 Ilse Wieland1*
Ilse Wieland1* Ina Schanze1Ina Marianti Felgendreher1Winfried Barthlen2
Ina Schanze1Ina Marianti Felgendreher1Winfried Barthlen2 Silke Vogelgesang3
Silke Vogelgesang3 Klaus Mohnike4
Klaus Mohnike4 Martin Zenker1
Martin Zenker1- 1Institute of Human Genetics, University Hospital Otto-von-Guericke- University Magdeburg, Magdeburg, Germany
- 2Department of Pediatric Surgery, Protestant Hospital of Bethel Foundation, University Hospital OWL, University of Bielefeld, Bielefeld, Germany
- 3University Medicine, Institute of Pathology, University of Greifswald, Greifswald, Germany
- 4Dept of Pediatrics, University Hospital Otto-von-Guericke-University Magdeburg, Magdeburg, Germany
Background: The focal form of CHI is caused by an autosomal recessive pathogenic variant affecting the paternal homologue of genes ABCC8 or KCNJ11 and a second somatic event specifically occurring in the affected islet of Langerhans. The approach of this study was to integrate the genetic changes occurring in pancreatic focal lesions of CHI at the genomic and transcriptional level.
Research Design and Methods: Patients receiving therapeutic surgery and with proven ABCC8 or KCNJ11 pathogenic variants were selected and analyzed for loss of heterozygosity (LOH), changes in copy number and uniparental disomy (UPD) on the short am of chromosome 11 by molecular microarray analysis and methylation-specific MLPA. Gene expression was analyzed by RT-PCR and Massive Analysis of cDNA Ends (MACE).
Results: Both genes, ABCC8 and KCNJ11, are located in proximity to the Beckwith-Wiedemann (BWS) imprinting control region on chromosome 11p15. Somatic paternal uniparental isodisomy (UPD) at chromosome 11p was identified as second genetic event in focal lesions resulting in LOH and monoallelic expression of the mutated ABCC8/KCNJ11 alleles. Of five patients with samples available for microarray analysis, the breakpoints of UPD on chromosome 11p were different. Samples of two patients were analyzed further for changes in gene expression. Profound downregulation of growth suppressing genes CDKN1 and H19 was detected in focal lesions whereas growth promoting gene ASCL2 and pancreatic transcription factors of the endocrine cell lineage were upregulated.
Conclusions: Paternal UPD on the short arm of chromosome 11 appears to be the major second genetic event specifically within focal lesions of CHI but no common breakpoint for UDP can be delineated. We show for the first time upregulation of growth promoting ASCL2 (achaete-scute homolog 2) suggestive of a driving factor in postnatal focal expansion in addition to downregulation of growth suppressing genes CDKN1C and H19.
Introduction
Congenital hyperinsulinism (CHI) causes persistent hypoglycemia due to uncontrolled insulin secretion in newborns and infants (1, 2). The most severe form of CHI is caused by inactivating pathogenic variants in the ABCC8 (MIM 600509) and KCNJ11 (MIM 600937) genes encoding subunits SUR1 and Kir6.2 of the ATP-sensitive K(+) channel (3, 4). This channel is primarily expressed in the pancreas and to a much lesser degree in other tissues. Genetic testing in several larger patient cohorts revealed more than 100 pathogenic variants in these genes. Most of them are small pathogenic variants detectable by standard sequence analysis (5). To improve patient care, genetic testing has now been implemented in clinical management of CHI (6, 7).
Two major clinical forms of CHI are known. The diffuse form affects all ß-cells in the pancreas and is mainly caused by biallelic recessive inheritance of inactivating pathogenic variants and less frequently by dominantly acting pathogenic variants. Medical therapy, frequent feeding and subtotal pancreatectomy are the current treatment regimens for the diffuse form (8). The focal form is characterized by ß-cell hyperplasia in an affected islet of Langerhans within the pancreas. Surgical resection of a focal lesion potentially cures the patient (9, 10). The focal form appears to be caused by an autosomal recessive pathogenic variant affecting the paternal homologue of either the ABCC8 or KCNJ11 gene combined with somatic loss of heterozygosity (LOH) in the lesion (11–13).
ABCC8 and KCNJ11 are neighboring genes and they are located on the short arm of chromosome 11 in region 11p15 proximal to the imprinting region that when disrupted causes imprinting disorders Beckwith-Wiedemann syndrome (BWS) and Russell-Silver syndrome (RSS). The critical genomic imprinting region is responsible for the expression of growth regulatory genes depending on the parental origin (14, 15). The IGF2 gene (MIM 147470) encoding insulin-like growth factor 2 is expressed from the paternally derived chromosome and functions as growth promoting factor during embryogenesis and fetal development. The non-coding RNA of the H19 gene (MIM 103280) is expressed from the maternally derived chromosome and it is a negative regulator of IGF2 and other genes (16–18). Likewise, the CDKN1C gene (MIM 600856) encoding the inhibitor of G1 cyclin dependent kinases p57kip2, is preferentially expressed from the maternal allele. In BWS the clinical features are variable manifestations of macrosomia, visceromegaly of intra-abdominal organs and additional features including hyperinsulinism in a small amount of patients. However, BWS patients usually do not carry pathogenic variants in either ABCC8 or KCNJ11 and the underlying mechanism responsible for hyperinsulinism in these patients is not known. In BWS, expression from the maternal chromosome 11p15.5 is compromised either by imprinting defects or by paternal uniparental isodisomy (UPD) in 25% of patients and copy number variations (CNVs) in 9% of patients (14, 19). The focal form of CHI appears to represent a highly restricted type of UPD11p15 somatic mosaicism. In focal lesions LOH at 11p15 was first described using microsatellites and was later confirmed by loss of the maternal allele (12, 20, 21). Further studies showed an imbalance of gene expression in focal lesions (22).
