 Linan Gong†
Linan Gong† Zanzan Wang
Zanzan Wang Zhiguo Zhang
Zhiguo Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 03 November 2021
Sec. Cellular Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.751020
This article is part of the Research TopicThe Impact of Adipose Tissue Dysfunction on Cardiovascular and Renal DiseaseView all 11 articles
Sestrin2 is a highly conserved protein that can be induced under a variety of stress conditions, including DNA damage, oxidative stress, endoplasmic reticulum (ER) stress, and metabolic stress. Numerous studies have shown that the AMP-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) signaling pathway has a crucial role in the regulation of metabolism. Sestrin2 regulates metabolism via a number of pathways, including activation of AMPK, inhibition of the mTOR complex 1 (mTORC1), activation of mTOR complex 2 (mTORC2), inhibition of ER stress, and promotion of autophagy. Therefore, modulation of Sestrin2 activity may provide a potential therapeutic target for the prevention of metabolic diseases such as insulin resistance, diabetes, obesity, non-alcoholic fatty liver disease, and myocardial ischemia/reperfusion injury. In this review, we examined the regulatory relationship between Sestrin2 and the AMPK/mTOR signaling pathway and the effects of Sestrin2 on energy metabolism.
Sestrins, a family of evolutionarily highly conserved stress-induced proteins, are upregulated under oxidative stress, genotoxic stress, hypoxia, and other stress conditions (1). As stress-induced metabolic modulators, Sestrins help cells adapt to diverse stress stimuli by activating catabolic reactions, stopping anabolic activities, and initiating cell repair mechanisms, to maintain cell homeostasis (2). In mammals, there are three members of the sestrin family, Sestrins1–3, which are encoded by three independent genes, while only one Sestrin ortholog has been identified in invertebrates (3–5). Sestrin1, also referred to as p53-activated gene 26 (PA26), was first identified by Velasco-Miguel et al. and is a growth arrest and DNA damage-inducing gene (5). In 2002, Sestrin2, also known as hypoxia-inducible gene 95 (Hi95), was reported by Budanov et al., is highly homologous to Sestrin1, and can be induced by prolonged hypoxia and DNA damage (6, 7). Sestrin3 is directly activated by forkhead box O (FOXO) transcriptional factors (8). These three Sestrin proteins have some shared mechanisms of action, including, but not limited to, inhibiting the production of reactive oxygen species (ROS), activating AMPK, and inhibiting mTORC1 (4, 9). However, there is growing evidence that the three Sestrins behave differently and promote different biological effects via AMPK/mTOR signaling because they are distributed differently in different organs (10, 11). To our knowledge, Sestrin1 has an antioxidant function that can activate the AMPK signal pathway while inhibiting the mechanistic target of the mTORC1 signal pathway (12). Furthermore, Sestrin1 can be activated in a p53-dependent manner under oxidative stress in skeletal muscle, kidney, brain, and lung (7). Recent studies suggest that Sestrin1 inhibits oxidized low-density lipoprotein-induced activation of NOD-like receptor protein 3 (NLRP3) inflammasome in macrophages in a murine atherosclerosis model (12). What is even more interesting is that in multiple mouse models, Sestrin1 influences plasma cholesterol and regulates cholesterol biosynthesis (13). Among these members, Sestrin2 is the most intensively studied since its discovery in 2002. As a p53 target gene, Sestrin2 (SESN2) can exert cytoprotective functions in the lung, heart, liver, adipose, and kidney through activation of AMPK and inhibition of mTORC1 (6, 11, 14, 15). Furthermore, Sestrin2 is able to suppress nitric oxide release and the production of classical pro-inflammatory cytokines in cardiomyocytes (16). Sestrin3 can inhibit mTORC1 and maintain the activity of protein kinase B (AKT) via activating the AMPK/TSC1/2 signaling pathway (8). Sestrin3 is largely expressed in skeletal muscle, intestine, adipose, colon, and brain (17).
Increasing evidence suggests that that Sestrin2 has two main biological functions. Through its own oxidoreductase activity or activation of antioxidant damage related pathways, Sestrin2 can reduce the damage of oxidative stress to protect cells and tissues and maintain redox homeostasis (18, 19). In addition to its redox activity, Sestrin2 can also inhibit the mammalian target of mTORC1 through AMPK-dependent or -independent pathways (20). These two activities of human Sestrin2 (hSestrin2) are supported through its two separate domains, which were determined from X-ray crystallographic studies. A recent study of the X-ray crystal structure of hSestrin2 showed that it consists of well-conserved Sesn-A, Sesn-B, and Sesn-C domains (11). Sesn-A and Sesn-C are structurally similar but functionally distinct from each other (21). Sestrin2 controls ROS and mTOR signaling through two separate functional domains (22). While Sesn-A reduces alkyl hydroperoxide radicals through its helix–turn–helix oxidoreductase motif, Sesn-C modifies this motif to accommodate physical interactions with GAP activity towards Rags 2 (GATOR2) and subsequent inhibition of mTORC1 (21, 23). Sestrin2 has a significant role in the inhibition of ER stress and the activation of autophagy and is considered to improve obesity-induced and age-related pathologies by inhibiting mTORC1 (15, 24). Therefore, Sestrin2 may represent a novel class of potential targets for the therapeutic intervention of metabolic diseases. In this review, we discuss the regulatory relationship between Sestrin2 and AMPK/mTOR signaling and the effects of Sestrin2 on energy metabolism.
