 Ananthi Rajamoorthi1
Ananthi Rajamoorthi1 Charles A. LeDuc
Charles A. LeDuc Vidhu V. Thaker
Vidhu V. Thaker- 1Department of Pediatrics, Columbia University Medical Center, New York, NY, United States
- 2The Naomi Berrie Diabetes Center, Columbia University IRVING Medical Center, New York, NY, United States
- 3Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, United States
Understanding the developmental origins of health and disease is integral to overcome the global tide of obesity and its metabolic consequences, including atherosclerotic cardiovascular disease, type 2 diabetes, hyperlipidemia, and nonalcoholic fatty liver disease. The rising prevalence of obesity has been attributed, in part, to environmental factors including the globalization of the western diet and unhealthy lifestyle choices. In this review we argue that how and when such exposures come into play from conception significantly impact overall risk of obesity and later health outcomes. While the laws of thermodynamics dictate that obesity is caused by an imbalance between caloric intake and energy expenditure, the drivers of each of these may be laid down before the manifestation of the phenotype. We present evidence over the last half-century that suggests that the temporospatial evolution of obesity from intrauterine life and beyond is, in part, due to the conditioning of physiological processes at critical developmental periods that results in maladaptive responses to obesogenic exposures later in life. We begin the review by introducing studies that describe an association between perinatal factors and later risk of obesity. After a brief discussion of the pathogenesis of obesity, including the systemic regulation of appetite, adiposity, and basal metabolic rate, we delve into the mechanics of how intrauterine, postnatal and early childhood metabolic environments may contribute to adult obesity risk through the process of metabolic conditioning. Finally, we detail the specific epigenetic pathways identified both in preclinical and clinical studies that synergistically “program” obesity.
Key Messages
1. Obesity is the result of a dynamic interplay between genetics, physiology, behavior, and environment, that accumulate over time from conception and can predispose to energy homeostasis perturbations throughout the lifespan.
2. Developmental plasticity allows for the conditioning or “programming” of obesity in response to the metabolic environments of intrauterine, postnatal, and early childhood, that may be responsible for resistance to weight loss efforts in later life.
3. The metabolic environments in the prenatal, postnatal, and early childhood periods, reflective of maternal nutrition, postnatal feeding, and early childhood adiposity, respectively, significantly contribute to adult health outcomes.
4. Emerging evidence suggests that in utero, postnatal and early childhood metabolic environments affect adult basal metabolic rate and, in turn, obesity risk through metabolic programming, that needs further exploration.
5. Altered maternal levels of metabolic substrates and hormones in obesity contribute to maternal insulin resistance, oxidative stress and inflammation, resulting in perturbations in glucose and lipid homeostasis and feeding circuitry in the perinatal period, contributing to long-term obesity risk.
6. The in utero environment of maternal obesity, and the concomitant short-term aberrations in energy homeostasis during the perinatal period, translate to long-term obesity risk through both central and peripheral epigenetic modifications that regulate energy homeostatic set points.
7. Epigenetic changes are not exclusive to the perinatal period; these changes may occur during childhood and beyond in response to behavioral and environmental factors.
8. Epigenetic pathways that contribute to the conditioning of obesity are highly dynamic and reversible. Currently known pathways include variations in methylation status of obesity-related genes, which occur in response to maternal, postnatal, and early childhood nutrition, and have been shown to be associated with differences in adiposity and body composition in infancy and beyond.
Introduction
Early life exposures “program” long-term obesity risk
The global impact of childhood obesity
Childhood obesity has become one of the most pervasive global health crises of this century. In 2020, 39 million children worldwide under the age of 5 years had overweight or obesity. The prevalence of overweight and obesity in children aged 5-19 years across genders quadrupled from 4% in 1975 to over 18% in 2016. Further, what was once considered to be an epidemic of the Western world, childhood obesity has infiltrated both low- and middle-income countries. Since 2000, there has been a 24% increase in the prevalence of overweight/obesity in children under 5 years in Africa, while 50% of all children under 5 years with overweight and obesity lived in Asia in 2019 (1).
Obesity impacts growth and development from infancy, and is associated with increased risk of body dissatisfaction, depressive symptoms and low self-esteem during childhood and adolescence (2, 3). Childhood obesity persists into adulthood and is associated with higher prevalence of cardiometabolic risks including atherosclerotic cardiovascular disease (ASCVD), type 2 diabetes (T2D), hyperlipidemia and nonalcoholic fatty liver disease (NAFLD) (4).
Systemic regulation of adiposity and metabolic rate
Many genes associated with body mass index (BMI) are expressed in the hypothalamus, which receives sensory inputs from peripheral organs including the gastrointestinal (GI) tract, liver, adipose tissue, pancreas, and skeletal muscle regarding the overall metabolic status of the organism, to regulate food intake, appetite, and body weight (5). The “gut-brain-axis” is critical for some of these inputs due to the presence of mechanosensors and chemoreceptors in the intestinal epithelial wall that sense both volume and nutrient content of ingested food. This information is communicated centrally via two mechanisms: vagal innervation and endocrine hormones. GI-derived hormones, such as glucagon like peptide-1 (GLP-1) and ghrelin, not only behave in a paracrine manner to affect the local absorption and metabolism of nutrients, but can also act centrally through signaling pathways to affect feeding behavior and whole-body energy homeostasis (6). Hormones produced by adipose tissue, pancreas, skeletal muscle, and liver also serve as sensory inputs for the hypothalamus, including leptin, cholecystokinin, peptide YY, insulin, fibroblast growth factor 21 (FGF21) and other “adipokines” and “myokines,” which regulate systemic lipid and carbohydrate homeostasis (7). The level of leptin, produced by adipose tissue, is directly proportional to its mass. Its functional roles during early stages of development are diverse (8). In adults, however, reduction and/or absence of leptin promotes food intake and decreases energy expenditure (9). Together with the corticolimbic system and hindbrain, the hypothalamus integrates peripherally derived inputs to regulate appetite and food intake.
In addition to food intake, energy expenditure (EE) is an important component of metabolic homeostasis and is largely a combination of basal metabolism, thermogenesis, and physical activity. While central neuronal networks play a role in the regulation of EE, the critical component of EE, basal metabolic rate (BMR), is primarily determined by fat-free mass. Fat-free or “lean” mass is composed of skeletal muscle, which together with other metabolically active organs including the liver, heart, brain and kidneys, contribute to BMR (10). Skeletal muscle is a major player for insulin mediated glucose disposal; and dysregulation of skeletal muscle metabolism can strongly influence glucose homeostasis and insulin sensitivity predisposing to obesity related metabolic diseases (11).
Based on the laws of thermodynamics, obesity is a consequence of an imbalance of food intake and energy expenditure. But how and when such perturbations take place, and in what circumstances and metabolic environments they are more likely to occur, plays a vital role in determining the overall risk of obesity which will be discussed in the following sections.
The role of “programming” in the pathogenesis and treatment of obesity
Obesity is the result of a dynamic interplay between genetics, physiology, behavior, and environment, that accumulate over time since conception and can predispose to perturbations in energy homeostasis throughout life. The most widely studied and accepted interventions for obesity are lifestyle modifications, including exercise and dietary changes. However, both the efficacy and sustainability of such interventions are questionable, as the majority of individuals with obesity are unable to maintain reduced weight despite effective weight loss (12).
Socioeconomic factors, including accessibility to resources, are important limitations to the overall success of lifestyle interventions. As obesity often affects populations of limited socioeconomic resources, lifestyle modifications are challenging to initiate and sustain in the populations that would most benefit from such interventions, often due to a lack of resources and low health literacy (13, 14).
Further, if obesity was simply driven by an imbalance between food intake and expenditure at a single time point, lifestyle interventions would result in more positive and consistent outcomes. However, despite many public health measures promoting healthy lifestyle habits and health care spending on the screening and prevention of obesity and metabolic syndrome, obesity related ASCVD remains the leading cause of morbidity and mortality in the US and worldwide (15, 16).
There are several physiological mechanisms that explain why sustained weight loss through lifestyle modifications remains difficult (7, 17). Caloric restriction, both in short- and long-term, reduces 24-hour EE (18–20). In addition, several neuroendocrine hormones fluctuate in response to either caloric restriction or acute weight loss, including reductions in leptin and cholecystokinin and an increase in ghrelin, which ultimately function to increase appetite and promote weight regain. Sumithran et al. found that these hormonal fluctuations and increased appetite following 10% weight loss over 8 weeks (and subsequent weight stabilization), persist at 1 year (7). What causes these acute and chronic fluctuations in neuroendocrine signaling that ultimately thwart intervention and promote obesity? While genetic predisposition, socioeconomic factors, lifestyle habits and environmental exposures all contribute to the risk of obesity, the metabolic environment during early development lays the critical foundation for the conditioning of obesity and long-term ASCVD risk (Figure 1).
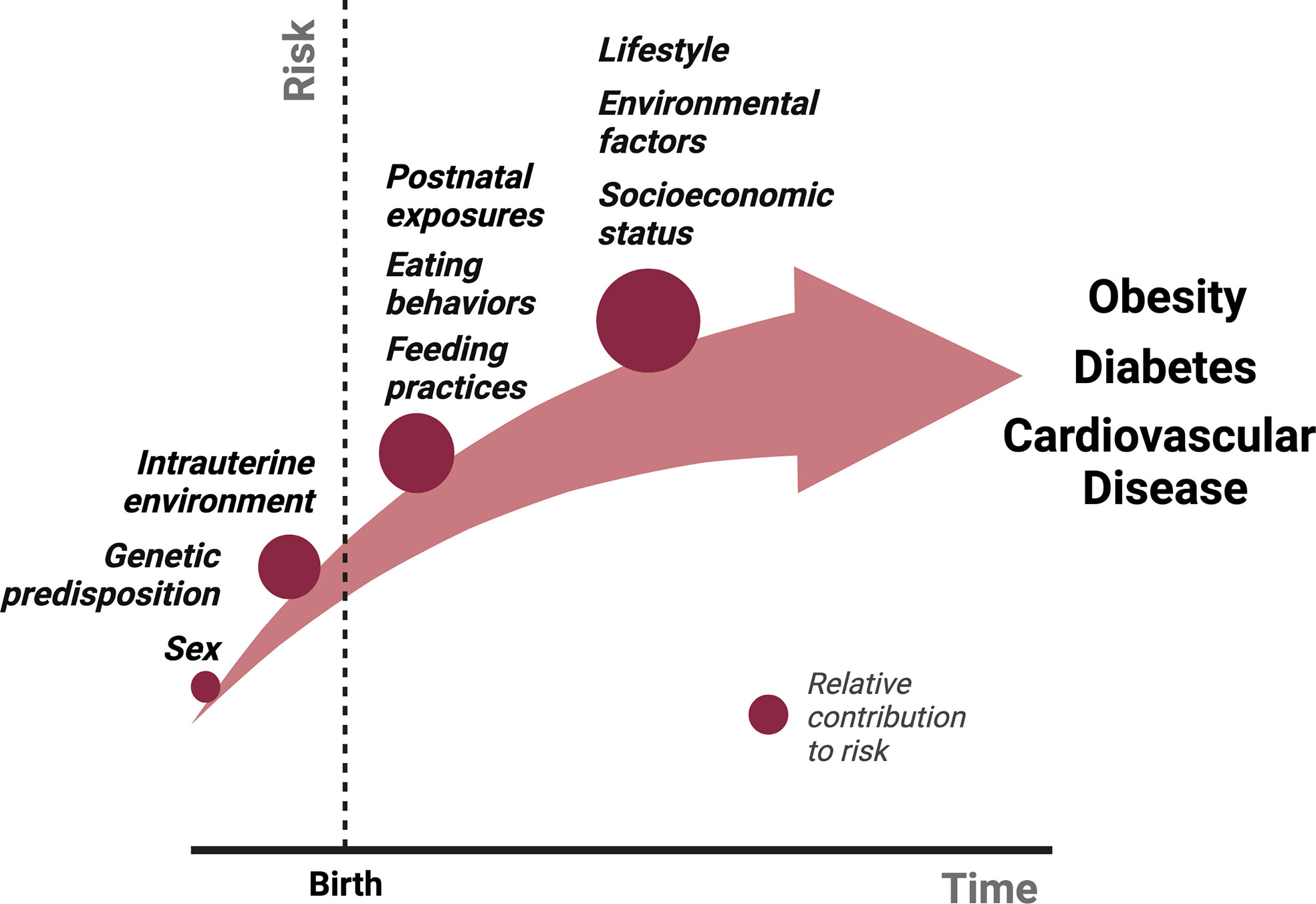
Figure 1 The relative contributions of various exposures to the cumulative risk for obesity over time from conception. Several factors contribute to the pathogenesis of obesity. We propose that intrauterine and postnatal exposures make relatively large contributions to the cumulative risk for obesity, in addition to the genetic predisposition. Studies described in this review have shown that postnatal exposures could potentially reverse the pathological events of intrauterine life in the setting of either maternal undernutrition or obesity, suggesting a more significant role for the postnatal environment in the “programming” obesity. We hypothesize that obesity risk acquired beyond early life, primarily in adolescence and adulthood through poor eating and lifestyle habits potentiated by socioeconomic status and other environmental factors, make an even greater contribution to obesity risk due to the sheer amount of time and opportunities available to accrue risk, although this is largely dependent on the individual.
Studies discussed in the following sections show a clear association between perinatal and childhood metabolic exposures including maternal under- and over-nutrition, maternal diabetes, breastfeeding, early infancy weight gain, and later health outcomes, including obesity and T2D. We hypothesize that developmental plasticity allows for the conditioning or “programming” of obesity in response to the metabolic environments of intrauterine, postnatal, and early childhood life, that play an important role in the resistance to weight loss efforts in later life. Further, if obesity is, in part, the result of conditioning or “programming” of certain physiologic processes established during critical developmental periods of early life, then lifestyle interventions initiated during adulthood will be insufficient to revert the initial “code” (21, 22). Addressing the risk for obesity in mothers prior to conception or during these critical developmental periods will likely produce favorable long-term outcomes.
The “developmental origins of health and disease” hypothesis and defining the phenomenon of metabolic programming
Metabolic programming (also called developmental programming) refers to the contribution of environmental exposures and nutrient imbalance from conception to early postnatal life – the critical times of organ development and maturation – to the risk of obesity and metabolic syndrome in adulthood, even after the absence or reversal of such exposures (17, 21–23). This is a more focused topic within a larger field known as the developmental origins of health and disease, or simply called “DOHaD.” Barker and other experts coined this concept as the “fetal origins of disease” in the 1990s (17, 23–26). Both terms refer to a similar phenomenon in which exposures during critical developmental periods affect long-term health outcomes.
Many experts argue that the term “programming” is not accurate or reflective of the reversible and dynamic nature of these physiologic yet nonadaptive responses and prefer the term “conditioning” instead (17). Throughout this review, accepting both the limitations of the word “programming” as well as its prevalent use in scientific literature, we will interchangeably use these terms referring to the same phenomenon.
Epidemiological studies supporting the role of metabolic programming of obesity
Risk factors for obesity acquired during different periods of development
Prenatal period
The metabolic environments in prenatal, postnatal, and early childhood, which are, in turn, reflective of maternal nutrition, postnatal feeding and early childhood adiposity, respectively, contribute to adult health outcomes. One of the earliest insights into the effects of the in utero environment on risk of obesity is from the evaluation of the Dutch “Hunger Winter:” the tragic food restriction imposed upon much of the Northern Holland population by Germany during World War II (27). Over a 6-month period (October 1944 to April 1945), caloric intake was markedly decreased from 1500 to 500 kcal, including that for pregnant women. Exposure to maternal undernutrition during the first or second trimesters increased the prevalence of obesity and cardiometabolic risk in the offspring at 18 years of age, when compared to those exposed in the third trimester.
