 Zoran Gluvic1*
Zoran Gluvic1* Milan Obradovic2*
Milan Obradovic2* Alan J. Stewart3
Alan J. Stewart3 Magbubah Essack4
Magbubah Essack4 Samantha J. Pitt3
Samantha J. Pitt3 Vladimir Samardzic1
Vladimir Samardzic1 Sanja Soskic2
Sanja Soskic2 Takashi Gojobori4
Takashi Gojobori4 Esma R. Isenovic2
Esma R. Isenovic2- 1Clinic for Internal Medicine, Department of Endocrinology and Diabetes, Zemun Clinical Hospital, School of Medicine, University of Belgrade, Belgrade, Serbia
- 2Department of Radiobiology and Molecular Genetics, VINČA Institute of Nuclear Sciences - National Institute of the Republic of Serbia, University of Belgrade, Belgrade, Serbia
- 3School of Medicine, University of St Andrews, St Andrews, United Kingdom
- 4King Abdullah University of Science and Technology (KAUST), Computer, Electrical, and Mathematical Sciences and Engineering (CEMSE) Division, Computational Bioscience Research Center (CBRC), Thuwal, Saudi Arabia
Levothyroxine (LT4) is used to treat frequently encountered endocrinopathies such as thyroid diseases. It is regularly used in clinical (overt) hypothyroidism cases and subclinical (latent) hypothyroidism cases in the last decade. Suppressive LT4 therapy is also part of the medical regimen used to manage thyroid malignancies after a thyroidectomy. LT4 treatment possesses dual effects: substituting new-onset thyroid hormone deficiency and suppressing the local and distant malignancy spreading in cancer. It is the practice to administer LT4 in less-than-high suppressive doses for growth control of thyroid nodules and goiter, even in patients with preserved thyroid function. Despite its approved safety for clinical use, LT4 can sometimes induce side-effects, more often recorded with patients under treatment with LT4 suppressive doses than in unintentionally LT4-overdosed patients. Cardiac arrhythmias and the deterioration of osteoporosis are the most frequently documented side-effects of LT4 therapy. It also lowers the threshold for the onset or aggravation of cardiac arrhythmias for patients with pre-existing heart diseases. To improve the quality of life in LT4-substituted patients, clinicians often prescribe higher doses of LT4 to reach low normal TSH levels to achieve cellular euthyroidism. In such circumstances, the risk of cardiac arrhythmias, particularly atrial fibrillation, increases, and the combined use of LT4 and triiodothyronine further complicates such risk. This review summarizes the relevant available data related to LT4 suppressive treatment and the associated risk of cardiac arrhythmia.
1 Introduction
Thyroidectomy is a surgical procedure, performed either as a standard open surgery or as an alternative approach surgery, such as minimally invasive video-assisted thyroidectomy (MIVAT) or robot-assisted transaxillary thyroidectomy, aiming to remove all or part of the thyroid gland (1). The procedure is commonly used to treat a range of thyroid-related disorders, including thyroid cancer, hyperthyroidism goiters, and thyroid nodules that can be obstructive and cause swallowing or breathing difficulties (2). The introduction of MIVAT improved the treatment options for some thyroid conditions. Despite superiority regarding patients’ satisfaction with faster recovery and decreased complications associated with standard open thyroidectomy (neck pain, voice problems, anxiety), it is confirmed as a reliable procedure in only strictly indicated cases (1). It is not suitable for patients with thyroiditis, large multinodular goiters, locally invasive thyroid carcinoma, or the presence of lateral neck compartment malignant lymph nodes. It evolves as standard procedure in the carefully selected cases with low- and intermediate-risk differentiated thyroid carcinoma (3, 4).
The thyroid gland produces the iodine-containing thyroid hormones, triiodothyronine (T3) and thyroxine (T4) in response to thyroid stimulation hormone (TSH) and the peptide hormone calcitonin, which is primarily regulated by serum calcium levels (5, 6). Together, these hormones regulate a wide range of metabolic and cardiovascular processes, including basal metabolic rate, appetite, gut motility, nutrient absorption, rate and strength of heart contractions, breathing, and oxygen consumption (7). Thyroid hormones also play a developmental role; they are essential for cell growth, while cells of the developing brain are a major target for T3 and T4 (8).
When the whole thyroid is extirpated, such gland surgery is referred to as total thyroidectomy. Knowing that thyroid hormones are essential for life, it is necessary to permanently replace the resultant deficiency with thyroxine after total thyroidectomy. Without replacement, a patient will develop signs and symptoms of hypothyroidism. Standard treatment in such instances is the long-term prescription of the synthetic thyroid hormone levothyroxine (LT4, a manufactured form of T4). In cancer cases, LT4 treatment after thyroidectomy can have the added advantage of suppressing local and distant malignancies from spreading. However, the LT4 dose must be carefully optimized to avoid potential adverse effects such as weight loss, sweating, anxiety, insomnia, osteoporosis (increased bone fracture risk), and an increased heart rate. Thus, LT4 treatment in individuals that have suffered a recent heart attack is cautiously recommended (9). It is not surprising that some individuals experience cardiovascular complications following LT4 treatment as thyroid hormones regulate cardiac functioning. Indeed, increased thyroid hormone levels are associated with an increased risk of developing heart arrhythmias (10). Here we examine the relevant literature related to LT4 treatment after thyroidectomy and the associated risk of cardiac arrhythmias in such patients.
2 Levothyroxine (LT4): Structure, Brief History, Pharmacokinetics, Pharmacodynamics, Dosing Regimens
Levothyroxine is a synthetic version of the secreted thyroid hormone thyroxine (T4) that completely mimics all physiologic effects of T4 (Figure 1). LT4 is used as replacement therapy in primary-thyroidal, secondary-pituitary, and tertiary-hypothalamic hypothyroidism (11, 12). Despite T4 being naturally present as a racemic mixture of the levo and dextro forms, LT4 is produced as a levo-isomer due to its greater physiological activity than the dextro form (13, 14).
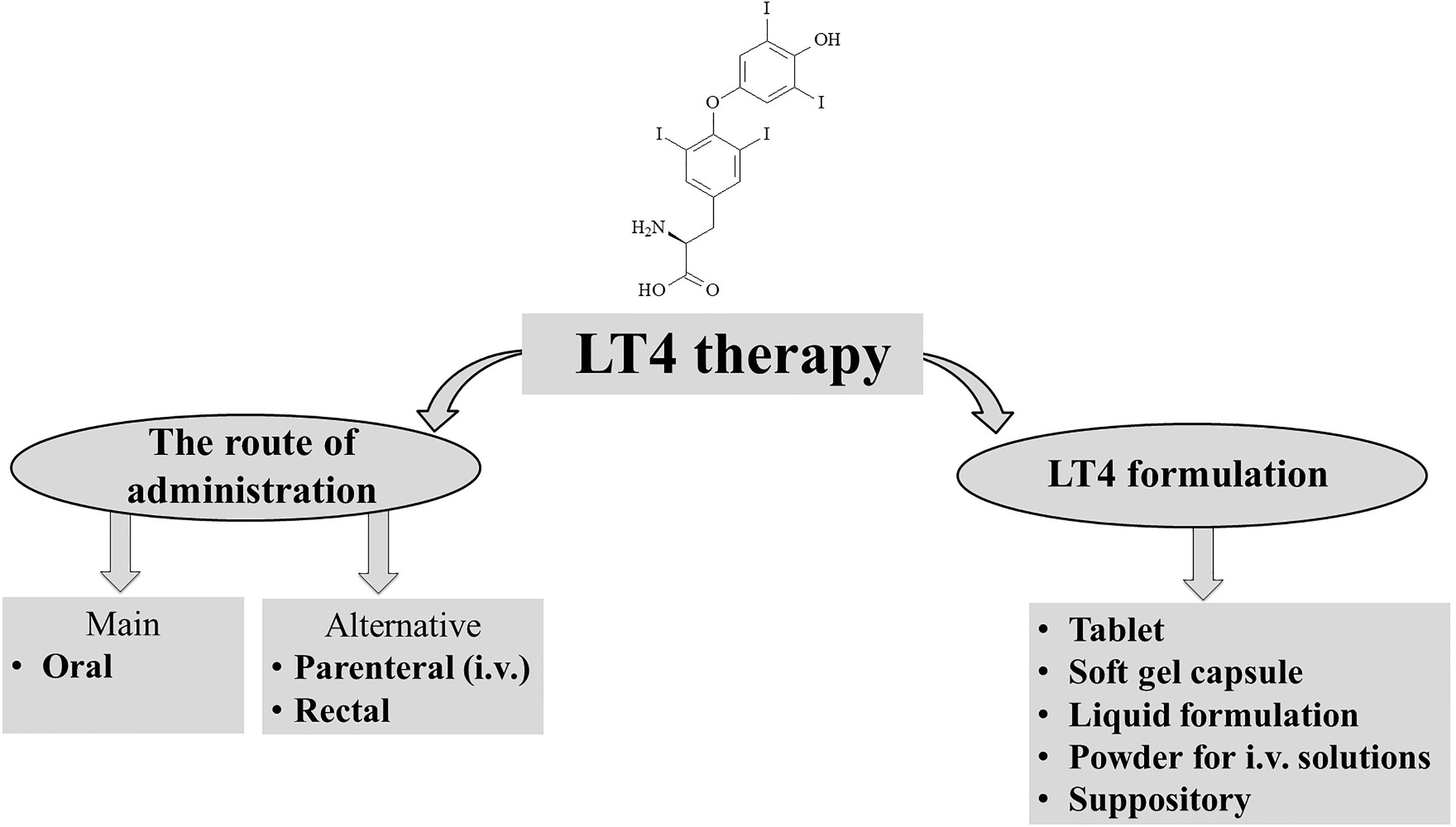
Figure 1 Levothyroxin (LT4) therapy.
The use of LT4 as a standard monotherapy came to the fore in the 1970s with evidence that T3 is predominantly produced by peripheral deiodination of T4. In patients treated with LT4 alone, thyroid function becomes normalized (15–17). Before that, combination therapy of synthetic LT4 and LT3 was the standard hormone replacement therapy in hypothyroid patients (18). Hypothyroidism treatment dates from the 6th century and Chinese medicine, where animal thyroid was used for therapy (17, 19). The same approach was applied in Europe but later in the 19th century (17, 19). In the 20th century, the discovery of thyroid hormones accelerated progress towards developing the current therapies (13, 17).
