 Sylvia Riedel
Sylvia Riedel Carmen Pheiffer
Carmen Pheiffer Rabia Johnson
Rabia Johnson Johan Louw
Johan Louw Christo J. F. Muller
Christo J. F. Muller- 1Biomedical Research and Innovation Platform, South African Medical Research Council, Tygerberg, South Africa
- 2Centre for Cardio-Metabolic Research in Africa, Division of Medical Physiology, Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, University of Stellenbosch, Tygerberg, South Africa
- 3Department of Obstetrics and Gynaecology, University of Pretoria, Pretoria, South Africa
- 4Department of Biochemistry and Microbiology, University of Zululand, KwaDlangezwa, South Africa
Noncommunicable diseases, such as type 2 diabetes (T2D), place a burden on healthcare systems worldwide. The rising prevalence of obesity, a major risk factor for T2D, is mainly attributed to the adoption of Westernized diets and lifestyle, which cause metabolic dysfunction and insulin resistance. Moreover, diet may also induce changes in the microbiota composition, thereby affecting intestinal immunity. The critical role of intestinal immunity and intestinal barrier function in the development of T2D is increasingly acknowledged, however, limited studies have investigated the link between intestinal function and metabolic disease. In this review, studies reporting specific roles of the intestinal immune system and intestinal epithelial cells (IECs) in metabolic disease are highlighted. Innate chemokine signaling, eosinophils, immunoglobulin A (IgA), T helper (Th) 17 cells and their cytokines were associated with obesity and/or dysregulated glucose homeostasis. Intestinal epithelial cells (IECs) emerged as critical modulators of obesity and glucose homeostasis through their effect on lipopolysaccharide (LPS) signaling and decontamination. Furthermore, IECs create a link between microbial metabolites and whole-body metabolic function. Future in depth studies of the intestinal immune system and IECs may provide new opportunities and targets to develop treatments and prevention strategies for obesity and T2D.
1 Introduction
Non-communicable diseases (NCDs) such as type 2 diabetes (T2D), are a leading cause of morbidity and mortality, with increasing incidence in the developing world (1). In 2019, approximately 463 million adults were living with diabetes worldwide, and this number is said to increase to 700 million by 2045 (2). Sub-Saharan Africa has the highest proportion of undiagnosed diabetes cases in the world (~60%) and it is estimated that 73% of deaths due to diabetes occur in individuals younger than 60 years. Projections indicate that diabetes prevalence in Africa will increase by 143% over the next 24 years (2), the most significant increase globally.
The rising T2D prevalence in Africa is mainly attributed to obesity and rapid urbanization, which is associated with the adoption of unhealthy lifestyles, characterized by energy-dense diets and physical inactivity. The fundamental role that diet plays in human health and well-being is appropriately expressed by the phrase “We are what we eat”, derived from the original quote in French “Dis-moi ce que tu manges et je te dirai qui tu es” (“Tell me what you eat: I tell you who you are”) that has been traced back to Jean Anthelme Brillat-Savarin and a book, which was first published in 1826 (3, 4). An unhealthy diet is arguably the greatest modifiable risk factor for obesity and T2D (5, 6). The westernized diet, which is a high caloric diet characterized by a high fat and high sugar content (7), leads to chronic subclinical inflammation, which, in turn, has been associated with obesity and T2D (8–11). Several lines of evidence demonstrate that overnutrition leads to adipocyte dysfunction and inflammation, which are considered primary mechanisms linking diet to metabolic disease (6, 12, 13).
In recent years, the role of the intestine in the development of obesity and T2D is increasingly recognized. Studies conducted in various animal models provide convincing evidence that inflammation originates in the intestine due to the modulation of gut barrier function leading to metabolic endotoxemia (14–18). Metabolic endotoxemia refers to a diet-induced 2-3 fold increase in gut-derived plasma lipopolysaccharide (LPS) level (14), which may result in low-grade systemic and tissue inflammation, contributing to a metabolic disease phenotype (14, 15, 19–22). Metabolic endotoxemia is indicative of decreased intestinal epithelial barrier function, also referred to as “leaky gut” syndrome (Figure 1), which allows undesirable luminal immunogens, such as LPS, but also bacterial DNA and RNA and to some degree viable bacteria, to cross into the blood and lymphatic system (14, 29). Accordingly, studies have detected high levels of gut-derived bacterial products in the blood of obese and diabetic patients (30, 31). The intestinal barrier has since proved to be altered by additional factors including food additives (such as emulsifiers and artificial sweeteners), and contaminants (such as mycotoxins), which may in addition affect and/or change microbiota composition (32, 33).
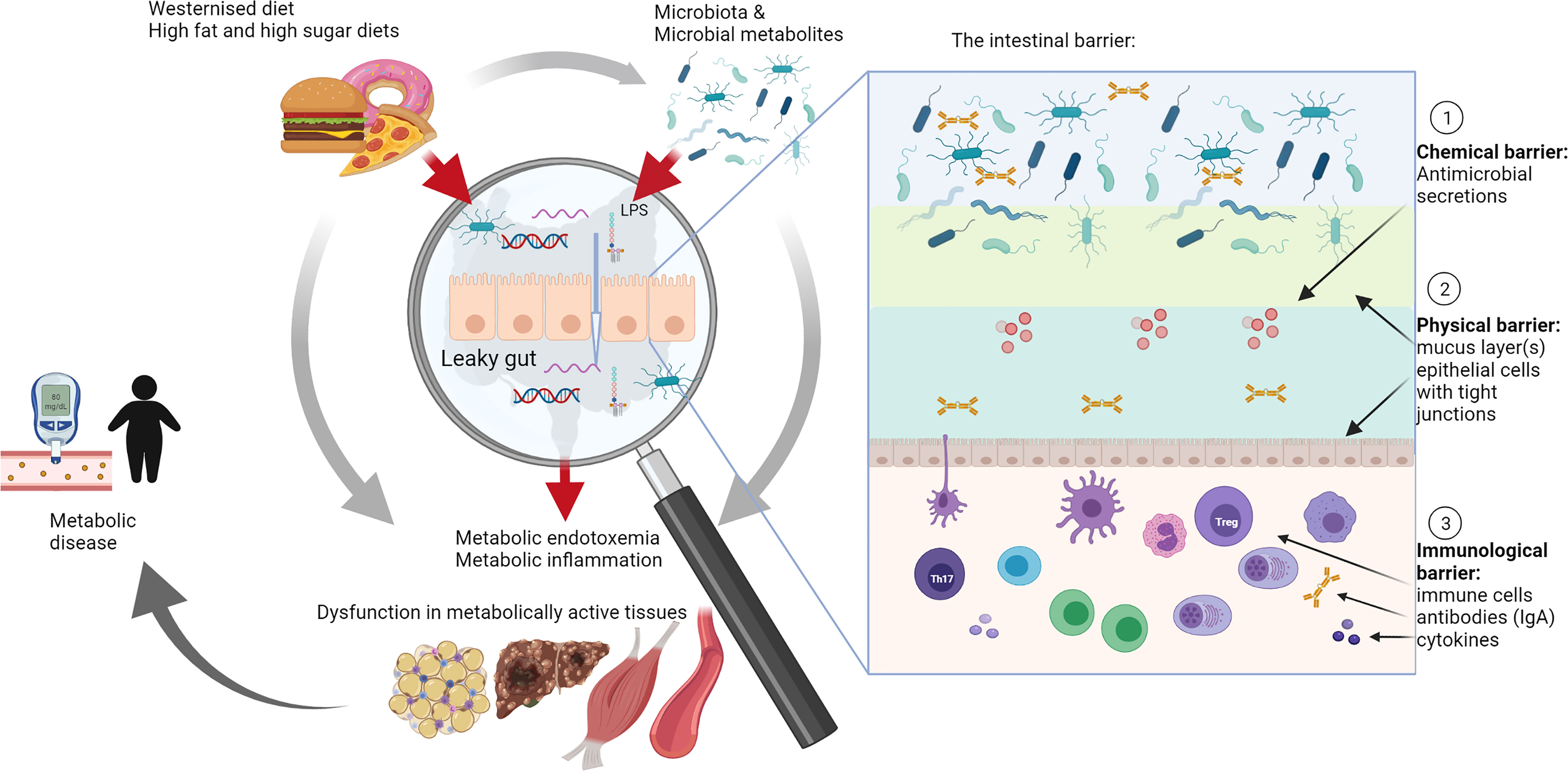
Figure 1 The role of intestinal barrier dysfunction and leaky gut syndrome in metabolic disease. A high fat and/or westernized diet affects and/or changes microbiota composition. Microbiota composition was associated with metabolic diseases in a large number of studies that investigated these in conjunction with metabolic outcomes, such as increased or decreased body weight, changes in glucose metabolism and effects on metabolically active tissues such as adipose tissue, liver and muscles (grey arrows) (18, 23–28). However, increasingly research is focusing on the role of the leaky gut, particularly investigating the role of the intestinal barrier and its different layers, including chemical, physical and immunological barriers (see insert), as mediators of interactions with diet and microbiota in the development of metabolic diseases (red arrows). Studies in this area are relatively limited, but this review summarizes compelling evidence for a leading role of the gut in development of metabolic disease through modulation of this complex intestinal barrier. IgA, immunoglobulin A; LPS, lipopolysaccharide; Th17, T helper cells 17; Treg, regulatory T cells.
Although many studies have reported on the role of microbiota in obesity and T2D, these mainly focused on characterization of microbiota composition and diversity in response to diet, disease and treatment (18, 23–28). Studies investigating causal relationships between microbiota and metabolic effects are lacking due to the complexity of host-microbe interactions (34–36). Emerging evidence, although scant to date, suggests that the mammalian host may shape and control their microbiota, a concept described as “ecosystem on a leash”, which is based on the evolutionary theory that host-microbe and microbe-microbe interactions play an essential role in the symbiotic relationship between hosts and their commensal microbiota (37). It has been postulated that host antimicrobial secretions, antibodies, such as immunoglobulin A (IgA), as well as gut antigen surveillance (e.g. through luminal antigen sampling by intestinal mucosal dendritic cells (DCs) or M cells), may exert this type of control on microbiota (37). Considering that the intestinal tract is exposed to an enormous antigen burden, from commensal microbiota to pathogens to food derived antigens, it is evident that the intestinal immune system is a critical role player in creating a tolerogenic milieu and preventing unwanted inflammatory responses. Thus, investigating the role of intestinal immune system and its potential in alleviating and preventing metabolic diseases through inflammatory, barrier-protective and other pathways have increasingly become focus of research, which was reviewed some years ago (38, 39).
Taken together, these studies suggest that gut health, barrier function and the intestinal immune system play an integral role in the development of metabolic disease and may place the intestines at the center and perhaps the beginning of the pathophysiology of obesity and T2D (Figure 1). The purpose of this review is to provide an overview of key studies that have afforded insight into how intestinal immune function, inflammation and barrier function impact on the development of obesity and T2D with specific focus on immune function and intestinal epithelial cells. Studies in animal models and humans that investigated intestinal immune cells, barrier function and/or inflammation in intestinal tissues together with effects on systemic glucose and/or insulin metabolism and/or weight gain during obesity and T2D were selected. Search terms included “intestinal barrier function”, “type 2 diabetes”, “intestinal immune system”, “obesity”, “lipopolysaccharides”, “leaky gut”, “intestinal epithelial cells”, “glucose intolerance”, “insulin resistance” and were used.
2 The Role of the Intestine in Metabolic Homoeostasis
Apart from its role in nutrient digestion and absorption, the gastrointestinal tract (GIT) functions as a physical barrier to prevent undesirable luminal components, such as pathogens, dietary antigens and microbiota from entering the body (40, 41). The intestinal barrier is complex and comprises multiple “layers” (see insert in Figure 1). A mucus layer covering the intestinal epithelium creates a physical barrier between the lumen and the tissues. Intestinal epithelial cells (IECs) secrete various antimicrobial compounds, such as lysozyme and defensins, which create a chemical barrier to prevent contact between microbiota and the tissues. The immunological barrier comprises secreted IgA, which is produced in plasma cells in the lamina propria, and a full complement of immune cells located in the lamina propria and gut-associated lymphoid tissues (42). Since the GIT is in constant contact with antigens and microbes, the gut-associated lymphoid tissues form a large part of the host immune system (43–45). Intestinal mononuclear phagocytes determine whether tolerance or an immune response is needed, depending on whether the antigens belong to commensal microbiota or pathogens (43). Antigen-presenting cells, such as tissue resident macrophages and DCs, shape the adaptive immune response (T and B cells) through secretion of cytokines and lipid mediators, which may have profound effects on the tissue microenvironment, including decreased epithelial barrier function and changing commensal microbiota composition (38, 39).
The role of the intestine in metabolic homeostasis has been mainly ascribed to enteroendocrine cells, that act as sensors for nutritional signals and produce and secrete key hormones, such as glucose-dependent insulinotropic polypeptide (GIP), glucacon-like peptide 1 (GLP-1) and peptide YY, into the circulation (46, 47). Of these hormones, GIP and GLP-1, commonly referred to as incretins, modulate postprandial glucose concentrations by inducing a rapid and strong insulinotropic effect after meal ingestion, which is blunted in patients with T2D (48) and has been widely explored as pharmaceutical target for T2D. Recently, enteroendocrine cells have been shown to sense microbial metabolites (49) and may thus serve as a link between microbiota and their metabolic effects (50). Further supporting a functional role of the intestinal tract in T2D is the phenomenon that compounds with low bioavailability, such as certain plant polyphenols or polyphenol rich plant extracts, can still elicit significant biological responses in vivo, such as anti-hyperglycemic effects (51–53). Accordingly, a recent review hypothesized that effects elicited in the gut might be the primary mechanism of action of polyphenols, including, for instance, modulating carbohydrate digestion and uptake, energy metabolism and interaction with microbiota (54). Similarly, a delayed-release preparation of the first-line drug for T2D, metformin was less bioavailable but as effective as “normal” metformin, suggesting an involvement of the distal bowel (ileum and colon) in its therapeutic efficacy, presumably via increased GLP-1 secretion from L cells and enhanced neural signaling (55).
