 Nadia Panera
Nadia Panera Claudia Mandato
Claudia Mandato Annalisa Crudele1
Annalisa Crudele1 Sara Bertrando
Sara Bertrando Pietro Vajro
Pietro Vajro Anna Alisi
Anna Alisi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 14 November 2022
Sec. Systems Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1006008
This article is part of the Research TopicGenetic Variants and Metabolic DiseasesView all 10 articles
Sedentary lifestyle and consumption of high-calorie foods have caused a relentless increase of overweight and obesity prevalence at all ages. Its presently epidemic proportion is disquieting due to the tight relationship of obesity with metabolic syndrome and several other comorbidities which do call for urgent workarounds. The usual ineffectiveness of present therapies and failure of prevention campaigns triggered overtime a number of research studies which have unveiled some relevant aspects of obesity genetic and epigenetic inheritable profiles. These findings are revealing extremely precious mainly to serve as a likely extra arrow to allow the clinician’s bow to achieve still hitherto unmet preventive goals. Evidence now exists that maternal obesity/overnutrition during pregnancy and lactation convincingly appears associated with several disorders in the offspring independently of the transmission of a purely genetic predisposition. Even the pre-conception direct exposure of either father or mother gametes to environmental factors can reprogram the epigenetic architecture of cells. Such phenomena lie behind the transfer of the obesity susceptibility to future generations through a mechanism of epigenetic inheritance. Moreover, a growing number of studies suggests that several environmental factors such as maternal malnutrition, hypoxia, and exposure to excess hormones and endocrine disruptors during pregnancy and the early postnatal period may play critical roles in programming childhood adipose tissue and obesity. A deeper understanding of how inherited genetics and epigenetics may generate an obesogenic environment at pediatric age might strengthen our knowledge about pathogenetic mechanisms and improve the clinical management of patients. Therefore, in this narrative review, we attempt to provide a general overview of the contribution of heritable genetic and epigenetic patterns to the obesity susceptibility in children, placing a particular emphasis on the mother-child dyad.
The steadily increasing prevalence of obesity in the general population it has now become a significant burden for human health (1). Worldwide estimates revealed more than 7% of children and adolescents had obesity in 2016 compared with less than 1% in 1975 (2). According to global health estimates provided by World Health Organization (WHO) 38.2 million of children under the age of 5 years were overweight or obese in 2019. Data also indicates that the prevalence of overweight and obesity in children and adolescents (from 5 to 19 years) has increased from approximately 4% in 1975 to more than 18% in 2016 noting that no significant differences emerge between girls and boys (3).
Obesity is a multifaceted disease widely regarded by now as the main risk condition for developing the metabolic syndrome in both children and adults (4, 5). It is precisely for this aspect that obesity is closely associated with a series of pathological manifestations such as insulin resistance, Type 2 diabetes (T2D) and non-alcoholic fatty liver disease (NAFLD) and sleep-related breathing disorder such as obstructive sleep apnea (OSA) (6, 7). In particular, over the past two decades, the mechanistic exploration of the nexus between obesity levels and metabolic diseases has highlighted a close relationship between nutrient excess, genetics, epigenetics, oxidative stress, and inflammatory signals (8, 9). It is widely accepted as a fact that the epidemic proportion reached by the obesity in pediatric population is mostly a consequence of the sedentary lifestyle and consumption of high-calorie junk foods, clearly indicating the crucial role of the current obesogenic environment in favoring obesity’s and comorbidities’ epidemics (10). However, the well-documented individual variation in body weight, central adiposity, and particularly the differences in the onset and severity of the obesity-associated metabolic diseases, has forced the researchers to search other driving causes, such as the genetic and epigenetic inheritable profiles, even if these mechanisms have not yet been fully explored (11). It is known that direct exposures of ancestral germline (sperm and eggs) to environmental factors and altered nutrition can reprogram the epigenome of the cells, thus transmitting the susceptibility for the obesity to future generations through a mechanism of epigenetic transgenerational inheritance (12). Moreover, maternal obesity is currently being studied to clarify the links between maternal overnutrition during gestation and breastfeeding period, and the occurrence of hepatic/metabolic/cardiovascular disorders in the resultant offspring independently of genetic predisposition. According to an increasing number of studies, environmental factors such as maternal malnutrition, hypoxia and exposure to excess hormones and endocrine disruptors, during gestation and early postnatal period, play a decisive role in the programming of adipose tissue and obesity in the childhood (13).
In this review, we provide an overview of contribution of heritable genetic and epigenetic causes in development of obesity in childhood and focus on the role of epigenetic transgenerational inheritance on the obesity susceptibility.
Here, we have summarized the existing literature that could be relevant to the purpose of our narrative review. Since we focused on the impact of heritable genetics and epigenetics factors on pediatric obesity we structured the article by two main subjects, including the role of gene mutations and epigenetic modifications. For these topics, literature search was performed in the PubMed and Scopus Databases with the following search terms: “polygenic obesity” OR “monogenic obesity”, AND “children”; “epigenetic”, AND “obesity”, AND “children”. We also reported the effects of parental and in utero epigenetic mechanisms on the obese phenotype and its comorbidities by extrapolating from the main search on transgenerational transmission of obesity. We searched for clinical studies, as well as reviews, published in the English language, until June of 2022. Eligible papers for our narrative review included studies that reported convincing data and theories supporting the hypothesis of genetics and epigenetics inheritance of obesity.
Obesity is a multifactorial disease where environmental factors and several genes play important pathogenetic roles (14). Indeed, excessive fat accumulation due to an excess intake of sugar-sweetened beverages, high-calorie foods, sedentary lifestyle and poor sleeping is a well-known leading cause to obesity (15). In addition, genetics can predispose people to obesity by affecting appetite regulation, body mass index (BMI), metabolism, body-fat distribution and by influencing food preferences and response to exercise (16). Regarding the diagnostic accuracy of BMI to diagnose obesity in children, it has been questioned as it may underestimate the prevalence of adolescents with overweight and obesity (17). Indeed, although obese children and adolescents are around five times more likely to be obese in adulthood than those who were not obese, a large proportion of obese adults (about 70%) were not classed as being obese in childhood or adolescence (17). This effect may be interpreted as due to the effects related to environmental factors linked to developmental origins of health and disease (DOHaD) (18).
Several studies were focused on the identification of genes associated with obesity (19). Indeed, genetic causes of obesity can be classified in different ways including monogenetic, polygenetic, or syndromic obesity (11).
Monogenetic obesity is rare and involves single-gene mutations or chromosomal deletions and the subjects usually exhibited a severe early-onset obesity associated with hyperphagia and endocrine disorders (20). Most of the gene mutations linked to monogenic obesity have been discovered by experimental studies on transgenic mice (21). Gene mutation are principally located in regions encoding for proteins belonging to the leptin-melanocortin pathway, which is responsible for maintaining energy balance through food consumption and energy expenditure (22) and follows a Mendelian pattern (23). The genetic determinants of monogenic obesity in children have been reported in Table 1. In particular, leptin was identified as the product of the obese (ob) gene following the characterization of monogenic obesity (ob/ob) mice model (24). Leptin is an anorexigenic hormone synthesized and secreted by white adipocytes that regulates the food intake and energy balance (25). Concentration of circulating leptin decreases during fasting (26), but increases during refeeding or overfeeding (27). Human congenital leptin deficiency was identified, for the first time, in two obese first-degree cousins from a Pakistani family, where a homozygous frame-shift mutation (c.398delG), leading to a non-secreted truncated leptin protein was genetically characterized (28). However, studies demonstrated that severe obesity develops from rare genetic mutations that affect genes of both leptin and its receptor (29), and lead to congenital leptin deficiency or leptin resistance (30). Other loci which have been implicated in severe childhood monogenic obesity include genes of the proopiomelanocortin (POMC), the melanocortin receptor 4 (MC4R), the protein convertase 1/3 (PC 1/3), the single-minded homolog 1 (SIM1), and the brain-derived neurotrophic factor (BDNF) (31). Obesity caused by mutations in POMC gene shows autosomal recessive inheritance, whereas those in MC4R are autosomal dominant (32). POMC deficiency results in the absence of its cleavage products, and inactivation of MC4R, thus causing hyperphagia, severe obesity, and red hair (33). Mutations in MC4R represent the most common cause of monogenic childhood obesity (34), and more than 150 different nonsynonymous variants of this gene have been described (35). Indeed, rs17782313 and rs12970134 in MC4R gene polymorphisms were associated with obesity in both European and Asian adults and children (36, 37). Also, the mutation in PC 1/3 gene has been correlated with severe early-onset obesity, endocrine disorders, intestinal dysfunctions, and impairment of glucose homeostasis (38, 39). SIM1 gene encode for proteins that play a key role in neuronal development and function of the paraventricular nucleus of hypothalamus which is a crucial region for food intake and control of energy homeostasis (40). Inactivating mutations in this gene have been associated with monogenic obesity in humans and early-onset obesity, although its contribution has only been investigated in a few populations (41, 42). Moreover, few point mutations in SIM1 have been recognized as a cause of monogenic obesity (43, 44). A novel SIM1 variant, p.D134N, potentially linked to monogenic pediatric obesity has been recently identified by Stanikova et al, in a cohort of children and adolescents (45). In addition, dominant forms of non-syndromic monogenic obesity in humans have been associated with mutations in BDNF gene, encoding for the protein involved in proliferation and survival of hypothalamic neurons (46). BDNF is essential for regulating energy balance and food intake and it has been found crucial in leptin down-stream signaling (47). The disruption of one copy of the BDNF gene as consequence of chromosomal rearrangement was described in a child affected by severe obesity and several cognitive alterations (48). While a full functional loss of one BDNF allele seems to cause severe obesity in humans, there is no consistent evidence about the pathogenic variants of this gene. Recent data showed the link of two rare single-nucleotide genetic variants (p.Ile231Val and p.Cys141Gly), to non-syndromic severe early-onset obesity (49). Very recently, BDNF p.Thr2Ile and p.Arg209Gln missense variants were found associated with severe obesity developed during childhood in a 35-year-old and a 46-year-old female patient respectively, thus suggesting a potential obesogenic role of this gene variant (50).
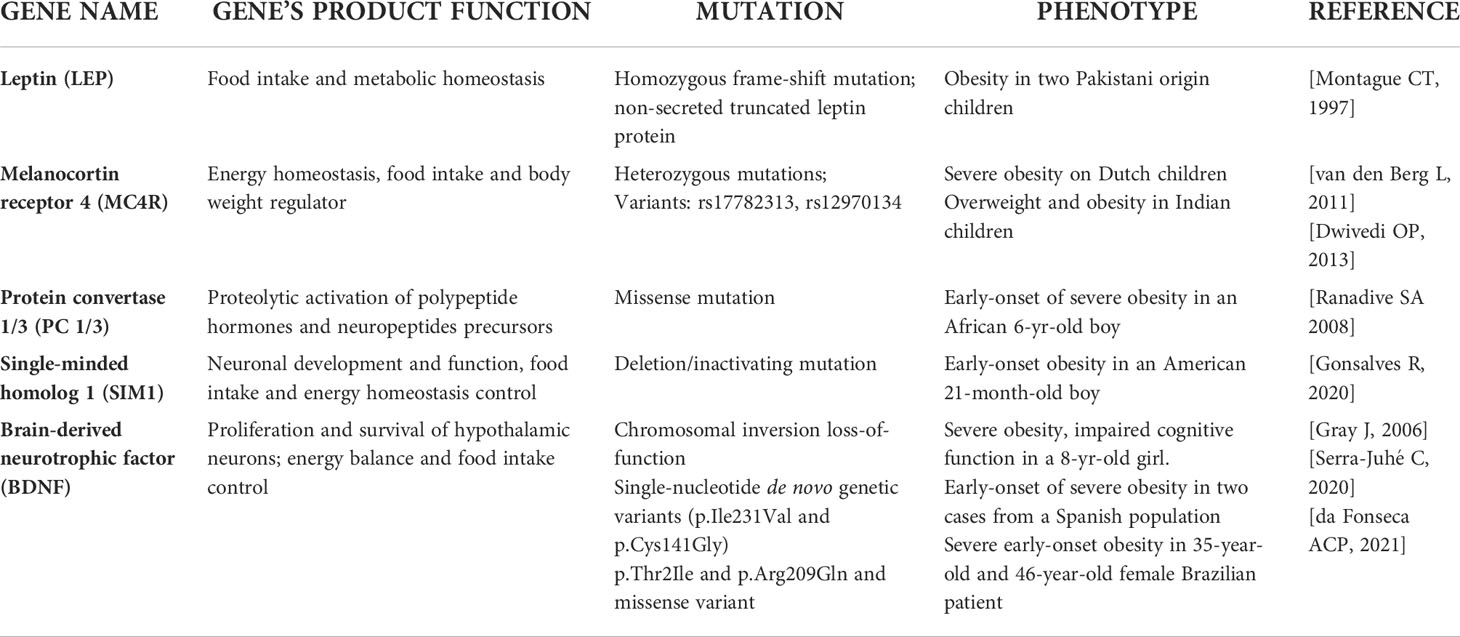
Table 1 Genetic determinants of monogenic obesity in children.