In this study, we performed an integrative analysis of LOH, copy number changes and methylation at the BWS/RSS region followed by changes in gene expression in focal pancreatic lesions of patients harboring pathogenic variants in ABCC8 and KCNJ11, respectively.
Materials and methods
Patients and pancreatic tissue samples
Patients were from the German Registry for Congenital Hyperinsulinism (23). Written informed consent was obtained from the parents of patients and in accordance to the approval by the local ethics committees. Patients were treated by surgical therapy because of the clinical and genetic indication of focal CHI and localization of lesions by imaging diagnostics (10). Histological examination of resected pancreatic tissue presented with a lobular structure. Focal accumulation of atypical islet cells of Langerhans showed only a small rim of adjacent exocrine parenchyma. The endocrine cells exhibited huge nuclei and a broad eosinophilic cytoplasm. By immunohistochemistry the endocrine cells showed a strong positive reaction with insulin, whereas no reaction with p57 antibody was observed.
Mutation/variant analysis
Following histological examination of resected pancreatic lesions DNA and RNA was simultaneously extracted from deep frozen tissues using the QIAamp DNeasy Blood & Tissue Kit and QIAmp RNeasy Mini Kit (QIAGEN GmbH, Hilden, Germany).
Pathogenic variants and LOH in genes ABCC8 and KCNJ11 were analyzed by PCR amplification of the corresponding exons followed by Sanger sequencing using the Big Dye Terminator Cycle Sequencing kit and ABI 3500XL sequencer (Applied Biosystems, Foster City, USA). Coding SNPs in ABCC8 (dbSNP, NCBI) rs1799858 (exon 14), rs1805036 (exon 21), rs1799859 (exon 31), rs757110 (exon 33) and in KCNJ11 rs5213, rs5215, rs5218, rs5219 were also included in LOH analysis.
Sequences were processed by SeqPilot software 4.2.1 (JSI Medical Systems GmbH, Ettenheim, Germany). The sequencing data were compared with reference sequence NM_001287174.1 (ENST00000302539; ABCC8) and NM_000525.3 (ENST00000339994; KCNJ11).
Methylation-specific MLPA
Methylation-specific (MS) Multiplex Ligation-dependent Probe amplification (MLPA) analysis was carried out using probemix ME030-C3 as described by the manufacturer (MRC Holland, Amsterdam, The Netherlands). Amplification products were identified and quantified by capillary electrophoresis on an ABI 3500XL genetic analyzer. MLPA profiles were analyzed with the module MLPA of SeqPilot (JSI Medical Systems GmbH, Ettenheim, Germany).
Molecular microarray analysis
SNP-based chromosomal microarray (CMA) analysis was performed using a CytoScan™ HD microarray and the Chromosome Analysis Suite v4.3.0.71 (Thermo Fisher Scientific, Waltham, MA USA). All genomic positions were according to the GRCh37/hg19 build of the human reference genome.
RT-PCR, massive analysis of cDNA ends (MACE)
Expression analysis of ABCC8 and KCNJ11 was performed by RT-PCR. The same pancreatic tissue samples were examined for genomic and RNA changes. Total RNA was reverse transcribed using the Super ScriptIII™ RT-PCR kit (Invitrogen, Carlsbad, USA) as recommended by the supplier. RT-PCR was performed with primers from the coding regions of the genes using internal exon-spanning primers for ABCC8. For the single exon gene KCNJ11 the RNA was treated with DNAse prior to reverse transcription in order to exclude amplification of residual genomic DNA in the reaction. For controlling specific transcript amplification of KCNJ11, coding SNPs of ABCC8 were analyzed in parallel by exon-spanning RT-PCR. RT-PCR products were analyzed by direct sequencing as outlined above.
MACE was performed on extracted RNA from focal lesions and adjacent pancreatic normal tissue of the same patients (Patient 3 and 8) and expression was compared within the same patient. Data were analyzed as described previously (24) by GenXPro GmbH, Frankfurt a.M., Germany.
Results
Pathogenic variants in KATP-channel genes ABCC8 and KCNJ11
Paternally transmitted heterozygous pathogenic variants in either ABCC8 or KCNJ11 were previously identified during molecular genetic analysis in blood cells of the patients and their parents and have been described by Mohnike et al. (23) and Barthlen et al. (10). Of the 10 patients included in this study, 6 harbored pathogenic variants in ABCC8 and 4 harbored pathogenic variants in KCNJ11 (Table 1).