Adults with newly diagnosed hypothyroidism and without other complications receive an initial dose of 1.6 µg/kg/day for a few months. After dose modification, it is recommended to check the TSH level every 6-8 weeks (11, 12). In adult hypothyroid patients, hypothyroid patients with pre-existing heart diseases or >65 years old, an initial dose of 25 µg/day of LT4 is given, followed by an adjusted dose of 12.5 to 25 mcg every 4-6 weeks (11, 12). In patients with severe hypothyroidism or myxedema coma, the initial dose may be 200 to 400 µg administered via nasogastric tube or intravenously, followed by a daily dose of 1.2 µg/kg/day. Older patients or patients with heart disease are recommended to use lower doses (11, 12). It is recommended to decrease the LT4 dose for elderly patients. Regarding suppressive treatment for the control of thyroid nodule growth, the LT4 dose is individually tailored to maintain TSH levels as low-normal or at a partial suppressive level. In the high-risk patients following thyroidectomy for well-differentiated thyroid cancer, the estimated LT4 full suppressive dose is ≥2 µg/kg body weight (11, 12, 20–23).
The level of thyroid-stimulating hormone (TSH) in serum is used as an indicator of monitoring and adjusting the dose of LT4 therapy in hypothyroid patients, except in patients with secondary or tertiary hypothyroidism, where the level of free or total T4 is used as a marker for the success of the therapy (11, 12, 24). The standard monitoring procedure requires determining TSH levels 6-8 weeks after the initial treatment with LT4 (11, 12). After achieving the correct dose of LT4, the level of TSH is monitored firstly at 4-6 months, and after that, every 12 months (11, 12).
LT4 toxicity is rare, but adverse effects due to inappropriate dosage (over-or under-dose) can occur, especially in patients with pre-existing comorbidities such as cardiovascular disease, uncorrected adrenal insufficiency, and elderly patients (25).
Absorption of orally administered LT4 from the gastrointestinal tract mainly occurs in the small intestine rather than the stomach (26, 27). The absorbed LT4 varies from 60% to 80%, with the maximum concentration in circulation achieved 3 hours after administration in hypothyroid subjects and slightly faster in euthyroid subjects, approximately 2 hours (28–30). Several factors influence LT4 absorption, including deranged small intestine physiology (e.g. bowel resection reduced absorption), fasting increased absorption, while different foods, drugs, and supplements can also disturb LT4 absorption (30, 31). All these factors indicate the need for permanent monitoring in individual approaches in LT4 replacement (32). The half-life for T4 is ~7.5 days in patients with primary hypothyroidism, with a daily turnover rate of ~10% for T4 and 50–70% for T3 (33). Contrary, in euthyroid subjects, the half-life for T4 is 6.2 days, and a little faster turnover rate (34–36). In addition, the estimated T3 half-life is 1.4 days in hypothyroid patients and 1.0 days for euthyroid individuals (34–36). The values reported for T4 clearance are very close in hypothyroid subjects (approx. 0.04-0.06 l/h) and normal control individuals (0.05-0.06 l/h) (33, 37).
The liver is the primary site of LT4 degradation (38, 39). Although T4 is catabolized via several routes, the major pathway of T4 catabolism is sequential deiodination in the presence of deiodinase enzymes (38–40). The removal of iodine from carbon 5 of the outer ring of T4 converts it to T3, while removing iodine from the inner ring of T4 leads to the formation of inactive reverse T3 (rT3) (41, 42). T3 and rT3 originate from T4 at an ~1:1 ratio, and about 80% of T3 in circulation stems from peripheral T4 (43, 44). Subsequently, T3 can be converted to both diiodothyronine (T2) and iodothyronine (T1), and rT3 to both rT2 and rT1 (45, 46).
3 Thyroid Diseases and Conditions Associated With LT4 Use
Long-term use of levothyroxine (LT4) has been demonstrated to be effective and safe. Initially, LT4 was used only in thyroxin (T4) deficiency cases, but LT4 usage has evolved and includes substitution and suppressive therapy (17). The aim of LT4 treatment differs according to the indication of use. In hypothyroid subjects, the goal of LT4 substitution is to establish a euthyroid rank of TSH while improving quality of life. The goal of LT4 treatment in controlling nodule growth is a low normal TSH level but avoiding LT4 overdose (47). After surgical management of malignant disease, treatment goals are to obtain suppressed levels of both TSH and thyroglobulin (48). Lower doses of LT4 are necessary to achieve substitution goals in elderly patients, while higher LT4 doses are required in patients undergoing total thyroidectomy (49). Long-term use of LT4 substitution reduces the risk of bradycardia, which is often associated with hypothyroidism (50). In patients with low-normal or suppressed TSH levels, an increased risk of cardiac arrhythmias, primarily atrial fibrillation (AF), is observed (10, 51, 52), as is an increased risk of osteoporosis (53, 54) and overall mortality (55). However, Flynn et al. (52) showed that patients with TSH levels between 0.04 to 0.4 mIU/ml did not experience an increased risk of cardiovascular disease, arrhythmias, or osteoporotic fractures (56).
The management with LT4 after MIVAT, either substitutive or suppressive modality, depends on the pathology of the partially or entirely extirpated thyroid gland. Each LT4 management modality must be individually tailored to the patient. MIVAT treated benign diseases require standard LT4 substitution therapy, while malignant disease (i.e. differentiated thyroid cancer) requires partial or complete TSH suppression depending on assigned risk. In high-risk patients, a complete suppressive LT4 regimen (TSH <0.1 mIU/ml) is recommended opposite to a partial LT4 suppressive regimen in lower-risk patients (TSH 0.1-0.4 mIU/ml) (48, 57).
In addition to hypothyroidism classification according to clinical presentation (subclinical or latent and clinical or overt), there is another classification based on the level of lesion-induced thyroid dysfunction. Primary hypothyroidism is the most frequently encountered in clinical practice (49). Secondary (at pituitary level) hypothyroidism, tertiary (at hypothalamus level) hypothyroidism, and thyroid hormone resistance account for less than 1% of overall hypothyroidism. They are mostly presented with symptoms and signs of mild hypothyroidism and sometimes with local effects (i.e., symptoms and signs of increased intracranial pressure). Besides the clinical presentation of hypothyroidism, thyroid hormone resistance syndromes can also present as mental health problems (49, 58, 59). LT4-dose tapering is more complex in patients with secondary and tertiary hypothyroidism as the fT4 levels, which is the basis of how the quality of LT4 dosing is assessed, are less flexible (49, 60). Whether iatrogenic or disease-induced, all forms of hypothyroidism (subclinical or clinical) unequivocally accelerate atherosclerosis processes, which can contribute to increased cardiovascular morbidity and mortality (61–66).
The prevalence of overt hypothyroidism in the general population is 0.3–3.7% in the USA (67) and 0.2–5.3% in Europe (68) depending on the definition of hypothyroidism. Hypothyroidism is more common in people over 65 years, women, and Caucasians. Among these, the most common cause of primary hypothyroidism in iodine-sufficient areas is chronic autoimmune thyroiditis. Although thyroid anti-peroxidase antibodies are of diagnostic significance, they also present in about 11% of people with no thyroid disease (49, 69).
4 LT4 Replacement Therapy in Cardiovascular Patients
LT4 replacement therapy compensates for endogenous thyroxine deficiency, whether the disease’s latent (subclinical) or manifested (clinical) form or post-procedural hypothyroidism. Excessive LT4 substitution in cardiovascular patients can have serious side effects, such as AF and osteoporosis, especially in postmenopausal women and elderly patients (49, 52). These complications are frequently observed in Hashimoto’s thyroiditis patients due to LT4 over-supplementation (54). In cases of long-term TSH suppression, an increase in left ventricular mass and consequent diastolic dysfunction may occur, which further contributes to cardiovascular morbidity, especially in those patients with already diagnosed cardiovascular disease (70). A study by Petersen et al. (71), suggested that the prevalence of ischemic heart disease in people over 65 years of age with suppressed TSH levels on LT4 substitution was increased relative to the general population. Other studies suggest that atherosclerosis acceleration can occur during subclinical hypothyroidism development (72, 73).
It is known that thyroid hormones, when present in excess, affect the cardiovascular (CV) system by increasing heart rate, myocardial contractility, left ventricular mass, and the predisposition to supraventricular arrhythmias (74). Lipophilic T3 binds to the thyroid hormone receptor (TR) upon entry into the cardiomyocyte nucleus. Activation of TR results in stimulating gene transcription of the heavy alpha chain of myosin, calcium ATPase, Na/K-ATPase, beta 1 adrenergic receptor, and atrial natriuretic peptide (75–77). Genomic and non-genomic effects of thyroid hormones on cardiomyocytes lead to increased myocardial contractility, which, among other hemodynamic effects, results in increased heart rate, increased circulating volume, left ventricular volume, ejection fraction, and cardiac output (74).
Subclinical thyroidopathies can harm the cardiovascular system and manifest as an increase in CV morbidity and mortality by 20-80% (78, 79). Regardless of whether it is of endogenous or exogenous origin, in subclinical hyperthyroidism type 2 (TSH <0.1mIU/ml), the risk of AF presence is higher (HR 2.54 vs. 1.63) than in type 1 (TSH 0.1-0.4mIU/ml) (80, 81). Subclinical hyperthyroidism is associated with an increased left ventricular mass that increases ejection fraction (EF) and diastolic dysfunction (80, 82, 83). Clinical hyperthyroidism is associated with a 16% increased risk of major CV events commonly manifested as worsening heart failure, including high-output heart failure (84). It is noteworthy that an increased supraventricular ectopic activity often accompanies hyperthyroidism (85).
5 Pathophysiology of Cardiac Rhythm Disorders
Abnormal Ca2+ handling within cardiomyocytes is central to many types of arrhythmias. Arrhythmia-related contractions begin with external Ca2+ entering the cell’s cytosol through the L-type calcium channels to signal the sarcoplasmic reticulum to release more Ca2+ via ryanodine receptor channels (RyRs) - specifically the RyR2 isoform (86, 87). RyRs facilitate downstream calcium-dependent processes throughout the cell, e.g., actin-myosin contraction (88, 89). Inositol 1,4,5-trisphosphate receptor (IP3R) also responds to cellular cues to release Ca2+ from the SR (90). Ca2+ release from the SR initiates contraction in cardiac myocytes, but removing Ca2+ from the cytosol following systole is equally vital. In diastole, sarco/endoplasmic reticulum calcium ATPase translocates cytosolic Ca2+ back into the SR. Diastole is a sensitive time window for Ca2+ clearance, and improper Ca2+ clearance can have significant arrhythmogenic consequences (87). Mutations or covalent modifications of RyR channels can cause Ca2+ leakage across the SR membrane during diastole, promoting arrhythmias (91, 92). Our recent work shows that Zn2+ regulates the open probability of RyR2 in a manner that regulates beat-to-beat contractions in cardiomyocytes (93). This finding suggests that aberrant intracellular zinc homeostasis could contribute to arrhythmogenic events. Premature Ca2+ flux from the SR results in untimely depolarization events known as early afterdepolarizations (EADs) and delayed afterdepolarizations (DADs) (87). Ventricular arrhythmias can be caused by abnormal Ca2+ handling, electrolyte imbalance, and/or myocardial scarring (94). Physical barriers can also promote arrhythmia (e.g., fibrotic tissue) developing from ischemia and subsequent scarring (95, 96). Although myocardial scarring is the common feature in fatal ventricular arrhythmias, metabolic abnormalities may also play a significant role.