It is widely accepted that obesity epidemic cannot be traced back to a single gene disorder, but rather to the interaction of a complex genetic background in combination with environment, where single nucleotide polymorphisms (SNPs) may contribute to development of the obese phenotype if they interact with epigenetic inputs. Indeed, polygenic obesity is the most common form of the childhood disease, which results from hundreds of SNPs and observes a pattern of heritability similar to that of other complex traits and diseases (51). Evidence shows that the expression of mutations causing monogenic obesity may, at least in part, be influenced by the individual’s polygenic susceptibility to obesity (52). A genetic study from 573 individuals with severe early onset obesity found 40 very rare predicted functional variants in 13 genes encoding Sema3s (Semaphorin 3 Ligands) and their receptors in severely obese cases compared to controls. Since Sema3 signaling is crucial for gonadotropin-releasing hormone development and function, the study highlighted that the critical perturbations in the development of hypothalamic melanocortin neurons are also crucial for body weight and/or fat mass control (53).
Genome-wide association studies (GWASs) are particularly effective for the identification and analysis of novel genetic variants associated with weight gain (54). The genetic determinants of polygenic obesity in children have been reported in Table 2.

Table 2 Genetic determinants of polygenic obesity in children.
Among the obesity-related genetic variants, those located in the fat mass and obesity associated (FTO) gene locus, were the first ones that showed a strong association with BMI. The FTO gene, expressed in the hypothalamus, is located on chromosome 16 and encodes a demethylation enzyme implicated in the control of energy homeostasis, food intake and energy expenditure (55). In particular, two studies demonstrated that the variants rs9939609 and rs9930506 in the first intron of the FTO gene are significantly associated with BMI, thus suggesting that the presence of these variants may increase the risk of obesity in both adults and children (56, 57). Accordingly, the rs9939609 variant was found to be associated with increased fat mass and BMI in Scottish children (58) and to obesity phenotype in Dutch children (59). A further study reported that the rs9939609 allele may predispose to pediatric obesity risk by acting on satiety responsiveness (60). An interesting study by Jacobsson et al. highlighted the rs9939609 variant was associated with obesity and gender in Swedish children (61). Moreover, in European adolescents with obesity the homozygosity for the rs9939609 FTO SNP was critically associated with trunk-weighted (62). Besides in the Caucasian population, the presence of the common risk allele of rs9939609 polymorphism has been significantly associated with BMI and obesity in large population-based survey performed in Chinese children, where the polymorphism was associated with weight, BMI, waist circumference, waist-to-height ratio and fat mass percentage (63). A meta-analysis collected and analyzed all these studies suggesting that the rs9939609 has to be considered as the major FTO gene variant associated with overweight/obesity across multiple pediatric populations (64). However, studies on the impact of rs9939609 variant were performed also in Brazilian children and adolescents (65, 66). Todendi et al. reported a correlation between AA genotype for the rs9939609 variant and alteration of waist circumference and overweight/obesity in both children and their parents, thus corroborating the parental contribution to obesity onset (65). A more recent longitudinal study by Reuter et al. have confirmed the association of rs9939609 variant and abdominal fat in Brazilian school children between 7 and 15 years old (66).
Of note, a line of evidence showed no association between FTO rs9939609 and BMI, thus highlighting that the ethnicity may be pivotal to determine the role of this variant on obesity risk (67–69). Other two FTO gene variants, the rs8050136 and the rs1421085, were found associated to obesity traits in different children population studies alone or in combination to the best investigated rs9939609 variant (67, 70–72). Although, the true role of these additional loci remain unknown, deserving further investigations in the next future, a very recent meta-analysis reported this and other variants in FTO genes and their association with pediatric obesity overall (73). Often FTO variants have been investigated with variants in the MC4R of which mutations can be positioned between monogenic obesity and the polygenic obesity (36, 73, 74).
Since there are several published studies reporting the critical pathogenic role of pro-inflammatory cytokines in obesity, further genetic association studies were focused on the influence of variants in genes encoding for these molecules in obesity (75). In obesity, increased production and secretion of a wide range of inflammatory molecules, including tumor necrosis factor (TNF)-α and interleukin-6 (IL-6) was reported in white adipose tissue (76). TNF-α was found to be involved in lipid metabolism leading to hypertriglyceridemia as a result of decreasing lipoprotein lipase activity and increasing hepatic de novo synthesis of fatty acids, while IL-6 is mainly associated with liver and adipose tissue inflammation (77). The rs1800629 variant in the TNF-α gene, associated with increased expression of the cytokine in adipose tissue, was found to be more frequent in obese children than in lean subjects (78, 79). However, to date the role of TNF-α rs1800629 on childhood obesity is not clear. A study on Portuguese obese adolescents revealed that only in males subjects, the circulating TNF-α levels were modulated by TNF-α rs1800629 (80). Instead, a study on Romanian children (aged 5–18 years), revealed that TNF-α rs1800629 variant was more frequent in normal weight than in overweight children (81). Few studies demonstrated that also a variant in IL-6 gene may correlate with the obese phenotype (82). In particular, the IL6 polymorphisms designated as rs1800795 as well, were associated with the risk of developing obesity in Egyptian children (83).
The mentioned SNPs only explain 6% of BMI elevation, therefore Khera et al., by incorporating the analysis from 2.1 million SNPs, proposed a polygenic risk score (PRS) that increased the prediction of BMI alteration up to 23%. Interestingly, this PRS was associated with adult BMI, but it showed a consistency in children too (84).
Recently, a GWAS identified nineteen significant genome regions associated with body fatness (85). From this study emerged, for the first time, the association of BDNF, Fragile Histidine Triad Diadenosine Triphosphatase (FHIT), and protein kinase D1 (PRKD1) gene regions with BMI in children.
Growing evidence showed that genetic variation (typically SNPs) can also affect epigenetic profiles independently of or in contribution with environmental factors. In particular, have been identified several variants in methylation quantitative trait loci (meQTLs) that are differentially distributed across the genome and are involved in obesity and metabolic traits (86). A meta-analysis showed that the rs17782313 variant may regulate the expression of MC4R gene by affecting its methylation levels, thus contributing to childhood obesity (87). Further studies by using the analysis of meQTLs with GWAS may help in understanding how genetic predisposition and environmental factors interact with each other to determine methylation status that predispose to develop an obese phenotype in children. Moreover, future research should attempt to verify whether methylome signals of prenatal exposure can be regulated by genetic variation/specific single polymorphisms in metabolic genes correlated also with children and adolescents’ obesity thus confirming the possibility of both a passive and active role of DNA methylation to regulatory interactions influencing gene expression (86, 88, 89).
Finally, very recently a group of studies have reported the relevance of rs10487505 variant of the region encoding for the long non-coding RNA lncOb, which was correlated with leptin levels and BMI in adults and children (90, 91).
Epigenetic changes in the human genome are defined as heritable regulatory mechanisms of gene expression overlaying the information enclosed in the DNA sequence. Epigenetic changes (also referred to as epigenetic marks) include mainly DNA methylation, post-translational modifications of histones, and ncRNAs (92, 93). DNA methylation, which is the most studied epigenetic mark, is a process occurring on the fifth carbon of cytosines mostly located in regions with symmetrical CpG dinucleotides, where the addition of a methyl group in the cytosine may cause transcriptional repression. This process is sustained by the activity of three types of DNA methyltransferases (DNMTs), including DNMT1, DNMT3A and DNMT3B (92). The term of post-translational modifications of histones comprises of different chemical changes, such as histone acetylation, histone methylation, histone phosphorylation, and histone ubiquitination. These modifications may be regulated by different enzymes, such as histone acetyltransferases, histone deacetylases, histone methyltransferases and demethylases, and may be associated with both transcriptional activation and repression (93). Commonly, ncRNAs are a group of RNAs that are classified according to their length in long ncRNAs (lncRNAs), and small ncRNAs including miRNAs, small nucleolar RNA (snoRNA), small nuclear RNA (snRNA), PIWI-interacting RNAs (piRNAs), and others, which may regulate gene expression at transcriptional, post-transcriptional, translational, and post-translational level in a variety of ways (94, 95).
There are several environmental and dietary factors (i.e. overnutrition) that may influence epigenetics, thus acting as regulators of the environment-gene interaction over the life course (96). A recent updated systematic review emphasized the association between epigenetic mechanisms and obesity in children (97). This systematic review describes studies reporting strong evidence of an epigenetic signature assessed by Epigenome-Wide Association Study (EWAS) in childhood obesity. Indeed, a recent EWAS by Robinson et al, based on the UK Avon Longitudinal study of Parents and Children (ALSPAC) cohort was able to identify for the first time the association between a differential DNA methylation profiles of blood samples in children with early life rapid weight gain. The candidate gene analysis on ALSPAC and in the replication cohort found a positive association between rapid growth gain and DNA methylation at the locus near the checkpoint with forkhead and ring finger domains (CHFR) (cg11531579) gene encoding for E3 ubiquitin- protein ligase, thus increasing the risk of subsequent pediatric overweight/obesity (98).
Xu et al. showed a substantial number of differentially methylated CpG sites between obese and non-obese adolescents (14–20 years old) and associated to gene previously linked to obesity and T2D, such as FTO, glucokinase (GCK), hepatocyte nuclear factor-1 α and β (HNF1A and HNF1B), peroxisome proliferator-activated receptor gamma (PPARγ), phosphatase and tensin homolog (PTEN), and transcription factor 7-like 2 (TCF7L2) (99). Subsequently, numerous studies have associated DNA methylation patterns with obesity, based on BMI or waist circumference (WC) in both adults and children (100, 101). Moreover, increasing evidence demonstrated that transgenerational inheritance of environmental exposures may be passed down to generations of offspring never exposed to the parental stimuli (12). All these findings support the emerging evidence that pre-pregnancy paternal exposure, and/or exposure during pregnancy to both maternal weight gain and obesogens could explain most of transgenerational epigenetic programming that explain the hereditary nature of obesity and its related comorbidities in children (102–104).
In the next paragraphs, we will report the literature evidence of transgenerational inheritance of epigenetics due to parental overnutrition or in utero exposure to factors that may promote an obese phenotype in children.
Increasing evidence links parental nutritional status to epigenetic programming in offspring explaining the hereditary nature of metabolic diseases such as obesity, thus supporting the emerging concept of DOHaD that emphasizes the role of environmental factors during the embryonic period in determining the development of lifestyle-related disorders in adulthood (105).
In the transgenerational inheritance the insult may alter the germline epigenomic signature during preconception and transmit the epimutations to the F2 generation without any exposure as shown in Figure 1 (106). One of the first studies that reported evidence on the role of human pre-conception parent nutritional status on epigenetic inheritance examined the Dutch famine birth cohort and its offspring. In particular, the study demonstrated that offspring of exposed F1 fathers were heavier and had a higher BMI than offspring of unexposed F1 fathers (107). Recent studies especially in animal models have demonstrated that both obesity and diabetes changed levels of DNA and histone methylation, histone acetylation, and ncRNAs, such as miRNAs in oocytes and sperm (108). In particular, it is well known that DNA methylation reprogramming is a crucial event that takes place immediately post-fertilization and during gametogenesis (109). DNA methylation was analyzed at imprinted regions which include genes with monoallelic expression and for this reason susceptible to the paternal or maternal modulation in the progeny. Insulin like growth factor 2 (IGF2) gene and H19 lncRNA are well-described imprinted genes associated with childhood obesity which are subject to epigenetic regulation. The IGF-2 gene is a paternally imprinted gene encoding the protein IGF-2 that presents a high affinity with the insulin receptor and is related to mitogenesis and mitochondrial biogenesis. On the other hand, H-19 is a maternally imprinted gene that also is related to muscle insulin sensitivity (110, 111).