Human beings exist in a constantly changing environment and face frequent challenges that threaten our survival and health. In response to stress, the body undergoes very subtle changes at the cellular and molecular level. Understanding how Sestrin2 is regulated under different stress conditions is very helpful for us in studying Sestrin2. Therefore, it is of great significance to study the regulatory mechanism of Sestrin2 expression under different types of stress conditions.
Reactive oxygen species and reactive nitrogen species (RNS) are generated continuously in the body through oxidative metabolism, biological functions of mitochondria, and immunologic functions (25). Physiological ROS are crucially important for intracellular and extracellular signal transduction (26). However, it is well-known that overloaded ROS and RNS can bind with and destroy most cellular biomolecules (lipids, enzymes, sugars, proteins, nucleic acids, and other small molecules) under oxidative stress (27–29). Oxidative stress is considered to be an imbalance in redox properties in certain cellular environments (30), and plays a crucial role in the development of numerous human diseases, including diabetes, obesity, and myocardial injury (31–33). Resistance to oxidative stress injury is one of the important functions of Sestrin2. In response to oxidative stress, the expression of Sesrin2 is regulated at the mRNA and protein level by various transcription factors, including nuclear factor kappa-B (NF-κB), activator protein-1 (AP-1), CCAAT-enhancer-binding protein beta (C/EBPβ), forkhead box O3 (FOXO3), and p53 (19, 24, 34–36). Sestrin2 has been suggested to maintain the balance of oxidative metabolism through two main biological functions. First, as an antioxidant enzyme, Sestrin2 is capable of directly reducing the accumulation of ROS (37). However, the intrinsic catalytic antioxidant activity of Sestrin2 remains elusive and limited. Second, recent studies have demonstrated that Sestrin2 inhibits ROS production and defends cells against oxidative stress, which is likely to be mainly attributed to its regulation of several signaling pathways related to oxidative stress: the Kelch-like Ech-associated protein 1 (KEAP1)/NF-E2 related factor-2 (NRF2) antioxidant signaling pathway (2) (Figure 1) and the AMPK and mTORC1 pathways (which will be described in detail later) (38). NRF2 is a transcription factor that can bind to antioxidant-responsive elements (AREs) to promote the expression of many antioxidant molecules to protect cells from oxidative insults (36). NRF2 is constitutively expressed in the cytoplasm under physiological conditions (39). Under normal conditions, KEAP1 binds to NRF2, preventing NRF2 translocation to the nucleus, promoting its ubiquitination and proteasome degradation, and maintaining free NRF2 in the cytoplasm at a low level (19). Under oxidative stress, NRF2 dissociates from KEAP1 and translocates to the nucleus. NRF2 binding to ARE activates the transcription of target genes PRX, SRX, superoxide dismutase (SOD), catalase (CAT), heme oxygenase 1 (HO1), and glutathione peroxidase 1 (GPX1) (40, 41). In cellular studies, it was found that Sestrin2 binds to unC-51-like kinase 1 (ULK1) and p62 to form functional complexes, and that Sestrin2 promotes the phosphorylation of p62, which further promotes KEAP1 degradation and NRF2 activation (42). In addition, in studies of liver damage caused by oxidative stress, Sestrin2 was shown to act as a scaffold protein to enhance the weak binding of KEAP1 to p62, thereby promoting KEAP1 autophagy degradation and preventing oxidative liver injury (43). More interestingly, NRF2 regulates the expression of Sestrin2 by binding to the ARE promoter of SESN2 under oxidative stress (37). A positive feedback loop is formed between Sestrin2 and NRF2 to promote the transcription and translation of antioxidant-related genes downstream of NRF2 and to protect cells from oxidative damage (36). Therefore, during oxidative stress, Sestrin2 is crucial to maintaining cellular homeostasis.
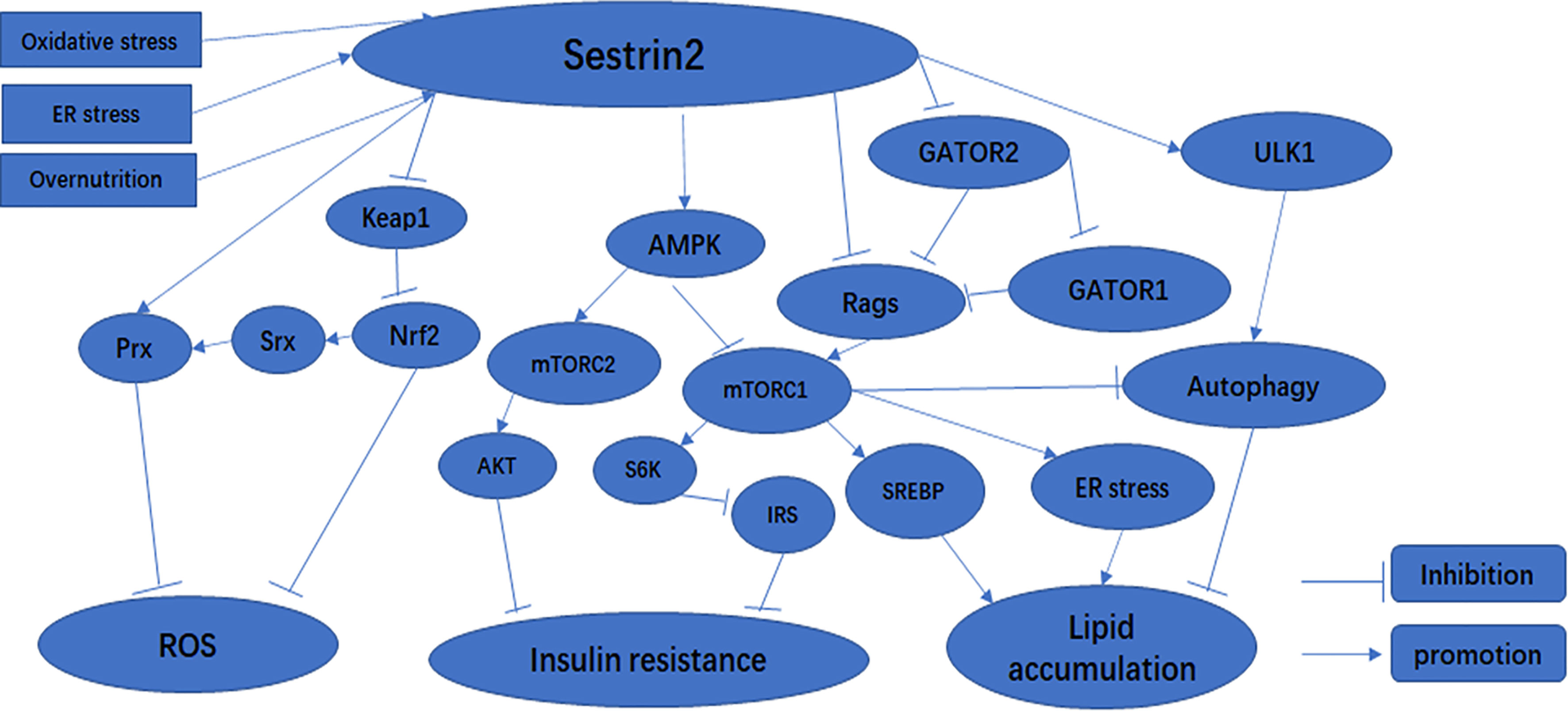
Figure 1 The effect of Sestrin2 on metabolic-related signaling pathways.