3 The Intestinal Immunological Barrier in Obesity and T2D
3.1 Modulation of LPS and Metabolic Endotoxemia
As a membrane component of gram-negative bacteria, LPS is found in copious quantities in the gut as part of commensal microbiota and pathogens. LPS has a broad spectrum of negative health effects in the body ranging from inflammation to cancer promotion (56–58). Its lipid components are responsible for inducing the strong immune activation (59). LPS signaling, which is facilitated by pathogen associated molecular pattern (PAMP) receptors, was identified as a critical feature in intestinal homeostasis (14). The LPS-induced toll-like receptor 4 (TLR4) signaling cascade involves the co-receptor cluster of differentiation (CD) 14 as well as intracellular adaptor molecules such as Myeloid differentiation primary response gene 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF) as key components (60–62). Metabolic endotoxemia, which refers to increased LPS levels in the blood, occurs when the intestinal barrier function is compromised (14). Several studies have reported strong associations between increased LPS levels due to intestinal barrier dysfunction and metabolic disease (63–66). In animal studies, increased LPS levels induced systemic and tissue inflammation similar to studies using high fat diet (HFD) model (14, 15). For example, when Cani et al. (14) mimicked HFD-induced endotoxemia by intraperitoneal infusion of LPS, they found significantly increased expression of inflammatory cytokines, such as interleukin (IL-) 1 in liver, adipose and muscle tissues. Furthermore, studies have associated metabolic endotoxemia with the development of non-alcoholic liver disease (67–69) as well as adipose tissue inflammation and dysfunction (58, 70), which are risk factors and/or precursors for the development of T2D. Accordingly, circulating LPS serves as a useful biomarker of compromised intestinal barrier function with augmented LPS levels often reported in obese and diabetic patients (71–73). It has been suggested that LPS plays an essential role in the onset of obesity and T2D development (14, 15). In contrast, Dalby et al. (74) showed that deletion of the TLR4 and co-receptor CD14, which are involved in LPS signaling (60, 61), did not prevent diet-induced obesity in mice, suggesting that obesity may develop independent of LPS and metabolic endotoxemia. Intriguingly, intestinal permeability and LPS levels were raised in obese TLR4 and CD14 knockout mice (74), suggesting that LPS could signal through an unidentified TLR4-independent pathway (75). Importantly, knockout of TLR4 in specific cell populations such as intestinal epithelial cells (IECs) rather than whole-body knockout may be required to induce metabolic effects (66). Lu et al. (66) recently showed that intestinal TLR4 could regulate the interaction between host and microbiota and thus affect metabolic syndrome. Using knockout of TLR4 specific to IECs or myeloid cells, these authors showed contrasting effects where deletion in IECs increased body weight and impaired glucose metabolism, whereas knockout in myeloid cells decreased body weight. Lu et al. (66) further showed that the metabolic effects of IEC-specific TLR4 knockout could be reversed by treating mice with antibiotics, suggesting that TLR4 expression in IECs play a central role in protective host-microbe interactions.
Another member of the TLR family, TLR5, plays a critical role in antigen recognition, specifically bacterial flagellin, while other roles include preserving barrier function in the gut (76). The important surveillance function of TLR5 in the intestines is supported by its location on the basolateral side of IECs, where it acts as a sensor for increased intestinal permeability (76). Vijay-Kumar et al. (77) showed that TLR5 knockout mice fed a standard chow diet developed colitis, mild inflammation and obesity. These mice also developed hypercholesterolemia, high blood pressure, hypertriglyceridemia, hyperinsulinemia and fasting hyperglycemia. TLR5 knockout mice developed T2D when these mice were fed an HFD, while food restriction prevented the increase in body weight although it could not improve glucose intolerance (77). These authors suggested that in TLR5 knockout mice, inflammatory mediators such as cytokines may inhibit the insulin signaling pathway, which may prevent a complete regain of glucose tolerance during food restriction. The important role of TLRs in obesity and metabolic disease is reiterated in that dietary fatty acids, especially saturated fatty acids supplied by HFDs, can activate TLRs in addition to LPS (78–80).
An enzyme intricately linked to endotoxemia is intestinal alkaline phosphatase (IAP), as it is involved in the detoxification of LPS (81). Knockout of IAP in mice leads to decreased intestinal barrier function and increased circulating levels of LPS and inflammatory markers (63). These mice also display glucose intolerance, insulin resistance and hyperinsulinemia. When IAP activity was diminished with a known inhibitor of IAP, such as dietary phenylalanine, impaired glucose tolerance was evident in mice fed an HFD when compared to HFD-fed control mice, while IAP knockout mice developed T2D when they were fed an HFD (63). On the other hand, IAP supplementation improved metabolic syndrome in mice and prevented LPS uptake via chylomicrons, leading to improved lipid profiles and liver steatosis scores (63). Malo (81) showed that fecal IAP levels are lower in diabetic compared to non-diabetic, obese patients. In contrast to Malo (81), de La Serre et al. (82) identified IAP as a key player in susceptibility to obesity. These authors showed that HFD-fed Sprague Dawley rats displayed altered microbiota composition. However, reduced IAP activity and increased endotoxemia, ileal TLR4 expression, and inflammation were observed in diet-induced obese rats compared to non-obese rats on an HFD. These findings suggest that decreased gut barrier and IAP activity are associated with obesity and that changes in the microbiota are independent of obesity, which agrees with other studies (83, 84). IAP levels decrease with age, and it was shown that supplementation of IAP in mice increased longevity and reduced frailty, supporting the notion that lower IAP activity may contribute to age-related diseases including metabolic syndrome (73).
3.2 Intestinal Inflammation
A healthy intestinal tract requires efficient tolerance signals to modulate and suppress inflammatory responses resulting from exposure to an overwhelming number of commensal bacteria, potential dietary antigens, and various ubiquitous enteric pathogens. This is accomplished through suppressor cells, whose tolerogenic and homeostatic functions are critical to facilitate effective barrier function and synergisms with intestinal microbiota. The intestinal immune system, therefore, requires a certain degree of isolation from the systemic immune system (85, 86). The effectiveness of this defense system is demonstrated by studies showing that early metabolic disease generates low-grade intestinal inflammation (87–89), while evidence of sub-clinical inflammatory changes, such as macrophage infiltration, has been reported (88, 90). Inflammatory signals play a crucial role in tight junction function and assembly (91), thus it is worthwhile to discuss selected key studies that specifically investigated intestinal inflammation below.
Kawano et al. (90) showed that in mice, inflammatory responses in the colon, including monocyte infiltration, loss of goblet cells as well as a decrease in crypt depth, preceded HFD-induced inflammatory changes in other tissues, such as the adipose tissue. Infiltrating colonic macrophages displayed a pro-inflammatory secretory profile after 4 weeks of HFD feeding, however, increased expression of inflammatory markers was only evident in liver and adipose tissue after 8 and 12 weeks, respectively, of HFD feeding (90). Oxidative stress was increased in the colon after 4 weeks of HFD feeding and occurred after 8 weeks in the epididymal fat depot. Intestinal inflammation preceded the development of obesity in HFD fed mice also in a study by Ding et al. (92). TNFα expression was increased in the ileum of these mice and was associated with higher body weight, fat mass and insulin resistance and 2 weeks of HFD feeding increased nuclear factor kappa B (NFκB) expression in these mice. In contrast, HFD feeding did not induce obesity and inflammation in germ-free mice, however, the inflammatory phenotype observed in conventional mice was transferrable to germ-free mice using fecal transplants (92). Taken together, these results suggest that the inflammatory effect observed in response to HFD feeding was associated with microbiota.
Changes in microbiota composition can affect LPS-induced signaling pathways including TLRs (15). In this regard, Kim et al. (19) showed that HFD-feeding in mice induced TLR4 expression, decreased intestinal barrier function and induced intestinal and systemic inflammation via increased pro-inflammatory cytokine production, increased cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase expression. That pro-inflammatory cytokine levels were not affected by the HFD in TLR4 deficient animals (19) provided evidence for a role of gut-derived LPS signaling in HFD-induced chronic inflammation.
Using 5-aminosalicylic (5-ASA, or mesalamine), a drug with intestine-specific anti-inflammatory properties, Luck et al. (88) showed that a 12 to 14 week treatment of obese mice ameliorated HFD-induced insulin resistance through anti-inflammatory and barrier protective properties. Although treatment with 5-ASA did not inhibit weight gain, it improved liver steatosis, reduced fasting plasma glucose concentrations and improved glucose tolerance (88). The effect of 5-ASA treatment was dependent on the intestinal immune system since it showed no effect in beta7 null mice, which are devoid of intestinal immune cells. Furthermore, 5-ASA treatment improved intestinal barrier leakage, as assessed using the fluorescein isothiocyanate (FITC)-dextran assay. Overall, Luck et al. (88) demonstrated that HFD-induced inflammation in the gut and treatment with a local acting anti-inflammatory drug improved metabolic parameters, which implicated the intestinal immune system as a potential mechanism to improve metabolic function. In addition, Liu et al. (93) showed that peroxisome-proliferator-activated receptor gamma (PPARγ) played a significant role in mitigating the inflammatory effects of the HFD feeding in the colon tissue of diet-induced obese mice. These authors showed that exercise produced anti-inflammatory effects in the colon of HFD-fed mice, which a PPARγ antagonist abrogated. PPARγ expression was previously found to be reduced by HFD feeding and its reduced expression is considered an indicator of inflammation in the colon (94).
3.3 A Role for the Innate Immune System in Metabolic Disease
While the role of the innate immune system in adipose tissue inflammation and insulin resistance is well documented (95–99), few studies have investigated the role of the intestinal innate immune system in the development of obesity, insulin resistance and T2D. The innate immune system, which includes DCs, macrophages, innate lymphoid cells and eosinophils, is the first line of defense against pathogens, especially in the intestines, but it is also essential for shaping the adaptive immune responses (100, 101).
Innate immune cells release monocyte chemoattractant protein-1 (MCP-1), also referred to as C-C motif chemokine ligand (CCL2), which is a critical chemoattractant that initiates recruitment of immune cells to sites of inflammation. CCL2 signals through the CCL2 receptor (CCR2) which is expressed on most innate immune cells and effector cells such as T cells (102, 103). Kawano et al. (90) showed that HFD feeding induced CCL2 expression and that knockout of IEC-specific CCL2 ameliorated HFD-induced insulin resistance, infiltration of pro-inflammatory macrophages in the colon mucosa, and adipose tissue inflammation. Furthermore, macrophage-specific knockout of CCR2 in the colon improved glucose tolerance and insulin resistance in mice. In the colon, CCR2 knockout was associated with immunological changes such as the decreased production of pro-inflammatory cytokines and barrier protection through increased expression of the tight junction protein, claudin 1. These changes resulted in improved barrier function and lowered systemic LPS levels as key events in improving metabolic disturbances. The CCL2/CCR2 signaling axis may therefore promote a pro-inflammatory environment in the colon which likely increases intestinal permeability with significant adverse effects on whole-body glucose homeostasis (90).
Another component of the innate immune system, eosinophils, are traditionally thought to be hallmarks of acute inflammatory, anti-parasitic and allergic responses, such as asthma (104, 105). However, eosinophils are also found in immune cell infiltrates in inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis, where eosinophil activation contributes to neutrophil recruitment and tissue ulcerations (106, 107). Findings by Johnson et al. (87) support a role for eosinophils in diet-induced metabolic disease. These authors showed that a short-term, 7-day HFD feeding regimen in mice decreased eosinophils levels in the small intestines, although no signs of inflammation, such as monocyte or macrophage infiltration, were observed. These changes were accompanied by increased permeability in the small intestine as measured by the FITC-dextran assay and fecal albumin levels, and, notably, occurred before the onset of metabolic dysfunction. Since ob/ob mice, which develop obesity through hyperphagia as they do not produce leptin due to a mutation, did not display similar eosinophil trafficking defects, Johnson et al. (87) concluded that this defect was caused by HFD feeding. While the significance of the HFD-induced reduction in the number of eosinophils in the intestines is not yet clear, new roles of eosinophils with focus on their involvement in intestinal tissue homeostasis are under investigation (108, 109). Emerging evidence also suggested that eosinophils may contribute to immunoglobulin A (IgA) expression (106, 110).
3.4 Adaptive Immune System
3.4.1 B Cells
IgA is the most abundant antibody subtype produced by B cells in the intestines and is generally considered the first line of defense (111). IgA is mainly secreted across mucosal surfaces, such as in the intestinal mucosa, where it fulfils several essential functions including maintaining gut homeostasis and microbial tolerance. Secretory IgA can bind to or “coat” microbiota, thereby inducing immune exclusion and neutralization by altering bacterial motility and gene expression, and facilitating antigen uptake (112–116). A recent study reported that fecal IgA levels did not differ between non-obese patients with T2D and healthy controls or patients with Crohn’s disease (117). However, these authors found a significant decrease in IgA expressing B cells in the intestinal mucosa of non-obese patients with T2D, specifically in mucosal CD19+CD20- cells, compared to healthy patients. These changes were accompanied by an increase in IgG expressing B cells, which may be linked to increased T cell-derived interferon-gamma (IFNγ) secretion. These authors suggest that plasma cells of non-obese T2D patients are more reactive to IFNγ compared to healthy controls, resulting in a pro-inflammatory environment and depletion of IgA+ cells in intestinal tissues (117).
In mice, IgA+ plasma cells were decreased in the large intestines of HFD-fed obese mice compared to normal-weight mice fed a standard diet, which was accompanied by increased insulin resistance and higher fasting plasma glucose concentrations (118). These results suggest a link between the abundance of IgA+ plasma cells and glucose metabolism in response to HFD feeding. Furthermore, lower intestinal IgA secretion was associated with altered microbiota composition and produced a microbiota signature eliciting a transplantable metabolic phenotype when transferred into germfree or antibiotic-treated mice. It was proposed that lower IgA levels and increased bacterial IgA binding affinity in response to HFD feeding stimulate or induce pathogenic bacteria to migrate across the intestinal barrier. Interestingly, metformin treatment or bariatric surgery improved IgA deficiency in this model (118). In a recent study by Sakamoto et al. (119), HFD feeding decreased IgA+ cells in the lamina propria of the small intestines, which was associated with increased body weight and fasting blood glucose concentrations compared to mice fed a standard chow diet. IgA, particularly in intestinal tissues, has not yet been the focus of in-depth studies investigating the pathophysiology of T2D. However, the highlighted effects on regulating glucose homeostasis and microbiota composition suggest that it could be an important target for prevention of metabolic diseases such as obesity and T2D. Further studies investigating its role in the pathophysiology of metabolic diseases are therefore warranted.