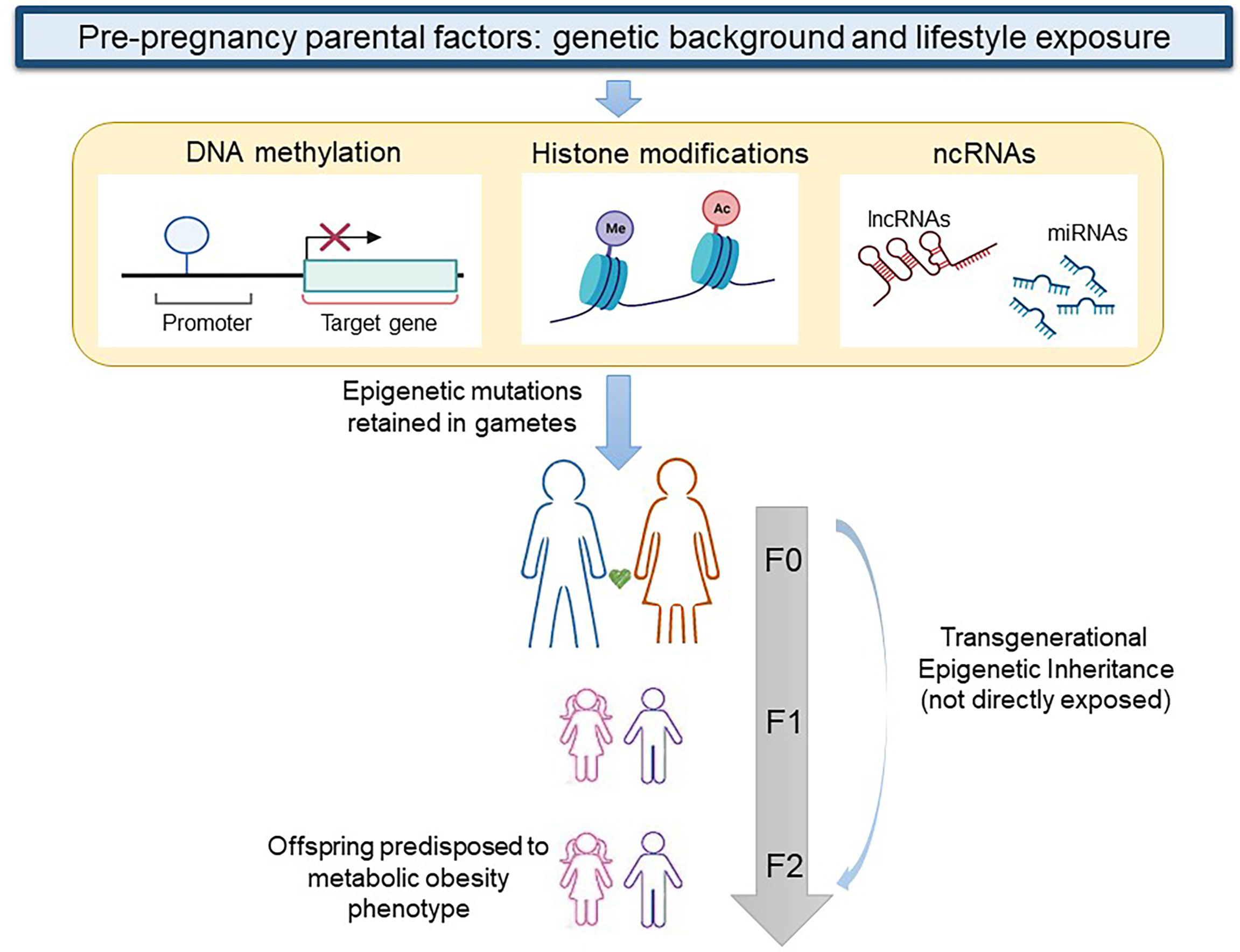
Figure 1 Transgenerational epigenetic inheritance of obesity. When genetic background and lifestyle exposure cause epigenetic modifications (DNA methylation, histone modifications, and changes in non-coding RNAs) in gametes of a woman or a man at F0, the first generation that exhibits a transgenerational epigenetic inheritance is the F2. lncRNA, long non coding RNA; ncRNA, non coding RNA; miRNA, micro RNA. Figure partially created with BioRender.com.
Unfortunately, the effect of epigenetic changes occurring in germline that may influence progeny obesity are poorly investigated because human sperms and oocytes are not available for research studies. Therefore, the negative effect on oocytes, and the link between obesity and epigenetics, have been performed mainly in animal models as reviewed by Ou et al. (108). In particular, animal models of diet, such as high-fat diet (HFD), or genetic-induced (i.e. ob/ob mice) obesity were found characterized by altered levels of DNA methylation in the oocytes (112, 113). HFD obese and ob/ob mice also exhibited a reduction of expression of STELLA protein (i.e., a maternal gene required for oogenesis and early embryogenesis) in oocytes that affected the levels of DNA methylation (114). Moreover, in the same experimental model Hou et al. reported modifications in the methylation levels of histone H3 lysine 9 and lysine 27 (113). Studies on ncRNAs and oocytes from individuals with obesity are still lacking.
Experimental studies by animal models also demonstrated a link between paternal obesity and changes in sperm cells DNA methylation profiles (112, 115). However, the interest for epigenetic inheritance of chronic metabolic disorders through the human paternal germ line is recently growing (116, 117). A previous human study demonstrated that babies born to fathers with obesity have altered DNA methylation at several regulatory regions of imprinted genes compared with children born to parents without obesity. The analysis of DNA methylation percentages at the differentially methylated regions (DMRs) from 92 umbilical cord blood leukocyte samples of newborns revealed that paternal obesity was significantly associated with lower methylation levels at the mesoderm specific transcript (MEST), paternally expressed 3 (PEG3), and neuronatin (NNAT) genes important for normal growth and development, suggesting the susceptibility for the next generation to develop chronic diseases in adulthood (118). Interestingly, a human study which compared sperm DNA methylation percentages, at 12 DMRs in 23 overweight/obese and 44 normal weight men showed that the obesity status is traceable in the sperm epigenome (119). In particular, the study reported a significant reduction of methylation levels in maternally expressed 3 (MEG3), necdin (NDN), small nuclear ribonucleoprotein polypeptide N (SNRPN), and epsilon sarcoglycan (SGCE)/paternally expressed 10 (PEG10) genes and a slightly increased of DNA methylation at the intergenic MEG3 and imprinted maternally expressed transcript (H19) DMRs in overweight/obese subjects compared to normal weight men (119). Recently, the impact of male obesity on DNA methylation reprogramming in sperm was also investigated by Keyhan et al. The author identified 3264 CpG sites in human sperm that are significantly associated with BMI in mature spermatozoa, then remarking the potential heritability of these DNA methylation alterations to the next generations (120).
Current research on how spermatozoa transmit acquired epigenetic changes affecting the metabolic health of the next generation revealed that both high-fat diets and low-protein diets may cause changes in spermatozoal content of ncRNA subtypes. In particular, altered sperm levels of snRNA due to high-fat diets in particular were associated with an altered phenotype in the offspring, with the offspring showing insulin resistance, altered body weight, and glucose intolerance (121). In men with obesity it was also observed an altered expression level of specific miRNAs, piRNAs, tRFs, and small nuclear RNA (snRNA) fragments in the spermatozoa with respect to lean men (122). Moreover, the authors reported also changes in DNA methylation patterns between obese and lean men in some genes, including neuropeptide Y (NPY), cannabinoid receptor type 1 (CR1), cocaine- and amphetamine-regulated transcript (CART), FTO, and carbohydrate sulfotransferase 8 (CHST8). Interestingly, the sperm pattern of DNA methylation was modulated by bariatric surgery, highlighting the relevance of the impact of weight loss and nutritional intake on the epigenome of human sperm (122).
Tissue and circulating miRNAs were extensively studied in the context of pediatric obesity (123, 124) but there are no studies on their pre-pregnancy modulation and effects on the obese phenotype on offspring. A work by López and colleagues showed that two miRNAs associated with inflammation and iron homeostasis (miR-155 and miR-122) were upregulated in sperm cells of obese subjects compared to subjects with normal weight (125). These findings support the hypothesis that differentially regulated microRNAs in sperms may potentially act upon DNA methylation post fertilization.
The interface between mother and fetus is the placenta, which regulates the flow of nutrients, oxygen, and hormonal supply. It follows that the placenta is involved in any abnormalities of fetal growth and of the resulting hepatometabolic and cardiovascular diseases in the offspring (126, 127). As depicted in Figure 2A, the placenta of pregnant women with obesity is subjected to increased inflammation and oxidative stress (128). Several data suggest that the chronic low-grade systemic inflammation in obese pregnancy and the accumulation of macrophages in the placenta cause uncontrolled local inflammation due to the production of pro-inflammatory cytokines that may alter maternal nutrient transport (129). A nonhuman primate model of excess nutrition in pregnancy provides evidence that a HFD decreases uterine blood flow and causes placental ischemia (130).
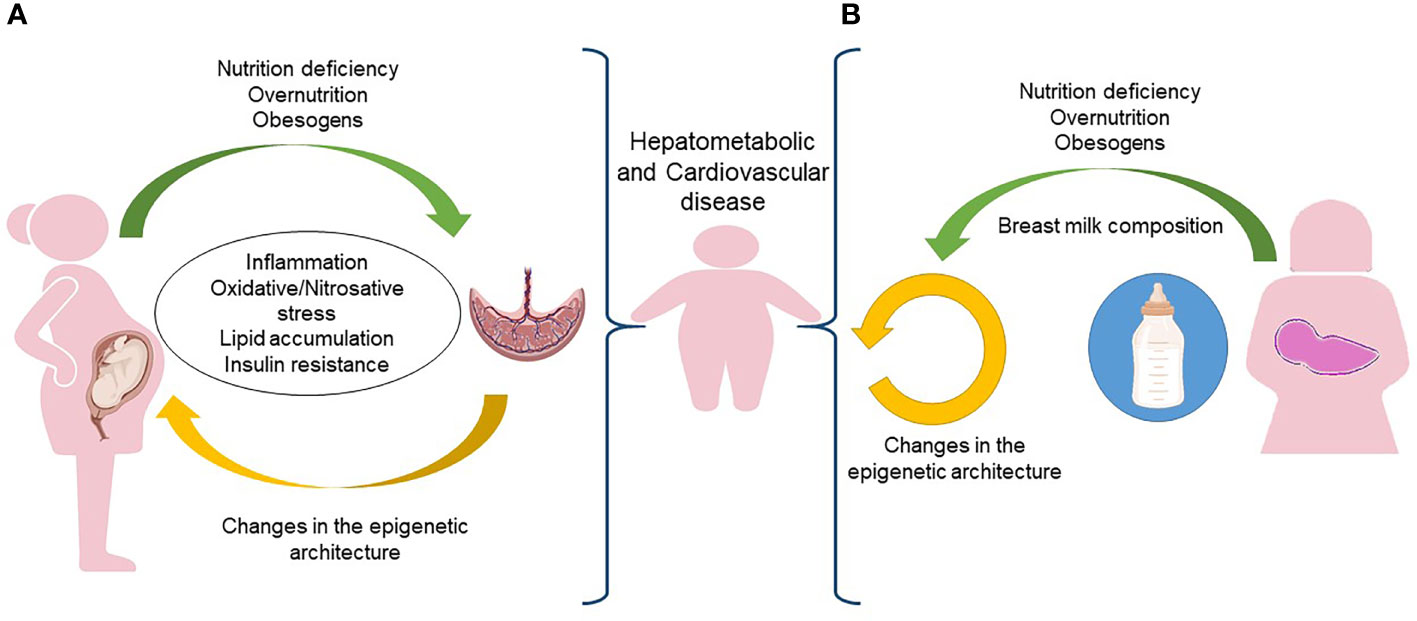
Figure 2 In utero and postnatal predisposition to obesity. (A) During pregnancy, the placenta of a woman exposed to nutrition deficiency, overnutrition or obesogens, is subjected to inflammation, oxidative/nitrosative stress, lipid accumulation and insulin resistance that increases the risk of obesity and its comorbidities in the newborn. (B) The same risk of obesity and comorbidities early in the life could be associated to epigenetic changes occurring in the breast milk. Figure partially created with BioRender.com.
In 2015 Sharp et al. analyzed the Avon Longitudinal Study of Parents and Children including a general population pregnancy cohort found that maternal pre-pregnancy BMI was positively associated with cord blood methylation at two CpG sites, at regions encoding for coiled-coil domain-containing 112 (CCDC112) and mucolipin TRP cation channel 3 (MCOLN3). Moreover, offspring of women with obesity had 28 CpG sites identified that were differentially methylated compared with offspring of women with normal weight. Four CpG sites in children peripheral blood were associated to maternal obesity. The study also reported a stronger association between offspring methylation and maternal obesity compared to paternal obesity, supporting an intrauterine mechanism (131).
The DNA methylation profile changes in the fetus can be influenced by the amount in the maternal diet of methyl donors from which depends the one carbon metabolism, including folate, methionine, betaine, or choline, thus leading to adverse metabolic effects during infancy and childhood. Maternal choline and betaine deficiencies also alter the methylation status of genes that participate in growth (Insulin-Like Growth Factor 2 Receptor, IGRF2, corticotropin releasing hormone, CRH, and nuclear receptor subfamily 3 group C member 1 (NR3C1) or vascular function (vascular endothelial growth factor C,VEFGC, and angiopoietin 2, ANGPT2), resulting in developmental abnormalities, blood vessel malformation, or increased predisposition for developing steatohepatitis in the offspring (132).
In the last ten years EWAS provided a useful and successful tool for longitudinal study designs of childhood obesity. Several studies highlighted the presence of a significant association between DNA methylation, obesity and some risk factors as birthweight and maternal BMI, supporting the emerging consensus on the heritability of BMI (133, 134).