ER stress occurs when unfolded or misfolded proteins accumulate in the endoplasmic network lumen due to adverse physiological conditions (44). During ER stress, cells can improve their protein folding ability, inhibit protein production and accumulation, induce ER stress-related gene transcription, and strengthen the self-repair ability of ER to restore protein-folding homeostasis and regulate ER homeostasis through a series of transduction pathways, including protein kinase R-like endoplasmic reticulum kinase-eukaryotic translation-initiation factor 2α (PERK-eIF2α), inositol-requiring enzyme 1a-X-box-binding protein 1 (IRE1-XBP1), and activating transcription factor 6-CREBH (ATF6-CREBH). These reactions are called the unfolded protein reaction (UPR) (45). If ER stress is too strong or lasts too long, these responses are not enough to restore ER homeostasis, and apoptosis is eventually induced (46). A growing body of research has demonstrated that expression of Sestrin2 can be upregulated under ER stress conditions (15, 35, 47). For instance, Park et al. (15) found that upregulated Sestrin2 is associated with an ER stress-activated transcription factor, CCAAT enhancer-binding protein beta (c/EBPb). Once induced, Sestrin2 in turn stops protein synthesis by inhibiting mTORC1. Recently, a study by H. Jeong Kim et al. (48) revealed that induction of Sestrin2-regulated genes can be connected via activation of the PERK/eIF2α/ATF4 pathway. Consistent with these findings, Jegal et al. (35) demonstrated that under ER stress, expression of Sestrin2 can be enhanced via activating transcription factor 6 in hepatocytes, and Sestrin2 decreases the phosphorylation of JNK and p38 as well as PARP cleavage, and blocks the cytotoxic effect of excessive ER stress so as to play a hepatoprotective role both in vitro and in vivo. Furthermore, Ding et al. (49) elucidated that upregulation of Sestrin2 expression is dependent on ATF4 and NRF2 but not p53 under ER stress induced by glucose starvation. To summarize, Sestrin2 might serve as an important regulator that exerts cell and tissue protection functions under excessive ER stress. However, the exact mechanism by which ER stress induces Sestrin2 expression remains poorly understood and requires further exploration.
Obesity is traditionally considered to be the excessive accumulation of fat in the body, which is a serious hazard to human health, and in clinical practice, obesity is usually assessed by the body mass index (BMI) (50). With the improvements in the general standard of living, the incidence of obesity has risen sharply (51). Obesity is a major contributor to the development of metabolic syndromes, including type 2 diabetes mellitus, hypertension, hyperlipidemia, and cardiovascular disease (52). Studies have shown that overnutrition and a sedentary lifestyle are the main causes of obesity (53). mTORC1 is a nutrient-sensitive protein kinase that has a fundamental role in maintaining metabolic homeostasis (54). Recent research has clarified that overnutrition can result in chronic mTORC1 activation (55). In response to persistent overnutrition, chronic mTORC1 activation can enhance protein and lipid biosynthesis and inhibit autophagic catabolism (56). Several studies confirmed that chronic mTORC1 activation mediated by stress responses such as overnutrition ultimately leads to overexpression of Sestrin2 (24, 57). Lee et al. (24) found that the expression of Drosophila Sestrin (dSesn) is upregulated upon chronic mTORC1 activation via the c-Jun N-terminal kinase (JNK) and FOXO signaling pathways. Loss of dSesn results in triglyceride accumulation and mitochondrial dysfunction. Furthermore, another study by Lee et al. (57) demonstrated that Sestrin2 is the only Sestrin protein that is induced by overnutrition and obesity and attenuates chronic mTORC1 activation via the mTORC1/S6K axis in mouse liver. In agreement with these conclusions, Kimball et al. (58) revealed that Sestrin2 expression was upregulated in the livers of rats fed with a high-fat diet. Thus, in a nutshell, Sestrin2 exerts important metabolic homeostatic functions.