3.4.2 T Cells
The role of different T cell subsets in intestinal immune homeostasis and metabolic diseases is the subject of intense research (85, 114, 120). T-helper (Th)17 cells, intraepithelial T lymphocytes, regulatory T (Treg) cells and the cytokines and cytokine receptors involved in their differentiation have emerged as targets of interest in metabolic disease. T cells set the inflammatory tone, with each subset of T-helper cells secreting distinct cytokine profiles. Monteiro-Sepulveda et al. (121) attempted to eliminate diet as a confounding factor by studying intestinal inflammation in metabolically healthy obese as well as obese patients with T2D. These authors showed that obesity-induced inflammation in the jejunum resulted in increased CD3+ T cells in the jejunal epithelium. The number of macrophages, CD3+ T cells and intraepithelial T cells was increased in the jejunum of obese patients, while mature DCs and natural killer cells were increased in obese patients with T2D (121). Increased epithelial T cell and innate immune cell populations were linked to intestinal and specifically jejunal inflammation in obesity and correlated with high dietary fat content. In addition, the IECs displayed insulin resistance due to T cell activation (121). The link between increased pro-inflammatory immune cell populations and reduced insulin sensitivity in IECs in obesity could provide new insight into the development of insulin resistance and T2D. Other studies have reported that diet-induced obesity is characterized by the increased presence of pro-inflammatory intestinal Th1 and CD8+ cells and reduced Treg cells (88). In the latter study, 3 weeks of HFD feeding changed the immune cell populations in the lamina propria of the colon towards a pro-inflammatory milieu, resulting in decreased Treg cells and increased IL-17 producing γδ T cells (88). After 14 weeks of HFD feeding these effects were also observed in the small intestine, with an increase in Th1 cells producing IFNγ and a decrease in CD4+/Forkhead box protein P3 (FoxP3)+ Treg cells. Reduced Treg cells and increased T-box protein expressing T cells (T-bet+ cells) and CD8+ cells were observed in the colon and small intestines of obese patients, while no histological signs of inflammation were apparent (88). Interestingly, Luck et al. (88) showed that beta7 null mice, which are devoid of intestinal immune cells, displayed improved glucose metabolism in response to a 12 week HFD-feeding regimen when compared to wild-type mice, without beneficial effects on body weight gain. The authors suggested that induction of disturbances in glucose metabolism and insulin resistance may therefore be dependent on gut immune system dysfunction (88). While Luck et al. (88) assessed T cell populations in the lamina propria of the small and large intestine, a recent study documented a critical role of intestinal intraepithelial αβ and γδ T cells in whole-body glucose and lipid metabolism (122). These authors showed that mice lacking integrin β7 were protected against diet-induced obesity and atherosclerosis with improved glucose tolerance and displayed reduced numbers of intraepithelial T cells. As these T cell subsets represent the major cell population expressing GLP-1 receptors, He et al. (122) were able to explain these positive metabolic effects in mice lacking integrin β7 through increased bioavailability of GLP-1 leading to increased glucose disposal.
While Th1 and Th2 cells are involved in shaping active immune responses, the main function of Treg cells is to create (self)tolerance, which is particularly important in the gut due to the high antigen load (123). Everard et al. (124) showed that FoxP3 expression was increased in the jejunum of MyD88 knockout mice fed an HFD compared to HFD-fed wild type mice, likely due to the key role of MyD88 in the TLR and LPS signaling pathway. MyD88 is an intracellular adapter protein that facilitates TLR and IL-1 receptor signaling and is, therefore, a critical link in the activation of pro-inflammatory transcription factors such as NFκB or mitogen-activated protein kinases (125). Compared to wild type mice, FoxP3 expression in Treg cells was increased in the colon of MyD88 knockout mice fed either a standard diet or HFD. Everard et al. (124) further showed that knockout of MyD88 in selected cell types such as in myeloid cells conferred no protection against obesity, while IEC-specific knockout of MyD88 played a pivotal role in development of T2D and obesity, likely through sensing diet-related stress (124). Epithelial MyD88 knockout also affected microbiota composition and transferring these microbiota to germfree mice conferred the positive metabolic traits of the MyD88 knockout, suggesting that microbiota can be shaped by the host (124).
Th17 cells, initially thought to induce pro-inflammatory responses, are now recognized to exert contrasting protective and pathological roles (126). Th17 cells may protect against certain infections (126) and in the small intestine, they have been implicated in maintaining microbiota homeostasis. A study by Hong et al. (127) showed that ob/ob mice and HFD fed mice displayed lower abundance of Th17 cells in their small intestines, which was accompanied by increased T-helper 1 (Th1) cell-derived IFNγ levels compared to Th17 cell-derived IL-17 and IL-22. However, increased abundance of Th17 cells improved metabolic status (127). Garidou et al. (128) showed that HFD feeding in mice influenced intestinal T cell populations. Feeding a diet consisting of 72% fat and devoid of carbohydrates for 10 and 30 days did not induce weight gain, but these animals developed glucose intolerance and insulin resistance with a concomitant loss of Th17 cells in the ileum, which preceded inflammation in adipose tissues. In the ileum and mesenteric lymph nodes, IL-17, IL-22 and IL-10 gene expression was reduced, while a loss of Th17 cells (i.e. IL-17 and IL-22 secreting) was detected in the colon and small intestinal tissues (128). Treg cell numbers were lower in the small intestinal lamina propria in HFD fed mice, with a proportional increase in Th1 cells. In a study using nucleotide oligomerization domain (NOD)2 knockout and wild type mice, Th17 responses were decreased in response to HFD feeding (129). Wild-type and NOD2 knockout mice displayed reduced neutrophil-dependent oxidative bursts against bacteria following HFD feeding, which could play a role in shaping microbiota composition (129). It seems that metabolic disease could be characterized by reduced Th17 responses in the intestines, in contrast to inflammatory bowel disease where increased IL-17 was associated with increased inflammation, disease severity and relapse (130, 131).
3.4.2.1 Role of Th17 Specific Cytokines
Th17 cells have been identified as a distinct cell lineage due to their cytokine secretion profiles, including IL-17, IL-22 and IL-23 (132), and were shown to fulfil critical functions in the defense against pathogens in the gut mucosa (133). The shaping of gut microbial populations is accomplished by producing antimicrobial peptides, which can be stimulated by Th17 cytokines such as IL-22 (134, 135). IL-22 is part of the IL-10 family and is expressed by Th17 cells, innate lymphoid cells and Th22 cells. Its protective and regenerative functions have been demonstrated in various tissues, including the gut (136, 137). IL-22 deficient mice develop metabolic syndrome when fed an HFD and treatment with an IL-22Fc fusion protein ameliorated body weight gain and glucose intolerance. However, continued treatment was required to maintain the therapeutic effect (138). Treatment with the IL-22Fc fusion protein also induced changes in microbiota composition, however, since this did not occur in mice fed the control diet, the effect of the IL-22Fc on microbiota was suggested to be HFD-dependent and secondary to metabolic changes (138).
To investigate intestinal inflammation, Gulhane et al. (65) used Winnie mice, who contain a mutation in the Muc2 gene, which results in misfolding of the mucin 2 glycoprotein, which is the main component of the intestinal mucus layer. Winnie mice are considered a model of endoplasmic reticulum stress-induced colitis, which constitutes an epithelial defect, while mice present with a normal immune system. HFD feeding aggravates colitis in Winnie mice, but when these mice were treated with IL-22, the intestinal mucosa produced lower cytokine levels in response to HFD, which increased claudin 1 protein expression, thereby improving tight junction function, and decreasing systemic LPS levels. IL-22 treatment further normalized the changes in the microbiota induced by HFD, for instance by increasing levels of Akkermansia muciniphila and decreasing levels of E. coli (65).
IL-23 is a member of the IL-12 family, which is associated with chronic inflammatory disease and is considered an important inducer of Th17 cells (139, 140). Interestingly, IL-23p19 knockout mice gained more weight compared to wild type mice in response to HFD feeding, as shown by total fat accumulation and a trend towards increased visceral adipose tissue (141). Furthermore, HFD feeding increased insulin resistance, glucose intolerance and fasting plasma glucose levels in IL-23 deficient animals compared to the wild type animals on HFD (141). These results suggest that IL-23p19 could have protective properties during diet-induced obesity.
4 Impact of Spatial Segregation in the Gut on Obesity and T2D
The intestinal barrier (compare insert in Figure 1) is a complex system comprising (i) an immunological barrier, which consists of immune cells, cytokines and IgA as discussed in the previous section, (ii) a chemical barrier, which is characterized by antimicrobial secretions and (iii) a physical barrier, which involves spatial segregation through mucus and epithelial cells that provide a barrier through intercellular tight junctions. The intestinal barrier and its permeability play an important role in the pathogenesis of obesity and T2D, however, it is still unclear whether increased intestinal permeability is a cause or consequence of metabolic disease, or perhaps both (34–36). Nevertheless, the critical role of the intestinal barrier in metabolic disease development and prevention is becoming increasingly recognized and mechanisms that prevent pathogens, microbiota and microbiota-derived products from gaining access to the circulation through breaching intestinal tissues will be discussed in the following section.
4.1 Chemical Barrier
Antimicrobial molecules produced by the intestinal epithelium are important defense mechanisms to prevent infection and dysbiosis of commensal microbiota. HFD feeding was shown to decrease the expression of antimicrobial peptides (124, 128, 142–145). The gene expression of the antimicrobial peptide regenerating islet-derived protein 3 gamma (RegIIIγ) was reduced in the jejunum and colon of HFD-fed wild-type mice, but not in HFD-fed MyD88 knockout mice (124). This is of interest as RegIIIγ has been suggested to modulate microbiota through spatial segregation, thereby preventing commensal microbiota from reaching the epithelium (124, 146, 147).
In contrast to studies that reported decreased antimicrobial peptides in response to HFD feeding (124, 128, 142–145), Gao et al. (148) showed that HFD feeding increased the expression of antimicrobial peptides in the colon, which was counteracted by their treatment with polyphenols derived from Pu-erh tea. This inconsistency between these studies regarding HFD-induced antimicrobial peptide expression may be due to diet composition. Variation in diet composition between studies poses a major challenge in microbiota research as differences in diet composition, particularly regarding fat and fiber content, can induce variable metabolic outcomes (149, 150). While both Everard et al. (124) and Gao et al. (148) provide information on fat content (60% and 45% of kcal as fat, respectively), the type of fat and fiber in the diets were not reported. It is tempting to speculate that the type of fat may affect intestinal barrier function and, although there is limited evidence, diets rich in anti-inflammatory omega 3 fatty acids are likely to display barrier protective effects and/or modulate microbiota composition (151–153). Dietary fiber is an important source for fermentation by selected microbiota. High fiber diets may, therefore, profoundly affect microbiota composition through selecting strains that produce beneficial microbial metabolites such as short chain fatty acids (SCFA), which can be used as energy sources by enterocytes. If a standard diet containing grain-based fiber is compared to an HFD lacking fiber, metabolic benefits cannot be directly compared (149, 150).
4.2 Physical Barrier
In addition to their critical function in nutrient absorption, IECs also form a physical barrier designed to segregate the internal system from the external luminal environment that is rich in antigens and microbes. The intestinal epithelium consists of a single layer of cells where the intercellular spaces are locked with tight junction complexes, which can be used to regulate permeability towards small solutes such as water, electrolytes and macromolecules (154). Tight junction complexes play a critical role in barrier function and modulation and have been extensively reviewed recently (155–157). HFD feeding alters tight junction function and increases intestinal permeability. Gulhane et al. (65) showed that the tight junction protein claudin 1 was significantly decreased after 11 weeks of HFD feeding, which was accompanied by increased mucosal permeability and circulating levels of LPS. Suzuki and Hara (158) showed that HFD feeding increased intestinal permeability in lean and obese rats, which was linked to decreased expression of the tight junction proteins claudin 1 and 3 as well as junctional adhesion molecule 1 (JAM-1). These authors used the obese Otsuka Long Evans Tokushima Fatty (OLETF) rat model, in which rats become hyperphagic due to knockout of the cholecystokinin receptor, which senses cholecystokinin that is released from I cells in the small intestine and regulates food intake and satiety through neuronal feedback to the brain (159). HFD feeding induced intestinal inflammation in OLETF rats, however, the role of endotoxemia as a driver of this inflammatory response was questioned as, although systemic LPS levels were increased, systemic cytokine levels remained unchanged (158).
Goblet cells are the main source of the mucus that forms a physical barrier, shielding the intestinal epithelium from luminal antigens and microbiota (160, 161). Everard et al. (162) reported that HFD feeding in mice decreases the intestinal mucus layer, which improved after supplementation with Akkermansia muciniphila, an interesting and well-researched beneficial member of the microbiota (163). Another study showed that dietary prebiotics, which provide nutrients for selected microbiota and therefore support growth of selected commensals, are able to increase the number of goblet cells and thus mucus layer thickness (164), which is an integral part of the healthy intestinal barrier (165). Goblet cell and barrier dysfunction as well as inflammation following HFD feeding was recently linked to non-esterified fatty acid (NEFA) levels, and specifically to palmitate-induced oxidative and endoplasmic reticulum (ER) stress in goblet cells (65). This resulted in the thinning of the mucus layer and increased expression of non-O-glycosylated mucin 2 precursors in the intestines.
4.2.1 Intestinal Epithelial Cells (IECs) and Their Functional Diversity
IECs play a central role in intestinal barrier function as they provide a single cell layer barrier with intercellular spaces tightly locked by tight junctions, as previously highlighted. IECs produce a variety of effector molecules, e.g. they are the main cell type to express IAP, which plays a critical role in ameliorating metabolic disease through modulating LPS levels (63). Everard et al. (124) characterized a role for IEC-specific MyD88 expression in glucose homeostasis as MyD88 is an important part of the LPS-signaling cascade resulting in inflammation, while Lu et al. (66) used a TLR4 knockout mouse model to show the critical role of this receptor in metabolic health. Kawano et al. (90) demonstrated the role of the CCL2/CCR2 signaling axis in IECs and showed the importance of immune crosstalk between IECs and underlying immune cell populations in whole-body glucose homeostasis (Figure 2). Additional functions of IECs explaining their pivotal role in metabolic disease are discussed in this section and summarized in Figure 2.