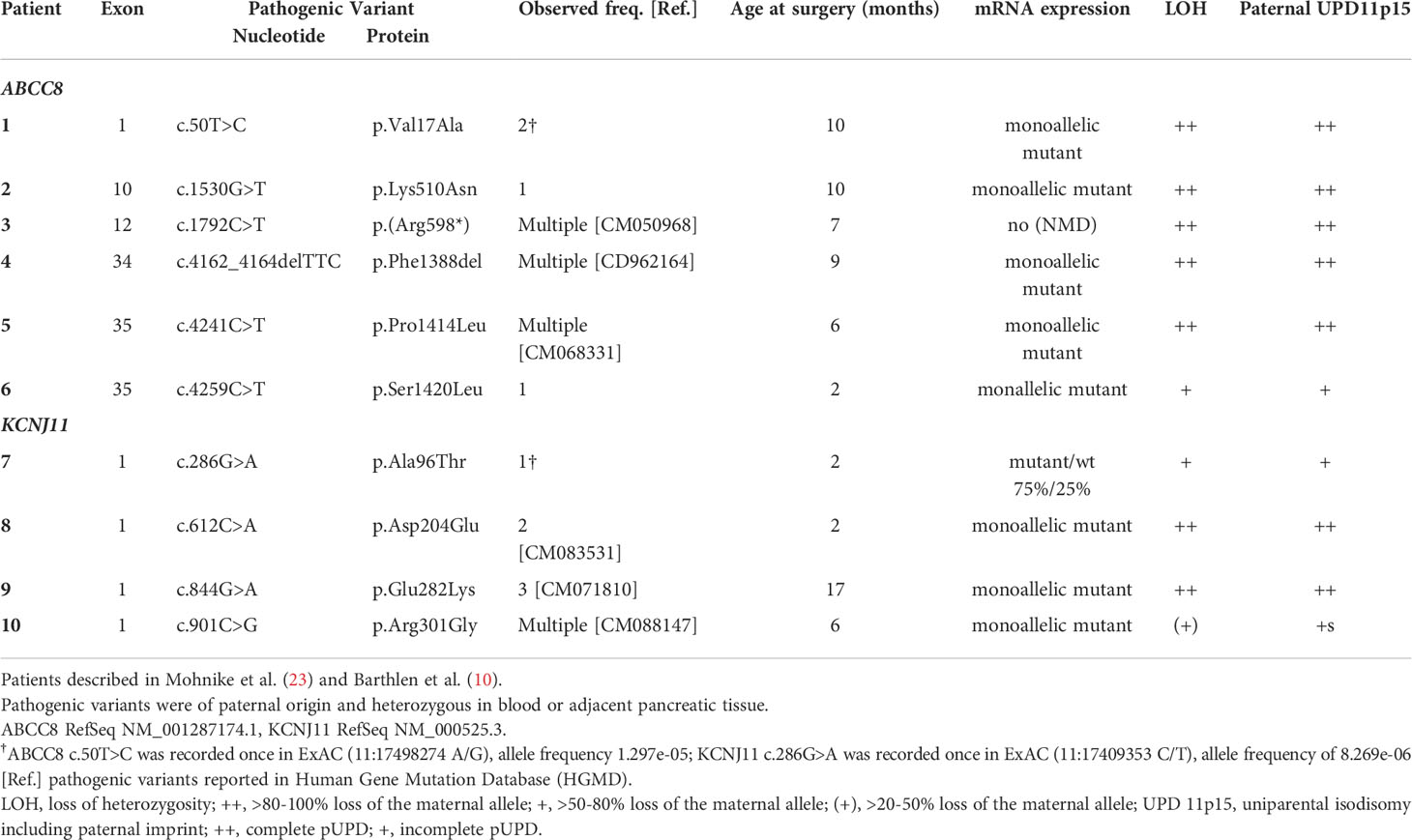
Table 1 Genetic and expression analysis of pancreatic lesions from focal CHI.
The DNA of pancreatic samples was analyzed for LOH following histological examination of frozen tissue biopsies. Focal lesions and adjacent normal pancreatic tissue if available were examined. LOH analysis performed at the pathogenic variant site in the respective sample in addition to informative intragenic single nucleotide polymorphisms (SNPs) of both, ABCC8 and KCNJ11, genes was close to 100% in 7 samples (Table 1).
Paternal uniparental isodisomy in focal lesions
Copy number-neutral LOH at the ABCC8/KCNJ11 locus as determined by MLPA suggested uniparental isodisomy (UPD) of chromosomal region 11p15 including the ABCC8 and KCNJ11 loci. At both imprinting centers of the BWS/RSS region on 11p15.5, an imbalance in methylation typically found in BWS patients with paternal UPD 11p15 was detected in 7 focal samples (Table 1; Figure 1). In 3 patients (Patient 6, 7, 10) with intermediate LOH in genomic DNA, paternal UPD 11p15 was similar in pattern but differences were less pronounced than in the other samples, thus suggesting a mosaic status for UPD 11p15 in these samples.