There is significant controversy on the association of birth weight and later obesity risk. In a series of studies conducted since 1989, Barker et al. showed that individuals from the UK with low birth weight had higher risk of coronary heart disease and other risk factors associated with metabolic syndrome including T2D and elevated blood pressure in adulthood (23–26). Based on these studies, Barker proposed that chronic, degenerative conditions of adulthood, including heart disease and T2D, may be triggered by malnutrition during the in utero period (fetal “programming”), remain latent for many years, and manifest later in life. Deng et al. found that children from Guangzhong, China with high birth weight had higher odds of overweight/obesity compared to infants with normal birth weight (Odds ratio [OR] 2.42, 95% confidence interval (CI): 1.56; 3.76) (28). Large scale studies have confirmed a U-shaped relationship between birth weight and long-term outcomes, with both higher and lower levels associated with increased risk of different magnitude (29, 30). Further, there is an additive interaction of high birth weight and insufficient physical activity, such that children with high birth weight and lack of physical activity were 3.75 times more likely of being overweight or obese at age 7-9 years compared to those with normal birth weight and sufficient activity (OR 3.75, 95% CI: 2.06; 6.83) (28).
But why such discrepancies? Birth weight, although can be considered a marker of fetal health, is not a holistic reflection of the dynamics of intrauterine metabolic environment over time. As shown in the Dutch famine study, the gestational age during which maternal caloric restriction occurs may affect later obesity risk differentially, suggesting the presence of critical developmental windows. Further, there are several paths that can lead to a low birth weight independent of maternal body composition, food/caloric intake, or nutritional status. Mild prematurity, as well as intrauterine growth restriction secondary to uteroplacental insufficiency in the setting of chronic or gestational hypertension, smoking exposure, et cetera may also contribute to low birth weight, although studies in the UK, USA and Australia have shown that the associations between low birth weight and cardiovascular disease risk are not the result of such confounding variables (31–34).
More importantly though, while Barker’s hypothesis on the effects of intrauterine life on later health outcomes has been confirmed, the “fetal origins of disease” hypothesis does not consider the metabolic environment in the postnatal period, that is also known to affect future obesity risk. Indeed, considering the exponential rise in the prevalence of ASCVD since Barker’s studies, individuals with birth weight across the spectrum are likely to be impacted.
Other prenatal factors such as maternal diabetes and obesity, have been shown to contribute to obesity risk in children both during infancy and later in life. A large population-based comprehensive cohort study evaluated the association between diabetes in the mother (diabetes with insulin treatment, non-insulin treated diabetes and gestational diabetes), stratified by maternal prepregnancy BMI, and large for gestational age (LGA) at birth (35). Pregnant mothers with increasing BMI in the absence of diabetes had increasing odds of having an LGA offspring (36, 37). The adjusted OR for mothers with prepregnancy BMI of 25-29, 30-34 and ≥ 35 having LGA infants were 1.91, 2.45 and 3.38, respectively (95% CI: 1.83-2.00; 2.29-2.62; 3.08-3.71). Risk for LGA in offspring of mothers with obesity doubled in the presence of gestational diabetes and roughly tripled with T2D. Mothers who had insulin-treated diabetes had the highest risk of having LGA offspring regardless of BMI (35).
The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study, designed to assess the impact of hyperglycemia on short- and long-term outcomes on the offspring, showed higher frequency of birth weight over 90th percentile, primary Caesarean section, and cord-blood serum C-peptide over 90th percentile (a marker of fetal hyperinsulinemia) with increasing hyperglycemia while adjusting for maternal prepregnancy BMI (38, 39). Further, children of mothers with hyperglycemia showed higher levels of obesity, glucose intolerance and insulin resistance at 10-14 years of age (40, 41). These data suggest that maternal hyperglycemia, even if not clinically considered gestational diabetes, is an important risk factor for perturbations in energy homeostasis and obesity in children, independent from maternal hyperinsulinemia, insulin resistance or obesity, perhaps driven by a hyperglycemic in utero environment.
The 1996 National Longitudinal Survey of Youth, Child and Young Adult in the US showed that children at 2-14 years of age born to mothers with obesity prior to pregnancy were approximately four times more likely of being obese (95% CI: 2.6; 6.4, P < 0.001) (42). Additionally, a meta-analysis of data acquired from birth cohort studies from Europe, North America and Australia showed that higher maternal prepregnancy BMI and gestational weight gain were associated with higher risk of childhood overweight/obesity, most significantly in late childhood (10-18 years) (43).
Postnatal period
In the postnatal period, breastfeeding is found to have a protective effect against obesity (44). In a systematic review, Horta et al. showed that children (age 1-9 years), adolescents (age 10-19 years) and adults (≥ 20 years) who were breastfed as infants, have a reduction in the prevalence of overweight or obesity by 26% (95% CI: 21%; 32%), 37% (95% CI: 27%; 46%) and 12% (95% CI: 6%; 18%) respectively (45). The protective effect was more substantial in youth compared to adults, likely due to the accumulation of other environmental factors that may dilute the impact of breastfeeding over time. Further, while most studies were from high-income settings, the effect was consistent across income classifications. Breastfeeding during infancy is also associated with reduced odds of T2D in subjects aged 10-19 years [pooled OR: 0.46 (95% CI: 0.33; 0.66)]. However, this effect was not statistically significant in subjects older than 20 years of age, re-emphasizing the impact of other contributors in adulthood (45).
Early childhood
Weight gain during the first two years of life, is often used as a surrogate marker for overall metabolic status and has been shown to affect persistent risk for obesity (46, 47). Rapid weight gain during early infancy, especially during the first 4-6 months of life, is associated with higher risk of obesity both in childhood (at 6-8 years of age) and in adulthood (at 20 years). Eid et al. showed that the prevalence of obesity was greater in infants with rapid weight gain (9.4%) in the first 6 months of life compared to those with slower weight gain (1.9%) as early as 1970 (48). In 2002, in a large-scale prospective cohort study, Stettler et al. demonstrated a 38% (95% CI: 32%-44%) increased risk of overweight status in children aged 7 years for each 100 gram per month increase in weight gain during the first 4 months of life, independent of birth weight (49). Over two dozen studies have since shown a similar association between rapid early childhood weight gain and later obesity risk (50–54).
Accelerated weight gain during infancy also increases the long-term risk of insulin resistance (55). Singhal et al. measured 32-33 split proinsulin, a serum marker of insulin resistance, in adolescent populations (age 13-16 years) with a history of prematurity who received either nutrient-enriched or lower-nutrient diet during infancy. Premature infants fed nutrient-enriched formula had greater levels of fasting 32-33 split proinsulin (7.2 pmol/L, 95% CI 6.4–8.1) in adolescence compared to those fed lower-nutrient diet (5.9 pmol/L, 95% CI 5.2–6.4), with a mean difference of 20.6% (p = 0.01). Furthermore, fasting 32-33 split proinsulin concentration was associated with greater weight gain in the first two weeks of life (13.2% [5.4–20.9] change per 100 g weight increase; p = 0.001). These data indicate that relative undernutrition during the postnatal period in premature infants may be protective against development of insulin resistance later in life.
The UK-based Avon longitudinal study of parents and children (ALSPAC), found a significant association between weight gain in infancy and seven other independently associated factors with the risk of obesity in 7-year-old children: birth weight, parental obesity, sleep duration, television viewing, size in early life, catch-up growth and early adiposity or BMI rebound (before 43 months) (56, 57). Children in the highest quartile for weight at age 8 months and 18 months were more likely to have obesity at age 7 years with an OR of 3.13 (95% CI: 1.43; 6.85, p = 0.004) and 2.65 (95% CI: 1.25; 5.59, p = 0.011), respectively. Children with a history of rapid catch-up growth between 0-2 years were approximately 2.6 times more likely to be obese at 7 years of age (OR 2.60, 95% CI: 1.09;6.16, p = 0.002). Adiposity rebound, which corresponds to the second rise in BMI that typically occurs between 5-7 years of age, was associated with later obesity risk. Specifically, early adiposity rebound (by 61 months) and very early adiposity rebound (by 43 months) were both associated with risk of obesity at 7 years, with ORs of 2.01 (95% CI: 0.81; 5.20, p < 0.001) and 15.00 (95% CI: 5.32; 42.30, p < 0.001) respectively.
Collectively, these studies describe associations between in utero, postnatal, and early infancy factors that affect one’s metabolic environment, predisposing not only to childhood obesity but also metabolic syndrome. Before taking a deeper dive into why such associations may exist, we will first define obesity and briefly summarize the factors that affect whole-body adiposity and metabolic rate, both of which contribute to the pathogenesis of obesity.
A word on BMI and other measures of obesity
In studies discussed throughout this review, various markers of obesity were used as proxy measures of fat accumulation. BMI (weight [in kg]/height2 [in m]) is often used as a marker of excessive fat accumulation and is the most common measure of obesity in epidemiological studies over the age of 2 years. The World Health Organization (WHO) defines obesity in adults as BMI ≥ 30 kg/m2. In children under 5 years, BMI-for-age, and weight-for-height measurements according to specific growth standards are frequently used (47). The WHO defines obesity as weight-for-height ≥ 3 standard deviations above the median WHO Child Growth standards, while the Centers for Disease Control (CDC) defines childhood obesity as BMI ≥ 95th percentile for sex and age as per the CDC 2000 growth charts.
There are several limitations to the use of BMI as a marker of fat accumulation. The calculation of BMI includes both body weight and height. However, body weight reflects both fat mass and fat-free mass. Thus, BMI is not an accurate depiction of one’s fat mass. Furthermore, several factors have been shown to affect BMI including age, race, physical training, et cetera. For example, BMI may not be an accurate measure of fat mass in various South Asian populations where the “thin yet fat” phenotype is prevalent (58), or in athletes where BMI overestimates adiposity (59).
Body mass and body composition are more accurate measures of adiposity. However, these are not easily accessible and expensive for large-scale studies. Nonetheless, various instrumentations have been used to measure lean and fat mass in humans and animals including dual energy X-ray absorptiometry (DEXA), bioelectrical impedance analysis (BIA), nuclear magnetic resonance (NMR), air displacement plethysmography, and doubly labeled isotope water, many of which are becoming more easily accessible in academic centers (47). Other methods to study adiposity include measuring waist and/or hip circumference, as markers of central obesity, and skinfold thickness, as a marker of subcutaneous or visceral adiposity (triceps, subscapular biceps, abdomen). However, the lack of accuracy and reproducibility of such measurements limits their use (47, 58). More accurate, cost-effective, and easily attainable and reproducible measures of adiposity should continue to be developed to improve the validity of these studies and should be considered an important limitation of studies of obesity in children and adults conducted thus far.
While the WHO and CDC have a clear definition of obesity from a macroscopic standpoint, on a cellular and physiologic level, obesity is an interplay of complex factors involving several organ systems. Importantly, although fat accumulation is reflective of the obese phenotype, a relative decrease in lean mass, which contributes to BMR, plays an important contribution to the development of obesity.
The role of fat free mass in the conditioning of obesity
While measures of body weight and adiposity in childhood and adulthood are associated with one’s perinatal metabolic environment, the role of fat-free mass and BMR in the conditioning and pathogenesis of obesity remains unclear.
It was initially hypothesized that birth weight, which contributes to obesity in adulthood, would be negatively associated with fat-free mass and adult BMR. However, several studies have shown contrary results (60–62). Weyer et al. showed that while birth weight is positively associated with both fat-free mass as well as 24-hour adult EE, it is negatively associated with sleeping metabolic rate (62). While the clinical significance of this finding with regards to the pathogenesis of obesity is unclear, more thorough investigation may be needed to assess the effects of intrauterine metabolic milieu on adult fat-free mass and BMR. Further, the use of birth weight itself as a proxy measure for intrauterine nutritional status, is highly contentious. Perhaps more appropriate, future studies will determine the association between postnatal factors such as breastfeeding and early infancy weight gain, and adult fat-free mass to gain a better understanding of the role of BMR in the pathogenesis of obesity.
Thus far we have (1) discussed the global impact of childhood obesity, (2) defined obesity and factors which contribute to its pathogenesis including central and peripheral regulation of appetite, adiposity and BMR, (3) introduced the concept of metabolic conditioning and its role in the pathogenesis of obesity, and (4) presented data that supports the phenomenon of programming, which show an association between in utero, postnatal and early infancy metabolic factors and long-term obesity risk. In the next section, we will discuss how early developmental metabolic conditioning contributes to adult obesity risk.
Mechanisms of metabolic conditioning of obesity and cardiovascular disease risk
The conditioning of obesity in response to maternal undernutrition vs maternal obesity
The concept of metabolic programming for obesity was first demonstrated preclinically in the 1960s, although the term “metabolic programming” would be coined a few decades later (46). Robert A. McCance, an English scientist, found that overfeeding rats during the early postnatal period increased their body size in adult life. A similar outcome was demonstrated in male infant baboons by Lewis et al, where feeding a nutrient-enriched formula, with 30% excess calories, resulted in increased mesenteric and omental fat depots at 5 years (46, 63).
The Dutch famine study linked the metabolic environment of the fetus and later obesity risk with the specific gestational period of maternal undernutrition. While human fetal adipose depots develop between 14-24 weeks of gestation, they undergo significant expansion only in the third trimester, which is dependent on fat cell replication (64, 65). On the other hand, hypothalamic neurons develop by 15-18 weeks’ gestation and hormone activity in the hypothalamic-pituitary-adrenal axis can be seen as early as 8-12 weeks’ gestation and continues to mature in subsequent weeks (66). In the cohort exposed to poor nutrition in the third trimester, it is possible that the lack of maternal caloric intake restricted expansion of fetal adipose tissue, resulting in the programming of adipocytes with decreased propensity for replication, even in the presence of sufficient caloric intake. On the other hand, children exposed to nutrient deprivation in the first two trimesters, were programmed for increased responsiveness to caloric cues, cravings, or appetite due to insufficient energy substrate availability during the critical development period of the hypothalamus.
Malnutrition during the first two trimesters results in a “thrifty” phenotype, commonly known as the “predictive adaptive response”. Such conditioning becomes maladaptive in the presence of sufficient or excess nutrition later in childhood, adolescence, or adulthood. It is this mismatch between developmental conditioning and paradoxical exposures later that ultimately lead to adverse health outcomes and the risk for non-communicable disease. When the environment changes, the conditioning becomes nonadaptive. We hypothesize that a more thorough understanding of the temporo-spatial progression of obesity from early development will aide in the discovery of therapeutic interventions to prevent or reverse these maladaptive processes.
The concept of mismatch has been studied more comprehensively in preclinical models (21, 67–69). Using a rat model, Vickers et al. showed that maternal undernutrition during pregnancy promotes obesity and metabolic syndrome in male and female offspring, that is worsened by high-fat diet (HFD) feeding in the postnatal period (68). Administering leptin in the early neonatal period of 3-13 days, a critical period characterized by a high degree of developmental plasticity, reversed this phenotype (68). Gluckman et al. showed that leptin treatment in this critical postnatal period modified the transcription and methylation of genes expressed in the liver that regulate whole-body energy homeostasis including peroxisomal proliferator-activated receptor alpha (PPARα); however, the metabolic “program” and direction of these changes depended on the initial perinatal environment (21). Using the same rat model of maternal undernutrition with postnatal hypercaloric nutrition, Vickers et al. also showed that IGF-1 treatment alleviated hyperphagia, obesity, hyperinsulinemia and hyperleptinemia in offspring from malnourished dams (69).