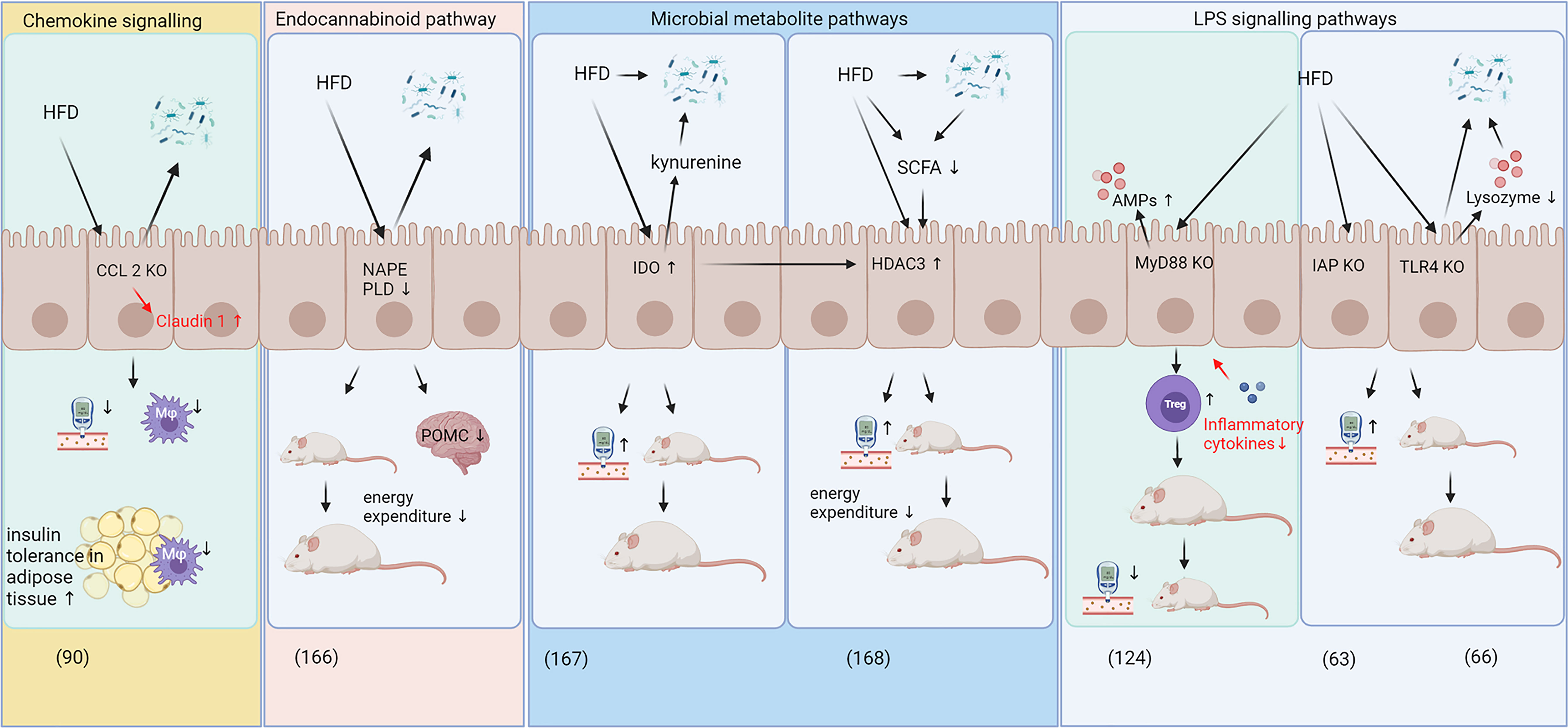
Figure 2 Specific and diverse roles of IECs in HFD-induced metabolic disease. The roles of intestinal epithelial cells (IECs) in body weight gain and glucose homeostasis can be categorized into immune crosstalk/chemokine signaling (90), pathways involving the endocannabinoid system (166), pathways involving microbial metabolites (167, 168) and signaling pathways related to LPS signaling (63, 66, 124). While most pathways exerted adverse effects on glucose metabolism and weight gain in response to HFD feeding, IEC specific knockout of CCL2 and MyD88 decreased body weight gain and improved glucose metabolism during high-fat diet feeding (90, 124). CCL2 and MyD88 knockout improved intestinal barrier function by increasing claudin 1 levels and decreased expression of inflammatory cytokines, respectively (highlighted in red). AMPs, antimicrobial peptides; CCL2, C-C motif chemokine ligand 2; HDAC3, histone deacetylase 3; HFD, high fat diet; IAP, intestinal alkaline phosphatase; IDO, Indolamine 2;3-dioxygenase; IEC, intestinal epithelial cells; KO, knockout; LPS, lipopolysaccharides; Mφ, macrophages; MyD88, myeloid differentiation primary response gene 88; NAPE PLD, N-acylphosphatidylethanolamine phospholipase D; POMC, pro-opiomelanocortin; SCFA, short chain fatty acids; TLR4, toll-like receptor 4; Treg, regulatory T cells.
Recent work has linked IECs with epigenetic mechanisms. Whitt et al. (168) showed that mice with histone deacetylase 3 (HDAC3) knockout in IECs were resistant to diet-induced obesity and showed improved glucose and lipid metabolism compared to wild-type mice, which may be due to increased energy expenditure, heat production and oxygen consumption (168). The study further showed that modulation of HDAC3 expression in IECs affected metabolically active tissues, such as the liver and adipose tissue and that an HFD reduces SCFAs (168), which are known to inhibit HDAC3 activity a (169, 170). The HFD feeding was suggested to induce HDAC3 activity, which was associated with increased body weight gain and blood glucose concentrations (Figure 2), while supplementation with butyrate, a SCFA, was able to reduce weight gain in HFD fed mice, presumably via HDAC3 dependent mechanisms. Taken together, these results suggest that HDAC3 may be implicated the development of obesity and T2D (168).
In addition to inhibiting HDAC3, microbiota-derived butyrate can also modulate indolamine 2,3-dioxygenase (IDO) activity in IECs (171). As a key enzyme in tryptophan catabolism, IDO converts tryptophane into kynurenine and is recognized as a modulator of immune function (172). IDO is expressed in macrophages, DCs and IECs and it contributes towards an immune suppressive and anti-inflammatory milieu (172) although a more complex role in intestinal homeostasis has been proposed recently, including adverse effects in metabolic disease (171, 173). Laurans et al. (167) demonstrated that IDO knockout mice were protected against HFD-induced body weight gain and displayed improved glucose tolerance. While Laurans et al. (167) did not use IEC-specific knockout, they demonstrated that IDO knockout in non-myeloid cells provides this metabolic benefit. These non-myeloid cells include IECs as the main cell type expressing IDO in the gut (171). In addition, Laurans et al. (167) showed that HFD feeding in wild-type mice increased IDO activity in the small and large intestines and this increase in kynurenine in the intestines may deplete indole derivatives that would otherwise be available for metabolism by the gut microbiota and may consequently exert a selective pressure on microbiota (Figure 2). The microbiota-derived indole-3-acetic acid is also an activator of the aryl hydrocarbon receptor (AHR), which, in addition, contributes to the protection against diet-induced obesity and intestinal barrier permeability (174). AHR plays a role in Treg cell differentiation (175) and a wide range of bioactive plant compounds, such as polyphenols, can act as AHR agonists and antagonists (176). Several reports have highlighted the role of the intestinal AHR, which is mainly expressed in IECs, in diet-induced obesity and metabolic disease (177–179).
IECs have been also investigated with regards to their role in the endocannabinoid system. Everard et al. (166) showed that knockout of N-acylphosphatidylethanolamine phospholipase D, a critical enzyme in endocannabinoid production, in IECs decreased energy expenditure and increased fat accumulation in response to HFD feeding without altering inflammatory status in adipose tissues or glucose metabolism. These authors showed that supplementation with A. muciniphila reversed most of the HFD-induced effects on N-acylphosphatidylethanolamine phospholipase D knockout mice. The endocannabinoid system plays a critical role in whole body energy metabolism (180), which was reviewed comprehensively by Veilleux et al. (181) and others (182, 183). Thus, these results suggest a significant role of endocannabinoids in intestinal tissues and in whole-body energy and glucose metabolism in relation to obesity and T2D development.
5 Intestinal Barrier and Immune System Play an Integral Part in the Pathophysiology of Obesity and T2D
Metabolic diseases, such as T2D, are complex diseases. Decades of dedicated research on T2D have advanced our understanding of the pathophysiology and keep producing advances in treatment options that improve lives. However, the complexity of disease mechanisms and the lack of a definitive cure for T2D suggest that our understanding of the disease is not yet complete. Figure 3 outlines the crosstalk between risk factors and key players in T2D development, which include diet, microbiota, intestines, metabolic inflammation and obesity. Two-way and indirect interactions between these key players add to the complexity of the disease:
1. Nutrient dense, high fat and high sugar diets likely exert a “one way effect” on glucose homeostasis as it is well established that chronic consumption of a western diet can lead to T2D (184). High fat diets also affect the intestinal immune system and barrier function, which can lead to metabolic inflammation (14, 15).
2. Changes in microbiota composition and diversity have been associated with obesity, dysregulated glucose metabolism and T2D, with metabolic inflammation (15, 23) and modify the intestinal immune and barrier system (152, 185). Conversely, microbiota may also affect the dietary intake through inducing behavioral changes and cravings (186, 187).
3. The intestines are known to shape microbiota through the intestinal immune system, antimicrobial and antibody secretions, which are critical components of the barrier function (37, 124, 129, 152). Barrier defects and dysfunction of the intestinal tissue can facilitate metabolic inflammation (14), but the intestines can also directly affect body weight and glucose homeostasis through the secretion of gut hormones (46, 47) and via activation of various inflammatory and pattern receptor signaling pathways in intestinal epithelial cells (63, 66, 82, 90, 142, 166–168).
4. Metabolic inflammation was shown to contribute to obesity (14, 18) and T2D development through inducing insulin resistance, which has been reviewed extensively (8–11). How metabolic inflammation may directly affect microbiota is not yet clear, however, systemic inflammation has negative effects on barrier function through the action of pro-inflammatory cytokines (154) and may therefore indirectly influence microbiota composition or diversity through its effect on the intestinal barrier and immune system.
5. That obesity contributes to metabolic inflammation is well established (95, 97, 99, 188), but obesity has also been associated with intestinal inflammation and barrier dysfunction (121, 189) and altered microbiota (27, 190). Obesity may influence diet and/or food intake through dysregulation in leptin and satiety levels (191). A more direct effect of obesity on T2D development occurs likely through increased release of free fatty acid from adipose tissue which can induce insulin resistance through oxidative stress and lipid metabolites (192).
6. T2D and dysregulated glucose homeostasis may affect obesity in the sense that insulin resistance and hyperglycemia lead to increased lipogenesis and lipid storage (192) although increased body weight may predominantly result from side effects of antidiabetic drugs. Hyperglycemia may induce metabolic inflammation directly via mechanisms related to increased oxidative stress (193) and indirectly through decreasing intestinal barrier function (194). Altered microbiota composition has been documented in type 2 diabetic patients (27).