5.1 Cardiac Arrhythmias Associated With Thyroid Hormones
Cardiac arrhythmias are defined as irregular heartbeats and range in severity. They are generally defined by the affected region of the heart and the type of defect: supraventricular arrhythmias (brady- and tachyarrhythmias including atrial premature complexes) and ventricular arrhythmias. AF is one of the most common types of chronic arrhythmia, recognized in an electrocardiogram as an irregular P–R interval and a missing P wave. AF occurs more frequently in obese people and is the most common arrhythmia associated with abnormal thyroid hormone levels (97). It is a highly prevalent arrhythmia promoting heart failure, embolic stroke, and death (98). Even short, subclinical episodes of AF are associated with an increased risk of stroke (99). Paroxysmal and sustained or permanent forms of AF confer a significant clinical burden and worsens the patient’s quality of life. AF is the most common cardiac complication of hyperthyroidism and LT4-induced thyrotoxicosis (97, 100) (Figure 2). Sinus tachycardia and atrial flutter are also commonly associated with hyperthyroidism (101). AF in thyrotoxicosis is associated with significant mortality and morbidity resulting from embolic events (97). The risk factors for AF in patients with hyperthyroidism are similar to those in the general population. They include age, male sex, and a history of ischemic, congestive, or valvular heart diseases (102).
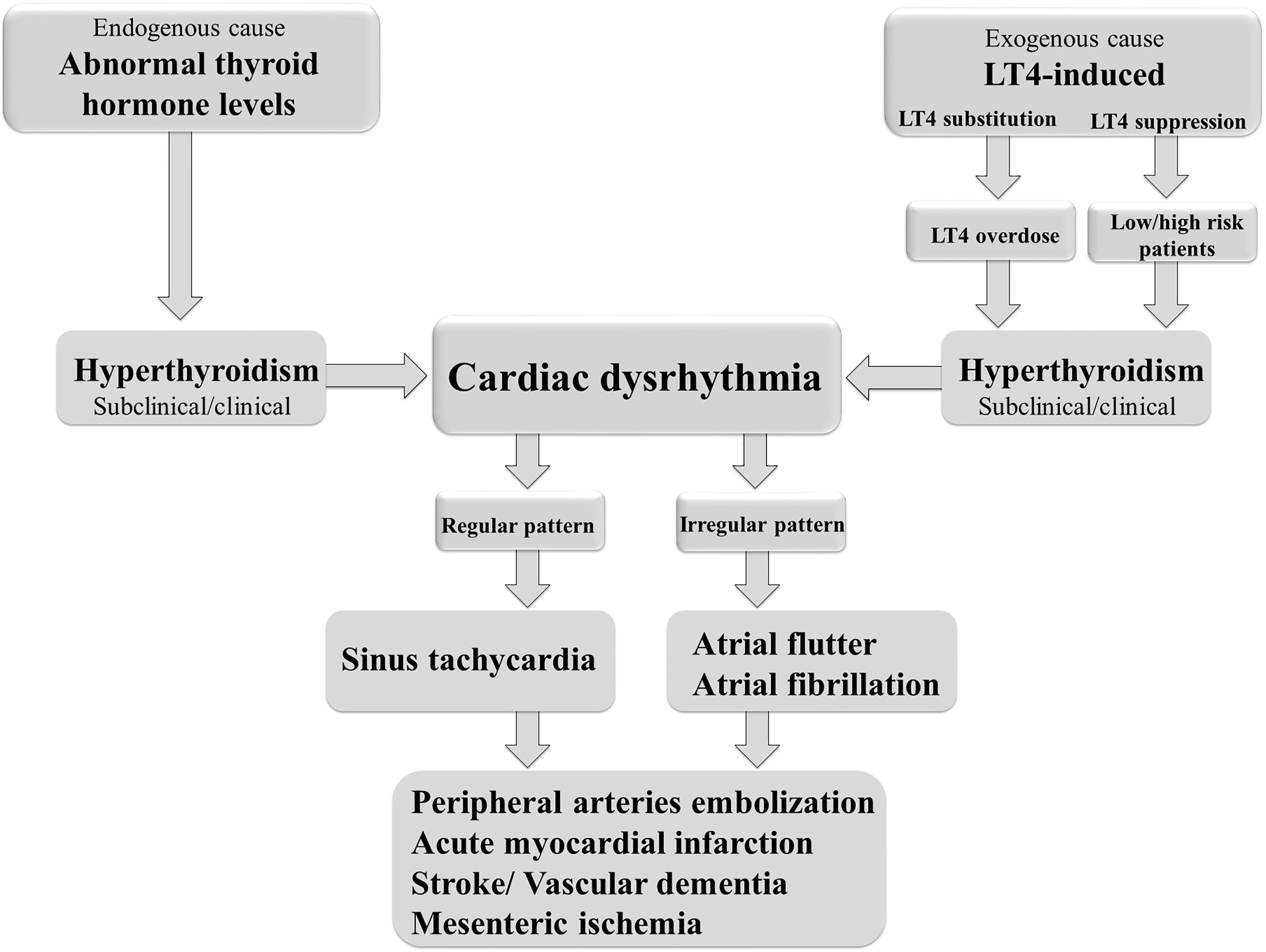
Figure 2 Thyroid dysfunction and the risk of cardiac arrhythmias.
AF occurs in up to 15% of patients with hyperthyroidism (103), compared with 4% in the general population (104). AF is more common in men and patients with T3 toxicosis (97). Also, subclinical hyperthyroidism is associated with an almost 3-fold increase in the risk of developing AF (103). Once initiated, AF alters the electrical and structural properties of the atria in a manner that affects its maintenance, increasing the risk of recurrence and can alter the response to antiarrhythmic drugs (97, 105). In addition, AF increases the risk of cerebrovascular stroke, peripheral embolization, and overall mortality (106–108). About 13-15% of individuals with newly developed AF have biochemical hyperthyroidism (103). The risk factors of developing AF in hyperthyroid individuals are age, pre-existing ischemic or valvular heart disease, or heart failure (85). Analysis of Framingham study results, related to the frequency of AF during a ten-year follow-up of patients >60 years, showed that AF occurred in 28% of those with subclinical hyperthyroidism, as opposed to 11% euthyroid patients. Furthermore, decreased TSH values, even with normal serum thyroid hormone values, were associated with a 3-fold increase in the frequency of AF (103). Heering et al. (Rotterdam study) showed a higher incidence of AF and sudden cardiac death in people >55 years with low normal TSH values and high normal FT4 levels (109). Studies using Mendeleev’s randomization to demonstrate an association between thyroid dysfunction and cardiovascular disease also reported an association between hyperthyroidism and AF (110, 111).
5.1.1 LT4 Therapy and Cardiac Arrhythmias
Effects of T3 such as acceleration of cardiac depolarization and repolarization, the shortening of the action potential duration, and the refractory period of the atrial myocardium and AV node are not observed in mono-LT4 therapy. Namely, the production of T3 in patients on LT4 substitution, primarily after thyroidectomy, is related to the peripheral deiodination of T4. Antithyroid therapy and beta-blockers affect heart rate control, even conversion to normal sinus rhythm in about 60% of patients (85, 112, 113). The most important factor influencing the conversion of AF to sinus rhythm in hyperthyroidism is the duration of AF (114). In cases where AF lasts for more than a year in the elderly and is resistant to antithyroid and beta-blocker therapy, AF is often associated with ischemic heart disease (115). In the cases where LT4 use (exogenous hyperthyroidism) induces TSH suppression, incidences of CV and arrhythmic events are increased compared to the general population (52). AF, provoked either by endogenous or exogenous hyperthyroidism, results in a significant increase in overall morbidity and mortality, mainly caused by the consequences of systemic embolism (97). Compared to euthyroid subjects, patients with suppressed TSH have increased sympathetic autonomic activity and decreased parasympathetic tone, resulting in increased heart rate variability and prolonged QT interval (116). The mentioned changes in the CV system in patients with subclinical or clinical hyperthyroidism may result in an increased frequency of cardiac arrhythmias, primarily AF (106), and a higher frequency of systolic and diastolic left ventricular dysfunction (74, 117).
In addition to AF, in patients on LT4 suppressive therapy, sinus tachycardia and shortening of the PR interval are often detected electrocardiographically as manifestations of accelerated atrioventricular conduction (118–120). The prolonged P wave is most often seen as a manifestation of impaired interatrial conduction, while a delay in intraventricular conduction often results in a right bundle branch block (121). Ventricular arrhythmias in patients on LT4 suppressive therapy are rare, and their presence should always arouse suspicion of pre-existing heart disease. It should be noted that the incidence of ventricular fibrillation (VF) attributed solely to the thyroid status imbalance is less frequent in humans compared to experimental animals, which are prone to both AF and VF in response to an excess of thyroid hormones (TH) (101, 122, 123).
Epidemiological studies have shown higher cardiovascular and all-cause mortality in patients with endogenous and exogenous hyperthyroidism than in the general population (55, 124, 125). Also, a study by Klein-Hesselink et al. reports 3.3 times higher mortality due to CV in patients with differentiated thyroid cancer (DTC). In comparison, mortality from all causes was 4.4 times higher than in the general population, regardless of age, gender, and cardiovascular risk factors. Furthermore, each 10-fold decrease in TSH levels increased the risk of cardiovascular death by 3-fold (126). A study by Suha et al. showed similar results, demonstrating a higher incidence of coronary heart disease (CVD) and cerebrovascular insult (CVI) in patients diagnosed with DTC, with the risk of CVD and CVI being directly proportional to the dose of LT4 administered (127).
Although the use of suppressive doses of LT4 can lead to a significant reduction in TN volume and diffuse atoxic goiter, the potential side effects of this therapy on the CV system and bone metabolism, especially in people over 60 years and postmenopausal women, are limiting factors for the routine use of this therapeutic approach (48). However, using supraphysiological LT4 doses in younger patients to suppress TSH levels reduces TN volume in one out of six patients without significant comorbidity (128).