Methylation status of leptin and its receptor (LEPR) gene promoters is one of the first evidence supporting the idea that epigenetic variations linked with intrauterine exposures and DNA methylation may influence childhood obesity (135). A further study demonstrated that a maternal poor weight gain during pregnancy was associated with increased methylation status of LEPR thus affecting the expression of the protein in umbilical vein of newborns (136).
A recent systematic review reported data from EWAS which investigated the DNA methylation levels, in peripheral blood or cord blood samples of children with or without obesity and/or overweight. Overall, 19 studies out of 23 found an association between childhood anthropometric parameters with at least one methylated CpG site or region. In four genes [histone deacetylase 4 (HDAC4), prolactin-releasing hormone receptor (PRLHR), tenascin XB (TNXB) genes, and PR domain containing 16 (PRDM16)] it was found the association of a DNA methylation profile with childhood obesity (97).
A meta-analysis study concerning data from EWAS of seven cohorts participating in the Pregnancy and Childhood Epigenetics (PACE) consortium found that maternal gestational diabetes mellitus (GDM) was associated with lower cord blood methylation levels within two regions. One region covering the promoter of OR2L13, a gene associated with autism spectrum disorder, and the other one covering the cytochrome P450 family 2 subfamily E member 1 (CYP2E1) gene, which is upregulated in type 1 and T2D (137).
A recent EWAS using maternal blood and cord blood samples from FinnGeDi (Finnish Gestational Diabetes) study cohort (298 mother-offspring pairs with GDM and 238 without) aimed to explore offspring-specific epigenetic effects in response to GDM exposure and maternal methylation levels (138). By a linear regression analysis, the authors found significant association of methylation at the transcription factor CP2 (TFCP2) gene in offspring of GDM exposure and maternal methylation. Furthermore, using the false discovery rate, the authors found seven significant DNA methylation differences positions between the mother’s methylation and GDM status. These loci included: DLG-Associated Protein 2 (DLGAP2) gene, H3 clustered histone 6 (H3C6) gene, family with sequence similarity 13 member A (FAM13A) gene, ubiquitin protein ligase E3C (UBE3C) gene, LOC127841, and two loci within intergenic regions. Interestingly, DLGAP2 and FAM13A, have previously been identified to have a role associated with diabetes and maternal insulin sensitivity in pregnancy, thus suggesting a possible function of their methylation in the future risk of obesity and T2D in offspring (139). Moreover, methylation changes in both FAM13A and DLGAP2 genes in newborns were also associated with environmental changes during pregnancy, including air pollution and smoking in pregnancy, respectively (140, 141).
The role of fetal programming in early infant weight gain due to maternal obesity has been recently further suggested by a study comparing the miRNA profile in peripheral blood mononuclear cell cultures of children from mothers who are normal weight or those who are overweight/obesity. The miRNA profile in mononuclear cells showed a downregulation of miR-155, miR-221, and miR-1301, as well as upregulation of miR-146a in the circulation of newborns from overweight and obese mothers. Moreover, the study showed that in the same samples from newborns, the RNA profile changes induced by overweight mothers and obesity were also associated to alteration in PPAR-γ gene expression, a master regulator of adipogenesis and altered cytokine pro-inflammatory levels in vitro blood mononuclear cell response to metabolic stimuli (142).
Also epigenetic regulation of intergenic regions appears to be important for obesity etiology. Indeed, Sasaki et al. (143) have recently shown that genomic loci associated with DMRs localized to intergenic regions and gene bodies most likely influence gene regulation. In particular, pathway analysis revealed that males, had a significant overrepresentation of genes involved in endocytosis and pathways in cancer, including IGF1R, which was previously shown to respond to diet-induced metabolic stress in animal models and in lymphocytes in the context of childhood obesity.
DNA methylation, considered as the key epigenetic mechanism by which maternal and paternal exposure to different dietary patterns, toxins or stressors causing phenotypical effects in unexposed future generations, may also explain some of the obesity-related comorbidities (132, 144). Studies reported that maternal obesity or overnutrition during pregnancy and/or lactation can modulate DNA methylation of different genes involved in energy metabolism, glucose homeostasis, insulin signaling and fat deposition, which support the role of DNA methylation in maternal obesity-induced risk of offspring NAFLD, obesity, and diabetes (145).
Several animal studies have shown that maternal nutrients and caloric regimen can impact fetal adiposity, interfering with long term metabolic homeostasis (146, 147). In murine models a HFD during pregnancy causes quite variable effects on fetal birth weight (130). These disparities in results are consistent with human observational studies showing increased rates of either large or small-for-gestational-age infants of mothers with obesity, although still both of these neonatal conditions are associated with risks of early onset childhood metabolic disease (148, 149) and histologically proven NAFLD as well (150, 151). Since obesity related NAFLD in pregnancy is independently associated also with hypertensive complications, postpartum hemorrhage and preterm birth, it should be considered a high-risk obstetric condition, with clinical implications for pre-conception counseling and pregnancy care (152).
In a mouse model, female offspring of mothers with obesity exposed to HFD was affected by obesity with high levels of inflammation in the adipose tissue in association with hypomethylation at certain inflammatory genes not only to the immediate offspring but also over three generations (153). Human studies suggested that social, environmental (e.g. education or cigarette smoking, respectively) and metabolic factors playing a role in the health of grandparents could influence the birth weight, the cardiovascular health, or even the neurodevelopmental outcomes of the grandchildren (154). This potential transgenerational inheritance is therefore now considered a relevant contributing factor to the epidemic levels of obesity and related comorbidities.
De Jesus et al. investigated the impact of paternal and/or maternal metabolic syndrome on the epigenetic reprogramming of metabolic homeostasis in offspring by using a non-dietary, genetically liver-specific insulin receptor deficient mice model of metabolic syndrome phenotype, including hyperglycemia, insulin resistance, and dyslipidemia. The experimental study found that parental metabolic syndrome induces downregulation of growth differentiation factor 15 gene in offspring by modulating the DNA methylation levels at the promoter region of the gene and boosting hepatic lipid accumulation in offspring (155). A study, investigating DMR in nondiabetic Pima Indians who were offspring of diabetic or nondiabetic mothers, supported the hypothesis that epigenetic changes may increase the risk of T2D in the offspring of mothers with diabetes during pregnancy (156). Moreover, a human study involving 128 offspring born at term to mothers with well-controlled GDM highlighted that this condition is associated with changes of DNA methylation at the alternative transcription start site zinc finger protein 696 (ZNF696) in cord blood cells, and changes of plasma glucose levels in newborns at 1 h after birth (157).
All in all, it emerges that the heritable epigenome, such as DNA methylation, histone modifications, and the expression of ncRNAs, could also predispose individuals to metabolic disorders and obesity phenotype. The epigenetic changes could be a target to control antepartum hyperglycemia, prevent gestational diabetes, and avoid excessive weight gain during pregnancy reducing childhood obesity and its related comorbidities. This proposed link between increased maternal insulin resistance during pregnancy and the development of T2D in the offspring should be taken into consideration for actively promoting changes in lifestyle before and during pregnancy in order to prevent the risk of developing obesity and obesity related metabolic diseases (157, 158).
Maternal serum free fatty acids reach also the placenta which represents a site of inflammatory and hypoxic changes. The unfolded protein response/endoplasmic reticulum stress (ERS) is, in fact, a source of reactive oxygen species. Upon disruption of mitochondria-associated membranes integrity, miscommunication directly or indirectly disturbs Ca2+ homeostasis and increases ERS and oxidative stress. Obesity-related maternal ERS and oxidative stress reaching the placenta may trigger inflammatory cytokines production and release. These molecules with maternal lipids may reach, through the umbilical cord vein and fetal portal vein, the fetal liver where they start to trigger hepatic lipogenesis and hepatocellular damage (159). In this scenario, the excess of maternal fatty acids could result in the accumulation of fetal ectopic fat. As the fetus subcutaneous adipose tissue (SAT) depot is not yet functioning, the accelerated adipogenesis during the perinatal window of SAT development underlies hepatometabolic and cardiovascular risk in offspring born to dams that are metabolically compromised (Figure 2). The exact mechanisms whereby these gestational events exacerbate the pathogenesis of fetal liver damage and also to cardiovascular risk are still poorly understood (150). Animal model studies showed that maternal obesity and HFD have a lasting cardiometabolic impact, and contribute to an increase in fetal liver inflammation and fatty liver in offspring (160–163).
In adult liver it appears quite well-established that NAFLD initiation and progression depend on several events. Among others, up-regulation of sterol regulatory element-binding protein-1c, a transcriptional activator of lipogenic enzymes such as stearoyl coenzyme-A desaturase1 and fatty acid synthase, which contributes to lipogenesis with an increased de novo synthesis of fatty acids, represent strong candidates for being involved also in fetal NAFLD (164, 165). NAFLD progression is also reflected by dysregulation of monocyte to macrophage differentiation-associated 2, the death decoy receptor TR6/DcR3 inhibiting T cell chemotaxis in vitro and in vivo, and chronic inhibition of nitric oxide synthase (166).
In addition to dietary composition, the exposure to chemical obesogens during pregnancy and early life is another mechanism that can strongly influence the subsequent predisposition to obesity. Obesogens are chemicals that inappropriately stimulate directly or indirectly adipogenesis and fat storage. According to animal models, the epigenetic effects of the exposure may persist on exposed animals and their offspring at least until generation F4 (102).
Exposure to mixtures of phthalates, parabens and other phenols is very common due to their ubiquitous prevalence in personal care products and plastics with also a risk of prenatal exposure. In a longitudinal study of mothers’ prenatal urinary concentration of phthalates and phenols was correlated with the z-score of BMI of the respective children (167).
Also the exposure to plastic derived endocrine disruptors EDCs (e.g. bisphenol) may affect the fetal growth and contribute to epigenetic and angiogenic changes, thus promoting the development of adult chronic diseases. Of note, these compounds may control the processes of epigenetic transgenerational inheritance of adult-onset diseases by modulating DNA methylation and epimutations in reproductive cells leading to the development of obesity and impaired glucose metabolism in the F2 generation (168). The endocrine disrupting effects of obesogens may directly or indirectly promote adipogenesis and obesity/obesity related metabolic conditions that drive metabolic syndrome through disturbance of several processes. Studies show that these effects are seen not only in individuals but also in their offspring because of their ability to epigenetically reprogram genetically inherited set points for the control of body weight and body composition at some critical stages of development, such as fetus, early life, and puberty (169, 170).
Promoting healthy early programming of adipose tissue is essential to implement new strategies that can control the spread of obesity and other metabolic disease (171, 172). However, the same obesogenic environment may have different effects on different individuals, highlighting an underlying individual susceptibility to obesity and fat distribution (173). In this scenario, the reversal of epigenetic changes could be used as an early and virtuous clinical intervention for reducing the risk of obesity in offspring (171). This goal is achievable through the adoption of healthy eating habits before and during critical periods of development, such as pregnancy and lactation (174). The particularly sensitive period capable of implementing epigenetic reprogramming strategies does not include only pregnancy, neonatal and infant life but encompasses a longer period defined the period of 1000 days, starting from conception and lasting the first 2 years of life (175). The milk of each mother is specific to her own baby, and the maternal-fetal dyad may affect the composition of breastfeeding and the baby’s tolerance to biologic breast milk (176). The identification of factors inducing epigenetic changes in breast milk, have led to the hypothesis of the possibility of transferring epigenetic information directly through breast milk (Figure 2B). Currently, studies in humans discovered more than 2000 miRNAs that can regulate collectively one third of the genes in the genome. The human milk provides abundant amounts of miRNA-148a, miR-152, miR-29b, and miR-21, which all target DNA methyl transferases (177, 178). The concept of milk siblings has been known for years (179, 180) and on this basis a study on rats showed that adoptive siblings, who had taken the same milk, had acquired the same hereditary epigenetic heritage (miRNAs). As in the consanguineous marriages, the children born from the marriage of these “milk siblings” may develop more serious pathologies (e.g., differentially expressed miRNAs were associated with pathways regulating metabolism, survival, and cancer development insulin signaling pathways).
Interpreting these results, we can deduce that the changes and strategies (nutrition, physical activity, and no-smoking) adopted during pregnancy and lactation can reverse the trend of a previous metabolic risk in the offspring. The lack of effective drugs easily available for the treatment of obesity and its comorbidities, such as obesity related-NAFLD, requires approaches with very timely changes in lifestyle (i.e. less sedentarism, more sleeping hours and exercise) and better eating habits. It has been shown that a normal pre-pregnancy maternal BMI and long duration of exclusive breastfeeding are favorable conditions not only for a direct effect on weight control, but also to prevent the development of NAFLD later in life (175).