mTOR is an evolutionally conserved protein that is a critical regulator of cell proliferation, proliferation, metabolism, and autophagy (37). mTOR promotes anabolic processes such as ribosome biogenesis and synthesis of proteins, nucleotides, fatty acids, and lipids, and inhibits catabolic processes such as autophagy (54). It is composed of two structurally and functionally distinct complexes, mTORC1 and mTORC2, which are characterized by the presence of Raptor and Rictor, respectively (59). mTORC1 consists of mTOR Raptor, PRAS40,and mLST8, while mTORC2 is composed of mTOR, Rictor, Sin1, Protor, and mLST8 (54). mTORC1 promotes protein and lipid synthesis through the phosphorylation of its distinctive substrates, such as ribosomal protein S6 kinase (S6K) and eukaryotic initiation factor 4E-binding protein 1 (4EBP1) (2). In addition, mTORC1 may also regulate adipogenesis through the regulation of the sterol regulatory element-binding proteins (60). Furthermore, mTORC1 can phosphorylate and suppress autophagy-initiating protein kinases unc-51-like kinase 1 (ULK1) to inhibit cellular autophagic catabolism (61). mTORC2 regulates metabolism and cytoskeletal tissue in response to growth factors through the activation of AGC family kinases, including AKT, SGK1, and PKCα (62). Recent studies have shown that mTORC2 in particular is a crucial controller of lipid metabolism that regulates adipogenesis in the liver (60).
AMPK, an important nutrient-sensing protein kinase, has a critical role in increased catabolism and decreased anabolism (63). AMPK can inhibit the phosphorylation of the acetyl-CoA carboxylases ACC1 and ACC2, HMG-CoA reductase, and the glycogen synthases GYS1 and GYS2 to regulate the biosynthesis of glycogen and lipids (63). It can also inhibit mTORC1 activity through the phosphorylation of its regulatory subunit Raptor (64) or through the phosphorylation of tuberous sclerosis complex 2 and inhibition of mTORC1-activating guanosine triphosphatase (GTPase) Rheb (4). In addition, AMPK restrains the transcriptional activity of sterol regulatory element binding protein (SREBP) through direct phosphorylation to decrease the expression of lipogenic genes (65).
Once induced by stress, Sestrin2 affects a variety of signaling pathways, thus upregulating stress adaptation mechanisms (23). When induced in response to oxidative stress, Sestrin2 inhibits mTORC1 through the activation of AMPK (66). Consequently, Sestrin2-deficient cells and tissues exhibit lower AMPK and higher mTORC1 activity under both normal and stressed conditions (6, 24, 66). It has been reported that Sestrin2 acts as a scaffold protein, promoting the binding of LKB1 to AMPK and subsequent AMPK phosphorylation and activation, and controls mTORC1 signaling as an inhibitor of guanine nucleotide dissociation in Rag GTPases (6, 67, 68). Sestrin2 can also activate AMPK through direct interaction with the α subunit of the AMPK complex (66).Recent studies have shown that Sestrin2 can inhibit mTORC1 through AMPK-dependent or -independent pathways (15, 20, 57, 68) (Figure 1). Sestrin2 can also modulate amino acid-stimulated mTORC1 activation through direct interactions with Rag A/B GTPases or GATOR2 complexes (68, 80). Sestrin2 binds to GATOR2 and releases GATOR1 from GATOR2-mediated inhibition. Released GATOR1 subsequently binds to and inactivates RagB, ultimately resulting in mTORC1 suppression (81) (Figure 1). In addition, Sestrin2 plays a critical role in the activation of autophagy through multiple mechanisms including activation of AMPK, inhibition of mTORC1, and activation of ULK1 (82) (Figure 1). Therefore, the AMPK/mTORC1 signaling pathway is critical for Sestrin2 in controlling cell metabolism and survival under stress conditions (Figure 1).
Mounting evidence has demonstrated that Sestrin2 is upregulated in response to diverse stress conditions, including oxidative stress, ER stress, and metabolic stress. Sestrin2 exerts a significant influence on the protection of human cells and tissues via related signal transduction pathways, and was shown to play a critical role against metabolic diseases, such as diabetes, obesity-related non-alcoholic fatty live, and myocardial I/R injury (Table 1).

Table 1 Summary of the role of Sestrin2 in metabolic diseases.