In DNA from pancreatic tissue specimens of patients 1, 3, 7, 8, and 9 we could demonstrate regions of copy neutral loss of heterozygosity (LOH) on the short arm of chromosome 11, suggesting segmental uniparental disomy (UPD) 11. All samples showed different dimensions of the UPD region with the smallest region in patient 3 (20,3Mb) and the largest region in patient 7 (43,2Mb) (Figure 1 and Table 2). In all patients the ABCC8 gene is located within the UPD region. Furthermore the samples showed different levels of mosaicism ranging from <50% to >75%.
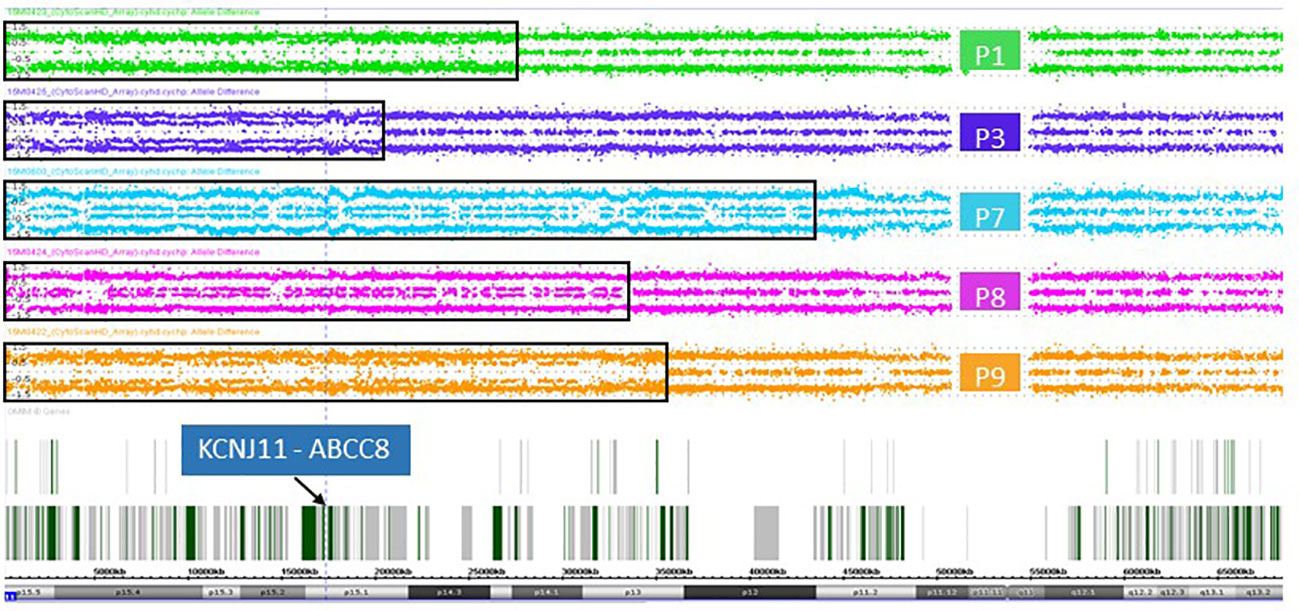
Figure 1 Allele difference plots of chromosome 11 on DNA extracted from pancreatic lesions of patient 1 (green panel), patient 3 (purple panel), patient 7 (blue panel), patient 8 (magenta panel) and patient 9 (orange panel) show a mosaic pattern of segmental uniparental disomy (UPD) of the short arm of chromosome 11 of different sizes. The extent of the UPD regions indicated by the black rectangles. The vertical dotted line and the arrow indicates the location of the genes KCNJ11 and ABCC8, which are located within the UPD region in all five patient samples.

Table 2 Molecular microarray analysis.
Monoallelic expression of ABCC8 and KCNJ11 transcripts from paternally transmitted mutant alleles
In normal pancreatic tissue biallelic expression of ABCC8 and KCNJ11 was found by sequencing and fragment analysis. In the normal pancreatic tissue of patient 3 only the ABCC8 transcript from the maternal wild-type allele was detectable (Figure 2A). This patient harbors a paternally inherited nonsense pathogenic variant in ABCC8 resulting in a premature termination codon that is likely to cause nonsense-mediated RNA decay (NMD). By RT-PCR the amount of mutant ABCC8 transcripts was below limit of detection despite apparent heterozygosity at the DNA level. Accordingly, there are no ABCC8 transcripts identified beyond background level in the focal lesion when the maternal wild-type allele is lost (Figure 2A, Patient 3). Monoallelic expression of the mutant transcripts encoded by the paternally inherited alleles was observed for all other focal lesions from the remaining patients in this series (Table 1; Figure 2A). Patient sample 7 showed residual expression of 10-25% of the wild-type transcript from the maternal allele concomitant with incomplete LOH in genomic DNA.