5.1.2 LT4 Combined With T3 Therapy and Cardiac Arrhythmias
The combined use of LT4 and LT3 has multiplied over the last decade. The impression is that such increases in use are rarely observed in the group of patients that strictly require combination therapy, but mainly result from patients wishing to have experience with its use or pharmaceutical companies’ pressure that favors its use in patients unsatisfied with LT4 treatment alone. The rationale for using T4 and T3 combination therapy is that defects in deiodinase enzymes that convert thyroxine to triiodothyronine could lead to persistent symptoms in patients despite being biochemically well-regulated on LT4 monotherapy (129). A lack of more extensive studies pointed out LT3+LT4 positive effects on patients’ wellbeing and the increase in the previously insufficient functional capacity of peripheral tissues. The real benefit of the LT3+LT4 combination could be expected in a relatively small number of hypothyroid patients. ETA suggests the LT3+LT4 combination as an experimental 3-month trial observed by an experienced endocrinologist in LT4 well-compliant patients persistently presented with hypothyroidism-associated complaints, despite normal TSH levels. If there is no improvement in LT3+LT4 treated patients after 3 months of use, it should be discontinued (130). LT3+LT4 combination is not recommended in patients with cardiac arrhythmias, as increased free T3 could act pro-arrythmically in prone patients (130, 131).
Additionally, Regalbuto et al. did not show any advantage of combined LT3+LT4 over LT4 monotherapy suppression in totally thyroidectomized patients for thyroid cancer regarding improved wellbeing and peripheral tissue response. Even though thyroid function tests suggested subclinical hyperthyroidism, the clinical syndrome of LT3 and LT4 excess was not registered (132). Similarly, Tariq et al. did not find any additional risk for atrial fibrillation and cardiovascular disease in patients on combined LT3+LT4 therapy (133).
6 Cardiac Electrical Remodeling Associated With Hyperthyroidism and LT4 Treatment
Electrophysiological studies reveal that TH modifies f-channel conductance in sinoatrial cells, which changes the diastolic depolarization rate (134–136). This finding suggests a direct effect on myocardial membrane-related electrogenesis. Moreover, it provides the potential mechanism behind bradycardia and sinus tachycardia’s association with hyperthyroidism.
Cardiac arrhythmia classification assumes disturbance of rhythm results from abnormal 1) impulse initiation and/or 2) intercellular impulse propagation (137). The abnormal impulse initiation is associated with abnormal automaticity and/or a triggered activity (induced by EAD or DAD). On the other hand, abnormal intercellular impulse propagation refers to a block of conduction and re-entry. Re-entry occurs when the propagating impulse persists due to continuous activity, after normal activation of the heart, instead of dying out, that re-excites the heart after the refractory period has ended (138).
TH effects on the development of AF and VF are more complex than chronotropic effects. There are several potential mechanisms by which TH can trigger arrhythmicity in the heart. Such effects are likely exerted through the same mechanisms as hyperthyroidism. One such mechanism is its direct involvement in controlling the transcription of genes encoding ion channels and other proteins involved in signal transduction. For example, TH regulates mRNA transcription of voltage-activated K+ channel genes, including those encoding Kv4.3, Kv.4.2 (which contribute to the transient outward potassium current) and Kv1.4, Kv1.5, and Kv1.2 (which contribute to the ultra-rapid delayed rectifier potassium current) (139–143), and about nine ion channel α- and β-subunits (144). Hyperthyroidism was found to up-regulate the expression of Kv1.5 mRNA, particularly in the atrium of the heart (145). Sunagawa et al. demonstrated decreased L-type Ca2+ channel expression in the atria and Kv1.2 and Kv1.4 in both atrial and ventricular tissue in LT4-treated rats (136). Interestingly, despite this decrease in L-type Ca2+ channel expression, the L-type Ca2+ current increased (146–148), most likely due to the TH-induced transcriptional regulation of myocardial Ca2+ cycling proteins.
TH has the potential to influence Ca2+ levels in cardiomyocytes through multiple mechanisms, including regulating the expression of sarcoplasmic reticulum Ca2+-ATPase (SERCA2) and RyR2 [Ca2+ cycling proteins; (149)], and down-regulating Na+/Ca2+ exchanger and phospholamban (150–153). Increased Ca2+ influx and efflux rates also characterize hyperthyroidism in ventricular cells (154) most likely due to altered activation of sarcolemmal Ca2+ channels and SERCA2 activity (148, 155). There are also rapid non-genomic TH responses that modulate the activity of Ca2+ cycling proteins and have consequent effects on intracellular Ca2+ currents (147, 148, 150, 156). It is expected that LT4 therapy can induce aberrant Ca2+ homeostasis through altered Ca2+ handling similar to hyperthyroidism/TH activity.
Another mechanism by which clinical use of LT4 may trigger arrhythmia is the modulation of cardiac connexins. Connexin-43 (Cx43) is expressed in the ventricles and atria of the heart and is responsible for gap junction formation and thus the transmission of electrical signals between cells. TH receptors bind to the Cx43 promoter, indicating that TH can modify the expression of Cx43 mRNA synthesis (157). For example, it has been shown that Cx43 protein levels increase in TH-treated neonatal cultured cardiomyocytes (158). Also, in the atria and ventricles of hyperthyroid rats, phosphorylation of Cx43 isoforms is reduced, compared to untreated controls (159, 160). This is interesting as VF (159) and AF (101) susceptibility increases in T3-treated rats when myocardial Cx43 phosphorylation decreases. Also, TH-treated rat liver epithelial cells stimulate gap junctional communication and Cx43 mRNA expression (161).
7 Conclusions
Patients who have undergone thyroidectomy exhibit compromised production of thyroid hormones, which warrant hormone replacement therapy to avoid developing hypothyroidism. To date, the standard treatment in such instances is a long-term LT4 treatment regimen. LT4 treatment after thyroidectomy to treat cancer has the added advantage of suppressing the local and distant malignancy from spreading. However, each patient’s LT4 dose must be optimized to avoid potential side effects such as weight loss, sweating, anxiety, insomnia, osteoporosis (increased bone fracture risk), and an increased heart rate. Thus, LT4 treatment is not recommended in recent heart attack patients.
Nonetheless, patients with no known CV issues can experience CV complications following LT4 treatment, which is not surprising given the role of TH in regulating cardiac functioning. It is associated with an increased risk of developing heart arrhythmias, primarily AF, as well as osteoporosis. Although, studies do show that LT4 treated patients with TSH levels from 0.04 to 0.4 mIU/ml had not experienced an increased risk of cardiovascular disease, arrhythmias, or osteoporotic fracture. Also, LT4 doses in younger patients to suppress TSH levels reduce TN volume in one out of six patients without significant comorbidities. Nevertheless, suppressive LT4 doses can lead to potential side effects of this therapy on the CV system and bone metabolism, especially in people over 60 years and postmenopausal women. Thus, there are limiting factors for the routine use of this therapeutic approach, but improving the approach to individualized medicine in the at-risk population (implementing less excessive LT4 treatment regimens and monitoring more LT4 arrhythmia triggers) may reduce cardiac arrhythmia risk and mortality.
Author Contributions
ZG, MO, and EI designed, wrote and supervised the manuscript. AJS, ME, SJP, VS, and SS wrote the manuscript. AJS and TG critically revised the manuscript. All authors contributed to manuscript revision, and approved the submitted version.
Funding
This work was funded by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Contract No#451-03-9/2021-14/200017) and KAUST grant OSR#4129 (awarded to EI and VBB), which also supported MO. ME has been supported by the KAUST Office of Sponsored Research (OSR) Award no. FCC/1/1976-17-01, and TG by the King Abdullah University of Science and Technology (KAUST) Base Research Fund (BAS/1/1059-01-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work is part of the collaboration between the Department of Radiobiology and Molecular Genetics, “VINČA” Institute of Nuclear Sciences - National Institute of the Republic of Serbia, University of Belgrade, Belgrade, Serbia, and Computational Bioscience Research Center (CBRC) at King Abdullah University of Science and Technology (KAUST).
References
1. Rossi L, Materazzi G, Bakkar S, Miccoli P. Recent Trends in Surgical Approach to Thyroid Cancer. Front Endocrinol (Lausanne) (2021) 12:699805. doi: 10.3389/fendo.2021.699805
2. Dotter K. Obstructive Dysphagia and Positional Dyspnea: Can You Identify the Cause? J Adv Practitioner Oncol (2018) 9:441–4. doi: 10.6004/jadpro.2018.9.4.8
3. Kania R, Kania R, Hammami H, Vérillaud B, Blancal JP, Sauvaget E, et al. Minimally Invasive Video-Assisted Thyroidectomy: Tips and Pearls for the Surgical Technique. Ann Otol Rhinol Laryngol (2014) 123:409–14. doi: 10.1177/0003489414526845
4. Miccoli P, Fregoli L, Rossi L, Papini P, Ambrosini CE, Bakkar S, et al. Minimally Invasive Video-Assisted Thyroidectomy (MIVAT). Gland Surg (2020) 9:S1–5. doi: 10.21037/gs.2019.12.05
5. Colledge NR, Walker BR, Ralston S, Davidson S. Davidson’s Principles and Practice of Medicine. Edinburgh: Churchill Livingstone/Elsevier (2010).
6. Xie J, Guo J, Kanwal Z, Wu M, Lv X, Ibrahim NA, et al. Calcitonin and Bone Physiology: In Vitro, In Vivo, and Clinical Investigations. Int J Endocrinol (2020) 2020:3236828. doi: 10.1155/2020/3236828
7. Hall JE, Guyton AC. Guyton and Hall Textbook of Medical Physiology (2011). Available at: http://www.clinicalkey.com/dura/browse/bookChapter/3-s2.0-C20090602506.
8. Rovet JF. The Role of Thyroid Hormones for Brain Development and Cognitive Function. Endocr Dev (2014) 26:26–43. doi: 10.1159/0003
9. Eghtedari B, Correa R. Levothyroxine. [Updated 2020 Oct 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing (2021). Available at: https://www.ncbi.nlm.nih.gov/books/NBK539808/.