Diabetes is the most common metabolic disease, and is a chronic disease characterized by persistent hyperglycemia (83). More than 90% of diabetics have type 2 diabetes, and insulin resistance is consistently found in patients with type 2 diabetes (84). Insulin resistance is an impaired biological response to insulin stimulation in target tissues, primarily liver, muscle, and adipose tissue (85). Insulin resistance impairs glucose processing, leading to a compensatory increase in beta cell insulin production and hyperinsulinemia. Sestrin2 is highly expressed in the liver (86). According to literature reports, two pathways of Sestrin2 affect cell signaling pathway transduction: one activates the AMPK pathway and the other downregulates the mTOR pathway (2, 87). AMPK is an enzyme activated in energy-deficient conditions (2). Sestrin2 is induced by oxidative stress through activation of the NRF2 and JNK/AP-1 signaling axes (4, 43, 88). In bacteria, AhpD is a critical member of the antioxidant defense system and regenerates peroxide AhpC, a bacterial peroxidant protein (Prx), through catalytic reduction. In mammalian cells, Sestrins interact with overoxidized PRX and promote its regeneration. Here, Sestrins act similarly to AhpD in bacteria (77). Sestrins have no direct catalytic activity leading to the reduction of PRX, but may regenerate PRX by promoting the activity of other oxidoreductases, such as thioredoxin (SRX) (4). Sestrins can increase SRX expression by activating NRF2 (6, 43) (Figure 1). Increased glucose downregulates Sestrin2 expression, thereby increasing mTOR activity and inhibiting AMPK (87, 89). Moreover, when treated with high levels of glucose, such as metformin (an AMPK agonist and mitochondrial respiratory inhibitor), Sestrin2 was upregulated, mTOR activity was significantly increased, and AMPK activity was decreased (6, 87). S6K is an effector of the mTOR pathway (90). By activating S6K, mTORC1 promotes insulin resistance by inhibiting phosphorylation of insulin receptor substrates (IRS) (Figure 1) and attenuating the insulin-induced phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway (6). mTORC1/S6K activity leads to serine phosphorylation and protein degradation of IRS, forming a negative feedback loop in which insulin signaling attenuates subsequent insulin action (89). Lack of amino acids, especially leucine, leads to rapid dephosphorylation of the mTORC1 effectors S6K and 4EBP1, which depend on mTORC1 for amino acid resynthesis (91). Sestrin2 is required to maintain insulin sensitivity in the liver in high-fat diet (HFD)-induced dietary obesity and Lepob mutation-induced inherited obesity (57). AMPK and mTORC1 are important protein kinases with complete antagonistic functions in metabolic homeostasis (92). Sestrin1 and Sestrin2 activate AMPK through direct interaction with the α subunit of the AMPK complex (66). Sestrin2 acts by activating AMPK and inhibiting various mechanisms of mTORC1. We know that AMPK and mTORC1 play critical roles in metabolism, and Sestrin2 is involved in many biological processes as an upstream regulator of AMPK and mTORC1 kinases (57) (Figure 1). Previous experiments in liver-specific Sesn3 transgenic mice and knockout mice showed that the transgenic mice were protected against insulin resistance induced by a high-fat diet, while the Sesn3 knockout mice showed metabolic defects such as insulin resistance and glucose intolerance (93, 94). Therefore, we can recognize that Sestrin2 is a potential insulin sensitizer, and that Sestrin deficiency and/or dysfunction may lead to insulin resistance, which can lead to the development of diabetes. Sestrin2 may be a potential therapeutic target for metabolic diseases such as diabetes (82).
With the global trend in obesity and its related metabolic syndromes, non-alcoholic fatty liver disease (NAFLD) has become an important cause of chronic liver disease in developed countries (95). NAFLD is the hepatic manifestation of metabolic syndrome characterized by intracellular excessive accumulation of lipids in hepatocytes, excluding alcohol and other damaging factors (95). NAFLD involves a range of liver pathological changes, including steatosis, steatohepatitis, advanced fibrosis, and cirrhosis (96). Existing studies have shown that NAFLD is closely associated with persistent ER stress, inhibition of autophagy, mitochondrial dysfunction, insulin resistance, lipotoxicity, and overnutrition (15, 71, 96, 97). Overnutrition and obesity give rise to excessive lipid accumulation in hepatocytes, known as hepatic steatosis (98). We have previously shown that overnutrition can lead to chronic mTORC1 activation (53). mTORC1 can intensify the transcriptional activity of sterol regulatory element binding protein (SREBP) and the expression of lipogenic genes to enhance lipid synthesis (Figure 1). It is evident that chronic mTORC1 activation along with persistent inhibition of autophagy attenuates clearance of liver lipid droplets, ultimately leading to hepatosteatosis (99). As a feedback inhibitor of mTORC1, Sestrin2 can partially ease the effect of chronic mTORC1 activation. For instance, loss of dSesn leads to moderate downregulation of AMPK and upregulation of dTORC1 in Drosophila, which contributes to the increased expression of liposomal enzyme genes and ultimately to the accumulation of triglycerides (24). Similarly, a study has confirmed that hepatosteatosis is more serious, and that the primary cause of hepatosteatosis is reduced lipid β-oxidation due to reduced autophagy or mitochondrial biogenesis, rather than increased adipogenesis in Sestrin2-deficient liver (57). Furthermore, Sestrin2 also reduces the susceptibility of the liver to oxidative damage via the NRF2/KEAP1 signaling pathway (43). In mice with Sestrin2 deficiency, cells continue to translate large amounts of proteins during ER stress, which subsequently leads to extensive liver damage, inflammation, and fibrosis (15). Accordingly, once induced by ER stress, Sestrin2 maintains endoplasmic reticulum homeostasis by inhibiting the AMPK/mTORC1 signaling pathway (Figure 1), thereby protecting against hepatosteatosis (15). Kim et al. found that carbon monoxide can induce Sestrin2 upregulation, and Sestrin2 protects against hepatosteatosis by activating autophagy through the AMPK/mTORC1 axis in a cellular model of NAFLD (48). More interestingly, Sestrin2 plays an important role in the protection against lipotoxicity-associated oxidative stress in the liver via suppression of JNKs (72). In summary, Sestrin2 has a significant impact on lipid metabolism and represents a potential therapeutic strategy for NAFLD.