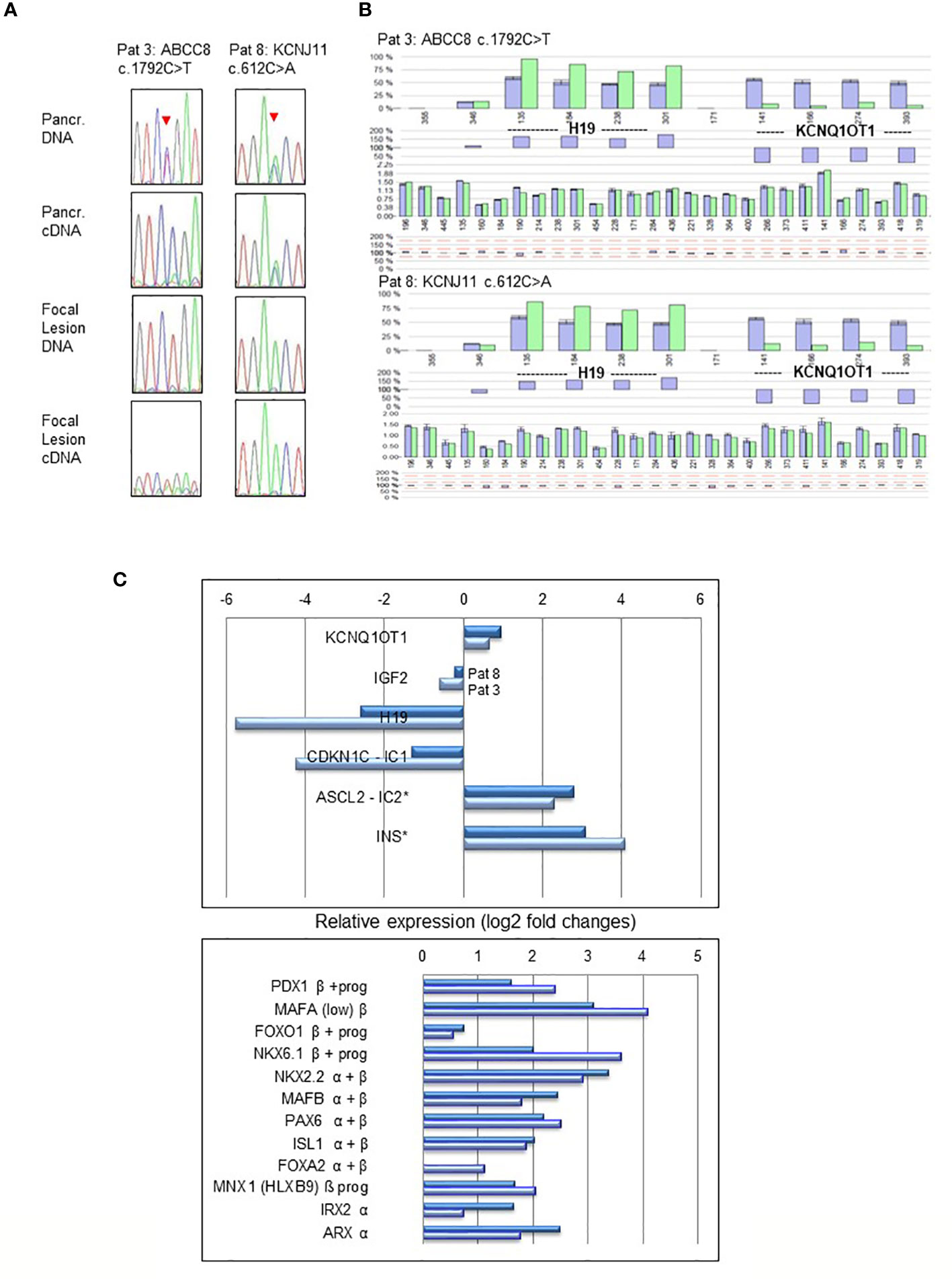
Figure 2 LOH and expression analysis performed in pancreatic normal tissue and focal lesions of Patient 3 (ABCC8 c.1792C>T) and Patient 8 (KCNJ11 c.612C>A). Heterozygosity of the pathogenic variants and biallelic expression (A) is evident in normal pancreatic tissue (upper panel), whereas focal lesions show monoallelic expression and LOH (lower panel). Sequencing profiles show the respective pathogenic variants in ABCC8 and KCNJ11 (indicated by an arrowhead) obtained from genomic DNA and reverse transcribed cDNA from focal lesion and pancreatic (Pancr.) normal tissue. In cDNA of Patients 3 pathogenic variant c.1792C>T causes NMD of the transcript resulting in lack of the respective ABCC8 transcripts. Epigenetic and copy number analysis (B) at the BWS/RSS imprinting region on 11p15.5 by MS-MLPA demonstrating paternal UPD11p15.5 in pancreatic focal lesions of Patient 3 (ABCC8 c.1792C>T) and Patient 8 (KCNJ11 c.612C>A). The pattern of hypermethylation of imprinting center 1 (IC1) at probes for H19 and hypomethylation of IC2 at probes for KCNQ1OT1 indicate paternal UPD11p15.5 typically found in BWS with UPD. Dark blue bars are results of three controls compared to the focal DNA (light green bars). Differences of controls and patient DNA shown below each profile. Relative gene expression (log2-fold changes) of BWS/RSS-Region 11p15 (upper panel) and pancreatic transcription factors (lower panel) by MACE analysis in focal lesions of Patient 3 (light blue) and Patient 8 (dark blue) compared to non-lesional tissue of the same patient (C). Genes ASCL2 located at imprinting center IC2 and INS are both not subject to genomic imprinting in humans. Pancreatic transcription factors expressed in progenitors (prog) of the endocrine lineage and in mature α- and β-cells are indicated.