10. Baumgartner C, da Costa BR, Collet TH, Feller M, Floriani C, Bauer DC, et al. Thyroid Function Within the Normal Range, Subclinical Hypothyroidism, and the Risk of Atrial Fibrillation. Circulation (2017) 136:2100–16. doi: 10.1161/CIRCULATIONAHA.117.028753
11. Garber JR, Cobin RH, Gharib H, Hennessey JV, Klein I, Mechanick JI, et al. Clinical Practice Guidelines for Hypothyroidism in Adults: Cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocr Pract (2012) 18:988–1028. doi: 10.4158/EP12280.GL
12. Jonklaas J, Bianco AC, Bauer AJ, Burman KD, Cappola AR, Celi FS, et al. Guidelines for the Treatment of Hypothyroidism: Prepared by the American Thyroid Association Task Force on Thyroid Hormone Replacement. Thyroid (2014) 24:1670–751. doi: 10.1089/thy.2014.0028
13. Harington CR, Barger G. Chemistry of Thyroxine: Constitution and Synthesis of Thyroxine. Biochem J (1927) 21:169–83. doi: 10.1042/bj0210169
14. Benvenga S, Carlé A. Levothyroxine Formulations: Pharmacological and Clinical Implications of Generic Substitution. Adv Ther (2019) 36:59–71. doi: 10.1007/s12325-019-01079-1
15. Surks MI, Schadlow AR, Oppenheimer JH. A New Radioimmunoassay for Plasma L-Triiodothyronine: Measurements in Thyroid Disease and in Patients Maintained on Hormonal Replacement. J Clin Invest (1972) 51:3104–13. doi: 10.1172/JCI107137
16. Surks MI, Schadlow AR, Stock JM, Oppenheimer JH. Determination of Iodothyronine Absorption and Conversion of L-Thyroxine (T 4) to L-Triiodothyronine (T 3) Using Turnover Rate Techniques. J Clin Invest (1973) 52:805–11. doi: 10.1172/JCI107244
17. Mateo RCI, Hennessey JV. Thyroxine and Treatment of Hypothyroidism: Seven Decades of Experience. Endocrine (2019) 66:10–7. doi: 10.1007/s12020-019-02006-8
18. Taylor S, Kapur M, Adie R. Combined Thyroxine and Triiodothyronine for Thyroid Replacement Therapy. Br Med J (1970) 2:270–1. doi: 10.1136/bmj.2.5704.270
19. Slater S. The Discovery of Thyroid Replacement Therapy. Part 3: A Complete Transformation. J R Soc Med (2011) 104:100–6. doi: 10.1258/jrsm.2010.10k052
20. Pearce SH, Brabant G, Duntas LH, Monzani F, Peeters RP, Razvi S, et al. 2013 ETA Guideline: Management of Subclinical Hypothyroidism. Eur Thyroid J (2013) 2:215–28. doi: 10.1159/000356507
21. Okosieme O, Gilbert J, Abraham P, Boelaert K, Dayan C, Gurnell M, et al. Management of Primary Hypothyroidism: Statement by the British Thyroid Association Executive Committee. Clin Endocrinol (Oxf) (2016) 84:799–808. doi: 10.1111/cen.12824
22. Bekkering GE, Agoritsas T, Lytvyn L, Heen AF, Feller M, Moutzouri E, et al. Thyroid Hormones Treatment for Subclinical Hypothyroidism: A Clinical Practice Guideline. Bmj (2019) 365:l2006. doi: 10.1136/bmj.l2006
23. NICE. Thyroid Disease: Assessment and Management, in: NICE Guideline [NG145] (2019). Available at: https://www.nice.org.uk/guidance/ng145/chapter/Recommendations (Accessed Jul 2020).
24. Duntas LH, Jonklaas J. Levothyroxine Dose Adjustment to Optimise Therapy Throughout a Patient’s Lifetime. Adv Ther (2019) 36:30–46. doi: 10.1007/s12325-019-01078-2
25. Bartalena L, Bogazzi F, Martino E. Adverse Effects of Thyroid Hormone Preparations and Antithyroid Drugs. Drug Safety (1996) 15:53–63. doi: 10.2165/00002018-199615010-00004
26. Hays MT. Thyroid Hormone and the Gut. Endocr Res (1988) 14:203–24. doi: 10.3109/07435808809032986
27. Hays MT. Localization of Human Thyroxine Absorption. Thyroid (1991) 1:241–8. doi: 10.1089/thy.1991.1.241
28. Read DG, Hays MT, Hershman JM. Absorption of Oral Thyroxine in Hypothyroid and Normal Man. J Clin Endocrinol Metab (1970) 30:798–9. doi: 10.1210/jcem-30-6-798
29. Hasselström K, Siersbaek-Nielsen K, Lumholtz IB, Faber J, Kirkegaard C, Friis T. The Bioavailability of Thyroxine and 3,5,3’-Triiodothyronine in Normal Subjects and in Hyper- and Hypothyroid Patients. Acta Endocrinol (Copenh) (1985) 110:483–6. doi: 10.1530/acta.0.1100483
30. Benvenga S, Bartolone L, Squadrito S, Lo Giudice F, Trimarchi F. Delayed Intestinal Absorption of Levothyroxine. Thyroid (1995) 5:249–53. doi: 10.1089/thy.1995.5.249
31. Stone E, Leiter LA, Lambert JR, Silverberg JD, Jeejeebhoy KN, Burrow GN. L-Thyroxine Absorption in Patients With Short Bowel. J Clin Endocrinol Metab (1984) 59:139–41. doi: 10.1210/jcem-59-1-139
32. Hoermann R, Midgley JEM, Larisch R, Dietrich JW. Individualised Requirements for Optimum Treatment of Hypothyroidism: Complex Needs, Limited Options. Drugs Context (2019) 8:212597–7. doi: 10.7573/dic.212597
33. Nicoloff JT, Low JC, Dussault JH, Fisher DA. Simultaneous Measurement of Thyroxine and Triiodothyronine Peripheral Turnover Kinetics in Man. J Clin Invest (1972) 51:473–83. doi: 10.1172/JCI106835
34. Braverman LE, Vagenakis A, Downs P, Foster AE, Sterling K, Ingbar SH. Effects of Replacement Doses of Sodium L-Thyroxine on the Peripheral Metabolism of Thyroxine and Triiodothyronine in Man. J Clin Invest (1973) 52:1010–7. doi: 10.1172/JCI107265
35. Colucci P, Yue CS, Ducharme M, Benvenga S. A Review of the Pharmacokinetics of Levothyroxine for the Treatment of Hypothyroidism. Eur Endocrinol (2013) 9:40–7. doi: 10.17925/EE.2013.09.01.40
36. Dayan C, Panicker V. Management of Hypothyroidism With Combination Thyroxine (T4) and Triiodothyronine (T3) Hormone Replacement in Clinical Practice: A Review of Suggested Guidance. Thyroid Res (2018) 11:1. doi: 10.1186/s13044-018-0045-x
37. Pittman CS, Shimizu T, Burger A, Chambers JB Jr. The Nondeiodinative Pathways of Thyroxine Metabolism: 3,5,3’,5-Tetraiodothyroacetic Acid Turnover in Normal and Fasting Human Subjects. J Clin Endocrinol Metab (1980) 50:712–6. doi: 10.1210/jcem-50-4-712
38. Mol JA, Visser TJ. Rapid and Selective Inner Ring Deiodination of Thyroxine Sulfate by Rat Liver Deiodinase. Endocrinology (1985) 117:8–12. doi: 10.1210/endo-117-1-8
39. Rutgers M, Heusdens FA, Visser TJ. Deiodination of Iodothyronine Sulfamates by Type I Iodothyronine Deiodinase of Rat Liver. Endocrinology (1991) 129:1375–81. doi: 10.1210/endo-129-3-1375
40. Galton VA, Larsen PR, Berry MJ. The Deiodinases: Their Identification and Cloning of Their Genes. Endocrinology (2021) 162:1–9. doi: 10.1210/endocr/bqab005
41. Braverman LE, Ingbar SH, Sterling K. Conversion of Thyroxine (T4) to Triiodothyronine (T3) in Athyreotic Human Subjects. J Clin Invest (1970) 49:855–64. doi: 10.1172/JCI106304
42. Sterling K, Brenner MA, Newman ES. Conversion of Thyroxine to Triiodothyronine in Normal Human Subjects. Science (1970) 169:1099–100. doi: 10.1126/science.169.3950.1099
43. Pittman CS, Chambers JB Jr., Read VH. The Extrathyroidal Conversion Rate of Thyroxine to Triiodothyronine in Normal Man. J Clin Invest (1971) 50:1187–96. doi: 10.1172/JCI106596
44. Inada M, Kasagi K, Kurata S, Kazama Y, Takayama H, Torizuka K, et al. Estimation of Thyroxine and Triiodothyronine Distribution and of the Conversion Rate of Thyroxine to Triiodothyronine in Man. J Clin Invest (1975) 55:1337–48. doi: 10.1172/JCI108053
45. Engler D, Merkelbach U, Steiger G, Burger AG. The Monodeiodination of Triiodothyronine and Reverse Triiodothyronine in Man: A Quantitative Evaluation of the Pathway by the Use of Turnover Rate Techniques. J Clin Endocrinol Metab (1984) 58:49–61. doi: 10.1210/jcem-58-1-49
46. Burger AG, O’Connell M, Scheidegger K, Woo R, Danforth E Jr. Monodeiodination of Triiodothyronine and Reverse Triiodothyronine During Low and High Calorie Diets. J Clin Endocrinol Metab (1987) 65:829–35. doi: 10.1210/jcem-65-5-829
47. Biondi B, Cooper DS. Thyroid Hormone Suppression Therapy. Endocrinol Metab Clin North Am (2019) 48:227–37. doi: 10.1016/j.ecl.2018.10.008
48. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for Adult Patients With Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid (2016) 26:1–133. doi: 10.1089/thy.2015.0020
49. Chaker L, Bianco AC, Jonklaas J, Peeters RP. Hypothyroidism. Lancet (2017) 390:1550–62. doi: 10.1016/S0140-6736(17)30703-1
50. Hylander B, Kennebäck G, Rosenqvist U, Edhag O. Long-Term ECG Recordings in Thyroxine-Substituted Hypothyroid Subjects. Acta Med Scand (1987) 222:429–32. doi: 10.1111/j.0954-6820.1987.tb10960.x
51. Cappola AR, Fried LP, Arnold AM, Danese MD, Kuller LH, Burke GL, et al. Thyroid Status, Cardiovascular Risk, and Mortality in Older Adults. Jama (2006) 295:1033–41. doi: 10.1001/jama.295.9.1033
52. Flynn RW, Bonellie SR, Jung RT, MacDonald TM, Morris AD, Leese GP. Serum Thyroid-Stimulating Hormone Concentration and Morbidity From Cardiovascular Disease and Fractures in Patients on Long-Term Thyroxine Therapy. J Clin Endocrinol Metab (2010) 95:186–93. doi: 10.1210/jc.2009-1625
53. Uzzan B, Campos J, Cucherat M, Nony P, Boissel JP, Perret GY. Effects on Bone Mass of Long Term Treatment With Thyroid Hormones: A Meta-Analysis. J Clin Endocrinol Metab (1996) 81:4278–89. doi: 10.1210/jcem.81.12.8954028
54. Mincer DL, Jialal I. Hashimoto Thyroiditis. In: StatPearls. Treasure Island (FL: StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC (2021).