This concept of developmental mismatch, however, does not apply when the exposure remains consistent. While maternal undernutrition may condition a “thrifty phenotype” resulting in offspring who develop obesity with exposure to high caloric diet later in life, how does maternal obesity result in obese offspring? One could hypothesize that like the evolutionary anticipation of possible future undernutrition in offspring born to malnourished mothers, offspring born to obese mothers should also develop a “predictive adaptive response” for future overnutrition and thus have reduced propensity for adipocyte replication and increased levels of satiety with feeding. However, several epidemiological studies, discussed previously, have shown the contrary: maternal obesity increases adiposity in offspring and programs later obesity possibly through obesogenic modifications in appetite regulation and energy metabolism (35–37, 42, 43). Preclinical studies have demonstrated the effects of maternal obesity on offspring adiposity phenotype (70–73). For example, Chen et al. noted that HFD feeding in female rats resulted in increased body weight and adiposity, hyperlipidemia and glucose intolerance in male pups at postnatal day 20 (71). White et al. showed that rat offspring at 18 weeks of age from dams fed a HFD prior to pregnancy, weighed significantly more than offspring from dams fed a normal diet (74).
Hanson and Gluckman in their comprehensive review of the DOHaD hypothesis, argue that maternal undernutrition (an integral thread throughout human history) has been recognized, evolutionarily, as harmful for normal development thus requiring a predictive adaptive response in order to produce fertile, viable offspring (17). Maternal obesity, on the other hand, is a relatively newer “stress” which is not yet recognized in our collective epigenetic or genetic code as maladaptive to development, since it was vanishingly rare only 10 generations ago, and the majority of offspring from mothers with obesity are viable into adulthood and capable of reproduction (17). In contrast to maternal undernutrition, maternal obesity likely plays a pathophysiological role in the development of obesity in offspring by altering the metabolic milieu of intrauterine life (Figure 2). We submit that altered maternal levels of metabolic substrates and hormones in the setting of obesity, which contribute to maternal insulin resistance, oxidative stress and inflammation, cause perturbations in glucose and lipid homeostasis and feeding circuitry in the perinatal period, ultimately contributing to long-term obesity risk. These pathophysiological factors will be discussed in detail in the next section.
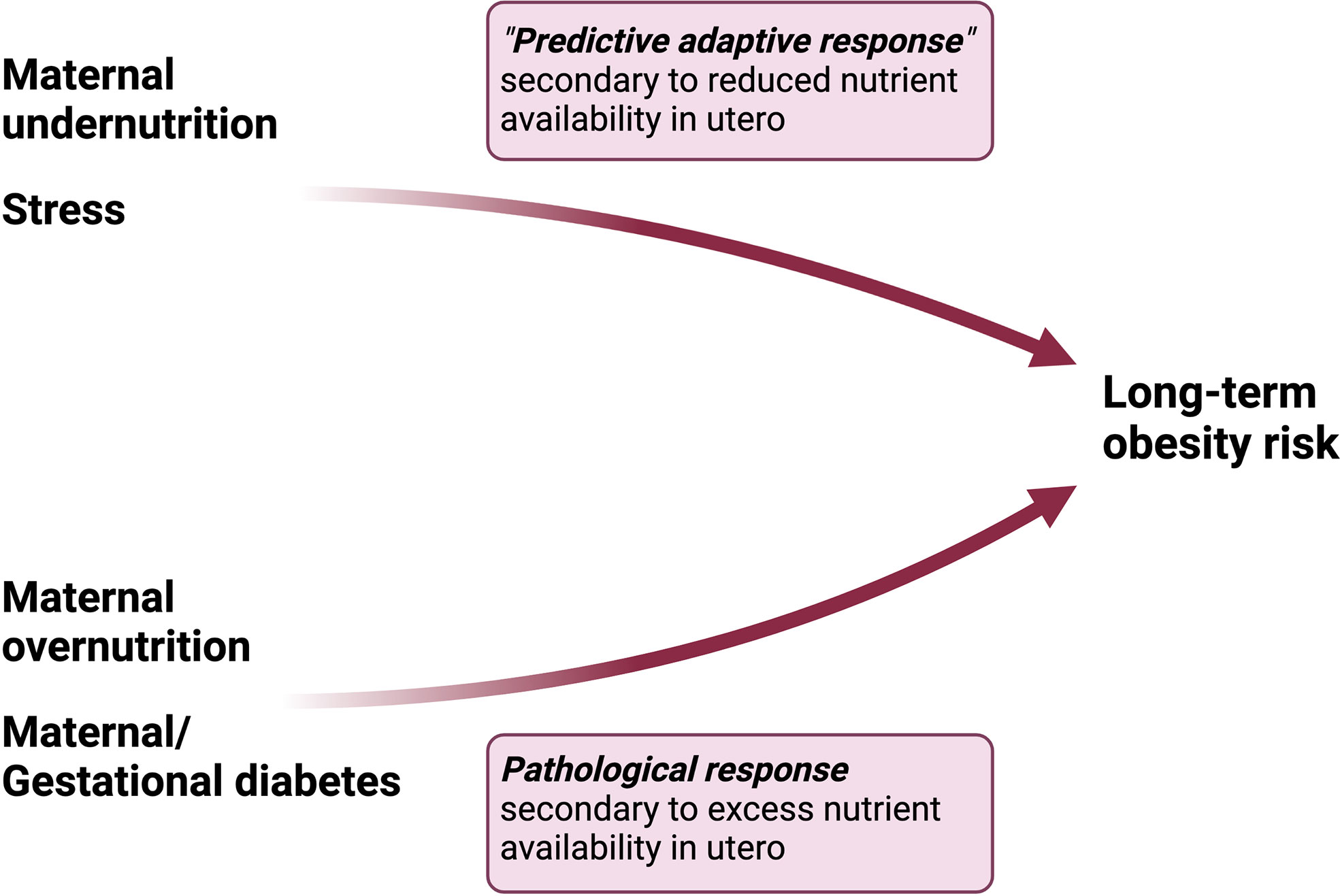
Figure 2 Differential pathways to obesity in response to maternal undernutrition vs overnutrition. Maternal undernutrition is hypothesized to condition obesity through a “predictive adaptive response,” in which the fetus programs its homeostatic set points in anticipation of persistent caloric deficit. However, the adaptive response becomes maladaptive in the setting of a mismatch between the anticipated environment and paradoxical exposures later in life. On the other hand, maternal overnutrition activates a pathological response in the fetus characterized by dysregulation in hormonal pathways involved in energy homeostasis.
Maternal obesogenic factors that mediate the conditioning of obesity
Circulating metabolic substrates
Maternal obesity is associated with elevated levels of metabolic substrates including free fatty acids (FFAs), glucose and altered lipoprotein profile. FFAs and glucose can independently contribute to fetal overgrowth due to increased availability of substrates (75). Development of maternal insulin resistance and a proinflammatory state in the setting of hyperglycemia and hyperlipidemia may also contribute to an abnormal metabolic transition in early postnatal life (75). Further, exposure to elevated metabolic substrates may cause direct glucotoxic and lipotoxic effects on placental proteins, which in turn have been shown to contribute to oxidative stress, endoplasmic reticulum (ER) stress and proinflammatory pathways affecting overall placental function (76). In response to increased placental transport of glucose to the fetus, fetal hyperinsulinemia develops which is hypothesized to contribute to obesity conditioning in the offspring (77). Intrahypothalamic administration of insulin to rat pups during early development of central feeding circuitry increases body weight, impairs glucose tolerance, and promotes development of obesity and diabetes later in life (78), emphasizing its role in body weight regulation in addition to being a potent growth factor (79).
Circulating hormones
Insulin and leptin
Maternal obesity is associated with hyperinsulinemia, however maternal insulin does not cross the placenta (75, 80). While the pregnant state is naturally associated with peripheral insulin resistance to maximize glucose availability for the developing fetus, pregnant women with obesity have 50-60% higher postprandial insulin levels and are more glucose intolerant than those without obesity. Regardless, the placenta remains normosensitive to insulin in women with obesity, thus, increased placental insulin activation, along with leptin, has been shown to promote mTOR signaling leading to increased glucose and amino acid transport across the placental barrier and fetal overgrowth (81).
It is unclear which components of abnormal glucose homeostasis associated with maternal obesity – maternal hyperglycemia, hyperinsulinemia, or insulin resistance – contribute to the dysregulation of metabolic homeostasis in offspring and future obesity risk. As discussed before, the HAPO study showed that maternal hyperglycemia, in the absence of overt diabetes, increases frequency of LGA and fetal hyperinsulinemia (38), aligning with the concept that increased glucose availability and placental uptake contributes to fetal overgrowth (75). Although insulin does not cross the placental barrier, maternal hyperinsulinemia may exert effects on the placenta allowing for increased fetal nutrient uptake. While difficult to isolate the effects of maternal insulin resistance without overt diabetes on outcomes in humans, using a heterozygous knockout mouse model for insulin receptor, Carmody et al. studied the effects of maternal insulin resistance with or without maternal HFD on progeny. Maternal insulin resistance alone did not affect body weight, body composition, glucose homeostasis or expression of hypothalamic neuropeptides that regulate feeding behavior, suggesting that the intersection of maternal insulin resistance with HFD feeding is required for the metabolic programming of obesity (80). Future clinical and preclinical studies are required to isolate the individual contributions of maternal hyperglycemia, hyperinsulinemia, and insulin resistance on perturbations in energy homeostasis and offspring obesity risk.
Leptin is produced by adipose tissue, and circulating levels in weight stable adults correspond to the degree of adiposity (82). At the placental level, leptin has been shown to increase system A amino acid transport activity and stimulates the release of proinflammatory cytokine IL-6, possibly contributing to abnormal placental function (75). In the hypothalamus, insulin and leptin modify expression of neuropeptides that regulate appetite and feeding behavior. Specifically, they increase expression of anorexigenic (appetite-suppressing) precursor of alpha melanocyte stimulating hormone (α−MSH) pro-opio melanocortin (POMC) and decrease expression of orexigenic (appetite-promoting) neuropeptide Y (NPY), overall functioning to suppress appetite (82). Thus, with worsening adiposity and hyperglycemia, increasing circulating levels of leptin and insulin aid to maintain the homeostatic set point by suppressing appetite. Ensuing central and peripheral resistance to these anorexigenic hormones both characterizes and contributes to obesity and metabolic syndrome. Although not extensively studied in models of metabolic programming, insulin and leptin resistance in offspring exposed to maternal HFD in utero may also contribute to deregulation in energy homeostasis (73, 83–85). In a rat model, Gupta et al. showed that maternal HFD feeding in utero increased expression of leptin long receptor (ObRb) and insulin receptor b-subunit (Ir-β) in the offspring. The downstream leptin and insulin signaling components including signal transducers and activators of transcription-3 (STAT3) and insulin receptor substrate-2 (Irs2), respectively, were decreased (73). Morris et al. found that maternal overnutrition during gestation in Sprague-Dawley rats significantly decreased expression of ObRb, without significant alteration in STAT3 expression (85). Isolated elevated developmental leptin exposure can alter adult weight homeostasis in mice. Using an inducible leptin mouse model, Skowronski et al. expressed leptin untethered to obesity and demonstrated that leptin overexpression during the weaning period (corresponding to the third trimester in human brain development) caused the mice to be overly suspectable to obesity when subsequently presented a highly palatable diet (86).
Preclinical studies have shown that HFD feeding in rats and mice promotes maternal obesity as well as maternal hyperinsulinemia and hyperleptinemia. Offspring of HFD-fed rats have elevated levels of insulin and leptin that alters expression of neuropeptide hormones Npy and Pomc and may exert long-term impact on the structure and function of hypothalamic feeding circuits (71–74, 85). Chen et al, for example, found that offspring of HFD-fed have increased mRNA expression of appetite-promoting Npy but lower expression of appetite-suppressing Pomc at 20 days of life, which may represent an adaptive response to obesity (71).
The contrasting role of leptin in maternal models of undernutrition vs HFD feeding may be perplexing at first glance. We previously discussed that leptin administration in pups exposed to undernutrition in utero reversed the obese phenotype. However, in offspring exposed to a HFD in utero, both leptin and insulin are initially elevated in early life. The leptin surge in the perinatal period is critical for the normal development of hypothalamic circuitry regulating energy homeostasis, appetite and feeding behaviors (87). Both the timing and magnitude of that surge appears to be critical and if either absent (as in states of maternal undernutrition) or inappropriately elevated over a prolonged period of time (as in states of maternal overnutrition), an obese, hyperphagic phenotype ensues (77, 88).
These studies suggest that insulin and leptin play critical roles in maintaining whole body energy homeostasis and perturbations in their expression during critical time periods of development may impair long-term regulation of appetite and feeding behaviors, ultimately contributing to an obese phenotype later in life.
Insulin-like growth factor
IGF-1 and IGF-2, which are predominantly synthesized in the liver, regulate fetal growth and development, as well as carbohydrate metabolism (75). Both IGF-1 and IGF-2 deficiency in mice demonstrate prenatal and postnatal growth failure (89, 90). In a human choriocarcinoma cell line, IGF-1 was shown to promote trophoblast proliferation and stimulate glucose and amino acid transport, and IGF-2 deletion reduced mouse placental growth and passive nutrient delivery, suggesting key roles for IGFs in promoting fetal overgrowth (75, 90, 91).
Adiponectin
Adiponectin inhibits insulin signaling in trophoblasts. Maternal obesity is associated with low levels of adiponectin, which is thought to promote placental insulin signaling, fetal nutrient transport and in turn fetal overgrowth (75). Adiponectin supplementation in pregnant mice normalizes maternal insulin sensitivity, placental insulin/mTORC1 signaling, nutrient transport and fetal growth, despite minimal changes in maternal visceral adiposity (92). A long-term study in the same mouse model showed in utero adiponectin treatment ameliorated the obesogenic phenotype in 14-week-old adult male offspring, with significant reductions in body weight, fat mass and normalization of insulin sensitivity and hepatic steatosis (93).
Placental signaling contributes to the effects of maternal obesity on offspring
Maternal circulating levels of metabolic substrates and hormones, as discussed above, significantly impact placental function, likely mediated by mTOR signaling on the placenta by activating amino acid and glucose transport as well as mitochondrial biogenesis and protein synthesis, ultimately contributing to excess fetal nutrient delivery with increased risk for fetal overgrowth in obese women (75, 76).
Maternal obesogenic factors could be conveyed into long-term obesity risk in offspring via epigenetic modifications
Several maternal obesogenic factors contribute to obesity in offspring, including glucose, FFAs, insulin, leptin, IGF-1 and adiponectin. Together, they promote maternal insulin resistance, hyperglycemia, hyperleptinemia, oxidative stress and inflammation in utero which perturb the development of central feeding circuitry and energy homeostasis during the perinatal transition. The ensuing fetal hyperleptinemia, insulinemia and transient neonatal hypoglycemia increase the risk for fetal overgrowth and LGA (Figure 3). But how does this short-term dysregulation in energy homeostasis during the perinatal period affect long-term risk for obesity? We posit that the in utero environment caused by maternal obesity, and the concomitant short-term perinatal aberrations in energy homeostasis, translate to long-term obesity risk through epigenetic modifications in both central and peripheral markers that regulate energy homeostatic set points.