Gene expression analysis
Expression analysis for genes located in 11p15 in focal lesions of patient 3 and 8 revealed downregulation of the imprinted gene H19 (log2: 5.8- and 2.6-fold, resulting from 690 766 and 568 794 reads in non-lesional pancreatic tissue as compared to 12 659 and 93 624 reads in focal lesions, respectively). Likewise, we found for the CDKN1 gene 50 766 and 27 339 reads in non-lesional pancreatic tissue compared to 2 685 and 11 102 reads in focal lesions of patients 3 and 8, respectively, resulting in changes of log2: 4.2- and 1.3-fold. On the other side, we found for ASCL2 read numbers of 7 002 and 3 417 in non-lesional pancreatic tissue versus 34 140 and 23 683 reads in focal lesions of patient 3 and 8, respectively (Figure 2C). This suggests a lower expression of ASCL2 in non-lesional tissue and upregulation of ASCL2 (achaete-scute homolog 2, MIM 601886) relative expression in focal lesions by more than log2: 2.3- and 2.8-fold, respectively. Upregulated gene expression was also observed for several pancreatic transcription factors involved in differentiation of the endocrine cell lineages of alpha and beta cells in both samples investigated (Figure 2C).
Discussion
Our comprehensive molecular analysis of native frozen pancreatic lesions from 10 focal CHI patients revealed monoallelic expression of the KATP-channel genes ABCC8 and KCNJ11 confined to focal lesions. The unaffected pancreatic tissue showed biallelic expression from both parental alleles at about similar ratios providing further evidence that both genes are not subject to parental imprinting in the pancreas. Our results also demonstrate that the recurrent nonsense pathogenic variant c.1792C>T (p.Arg598*) leads to loss of the mutated ABCC8 transcript independent of the second genetic event presumably due to nonsense-mediated mRNA decay [NMD; (25)] as shown for patient 3. Monoallelic expression in focal lesions coincided with LOH at the DNA level. This agrees well with the concept of somatic mosaicism in focal CHI that occurred in an islet by loss of the maternal allele in a progenitor cell and subsequent massive clonal expansion of the progeny cells (22). Incomplete LOH at the DNA level in 3 samples may be explained by admixture of a small cell population with features of non-lesional pancreatic cells residing within that lesion.
In all samples showing LOH, no imbalance in copy numbers was observed on 11p15 by MLPA. This suggests that neither deletion nor duplication at 11p15 is involved in focal CHI. Methylation-sensitive MLPA, however, showed an imprinting pattern of paternal UPD. These results further support the proposed paternal isodisomic UPD 11p15 as the major second genetic event causing LOH in focal CHI (21). Breakpoint-mapping in pancreatic lesions revealed no common breakpoints but encompassed the ABCC8 and KCNJ11 genes in the recombination interval as expected (11, 26). Somatic segmental UPD also occurs in many types of cancers and may convey a permissive growth advantage in light of Knudson’s two-hit hypothesis. A possible mechanism proposed in formation of segmental UPD in cancerous cells has been mitotic recombination events of homologous non-sister chromatids. Alternatively, an initial deletion may be compensated by re-duplication of the homologous region from the remaining chromosomal region of the other chromosome (27). In non-neoplastic tissue, less information exists on tissue-restricted UPD contributing to disease development. The focal form of CHI represents one of few examples of a cell-type restricted segmental paternal UPD 11p resembling a BWS micromosaicism. Based on the prevalence of focal compared to diffuse CHI, the risk of focal CHI in a child who carries a paternally inherited recessive pathogenic variant in ABCC8 or KCNJ11 was estimated to be around 1:270 (28). This may suggest that cell and tissue restricted segmental UPD are not rare somatic genetic events in pancreatic and possibly in other tissues. In fact, late onset ß-thalassemia and sickle cell anemia are other diseases with a similar mechanism of mosaic segmental paternal isodisomy at 11p15 unmasking a pathogenic variant in the HBB gene followed by clonal selection of hematopoetic progenitor cells due to enhanced proliferation (29, 30).
In focal pancreatic lesions the growth suppressing imprinted genes H19 and CDKN1C located in 11p15 are downregulated, while no substantial change in growth promoting IGF2 expression was detected. However, we observed upregulation of ASCL2 (achaete-scute homolog 2) also located in 11p15 but not subject to imprinting in humans. The gene encodes a basic helix-loop-helix transcription factor, which is target of WNT signaling in intestinal stem cells and exerts oncogenic function in cell culture (31). In a mouse model, transgenic rescue of Ascl2 expression leads to placentomegaly associated with BWS indicating a critical role of Ascl2 in placental overgrowth (32). Our results suggest that in fact upregulated ASCL2 is a driver in focus formation in postnatal CHI in addition to downregulated H19 and CDKN1. Currently, it is not known whether downregulated expression of H19 or additional components are responsible for upregulation of ASCL2 in focal lesions of the pancreas. Concomitantly, several key transcription factors of the endocrine pancreatic linages including premature stages (33) were upregulated in focal lesions.