Due to the lack of convincing data of efficacy and safety for both pregnant women with obesity and their offspring after pharmacological interventions during gestation with available medications (i.e. metformin) (181–184), the epigenetic modifiers may represent a promising approach (145, 185). Interestingly, maternal diet supplemented with methyl donors during lactation has been shown to prevent the development of an obese phenotype and fatty liver of offspring in mice (186). Similarly, recent studies in humans found that both paternal and maternal diet supplemented with choline, betaine, folate, and methionine, can affect offspring’s DNA methylation of genes involved in the regulation of metabolism, growth, and appetite (187, 188).
Although these findings suggest that a substantial component of obesity and metabolic disease risk has a prenatal developmental basis, the question remains unsolved because its complexity. In fact, in human studies it appears that there is no simple correlation between maternal intake of methyl group donors, higher or lower offspring DNA methylation and subsequent anthropometric outcomes (188). These effects could depend on the different tested developmental ages (170, 188). Alternatively, it has been suggested that the involvement of adjacent or nearby CpGs within the same promoter showing different strengths of association with child’s adiposity may imply highly specific changes in the transcriptional regulation of these genes induced by the developmental environment, rather than generalized changes in promoter methylation (170, 188). All in all, it appears that epigenetic reprogramming may represent an adaptable response through which the developing offspring modify their physiology to the postnatal environment (189).
The growing number of studies in the last few decades makes it clear that obesity is influenced by complex interactions between genetic, developmental, and environmental pressures. All these factors in turn create a network of relationships which are the basis for the concept of the multifactorial pathogenesis of obesity and its comorbidities.
It is obesogens evident that genetic factors play a relevant role, even though it has to be well dissected.
Purely monogenic forms of obesity presenting with severe early-onset obesity associated with hyperphagia and endocrine disruption in fact are so rare that the obesity epidemic cannot depend on single gene disorders. Therefore, the interaction of a more complex (polygenic) genetic background with the environment, where hundreds of SNPs exhibiting a heritability pattern similar to other complex traits and diseases can contribute to the development of the obese phenotype, is a more plausible explanation (20, 51).
Moreover, since environmental influences may cause epigenetic changes already in the gamete cell stage of both parents, but also during pregnancy and the early postnatal period including lactation with a lasting effect on the metabolism of the offspring, some inherent considerations follow. First, it appears now paramount the need of implementing the lifestyle and health status of both parents very early, even before conception (160, 190). Second, specific supplementation of the maternal diet with methyl donors during early gestation and during breastfeeding could prevent the development of an obese phenotype and fatty liver of offspring (186, 188). Further studies in this regard are however necessary to clarify relevant issues such as timing and dosage of methyl donors supplementation in pregnant and breastfeeding women (188, 191, 192).
However, the entire scenario of how parental diet and/or exposure to hormones and other environmental factors (e.g, endocrine disruptors) act transgenerationally on the epigenetic landscape and consequently on development of the offspring’s human adipose tissue and its plasticity over the life course requires further investigation (13).
Currently, epigenetic changes appear to be correctable by targeted interventions such as body weight reduction through virtuous dietary changes, exercise, or even metabolic surgery, all of them regularly affecting DNA methylation. However, targeted interventions with specific epigenetic modifiers to reprogram epigenetic changes is an extremely interesting future playground which has already started to be explored in some other fields. Cancer studies have in fact shown that a number of natural phytochemical substances (e.g. polyphenols) can target the epigenetic machinery with potential benefit due to reverting effects in the epigenetic modifications of relevant genes and restoring their protein expression levels (193). Similar new frontiers studies of cells reprogramming will likely prove meaningful if applicable to adipocytes also in obesity and related conditions as well.
In conclusion, although more evidence is needed, the reported literature strongly suggests that the analysis of the epigenetic architecture at the interface between gene expression and the epigenetic environment could be relevant for a better understanding of obesity and its associated comorbidities. Moreover, further studies addressing the potential intertwining between genetic and epigenetics in childhood obesity could provide the hint to improve prevention and therapeutic management.
NP, CM, AA, and PV contributed to the conception and design of the manuscript. All authors wrote sections of the manuscript and contributed to design of Figures and Tables. All authors approved the submitted version.
This study was supported by 5‰ 2022 assigned to AA by the Italian Ministry of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gregory JW. Prevention of obesity and metabolic syndrome in children. Front Endocrinol (2019) 10:669. doi: 10.3389/fendo.2019.00669
2. NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet (2017) 390:2627–42. doi: 10.1016/S0140-6736(17)32129-3
3. World Health Organization. Obesity and overweight (2018). Available at: https://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight (Accessed March 15, 2022).
4. Lloyd LJ, Langley-Evans SC, McMullen S. Childhood obesity and risk of the adult metabolic syndrome: a systematic review. Int J Obes (Lond) (2012) 36:1–11. doi: 10.1038/ijo.2011.186
5. Nehus E, Mitsnefes M. Childhood obesity and the metabolic syndrome. Pediatr Clin North Am (2019) 66:31–43. doi: 10.1016/j.pcl.2018.08.004
6. Drozdz D, Alvarez-Pitti J, Wójcik M, Borghi C, Gabbianelli R, Mazur A, et al. Obesity and cardiometabolic risk factors: From childhood to adulthood. Nutrients (2021) 13:4176. doi: 10.3390/nu13114176
7. Younossi ZM, Henry L. Fatty liver through the ages: Nonalcoholic steatohepatitis. Endocr Pract (2022) 28:204–13. doi: 10.1016/j.eprac.2021.12.010
8. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest (2017) 127:1–4. doi: 10.1172/JCI92035
9. Panera N, Barbaro B, Della Corte C, Mosca A, Nobili V, Alisi A. A review of the pathogenic and therapeutic role of nutrition in pediatric nonalcoholic fatty liver disease. Nutr Res (2018) 58:1–16. doi: 10.1016/j.nutres.2018.05.002
10. Weihrauch-Blüher S, Wiegand S. Risk factors and implications of childhood obesity. Curr Obes Rep (2018) 7:254–59. doi: 10.1007/s13679-018-0320-0
11. Thaker VV. Genetic and epigenetic causes of obesity. Adolesc Med State Art Rev (2017) 28:379–405.
12. King SE, Skinner MK. Epigenetic transgenerational inheritance of obesity susceptibility. Trends Endocrinol Metab (2020) 31:478–94. doi: 10.1016/j.tem.2020.02.009
13. Rodgers A, Sferruzzi-Perri AN. Developmental programming of offspring adipose tissue biology and obesity risk. Int J Obes (Lond) (2021) 45:1170–92. doi: 10.1038/s41366-021-00790-w
14. Sheikh AB, Nasrullah A, Haq S, Akhtar A, Ghazanfar H, Nasir A, et al. The interplay of genetics and environmental factors in the development of obesity. Cureus (2017) 9:e1435. doi: 10.7759/cureus.1435
15. Hruby A, Manson JE, Qi L, Malik VS, Rimm EB, Sun Q, et al. Determinants and consequences of obesity. Am J Public Health (2016) 106:1656–62. doi: 10.2105/AJPH.2016.303326
16. Elks CE, den Hoed M, Zhao JH, Sharp SJ, Wareham NJ, Loos RJ, et al. Variability in the heritability of body mass index: a systematic review and meta-regression. Front Endocrinol (2012) 3:29. doi: 10.3389/fendo.2012.00029
17. Karchynskaya V, Kopcakova J, Klein D, Gába A, Madarasova-Geckova A, van Dijk JP, et al. Is BMI a valid indicator of overweight and obesity for adolescents? Int J Environ Res Public Health (2020) 17:4815. doi: 10.3390/ijerph17134815
18. Bateson P, Barker D, Clutton-Brock T, Deb D, D'Udine B, Foley RA, et al. Developmental plasticity and human health. Nature (2004) 430:419–21. doi: 10.1038/nature02725
19. Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, Argyropoulos G, Walts B, et al. The human obesity gene map: the 2005 update. Obesity (2006) 14:529–44. doi: 10.1038/oby.2006.71
20. Huvenne H, Dubern B, Clément K, Poitou C. Rare genetic forms of obesity: Clinical approach and current treatments in 2016. Obes Facts (2016) 9:158–73. doi: 10.1159/000445061
21. Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Hered (1950) 41:317–18. doi: 10.1093/oxfordjournals.jhered.a106073
22. Koves IH, Roth C. Genetic and syndromic causes of obesity and its management. Indian J Pediatr (2018) 85:478–85. doi: 10.1007/s12098-017-2502-2
23. Farooqi S, O'Rahilly S. Genetics of obesity in humans. Endocr Rev (2006) 27:710–18. doi: 10.1210/er.2006-0040
24. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi: 10.1038/372425a0
25. Obradovic M, Sudar-Milovanovic E, Soskic S, Essack M, Arya S, Stewart AJ, et al. Leptin and obesity: Role and clinical implication. Front Endocrinol (Lausanne) (2021) 12:585887. doi: 10.3389/fendo.2021.585887
26. Boden G, Chen X, Mozzoli M, Ryan I. Effect of fasting on serum leptin in normal human subjects. J Clin Endocrinol Metab (1996) 81:3419–23. doi: 10.1210/jcem.81.9.8784108
27. Kolaczynski JW, Considine RV, Ohannesian J, Marco C, Opentanova I, Nyce MR, et al. Responses of leptin to short-term fasting and refeeding in humans: a link with ketogenesis but not ketones themselves. Diabetes (1996) 45:1511–15. doi: 10.2337/diab.45.11.1511
28. Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature (1997) 387:903–8. doi: 10.1038/43185
29. Farr OM, Gavrieli A, Mantzoros CS. Leptin applications in 2015: What have we learned about leptin and obesity? Curr Opin Endocrinol Diabetes Obes (2015) 22:353–9. doi: 10.1097/MED.0000000000000184
30. Dubern B, Clement K. Leptin and leptin receptor-related monogenic obesity. Biochimie (2012) 94:2111–5. doi: 10.1016/j.biochi.2012.05.010
31. Chiurazzi M, Cozzolino M, Orsini RC, Di Maro M, Di Minno MND, Colantuoni A. Impact of genetic variations and epigenetic mechanisms on the risk of obesity. Int J Mol Sci (2020) 21:9035. doi: 10.3390/ijms21239035
32. Crocker MK, Yanovski JA. Pediatric obesity: etiology and treatment. Endocrinol Metab Clin North Am (2009) 38:525–48. doi: 10.1016/j.ecl.2009.06.007
33. Krude H, Grüters A. Implications of proopiomelanocortin (POMC) mutations in humans: the POMC deficiency syndrome. Trends Endocrinol Metab (2000) 11:15–22. doi: 10.1016/s1043-2760(99)00213-1
34. Chung WK. An overview of mongenic and syndromic obesities in humans. Pediatr Blood Cancer (2012) 58:122–8. doi: 10.1002/pbc.23372
35. van den Berg L, van Beekum O, Heutink P, Felius BA, van de Heijning MP, Strijbis S, et al. Melanocortin-4 receptor gene mutations in a Dutch cohort of obese children. Obesity (2011) 19:604–11. doi: 10.1038/oby.2010.259