Coronary heart disease, also known as ischemic heart disease (IHD), refers to the interruption of blood flow to the heart muscle due to atherosclerosis, coronary thrombosis, and narrowing of the small arteries of the heart, which remains the leading cause of death worldwide because the incidence of IHD increases with age (100, 101). After an acute myocardial infarction, although early and successful myocardial reperfusion through thrombolytic or percutaneous coronary intervention is the most effective way to rescue the ischemic heart and improve the clinical outcome, the recovery of blood flow can result in myocardial injury, which reduces the efficacy of myocardial reperfusion, namely ischemia/reperfusion (I/R) injury (78, 102). Myocardial I/R injury is closely related to ROS, calcium overload, energy metabolism disorders, acidosis, and inflammation (102). Some studies have reported that the I/R process usually results in elevated levels of ROS production, especially in the early stages of reperfusion, directly causing myocardial injury (103). Moreover, excessive ROS leads to programmed cell death through the activation of the mitogen-activated protein kinase signaling pathway (104). Mitochondria have an important role in ROS degradation, and dysfunctional mitochondria are the main sources of pathological ROS (105, 106). AMPK can protect mitochondria and play an antioxidant role during the I/R process (78). Furthermore, AMPK has an essential role in the activation of glucose uptake in the ischemic heart (107–109). AMPK also activates 6-phosphofructo 2-kinase, which leads to the production of fructose 2, 6-bisphosphate, further promoting glucose utilization in the ischemic heart (75, 110, 111). Therefore, AMPK is a protein kinase with significant cardiac protection against myocardial I/R injury (74). Sestrin2 has been shown to increase the activation of AMPK via interactions with LKB1 to improve myocardial substrate metabolism under I/R stress (75). Sestrin2 was originally characterized as a critical antioxidant protein that contributes to cycling of peroxiredoxins (77). Independent of this redox-regulating activity, Sestrin2 can modulate the activation of AMPK to maintain the integrity of mitochondrial function and reduce the generation of ROS (14, 66, 74, 112). A study by Quan et al. (76) revealed that Sestrin2 greatly reduces myocardial damage by modulating inflammation and redox homeostasis in mouse hearts during I/R stress. Hence, Sestrin2 provides cardioprotection by repressing ROS during I/R injury. Furthermore, Quan et al. (73) found that the decreased Sestrin2 levels in aging and Sesn2-knockout mice led to increased sensitivity to ischemic insults and areas of myocardial injury, which aggravated worsened cardiac dysfunction. Sestrin2 protects mitochondrial function by activating the AMPK/PGC-1α signaling pathway during myocardial ischemia (73). Sestrin2 has been shown to be upregulated under anoxic and ischemic conditions and has a protective role against myocardial ischemia (7, 74, 78). The loss of Sestrin2 aggravates the accumulation of fatty acids, thereby altering substrate metabolism in the heart and increasing the production of ROS (37, 78). Inactivation of the SESN gene in invertebrates can lead to a variety of metabolic diseases such as muscle degeneration, oxidative damage, fat accumulation, and mitochondrial dysfunction (4). Existing studies have reported that Sestrin2 is involved in the protection of cardiovascular disease by regulating the AMPK signaling pathway (38). Sestrin2 protein accumulates in the heart during myocardial ischemia (17), and the myocardial infarction area in Sesn2 knockout mice was significantly larger than that in wild-type mice when myocardial ischemia reperfusion occurred (74). In conclusion, Sestrin2 has an influential role in cardioprotection during myocardial I/R injury. Therefore, Sestrin2 may be a therapeutic target for cardiovascular disease, potentially revealing a new avenue of investigation for the treatment of cardiovascular diseases.
Sestrin2 is a critical intracellular sensor that activates AMPK and inhibits mTORC1 to regulate autophagy, ER stress, inflammation, metabolic homeostasis, and oxidative stress. Clearly, the AMPK/mTORC1 axis is regulated by Sestrin2 and it provides the main channel for its function. Sestrin2 regulates metabolism-related signaling pathways, as summarized in Figure 1. However, despite their physiological relevance, the exact mechanism by which Sestrin2 promotes AMPK activation remains unclear. Therefore, further studies are needed to determine the detailed molecular function of Sestrin2.
Evidence suggests that Sestrin2 has an important clinical function in responding to a variety of metabolic diseases, such as diabetes mellitus, insulin resistance, and lipid metabolism disorders. In recent studies, serum Sestrin2 levels were significantly reduced in obese children and patients with diabetic nephropathy (113, 114). This suggests that the expression or secretion of Sestrin2 is somewhat obstructed in the disease state. Furthermore, a study by Kim et al. revealed that in NAFLD cell models, carbon monoxide protects the liver against steatosis by inducing upregulation of Sestrin2, which activates autophagy through the AMPK/mTORC1 axis (48). Consistent with this view, as a glucagon-like peptide 1 (GLP-1) analog, liraglutide could reverse NAFLD by enhancing the level of Sestrin2 protein and the Sestrin2-mediated NRF2/HO-1 pathway (71). Therefore, we hypothesized that upregulation of Sestrin2 expression could ameliorate metabolism-related diseases. Sestrin2 shows great potential as a good prognostic marker and a viable therapeutic target in a variety of diseases. However, how to induce Sestrin2 upregulation remains elusive under different disease conditions. To design therapeutic strategies to upregulate Sestrin2, it is important to further study the upstream and downstream pathways of the multipotent beneficial effects of Sestrin2. Future studies should use transgenic animal models with conditional organ-specific knockout of Sesn2 and attempt to link Sestrin2 levels to disease progression, which will help us identify biochemical pathways regulated by Sestrin2 in specific diseases.