Conclusion
In conclusion, our results support the hypothesis of paternal UPD 11p15 in focal CHI that leads to monoallelic expression of the mutated channel genes and appears to be the major second genetic event specifically in the pancreatic endocrine lineage. Clonal expansion of focal lesions appears to be driven by upregulation of growth promoting ASCL2 (achaete-scute homolog 2) in addition to downregulation of growth suppressing genes CDKN1 and H19.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, upon request.
Ethics statement
The studies involving human participants were reviewed and approved by University Hospital Magdeburg. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
IW, MZ planned this study. IW, IS, IF, SV, MZ performed, analyzed and interpreted the data. WB, KM recruited and collected clinical samples. IW, IS, MZ participated in writing. All authors approved the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the participants of the German Registry for Congenital Hyperinsulinism for their cooperation. Part of this work is contained in the doctoral thesis of IF.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arnoux JB, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V, et al. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis (2011) 6:63. doi: 10.1186/1750-1172-6-63
2. Stanley CA, De León DD. Frontiers in diabetes. In: Monogenic hyperinsulinemic hypoglycemia disorders, vol. Vol 21. (Basel, CH: Karger) (2012). doi: 10.1159/isbn.978-3-8055-9944-3
3. Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science (1995) 268:426–9. doi: 10.1126/science.7716548
4. Nestorowicz A, Inagaki N, Gonoi T, Schoor KP, Wilson BA, Glaser B, et al. A nonsense mutation in the inward rectifier potassium channel gene, Kir6.2, is associated with familial hyperinsulinism. Diabetes (1997) 46:1743–8. doi: 10.2337/diab.46.11.1743
5. De Franco E, Saint-Martin C, Brusgaard K, Knight Johnson AE, Aguilar-Bryan L, Bowman P, et al. Update of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum Mutat (2020) 41:884–905. doi: 10.1002/humu.23995
6. Banerjee I, Avatapalle B, Padidela R, Stevens A, Cosgrove KE, Clayton PE, et al. Integrating genetic and imaging investigations into the clinical management of congenital hyperinsulinism. Clin Endocrinol (2013) 78:803–13. doi: 10.1111/cen.12153
7. Maiorana A, Barbetti F, Boiani A, Rufini V, Pizzoferro M, Francalanci P, et al. Focal congenital hyperinsulinism managed by medical treatment: A diagnostic algorithm based on molecular genetic screening. Clin Endocrinol (2014) 81:679–88. doi: 10.1111/cen.12400
8. Lord K, Dzata E, Snider KE, Gallagher PR, De León DD. Clinical presentation and management of children with diffuse and focal hyperinsulinism: a review of 223 cases. J Clin Endocrinol Metab (2013) 98:E1786–1789. doi: 10.1210/jc.2013-2094
9. Laje P, Stanley CA, Palladino AA, Becker SA, Adzick NS. Pancreatic head resection and roux-en-Y pancreaticojejunostomy for the treatment of the focal form of congenital hyperinsulinism. J Pediatr Surg (2012) 47:130–5. doi: 10.1016/j.jpedsurg.2011.10.032
10. Barthlen W, Varol E, Emptin S, Wieland I, Zenker M, Mohnike W, et al. Surgery in focal congenital hyperinsulinism (CHI) – the “Hyperinsulinism Germany international” experience in 30 children. Pediatr Endocrinol Rev (2016) 14:48–56. doi: 10.17458/PER.2016.BVE
11. Giurgea I, Sempoux C, Bellanné-Chantelot C, Ribeiro M, Hubert L, Boddaert N, et al. The knudson’s two-hit model and timing of somatic mutation may account for the phenotypic diversity of focal congenital hyperinsulinism. J Clin Endocrinol Metab (2006) 91:4118–23. doi: 10.1210/jc.2006-0397
12. Damaj L, le Lorch M, Verkarre V, Werl C, Hubert L, Nihoul-Fétéké C, et al. Chromosome 11p15 paternal isodisomy in focal forms of neonatal hyperinsulinism. J Clin Endocrinol Metab (2008) 93:4941–7. doi: 10.1210/jc.2008-0673
13. Gillis D. Familial hyperinsulinism. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. (Seattle, USA: University of Washington) (2019). p. 1993–2020.