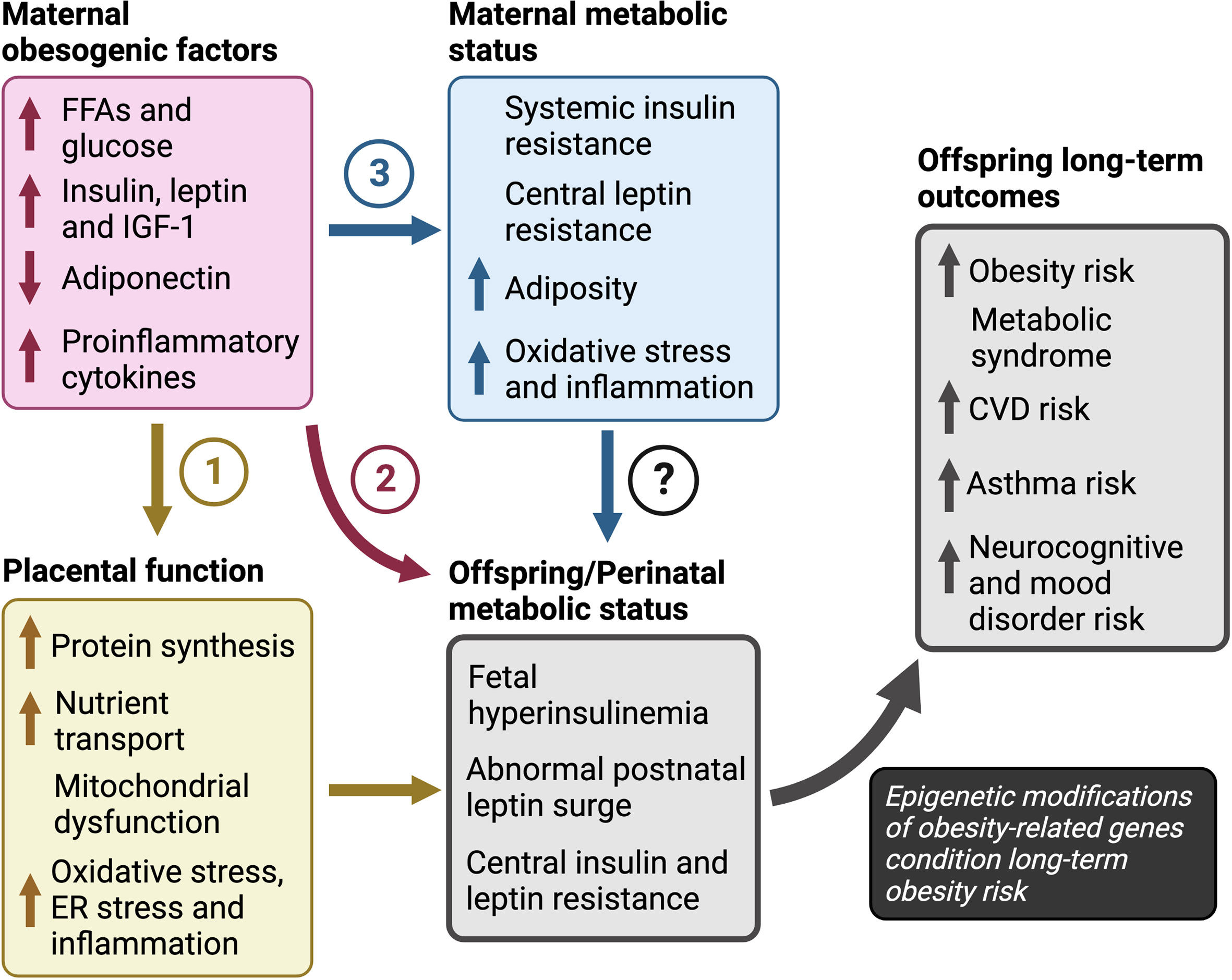
Figure 3 Pathways to obesity in children in response to maternal overnutrition The pathways of maternal obesogenic factors towards long-term obesity risk include: (1) dysregulation of placental function in which excess nutrients promote placental protein synthesis, mitochondrial dysfunction and increased nutrient transport, (2) increased nutrient availability, that may have a direct effect on fetal growth and development independent of changes in placental function, and (3) altered maternal sensitivity to hormones leptin and insulin. However, the roles of maternal insulin and leptin resistance, independent of maternal hyper-insulinemia or -leptinemia, are unclear and further investigation is required to determine their impact on the perinatal metabolic transition to life and outcomes in offspring. Finally, we propose that increased risk for obesity, metabolic syndrome, and atherosclerotic cardiovascular disease, depends on an abnormal metabolic status during the perinatal period in response to maternal factors. CVD; cardiovascular disease, ER; endoplasmic reticulum, FFAs; free fatty acids, IGF-1; insulin-like growth factor-1.
Epigenetics is the study of heritable changes in gene expression secondary to chromatin modifications such as DNA methylation, as well as histone and miRNA modifications, without an actual change in DNA sequence (94). DNA methylation contributes to gene expression and is considered one of the primary mechanisms of cellular memory. Specifically, methylation of the 5’ position of cytosine by DNA methyltransferases to form 5-methylcytosine (5mC) impedes binding of transcription factors, which ultimately represses transcription of the gene. On the other hand, hypermethylation of CpG islands can promote transcriptional activation (95). Such epigenetic changes to gene expression are critical for normal cell differentiation and development and maturation of organ systems early in life (96, 97). In response to an abnormal metabolic environment in utero and in the perinatal period, alterations in epigenetic modifications of obesity-related genes can have lasting effects on gene expression, ultimately contributing to obesity risk (21, 98–100).
Many epigenetic modifications have been identified to play a role in the pathogenesis of obesity. For example, altered methylation patterns have been seen in genes that regulate systemic energy homeostasis such as HIF3A (hypoxia-inducible factor 3A), LEP (leptin) and ADP (adiponectin), as well as POMC, PPARα, PGC1α, IGF-2, IRS-1, IL-6 (other genes involved in whole-body lipid and carbohydrate metabolism, insulin signaling and inflammation). Hypermethylation patterns have also been identified in genes related to the circadian rhythm –CLOCK, BMAL1 (in leukocytes and adipocytes). Histone modifications of genes related to adipogenesis and adipocyte differentiation include PREF-1, C/EBPα/β, PPARγ and aP2. Finally, expression of certain miRNAs are associated with increased fat storage in adipocytes and may upregulate adipogenesis in obese states. For example, miR-26b promotes proliferation of preadipocytes and expression of PPARγ (94, 95). Epigenetic changes can be stable in an organism from one cell to its progeny through cellular division and mitosis, but also across generations and, are thus, considered inheritable. For example, the DNA methylation status of the Agouti focus, responsible in determining fur coloration, is passed on transgenerationally through maternal inheritance (94, 95).
An elegant study performed by Masuyama et al. showed that exposure to a HFD in utero results in an obese phenotype in offspring over three generations through the maternal line along with elevated plasma and tissue level expression of leptin and reduction in adiponectin (101). The adiponectin level was driven by increased acetylation and decreased methylation of H3K9 at the promoter region of adiponectin gene in offspring of HFD-fed mice. In contrast, monomethyl H4K20 levels were increased in the Lep promoter region of these same offspring. Reversal of the phenotype and epigenome was only possible by chow fed diet feeding in three subsequent generations (101).
Obesity conditioning occurs primarily during critical developmental periods
The critical periods of metabolic programming extend from fetal, perinatal, as well as early postnatal stages (17, 102). During the fetal stage and metabolic transition to postnatal life, there is a high degree of plasticity and epigenetic modifications have been demonstrated in key regulatory genes which affect the development of the hypothalamus and its peripheral connections (66, 94, 95). Further in mammals, significant structural and functional development of adipose tissue, liver and gut occurs after birth. Thus, the postnatal period serves as an important continuation of the fetal phase (102).
For example, in rats and mice, the hypothalamic-pituitary axis is immature at the time of birth and undergoes maturation in the first 2 weeks of life (66, 87, 94, 103). Variations in DNA methylation between hypothalamic and non-neuronal cells are established in the postnatal period (104). In humans, DNA methylation of prefrontal cortex involved in appetite control, satiation, and food craving, increases steadily overtime, exhibiting prolonged postnatal maturation (94, 102–104).
In rodents, white adipose tissue (WAT) is minimal at the time of birth and undergoes postnatal maturation (102). Epididymal WAT is composed of progenitor cells that lack the capacity to differentiate from postnatal days 1 to 4, following which dividing cells are observed. In humans, although WAT is present from the second trimester of gestation, significant growth occurs from birth to 6 weeks of age, during which time body fat doubles from approximately 10% to 20%. After birth, both adipocyte size and number increases accounting for the large adipose tissue expansion (64, 65, 94, 102).
Finally, the liver acquires its metabolic functions and undergoes most of its methylation postnatally (102). Indeed, regulatory regions, including promoters and enhancers of genes involved in lipid and glucose metabolism in the liver, undergo programmed active DNA demethylation in a time-dependent manner after birth. This process has been shown to be partly dependent on the activity of ten-eleven translocation dioxygenases, otherwise known as “TET” enzymes (Tet2 and Tet3), which promote DNA demethylation (105).
Despite the high degree of plasticity during the perinatal period, several other factors may result in epigenetic modifications that alter the long-term ability to mobilize fat beyond these critical developmental periods. These include environmental toxins known as “obesogens,” changes in gut microbiota and excess dietary intake during early childhood (94, 102). We argue that epigenetic changes are not exclusive to the perinatal period and that these changes may occur along any time in response to various behavioral and environmental factors.
Epigenetic modifications, in response to early environmental exposures, are known to persist into adulthood. For example, the cohort of children from the Dutch Hunger Winter exposed to maternal caloric restriction in the first trimester of gestation had less DNA methylation of the IGF2 gene compared with their unexposed siblings six decades later (94, 106). However, epigenetic changes are not confined by longevity or permanence. Indeed, contrary to the initial understanding of epigenetic mechanisms in the programming of obesity, we now know that these changes are highly dynamic and reversible.
If one’s metabolic milieu is altered in favor of weight loss, the epigenetic alterations in DNA methylation may be reversible (94). As discussed previously, the methylation patterns of the promoter regions of leptin and adiponectin genes in mice exposed to a HFD in utero, were reversed in three generations upon transitioning to a normal chow diet (101). Further, dietary changes, exercise, and surgical interventions may reverse epigenetic changes. For example, maternal bariatric surgery and associated weight loss improved the metabolic profile of children during their adolescent period compared to their siblings born prior to weight loss surgery, with decreased birth weight, obesity incidence, blood pressure and adiposity, as well as improved insulin sensitivity and lipid profile (107, 108). Siblings born after weight loss surgery exhibited differences in epigenetic modifications in genes related to glucose homeostasis, insulin resistance, inflammation, and vascular disease (99).
The discovery of ten-eleven translocation dioxygenases enzymes, which reverse DNA methylation, has made the mechanistic underpinnings of methylation reversal a possibility (102, 105). “Ten-Eleven Translocation dioxygenases” or TETs convert 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), that can then undergo serial oxidation resulting in active DNA demethylation via removal and substitution with unmethylated cytosine via base excision repair. Indeed, 5hmC is detected in several different cell types near regulatory regions (promoters/enhancers) and positively correlates with gene expression (105).
Epigenetic modifications during different time periods of development affect later obesity risk
Prenatal period
The currently known epigenetic pathways that contribute to the conditioning of obesity include variations in methylation status of obesity-related genes associated with differences in adiposity and body composition in infancy and beyond. An elegant longitudinal clinical study measured the methylation status of CpGs in the promoters of candidate genes from umbilical cord tissue of neonates obtained at birth along with clinical measures of adiposity at 9 years of age (98). Higher methylation level of retinoid X receptor-alpha (RXRα) at birth was strongly correlated with adiposity in childhood. RXRα is a transcription factor that regulates expression of genes including PPARα involved in whole-body lipid and glucose metabolism, insulin sensitivity and adipogenesis, suggesting an association between early life epigenetic modifications and later obesity risk.
Maternal weight loss surgery is known to reduce the risk of offspring obesity without large maternal weight loss (100, 108). Children born to mothers who underwent biliopancreatic diversion prior to pregnancy had significantly reduced odds of developing obesity from 2-18 years of age, even if the mothers remained overweight after surgery. Altered methylation patterns and expression of genes involved in glucose metabolism, insulin sensitivity, immune and inflammatory-related functions was noted in the cord blood after the surgery (99). We hypothesize that overall improved nutritional status during gestation with concomitant decreased maternal insulin resistance, hyperglycemia, hyperlipidemia, and hyperinsulinemia contribute to a healthy in utero environment translating to better offspring outcomes.
Preclinical studies have also shown that variations in the methylation status of obesity-related genes, in response to abnormal maternal diet in utero, are associated with weight gain and obesity in the offspring (109–112). Jousse et al. found that maternal undernutrition in mice during gestation and lactation produces hyperphagic offspring with higher food intake throughout life (112). Mice exposed to maternal undernutrition showed persistent changes in serum levels, mRNA expression and methylation patterns of leptin. Fetal malnutrition results in lower leptin levels with corresponding decreased adipose-specific mRNA expression, as well as long-term changes in methylation of the leptin promoter (112).
Lillycrop et al. found that maternal protein restriction in female rats during pregnancy decreased methylation of hepatic glucocorticoid receptor (Gr) and Pparα, and increased expression of mRNA transcripts in pups at 6 days of life (109). These epigenetic changes persisted after weaning (while pups were no longer on decreased protein intake). A follow-up study showed persistence of changes in methylation of Pparα at 34 and 80 days, corresponding to the adolescent and adult periods respectively (113). Further, folic acid supplementation either during gestation or in adolescence prevented or reversed these epigenetic modifications respectively (109, 114). As DNA methylation requires S-adenosylmethionine as a methyl donor, which is dependent upon serine, glycine, folate, and vitamin B12, epigenetic modifications may be altered by availability of these amino acids and micronutrients in utero. These data suggest that maternal nutritional status alters the fetal epigenome and the expression of energy homeostasis genes throughout lifespan (109, 110, 113).
Gluckman et al. showed that maternal protein restriction and post-weaning hypercaloric nutrition followed by leptin treatment from days 3 to 10 of life, reversed the obese phenotype in offspring described previously, and resulted in stable and long-term changes in the methylation patterns and expression of various markers later in life (day 170) (21). Methylation of the Pparα promoter was either (1) decreased by leptin treatment in offspring exposed to maternal undernutrition in utero, corresponding to enhanced transcript expression, or (2) increased by leptin treatment in offspring without exposure, corresponding to decreased transcript expression. Offspring transitioned to a HFD post-weaning obscured the effects of leptin on Pparα expression. Methylation of the Gr promoter, on the other hand, was elevated by leptin in maternally well-nourished offspring but unaffected in offspring exposed to maternal protein restriction in utero. Finally, leptin also had bidirectional effects on the expression of hepatic 11β-hydroxysteroid dehydrogenase type 2 (11β-Hsd2), which is an enzyme that inactivates glucocorticoids. While neonatal leptin treatment decreased 11β-Hsd2 in offspring from undernourished mothers, it increased 11β-Hsd2 in offspring from well-nourished mothers (21). Using a rat model of overfeeding in which mothers developed obesity preconception, Borengasser et al. showed altered methylation patterns and corresponding expression of adipogenic genes in white adipose tissue of offspring (115). Offspring of obese dams showed decreased methylation of C/ebp-β, Zfp324 and Pparγ, which are involved in the development of adipose tissue, as well as corresponding increased expression of their downstream targets, ultimately resulting in enhanced adipogenic differentiation and greater adiposity in offspring. Fernandez-Twinn et al. showed that HFD-feeding during gestation in a mouse model produced offspring with unchanged body composition, energy expenditure, activity levels or glucose tolerance at 8 weeks of age; however, fasting insulin levels were significantly higher compared to mice from chow-fed mothers (116). Interestingly, protein expression of Irβ was downregulated in epididymal fat in offspring from HFD-fed mothers, in addition to further downstream markers of insulin signaling including Irs-1, Pi3k, Akt1 and Akt2. Further miR-126 was found to regulate the expression of Irs-1, with a potential role in the epigenetic programming of insulin resistance.