LG and ZZW worked out the theme and content of the article. ZGW completed the production of charts. ZZ reviewed and revised the full text. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kumar A, Dhiman D, Shaha C. Sestrins: Darkhorse in the Regulation of Mitochondrial Health and Metabolism. Mol Biol Rep (2020) 47(10):8049–60. doi: 10.1007/s11033-020-05769-w
2. Pasha M, Eid AH, Eid AA, Gorin Y, Munusamy S. Sestrin2 as a Novel Biomarker and Therapeutic Target for Various Diseases. Oxid Med Cell Longev (2017) 2017:3296294. doi: 10.1155/2017/3296294
3. Budanov AV. Stress-Responsive Sestrins Link P53 With Redox Regulation and Mammalian Target of Rapamycin Signaling. Antioxid Redox Signal (2011) 15(6):1679–90. doi: 10.1089/ars.2010.3530
4. Lee JH, Budanov AV, Karin M. Sestrins Orchestrate Cellular Metabolism to Attenuate Aging. Cell Metab (2013) 18(6):792–801. doi: 10.1016/j.cmet.2013.08.018
5. Velasco-Miguel S, Buckbinder L, Jean P, Gelbert L, Talbott R, Laidlaw J, et al. PA26, a Novel Target of the P53 Tumor Suppressor and Member of the GADD Family of DNA Damage and Growth Arrest Inducible Genes. Oncogene (1999) 18(1):127–37. doi: 10.1038/sj.onc.1202274
6. Sun W, Wang Y, Zheng Y, Quan N. The Emerging Role of Sestrin2 in Cell Metabolism, and Cardiovascular and Age-Related Diseases. Aging Dis (2020) 11(1):154–63. doi: 10.14336/AD.2019.0320
7. Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, et al. Identification of a Novel Stress-Responsive Gene Hi95 Involved in Regulation of Cell Viability. Oncogene (2002) 21(39):6017–31. doi: 10.1038/sj.onc.1205877
8. Chen CC, Jeon SM, Bhaskar PT, Nogueira V, Sundararajan D, Tonic I, et al. FoxOs Inhibit Mtorc1 and Activate Akt by Inducing the Expression of Sestrin3 and Rictor. Dev Cell (2010) 18(4):592–604. doi: 10.1016/j.devcel.2010.03.008
9. Parmigiani A, Budanov AV. Sensing the Environment Through Sestrins: Implications for Cellular Metabolism. Int Rev Cell Mol Biol (2016) 327:1–42. doi: 10.1016/bs.ircmb.2016.05.003
10. Wang LX, Zhu XM, Yao YM. Sestrin2: Its Potential Role and Regulatory Mechanism in Host Immune Response in Diseases. Front Immunol (2019) 10:2797. doi: 10.3389/fimmu.2019.02797
11. Ro SH, Fay J, Cyuzuzo CI, Jang Y, Lee N, Song HS, et al. SESTRINs: Emerging Dynamic Stress-Sensors in Metabolic and Environmental Health. Front Cell Dev Biol (2020) 8:603421. doi: 10.3389/fcell.2020.603421
12. Keping Y, Yunfeng S, Pengzhuo X, Liang L, Chenhong X, Jinghua M. Sestrin1 Inhibits Oxidized Low-Density Lipoprotein-Induced Activation of NLRP3 Inflammasome in Macrophages in a Murine Atherosclerosis Model. Eur J Immunol (2020) 50(8):1154–66. doi: 10.1002/eji.201948427
13. Li Z, Votava JA, Zajac GJM, Nguyen JN, Leyva Jaimes FB, Ly SM, et al. Integrating Mouse and Human Genetic Data to Move Beyond GWAS and Identify Causal Genes in Cholesterol Metabolism. Cell Metab (2020) 31(4):741–54.e5. doi: 10.1016/j.cmet.2020.02.015
14. Shi X, Xu L, Doycheva DM, Tang J, Yan M, Zhang JH. Sestrin2, as a Negative Feedback Regulator of mTOR, Provides Neuroprotection by Activation AMPK Phosphorylation in Neonatal Hypoxic-Ischemic Encephalopathy in Rat Pups. J Cereb Blood Flow Metab (2017) 37(4):1447–60. doi: 10.1177/0271678X16656201
15. Park HW, Park H, Ro SH, Jang I, Semple IA, Kim DN, et al. Hepatoprotective Role of Sestrin2 Against Chronic ER Stress. Nat Commun (2014) 5:4233. doi: 10.1038/ncomms5233
16. Yang JH, Kim KM, Kim MG, Seo KH, Han JY, Ka SO, et al. Role of Sestrin2 in the Regulation of Proinflammatory Signaling in Macrophages. Free Radic Biol Med (2015) 78:156–67. doi: 10.1016/j.freeradbiomed.2014.11.002
17. Peeters H, Debeer P, Bairoch A, Wilquet V, Huysmans C, Parthoens E, et al. PA26 is a Candidate Gene for Heterotaxia in Humans: Identification of a Novel PA26-Related Gene Family in Human and Mouse. Hum Genet (2003) 112(5-6):573–80. doi: 10.1007/s00439-003-0917-5
18. Budanov AV, Lee JH, Karin M. Stressin’ Sestrins Take an Aging Fight. EMBO Mol Med (2010) 2(10):388–400. doi: 10.1002/emmm.201000097
19. Liu Y, Li M, Du X, Huang Z, Quan N. Sestrin 2, a Potential Star of Antioxidant Stress in Cardiovascular Diseases. Free Radic Biol Med (2021) 163:56–68. doi: 10.1016/j.freeradbiomed.2020.11.015