14. Feinberg AP. Phenotypic plasticity and epigenetics of human disease. Nature (2007) 447:433–40. doi: 10.1038/nature05919
15. Demars J, Gicquel C. Epigenetic and genetic disturbance of the imprinted 11p15 region in beckwith-wiedemann and silver-russel snydromes. Clin Genet (2012) 81:350–61. doi: 10.1111/j.1399-0004.2011.01822.x
16. Gabory A, Ripoche M-A, Le Digarcher A, Watrin F, Ziyyat A, Forné T, et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development (2009) 136:3413–21. doi: 10.1242/dev.036061
17. Monnier P, Martinet C, Pontis J, Stancheva I, Ait-Si-Ali S, Dandolo L. H19 lncRNA controls gene expression of the imprinted gene network by recruiting MBD1. Proc Natl Acad Sci USA (2013) 110:20693–8. doi: 10.1073/pnas.1310201110
18. Wang B, Suen CW, Ma H, Wang Y, Kong L, Qin D, et al. The roles of H19 in regulating inflammation and aging. Front Immunol (2020) 11:579687. doi: 10.3389/fimmu.2020.579687
19. Begemann M, Spengler S, Gogiel M, Grasshoff U, Bonin M, Betz R. Clinical significance of copy number variations in the 11p15.5 imprinting control regions: new cases and review of the literature. J Med Genet (2012) 49:547–53. doi: 10.1136/jmedgenet-2012-100967
20. De Lonlay P, Fournet JC, Rahier J, Gross-Morand MS, Poggi-Travert F, Foussier V, et al. Somatic deletion of the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J Clin Invest (1997) 100:802–7. doi: 10.1172/JCI119594
21. Verkarre V, Fournet JC, de Lonlay P, Gross-Morand MS, Devillers M, Rahier J, et al. Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinism in focal adenomatous hyperplasia. J Clin Invest (1998) 102:1286–91. doi: 10.1172/JCI4495
22. Fournet JC, Mayaud C, de Lonlay P, Gross-Morand MS, Verkarre V, Castanet M, et al. Unbalanced expression of 11p15 imprinted genes in focal forms of congenital hyperinsulinism: association with a reduction to homozygosity of a mutation in ABCC8 or KCNJ11. Am J Pathol (2001) 158:2177–84. doi: 10.1016/S0002-9440(10)64689-5
23. Mohnike K, Wieland I, Barthlen W, Vogelgesang S, Empting S, Mohnike W, et al. Clinical and genetic evaluation of patients with KATP channel mutations from the German registry for congenital hyperinsulinism. Horm Res Paediatr (2014) 81:156–68. doi: 10.1159/000356905
24. Zawada AM, Rogacev KS, Müller S, Rotter B, Winter P, Fliser D, et al. Massive analysis of cDNA ends (MACE) and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease. Epigenetics (2014) 9:161–72. doi: 10.4161/epi.26931
25. Popp MW, Maquat LE. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu Rev Genet (2013) 47:139–65. doi: 10.1146/annurev-genet-111212-133424
26. Lee BH, Lee J, Kim J-M, Kang M, Kim G-H, Choi J-H, et al. Three novel pathogenic mutations in KATP channel genes and somatic imprinting alterations of the 11p15 region in pancreatic tissue in patients with congenital hyperinsulinism. Horm Res Paediatr (2015) 83:204–10. doi: 10.1159/000371445
27. Makishima H, Maciejewski JP. Pathogenesis and consequences of uniparental disomy in cancer. Clin Cancer Res (2011) 17:3913–23. doi: 10.1158/1078-0432.CCR-10-2900
28. Glaser B, Blech I, Krakinovsky Y, Ekstein J, Gillis D, Mazor-Aronovitch K, et al. ABCC8 mutation allele frequency in the ashkenazi Jewish population and risk of focal hyperinsulinemic hypoglycemia. Genet Med (2011) 13:891–4. doi: 10.1097/GIM.0b013e31821fea33
29. Badens C, Mattei MG, Imbert AM, Lapouméroulie C, Martini N, Michel G, et al. A novel mechansims for thalassemia intermedia. Lancet (2002) 359:132–3. doi: 10.1016/s0140-6736(02)07338-5
30. Vinatier I, Martin X, Costa JM, Bazin A, Giraudier S, Joly P. A late onset sickle cell disease reveals a mosaic segmental uniparental isodisomy of chromosome 11p15. Blood Cells Mol Dis (2015) 54:53–5. doi: 10.1016/j.bcmd.2014.07.021
31. Jubb AM, Chalasani S, Frantz GD, Smits R, Grasch HI, Kavi V, et al. Achaete-scute like 2 (ascl2) is a target of wnt signaling and is upregulated in intestinal neoplasia. Oncogene (2006) 25:3445–57. doi: 10.1038/sj.onc.1209382
32. Tunster SJ, Van de Pette M, Creeth HDJ, Lefebvre L, John RM. Fetal growth restriction in a genetic model of sporadic beckwith-wiedemann syndrome. Dis Model Mech (2018) 11:pii: dmm035832. doi: 10.1242/dmm.035832
Keywords: ABCC8, Achaete-scute complex homolog 2, CHI, KCNJ11, UPD 11p
Citation: Wieland I, Schanze I, Felgendreher IM, Barthlen W, Vogelgesang S, Mohnike K and Zenker M (2022) Integration of genomic analysis and transcript expression of ABCC8 and KCNJ11 in focal form of congenital hyperinsulinism. Front. Endocrinol. 13:1015244. doi: 10.3389/fendo.2022.1015244
Received: 09 August 2022; Accepted: 03 October 2022;
Published: 21 October 2022.
Edited by:
Violeta Iotova, University Hospital St. Marina, BulgariaReviewed by:
Dongsheng Guo, University of Massachusetts Medical School, United StatesSenthil Senniappan, Alder Hey Children’s Hospital, United Kingdom
Sarah Flanagan, University of Exeter, United Kingdom
Dinesh Giri, Alder Hey Children’s Hospital, United Kingdom
Copyright © 2022 Wieland, Schanze, Felgendreher, Barthlen, Vogelgesang, Mohnike and Zenker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilse Wieland, aWxzZS53aWVsYW5kQG1lZC5vdmd1LmRl