55. Parle JV, Maisonneuve P, Sheppard MC, Boyle P, Franklyn JA. Prediction of All-Cause and Cardiovascular Mortality in Elderly People From One Low Serum Thyrotropin Result: A 10-Year Cohort Study. Lancet (2001) 358:861–5. doi: 10.1016/S0140-6736(01)06067-6
56. Toft AD, Beckett GJ. Thyroid Function Tests and Hypothyroidism. Bmj (2003) 326:295–6. doi: 10.1136/bmj.326.7384.295
57. Perros P, Boelaert K, Colley S, Evans C, Evans RM, Gerrard Ba G, et al. Guidelines for the Management of Thyroid Cancer. Clin Endocrinol (Oxf) (2014) 81 Suppl 1:1–122. doi: 10.1111/cen.12515
58. Barbesino G, Sluss PM, Caturegli P. Central Hypothyroidism in a Patient With Pituitary Autoimmunity: Evidence for TSH-Independent Thyroid Hormone Synthesis. J Clin Endocrinol Metab (2012) 97:345–50. doi: 10.1210/jc.2011-1591
59. Pappa T, Refetoff S. Resistance to Thyroid Hormone Beta: A Focused Review. Front Endocrinol (Lausanne) (2021) 12:656551. doi: 10.3389/fendo.2021.656551
60. Persani L. Clinical Review: Central Hypothyroidism: Pathogenic, Diagnostic, and Therapeutic Challenges. J Clin Endocrinol Metab (2012) 97:3068–78. doi: 10.1210/jc.2012-1616
61. Kharbanda RK, Walton B, Allen M, Klein N, Hingorani AD, MacAllister RJ, et al. Prevention of Inflammation-Induced Endothelial Dysfunction: A Novel Vasculo-Protective Action of Aspirin. Circulation (2002) 105:2600–4. doi: 10.1161/01.CIR.0000017863.52347.6C
62. Taddei S, Caraccio N, Virdis A, Dardano A, Versari D, Ghiadoni L, et al. Impaired Endothelium-Dependent Vasodilatation in Subclinical Hypothyroidism: Beneficial Effect of Levothyroxine Therapy. J Clin Endocrinol Metab (2003) 88:3731–7. doi: 10.1210/jc.2003-030039
63. Schuett H, Luchtefeld M, Grothusen C, Grote K, Schieffer B. How Much Is Too Much? Interleukin-6 and Its Signalling in Atherosclerosis. Thromb Haemost (2009) 102:215–22. doi: 10.1160/TH09-05-0297
64. Türemen EE, Çetinarslan B, Şahin T, Cantürk Z, Tarkun İ. Endothelial Dysfunction and Low Grade Chronic Inflammation in Subclinical Hypothyroidism Due to Autoimmune Thyroiditis. Endocr J (2011) 58:349–54. doi: 10.1507/endocrj.K10E-333
65. Obradovic M, Gluvic Z, Sudar-Milovanovic E, Panic A, Trebaljevac J, Bajic V, et al. Nitric Oxide as a Marker for Levo-Thyroxine Therapy in Subclinical Hypothyroid Patients. Curr Vasc Pharmacol (2016) 14:266–70. doi: 10.2174/1570161114666160208143537
66. Gluvic ZM, Obradovic MM, Sudar-Milovanovic EM, Zafirovic SS, Radak DJ, Essack MM, et al. Regulation of Nitric Oxide Production in Hypothyroidism. BioMed Pharmacother (2020) 124:109881. doi: 10.1016/j.biopha.2020.109881
67. Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado Thyroid Disease Prevalence Study. Arch Intern Med (2000) 160:526–34. doi: 10.1001/archinte.160.4.526
68. Garmendia Madariaga A, Santos Palacios S, Guillén-Grima F, Galofré JC. The Incidence and Prevalence of Thyroid Dysfunction in Europe: A Meta-Analysis. J Clin Endocrinol Metab (2014) 99:923–31. doi: 10.1210/jc.2013-2409
69. Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, et al. T(4), and Thyroid Antibodies in the United States Population 1998 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab (2002) 87:489–99. doi: 10.1210/jcem.87.2.8182
70. Fazio S, Biondi B, Carella C, Sabatini D, Cittadini A, Panza N, et al. Diastolic Dysfunction in Patients on Thyroid-Stimulating Hormone Suppressive Therapy With Levothyroxine: Beneficial Effect of Beta-Blockade. J Clin Endocrinol Metab (1995) 80:2222–6. doi: 10.1210/jcem.80.7.7608283
71. Petersen K, Bengtsson C, Lapidus L, Lindstedt G, Nyström E. Morbidity, Mortality, and Quality of Life for Patients Treated With Levothyroxine. Arch Intern Med (1990) 150:2077–81. doi: 10.1001/archinte.1990.00390210063015
72. Razvi S, Ingoe L, Keeka G, Oates C, McMillan C, Weaver JU. The Beneficial Effect of L-Thyroxine on Cardiovascular Risk Factors, Endothelial Function, and Quality of Life in Subclinical Hypothyroidism: Randomized, Crossover Trial. J Clin Endocrinol Metab (2007) 92:1715–23. doi: 10.1210/jc.2006-1869
73. Razvi S, Jabbar A, Pingitore A, Danzi S, Biondi B, Klein I, et al. Thyroid Hormones and Cardiovascular Function and Diseases. J Am Coll Cardiol (2018) 71:1781–96. doi: 10.1016/j.jacc.2018.02.045
74. Klein I, Danzi S. Thyroid Disease and the Heart. Circulation (2007) 116:1725–35. doi: 10.1161/CIRCULATIONAHA.106.678326
75. Izumo S, Nadal-Ginard B, Mahdavi V. All Members of the MHC Multigene Family Respond to Thyroid Hormone in a Highly Tissue-Specific Manner. Science (1986) 231:597–600. doi: 10.1126/science.3945800
76. Morkin E. Regulation of Myosin Heavy Chain Genes in the Heart. Circulation (1993) 87:1451–60. doi: 10.1161/01.CIR.87.5.1451
77. Zarain-Herzberg A, Marques J, Sukovich D, Periasamy M. Thyroid Hormone Receptor Modulates the Expression of the Rabbit Cardiac Sarco (Endo) Plasmic Reticulum Ca(2+)-ATPase Gene. J Biol Chem (1994) 269:1460–7. doi: 10.1016/S0021-9258(17)42279-4
78. Larsen PR. Thyroid-Pituitary Interaction: Feedback Regulation of Thyrotropin Secretion by Thyroid Hormones. N Engl J Med (1982) 306:23–32. doi: 10.1056/NEJM198201073060107
79. Jabbar A, Pingitore A, Pearce SH, Zaman A, Iervasi G, Razvi S. Thyroid Hormones and Cardiovascular Disease. Nat Rev Cardiol (2017) 14:39–55. doi: 10.1038/nrcardio.2016.174
80. Biondi B, Palmieri EA, Lombardi G, Fazio S. Effects of Thyroid Hormone on Cardiac Function: The Relative Importance of Heart Rate, Loading Conditions, and Myocardial Contractility in the Regulation of Cardiac Performance in Human Hyperthyroidism. J Clin Endocrinol Metab (2002) 87:968–74. doi: 10.1210/jcem.87.3.8302
81. Collet TH, Gussekloo J, Bauer DC, den Elzen WP, Cappola AR, Balmer P, et al. Subclinical Hyperthyroidism and the Risk of Coronary Heart Disease and Mortality. Arch Intern Med (2012) 172:799–809. doi: 10.1001/archinternmed.2012.402
82. Shapiro LE, Sievert R, Ong L, Ocampo EL, Chance RA, Lee M, et al. Minimal Cardiac Effects in Asymptomatic Athyreotic Patients Chronically Treated With Thyrotropin-Suppressive Doses of L-Thyroxine. J Clin Endocrinol Metab (1997) 82:2592–5. doi: 10.1210/jc.82.8.2592
83. Biondi B, Fazio S, Palmieri EA, Carella C, Panza N, Cittadini A, et al. Left Ventricular Diastolic Dysfunction in Patients With Subclinical Hypothyroidism. J Clin Endocrinol Metab (1999) 84:2064–7. doi: 10.1210/jcem.84.6.5733
84. Selmer C, Olesen JB, Hansen ML, von Kappelgaard LM, Madsen JC, Hansen PR, et al. Subclinical and Overt Thyroid Dysfunction and Risk of All-Cause Mortality and Cardiovascular Events: A Large Population Study. J Clin Endocrinol Metab (2014) 99:2372–82. doi: 10.1210/jc.2013-4184
85. Bielecka-Dabrowa A, Mikhailidis DP, Rysz J, Banach M. The Mechanisms of Atrial Fibrillation in Hyperthyroidism. Thyroid Res (2009) 2:4. doi: 10.1186/1756-6614-2-4
86. Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb Perspect Biol (2010) 2:a003996. doi: 10.1101/cshperspect.a003996
87. Wakili R, Voigt N, Kääb S, Dobrev D, Nattel S. Recent Advances in the Molecular Pathophysiology of Atrial Fibrillation. J Clin Invest (2011) 121:2955–68. doi: 10.1172/JCI46315
88. Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, et al. Structural Basis for the Gating Mechanism of the Type 2 Ryanodine Receptor Ryr2. Science (2016) 354:aah5324. doi: 10.1126/science.aah5324
89. Santulli G, Lewis D, des Georges A, Marks AR, Frank J. Ryanodine Receptor Structure and Function in Health and Disease. Subcell Biochem (2018) 87:329–52. doi: 10.1007/978-981-10-7757-9_11
90. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-Dependent Protein Kinase Modulates Cardiac Ryanodine Receptor Phosphorylation and Sarcoplasmic Reticulum Ca2+ Leak in Heart Failure. Circ Res (2005) 97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89
91. Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A, et al. Calcium Leak Through Ryanodine Receptors Leads to Atrial Fibrillation in 3 Mouse Models of Catecholaminergic Polymorphic Ventricular Tachycardia. Circ Res (2012) 111:708–17. doi: 10.1161/CIRCRESAHA.112.273342
92. Uehara A, Murayama T, Yasukochi M, Fill M, Horie M, Okamoto T, et al. Extensive Ca2+ Leak Through K4750Q Cardiac Ryanodine Receptors Caused by Cytosolic and Luminal Ca2+ Hypersensitivity. J Gen Physiol (2017) 149:199–218. doi: 10.1085/jgp.201611624
93. Woodier J, Rainbow RD, Stewart AJ, Pitt SJ. Intracellular Zinc Modulates Cardiac Ryanodine Receptor-Mediated Calcium Release. J Biol Chem (2015) 290:17599–610. doi: 10.1074/jbc.M115.661280
94. Skogestad J, Aronsen JM. Hypokalemia-Induced Arrhythmias and Heart Failure: New Insights and Implications for Therapy. Front Physiol (2018) 9:1500–0. doi: 10.3389/fphys.2018.01500
95. Homan EA, Reyes MV, Hickey KT, Morrow JP. Clinical Overview of Obesity and Diabetes Mellitus as Risk Factors for Atrial Fibrillation and Sudden Cardiac Death. Front Physiol (2018) 9:1847. doi: 10.3389/fphys.2018.01847