In a genetic mouse model of maternal obesity and diabetes, offspring at 12 weeks of age displayed elevated liver lipid contents, as well as elevated serum leptin (117). Offspring exposed to maternal obesity and diabetes and subsequently challenged with a western-style diet gained significantly more weight and demonstrated glucose intolerance and insulin resistance, compared to offspring from control mice. These phenotypic changes corresponded to variations in hepatic gene expression, specifically those regulating mitochondrial activity such as Atpase6 and Cytb. The epigenetic alterations were not restricted to genes involved in energy homeostasis but were widespread relating to embryonic and tissue/organ development.
In another mouse model of diet-induced maternal obesity, significant alterations in histone acetylation and methylation of hepatic transcripts involved in lipid metabolism were observed in offspring exposed to maternal overnutrition during gestation, which persisted into adulthood (5 weeks of age) (118). Specifically, Sirt1, which encodes a histone deacetylase with functions in lipid metabolism and obesity (serving as a marker for cellular energy levels), was significantly reduced in offspring exposed to maternal obesity and persisted into adulthood. Further, differential histone modifications were also seen in Pparα, Pparγ, Rora and Rxrα in the offspring from obese mothers that were not sustained at 5 weeks.
Postnatal period
Several human studies show a differential methylation pattern of LEP promoter in breast fed infants (102, 119–122). A cross-sectional study in 120 Dutch children found that longer breastfeeding duration reduced the average CpG methylation of LEP promoter in peripheral blood and was negatively associated with plasma leptin and infant BMI at an average age of 1.4 years (120). In a prospective cohort study, Pauwels et al. noted that every extra month of breastfeeding was associated with a 0.217% increase in RXRα CpG2 methylation (95% CI: 0.103, 0.330; p < 0.001). Similarly, increased levels of CpG3 methylation of LEP promoter was seen with 7-9 months of breastfeeding (6.1%) compared to 1-3 months (4.3%; p = 0.007) that did not persist at 10-12 months. However, infant weight and BMI-for-age at 1 year was significantly lower in children who were breastfed for 10-12 months, suggesting that breastfeeding-mediated epigenetic modifications in RXRα and LEP may have a role in childhood obesity. In a longer prospective cohort study, Sherwood et al. demonstrated that LEP methylation at four CpG sites at 10 years of age was associated with exclusive breastfeeding (121).
Rodent models also show an association between postnatal feeding and differential methylation patterns of obesity-related genes. Rats overnourished during the breastfeeding period by reduction in litter size have increased growth rate, hyperinsulinemia and hyperleptinemia, and overweight throughout life with central leptin and insulin resistance in adulthood (123–125). In another rat model of postnatal overnutrition, Mahmood et al. demonstrated hyperinsulinemia persistent in the post-weaning period in newborn rats fed a high-carbohydrate milk formula (126–128). These animals developed hyperphagic obesity in adult life on a standard chow diet along with associated epigenetic alterations in Npy and Pomc (128). While Npy mRNA expression was increased in the hypothalami of both 16- and 100-day old high-carb fed rats, with increased methylation of specific CpG positions and histone acetylation, Pomc expression was reduced, accompanied by a decrease in histone acetylation.
Postnatal exposures can reverse the gestational programming of obesity
Several studies suggest that postnatal exposures may have a greater impact compared to in utero exposures (100, 129). Using a rat model of diet-induced obesity in which Sprague-Dawley rats were selectively bred for diet-induced obesity and resistance, Gorski et al, found that obesity-prone pups fostered to lean dams at birth remained obese in spite of reduction in food intake and gradual improvement in insulin sensitivity compared to obesity-prone pups fostered to obese dams (129). They noted an upregulation in hypothalamic expression of leptin receptor (Lepr-b) and insulin receptor (Ir), suggesting improved leptin sensitivity. On the other hand, lean pups fostered to obese dams during lactation develop obesity and insulin resistance when fed high-caloric diet, with a decrease in expression of Lepr-b and Insr.
Obesity programming may also have a role in the development of the neuronal projections relevant to energy homeostasis. In a study of offspring born to dams fed a HFD during lactation, Vogt et al. observed no differences in the expression of Pomc, Npy and agouti-related peptide (AgRP). However, they noted impaired Pomc- and Agrp- neuronal projections to target sites within the hypothalamus (130).
Finally, as discussed previously, rat models of maternal food restriction during pregnancy cause intrauterine growth restriction in offspring who later show rapid catch-up growth with adult obesity and metabolic syndrome (21, 109, 111–113). This phenotype can be prevented through continued maternal undernutrition during the lactation period, where availability of nutrients is also limited to the newborn (131, 132). Tosh et al. showed that maternal food restriction during both pregnancy and lactation decreased hepatic mRNA and protein expression of Igf1, that persisted at 9 months of age (132). Transitioning the dams to a regular diet during the lactation period rescued the expression of Igf1 in the adult offspring, both in transcript expression and serum levels.
Discussion
Future directions to prevent the evolution of obesity from conception
Summary and additional remarks
Early exposure to maternal obesogenic factors from conception “programs” long-term obesity risk through epigenetic modifications in genes involved in energy homeostasis. While there are several risk factors for childhood obesity, maternal overnutrition during pregnancy has consistently been shown to alter the fetal epigenome. The effects of epigenetic alterations often persist into adulthood, contributing to the long-term obesity risk in offspring. Observational studies suggest that maternal diet, rather than maternal weight in isolation, may be the driver of the epigenetic modifications during the perinatal period. Previously described study of siblings born before and after maternal bariatric surgery found that significant weight reduction or normalization of BMI was not necessary to reduce the risk of obesity in offspring, although the systemic effects of bariatric surgery on programming of feeding behavior and whole-body metabolism in offspring remain unclear (108). Further, the Dutch famine study and studies on rodent models have shown that maternal undernutrition also predisposes to longer-term obesity in offspring (21, 82, 106, 109, 111–113). Thus, we hypothesize that the in utero metabolic environment, which is significantly influenced by maternal diet, determines the predisposition to obesity in offspring.
In this review, we have described several markers that have been shown to be susceptible to epigenetic modifications during critical developmental periods to promote an obesogenic phenotype during lifespan. These obesity-related genes are primarily expressed in the hypothalamus, liver, adipose tissue, and pancreas and serve as potential therapeutic targets for both the treatment and prevention of obesity, summarized in Table 1.
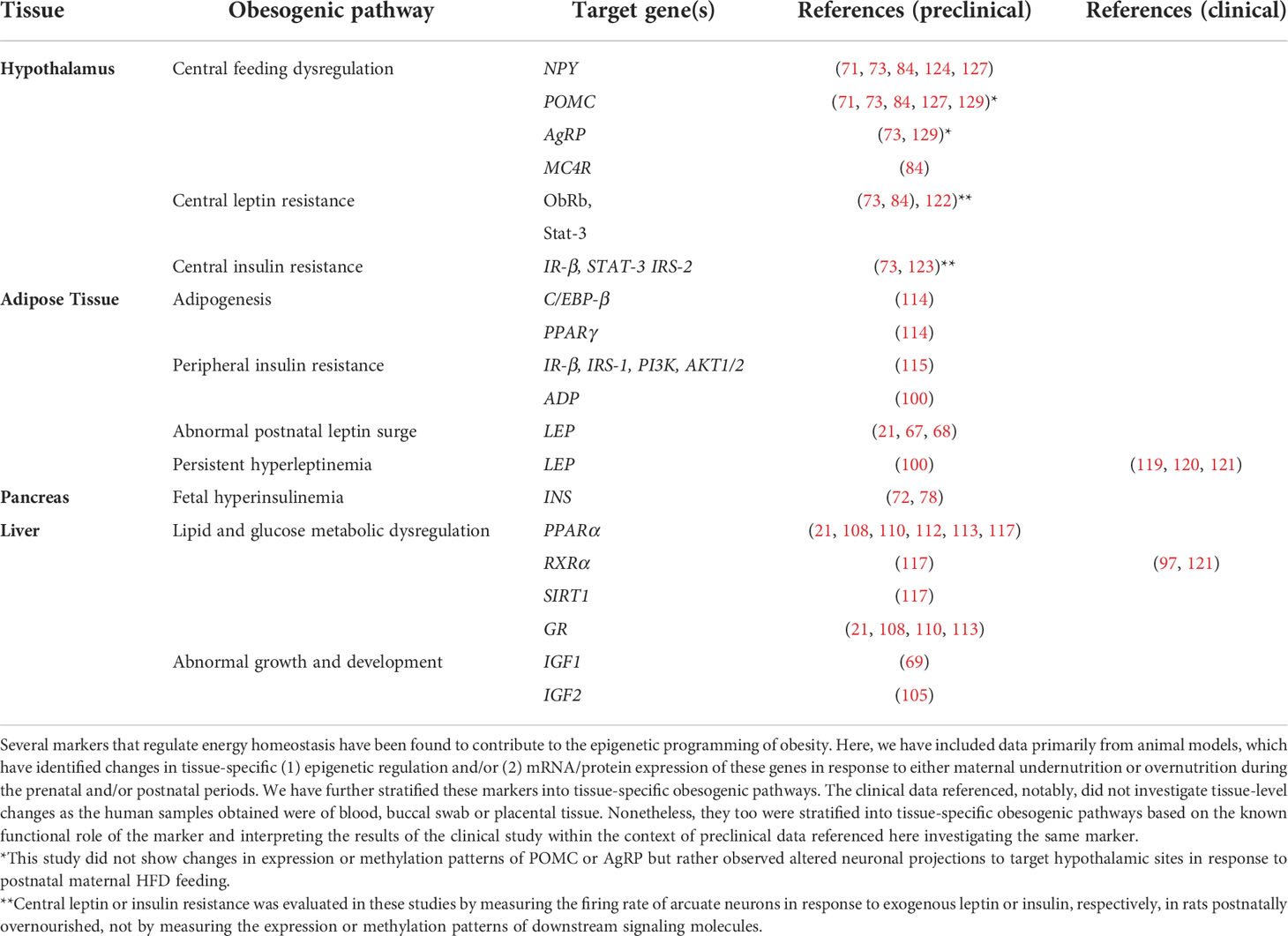
Table 1 Tissue-specific pathways and genes involved in the metabolic programming of obesity.
A preclinical study on the long-term effects of epigenetic changes in offspring in response to maternal undernutrition showed that some epigenetic modifications do not persist into adulthood (121). While it is unclear why some genes are more susceptible to reversal of epigenetic modifications compared to others, we argue that the effects of those initial epigenetic alterations likely contribute to the cumulative risk for obesity throughout life. Epigenetic alterations sustained during early critical developmental time periods can be considered as the “first-hit” (of many) in the pathogenesis of obesity. While, in the setting of maternal overnutrition, hypothalamic circuitry has been re-programmed towards lower levels of satiety and increased appetite, peripheral metabolism has been conditioned towards increased lipid storage and insulin resistance. Thus, we argue that the homeostatic set points which are established in early life initiate the temporospatial evolution of obesity and contribute to cumulative risk of metabolic syndrome, even if some epigenetic modifications which conditioned those set points may not persist. The mechanisms behind this hypothesis are unclear, and further investigation is required to understand the factors mediating the changes caused by these epigenetic modifications throughout lifespan.
Transgenerational transfer of metabolic risk
We have outlined evidence from human and animal studies on the metabolic risk caused by intrauterine and maternal exposures (F0 generation) to the offspring (F1 generation). The transmission of this risk to the subsequent generations resulting in transgenerational risk of metabolic disease into F2/F3 generation and beyond is possible. Epidemiological evidence suggests that exposure to maternal obesity and T2D results in higher prevalence of obesity, T2D (133, 134) and cardiometabolic risk in adult children (135), that may potentially remain in subsequent generation(s). Animal studies, that allow isolation of the gestational and the postnatal environment have established that the transmission of the risks conferred by maternal/gestational exposures may persist for up to 4 generations through both maternal and paternal germlines (136, 137). Evidence for such transmission is available in many animal species by environmental exposures. As an example, sex-specific inheritance of impact of high fat diet induced changes was demonstrated in females in F3 generation via paternal transmission in mice (138) that could be abolished by normal diet for three generations (101). Many similar examples are available in the review by King et al. (136) and Mohajer et al. (137) emphasizing the need and urgency of interrupting the cycle of transgenerational transfer of metabolic risk.
Reversing the epigenetic modifications which “program” obesity
We previously described both clinical and preclinical studies that showed a reversal of epigenetic modifications of obesity-related genes with improvement in metabolic status in utero or in later life (94, 99, 101). The discovery of TET enzymes made the mechanistic reversal of DNA methylation a possibility, however, how this process is activated, and under what conditions, remains unknown.
Dietary supplementation with folic acid has been shown to modulate the epigenetic modifications by altering availability of substrates for methylation of DNA, thus potentially reversing the epigenetic alterations of obesity-related genes (109). Natural compounds from dietary sources such as quercetin, curcumin, genistein, resveratrol and lycopene, may act as epigenetic modifiers (139). These compounds have been studied extensively in cancer biology, as they simultaneously possess anti-inflammatory, antioxidant and anticancer properties and may have a potential benefit in targeting the conditioning of obesity (140, 141).
Cancer and immunology research have further provided data for the use of “epigenetic drugs” (141, 142). Certain classes of epigenetic modifiers, such as histone deacetylase inhibitors (HDACi), histone acetyltransferase inhibitors (HATi), DNA methyltransferase inhibitors (DNMTis), histone demtheylating inhibitors (HDMis) and sirtuin-activating compounds (STACs), are currently being used for the treatment of cancer, urea cycle disorders, epilepsy, and hypertension. It is possible that these or other similar drugs may have a potential for the treatment and prevention of metabolic diseases such as obesity and diabetes. Early studies of synthetic and natural compounds including resveratrol (STAC), curcumin (HATi), hydralazine (DNMTi), valproic acid (HDAi) and sodium phenylbutyrate (HDAi) in obesity and diabetes are promising (140).
Limitations of epigenetic studies
Evaluating a cause-and-effect relationship between early life epigenetic modifications in response to maternal exposures and later obesity risk in humans is challenging for two primary reasons. First, most clinical studies investigating the epigenetic programming of obesity use blood, easy-to-obtain epithelial (i.e., buccal swabs) or placental samples (i.e., cord blood or tissue) to measure epigenetic modifications during the perinatal period and later in childhood. There is, for obvious reasons, limited access to central and peripheral tissues including the hypothalamus, adipose tissue, liver, and skeletal muscle which would otherwise help to more thoroughly understand the functional roles of obesity-related genes that undergo epigenetic alterations during critical time periods of development and condition obesity. Discovery of circulating epigenetic markers that more accurately reflect tissue-specific changes will be important moving forward in this field.
The second limitation of both preclinical and clinical studies investigating the role of epigenetics in the conditioning of obesity is simply the vast number of epigenetic modifications possible for one gene, which synergistically affect its expression. In other words, while most studies discussed in this review have focused on measuring differential methylation patterns of gene promoters or CpG islands, several other DNA and histone modifications shape the expression patterns of genes including histone acetylation and non-coding RNAs (i.e. microRNAs). While it is time-consuming and challenging to measure multiple epigenetic modifications, it is also unclear whether long-term expression of genes can be extrapolated based on single or few epigenetic changes that can be currently studied.
Lifestyle modifications remain vital in the prevention and treatment of childhood obesity
This and other prior reviews regarding the epigenetic conditioning of obesity discuss data on the contribution of in utero exposures on long-term obesity risk due to the abnormal “programming” of energy homeostatic set points likely making it more challenging to initiate and maintain weight loss. We hypothesize that lifestyle modifications may lead to better outcomes when applied earlier in life during periods of developmental plasticity. Preclinical studies suggest that improved nutrition during lactation can reverse and/or prevent the consequences of poor maternal diet in utero, although how maternal diet and exposures during postnatal feeding affect breastmilk content requires further investigation. Nevertheless, although lifestyle modifications in adults are not successful in promoting long-term maintenance of weight loss, we hypothesize that maternal lifestyle modifications either before pregnancy, during gestation and/or in the postnatal period, will improve long-term outcomes in children and reduce their risk for obesity in later life.