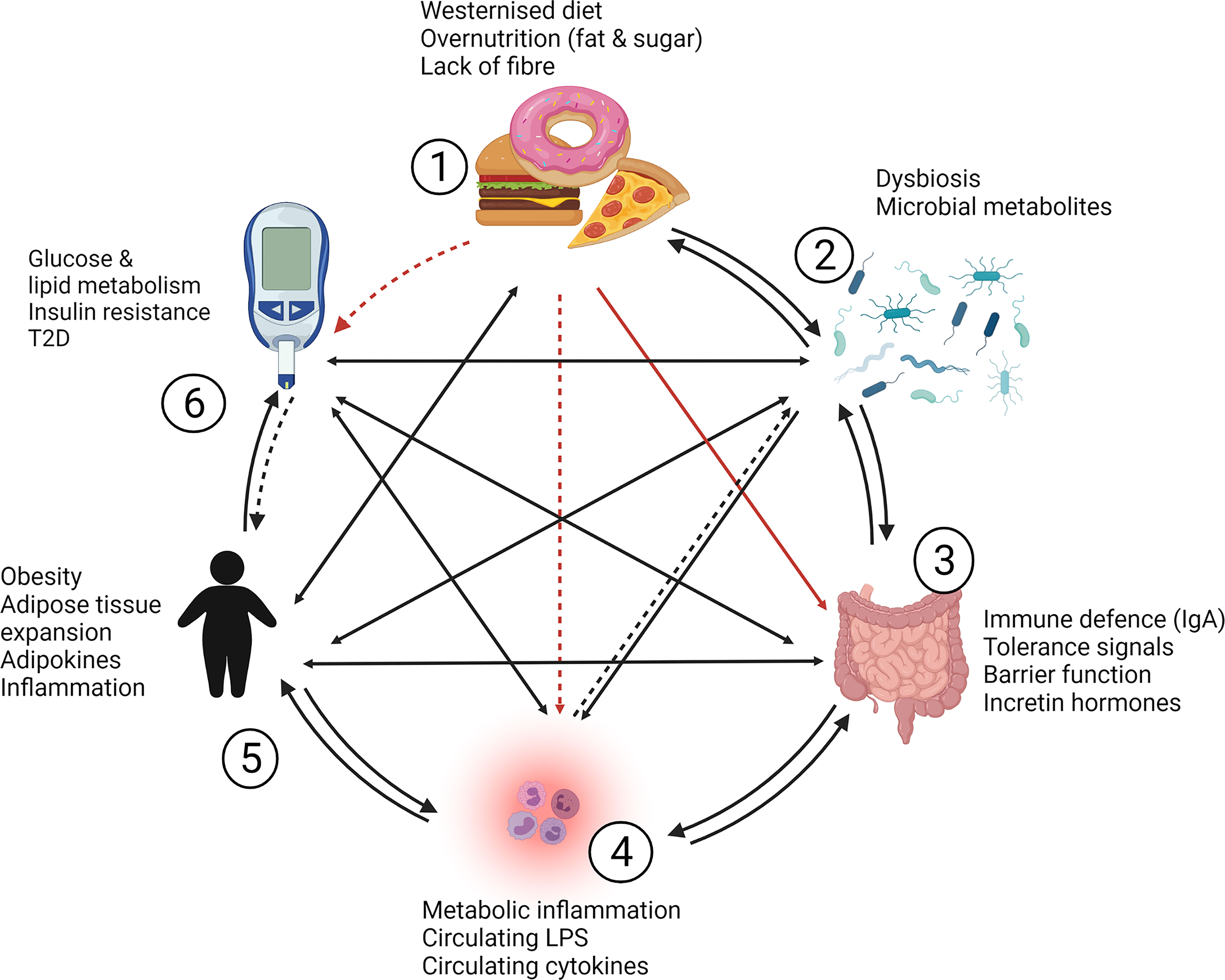
Figure 3 Complexity and cross-talk between contributors to obesity and T2D. Westernized diets [1], gut microbiota [2], the intestinal barrier and immune system [3] and metabolic inflammation [4] play a significant role in the development of obesity [5] and type 2 diabetes [6]. [1] Diet likely exerts a “one way effect” (indicated by red arrows) on glucose homeostasis, while high fat diets also affect the intestinal immune system and barrier function which can lead to [4] metabolic inflammation (indirect effects are indicated with dashed arrows). [2] Changes in microbiota composition and diversity have been associated with obesity, dysregulated glucose metabolism and T2D, with metabolic inflammation, and the intestinal immune and barrier system. Conversely, microbiota may also affect the dietary intake through inducing behavioral changes and cravings. [3] The intestines are known to shape microbiota and facilitate metabolic inflammation through barrier defects, but the intestines can also directly affect body weight and glucose homeostasis through gut hormone production and via expression of effector molecules and pathways in intestinal epithelial cells. [4] Metabolic inflammation contributes to obesity and T2D development through inducing insulin resistance. How metabolic inflammation may directly affect microbiota is not yet clear, but may occur indirectly through effects on intestinal barrier and immune function. [5] Obesity contributes to metabolic inflammation, but is associated with intestinal inflammation, barrier dysfunction and altered microbiota. Obesity may also influence diet and/or food intake through dysregulation in signaling related to satiety. A potentially direct effect of obesity on T2D development may involve the increased release of free fatty acid from adipose tissue which can induce insulin resistance through oxidative stress and lipid metabolites. [6] Type 2 diabetes and dysregulated glucose homeostasis may induce obesity in the sense that insulin resistance and hyperglycemia lead to increased lipogenesis and lipid storage but a direct pathway to increased body weight is likely the result of anti-diabetic drugs and their side effects. Hyperglycemia may induce metabolic inflammation directly via increased oxidative stress and indirectly through decreasing intestinal barrier function and altered microbiota composition. IgA, immunoglobulin A; LPS, lipopolysaccharide; T2D, type 2 diabetes.
Overall, this complex interactive system between organ structures and risk factors could provide an explanation why causal relationships between changes in microbiota composition, intestinal tissue and barrier effects and metabolic health have been difficult to attain.
6 Summary and Conclusion
There is a paucity of knowledge about the role of intestinal immunity and intestinal barrier function in the development of metabolic disease. This review highlights that the intestinal immune and barrier system can be a powerful modulator of diet-induced inflammation, glucose homeostasis, obesity and T2D development. Given the importance of the intestinal system as the “first responder” for dietary nutrients, antigens, commensals and toxins, future research on intestinal tissue and immune homeostasis should address changes in the intestinal immune system, including Treg cells, Th17 cells, as well as IgA producing plasma cells and their secretory profiles. Studies on innate and adaptive immune cell populations have provided compelling evidence for the contribution of these cell populations in the pathogenesis of metabolic disease (87, 88, 90, 117, 118, 127, 128), however, many questions still remain unanswered.
This review also highlighted the complexity of interconnected risk factors, organ systems and their cross-talk in metabolic disease, summarized in Figure 3. Host immune cells can shape commensal microbiota (37, 124) and inflammatory tone. The subtle inflammatory changes in the intestines in the early stages of metabolic disease development (87–89) suggest that inflammation in the intestinal sense could be viewed as shift in immune cell populations whose cytokine profiles set either an inflammatory or tolerant “tone”, e.g. by recruiting regulatory T cells (88, 90, 127). IgA represents the initial defense that the body offers against invading pathogens and dysbiosis (195) and studies have associated IgA producing cells with microbiota composition and metabolic effects (117, 118), future research should characterize the role of IgA in obesity metabolic disease in more detail.
One way to improve intestinal barrier function may be through focusing on IECs, as they possess more diverse and vital functions in addition to nutrient and electrolyte transport than previously known. In Figure 2, some of the recently discovered underlying mechanisms of how IECs contribute to metabolic disease have been highlighted. Exactly how this crosstalk between IECs, the intestinal immune system and metabolically active tissues is facilitated and translates into improved metabolic phenotype is not yet fully elucidated, but the intestines may play a pivotal role in the development of metabolic diseases and deserve further investigation. This may indeed place the intestines, including the intestinal immune system and IECs, at the center of metabolic disease development and provide a new focus area to develop improved treatments for obesity and T2D in future.
Author Contributions
Conceptualization, SR, CP, and RJ. Writing—original draft preparation, SR. Writing—review and editing, SR, CP, RJ, JL, and CM. All authors have read and agreed to the published version of the manuscript.
Funding
SR received funding from the South African Medical Research Council (baseline funding), the South African Rooibos Council and the National Research Foundation (NRF) of South Africa, grant UID 121919. CM received funding from the NRF of South Africa under the IRG-Taiwan/South African Research Cooperation Programme, grant UID 121232.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Figures were created with BioRender.com.
References
1. WHO. WHO | World Health Statistics 2018. Geneva: WHO (2018). Available at: https://www.who.int/gho/publications/world_health_statistics/2018/en/.
2. International Diabetes Federation. IDF Diabetes Atlas. 9th Edition. Brussels, Belgium: International Diabetes Federation (2019). Available at: https://www.diabetesatlas.org/en/.
3. CulinaryLore. Who First Said “You Are What You Eat? (2016). Available at: https://culinarylore.com/food-history:who-first-said-youare-what-you-eat/ [Accessed October 6, 2021].
4. Anon. Physiologie Du Goût, Ou Méditations De Gastronomie Transcendante, Ouvrage Théorique, Historique Et À L’ordre Du Jour, Dédié Aux Gastronomes Parisiens Par Un Professeur, Membre De Plusieurs Sociétés Littéraires Et Savantes. Paris: A. Sautelet et Cie (1826).
5. Weickert MO. What Dietary Modification Best Improves Insulin Sensitivity and Why? Clin Endocrinol (Oxf) (2012) 77:508–12. doi: 10.1111/j.1365-2265.2012.04450.x
6. Rogero MM, Calder PC. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients (2018) 10:432. doi: 10.3390/nu10040432
7. Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, et al. Origins and Evolution of the Western Diet: Health Implications for the 21st Century. Am J Clin Nutr (2005) 81:341–54. doi: 10.1093/ajcn.81.2.341
8. Shoelson SE, Lee J, Goldfine AB. Inflammation and Insulin Resistance. J Clin Invest (2006) 116:1793–801. doi: 10.1172/JCI29069
9. Hotamisligil GS. Inflammation and Metabolic Disorders. Nature (2006) 444:860–7. doi: 10.1038/nature05485
10. Saltiel AR, Olefsky JM. Inflammatory Mechanisms Linking Obesity and Metabolic Disease. J Clin Invest (2017) 127:1–4. doi: 10.1172/JCI92035
11. Armstrong H, Bording-Jorgensen M, Dijk S, Wine E. The Complex Interplay Between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent it. Cancers (2018) 10:83. doi: 10.3390/cancers10030083
12. Calle MC, Fernandez ML. Inflammation and Type 2 Diabetes. Diabetes Metab (2012) 38:183–91. doi: 10.1016/j.diabet.2011.11.006
13. Torres S, Fabersani E, Marquez A, Gauffin-Cano P. Adipose Tissue Inflammation and Metabolic Syndrome. The Proactive Role of Probiotics. Eur J Nutr (2019) 58:27–43. doi: 10.1007/s00394-018-1790-2
14. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes (2007) 56:1761–72. doi: 10.2337/db06-1491
15. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes (2008) 57:1470–81. doi: 10.2337/db07-1403
16. Boutagy NE, McMillan RP, Frisard MI, Hulver MW. Metabolic Endotoxemia With Obesity: Is it Real and is it Relevant? Biochimie (2016) 124:11–20. doi: 10.1016/j.biochi.2015.06.020
17. Dey P, Sasaki GY, Wei P, Li J, Wang L, Zhu J, et al. Green Tea Extract Prevents Obesity in Male Mice by Alleviating Gut Dysbiosis in Association With Improved Intestinal Barrier Function That Limits Endotoxin Translocation and Adipose Inflammation. J Nutr Biochem (2019) 67:78–89. doi: 10.1016/j.jnutbio.2019.01.017
18. Fuke N, Nagata N, Suganuma H, Ota T. Regulation of Gut Microbiota and Metabolic Endotoxemia With Dietary Factors. Nutrients (2019) 11:E2277. doi: 10.3390/nu11102277
19. Kim K-A, Gu W, Lee I-A, Joh E-H, Kim D-H. High Fat Diet-Induced Gut Microbiota Exacerbates Inflammation and Obesity in Mice via the TLR4 Signaling Pathway. PloS One (2012) 7:e47713. doi: 10.1371/journal.pone.0047713
20. Million M, Maraninchi M, Henry M, Armougom F, Richet H, Carrieri P, et al. Obesity-Associated Gut Microbiota Is Enriched in Lactobacillus Reuteri and Depleted in Bifidobacterium Animalis and Methanobrevibacter Smithii. Int J Obes 2005 (2012) 36:817–25. doi: 10.1038/ijo.2011.153
21. Lecomte V, Kaakoush NO, Maloney CA, Raipuria M, Huinao KD, Mitchell HM, et al. Changes in Gut Microbiota in Rats Fed a High Fat Diet Correlate With Obesity-Associated Metabolic Parameters. PloS One (2015) 10:e0126931. doi: 10.1371/journal.pone.0126931
22. Mu Q, Kirby J, Reilly CM, Luo XM. Leaky Gut As a Danger Signal for Autoimmune Diseases. Front Immunol (2017) 8:598. doi: 10.3389/fimmu.2017.00598
23. He M, Shi B. Gut Microbiota as a Potential Target of Metabolic Syndrome: The Role of Probiotics and Prebiotics. Cell Biosci (2017) 7:54. doi: 10.1186/s13578-017-0183-1
24. Madsen L, Myrmel LS, Fjære E, Liaset B, Kristiansen K. Links Between Dietary Protein Sources, the Gut Microbiota, and Obesity. Front Physiol (2017) 8:1047. doi: 10.3389/fphys.2017.01047
25. Kang C, Wang B, Kaliannan K, Wang X, Lang H, Hui S, et al. Gut Microbiota Mediates the Protective Effects of Dietary Capsaicin Against Chronic Low-Grade Inflammation and Associated Obesity Induced by High-Fat Diet. mBio (2017) 8:e00470–17. doi: 10.1128/mBio.00470-17
26. Uhr GT, Dohnalová L, Thaiss CA. The Dimension of Time in Host-Microbiome Interactions. mSystems (2019) 4:00216–18. doi: 10.1128/mSystems.00216-18
27. Caesar R. Pharmacologic and Nonpharmacologic Therapies for the Gut Microbiota in Type 2 Diabetes. Can J Diabetes (2019) 43:224–31. doi: 10.1016/j.jcjd.2019.01.007
28. Zhang Z, Xu H, Zhao H, Geng Y, Ren Y, Guo L, et al. Edgeworthia Gardneri (Wall.) Meisn. Water Extract Improves Diabetes and Modulates Gut Microbiota. J Ethnopharmacol (2019) 239:111854. doi: 10.1016/j.jep.2019.111854
29. Massier L, Blüher M, Kovacs P, Chakaroun RM. Impaired Intestinal Barrier and Tissue Bacteria: Pathomechanisms for Metabolic Diseases. Front Endocrinol (2021) 12:616506. doi: 10.3389/fendo.2021.616506
30. Amar J, Serino M, Lange C, Chabo C, Iacovoni J, Mondot S, et al. Involvement of Tissue Bacteria in the Onset of Diabetes in Humans: Evidence for a Concept. Diabetologia (2011) 54:3055–61. doi: 10.1007/s00125-011-2329-8
31. Gérard C, Vidal H. Impact of Gut Microbiota on Host Glycemic Control. Front Endocrinol (2019) 10:29. doi: 10.3389/fendo.2019.00029
32. Akbari P, Braber S, Varasteh S, Alizadeh A, Garssen J, Fink-Gremmels J. The Intestinal Barrier as an Emerging Target in the Toxicological Assessment of Mycotoxins. Arch Toxicol (2017) 91:1007–29. doi: 10.1007/s00204-016-1794-8
33. Pereira MT, Malik M, Nostro JA, Mahler GJ, Musselman LP. Effect of Dietary Additives on Intestinal Permeability in Both Drosophila and a Human Cell Co-Culture. Dis Model Mech (2018) 11:dmm034520. doi: 10.1242/dmm.034520
34. Bell DSH. Changes Seen in Gut Bacteria Content and Distribution With Obesity: Causation or Association? Postgrad Med (2015) 127:863–8. doi: 10.1080/00325481.2015.1098519
35. Lam YY, Zhang C, Zhao L. Causality in Dietary Interventions-Building a Case for Gut Microbiota. Genome Med (2018) 10:62. doi: 10.1186/s13073-018-0573-y
36. Cani PD, Van Hul M. Gut Microbiota and Obesity: Causally Linked? Expert Rev Gastroenterol Hepatol (2020) 14:401–3. doi: 10.1080/17474124.2020.1758064
37. Foster KR, Schluter J, Coyte KZ, Rakoff-Nahoum S. The Evolution of the Host Microbiome as an Ecosystem on a Leash. Nature (2017) 548:43–51. doi: 10.1038/nature23292
38. Winer DA, Luck H, Tsai S, Winer S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab (2016) 23:413–26. doi: 10.1016/j.cmet.2016.01.003
39. Winer DA, Winer S, Dranse HJ, Lam TKT. Immunologic Impact of the Intestine in Metabolic Disease. J Clin Invest (2017) 127:33–42. doi: 10.1172/JCI88879
40. Groschwitz KR, Hogan SP. Intestinal Barrier Function: Molecular Regulation and Disease Pathogenesis. J Allergy Clin Immunol (2009) 124:3–20; quiz 21–22. doi: 10.1016/j.jaci.2009.05.038