96. Gowen BH, Reyes MV, Joseph LC, Morrow JP. Mechanisms of Chronic Metabolic Stress in Arrhythmias. Antioxid (Basel Switzerland) (2020) 9:1012. doi: 10.3390/antiox9101012
97. Staffurth JS, Gibberd MC, Fui SN. Arterial Embolism in Thyrotoxicosis With Atrial Fibrillation. Br Med J (1977) 2:688–90. doi: 10.1136/bmj.2.6088.688
98. Packer M. Effect of Catheter Ablation on Pre-Existing Abnormalities of Left Atrial Systolic, Diastolic, and Neurohormonal Functions in Patients With Chronic Heart Failure and Atrial Fibrillation. Eur Heart J (2019) 40:1873–9. doi: 10.1093/eurheartj/ehz284
99. Mahajan R, Perera T, Elliott AD, Twomey DJ, Kumar S, Munwar DA, et al. Subclinical Device-Detected Atrial Fibrillation and Stroke Risk: A Systematic Review and Meta-Analysis. Eur Heart J (2018) 39:1407–15. doi: 10.1093/eurheartj/ehx731
100. Gong IY, Atzema CL, Lega IC, Austin PC, Na Y, Rochon PA, et al. Levothyroxine Dose and Risk of Atrial Fibrillation: A Nested Case-Control Study. Am Heart J (2021) 232:47–56. doi: 10.1016/j.ahj.2020.09.016
101. Tribulova N, Knezl V, Shainberg A, Seki S, Soukup T. Thyroid Hormones and Cardiac Arrhythmias. Vasc Pharmacol (2010) 52:102–12. doi: 10.1016/j.vph.2009.10.001
102. Frost L, Vestergaard P, Mosekilde L. Hyperthyroidism and Risk of Atrial Fibrillation or Flutter: A Population-Based Study. Arch Intern Med (2004) 164:1675–8. doi: 10.1001/archinte.164.15.1675
103. Sawin CT, Geller A, Wolf PA, Belanger AJ, Baker E, Bacharach P, et al. Low Serum Thyrotropin Concentrations as a Risk Factor for Atrial Fibrillation in Older Persons. New Engl J Med (1994) 331:1249–52. doi: 10.1056/NEJM199411103311901
104. Forfar JC, Miller HC, Toft AD. Occult Thyrotoxicosis: A Correctable Cause of “Idiopathic” Atrial Fibrillation. Am J Cardiol (1979) 44:9–12. doi: 10.1016/0002-9149(79)90243-1
105. Mariscalco G, Klersy C, Zanobini M, Banach M, Ferrarese S, Borsani P, et al. Atrial Fibrillation After Isolated Coronary Surgery Affects Late Survival. Circulation (2008) 118:1612–8. doi: 10.1161/CIRCULATIONAHA.108.777789
106. Wolf PA, Abbott RD, Kannel WB. Atrial Fibrillation as an Independent Risk Factor for Stroke: The Framingham Study. Stroke (1991) 22:983–8. doi: 10.1161/01.STR.22.8.983
107. Frost L, Engholm G, Johnsen S, Møller H, Henneberg EW, Husted S. Incident Thromboembolism in the Aorta and the Renal, Mesenteric, Pelvic, and Extremity Arteries After Discharge From the Hospital With a Diagnosis of Atrial Fibrillation. Arch Intern Med (2001) 161:272–6. doi: 10.1001/archinte.161.2.272
108. Wong CL, Tam HV, Fok CV, Lam PE, Fung LM. Thyrotoxic Atrial Fibrillation: Factors Associated With Persistence and Risk of Ischemic Stroke. J Thyroid Res (2017) 2017:4259183. doi: 10.1155/2017/4259183
109. Heeringa J, van der Kuip DA, Hofman A, Kors JA, van Herpen G, Stricker BH, et al. Prevalence, Incidence and Lifetime Risk of Atrial Fibrillation: The Rotterdam Study. Eur Heart J (2006) 27:949–53. doi: 10.1093/eurheartj/ehi825
110. Ellervik C, Roselli C, Christophersen IE, Alonso A, Pietzner M, Sitlani CM, et al. Assessment of the Relationship Between Genetic Determinants of Thyroid Function and Atrial Fibrillation: A Mendelian Randomization Study. JAMA Cardiol (2019) 4:144–52. doi: 10.1001/jamacardio.2018.4635
111. Larsson SC, Allara E, Mason AM, Michaëlsson K, Burgess S. Thyroid Function and Dysfunction in Relation to 16 Cardiovascular Diseases. Circ Genom Precis Med (2019) 12:e002468. doi: 10.1161/CIRCGEN.118.002468
112. Nakazawa HK, Sakurai K, Hamada N, Momotani N, Ito K. Management of Atrial Fibrillation in the Post-Thyrotoxic State. Am J Med (1982) 72:903–6. doi: 10.1016/0002-9343(82)90850-6
113. Biondi B, Bartalena L, Cooper DS, Hegedüs L, Laurberg P, Kahaly GJ. The 2015 European Thyroid Association Guidelines on Diagnosis and Treatment of Endogenous Subclinical Hyperthyroidism. Eur Thyroid J (2015) 4:149–63. doi: 10.1159/000438750
114. Banach M, Mariscalco G, Ugurlucan M, Mikhailidis DP, Barylski M, Rysz J. The Significance of Preoperative Atrial Fibrillation in Patients Undergoing Cardiac Surgery: Preoperative Atrial Fibrillation–Still Underestimated Opponent. Europace (2008) 10:1266–70. doi: 10.1093/europace/eun273
115. Shimizu T, Koide S, Noh JY, Sugino K, Ito K, Nakazawa H. Hyperthyroidism and the Management of Atrial Fibrillation. Thyroid (2002) 12:489–93. doi: 10.1089/105072502760143863
116. Liu C, Lv H, Li Q, Fu S, Tan J, Wang C, et al. Effect of Thyrotropin Suppressive Therapy on Heart Rate Variability and QT Dispersion in Patients With Differentiated Thyroid Cancer. Med (Baltimore) (2020) 99:e21190. doi: 10.1097/MD.0000000000021190
117. Ikram H. The Nature and Prognosis of Thyrotoxic Heart Disease. Q J Med (1985) 54:19–28. doi: 10.1093/oxfordjournals.qjmed.a067825
118. Iskandar SB, Jordan RM, Peiris AN. Electrocardiographic Abnormalities and Endocrine Diseases–Part a: Hyperthyroidism. Tenn Med (2007) 100, 47:50.
119. Zhang Y, Post WS, Cheng A, Blasco-Colmenares E, Tomaselli GF, Guallar E. Thyroid Hormones and Electrocardiographic Parameters: Findings From the Third National Health and Nutrition Examination Survey. PloS One (2013) 8:e59489. doi: 10.1371/journal.pone.0059489
120. Baladi IH, Rai AA, Ahmed SM. ECG Changes in Patients With Primary Hyperthyroidism. Pan Afr Med J (2018) 30:246. doi: 10.11604/pamj.2018.30.246.12244
121. von Olshausen K, Bischoff S, Kahaly G, Mohr-Kahaly S, Erbel R, Beyer J, et al. Cardiac Arrhythmias and Heart Rate in Hyperthyroidism. Am J Cardiol (1989) 63:930–3. doi: 10.1016/0002-9149(89)90142-2
122. Szeiffová Bačová B, Egan Beňová T, Viczenczová C, Soukup T, Rauchová H, Pavelka S, et al. Cardiac Connexin-43 and PKC Signaling in Rats With Altered Thyroid Status Without and With Omega-3 Fatty Acids Intake. Physiol Res (2016) 65 Suppl 1:S77–90. doi: 10.33549/physiolres.933413
123. Bačová BS, Vinczenzová C, Žurmanová J, Kašparová D, Knezl V, Beňová TE, et al. Altered Thyroid Status Affects Myocardial Expression of Connexin-43 and Susceptibility of Rat Heart to Malignant Arrhythmias That Can be Partially Normalized by Red Palm Oil Intake. Histochem Cell Biol (2017) 147:63–73. doi: 10.1007/s00418-016-1488-6
124. Osman F, Franklyn JA, Holder RL, Sheppard MC, Gammage MD. Cardiovascular Manifestations of Hyperthyroidism Before and After Antithyroid Therapy: A Matched Case-Control Study. J Am Coll Cardiol (2007) 49:71–81. doi: 10.1016/j.jacc.2006.08.042
125. Brandt F, Green A, Hegedüs L, Brix TH. A Critical Review and Meta-Analysis of the Association Between Overt Hyperthyroidism and Mortality. Eur J Endocrinol (2011) 165:491–7. doi: 10.1530/EJE-11-0299
126. Klein Hesselink EN, Klein Hesselink MS, de Bock GH, Gansevoort RT, Bakker SJ, Vredeveld EJ, et al. Long-Term Cardiovascular Mortality in Patients With Differentiated Thyroid Carcinoma: An Observational Study. J Clin Oncol (2013) 31:4046–53. doi: 10.1200/JCO.2013.49.1043
127. Suh B, Shin DW, Park Y, Lim H, Yun JM, Song SO, et al. Increased Cardiovascular Risk in Thyroid Cancer Patients Taking Levothyroxine: A Nationwide Cohort Study in Korea. Eur J Endocrinol (2019) 180:11–20. doi: 10.1530/EJE-18-0551
128. Hegedüs L, Bonnema SJ, Bennedbaek FN. Management of Simple Nodular Goiter: Current Status and Future Perspectives. Endocr Rev (2003) 24:102–32. doi: 10.1210/er.2002-0016
129. Michaelsson LF, Medici BB, la Cour JL, Selmer C, Røder M, Perrild H, et al. Treating Hypothyroidism With Thyroxine/Triiodothyronine Combination Therapy in Denmark: Following Guidelines or Following Trends? Eur Thyroid J (2015) 4:174–80. doi: 10.1159/000437262
130. Wiersinga WM, Duntas L, Fadeyev V, Nygaard B, Vanderpump MP. 2012 ETA Guidelines: The Use of L-T4 + L-T3 in the Treatment of Hypothyroidism. Eur Thyroid J (2012) 1:55–71. doi: 10.1159/000339444
131. Bahn Chair RS, Burch HB, Cooper DS, Garber JR, Greenlee MC, Klein I, et al. Hyperthyroidism and Other Causes of Thyrotoxicosis: Management Guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid (2011) 21:593–646. doi: 10.1089/thy.2010.0417
132. Regalbuto C, Maiorana R, Alagona C, Paola RD, Cianci M, Alagona G, et al. Effects of Either LT4 Monotherapy or LT4/LT3 Combined Therapy in Patients Totally Thyroidectomized for Thyroid Cancer. Thyroid (2007) 17:323–31. doi: 10.1089/thy.2006.0084
133. Tariq A, Wert Y, Cheriyath P, Joshi R. Effects of Long-Term Combination LT4 and LT3 Therapy for Improving Hypothyroidism and Overall Quality of Life. South Med J (2018) 111:363–9. doi: 10.14423/SMJ.0000000000000823
134. Renaudon B, Lenfant J, Decressac S, Bois P. Thyroid Hormone Increases the Conductance Density of F-Channels in Rabbit Sino-Atrial Node Cells. Recept Channels (2000) 7:1–8.