Further, although the perinatal period likely has the highest degree of central and peripheral plasticity; central feeding circuitry, peripheral innervations and metabolically active tissues are still susceptible to epigenetic modifications, and the consequences of in utero exposures may likely be reversed when lifestyle modifications are applied during childhood and even the adolescent period.
Proven to be more efficacious and successful than lifestyle modifications, maternal bariatric surgery prior to pregnancy has been shown to reduce long-term obesity risk in children; however, is typically only performed in patients with significant obesity (BMI ≥ 40 or BMI ≥ 35 in patients with significant comorbidities) (108, 143). Although less efficacious than bariatric surgery, several pharmaceutical interventions have been approved by the Food and Drug Administration (FDA) for the treatment of obesity (143–148). Of note, almost all current drug therapies are more efficacious in weight loss when combined with lifestyle modifications. Treatments such as GLP-1 receptor agonists, with or without other peptides, significantly improve overall metabolic status by increasing insulin sensitivity and modifying absorption of carbohydrates (149). As discussed before, improvement in maternal metabolic status appears to be more important than reduction of maternal adiposity alone to reduce long-term risk for obesity in children, although, clinically, adiposity and insulin resistance are often interdependent especially in the setting of maternal overnutrition.
We hypothesize that treatments targeting adiposity and insulin resistance simultaneously, in patients with or without overt diabetes, may be efficacious in both treating obesity and reducing offspring risk for obesity and diabetes. Increasing number of drugs, including GLP-1 receptor analogues, have been approved for treatment of obesity and diabetes in youth 12 years and older. Further, targeted drug therapy for specific genetic variants involved in the leptin melanocortin pathway are approved for children 6 years and older; ongoing clinical trials may expand this armamentarium in the future (150).
Clinical practice recommendations to reduce long-term risk of obesity in children
Based on data presented in this review, we recommend mothers with obesity to incorporate lifestyle modifications including consuming a well-balanced diet and physical activity prior to conception, during gestation (within reasonable limits) as well as during lactation to reduce long-term obesity risk in children.
Rapid weight gain during early infancy has been shown to significantly increase the risk for obesity later in life. We propose that breastfeeding should be encouraged in all infants, especially those of mothers with obesity and formula supplementation should be limited in breastfed infants to reduce risk of overfeeding. Further, based on the study by Huh et al. (151), the introduction of solid foods should be withheld prior to 4 months of age to avoid overnutrition in the first few months of life – a critical window of development during maturation of energy homeostatic systems setting the course for future feeding behavior and basal metabolism.
Below is a summary of clinical practices based on both human and animal studies regarding the metabolic programming of obesity, to reduce the long-term risk of obesity in children. Many of these have been recommended by professional organizations including the International Federation of Gynecology and Obstetrics (FIGO), American College of Obstetricians and Gynecologists (ACOG), American Academy of Pediatrics (AAP) and National Institutes of Health (NIH) (152). Figure 4 incorporates these recommendations and reviews current therapeutic strategies to target obesity, as well as epigenetic modifiers and potential pathways to target based on preclinical and clinical studies discussed in this review, both for the prevention and treatment of childhood obesity.
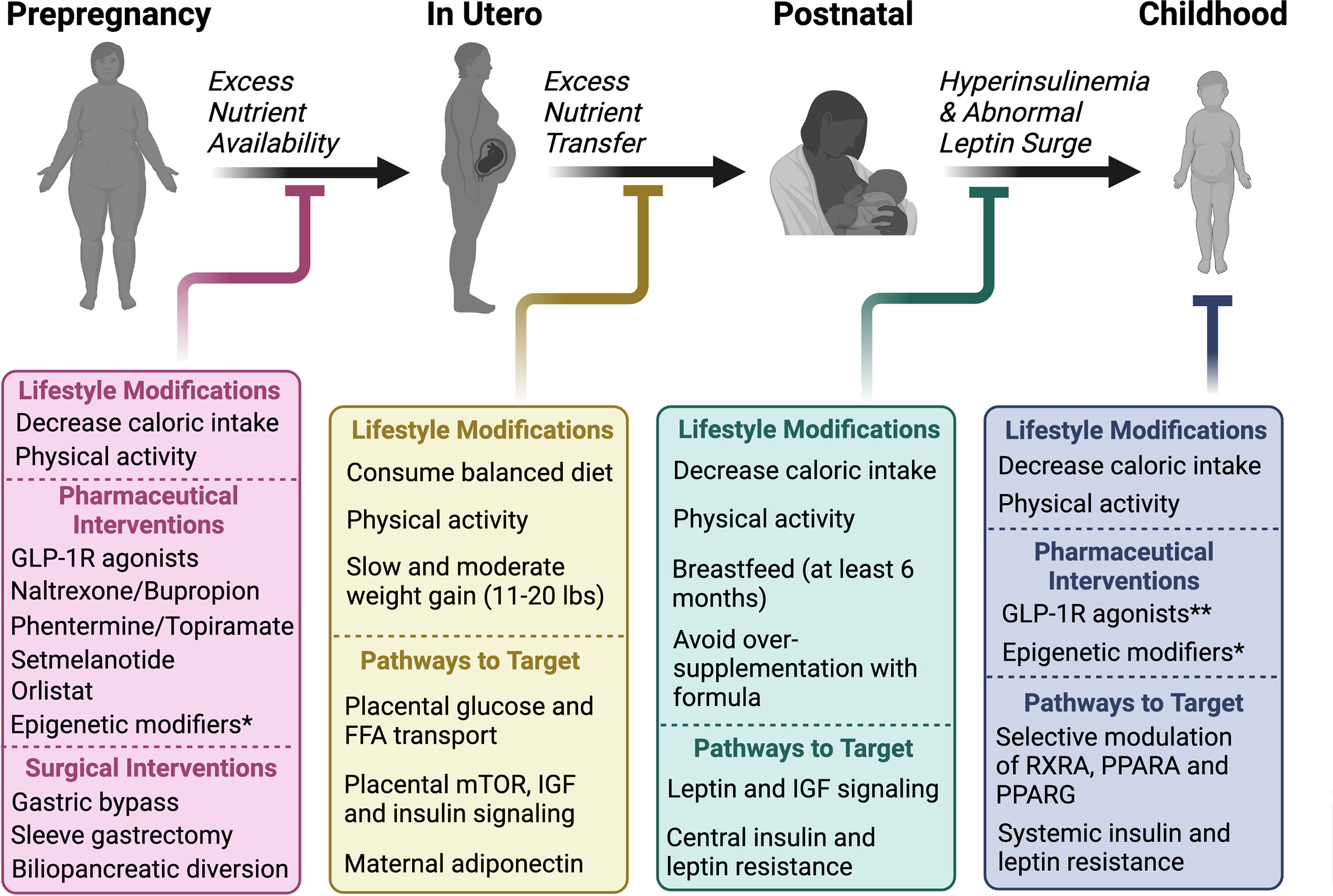
Figure 4 Time-dependent interventions to prevent the temporospatial evolution of obesity from prepregnancy to childhood Maternal lifestyle modifications, despite lack of effectiveness in long-term weight loss maintenance in adults, remain critical for the prevention of obesity in children. Excess nutrient availability, which concomitantly leads to an over-nourished in utero environment, is the initial insult in maternal obesity that predisposes children to obesity. Lifestyle modifications including a reduction in caloric intake and physical activity, as well as surgical and pharmaceutical interventions are available to ameliorate excess nutrient availability to a potential fetus. Although weight loss is not recommended while pregnant, methods to target excess nutrient transfer to the fetus include consuming a balanced diet and maintaining a moderate level of physical activity with slow weight gain (a maximum of 11-20 lbs is recommended by the NIH for pregnant women with obesity). In addition, research efforts should be focused on targeting pathways that promote nutrient transport to the fetus including energy substrate transporters, as well as mTOR, IGF and insulin signaling. Maternal adiponectin treatment in rodent models has been shown to improve metabolic status in offspring and is another possible therapeutic option in humans during this period. After birth, maternal lifestyle modifications again are critical although how these affect breastmilk content remains unclear. Nonetheless, breastfeeding for at least 6 months has been shown to reduce risk for obesity in children. Further, avoiding overfeeding and rapid weight gain during infancy is critical in reducing long-term risk for metabolic syndrome. Finally, in children with obesity, current interventions remain limited. Based on preclinical and clinical data discussed, we hypothesize that obesity treatments, which may help to reverse the epigenetic programming of obesity, may be more effective during childhood vs adulthood as obesity-related genes and signaling pathways regulating central feeding circuitry and basal metabolism are likely more susceptible to reversal of epigenetic alterations during this time. In addition to epigenetic modifiers, other pathways to target include systemic insulin and leptin, as well as RXRA, PPARA and PPARG signaling which regulate feeding behavior, as well as systemic lipid and glucose homeostasis, respectively.*Epigenetic modifiers (both natural and synthetic compounds) are not currently approved for the treatment or prevention of obesity but have excellent therapeutic potential based on data discussed in this review.**Several drugs are approved for use in youth ≥ 12 years of age and others in clinical trials.
Conclusion
Here, we have summarized the mechanistic underpinnings of the metabolic programming of obesity, as well as the epigenetic pathways that “program” obesity, which should be considered as potential therapeutic targets both for the prevention and treatment of childhood obesity. We hope that this review challenges the commonly held belief that obesity is the result of a lack of motivation or conviction to practice healthy lifestyle choices, both in the individual and as a community. Several factors resist changes in feeding behavior and adiposity, some of which have been described in this review. We hope that further understanding of the metabolic conditioning of obesity both in the clinical and research arenas will help us collectively address the global epidemic of obesity.
Author contributions
AR, CL, and VT conceived the manuscript and undertook research. AR wrote the first draft of the manuscript. VVT is responsible for the integrity of the content. All authors contributed to the article and approved the submitted version.
Funding
Research reported in this publication was supported by National Institute of Diabetes and Digestive and Kidney DIseases of the National Institutes of Health under award number: K23DK110539 and P30DK026687. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
The authors would like to thank Drs. Alicja A. Skowronski and Rudy Leibel for the helpful discussions about metabolic programming. The figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. World Health Organization. Obesity and overweight: World health organization (2021). Available at: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight.
2. Paxton SJ, Damiano SR. The development of body image and weight bias in childhood. Adv Child Dev Behav (2017) 52:269–98. doi: 10.1016/bs.acdb.2016.10.006
3. Sagar R, Gupta T. Psychological aspects of obesity in children and adolescents. Indian J Pediatr (2018) 85(7):554–9. doi: 10.1007/s12098-017-2539-2
4. Rolland-Cachera MF, Deheeger M, Maillot M, Bellisle F. Early adiposity rebound: Causes and consequences for obesity in children and adults. Int J Obes (Lond). (2006) 30 Suppl 4:S11–7. doi: 10.1038/sj.ijo.0803514
5. Berthoud HR, Munzberg H, Morrison CD. Blaming the brain for obesity: Integration of hedonic and homeostatic mechanisms. Gastroenterology (2017) 152(7):1728–38. doi: 10.1053/j.gastro.2016.12.050
6. Decarie-Spain L, Kanoski SE. Ghrelin and glucagon-like peptide-1: A gut-brain axis battle for food reward. Nutrients (2021) 3(3):977. doi: 10.3390/nu13030977
7. Sumithran P, Prendergast LA, Delbridge E, Purcell K, Shulkes A, Kriketos A, et al. Long-term persistence of hormonal adaptations to weight loss. N. Engl J Med (2011) 365(17):1597–604. doi: 10.1056/NEJMoa1105816
8. LeDuc CA, Skowronski AA, Rosenbaum M. The role of leptin in the development of energy homeostatic systems and the maintenance of body weight. Front Physiol (2021) 12:789519. doi: 10.3389/fphys.2021.789519
9. Rosenbaum M, Leibel RL. 20 years of leptin: Role of leptin in energy homeostasis in humans. J Endocrinol (2014) 223(1):T83–96. doi: 10.1530/JOE-14-0358
10. Zurlo F, Larson K, Bogardus C, Ravussin E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest (1990) 86(5):1423–7. doi: 10.1172/JCI114857
11. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest (2017) 127(1):43–54. doi: 10.1172/JCI88880
12. Wadden TA. Treatment of obesity by moderate and severe caloric restriction. results of clinical research trials. Ann Intern Med (1993) 119(7 Pt 2):688–93. doi: 10.7326/0003-4819-119-7_part_2-199310011-00012
13. Trust for America’s Health. The state of obesity 2021. Available at: https://www.tfah.org/wp-content/uploads/2021/09/2021ObesityReport_Fnl.pdf.