41. Schulz O, Pabst O. Antigen Sampling in the Small Intestine. Trends Immunol (2013) 34:155–61. doi: 10.1016/j.it.2012.09.006
42. Scaldaferri F, Pizzoferrato M, Gerardi V, Lopetuso L, Gasbarrini A. The Gut Barrier: New Acquisitions and Therapeutic Approaches. J Clin Gastroenterol (2012) 46 Suppl:S12–7. doi: 10.1097/MCG.0b013e31826ae849
43. Bain CC, Mowat AM. The Monocyte-Macrophage Axis in the Intestine. Cell Immunol (2014) 291:41–8. doi: 10.1016/j.cellimm.2014.03.012
44. Gross M, Salame T-M, Jung S. Guardians of the Gut - Murine Intestinal Macrophages and Dendritic Cells. Front Immunol (2015) 6:254. doi: 10.3389/fimmu.2015.00254
45. Ahluwalia B, Magnusson MK, Öhman L. Mucosal Immune System of the Gastrointestinal Tract: Maintaining Balance Between the Good and the Bad. Scand J Gastroenterol (2017) 52:1185–93. doi: 10.1080/00365521.2017.1349173
46. Martin AM, Sun EW, Keating DJ. Mechanisms Controlling Hormone Secretion in Human Gut and its Relevance to Metabolism. J Endocrinol (2019) 244:R1–15. doi: 10.1530/JOE-19-0399
47. Makaronidis JM, Batterham RL. The Role of Gut Hormones in the Pathogenesis and Management of Obesity. Curr Opin Physiol (2019) 12:1–11. doi: 10.1016/j.cophys.2019.04.007
48. Nauck MA, Meier JJ. Incretin Hormones: Their Role in Health and Disease. Diabetes Obes Metab (2018) 20 Suppl 1:5–21. doi: 10.1111/dom.13129
49. Lund ML, Egerod KL, Engelstoft MS, Dmytriyeva O, Theodorsson E, Patel BA, et al. Enterochromaffin 5-HT Cells - A Major Target for GLP-1 and Gut Microbial Metabolites. Mol Metab (2018) 11:70–83. doi: 10.1016/j.molmet.2018.03.004
50. Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Muccioli GM, et al. Responses of Gut Microbiota and Glucose and Lipid Metabolism to Prebiotics in Genetic Obese and Diet-Induced Leptin-Resistant Mice. Diabetes (2011) 60:2775–86. doi: 10.2337/db11-0227
51. Muller CJF, Joubert E, de Beer D, Sanderson M, Malherbe CJ, Fey SJ, et al. Acute Assessment of an Aspalathin-Enriched Green Rooibos (Aspalathus Linearis) Extract With Hypoglycemic Potential. Phytomed Int J Phytother Phytopharm (2012) 20:32–9. doi: 10.1016/j.phymed.2012.09.010
52. Son MJ, Minakawa M, Miura Y, Yagasaki K. Aspalathin Improves Hyperglycemia and Glucose Intolerance in Obese Diabetic Ob/Ob Mice. Eur J Nutr (2013) 52:1607–19. doi: 10.1007/s00394-012-0466-6
53. Bowles S, Joubert E, de Beer D, Louw J, Brunschwig C, Njoroge M, et al. Intestinal Transport Characteristics and Metabolism of C-Glucosyl Dihydrochalcone, Aspalathin. Mol Basel Switz (2017) 22:554. doi: 10.3390/molecules22040554
54. Villa-Rodriguez JA, Ifie I, Gonzalez-Aguilar GA, Roopchand DE. The Gastrointestinal Tract as Prime Site for Cardiometabolic Protection by Dietary Polyphenols. Adv Nutr Bethesda Md (2019) 10:999–1011. doi: 10.1093/advances/nmz038
55. Buse JB, DeFronzo RA, Rosenstock J, Kim T, Burns C, Skare S, et al. The Primary Glucose-Lowering Effect of Metformin Resides in the Gut, Not the Circulation: Results From Short-Term Pharmacokinetic and 12-Week Dose-Ranging Studies. Diabetes Care (2016) 39:198–205. doi: 10.2337/dc15-0488
56. Dapito DH, Mencin A, Gwak G-Y, Pradere J-P, Jang M-K, Mederacke I, et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell (2012) 21:504–16. doi: 10.1016/j.ccr.2012.02.007
57. Fedirko V, Tran HQ, Gewirtz AT, Stepien M, Trichopoulou A, Aleksandrova K, et al. Exposure to Bacterial Products Lipopolysaccharide and Flagellin and Hepatocellular Carcinoma: A Nested Case-Control Study. BMC Med (2017) 15:72. doi: 10.1186/s12916-017-0830-8
58. Hersoug L-G, Møller P, Loft S. Role of Microbiota-Derived Lipopolysaccharide in Adipose Tissue Inflammation, Adipocyte Size and Pyroptosis During Obesity. Nutr Res Rev (2018) 31:153–63. doi: 10.1017/S0954422417000269
59. Rhee SH. Lipopolysaccharide: Basic Biochemistry, Intracellular Signaling, and Physiological Impacts in the Gut. Intest Res (2014) 12:90–5. doi: 10.5217/ir.2014.12.2.90
60. Akira S, Takeda K. Toll-Like Receptor Signalling. Nat Rev Immunol (2004) 4:499–511. doi: 10.1038/nri1391
61. Abreu MT. Toll-Like Receptor Signalling in the Intestinal Epithelium: How Bacterial Recognition Shapes Intestinal Function. Nat Rev Immunol (2010) 10:131–44. doi: 10.1038/nri2707
62. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
63. Kaliannan K, Hamarneh SR, Economopoulos KP, Nasrin Alam S, Moaven O, Patel P, et al. Intestinal Alkaline Phosphatase Prevents Metabolic Syndrome in Mice. Proc Natl Acad Sci USA (2013) 110:7003–8. doi: 10.1073/pnas.1220180110
64. Taira R, Yamaguchi S, Shimizu K, Nakamura K, Ayabe T, Taira T. Bacterial Cell Wall Components Regulate Adipokine Secretion From Visceral Adipocytes. J Clin Biochem Nutr (2015) 56:149–54. doi: 10.3164/jcbn.14-74
65. Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation That is Reversed by IL-22. Sci Rep (2016) 6:28990. doi: 10.1038/srep28990
66. Lu P, Sodhi CP, Yamaguchi Y, Jia H, Prindle T, Fulton WB, et al. Intestinal Epithelial Toll-Like Receptor 4 Prevents Metabolic Syndrome by Regulating Interactions Between Microbes and Intestinal Epithelial Cells in Mice. Mucosal Immunol (2018) 11:727–40. doi: 10.1038/mi.2017.114
67. Taylor R. Pathogenesis of Type 2 Diabetes: Tracing the Reverse Route From Cure to Cause. Diabetologia (2008) 51:1781–9. doi: 10.1007/s00125-008-1116-7
68. Delzenne NM, Knudsen C, Beaumont M, Rodriguez J, Neyrinck AM, Bindels LB. Contribution of the Gut Microbiota to the Regulation of Host Metabolism and Energy Balance: A Focus on the Gut-Liver Axis. Proc Nutr Soc (2019) 78:319–28. doi: 10.1017/S0029665118002756
69. Ding Y, Yanagi K, Cheng C, Alaniz RC, Lee K, Jayaraman A. Interactions Between Gut Microbiota and non-Alcoholic Liver Disease: The Role of Microbiota-Derived Metabolites. Pharmacol Res (2019) 141:521–9. doi: 10.1016/j.phrs.2019.01.029
70. Geurts L, Neyrinck AM, Delzenne NM, Knauf C, Cani PD. Gut Microbiota Controls Adipose Tissue Expansion, Gut Barrier and Glucose Metabolism: Novel Insights Into Molecular Targets and Interventions Using Prebiotics. Benef Microbes (2014) 5:3–17. doi: 10.3920/BM2012.0065
71. Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez-Humarán LG, et al. Intestinal Mucosal Adherence and Translocation of Commensal Bacteria at the Early Onset of Type 2 Diabetes: Molecular Mechanisms and Probiotic Treatment. EMBO Mol Med (2011) 3:559–72. doi: 10.1002/emmm.201100159
72. Shen L, Ao L, Xu H, Shi J, You D, Yu X, et al. Poor Short-Term Glycemic Control in Patients With Type 2 Diabetes Impairs the Intestinal Mucosal Barrier: A Prospective, Single-Center, Observational Study. BMC Endocr Disord (2019) 19:29. doi: 10.1186/s12902-019-0354-7
73. Kühn F, Adiliaghdam F, Cavallaro PM, Hamarneh SR, Tsurumi A, Hoda RS, et al. Intestinal Alkaline Phosphatase Targets the Gut Barrier to Prevent Aging. JCI Insight (2020) 5:e134049. doi: 10.1172/jci.insight.134049
74. Dalby MJ, Aviello G, Ross AW, Walker AW, Barrett P, Morgan PJ. Diet Induced Obesity is Independent of Metabolic Endotoxemia and TLR4 Signalling, But Markedly Increases Hypothalamic Expression of the Acute Phase Protein, SerpinA3N. Sci Rep (2018) 8:15648. doi: 10.1038/s41598-018-33928-4
75. Zamora R, Chavan S, Zanos T, Simmons RL, Billiar TR, Vodovotz Y. Spatiotemporally Specific Roles of TLR4, TNF, and IL-17A in Murine Endotoxin-Induced Inflammation Inferred From Analysis of Dynamic Networks. Mol Med Camb Mass (2021) 27:65. doi: 10.1186/s10020-021-00333-z
76. Yang J, Yan H. TLR5: Beyond the Recognition of Flagellin. Cell Mol Immunol (2017) 14:1017–9. doi: 10.1038/cmi.2017.122
77. Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic Syndrome and Altered Gut Microbiota in Mice Lacking Toll-Like Receptor 5. Science (2010) 328:228–31. doi: 10.1126/science.1179721
78. Volpe CMO, Nogueira-Machado JA. The Dual Role of Free Fatty Acid Signaling in Inflammation and Therapeutics. Recent Pat Endocr Metab Immune Drug Discovery (2013) 7:189–97. doi: 10.2174/18715303113139990041
79. Snodgrass RG, Huang S, Choi I-W, Rutledge JC, Hwang DH. Inflammasome-Mediated Secretion of IL-1β in Human Monocytes Through TLR2 Activation; Modulation by Dietary Fatty Acids. J Immunol Baltim Md 1950 (2013) 191:4337–47. doi: 10.4049/jimmunol.1300298
80. Litvak Y, Byndloss MX, Bäumler AJ. Colonocyte Metabolism Shapes the Gut Microbiota. Science (2018) 362:eaat9076. doi: 10.1126/science.aat9076
81. Malo MS. A High Level of Intestinal Alkaline Phosphatase Is Protective Against Type 2 Diabetes Mellitus Irrespective of Obesity. EBioMedicine (2015) 2:2016–23. doi: 10.1016/j.ebiom.2015.11.027
82. de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to High-Fat Diet-Induced Obesity in Rats is Associated With Changes in the Gut Microbiota and Gut Inflammation. Am J Physiol Gastrointest Liver Physiol (2010) 299:G440–8. doi: 10.1152/ajpgi.00098.2010
83. Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen Y-Y, et al. High-Fat Diet Determines the Composition of the Murine Gut Microbiome Independently of Obesity. Gastroenterology (2009) 137:1716–24.e1–2. doi: 10.1053/j.gastro.2009.08.042
84. Carmody RN, Gerber GK, Luevano JM, Gatti DM, Somes L, Svenson KL, et al. Diet Dominates Host Genotype in Shaping the Murine Gut Microbiota. Cell Host Microbe (2015) 17:72–84. doi: 10.1016/j.chom.2014.11.010
85. Chewning JH, Weaver CT. Development and Survival of Th17 Cells Within the Intestines: The Influence of Microbiome- and Diet-Derived Signals. J Immunol Baltim Md 1950 (2014) 193:4769–77. doi: 10.4049/jimmunol.1401835
86. Bain CC, Schridde A. Origin, Differentiation, and Function of Intestinal Macrophages. Front Immunol (2018) 9:2733. doi: 10.3389/fimmu.2018.02733
87. Johnson AMF, Costanzo A, Gareau MG, Armando AM, Quehenberger O, Jameson JM, et al. High Fat Diet Causes Depletion of Intestinal Eosinophils Associated With Intestinal Permeability. PloS One (2015) 10:e0122195. doi: 10.1371/journal.pone.0122195
88. Luck H, Tsai S, Chung J, Clemente-Casares X, Ghazarian M, Revelo XS, et al. Regulation of Obesity-Related Insulin Resistance With Gut Anti-Inflammatory Agents. Cell Metab (2015) 21:527–42. doi: 10.1016/j.cmet.2015.03.001
89. Nascimento JC, Matheus VA, Oliveira RB, Tada SFS, Collares-Buzato CB. High-Fat Diet Induces Disruption of the Tight Junction-Mediated Paracellular Barrier in the Proximal Small Intestine Before the Onset of Type 2 Diabetes and Endotoxemia. Dig Dis Sci (2021) 66:3359–74. doi: 10.1007/s10620-020-06664-x
90. Kawano Y, Nakae J, Watanabe N, Kikuchi T, Tateya S, Tamori Y, et al. Colonic Pro-Inflammatory Macrophages Cause Insulin Resistance in an Intestinal Ccl2/Ccr2-Dependent Manner. Cell Metab (2016) 24:295–310. doi: 10.1016/j.cmet.2016.07.009
91. Turner JR, Buschmann MM, Romero-Calvo I, Sailer A, Shen L. The Role of Molecular Remodeling in Differential Regulation of Tight Junction Permeability. Semin Cell Dev Biol (2014) 36:204–12. doi: 10.1016/j.semcdb.2014.09.022
92. Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NMJ, Magness S, et al. High-Fat Diet: Bacteria Interactions Promote Intestinal Inflammation Which Precedes and Correlates With Obesity and Insulin Resistance in Mouse. PloS One (2010) 5:e12191. doi: 10.1371/journal.pone.0012191
93. Liu W-X, Wang T, Zhou F, Wang Y, Xing J-W, Zhang S, et al. Voluntary Exercise Prevents Colonic Inflammation in High-Fat Diet-Induced Obese Mice by Up-Regulating PPAR-γ Activity. Biochem Biophys Res Commun (2015) 459:475–80. doi: 10.1016/j.bbrc.2015.02.047
94. Delage B, Bairras C, Buaud B, Pallet V, Cassand P. A High-Fat Diet Generates Alterations in Nuclear Receptor Expression: Prevention by Vitamin A and Links With Cyclooxygenase-2 and Beta-Catenin. Int J Cancer (2005) 116:839–46. doi: 10.1002/ijc.21108
95. Gregor MF, Hotamisligil GS. Inflammatory Mechanisms in Obesity. Annu Rev Immunol (2011) 29:415–45. doi: 10.1146/annurev-immunol-031210-101322
96. Fernández-Real JM, Pérez del Pulgar S, Luche E, Moreno-Navarrete JM, Waget A, Serino M, et al. CD14 Modulates Inflammation-Driven Insulin Resistance. Diabetes (2011) 60:2179–86. doi: 10.2337/db10-1210
97. Ferrante AW. Macrophages, Fat, and the Emergence of Immunometabolism. J Clin Invest (2013) 123:4992–3. doi: 10.1172/JCI73658
98. McNelis JC, Olefsky JM. Macrophages, Immunity, and Metabolic Disease. Immunity (2014) 41:36–48. doi: 10.1016/j.immuni.2014.05.010
99. Francisco V, Pino J, Campos-Cabaleiro V, Ruiz-Fernández C, Mera A, Gonzalez-Gay MA, et al. Obesity, Fat Mass and Immune System: Role for Leptin. Front Physiol (2018) 9:640. doi: 10.3389/fphys.2018.00640
100. Fan H, Wang A, Wang Y, Sun Y, Han J, Chen W, et al. Innate Lymphoid Cells: Regulators of Gut Barrier Function and Immune Homeostasis. J Immunol Res (2019) 2019:2525984. doi: 10.1155/2019/2525984
101. Muller PA, Matheis F, Mucida D. Gut Macrophages: Key Players in Intestinal Immunity and Tissue Physiology. Curr Opin Immunol (2020) 62:54–61. doi: 10.1016/j.coi.2019.11.011
102. Boring L, Gosling J, Chensue S, Kunkel S, Farese R, Broxmeyer H, et al. Impaired Monocyte Migration and Reduced Type 1 (Th1) Cytokine Responses in C-C Chemokine Receptor 2 Knockout Mice. J Clin Invest (1997) 100:2552–61. doi: 10.1172/JCI119798
103. Frade JM, Mellado M, del Real G, Gutierrez-Ramos JC, Lind P, Martinez-A C. Characterization of the CCR2 Chemokine Receptor: Functional CCR2 Receptor Expression in B Cells. J Immunol Baltim Md 1950 (1997) 159:5576–84.