135. Sun Z-Q, Ojamaa K, Nakamura TY, Artman M, Klein I, Coetzee WA. Thyroid Hormone Increases Pacemaker Activity in Rat Neonatal Atrial Myocytes. J Mol Cell Cardiol (2001) 33:811–24. doi: 10.1006/jmcc.2001.1353
136. Sunagawa M, Yamakawa M, Shimabukuro M, Higa N, Takasu N, Kosugi T. Electrophysiologic Characteristics of Atrial Myocytes in Levo-Thyroxine-Treated Rats. Thyroid (2005) 15:3–11. doi: 10.1089/thy.2005.15.3
137. Hoffman BF, Rosen MR. Cellular Mechanisms for Cardiac Arrhythmias. Circ Res (1981) 49:1–15. doi: 10.1161/01.RES.49.1.1
138. Tribulova N, Kurahara LH, Hlivak P, Hirano K, Szeiffova Bacova B. Pro-Arrhythmic Signaling of Thyroid Hormones and Its Relevance in Subclinical Hyperthyroidism. Int J Mol Sci (2020) 21:2844. doi: 10.3390/ijms21082844
139. Shimoni Y, Severson D, Giles W. Thyroid Status and Diabetes Modulate Regional Differences in Potassium Currents in Rat Ventricle. J Physiol (1995) 488(Pt 3):673–88. doi: 10.1113/jphysiol.1995.sp020999
140. Shimoni Y, Fiset C, Clark RB, Dixon JE, McKinnon D, Giles WR. Thyroid Hormone Regulates Postnatal Expression of Transient K+ Channel Isoforms in Rat Ventricle. J Physiol (1997) 500(Pt 1):65–73. doi: 10.1113/jphysiol.1997.sp021999
141. Wickenden AD, Kaprielian R, Parker TG, Jones OT, Backx PH. Effects of Development and Thyroid Hormone on K+ Currents and K+ Channel Gene Expression in Rat Ventricle. J Physiol (1997) 504(Pt 2):271–86. doi: 10.1111/j.1469-7793.1997.271be.x
142. Ojamaa K, Sabet A, Kenessey A, Shenoy R, Klein I. Regulation of Rat Cardiac Kv1.5 Gene Expression by Thyroid Hormone Is Rapid and Chamber Specific. Endocrinology (1999) 140:3170–6. doi: 10.1210/endo.140.7.6776
143. Ma ML, Watanabe K, Watanabe H, Hosaka Y, Komura S, Aizawa Y, et al. Different Gene Expression of Potassium Channels by Thyroid Hormone and an Antithyroid Drug Between the Atrium and Ventricle of Rats. Jpn Heart J (2003) 44:101–10. doi: 10.1536/jhj.44.101
144. Le Bouter S, Demolombe S, Chambellan A, Bellocq C, Aimond F, Toumaniantz G, et al. Microarray Analysis Reveals Complex Remodeling of Cardiac Ion Channel Expression With Altered Thyroid Status: Relation to Cellular and Integrated Electrophysiology. Circ Res (2003) 92:234–42. doi: 10.1161/01.RES.0000053185.75505.8E
145. Watanabe H, Ma M, Washizuka T, Komura S, Yoshida T, Hosaka Y, et al. Thyroid Hormone Regulates mRNA Expression and Currents of Ion Channels in Rat Atrium. Biochem Biophys Res Commun (2003) 308:439–44. doi: 10.1016/S0006-291X(03)01420-7
146. Mager S, Palti Y, Binah O. Mechanism of Hyperthyroidism-Induced Modulation of the L-Type Ca2+ Current in Guinea Pig Ventricular Myocytes. Pflugers Arch (1992) 421:425–30. doi: 10.1007/BF00370252
147. Gøtzsche LB. L-Triiodothyronine Acutely Increases Ca2+ Uptake in the Isolated, Perfused Rat Heart. Changes in L-Type Ca2+ Channels and Beta-Receptors During Short- and Long-Term Hyper- and Hypothyroidism. Eur J Endocrinol (1994) 130:171–9. doi: 10.1530/eje.0.1300171
148. Watanabe H, Washizuka T, Komura S, Yoshida T, Hosaka Y, Hatada K, et al. Genomic and Non-Genomic Regulation of L-Type Calcium Channels in Rat Ventricle by Thyroid Hormone. Endocr Res (2005) 31:59–70. doi: 10.1080/07435800500229227
149. Campanha F, Perone D, de Campos D, Luvizotto R, de Síbio M, de Oliveira M, et al. Thyroxine Increases Serca2 and Ryr2 Gene Expression in Heart Failure Rats With Euthyroid Sick Syndrome. Arch Endocrinol Metab (2013) 60:582–6. doi: 10.1590/2359-3997000000208
150. Connelly TJ, el-Hayek R, Sukhareva M, Coronado R. L-Thyroxine Activates the Intracellular Ca2+ Release Channel of Skeletal Muscle Sarcoplasmic Reticulum. Biochem Mol Biol Int (1994) 32:441–8.
151. Kiss E, Jakab G, Kranias EG, Edes I. Thyroid Hormone-Induced Alterations in Phospholamban Protein Expression. Regulatory Effects on Sarcoplasmic Reticulum Ca2+ Transport and Myocardial Relaxation. Circ Res (1994) 75:245–51. doi: 10.1161/01.RES.75.2.245
152. Cernohorský J, Kolár F, Pelouch V, Korecky B, Vetter R. Thyroid Control of Sarcolemmal Na+/Ca2+ Exchanger and SR Ca2+-ATPase in Developing Rat Heart. Am J Physiol (1998) 275:H264–73. doi: 10.1152/ajpheart.1998.275.1.H264
153. Shenoy R, Klein I, Ojamaa K. Differential Regulation of SR Calcium Transporters by Thyroid Hormone in Rat Atria and Ventricles. Am J Physiol Heart Circ Physiol (2001) 281:H1690–6. doi: 10.1152/ajpheart.2001.281.4.H1690
154. Kim D, Smith TW. Effects of Thyroid Hormone on Calcium Handling in Cultured Chick Ventricular Cells. J Physiol (1985) 364:131–49. doi: 10.1113/jphysiol.1985.sp015735
155. Mylotte KM, Cody V, Davis PJ, Davis FB, Blas SD, Schoenl M. Milrinone and Thyroid Hormone Stimulate Myocardial Membrane Ca2+-ATPase Activity and Share Structural Homologies. Proc Natl Acad Sci USA (1985) 82:7974–8. doi: 10.1073/pnas.82.23.7974
156. Segal J, Masalha S, Schwalb H, Merin G, Borman JB, Uretzky G. Acute Effect of Thyroid Hormone in the Rat Heart: Role of Calcium. J Endocrinol (1996) 149:73–80. doi: 10.1677/joe.0.1490073
157. Stock A, Sies H. Thyroid Hormone Receptors Bind to an Element in the Connexin43 Promoter. Biol Chem (2000) 381:973–9. doi: 10.1515/BC.2000.120
158. Tribulova N, Shneyvays V, Mamedova LK, Moshel S, Zinman T, Shainberg A, et al. Enhanced Connexin-43 and Alpha-Sarcomeric Actin Expression in Cultured Heart Myocytes Exposed to Triiodo-L-Thyronine. J Mol Histol (2004) 35:463–70. doi: 10.1023/B:HIJO.0000045945.16046.b5
159. Lin H, Mitasikova M, Dlugosova K, Okruhlicova L, Imanaga I, Ogawa K, et al. Thyroid Hormones Suppress Epsilon-PKC Signalling, Down-Regulate Connexin-43 and Increase Lethal Arrhythmia Susceptibility in Non-Diabetic and Diabetic Rat Hearts. J Physiol Pharmacol (2008) 59:271–85.
160. Mitašíková M, Lin H, Soukup T, Imanaga I, Tribulová N. Diabetes and Thyroid Hormones Affect Connexin-43 and PKC-Epsilon Expression in Rat Heart Atria. Physiol Res (2009) 58:211–7. doi: 10.33549/physiolres.931425
Keywords: levothyroxine, thyroid, hypothyroidism, cardiac arrhythmias, osteoporosis, replacement therapy
Citation: Gluvic Z, Obradovic M, Stewart AJ, Essack M, Pitt SJ, Samardzic V, Soskic S, Gojobori T and Isenovic ER (2021) Levothyroxine Treatment and the Risk of Cardiac Arrhythmias – Focus on the Patient Submitted to Thyroid Surgery. Front. Endocrinol. 12:758043. doi: 10.3389/fendo.2021.758043
Received: 13 August 2021; Accepted: 11 October 2021;
Published: 04 November 2021.
Edited by:
Melvin Khee Shing Leow, Tan Tock Seng Hospital, SingaporeReviewed by:
Laura Sterian Ward, State University of Campinas, BrazilWeiping Teng, First Affiliated Hospital of Anhui Medical University, China
Copyright © 2021 Gluvic, Obradovic, Stewart, Essack, Pitt, Samardzic, Soskic, Gojobori and Isenovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zoran Gluvic, em9yYW5nbHV2aWNAeWFob28uY29t; Milan Obradovic, b2JyYWRvdmljbWlsYW5AaG90bWFpbC5jb20=