14. Chrissini MK, Panagiotakos DB. Health literacy as a determinant of childhood and adult obesity: A systematic review. Int J Adolesc Med Health (2021) 33(3):9–39. doi: 10.1515/ijamh-2020-0275
15. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: A report from the American heart association. Circulation (2019) 139(10):e56–e528. doi: 10.1161/CIR.0000000000000659
16. Mensah GA, Roth GA, Fuster V. The global burden of cardiovascular diseases and risk factors: 2020 and beyond. J Am Coll Cardiol (2019) 74(20):2529–32. doi: 10.1016/j.jacc.2019.10.009
17. Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: Physiology or pathophysiology? Physiol Rev (2014) 94(4):1027–76. doi: 10.1152/physrev.00029.2013
18. Aronne LJ, Hall KD, Jakicic MJ, Leibel RL, Lowe MR, Rosenbaum M, et al. Describing the weight-reduced state: Physiology, behavior, and interventions. Obes (Silver. Spring). (2021) 29 Suppl 1:S9–s24. doi: 10.1002/oby.23086
19. Ravussin E, Smith SR, Ferrante AW Jr. Physiology of energy expenditure in the weight-reduced state. Obes (Silver. Spring). (2021) 29 Suppl 1:S31–s8. doi: 10.1002/oby.23095
20. Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N. Engl J Med (1995) 332(10):621–8. doi: 10.1056/NEJM199503093321001
21. Gluckman PD, Lillycrop KA, Vickers MH, Pleasants AB, Phillips ES, Beedle AS, et al. Metabolic plasticity during mammalian development is directionally dependent on early nutritional status. Proc Natl Acad Sci U.S.A. (2007) 104(31):12796–800. doi: 10.1073/pnas.0705667104
22. Gluckman PD, Hanson M, Zimmet P, Forrester T. Losing the war against obesity: The need for a developmental perspective. Sci Transl Med (2011) 3(93):93cm19. doi: 10.1126/scitranslmed.3002554
23. Barker DJ, Eriksson JG, Forsen T, Osmond C. Fetal origins of adult disease: Strength of effects and biological basis. Int J Epidemiol (2002) 31(6):1235–9. doi: 10.1093/ije/31.6.1235
24. Barker DJ. Fetal origins of coronary heart disease. Br Heart J (1993) 69(3):195–6. doi: 10.1136/hrt.69.3.195
25. Barker DJ, Fall CH. Fetal and infant origins of cardiovascular disease. Arch Dis Child (1993) 68(6):797–9. doi: 10.1136/adc.68.6.797
26. Barker DJP. Fetal origins of cardiovascular disease. Ann Med (1999) 31(sup1):3–6. doi: 10.1080/07853890.1999.11904392
27. Dietz WH. Periods of risk in childhood for the development of adult obesity–what do we need to learn? J Nutr (1997) 127(9):1884S–6S. doi: 10.1093/jn/127.9.1884S
28. Deng JR, Tan WQ, Yang SY, Ao LP, Liang JP, Li LX, et al. High birth weight and its interaction with physical activity influence the risk of obesity in early school-aged children. World J Pediatr (2020) 16(4):385–92. doi: 10.1007/s12519-020-00336-6
29. Qiao Y, Ma J, Wang Y, Li W, Katzmarzyk PT, Chaput JP, et al. Birth weight and childhood obesity: A 12-country study. Int J Obes Suppl (2015) 5(Suppl 2):S74–9. doi: 10.1038/ijosup.2015.23
30. Kapral N, Miller SE, Scharf RJ, Gurka MJ, DeBoer MD. Associations between birthweight and overweight and obesity in school-age children. Pediatr Obes (2018) 13(6):333–41. doi: 10.1111/ijpo.12227
31. Liang J, Xu C, Liu Q, Fan X, Xu J, Zhang L, et al. Association between birth weight and risk of cardiovascular disease: Evidence from uk biobank. Nutr Metab Cardiovasc Dis (2021) 31(9):2637–43. doi: 10.1016/j.numecd.2021.05.017
32. Arnold L, Hoy W, Wang Z. Low birthweight increases risk for cardiovascular disease hospitalisations in a remote indigenous Australian community–a prospective cohort study. Aust N. Z. J Public Health (2016) 40 Suppl 1:S102–6. doi: 10.1111/1753-6405.12426
33. Sjoholm P, Pahkala K, Davison B, Niinikoski H, Raitakari O, Juonala M, et al. Birth weight for gestational age and later cardiovascular health: A comparison between longitudinal Finnish and indigenous Australian cohorts. Ann Med (2021) 53(1):2060–71. doi: 10.1080/07853890.2021.1999491
34. Wang YX, Li Y, Rich-Edwards JW, Florio AA, Shan Z, Wang S, et al. Associations of birth weight and later life lifestyle factors with risk of cardiovascular disease in the USA: A prospective cohort study. EClinicalMedicine (2022) 51:101570. doi: 10.1016/j.eclinm.2022.101570
35. Kong L, Nilsson IAK, Gissler M, Lavebratt C. Associations of maternal diabetes and body mass index with offspring birth weight and prematurity. JAMA Pediatr (2019) 173(4):371–8. doi: 10.1001/jamapediatrics.2018.5541
36. Kim SY, Sharma AJ, Sappenfield W, Wilson HG, Salihu HM. Association of maternal body mass index, excessive weight gain, and gestational diabetes mellitus with Large-for-Gestational-Age births. Obstet. Gynecol. (2014) 123(4):737–44. doi: 10.1097/AOG.0000000000000177
37. Bowers K, Laughon SK, Kiely M, Brite J, Chen Z, Zhang C. Gestational diabetes, pre-pregnancy obesity and pregnancy weight gain in relation to excess fetal growth: Variations by Race/Ethnicity. Diabetologia (2013) 56(6):1263–71. doi: 10.1007/s00125-013-2881-5
38. Group HSCR, Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl J Med (2008) 358(19):1991–2002. doi: 10.1056/NEJMoa0707943
39. Group HSCR. Hyperglycemia and adverse pregnancy outcome (Hapo) study: Associations with neonatal anthropometrics. Diabetes (2009) 58(2):453–9. doi: 10.2337/db08-1112
40. Lowe WL Jr., Lowe LP, Kuang A, Catalano PM, Nodzenski M, Talbot O, et al. Maternal glucose levels during pregnancy and childhood adiposity in the hyperglycemia and adverse pregnancy outcome follow-up study. Diabetologia (2019) 62(4):598–610. doi: 10.1007/s00125-018-4809-6
41. Scholtens DM, Kuang A, Lowe LP, Hamilton J, Lawrence JM, Lebenthal Y, et al. Hyperglycemia and adverse pregnancy outcome follow-up study (Hapo fus): Maternal glycemia and childhood glucose metabolism. Diabetes Care (2019) 42(3):381–92. doi: 10.2337/dc18-2021
42. Yu Z, Han S, Zhu J, Sun X, Ji C, Guo X. Pre-pregnancy body mass index in relation to infant birth weight and offspring Overweight/Obesity: A systematic review and meta-analysis. PloS One (2013) 8(4):e61627. doi: 10.1371/journal.pone.0061627
43. Voerman E, Santos S, Patro Golab B, Amiano P, Ballester F, Barros H, et al. Maternal body mass index, gestational weight gain, and the risk of overweight and obesity across childhood: An individual participant data meta-analysis. PloS Med (2019) 16(2):e1002744. doi: 10.1371/journal.pmed.1002744
44. Victora CG, Bahl R, Barros AJ, Franca GV, Horton S, Krasevec J, et al. Breastfeeding in the 21st century: Epidemiology, mechanisms, and lifelong effect. Lancet (2016) 387(10017):475–90. doi: 10.1016/S0140-6736(15)01024-7
45. Horta BL, Loret de Mola C, Victora CG. Long-term consequences of breastfeeding on cholesterol, obesity, systolic blood pressure and type 2 diabetes: A systematic review and meta-analysis. Acta Paediatr (2015) 104(467):30–7. doi: 10.1111/apa.13133
46. Singhal A. Does weight gain in infancy influence the later risk of obesity? J Pediatr Gastroenterol Nutr (2010) 51 Suppl 3:S119–20. doi: 10.1097/MPG.0b013e3181efd077
47. Yang Z, Huffman SL. Nutrition in pregnancy and early childhood and associations with obesity in developing countries. Matern. Child Nutr (2013) 9 Suppl 1:105–19. doi: 10.1111/mcn.12010
48. Eid EE. Follow-up study of physical growth of children who had excessive weight gain in first six months of life. Br Med J (1970) 2(5701):74–6. doi: 10.1136/bmj.2.5701.74
49. Stettler N, Zemel BS, Kumanyika S, Stallings VA. Infant weight gain and childhood overweight status in a multicenter, cohort study. Pediatrics (2002) 109(2):194–9. doi: 10.1542/peds.109.2.194
50. Singh AS, Mulder C, Twisk JW, van Mechelen W, Chinapaw MJ. Tracking of childhood overweight into adulthood: A systematic review of the literature. Obes Rev (2008) 9(5):474–88. doi: 10.1111/j.1467-789X.2008.00475.x
51. Aris IM, Rifas-Shiman SL, Li LJ, Kleinman KP, Coull BA, Gold DR, et al. Patterns of body mass index milestones in early life and cardiometabolic risk in early adolescence. Int J Epidemiol (2019) 48(1):157–67. doi: 10.1093/ije/dyy286
52. Arisaka O, Ichikawa G, Koyama S, Sairenchi T. Childhood obesity: Rapid weight gain in early childhood and subsequent cardiometabolic risk. Clin Pediatr Endocrinol (2020) 29(4):135–42. doi: 10.1297/cpe.29.135
53. Lu Y, Pearce A, Li L. Weight gain in early years and subsequent body mass index trajectories across birth weight groups: A prospective longitudinal study. Eur J Public Health (2020) 30(2):316–22. doi: 10.1093/eurpub/ckz232
54. Woo JG. Infant growth and long-term cardiometabolic health: A review of recent findings. Curr Nutr Rep (2019) 8(1):29–41. doi: 10.1007/s13668-019-0259-0
55. Singhal A, Fewtrell M, Cole TJ, Lucas A. Low nutrient intake and early growth for later insulin resistance in adolescents born preterm. Lancet (2003) 361(9363):1089–97. doi: 10.1016/S0140-6736(03)12895-4
56. Baird J, Fisher D, Lucas P, Kleijnen J, Roberts H, Law C. Being big or growing fast: Systematic review of size and growth in infancy and later obesity. BMJ (2005) 331(7522):929. doi: 10.1136/bmj.38586.411273.E0
57. Reilly JJ, Armstrong J, Dorosty AR, Emmett PM, Ness A, Rogers I, et al. Early life risk factors for obesity in childhood: Cohort study. BMJ (2005) 330(7504):1357. doi: 10.1136/bmj.38470.670903.E0
58. Kuriyan R. Body composition techniques. Indian J Med Res (2018) 148(5):648–58. doi: 10.4103/ijmr.IJMR_1777_18
59. Ode JJ, Pivarnik JM, Reeves MJ, Knous JL. Body mass index as a predictor of percent fat in college athletes and nonathletes. Med Sci Sports. Exerc. (2007) 39(3):403–9. doi: 10.1249/01.mss.0000247008.19127.3e
60. Eriksson J, Forsen T, Tuomilehto J, Osmond C, Barker D. Size at birth, fat-free mass and resting metabolic rate in adult life. Horm Metab Res (2002) 34(2):72–6. doi: 10.1055/s-2002-20518
61. Sandboge S, Moltchanova E, Blomstedt PA, Salonen MK, Kajantie E, Osmond C, et al. Birth-weight and resting metabolic rate in adulthood - sex-specific differences. Ann Med (2012) 44(3):296–303. doi: 10.3109/07853890.2010.549147
62. Weyer C, Pratley RE, Lindsay RS, Tataranni PA. Relationship between birth weight and body composition, energy metabolism, and sympathetic nervous system activity later in life. Obes Res (2000) 8(8):559–65. doi: 10.1038/oby.2000.72
63. Lewis DS, Bertrand HA, McMahan CA, McGill HC Jr., Carey KD, Masoro EJ. Preweaning food intake influences the adiposity of young adult baboons. J Clin Invest (1986) 78(4):899–905. doi: 10.1172/JCI112678
64. Poissonnet CM, Burdi AR, Garn SM. The chronology of adipose tissue appearance and distribution in the human fetus. Early. Hum Dev (1984) 10(1-2):1–11. doi: 10.1016/0378-3782(84)90106-3
65. Enzi G, Zanardo V, Caretta F, Inelmen EM, Rubaltelli F. Intrauterine growth and adipose tissue development. Am J Clin Nutr (1981) 34(9):1785–90. doi: 10.1093/ajcn/34.9.1785
66. Ng PC. The fetal and neonatal hypothalamic-Pituitary-Adrenal axis. Arch Dis Child Fetal. Neonatal. Ed. (2000) 82(3):F250–4. doi: 10.1136/fn.82.3.f250
67. Ellis PJ, Morris TJ, Skinner BM, Sargent CA, Vickers MH, Gluckman PD, et al. Thrifty metabolic programming in rats is induced by both maternal undernutrition and postnatal leptin treatment, but masked in the presence of both: Implications for models of developmental programming. BMC Genomics (2014) 15:49. doi: 10.1186/1471-2164-15-49
68. Vickers MH, Gluckman PD, Coveny AH, Hofman PL, Cutfield WS, Gertler A, et al. Neonatal leptin treatment reverses developmental programming. Endocrinology (2005) 146(10):4211–6. doi: 10.1210/en.2005-0581
69. Vickers MH, Ikenasio BA, Breier BH. Igf-I treatment reduces hyperphagia, obesity, and hypertension in metabolic disorders induced by fetal programming. Endocrinology (2001) 142(9):3964–73. doi: 10.1210/endo.142.9.8390
70. Schoonejans JM, Ozanne SE. Developmental programming by maternal obesity: Lessons from animal models. Diabetes Med (2021) 38(12):e14694. doi: 10.1111/dme.14694
71. Chen H, Simar D, Lambert K, Mercier J, Morris MJ. Maternal and postnatal overnutrition differentially impact appetite regulators and fuel metabolism. Endocrinology (2008) 149(11):5348–56. doi: 10.1210/en.2008-0582
72. Srinivasan M, Katewa SD, Palaniyappan A, Pandya JD, Patel MS. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am J Physiol Endocrinol Metab (2006) 291(4):E792–9. doi: 10.1152/ajpendo.00078.2006
73. Gupta A, Srinivasan M, Thamadilok S, Patel MS. Hypothalamic alterations in fetuses of high fat diet-fed obese female rats. J Endocrinol (2009) 200(3):293–300. doi: 10.1677/JOE-08-0429
74. White CL, Purpera MN, Morrison CD. Maternal obesity is necessary for programming effect of high-fat diet on offspring. Am J Physiol Regul Integr Comp Physiol (2009) 296(5):R1464–72. doi: 10.1152/ajpregu.91015.2008
75. Kelly AC, Powell TL, Jansson T. Placental function in maternal obesity. Clin Sci (Lond). (2020) 134(8):961–84. doi: 10.1042/CS20190266
76. Brombach C, Tong W, Giussani DA. Maternal obesity: New placental paradigms unfolded. Trends Mol Med (2022) 28(10):823–835. doi: 10.1016/j.molmed.2022.05.013
77. Sullivan EL, Grove KL. Metabolic imprinting in obesity. Forum Nutr (2010) 63:186–94. doi: 10.1159/000264406
78. Plagemann A, Heidrich I, Gotz F, Rohde W, Dorner G. Lifelong enhanced diabetes susceptibility and obesity after temporary intrahypothalamic hyperinsulinism during brain organization. Exp Clin Endocrinol (1992) 99(2):91–5. doi: 10.1055/s-0029-1211143
79. Hill DJ, Milner RDG. Insulin as a growth factor. Pediatr Res (1985) 19(9):879–86. doi: 10.1203/00006450-198509000-00001
80. Carmody JS, Wan P, Accili D, Zeltser LM, Leibel RL. Respective contributions of maternal insulin resistance and diet to metabolic and hypothalamic phenotypes of progeny. Obesity (2011) 19(3):492–9. doi: 10.1038/oby.2010.245
81. Castillo-Castrejon M, Jansson T, Powell TL. No evidence of attenuation of placental insulin-stimulated akt phosphorylation and amino acid transport in maternal obesity and gestational diabetes mellitus. Am J Physiol Endocrinol Metab (2019) 317(6):E1037–E49. doi: 10.1152/ajpendo.00196.2019
82. Ahima RS, Saper CB, Flier JS, Elmquist JK. Leptin regulation of neuroendocrine systems. Front Neuroendocr. (2000) 21(3):263–307. doi: 10.1006/frne.2000.0197
83. Levin BE, Dunn-Meynell AA. Reduced central leptin sensitivity in rats with diet-induced obesity. Am J Physiol Regul Integr Comp Physiol (2002) 283(4):R941–8. doi: 10.1152/ajpregu.00245.2002
84. Lustig RH, Sen S, Soberman JE, Velasquez-Mieyer PA. Obesity, leptin resistance, and the effects of insulin reduction. Int J Obes Relat Metab Disord (2004) 28(10):1344–8. doi: 10.1038/sj.ijo.0802753
85. Morris MJ, Chen H. Established maternal obesity in the rat reprograms hypothalamic appetite regulators and leptin signaling at birth. Int J Obes (2009) 33(1):115–22. doi: 10.1038/ijo.2008.213
86. Skowronski AA, LeDuc CA, Foo KS, Goffer Y, Burnett LC, Egli D, et al. Physiological consequences of transient hyperleptinemia during discrete developmental periods on body weight in mice. Sci Transl Med (2020) 12(524). doi: 10.1126/scitranslmed.aax6629
87. Skowronski AA, Shaulson ED, Leibel RL, LeDuc CA. The postnatal leptin surge in mice is variable in both time and intensity and reflects nutritional status. Int J Obes (2022) 46(1):39–49. doi: 10.1038/s41366-021-00957-5
88. Amitani M, Asakawa A, Amitani H, Inui A. The role of leptin in the control of insulin-glucose axis. Front Neurosci (2013) 7:51. doi: 10.3389/fnins.2013.00051
89. DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor ii gene disrupted by targeting. Nature (1990) 345(6270):78–80. doi: 10.1038/345078a0
90. Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, et al. Placental-specific igf-ii is a major modulator of placental and fetal growth. Nature (2002) 417(6892):945–8. doi: 10.1038/nature00819
91. Kniss DA, Shubert PJ, Zimmerman PD, Landon MB, Gabbe SG. Insulinlike growth factors. their regulation of glucose and amino acid transport in placental trophoblasts isolated from first-trimester chorionic villi. J Reprod Med (1994) 39(4):249–56.