104. Kraneveld AD, Folkerts G, Van Oosterhout AJ, Nijkamp FP. Airway Hyperresponsiveness: First Eosinophils and Then Neuropeptides. Int J Immunopharmacol (1997) 19:517–27. doi: 10.1016/s0192-0561(97)00085-4
105. Klion AD, Nutman TB. The Role of Eosinophils in Host Defense Against Helminth Parasites. J Allergy Clin Immunol (2004) 113:30–7. doi: 10.1016/j.jaci.2003.10.050
106. Lee JJ, Jacobsen EA, McGarry MP, Schleimer RP, Lee NA. Eosinophils in Health and Disease: The LIAR Hypothesis. Clin Exp Allergy J Br Soc Allergy Clin Immunol (2010) 40:563–75. doi: 10.1111/j.1365-2222.2010.03484.x
107. Dent G, Loweth SC, Hasan AM, Leslie FM. Synergic Production of Neutrophil Chemotactic Activity by Colonic Epithelial Cells and Eosinophils. Immunobiology (2014) 219:793–7. doi: 10.1016/j.imbio.2014.06.008
108. Svensson-Frej M. Immunobiology of Intestinal Eosinophils - a Dogma in the Changing? J Innate Immun (2011) 3:565–76. doi: 10.1159/000328799
109. Arnold IC, Artola-Borán M, Tallón de Lara P, Kyburz A, Taube C, Ottemann K, et al. Eosinophils Suppress Th1 Responses and Restrict Bacterially Induced Gastrointestinal Inflammation. J Exp Med (2018) 215:2055–72. doi: 10.1084/jem.20172049
110. Chu VT, Beller A, Rausch S, Strandmark J, Zänker M, Arbach O, et al. Eosinophils Promote Generation and Maintenance of Immunoglobulin-A-Expressing Plasma Cells and Contribute to Gut Immune Homeostasis. Immunity (2014) 40:582–93. doi: 10.1016/j.immuni.2014.02.014
111. Fagarasan S. Evolution, Development, Mechanism and Function of IgA in the Gut. Curr Opin Immunol (2008) 20:170–7. doi: 10.1016/j.coi.2008.04.002
112. Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive Immune Regulation in the Gut: T Cell-Dependent and T Cell-Independent IgA Synthesis. Annu Rev Immunol (2010) 28:243–73. doi: 10.1146/annurev-immunol-030409-101314
113. Pabst O. New Concepts in the Generation and Functions of IgA. Nat Rev Immunol (2012) 12:821–32. doi: 10.1038/nri3322
114. Shi Y, Mu L. An Expanding Stage for Commensal Microbes in Host Immune Regulation. Cell Mol Immunol (2017) 14:339–48. doi: 10.1038/cmi.2016.64
115. Bunker JJ, Bendelac A. IgA Responses to Microbiota. Immunity (2018) 49:211–24. doi: 10.1016/j.immuni.2018.08.011
116. Pabst O, Slack E. IgA and the Intestinal Microbiota: The Importance of Being Specific. Mucosal Immunol (2020) 13:12–21. doi: 10.1038/s41385-019-0227-4
117. Zhou J, Wang Y, He Y, Gao Y, Wan R, Cai M, et al. Non-Obese Type 2 Diabetes Patients Present Intestinal B Cell Dysregulations Associated With Hyperactive Intestinal Tfh Cells. Mol Immunol (2018) 97:27–32. doi: 10.1016/j.molimm.2018.03.008
118. Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S, et al. Gut-Associated IgA+ Immune Cells Regulate Obesity-Related Insulin Resistance. Nat Commun (2019) 10:3650. doi: 10.1038/s41467-019-11370-y
119. Sakamoto Y, Niwa M, Muramatsu K, Shimo S. High-Fat Diet and Age-Dependent Effects of IgA-Bearing Cell Populations in the Small Intestinal Lamina Propria in Mice. Int J Mol Sci (2021) 22:1165. doi: 10.3390/ijms22031165
120. Stockinger B, Omenetti S. The Dichotomous Nature of T Helper 17 Cells. Nat Rev Immunol (2017) 17:535–44. doi: 10.1038/nri.2017.50
121. Monteiro-Sepulveda M, Touch S, Mendes-Sá C, André S, Poitou C, Allatif O, et al. Jejunal T Cell Inflammation in Human Obesity Correlates With Decreased Enterocyte Insulin Signaling. Cell Metab (2015) 22:113–24. doi: 10.1016/j.cmet.2015.05.020
122. He S, Kahles F, Rattik S, Nairz M, McAlpine CS, Anzai A, et al. Gut Intraepithelial T Cells Calibrate Metabolism and Accelerate Cardiovascular Disease. Nature (2019) 566:115–9. doi: 10.1038/s41586-018-0849-9
123. Sakaguchi S, Powrie F. Emerging Challenges in Regulatory T Cell Function and Biology. Science (2007) 317:627–9. doi: 10.1126/science.1142331
124. Everard A, Geurts L, Caesar R, Van Hul M, Matamoros S, Duparc T, et al. Intestinal Epithelial MyD88 is a Sensor Switching Host Metabolism Towards Obesity According to Nutritional Status. Nat Commun (2014) 5:5648. doi: 10.1038/ncomms6648
125. Deguine J, Barton GM. MyD88: A Central Player in Innate Immune Signaling. F1000prime Rep (2014) 6:97. doi: 10.12703/P6-97
126. Hoe E, Anderson J, Nathanielsz J, Toh ZQ, Marimla R, Balloch A, et al. The Contrasting Roles of Th17 Immunity in Human Health and Disease. Microbiol Immunol (2017) 61:49–56. doi: 10.1111/1348-0421.12471
127. Hong C-P, Park A, Yang B-G, Yun CH, Kwak M-J, Lee G-W, et al. Gut-Specific Delivery of T-Helper 17 Cells Reduces Obesity and Insulin Resistance in Mice. Gastroenterology (2017) 152:1998–2010. doi: 10.1053/j.gastro.2017.02.016
128. Garidou L, Pomié C, Klopp P, Waget A, Charpentier J, Aloulou M, et al. The Gut Microbiota Regulates Intestinal CD4 T Cells Expressing Rorγt and Controls Metabolic Disease. Cell Metab (2015) 22:100–12. doi: 10.1016/j.cmet.2015.06.001
129. Cavallari JF, Denou E, Foley KP, Khan WI, Schertzer JD. Different Th17 Immunity in Gut, Liver, and Adipose Tissues During Obesity: The Role of Diet, Genetics, and Microbes. Gut Microbes (2016) 7:82–9. doi: 10.1080/19490976.2015.1127481
130. Jiang W, Su J, Zhang X, Cheng X, Zhou J, Shi R, et al. Elevated Levels of Th17 Cells and Th17-Related Cytokines are Associated With Disease Activity in Patients With Inflammatory Bowel Disease. Inflammation Res Off J Eur Histamine Res Soc Al (2014) 63:943–50. doi: 10.1007/s00011-014-0768-7
131. Ueno A, Jeffery L, Kobayashi T, Hibi T, Ghosh S, Jijon H. Th17 Plasticity and its Relevance to Inflammatory Bowel Disease. J Autoimmun (2018) 87:38–49. doi: 10.1016/j.jaut.2017.12.004
132. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, Cytokine Profile and Function of Human Interleukin 17-Producing Helper T Cells. Nat Immunol (2007) 8:950–7. doi: 10.1038/ni1497
133. Blaschitz C, Raffatellu M. Th17 Cytokines and the Gut Mucosal Barrier. J Clin Immunol (2010) 30:196–203. doi: 10.1007/s10875-010-9368-7
134. Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are Coexpressed by Th17 Cells and Cooperatively Enhance Expression of Antimicrobial Peptides. J Exp Med (2006) 203:2271–9. doi: 10.1084/jem.20061308
135. Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 Links IL-22 Signaling in Intestinal Epithelial Cells to Mucosal Wound Healing. J Exp Med (2009) 206:1465–72. doi: 10.1084/jem.20082683
136. Parks OB, Pociask DA, Hodzic Z, Kolls JK, Good M. Interleukin-22 Signaling in the Regulation of Intestinal Health and Disease. Front Cell Dev Biol (2015) 3:85. doi: 10.3389/fcell.2015.00085
137. Keir ME, Yi T, Lu TT, Ghilardi N. The Role of IL-22 in Intestinal Health and Disease. J Exp Med (2020) 217:e20192195. doi: 10.1084/jem.20192195
138. Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, et al. Interleukin-22 Alleviates Metabolic Disorders and Restores Mucosal Immunity in Diabetes. Nature (2014) 514:237–41. doi: 10.1038/nature13564
139. Bianchi E, Rogge L. The IL-23/IL-17 Pathway in Human Chronic Inflammatory Diseases-New Insight From Genetics and Targeted Therapies. Genes Immun (2019) 20:415–25. doi: 10.1038/s41435-019-0067-y
140. Khader SA, Thirunavukkarasu S. The Tale of IL-12 and IL-23: A Paradigm Shift. J Immunol Baltim Md 1950 (2019) 202:629–30. doi: 10.4049/jimmunol.1801603
141. Martins LMS, Perez MM, Pereira CA, Costa FRC, Dias MS, Tostes RC, et al. Interleukin-23 Promotes Intestinal T Helper Type17 Immunity and Ameliorates Obesity-Associated Metabolic Syndrome in a Murine High-Fat Diet Model. Immunology (2018) 154:624–36. doi: 10.1111/imm.12946
142. Everard A, Lazarevic V, Gaïa N, Johansson M, Ståhlman M, Backhed F, et al. Microbiome of Prebiotic-Treated Mice Reveals Novel Targets Involved in Host Response During Obesity. ISME J (2014) 8:2116–30. doi: 10.1038/ismej.2014.45
143. Tomas J, Mulet C, Saffarian A, Cavin J-B, Ducroc R, Regnault B, et al. High-Fat Diet Modifies the PPAR-γ Pathway Leading to Disruption of Microbial and Physiological Ecosystem in Murine Small Intestine. Proc Natl Acad Sci USA (2016) 113:E5934–43. doi: 10.1073/pnas.1612559113
144. Olivares M, Neyrinck AM, Pötgens SA, Beaumont M, Salazar N, Cani PD, et al. The DPP-4 Inhibitor Vildagliptin Impacts the Gut Microbiota and Prevents Disruption of Intestinal Homeostasis Induced by a Western Diet in Mice. Diabetologia (2018) 61:1838–48. doi: 10.1007/s00125-018-4647-6
145. Puértolas-Balint F, Schroeder BO. Does an Apple a Day Also Keep the Microbes Away? The Interplay Between Diet, Microbiota, and Host Defense Peptides at the Intestinal Mucosal Barrier. Front Immunol (2020) 11:1164. doi: 10.3389/fimmu.2020.01164
146. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The Antibacterial Lectin RegIIIgamma Promotes the Spatial Segregation of Microbiota and Host in the Intestine. Science (2011) 334:255–8. doi: 10.1126/science.1209791
147. Frantz AL, Rogier EW, Weber CR, Shen L, Cohen DA, Fenton LA, et al. Targeted Deletion of MyD88 in Intestinal Epithelial Cells Results in Compromised Antibacterial Immunity Associated With Downregulation of Polymeric Immunoglobulin Receptor, Mucin-2, and Antibacterial Peptides. Mucosal Immunol (2012) 5:501–12. doi: 10.1038/mi.2012.23
148. Gao X, Xie Q, Kong P, Liu L, Sun S, Xiong B, et al. Polyphenol- and Caffeine-Rich Postfermented Pu-Erh Tea Improves Diet-Induced Metabolic Syndrome by Remodeling Intestinal Homeostasis in Mice. Infect Immun (2018) 86:e00601–17. doi: 10.1128/IAI.00601-17
149. Dalby MJ, Ross AW, Walker AW, Morgan PJ. Dietary Uncoupling of Gut Microbiota and Energy Harvesting From Obesity and Glucose Tolerance in Mice. Cell Rep (2017) 21:1521–33. doi: 10.1016/j.celrep.2017.10.056
150. Pellizzon MA, Ricci MR. The Common Use of Improper Control Diets in Diet-Induced Metabolic Disease Research Confounds Data Interpretation: The Fiber Factor. Nutr Metab (2018) 15:3. doi: 10.1186/s12986-018-0243-5
151. Shen W, Gaskins HR, McIntosh MK. Influence of Dietary Fat on Intestinal Microbes, Inflammation, Barrier Function and Metabolic Outcomes. J Nutr Biochem (2014) 25:270–80. doi: 10.1016/j.jnutbio.2013.09.009