36. Xi B, Chandak GR, Shen Y, Wang Q, Zhou D. Association between common polymorphism near the MC4R gene and obesity risk: a systematic review and meta-analysis. PloS One (2012) 7:e45731. doi: 10.1371/journal.pone.0045731
37. Dwivedi OP, Tabassum R, Chauhan G, Kaur I, Ghosh S, Marwaha RK, et al. Strong influence of variants near MC4R on adiposity in children and adults: a cross-sectional study in Indian population. J Hum Genet (2013) 58:27–32. doi: 10.1038/jhg.2012.129
38. Farooqi IS, Volders K, Stanhope R, Heuschkel R, White A, Lank E, et al. Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab (2007) 92:3369–73. doi: 10.1210/jc.2007-0687
39. Ranadive SA, Vaisse C. Lessons from extreme human obesity: monogenic disorders. Endocrinol Metab Clin North Am (2008) 37:733–51. doi: 10.1016/j.ecl.2008.07.003
40. Xi D, Gandhi N, Lai M, Kublaoui BM. Ablation of Sim1 neurons causes obesity through hyperphagia and reduced energy expenditure. PloS One (2012) 7:e36453. doi: 10.1371/journal.pone.0036453
41. Gonsalves R, Aleck K, Newbern D, Shaibi G, Kapadia C, Oatman O. Severe early onset obesity and hypopituitarism in a child with a novel SIM1 gene mutation. Endocrinol Diabetes Metab Case Rep (2020) 2020:20–0042. doi: 10.1530/EDM-20-0042
42. Niazi RK, Gjesing AP, Hollensted M, Have CT, Borisevich D, Grarup N, et al. Screening of 31 genes involved in monogenic forms of obesity in 23 Pakistani probands with early-onset childhood obesity: a case report. BMC Med Genet (2019) 20:152. doi: 10.1186/s12881-019-0886-8
43. Bonnefond A, Raimondo A, Stutzmann F, Ghoussaini M, Ramachandrappa S, Bersten DC, et al. Loss-of-function mutations in SIM1 contribute to obesity and prader-willi-like features. J Clin Invest (2013) 123:3037–41. doi: 10.1172/JCI68035
44. Zegers D, Beckers S, Hendrickx R, Van Camp JK, de Craemer V, Verrijken A, et al. Mutation screen of the SIM1 gene in pediatric patients with early-onset obesity. Int J Obes (Lond) (2014) 38:1000–4. doi: 10.1038/ijo.2013.188
45. Stanikova D, Buzga M, Krumpolec P, Skopkova M, Surova M, Ukropcova B, et al. Genetic analysis of single-minded 1 gene in early-onset severely obese children and adolescents. PloS One (2017) 12:e0177222. doi: 10.1371/journal.pone.0177222
46. Tapia-Arancibia L, Rage F, Givalois L, Arancibia S. Physiology of BDNF: focus on hypothalamic function. Front Neuroendoc (2004) 25:77–107. doi: 10.1016/j.yfrne.2004.04.001
47. Wang P, Loh KH, Wu M, Morgan DA, Schneeberger M, Yu X, et al. A leptin-BDNF pathway regulating sympathetic innervation of adipose tissue. Nature (2020) 583:839–44. doi: 10.1038/s41586-020-2527-y
48. Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes (2006) 55:3366–71. doi: 10.2337/db06-0550
49. Serra-Juhé C, Martos-Moreno GÁ, Bou de Pieri F, Flores R, Chowen JA, Pérez-Jurado LA, et al. Heterozygous rare genetic variants in non-syndromic early-onset obesity. Int J Obes (2020) 44:830–41. doi: 10.1038/s41366-019-0357-5
50. da Fonseca ACP, Abreu GM, Palhinha L, Zembrzuski VM, Campos Junior M, Carneiro JRI, et al. A rare potential pathogenic variant in the BDNF gene is found in a Brazilian patient with severe childhood-onset obesity. Diabetes Metab Syndr Obes (2021) 14:11–22. doi: 10.2147/DMSO.S267202
51. Crouch DJM, Bodmer WF. Polygenic inheritance, GWAS, polygenic risk scores, and the search for functional variants. Proc Natl Acad Sci U.S.A. (2020) 117:18924–33. doi: 10.1073/pnas.2005634117
52. Chami N, Preuss M, Walker RW, Moscati A, Loos RJF. The role of polygenic susceptibility to obesity among carriers of pathogenic mutations in MC4R in the UK biobank population. PloS Med (2020) 17:e1003196. doi: 10.1371/journal.pmed.1003196
53. van der Klaauw AA, Croizier S, Mendes de Oliveira E, Stadler LKJ, Park S, Kong Y, et al. Human semaphorin 3 variants link melanocortin circuit development and energy balance. Cell (2019) 176:729–42.e18. doi: 10.1016/j.cell.2018.12.009
54. Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet (2018) 27:3641–49. doi: 10.1093/hmg/ddy271
55. Larder R, Cheung MK, Tung YC, Yeo GS, Coll AP. Where to go with FTO? Trends Endocrinol Metab (2011) 22:53–9. doi: 10.1016/j.tem.2010.11.001
56. Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science (2007) 316:889–94. doi: 10.1126/science.1141634
57. Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PloS Genet (2007) 3:e115. doi: 10.1371/journal.pgen.0030115
58. Cecil JE, Tavendale R, Watt P, Hetherington MM, Palmer CN. An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med (2008) 359:2558–66. doi: 10.1056/NEJMoa0803839
59. Rutters F, Nieuwenhuizen AG, Bouwman F, Mariman E, Westerterp-Plantenga MS. Associations between a single nucleotide polymorphism of the FTO gene (rs9939609) and obesity-related characteristics over time during puberty in a Dutch children cohort. J Clin Endocrinol Metab (2011) 96:E939–42. doi: 10.1210/jc.2010-2413
60. Wardle J, Carnell S, Haworth CM, Farooqi IS, O'Rahilly S, Plomin R. Obesity associated genetic variation in FTO is associated with diminished satiety. J Clin Endocrinol Metab (2008) 93:3640–3. doi: 10.1210/jc.2008-0472
61. Jacobsson JA, Danielsson P, Svensson V, Klovins J, Gyllensten U, Marcus C, et al. Major gender difference in association of FTO gene variant among severely obese children with obesity and obesity related phenotypes. Biochem Biophys Res Commun (2008) 368:476–82. doi: 10.1016/j.bbrc.2008.01.087
62. Mangge H, Renner W, Almer G, Weghuber D, Möller R, Horejsi R. Rs9939609 variant of the fat mass and obesity-associated gene and trunk obesity in adolescents. J Obes (2011) 2011:186368. doi: 10.1155/2011/186368
63. Xi B, Shen Y, Zhang M, Liu X, Zhao X, Wu L, et al. The common rs9939609 variant of the fat mass and obesity-associated gene is associated with obesity risk in children and adolescents of Beijing, China. BMC Med Genet (2010) 11:107. doi: 10.1186/1471-2350-11-107
64. Liu C, Mou S, Cai Y. FTO gene variant and risk of overweight and obesity among children and adolescents: a systematic review and meta-analysis. PloS One (2013) 8:e82133. doi: 10.1371/journal.pone.0082133
65. Todendi PF, Martínez JA, Reuter CP, Klinger EI, Fiegenbaum M, Rosane de Moura Valim A. Influence of FTO (Fat mass and obesity) gene and parental obesity on Brazilian children and adolescents adiposity. J Pediatr Endocrinol Metab (2020):975–82. doi: 10.1515/jpem-2019-0594
66. Reuter ÉM, Reuter CP, de Castro Silveira JF, Carroll S, Hobkirk JP, Todendi PF, et al. FTO gene polymorphism and longitudinal changes in nutritional/obesity status in children and adolescents: Schoolchildren's health cohort study. Eur J Pediatr (2021) 180:3325–33. doi: 10.1007/s00431-021-04120-0
67. Grant SF, Li M, Bradfield JP, Kim CE, Annaiah K, Santa E, et al. Association analysis of the FTO gene with obesity in children of Caucasian and African ancestry reveals a common tagging SNP. PloS One (2008) 3:e1746. doi: 10.1371/journal.pone.0001746
68. Pereira P de A, Alvim-Soares AM, Sandrim VC, Moreira Lanna CM, Souza-Costa DC, de Almeida Belo V, et al. Lack of association between genetic polymorphism of FTO, AKT1 and AKTIP in childhood overweight and obesity. J Pediatr (2016) 92:521–7. doi: 10.1016/j.jped.2015.12.007
69. López-Rodríguez G, Estrada-Neria A, Suárez-Diéguez T, Tejero ME, Fernández JC, Galván M. Common polymorphisms in MC4R and FTO genes are associated with BMI and metabolic indicators in Mexican children: Differences by sex and genetic ancestry. Gene (2020) 754:144840. doi: 10.1016/j.gene.2020.144840
70. Mejía-Benítez A, Klünder-Klünder M, Yengo L, Meyre D, Aradillas C, Cruz E, et al. Analysis of the contribution of FTO, NPC1, ENPP1, NEGR1, GNPDA2 and MC4R genes to obesity in Mexican children. BMC Med Genet (2013) 14:21. doi: 10.1186/1471-2350-14-21
71. Albuquerque D, Nóbrega C, Manco L. Association of FTO polymorphisms with obesity and obesity-related outcomes in Portuguese children. PloS One (2013) 8:e54370. doi: 10.1371/journal.pone.0054370
72. Inandiklioğlu N, Yaşar A. Association between rs1421085 and rs9939609 polymorphisms of fat mass and obesity-associated gene with high-density lipoprotein cholesterol and triglyceride in obese Turkish children and adolescents. J Pediatr Genet (2021) 10:9–15. doi: 10.1055/s-0040-1713154009-15
73. Dastgheib SA, Bahrami R, Setayesh S, Salari S, Mirjalili SR, Noorishadkam M, et al. Evidence from a meta-analysis for association of MC4R rs17782313 and FTO rs9939609 polymorphisms with susceptibility to obesity in children. Diabetes Metab Syndr (2021) 15:102234. doi: 10.1016/j.dsx.2021.102234
74. Almeida SM, Furtado JM, Mascarenhas P, Ferraz ME, Ferreira JC, Monteiro MP, et al. Association between LEPR, FTO, MC4R, and PPARG-2 polymorphisms with obesity traits and metabolic phenotypes in school-aged children. Endocrine (2018) 60:466–78. doi: 10.1007/s12020-018-1587-3
75. Moghbeli M, Khedmatgozar H, Yadegari M, Avan A, Ferns GA, Ghayour Mobarhan M. Cytokines and the immune response in obesity-related disorders. Adv Clin Chem (2021) 101:135–68. doi: 10.1016/bs.acc.2020.06.004
76. Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr (2004) 92:347–55. doi: 10.1079/bjn20041213
77. Panera N, Della Corte C, Crudele A, Stronati L, Nobili V, Alisi A. Recent advances in understanding the role of adipocytokines during non-alcoholic fatty liver disease pathogenesis and their link with hepatokines. Expert Rev Gastroenterol Hepatol (2016) 10:393–403. doi: 10.1586/17474124.2016.1110485
78. Brand E, Schorr U, Kunz I, Kertmen E, Ringel J, Distler A, et al. Tumor necrosis factor-alpha–308 G/A polymorphism in obese caucasians. Int J Obes Relat Metab Disord (2001) 25:581–5. doi: 10.1038/sj.ijo.0801576
79. Popko K, Górska E, Pyrzak B, Telmaszczyk-Emmel A, Wisniewska A, Majcher A, et al. Influence of proinflammatory cytokine gene polymorphism on childhood obesity. Eur J Med Res (2009) Suppl 4:59–62. doi: 10.1186/2047-783x-14-s4-59
80. Nascimento H, Vieira E, Coimbra S, Catarino C, Costa E, Bronze-da-Rocha E, et al. Adipokine gene single-nucleotide polymorphisms in Portuguese obese adolescents: Associations with plasma concentrations of adiponectin, resistin, IL-6, IL-1β, and TNF-α. Child Obes (2016) 12:300–13. doi: 10.1089/chi.2015.0235
81. Mărginean CO, Mărginean C, Iancu M, Moldovan VG, Melit LE, Bănescu C. The impact of TNF-α 308G>A gene polymorphism on children's overweight risk and an assessment of biochemical variables: A cross-sectional single-center experience. Pediatr Neonatol (2019) 60:19–27. doi: 10.1016/j.pedneo.2018.03.003
82. Mǎrginean CO, Mǎrginean C, Meliţ LE. New insights regarding genetic aspects of childhood obesity: A minireview. Front Pediatr (2018) 6:271. doi: 10.3389/fped.2018.00271
83. Ibrahim OM, Gabre AA, Sallam SF, El-Alameey IR, Sabry RN, Galal EM, et al. Influence of interleukin-6 (174G/C) gene polymorphism on obesity in Egyptian children. Open Access Maced J Med Sci (2017) 5:831–35. doi: 10.3889/oamjms.2017.175
84. Khera AV, Chaffin M, Wade KH, Zahid S, Brancale J, Xia R, et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell (2019) 177:587–96.e9. doi: 10.1016/j.cell.2019.03.028
85. Warner ET, Jiang L, Adjei DN, Turman C, Gordon W, Wang L, et al. A genome-wide association study of childhood body fatness. Obes (Silver Spring) (2021) 29:446–53. doi: 10.1002/oby.23070
86. Villicaña S, Bell JT. Genetic impacts on DNA methylation: research findings and future perspectives. Genome Biol (2021) 22:127. doi: 10.1186/s13059-021-02347-6