20. Quan N, Li X, Zhang J, Han Y, Sun W, Ren D, et al. Substrate Metabolism Regulated by Sestrin2-Mtorc1 Alleviates Pressure Overload-Induced Cardiac Hypertrophy in Aged Heart. Redox Biol (2020) 36:101637. doi: 10.1016/j.redox.2020.101637
21. Kim H, An S, Ro SH, Teixeira F, Park GJ, Kim C, et al. Janus-Faced Sestrin2 Controls ROS and mTOR Signalling Through Two Separate Functional Domains. Nat Commun (2015) 6:10025. doi: 10.1038/ncomms10025
22. Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T, et al. Structural Basis for Leucine Sensing by the Sestrin2-Mtorc1 Pathway. Science (2016) 351(6268):53–8. doi: 10.1126/science.aad2087
23. Kim M, Kowalsky AH, Lee JH. Sestrins in Physiological Stress Responses. Annu Rev Physiol (2021) 83:381–403. doi: 10.1146/annurev-physiol-031620-092317
24. Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, et al. Sestrin as a Feedback Inhibitor of TOR That Prevents Age-Related Pathologies. Science (2010) 327(5970):1223–8. doi: 10.1126/science.1182228
25. Tan BL, Norhaizan ME, Liew WP. Nutrients and Oxidative Stress: Friend or Foe? Oxid Med Cell Longev (2018) 2018:9719584. doi: 10.1155/2018/9719584
26. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-Mediated Cellular Signaling. Oxid Med Cell Longev (2016) 2016:4350965. doi: 10.1155/2016/4350965
27. Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, et al. Oxidative Stress, Aging, and Diseases. Clin Interv Aging (2018) 13:757–72. doi: 10.2147/CIA.S158513
28. Negre-Salvayre A, Auge N, Ayala V, Basaga H, Boada J, Brenke R, et al. Pathological Aspects of Lipid Peroxidation. Free Radic Res (2010) 44(10):1125–71. doi: 10.3109/10715762.2010.498478
29. Roberts RA, Smith RA, Safe S, Szabo C, Tjalkens RB, Robertson FM. Toxicological and Pathophysiological Roles of Reactive Oxygen and Nitrogen Species. Toxicology (2010) 276(2):85–94. doi: 10.1016/j.tox.2010.07.009
30. Marrocco I, Altieri F, Peluso I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid Med Cell Longev (2017) 2017:6501046. doi: 10.1155/2017/6501046
31. Yaribeygi H, Sathyapalan T, Atkin SL, Sahebkar A. Molecular Mechanisms Linking Oxidative Stress and Diabetes Mellitus. Oxid Med Cell Longev (2020) 2020:8609213. doi: 10.1155/2020/8609213
32. McMurray F, Patten DA, Harper ME. Reactive Oxygen Species and Oxidative Stress in Obesity-Recent Findings and Empirical Approaches. Obes (Silver Spring) (2016) 24(11):2301–10. doi: 10.1002/oby.21654
33. Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J Am Coll Cardiol (2017) 70(2):212–29. doi: 10.1016/j.jacc.2017.05.035
34. Kim MG, Yang JH, Kim KM, Jang CH, Jung JY, Cho IJ, et al. Regulation of Toll-Like Receptor-Mediated Sestrin2 Induction by AP-1, Nrf2, and the Ubiquitin-Proteasome System in Macrophages. Toxicol Sci (2015) 144(2):425–35. doi: 10.1093/toxsci/kfv012
35. Jegal KH, Park SM, Cho SS, Byun SH, Ku SK, Kim SC, et al. Activating Transcription Factor 6-Dependent Sestrin 2 Induction Ameliorates ER Stress-Mediated Liver Injury. Biochim Biophys Acta Mol Cell Res (2017) 1864(7):1295–307. doi: 10.1016/j.bbamcr.2017.04.010
36. Shin BY, Jin SH, Cho IJ, Ki SH. Nrf2-ARE Pathway Regulates Induction of Sestrin-2 Expression. Free Radic Biol Med (2012) 53(4):834–41. doi: 10.1016/j.freeradbiomed.2012.06.026
37. Liu Y, Du X, Huang Z, Zheng Y, Quan N. Sestrin 2 Controls the Cardiovascular Aging Process via an Integrated Network of Signaling Pathways. Ageing Res Rev (2020) 62:101096. doi: 10.1016/j.arr.2020.101096
38. Gao A, Li F, Zhou Q, Chen L. Sestrin2 as a Potential Therapeutic Target for Cardiovascular Diseases. Pharmacol Res (2020) 159:104990. doi: 10.1016/j.phrs.2020.104990
39. Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) Signaling in Oxidative Stress. Free Radic Biol Med (2009) 47(9):1304–9. doi: 10.1016/j.freeradbiomed.2009.07.035
40. Silva-Palacios A, Königsberg M, Zazueta C. Nrf2 Signaling and Redox Homeostasis in the Aging Heart: A Potential Target to Prevent Cardiovascular Diseases? Ageing Res Rev (2016) 26:81–95. doi: 10.1016/j.arr.2015.12.005
41. Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol