and persistent changes in chromatin structure accompany activation of a glucocorticoid-dependent enhancer element. Cell (1984) 38:29–38. doi: 10.1002/j.1460-2075.1987.tb02507.x
83. Richard-Foy H, Hager GL. Sequence-specific positioning of nucleosomes over the steroid-inducible MMTV promoter. EMBO J (1987) 6:2321–8. doi: 10.1016/j.molcel.2008.02.010
84. John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, et al. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol Cell (2008) 29:611–24. doi: 10.1038/ng.759
85. John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet (2011) 43:264–8. doi: 10.1093/nar/gkw1163
86. Love MI, Huska MR, Jurk M, Schopflin R, Starick SR, Schwahn K, et al. Role of the chromatin landscape and sequence in determining cell type-specific genomic glucocorticoid receptor binding and gene regulation. Nucleic Acids Res (2017) 45:1805–19. doi: 10.1016/j.cels.2018.06.007
87. D'Ippolito AM, McDowell IC, Barrera A, Hong LK, Leichter SM, Bartelt LC, et al. Pre-established chromatin interactions mediate the genomic response to glucocorticoids. Cell Syst (2018) 7:146–160.e7. doi: 10.1093/nar/gkx1044
88. Johnson TA, Chereji RV, Stavreva DA, Morris SA, Hager GL, Clark DJ. Conventional and pioneer modes of glucocorticoid receptor interaction with enhancer chromatin in vivo. Nucleic Acids Res (2018) 46:203–14. doi: 10.1016/j.molcel.2011.06.016
89. Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell (2011) 43:145–55. doi: 10.1038/emboj.2013.106
90. Grontved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, et al. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J (2013) 32:1568–83. doi: 10.1038/nsmb.2718
91. Morris SA, Baek S, Sung MH, John S, Wiench M, Johnson TA, et al. Overlapping chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions. Nat Struct Mol Biol (2014) 21:73–81. doi: 10.1002/bies.201600137
92. Swinstead EE, Paakinaho V, Presman DM, Hager GL. Pioneer factors and ATP-dependent chromatin remodeling factors interact dynamically: A new perspective: Multiple transcription factors can effect chromatin pioneer functions through dynamic interactions with ATP-dependent chromatin remodeling factors. BioEssays News Rev molecular Cell Dev Biol (2016) 38:1150–7. doi: 10.1007/s00709-016-1063-y
93. Grbesa I, Hakim O. Genomic effects of glucocorticoids. Protoplasma (2017) 254:1175–85. doi: 10.1371/journal.pbio.1001813
94. Presman DM, Ogara MF, Stortz M, Alvarez LD, Pooley JR, Schiltz RL, et al. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PloS Biol (2014) 12:e1001813. doi: 10.1126/science.1164265
95. Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA Binding site sequence directs glucocorticoid receptor structure and activity. Science (2009) 324:407–10. doi: 10.1038/nsmb.2595
96. Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol (2013) 20:876–83. doi: 10.1101/gr.244814.118
97. Paakinaho V, Johnson TA, Presman DM, Hager GL. Glucocorticoid receptor quaternary structure drives chromatin occupancy and transcriptional outcome. Genome Res (2019) 29:1223–34. doi: 10.1007/s11010-016-2810-2
98. Hegde SM, Kumar MN, Kavya K, Kumar KM, Nagesh R, Patil RH, et al. Interplay of nuclear receptors (ER, PR, and GR) and their steroid hormones in MCF-7 cells. Mol Cell Biochem (2016) 422:109–20. doi: 10.1158/1541-7786.MCR-15-0433
99. West DC, Pan D, Tonsing-Carter EY, Hernandez KM, Pierce CF, Styke SC, et al. GR and ER coactivation alters the expression of differentiation genes and associates with improved ER+ breast cancer outcome. Mol Cancer Res MCR (2016) 14:707–19. doi: 10.1093/nar/gkt100
100. Bolt MJ, Stossi F, Newberg JY, Orjalo A, Johansson HE, Mancini MA. Coactivators enable glucocorticoid receptor recruitment to fine-tune estrogen receptor transcriptional responses. Nucleic Acids Res (2013) 41:4036–48. doi: 10.1038/s41586-022-04875-y
101. Diamantopoulou Z, Castro-Giner F, Schwab FD, Foerster C, Saini M, Budinjas S, et al. The metastatic spread of breast cancer accelerates during sleep. Nature (2022) 607:156–62. doi: 10.1210/er.13.2.146
102. Horwitz KB. The molecular biology of RU486 is there a role for antiprogestins in the treatment of breast cancer? Endo Rev (1992) 13:146–63. doi: 10.1210/mend.16.6.0848
103. Wan Y, Nordeen SK. Overlapping but distinct gene regulation profiles by glucocorticoids and progestins in human breast cancer cells. Mol Endocrinol (2002) 16:1204–14. doi: 10.1007/978-0-387-78818-0_7
104. Lange CA, Sartorius CA, Abdel-Hafiz H, Spillman MA, Horwitz KB, Jacobsen BM. Progesterone receptor action: translating studies in breast cancer models to clinical insights. Adv Exp Med Biol (2008) 630:94–111.
105. Goya L, Maiyar AC, Ge Y, Firestone GL. Glucocorticoids induce a G1/G0 cell cycle arrest of Con8 rat mammary tumor cells that is synchronously reversed by steroid withdrawal or addition of transforming growth factor-alpha. Mol Endocrinol (1993) 7:1121–32. doi: 10.1016/j.bcp.2014.04.006
106. Orqueda AJ, Dansey MV, Espanol A, Veleiro AS, Bal de Kier Joffe E, Sales ME, et al. The rigid steroid 21-hydroxy-6,19-epoxyprogesterone (21OH-6,19OP) is a dissociated glucocorticoid receptor modulator potentially useful as a novel coadjuvant in breast cancer chemotherapy. Biochem Pharmacol (2014) 89:526–35. doi: 10.1210/mend-3-8-1270
107. Nordeen SK, Kuhnel B, Lawler-Heavner J, Barber DA, Edwards DP. A quantitative comparison of dual control of a hormone response element by progestins and glucocorticoids in the same cell line. Mol Endocrinol (1989) 3:1270–8. doi: 10.1210/mend-2-11-1064
108. Arteaga CL, Coronado E, Osborne CK. Blockade of the epidermal growth factor receptor inhibits transforming growth factor alpha-induced but not estrogen-induced growth of hormone-dependent human breast cancer. Mol Endocrinol (1988) 2:1064–9. doi: 10.1074/jbc.M506147200
Keywords: glucocorticoid receptor, progesterone receptor, mammary epithelial cell proliferation, chromatin, gene expression-regulation
Citation: Pecci A, Ogara MF, Sanz RT and Vicent GP (2022) Choosing the right partner in hormone-dependent gene regulation: Glucocorticoid and progesterone receptors crosstalk in breast cancer cells. Front. Endocrinol. 13:1037177. doi: 10.3389/fendo.2022.1037177
Received: 05 September 2022; Accepted: 06 October 2022;
Published: 04 November 2022.
Edited by:
Anton M. Jetten, National Institute of Environmental Health Sciences (NIH), United StatesCopyright © 2022 Pecci, Ogara, Sanz and Vicent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adali Pecci, YXBlY2NpQHFiLmZjZW4udWJhLmFy; Guillermo Pablo Vicent, Z3ZtYm1jQGlibWIuY3NpYy5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.