87. Tang Y, Jin B, Zhou L, Lu W. MeQTL analysis of childhood obesity links epigenetics with a risk SNP rs17782313 near MC4R from meta-analysis. Oncotarget (2017) 8:2800–6. doi: 10.18632/oncotarget.13742
88. Gutierrez-Arcelus M, Lappalainen T, Montgomery SB, Buil A, Ongen H, Yurovsky A, et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife (2013) 2:e00523. doi: 10.7554/eLife.00523
89. Jones MJ, Fejes AP, Kobor MS. DNA Methylation, genotype and gene expression: who is driving and who is along for the ride? Genome Biol (2013) 14:126. doi: 10.1186/gb-2013-14-7-126
90. Dallner OS, Marinis JM, Lu YH, Birsoy K, Werner E, Fayzikhodjaeva G, et al. Dysregulation of a long noncoding RNA reduces leptin leading to a leptin-responsive form of obesity. Nat Med (2019) 25:507–16. doi: 10.1038/s41591-019-0370-1
91. Manco M, Crudele A, Mosca A, Caccamo R, Braghini MR, De Vito R, et al. LncOb rs10487505 variant is associated with leptin levels in pediatric non-alcoholic fatty liver disease. Pediatr Res (2022) Epub ahead of print. doi: 10.1038/s41390-022-02032-9
92. Greenberg MVC, Bourc'his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol (2019) 20:590–607. doi: 10.1038/s41580-019-0159-6
93. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res (2011) 21:381–95. doi: 10.1038/cr.2011.22
94. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature (2010) 466:835–40. doi: 10.1038/nature09267
95. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell (2018) 172:393–407. doi: 10.1016/j.cell.2018.01.011
96. Mahmoud AM. An overview of epigenetics in obesity: The role of lifestyle and therapeutic interventions. Int J Mol Sci (2022) 23:1341. doi: 10.3390/ijms23031341
97. Alfano R, Robinson O, Handakas E, Nawrot TS, Vineis P, Plusquin M. Perspectives and challenges of epigenetic determinants of childhood obesity: A systematic review. Obes Rev (2022) 23:e13389. doi: 10.1111/obr.13389
98. Robinson N, Brown H, Antoun E, Godfrey KM, Hanson MA, Lillycrop KA, et al. Childhood DNA methylation as a marker of early life rapid weight gain and subsequent overweight. Clin Epigenet (2021) 13:8. doi: 10.1186/s13148-020-00952-z
99. Xu X, Su S, Barnes VA, De Miguel C, Pollock J, Ownby D, et al. A genome-wide methylation study on obesity: differential variability and differential methylation. Epigenetics (2013) 8:522–33. doi: 10.4161/epi.24506
100. Huang RC, Garratt ES, Pan H, Wu Y, Davis EA, Barton SJ, et al. Genome-wide methylation analysis identifies differentially methylated CpG loci associated with severe obesity in childhood. Epigenetics (2015) 10:995–1005. doi: 10.1080/15592294.2015.1080411
101. Sayols-Baixeras S, Subirana I, Fernández-Sanlés A, Sentí M, Lluís-Ganella C, Marrugat J, et al. DNA Methylation and obesity traits: An epigenome-wide association study. REGICOR study. Epigenet (2017) 12:909–16. doi: 10.1080/15592294.2017.1363951
102. Lee MK, Blumberg B. Transgenerational effects of obesogens. Basic Clin Pharmacol Toxicol (2019) 125 Suppl 3:44–57. doi: 10.1111/bcpt.13214
103. Mohajer N, Joloya EM, Seo J, Shioda T, Blumberg B. Epigenetic transgenerational inheritance of the effects of obesogen exposure. Front Endocrinol (2021) 12:787580. doi: 10.3389/fendo.2021.787580
104. Opsahl JO, Moen GH, Qvigstad E, Böttcher Y, Birkeland KI, Sommer C. Epigenetic signatures associated with maternal body mass index or gestational weight gain: a systematic review. J Dev Orig Health Dis (2021) 12:373–83. doi: 10.1017/S2040174420000811
105. Lopomo A, Burgio E, Migliore L. Epigenetics of obesity. Prog Mol Biol Transl Sci (2016) 140:151–84. doi: 10.1016/bs.pmbts.2016.02.002
106. Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet (2007) 8:253–62. doi: 10.1038/nrg2045
107. Veenendaal MV, Painter RC, de Rooij SR, Bossuyt PM, van der Post JA, Gluckman PD, et al. Transgenerational effects of prenatal exposure to the 1944-45 Dutch famine. BJOG (2013) 120:548–53. doi: 10.1111/1471-0528.12136
108. Ou XH, Zhu CC, Sun SC. Effects of obesity and diabetes on the epigenetic modification of mammalian gametes. J Cell Physiol (2019) 234:7847–55. doi: 10.1002/jcp.27847
109. Ly L, Chan D, Trasler JM. Developmental windows of susceptibility for epigenetic inheritance through the male germline. Semin Cell Dev Biol (2015) 43:96–105. doi: 10.1016/j.semcdb.2015.07.006
110. Wang MJ, Chen F, Liu QG, Liu CC, Yao H, Yu B, et al. Insulin-like growth factor 2 is a key mitogen driving liver repopulation in mice. Cell Death Dis (2018) 9:26. doi: 10.1038/s41419-017-0186-1
111. Geng T, Liu Y, Xu Y, Jiang Y, Zhang N, Wang Z, et al. H19 lncRNA promotes skeletal muscle insulin sensitivity in part by targeting AMPK. Diabetes (2018) 67:2183–98. doi: 10.2337/db18-0370
112. Ge ZJ, Luo SM, Lin F, Liang QX, Huang L, Wei YC, et al. DNA Methylation in oocytes and liver of female mice and their offspring: effects of high-fat-diet-induced obesity. Environ Health Perspect (2014) 122:159–64. doi: 10.1289/ehp.1307047
113. Hou YJ, Zhu CC, Duan X, Liu HL, Wang Q, Sun SC. Both diet and gene mutation induced obesity affect oocyte quality in mice. Sci Rep (2016) 6:18858. doi: 10.1038/srep18858
114. Han L, Ren C, Li L, Li X, Ge J, Wang H, et al. Publisher correction: Embryonic defects induced by maternal obesity in mice derive from Stella insufficiency in oocytes. Nat Genet (2018) 50:768. doi: 10.1038/s41588-018-0077-0
115. Youngson NA, Lecomte V, Maloney CA, Leung P, Liu J, Hesson LB, et al. Obesity-induced sperm DNA methylation changes at satellite repeats are reprogrammed in rat offspring. Asian J Androl (2016) 18:930–36. doi: 10.4103/1008-682X.163190
116. Illum LRH, Bak ST, Lund S, Nielsen AL. DNA Methylation in epigenetic inheritance of metabolic diseases through the male germ line. J Mol Endocrinol (2018) 60:R39–56. doi: 10.1530/JME-17-0189
117. Raad G, Hazzouri M, Bottini S, Trabucchi M, Azoury J, Grandjean V. Paternal obesity: how bad is it for sperm quality and progeny health? Basic Clin Androl (2017) 27:20. doi: 10.1186/s12610-017-0064-9
118. Soubry A, Murphy SK, Wang F, Huang Z, Vidal AC, Fuemmeler BF, et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes (Lond) (2015) 39:650–7. doi: 10.1038/ijo.2013.193
119. Soubry A, Guo L, Huang Z, Hoyo C, Romanus S, Price T, et al. Obesity-related DNA methylation at imprinted genes in human sperm: Results from the TIEGER study. Clin Epigenet (2016) 8:51. doi: 10.1186/s13148-016-0217-2
120. Keyhan S, Burke E, Schrott R, Huang Z, Grenier C, Price T, et al. Male Obesity impacts DNA methylation reprogramming in sperm. Clin Epigenet (2021) 13:17. doi: 10.1186/s13148-020-00997-0
121. Klastrup LK, Bak ST, Nielsen AL. The influence of paternal diet on sncRNA-mediated epigenetic inheritance. Mol Genet Genomics (2019) 294:1–11. doi: 10.1007/s00438-018-1492-8
122. Donkin I, Versteyhe S, Ingerslev LR, Qian K, Mechta M, Nordkap L, et al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab (2016) 23:369–78. doi: 10.1016/j.cmet.2015.11.004
123. Hutny M, Hofman J, Zachurzok A, Matusik P. MicroRNAs as the promising markers of comorbidities in childhood obesity-a systematic review. Pediatr Obes (2022) 17:e12880. doi: 10.1111/ijpo.12880
124. Ji C, Guo X. The clinical potential of circulating microRNAs in obesity. Nat Rev Endocrinol (2019) 15:731–43. doi: 10.1038/s41574-019-0260-0
125. López P, Castro A, Flórez M, Miranda K, Aranda P, Sánchez-González C, et al. miR-155 and miR-122 expression of spermatozoa in obese subjects. Front Genet (2018) 9:175. doi: 10.3389/fgene.2018.00175
126. Roberts DJ, Post MD. The placenta in pre-eclampsia and intrauterine growth restriction. J Clin Pathol (2008) 61:1254–60. doi: 10.1136/jcp.2008.055236
127. Amir H, Weintraub A, Aricha-Tamir B, Apel-Sarid L, Holcberg G, Sheiner E. A piece in the puzzle of intrauterine fetal death: pathological findings in placentas from term and preterm intrauterine fetal death pregnancies. J Matern Fetal Neonatal Med (2009) 22:759–64. doi: 10.3109/14767050902929396
128. Bedell S, Hutson J, de Vrijer B, Eastabrook. Effects of maternal obesity and gestational diabetes mellitus on the placenta: Current knowledge and targets for therapeutic interventions. Curr Vasc Pharmacol (2021) 19:176–92. doi: 10.2174/1570161118666200616144512
129. Corrales P, Vidal-Puig A, Medina-Gómez G. Obesity and pregnancy, the perfect metabolic storm. Eur J Clin Nutr (2021) 75:1723–34. doi: 10.1038/s41430-021-00914-5
130. Frias AE, Grove KL. Obesity: a transgenerational problem linked to nutrition during pregnancy. Semin Reprod Med (2012) 30:472–8. doi: 10.1055/s-0032-1328875
131. Sharp GC, Lawlor DA, Richmond RC, Fraser A, relton A, Suderman M, et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: findings from the Avon longitudinal study of parents and children. Int J Epidemiol (2015) 44:1288–304. doi: 10.1093/ije/dyv042
132. Samblas M, Milagro FI, Martínez A. DNA Methylation markers in obesity, metabolic syndrome, and weight loss. Epigenetics (2019) 14:421–44. doi: 10.1080/15592294.2019.1595297
133. Küpers LK, Monnereau C, Sharp GC, Yousefi P, Salas LA, Ghantous A, et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat Commun (2019) 10:1893. doi: 10.1038/s41467-019-09671-3
134. Sharp GC, Salas LA, Monnereau C, Allard C, Yousefi P, Everson TM, et al. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum Mol Genet (2017) 26:4067–85. doi: 10.1093/hmg/ddx290
135. García-Cardona MC, Huang F, García-Vivas JM, López-Camarillo C, Del Río Navarro BE, Navarro Olivos E, et al. DNA Methylation of leptin and adiponectin promoters in children is reduced by the combined presence of obesity and insulin resistance. Int J Obes (Lond) (2014) 38:1457–65. doi: 10.1038/ijo.2014.30
136. Chavira-Suárez E, Ramírez-Mendieta AJ, Martínez-Gutiérrez S, Zárate-Segura P, Beltrán-Montoya J, Espinosa-Maldonado NC, et al. Influence of pre-pregnancy body mass index (p-BMI) and gestational weight gain (GWG) on DNA methylation and protein expression of obesogenic genes in umbilical vein. PloS One (2019) 14:e0226010. doi: 10.1371/journal.pone.0226010
137. Howe CG, Cox B, Fore R, Jungius J, Kvist T, Lent S, et al. Maternal gestational diabetes mellitus and newborn DNA methylation: Findings from the pregnancy and childhood epigenetics consortium. Diabetes Care (2020) 43:98–105. doi: 10.2337/dc19-0524
138. Canouil M, Khamis A, Keikkala E, Hummel S, Lobbens S, Bonnefond A, et al. Epigenome-wide association study reveals methylation loci associated with offspring gestational diabetes mellitus exposure and maternal methylome. Diabetes Care (2021) 44:1992–99. doi: 10.2337/dc20-2960
139. Hivert MF, Cardenas A, Allard C, Doyon M, Powe CE, Catalano PM, et al. Interplay of placental DNA methylation and maternal insulin sensitivity in pregnancy. Diabetes (2020) 69:484–92. doi: 10.2337/db19-0798
140. Gruzieva O, Xu CJ, Yousefi P, Relton C, Merid SK, Breton CV, et al. Prenatal particulate air pollution and DNA methylation in newborns: An epigenome-wide meta-analysis. Environ Health Perspect (2019) 127:57012. doi: 10.1289/EHP4522
141. Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, et al. DNA Methylation in newborns and maternal smoking in pregnancy: genome-wide consortium metaanalysis. Am J HumGenet (2016) 98:680–96. doi: 10.1016/j.ajhg.2016.02.019
142. Gaytán-Pacheco N, Lima-Rogel V, Méndez-Mancilla A, Escalante-Padrón F, Toro-Ortíz JC, Jiménez-Capdeville ME, et al. Changes in PPAR-γ expression are associated with microRNA profiles during fetal programming due to maternal overweight and obesity. Gynecol Obstet Invest (2021) 86:415–26. doi: 10.1159/000517116