152. Kaliannan K, Wang B, Li X-Y, Kim K-J, Kang JX. A Host-Microbiome Interaction Mediates the Opposing Effects of Omega-6 and Omega-3 Fatty Acids on Metabolic Endotoxemia. Sci Rep (2015) 5:11276. doi: 10.1038/srep11276
153. Murakami Y, Tanabe S, Suzuki T. High-Fat Diet-Induced Intestinal Hyperpermeability is Associated With Increased Bile Acids in the Large Intestine of Mice. J Food Sci (2016) 81:H216–22. doi: 10.1111/1750-3841.13166
154. Odenwald MA, Turner JR. Intestinal Permeability Defects: Is it Time to Treat? Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc (2013) 11:1075–83. doi: 10.1016/j.cgh.2013.07.001
155. Zuo L, Kuo W-T, Turner JR. Tight Junctions as Targets and Effectors of Mucosal Immune Homeostasis. Cell Mol Gastroenterol Hepatol (2020) 10:327–40. doi: 10.1016/j.jcmgh.2020.04.001
156. Suzuki T. Regulation of the Intestinal Barrier by Nutrients: The Role of Tight Junctions. Anim Sci J Nihon Chikusan Gakkaiho (2020) 91:e13357. doi: 10.1111/asj.13357
157. Ghosh S, Whitley CS, Haribabu B, Jala VR. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell Mol Gastroenterol Hepatol (2021) 11:1463–82. doi: 10.1016/j.jcmgh.2021.02.007
158. Suzuki T, Hara H. Dietary Fat and Bile Juice, But Not Obesity, are Responsible for the Increase in Small Intestinal Permeability Induced Through the Suppression of Tight Junction Protein Expression in LETO and OLETF Rats. Nutr Metab (2010) 7:19. doi: 10.1186/1743-7075-7-19
159. Bi S, Moran TH. Obesity in the Otsuka Long Evans Tokushima Fatty Rat: Mechanisms and Discoveries. Front Nutr (2016) 3:21. doi: 10.3389/fnut.2016.00021
160. Kim YS, Ho SB. Intestinal Goblet Cells and Mucins in Health and Disease: Recent Insights and Progress. Curr Gastroenterol Rep (2010) 12:319–30. doi: 10.1007/s11894-010-0131-2
161. Ermund A, Schütte A, Johansson MEV, Gustafsson JK, Hansson GC. Studies of Mucus in Mouse Stomach, Small Intestine, and Colon. I. Gastrointestinal Mucus Layers Have Different Properties Depending on Location as Well as Over the Peyer’s Patches. Am J Physiol Gastrointest Liver Physiol (2013) 305:G341–347. doi: 10.1152/ajpgi.00046.2013
162. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-Talk Between Akkermansia Muciniphila and Intestinal Epithelium Controls Diet-Induced Obesity. Proc Natl Acad Sci USA (2013) 110:9066–71. doi: 10.1073/pnas.1219451110
163. Cani PD, de Vos WM. Next-Generation Beneficial Microbes: The Case of Akkermansia Muciniphila. Front Microbiol (2017) 8:1765. doi: 10.3389/fmicb.2017.01765
164. Kleessen B, Hartmann L, Blaut M. Fructans in the Diet Cause Alterations of Intestinal Mucosal Architecture, Released Mucins and Mucosa-Associated Bifidobacteria in Gnotobiotic Rats. Br J Nutr (2003) 89:597–606. doi: 10.1079/BJN2002827
165. Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, et al. Mucus Enhances Gut Homeostasis and Oral Tolerance by Delivering Immunoregulatory Signals. Science (2013) 342:447–53. doi: 10.1126/science.1237910
166. Everard A, Plovier H, Rastelli M, Van Hul M, de Wouters d’Oplinter A, Geurts L, et al. Intestinal Epithelial N-Acylphosphatidylethanolamine Phospholipase D Links Dietary Fat to Metabolic Adaptations in Obesity and Steatosis. Nat Commun (2019) 10:457. doi: 10.1038/s41467-018-08051-7
167. Laurans L, Venteclef N, Haddad Y, Chajadine M, Alzaid F, Metghalchi S, et al. Genetic Deficiency of Indoleamine 2,3-Dioxygenase Promotes Gut Microbiota-Mediated Metabolic Health. Nat Med (2018) 24:1113–20. doi: 10.1038/s41591-018-0060-4
168. Whitt J, Woo V, Lee P, Moncivaiz J, Haberman Y, Denson L, et al. Disruption of Epithelial HDAC3 in Intestine Prevents Diet-Induced Obesity in Mice. Gastroenterology (2018) 155:501–13. doi: 10.1053/j.gastro.2018.04.017
169. Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of Histone-Deacetylase Activity by Short-Chain Fatty Acids and Some Polyphenol Metabolites Formed in the Colon. J Nutr Biochem (2008) 19:587–93. doi: 10.1016/j.jnutbio.2007.08.002
170. Sanchez HN, Moroney JB, Gan H, Shen T, Im JL, Li T, et al. B Cell-Intrinsic Epigenetic Modulation of Antibody Responses by Dietary Fiber-Derived Short-Chain Fatty Acids. Nat Commun (2020) 11:60. doi: 10.1038/s41467-019-13603-6
171. Martin-Gallausiaux C, Larraufie P, Jarry A, Béguet-Crespel F, Marinelli L, Ledue F, et al. Butyrate Produced by Commensal Bacteria Down-Regulates Indolamine 2,3-Dioxygenase 1 (IDO-1) Expression. via Dual Mech Hum Intestinal Epithelial Cells Front Immunol (2018) 9:2838. doi: 10.3389/fimmu.2018.02838
172. Cherayil BJ. Indoleamine 2,3-Dioxygenase in Intestinal Immunity and Inflammation. Inflamm Bowel Dis (2009) 15:1391–6. doi: 10.1002/ibd.20910
173. Metghalchi S, Ponnuswamy P, Simon T, Haddad Y, Laurans L, Clément M, et al. Indoleamine 2,3-Dioxygenase Fine-Tunes Immune Homeostasis in Atherosclerosis and Colitis Through Repression of Interleukin-10 Production. Cell Metab (2015) 22:460–71. doi: 10.1016/j.cmet.2015.07.004
174. Postal BG, Ghezzal S, Aguanno D, André S, Garbin K, Genser L, et al. AhR Activation Defends Gut Barrier Integrity Against Damage Occurring in Obesity. Mol Metab (2020) 39:101007. doi: 10.1016/j.molmet.2020.101007
175. Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S, et al. The Aryl Hydrocarbon Receptor Preferentially Marks and Promotes Gut Regulatory T Cells. Cell Rep (2017) 21:2277–90. doi: 10.1016/j.celrep.2017.10.114
176. Lamas B, Natividad JM, Sokol H. Aryl Hydrocarbon Receptor and Intestinal Immunity. Mucosal Immunol (2018) 11:1024–38. doi: 10.1038/s41385-018-0019-2
177. Natividad JM, Agus A, Planchais J, Lamas B, Jarry AC, Martin R, et al. Impaired Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is a Key Factor in Metabolic Syndrome. Cell Metab (2018) 28:737–749.e4. doi: 10.1016/j.cmet.2018.07.001
178. Lin Y-H, Luck H, Khan S, Schneeberger PHH, Tsai S, Clemente-Casares X, et al. Aryl Hydrocarbon Receptor Agonist Indigo Protects Against Obesity-Related Insulin Resistance Through Modulation of Intestinal and Metabolic Tissue Immunity. Int J Obes 2005 (2019) 43:2407–21. doi: 10.1038/s41366-019-0340-1
179. Biagioli M, Carino A, Fiorucci C, Annunziato G, Marchianò S, Bordoni M, et al. The Aryl Hydrocarbon Receptor (AhR) Mediates the Counter-Regulatory Effects of Pelargonidins in Models of Inflammation and Metabolic Dysfunctions. Nutrients (2019) 11:E1820. doi: 10.3390/nu11081820
180. Gatta-Cherifi B, Cota D. New Insights on the Role of the Endocannabinoid System in the Regulation of Energy Balance. Int J Obes 2005 (2016) 40:210–9. doi: 10.1038/ijo.2015.179
181. Veilleux A, Di Marzo V, Silvestri C. The Expanded Endocannabinoid System/Endocannabinoidome as a Potential Target for Treating Diabetes Mellitus. Curr Diabetes Rep (2019) 19:117. doi: 10.1007/s11892-019-1248-9
182. Gruden G, Barutta F, Kunos G, Pacher P. Role of the Endocannabinoid System in Diabetes and Diabetic Complications. Br J Pharmacol (2016) 173:1116–27. doi: 10.1111/bph.13226
183. Doris JM, Millar SA, Idris I, O’Sullivan SE. Genetic Polymorphisms of the Endocannabinoid System in Obesity and Diabetes. Diabetes Obes Metab (2019) 21:382–7. doi: 10.1111/dom.13504
184. Kopp W. How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab Syndr Obes Targets Ther (2019) 12:2221–36. doi: 10.2147/DMSO.S216791
185. Allaire JM, Crowley SM, Law HT, Chang S-Y, Ko H-J, Vallance BA. The Intestinal Epithelium: Central Coordinator of Mucosal Immunity. Trends Immunol (2018) 39:677–96. doi: 10.1016/j.it.2018.04.002
186. Alcock J, Maley CC, Aktipis CA. Is Eating Behavior Manipulated by the Gastrointestinal Microbiota? Evolutionary Pressures and Potential Mechanisms. BioEssays News Rev Mol Cell Dev Biol (2014) 36:940–9. doi: 10.1002/bies.201400071
187. Kim N, Yun M, Oh YJ, Choi H-J. Mind-Altering With the Gut: Modulation of the Gut-Brain Axis With Probiotics. J Microbiol Seoul Korea (2018) 56:172–82. doi: 10.1007/s12275-018-8032-4
188. Lumeng CN, Saltiel AR. Inflammatory Links Between Obesity and Metabolic Disease. J Clin Invest (2011) 121:2111–7. doi: 10.1172/JCI57132
189. Zhou W, Davis EA, Dailey MJ. Obesity, Independent of Diet, Drives Lasting Effects on Intestinal Epithelial Stem Cell Proliferation in Mice. Exp Biol Med Maywood NJ (2018) 243:826–35. doi: 10.1177/1535370218777762
190. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity Alters Gut Microbial Ecology. Proc Natl Acad Sci USA (2005) 102:11070–5. doi: 10.1073/pnas.0504978102
191. Di Marzo V, Goparaju SK, Wang L, Liu J, Bátkai S, Járai Z, et al. Leptin-Regulated Endocannabinoids are Involved in Maintaining Food Intake. Nature (2001) 410:822–5. doi: 10.1038/35071088
192. Boden G. Obesity, Insulin Resistance and Free Fatty Acids. Curr Opin Endocrinol Diabetes Obes (2011) 18:139–43. doi: 10.1097/MED.0b013e3283444b09
193. Corkey BE. Diabetes: Have We Got it All Wrong? Insulin Hypersecretion and Food Additives: Cause of Obesity and Diabetes? Diabetes Care (2012) 35:2432–7. doi: 10.2337/dc12-0825
194. Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, et al. Hyperglycemia Drives Intestinal Barrier Dysfunction and Risk for Enteric Infection. Science (2018) 359:1376–83. doi: 10.1126/science.aar3318
Keywords: intestinal immune system, intestinal barrier function, type 2 diabetes, obesity, leaky gut, intestinal epithelial cells
Citation: Riedel S, Pheiffer C, Johnson R, Louw J and Muller CJF (2022) Intestinal Barrier Function and Immune Homeostasis Are Missing Links in Obesity and Type 2 Diabetes Development. Front. Endocrinol. 12:833544. doi: 10.3389/fendo.2021.833544
Received: 11 December 2021; Accepted: 27 December 2021;
Published: 25 January 2022.
Edited by:
Katsumi Iizuka, Fujita Health University, JapanReviewed by:
Zan Huang, Nanjing Agricultural University, ChinaMaude Le Gall, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2022 Riedel, Pheiffer, Johnson, Louw and Muller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvia Riedel, c3lsdmlhLnJpZWRlbEBtcmMuYWMuemE=