92. Aye IL, Rosario FJ, Powell TL, Jansson T. Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc Natl Acad Sci U.S.A. (2015) 112(41):12858–63. doi: 10.1073/pnas.1515484112
93. Paulsen ME, Rosario FJ, Wesolowski SR, Powell TL, Jansson T. Normalizing adiponectin levels in obese pregnant mice prevents adverse metabolic outcomes in offspring. FASEB J (2019) 33(2):2899–909. doi: 10.1096/fj.201801015R
94. Lopomo A, Burgio E, Migliore L. Epigenetics of obesity. Prog Mol Biol Transl Sci (2016) 140:151–84. doi: 10.1016/bs.pmbts.2016.02.002
95. van Dijk SJ, Tellam RL, Morrison JL, Muhlhausler BS, Molloy PL. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin Epigenet (2015) 7:66. doi: 10.1186/s13148-015-0101-5
96. Razin A, Shemer R. DNA Methylation in early development. Hum Mol Genet (1995) 4:1751–5. doi: 10.1093/hmg/4.suppl_1.1751
97. Ehrlich M. Expression of various genes is controlled by DNA methylation during mammalian development. J Cell Biochem (2003) 88(5):899–910. doi: 10.1002/jcb.10464
98. Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes (2011) 60(5):1528–34. doi: 10.2337/db10-0979
99. Guénard F, Deshaies Y, Cianflone K, Kral JG, Marceau P, Vohl MC. Differential methylation in glucoregulatory genes of offspring born before vs. after maternal gastrointestinal bypass surgery. Proc Natl Acad Sci U.S.A. (2013) 110(28):11439–44. doi: 10.1073/pnas.1216959110
100. Dearden L, Ozanne SE. Early life origins of metabolic disease: Developmental programming of hypothalamic pathways controlling energy homeostasis. Front Neuroendocr. (2015) 39:3–16. doi: 10.1016/j.yfrne.2015.08.001
101. Masuyama H, Mitsui T, Nobumoto E, Hiramatsu Y. The effects of high-fat diet exposure in utero on the obesogenic and diabetogenic traits through epigenetic changes in adiponectin and leptin gene expression for multiple generations in female mice. Endocrinology (2015) 156(7):2482–91. doi: 10.1210/en.2014-2020
102. Marousez L, Lesage J, Eberle D. Epigenetics: Linking early postnatal nutrition to obesity programming? Nutrients (2019) 11(12):2966. doi: 10.3390/nu11122966
103. Zeltser LM. Feeding circuit development and early-life influences on future feeding behaviour. Nat Rev Neurosci (2018) 19(5):302–16. doi: 10.1038/nrn.2018.23
104. Howland MA, Sandman CA, Glynn LM. Developmental origins of the human hypothalamic-Pituitary-Adrenal axis. Expert Rev Endocrinol Metab (2017) 12(5):321–39. doi: 10.1080/17446651.2017.1356222
105. Wu X, Zhang Y. Tet-mediated active DNA demethylation: Mechanism, function and beyond. Nat Rev Genet (2017) 18(9):517–34. doi: 10.1038/nrg.2017.33
106. Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DI, Roseboom TJ. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG (2008) 115(10):1243–9. doi: 10.1111/j.1471-0528.2008.01822.x
107. Smith J, Cianflone K, Biron S, Hould FS, Lebel S, Marceau S, et al. Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J Clin Endocrinol Metab (2009) 94(11):4275–83. doi: 10.1210/jc.2009-0709
108. Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S, et al. Large Maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics (2006) 118(6):e1644–9. doi: 10.1542/peds.2006-1379
109. Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr (2005) 135(6):1382–6. doi: 10.1093/jn/135.6.1382
110. Wu G, Bazer FW, Cudd TA, Meininger CJ, Spencer TE. Maternal nutrition and fetal development. J Nutr (2004) 134(9):2169–72. doi: 10.1093/jn/134.9.2169
111. Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr (2007) 97(6):1064–73. doi: 10.1017/S000711450769196X
112. Jousse C, Parry L, Lambert-Langlais S, Maurin AC, Averous J, Bruhat A, et al. Perinatal undernutrition affects the methylation and expression of the leptin gene in adults: Implication for the understanding of metabolic syndrome. FASEB J (2011) 25(9):3271–8. doi: 10.1096/fj.11-181792
113. Lillycrop KA, Phillips ES, Torrens C, Hanson MA, Jackson AA, Burdge GC. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic ppar alpha promoter of the offspring. Br J Nutr (2008) 100(2):278–82. doi: 10.1017/S0007114507894438
114. Burdge GC, Lillycrop KA, Phillips ES, Slater-Jefferies JL, Jackson AA, Hanson MA. Folic acid supplementation during the juvenile-pubertal period in rats modifies the phenotype and epigenotype induced by prenatal nutrition. J Nutr (2009) 139(6):1054–60. doi: 10.3945/jn.109.104653
115. Borengasser SJ, Zhong Y, Kang P, Lindsey F, Ronis MJ, Badger TM, et al. Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in Male rat offspring. Endocrinology (2013) 154(11):4113–25. doi: 10.1210/en.2012-2255
116. Fernandez-Twinn DS, Alfaradhi MZ, Martin-Gronert MS, Duque-Guimaraes DE, Piekarz A, Ferland-McCollough D, et al. Downregulation of irs-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol Metab (2014) 3(3):325–33. doi: 10.1016/j.molmet.2014.01.007
117. Li CC, Young PE, Maloney CA, Eaton SA, Cowley MJ, Buckland ME, et al. Maternal obesity and diabetes induces latent metabolic defects and widespread epigenetic changes in isogenic mice. Epigenetics (2013) 8(6):602–11. doi: 10.4161/epi.24656
118. Suter MA, Ma J, Vuguin PM, Hartil K, Fiallo A, Harris RA, et al. In utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. Am J Obstet. Gynecol. (2014) 210(5):463 e1– e11. doi: 10.1016/j.ajog.2014.01.045
119. Hartwig FP, Loret de Mola C, Davies NM, Victora CG, Relton CL. Breastfeeding effects on DNA methylation in the offspring: A systematic literature review. PloS One (2017) 12(3):e0173070. doi: 10.1371/journal.pone.0173070
120. Obermann-Borst SA, Eilers PH, Tobi EW, de Jong FH, Slagboom PE, Heijmans BT, et al. Duration of breastfeeding and gender are associated with methylation of the leptin gene in very young children. Pediatr Res (2013) 74(3):344–9. doi: 10.1038/pr.2013.95
121. Sherwood WB, Bion V, Lockett GA, Ziyab AH, Soto-Ramirez N, Mukherjee N, et al. Duration of breastfeeding is associated with leptin (Lep) DNA methylation profiles and bmi in 10-Year-Old children. Clin Epigenet (2019) 11(1):128. doi: 10.1186/s13148-019-0727-9
122. Pauwels S, Symons L, Vanautgaerden EL, Ghosh M, Duca RC, Bekaert B, et al. The influence of the duration of breastfeeding on the infant's metabolic epigenome. Nutrients (2019) 11(6):1408. doi: 10.3390/nu11061408
123. Davidowa H, Plagemann A. Decreased inhibition by leptin of hypothalamic arcuate neurons in neonatally overfed young rats. Neuroreport (2000) 11(12):2795–8. doi: 10.1097/00001756-200008210-00037
124. Davidowa H, Plagemann A. Insulin resistance of hypothalamic arcuate neurons in neonatally overfed rats. Neuroreport (2007) 18(5):521–4. doi: 10.1097/WNR.0b013e32805dfb93
125. Plagemann A, Harder T, Rake A, Waas T, Melchior K, Ziska T, et al. Observations on the orexigenic hypothalamic neuropeptide y-system in neonatally overfed weanling rats. J Neuroendocr. (1999) 11(7):541–6. doi: 10.1046/j.1365-2826.1999.00357.x
126. Srinivasan M, Patel MS. Metabolic programming in the immediate postnatal period. Trends Endocrinol Metab (2008) 19(4):146–52. doi: 10.1016/j.tem.2007.12.001
127. Patel MS, Srinivasan M. Metabolic programming in the immediate postnatal life. Ann Nutr Metab (2011) 58 Suppl 2:18–28. doi: 10.1159/000328040
128. Mahmood S, Smiraglia DJ, Srinivasan M, Patel MS. Epigenetic changes in hypothalamic appetite regulatory genes may underlie the developmental programming for obesity in rat neonates subjected to a high-carbohydrate dietary modification. J Dev Orig. Health Dis (2013) 4(6):479–90. doi: 10.1017/S2040174413000238
129. Gorski JN, Dunn-Meynell AA, Hartman TG, Levin BE. Postnatal environment overrides genetic and prenatal factors influencing offspring obesity and insulin resistance. Am J Physiol Regul Integr Comp Physiol (2006) 291(3):R768–78. doi: 10.1152/ajpregu.00138.2006
130. Vogt MC, Paeger L, Hess S, Steculorum SM, Awazawa M, Hampel B, et al. Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell (2014) 156(3):495–509. doi: 10.1016/j.cell.2014.01.008
131. Desai M, Gayle D, Babu J, Ross MG. Programmed obesity in intrauterine growth-restricted newborns: Modulation by newborn nutrition. Am J Physiol Regul Integr Comp Physiol (2005) 288(1):R91–6. doi: 10.1152/ajpregu.00340.2004
132. Tosh DN, Fu Q, Callaway CW, McKnight RA, McMillen IC, Ross MG, et al. Epigenetics of programmed obesity: Alteration in iugr rat hepatic Igf1 mrna expression and histone structure in rapid vs. delayed postnatal catch-up growth. Am J Physiol Gastroint. Liver. Physiol (2010) 299(5):G1023–9. doi: 10.1152/ajpgi.00052.2010
133. Dabelea D, Crume T. Maternal environment and the transgenerational cycle of obesity and diabetes. Diabetes (2011) 60(7):1849–55. doi: 10.2337/db11-0400
134. Dabelea D, Mayer-Davis EJ, Lamichhane AP, D'Agostino RB Jr., Liese AD, Vehik KS, et al. Association of intrauterine exposure to maternal diabetes and obesity with type 2 diabetes in youth: The search case-control study. Diabetes Care (2008) 31(7):1422–6. doi: 10.2337/dc07-2417
135. Kankowski L, Ardissino M, McCracken C, Lewandowski AJ, Leeson P, Neubauer S, et al. The impact of maternal obesity on offspring cardiovascular health: A systematic literature review. Front Endocrinol (Lausanne). (2022) 13:868441. doi: 10.3389/fendo.2022.868441
136. King SE, Skinner MK. Epigenetic transgenerational inheritance of obesity susceptibility. Trends Endocrinol Metab (2020) 31(7):478–94. doi: 10.1016/j.tem.2020.02.009
137. Mohajer N, Joloya EM, Seo J, Shioda T, Blumberg B. Epigenetic transgenerational inheritance of the effects of obesogen exposure. Front Endocrinol (Lausanne). (2021) 12:787580. doi: 10.3389/fendo.2021.787580
138. Dunn GA, Bale TL. Maternal high-fat diet effects on third-generation female body size Via the paternal lineage. Endocrinology (2011) 152(6):2228–36. doi: 10.1210/en.2010-1461
139. Aggarwal R, Jha M, Shrivastava A, Jha AK. Natural compounds: Role in reversal of epigenetic changes. Biochem (Mosc). (2015) 80(8):972–89. doi: 10.1134/S0006297915080027
140. Arguelles AO, Meruvu S, Bowman JD, Choudhury M. Are epigenetic drugs for diabetes and obesity at our door step? Drug Discovery Today (2016) 21(3):499–509. doi: 10.1016/j.drudis.2015.12.001
141. Ghasemi S. Cancer's epigenetic drugs: Where are they in the cancer medicines? Pharmacogen. J (2020) 20(3):367–79. doi: 10.1038/s41397-019-0138-5
142. Abhimanyu, Ontiveros CO, Guerra-Resendez RS, Nishiguchi T, Ladki M, Hilton IB, et al. Reversing post-infectious epigenetic-mediated immune suppression. Front Immunol (2021) 12:688132. doi: 10.3389/fimmu.2021.688132
143. Jackson VM, Breen DM, Fortin JP, Liou A, Kuzmiski JB, Loomis AK, et al. Latest approaches for the treatment of obesity. Expert Opin Drug Discovery (2015) 10(8):825–39. doi: 10.1517/17460441.2015.1044966
144. Dragano NRV, Ferno J, Dieguez C, Lopez M, Milbank E. Recent updates on obesity treatments: Available drugs and future directions. Neuroscience (2020) 437:215–39. doi: 10.1016/j.neuroscience.2020.04.034
145. Knudsen LB, Lau J. The discovery and development of liraglutide and semaglutide. Front Endocrinol (Lausanne). (2019) 10:155. doi: 10.3389/fendo.2019.00155
146. Sherman MM, Ungureanu S, Rey JA. Naltrexone/Bupropion er (Contrave): Newly approved treatment option for chronic weight management in obese adults. P. T. (2016) 41(3):164–72.
147. Bays H. Phentermine, topiramate and their combination for the treatment of adiposopathy ('Sick fat') and metabolic disease. Expert Rev Cardiovasc Ther (2010) 8(12):1777–801. doi: 10.1586/erc.10.125
148. Sharma S, Garfield AS, Shah B, Kleyn P, Ichetovkin I, Moeller IH, et al. Current mechanistic and pharmacodynamic understanding of melanocortin-4 receptor activation. Molecules (2019) 24(10):1892. doi: 10.3390/molecules24101892
149. Ryan PM, Seltzer S, Hayward NE, Rodriguez DA, Sless RT, Hawkes CP. Safety and efficacy of glucagon-like peptide-1 receptor agonists in children and adolescents with obesity: A meta-analysis. J Pediatr (2021) 236:137–47 e13. doi: 10.1016/j.jpeds.2021.05.009
150. Rosenbaum M, Thaker V. Special considerations relevant to pediatric obesity. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, et al, editors. Endotext. South Dartmouth (MA: MDText.com, Inc (2022).
151. Huh SY, Rifas-Shiman SL, Taveras EM, Oken E, Gillman MW. Timing of solid food introduction and risk of obesity in preschool-aged children. Pediatrics (2011) 127(3):e544–51. doi: 10.1542/peds.2010-0740
152. McAuliffe FM, Killeen SL, Jacob CM, Hanson MA, Hadar E, McIntyre HD, et al. Management of prepregnancy, pregnancy, and postpartum obesity from the figo pregnancy and non-communicable diseases committee: A figo (International federation of gynecology and obstetrics) guideline. Int J Gynaecol. Obstet. (2020) 151 Suppl 1:16–36. doi: 10.1002/ijgo.13334
Keywords: childhood obesity, metabolic programing, epigenetics, in utero environment, cardiometabolic risk, postnatal conditions
Citation: Rajamoorthi A, LeDuc CA and Thaker VV (2022) The metabolic conditioning of obesity: A review of the pathogenesis of obesity and the epigenetic pathways that “program” obesity from conception. Front. Endocrinol. 13:1032491. doi: 10.3389/fendo.2022.1032491
Received: 30 August 2022; Accepted: 29 September 2022;
Published: 18 October 2022.
Edited by:
Artur Mazur, University of Rzeszow, PolandReviewed by:
Sabina Galiniak, University of Rzeszow, PolandŁukasz Dembiński, Medical University of Warsaw, Poland
Copyright © 2022 Rajamoorthi, LeDuc and Thaker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vidhu V. Thaker, dnZ0MjExNEBjdW1jLmNvbHVtYmlhLmVkdQ==