143. Sasaki A, Murphy KE, Briollais L, McGowan PO, Matthews SG. DNA Methylation profiles in the blood of newborn term infants born to mothers with obesity. PloS One (2022) 17:e0267946. doi: 10.1371/journal.pone.0267946
144. Sharma A. Transgenerational epigenetics: integrating soma to germline communication with gametic inheritance. Mech Ageing Dev (2017) 163:15–22. doi: 10.1016/j.mad.2016.12.015
145. Li Y, Pollock CA, Saad S. Aberrant DNA methylation mediates the transgenerational risk of metabolic and chronic disease due to maternal obesity and overnutrition. Genes (Basel) (2021) 12:1653. doi: 10.3390/genes12111653
146. Bispham J, Gardner DS, Gnanalingham MG, Stephenson T, Symonds ME, Budge H. Maternal nutritional programming of fetal adipose tissue development: differential effects on messenger ribonucleic acid abundance for uncoupling proteins and peroxisome proliferator-activated and prolactin receptors. Endocrinology (2005) 146:3943–9. doi: 10.1210/en.2005-0246
147. Long NM, Tousley CB, Underwood KR, Paisley SI, Means WJ, Hess BW, et al. Effects of early- to mid-gestational under- nutrition with or without protein supplementation on offspring growth, carcass characteristics, and adipocyte size in beef cattle1. J Anim Sci (2012) 90:197–206. doi: 10.2527/jas.2009-2568
148. Perlow JH, Morgan MA, Montgomery D, Towers CV, Porto M. Perinatal outcome in pregnancy complicated by massive obesity. Am J Obstet Gynecol (1992) 167:958–62. doi: 10.1016/s0002-9378(12)80019-6
149. Ehrenberg HM, Mercer BM, Catalano PM. The influence of obesity and diabetes on the prevalence of macrosomia. Am J Obstet Gynecol (2004) 191:964–8. doi: 10.1016/j.ajog.2004.05.052
150. Spradley FT, Smith JA, Alexander BT, Anderson CD. Developmental origins of nonalcoholic fatty liver disease as a risk factor for exaggerated metabolic and cardiovascular-renal disease. Am J Physiol Endocrinol Metab (2018) 315:E795–814. doi: 10.1152/ajpendo.00394.2017
151. Mosca A, Panera N, Maggiore G, Alisi A. From pregnant women to infants: Non-alcoholic fatty liver disease is a poor inheritance. J Hepatol (2020) 73:1590–2. doi: 10.1016/j.jhep.2020.06.043
152. Sarkar M, Grab J, Dodge JL, Gunderson EP, Rubin J, Irani RA, et al. Non-alcoholic fatty liver disease in pregnancy is associated with adverse maternal and perinatal outcomes. J Hepatol (2020) 73:516–22. doi: 10.1016/j.jhep.2020.03.049
153. Ding Y, Li J, Liu S, Zhang L, Xiao H, Li J, et al. DNA Hypomethylation of inflammation-associated genes in adipose tissue of female mice after multigenerational high fat diet feeding. Int J Obes (Lond) (2014) 38:198–204. doi: 10.1038/ijo.2013.98
154. Breton CV, Landon R, Kahn LG, Enlow MB, Peterson AK, Bastain T, et al. Exploring the evidence for epigenetic regulation of environmental influences on child health across generations. Commun Biol (2021) 4:769. doi: 10.1038/s42003-021-02316-6
155. De Jesus DF, Orime K, Kaminska D, Kimura T, Basile G, Wang CH, et al. Parental metabolic syndrome epigenetically reprograms offspring hepatic lipid metabolism in mice. J Clin Invest (2020) 130:2391–407. doi: 10.1172/JCI127502
156. del Rosario MC, Ossowski V, Knowler WC, Bogardus C, Baier LJ, Hanson RL. Potential epigenetic dysregulation of genes associated with MODY and type 2 diabetes in humans exposed to a diabetic intrauterine environment: an analysis of genome-wide DNA methylation. Metabolism (2014) 63:654–60. doi: 10.1016/j.metabol.2014.01.007
157. Kasuga Y, Kawai T, Miyakoshi K, Saisho Y, Tamagawa M, Hasegawa K, et al. Epigenetic changes in neonates born to mothers with gestational diabetes mellitus may be associated with neonatal hypoglycaemia. Front Endocrinol (Lausanne) (2021) 12:690648. doi: 10.3389/fendo.2021.690648
158. Reichetzeder C. Overweight and obesity in pregnancy: their impact on epigenetics. Eur J Clin Nutr (2021) 75:1710–22. doi: 10.1038/s41430-021-00905-6
159. Trivett C, Lees ZJ, Freeman DJ. Adipose tissue function in healthy pregnancy, gestational diabetes mellitus and pre-eclampsia. Eur J Clin Nutr (2021) 75:1745–56. doi: 10.1038/s41430-021-00948-9
160. Strauss A. Obesity in pregnant women: maternal, fetal, and transgenerational consequences. Eur J Clin Nutr (2021) 75:1681–3. doi: 10.1038/s41430-021-01015-z
161. Delli Bovi AP, Marciano F, Mandato C, Siano MA, Savoia M, Vajro P. Oxidative stress in non-alcoholic fatty liver disease. an updated mini review. Front Med (Lausanne) (2021), 8:595371. doi: 10.3389/fmed.2021.595371
162. Kislal S, Shook LL, Edlow AG. Perinatal exposure to maternal obesity: Lasting cardiometabolic impact on offspring. Prenat Diagn (2020) 40:1109–25. doi: 10.1002/pd.5784
163. Mikolajczak A, Sallam NA, Singh RD, Scheidl TB, Walsh EJ, Larion S, et al. Accelerated developmental adipogenesis programs adipose tissue dysfunction and cardiometabolic risk in offspring born to dams with metabolic dysfunction. Am J Physiol Endocrinol Metab (2021) 321:E581–91. doi: 10.1152/ajpendo.00229.2021
164. Kohjima M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med (2008) 21:507–11. doi: 10.3892/ijmm.21.4.507
165. Kim YR, Lee EJ, Shin KO, Kim MH, Pewzner-Jung Y, Lee YM, et al. Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Exp Mol Med (2019) 51:1–16. doi: 10.1038/s12276-019-0340-1
166. Sheldon RD, Padilla J, Jenkins NT, Laughlin MH, Rector RS. Chronic NOS inhibition accelerates NAFLD progression in an obese rat model. Am J Physiol Gastroint Liver Physiol (2015) 308:G540–9. doi: 10.1152/ajpgi.00247.2014
167. Berger K, Hyland C, Ames JL, Mora AM, Huen K, Eskenazi B, et al. Prenatal exposure to mixtures of phthalates, parabens, and other phenols and obesity in five-Year-Olds in the CHAMACOS cohort. Int J Environ Res Public Health (2021) 18:1796. doi: 10.3390/ijerph18041796
168. Basak S, Das MK, Duttaroy AK. Plastics derived endocrine-disrupting compounds and their effects on early development. Birth Defects Res (2020) 112:1308–25. doi: 10.1002/bdr2.1741
169. Kladnicka I, Bludovska M, Plavinova I, Muller L, Mullerova D. Obesogens in foods. Biomolecules (2022) 12:680. doi: 10.3390/biom12050680
170. Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes (2011) 60:1528–34. doi: 10.2337/db10-0979
171. Moreno-Mendez E, Quintero-Fabian S, Fernandez-Mejia C, Lazo-de-la-Vega-Monroy ML. Early-life programming of adipose tissue. Nutr Res Rev (2020) 33:244–59.doi: 10.1017/S0954422420000037
172. Maude H, Sanchez-Cabanillas C, Cebola I. Epigenetics of hepatic insulin resistance. Front Endocrinol (Lausanne) (2021) 12:681356. doi: 10.3389/fendo.2021.681356
173. Herrera BM, Keildson S, Lindgren CM. Genetics and epigenetics of obesity. Maturitas (2011) 69:41–9. doi: 10.1016/j.maturitas.2011.02.018
174. Haire-Joshu D, Tabak R. Preventing obesity across generations: Evidence for early life intervention. Annu Rev Public Health (2016) 37:253–71. doi: 10.1146/annurev-publhealth-032315-021859
175. Linner A, Almgren M. Epigenetic programming-the important first 1000 days. Acta Paediatr (2020) 109:443–452. doi: 10.1111/apa.15050
176. Twigger AJ, Hepworth AR, Lai CT, Chetwynd E, Stuebe AM, Blancafort P, et al. Gene expression in breastmilk cells is associated with maternal and infant characteristics. Sci Rep (2015) 5:12933. doi: 10.1038/srep12933
177. Melnik BC, Schmitz G. Milk’s role as an epigenetic regulator in health and disease. Diseases (2017) 5:12. doi: 10.3390/diseases5010012
178. Melnik BC, Gerd S. Milk exosomes and microRNAs: potential epigenetic regulators. In: Patel V, editor. Handbook of nutrition, diet, and epigenetics. Cham: Springer (2017). p. p1–28. doi: 10.1007/978-3-319-31143-2_86-1
179. Ozkan H, Tuzun F, Kumral A, Duman N. Milk kinship hypothesis in light of epigenetic knowledge. Clin Epigenet (2012) 4:14. doi: 10.1186/1868-7083-4-14
180. Ozkan H, Tuzun F, Taheri S, Korhan P, Akokay P, Yılmaz O, et al. Epigenetic programming through breast milk and its impact on milk-siblings mating. Front Genet (2020) 11:569232. doi: 10.3389/fgene.2020.569232
181. Hufnagel A, Fernandez-Twinn DS, Blackmore HL, Ashmore TJ, Heaton RA, Jenkins B, et al. Maternal but not fetoplacental health can be improved by metformin in a murine diet-induced model of maternal obesity and glucose intolerance. J Physiol (2022) 600:903–19. doi: 10.1113/JP281902
182. Huang SW, Ou YC, Tang KS, Yu HR, Huang LT, Tain YL, et al. Metformin ameliorates maternal high-fat diet-induced maternal dysbiosis and fetal liver apoptosis. Lipids Health Dis (2021) 20:100. doi: 10.1186/s12944-021-01521-w
183. Zheng J, Woo SL, Hu X, Botchlett R, Chen L, Huo Y, et al. Metformin and metabolic diseases: a focus on hepatic aspects. Front Med (2015) 9:173–86. doi: 10.1007/s11684-015-0384-0
184. Hur KY, Lee MS. New mechanisms of metformin action: Focusing on mitochondria and the gut. J Diabetes Investig (2015) 6:600–9. doi: 10.1111/jdi.12328
185. Sodum N, Kumar G, Bojja SL, Kumar N, Rao CM. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol Res (2021) 167:105484. doi: 10.1016/j.phrs.2021.105484
186. Cordero P, Milagro FI, Campion J, Martinez JA. Supplementation with methyl donors during lactation to high-fat- sucrose-fed dams protects offspring against liver fat accumulation when consuming an obesogenic diet. J Dev Orig Health Dis (2014) 5:385–95. doi: 10.1017/S204017441400035X
187. Pauwels S, Truijen I, Ghosh M, Duca RC, Langie SAS, Bekaert B, et al. The effect of paternal methyl-group donor intake on offspring DNA methylation and birth weight. J Dev Orig Health Dis (2017) 8:311–21. doi: 10.1017/S2040174417000046
188. Pauwels S, Ghosh M, Duca RC, Bekaert B, Freson K, Huybrechts I, et al. Maternal intake of methyl-group donors affects DNA methylation of metabolic genes in infants. Clin Epigenet (2017) 9:16. doi: 10.1186/s13148-017-0321-y
189. Jiménez-Chillarón JC, Díaz R, Martínez D, Pentinat T, Ramón-Krauel M, Ribó S, et al. The role of nutrition on epigenetic modifications and their implications on health. Biochimie (2012) 94:2242–63. doi: 10.1016/j.biochi.2012.06.012
190. Zeisel SH, da Costa KA. Choline: an essential nutrient for public health. Nutr Rev (2009) 67:615–23. doi: 10.1111/j.1753-4887.2009.00246.x
191. Ilcol YO, Ozbek R, Hamurtekin E, Ulus IH. Choline status in newborns, infants, children, breast-feeding women, breast-fed infants and human breast milk. J Nutr Biochem (2005) 16:489–99. doi: 10.1016/j.jnutbio.2005.01.011
192. Alisi A, Vajro P. Pre-natal and post-natal environment monitoring to prevent non-alcoholic fatty liver disease development. J Hepatol (2017) 67:451–3. doi: 10.1016/j.jhep.2017.04.016
Keywords: obesity, pregnancy, gestation, transgenerational transmission, genetics, epigenetics, polymorphisms, methylation
Citation: Panera N, Mandato C, Crudele A, Bertrando S, Vajro P and Alisi A (2022) Genetics, epigenetics and transgenerational transmission of obesity in children. Front. Endocrinol. 13:1006008. doi: 10.3389/fendo.2022.1006008
Received: 28 July 2022; Accepted: 27 October 2022;
Published: 14 November 2022.
Edited by:
Deanne Helena Hryciw, Griffith University, AustraliaReviewed by:
Rebecca Jean Ryznar, Rocky Vista University, United StatesCopyright © 2022 Panera, Mandato, Crudele, Bertrando, Vajro and Alisi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Alisi, YW5uYS5hbGlzaUBvcGJnLm5ldA==; Claudia Mandato, Y21hbmRhdG9AdW5pc2EuaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.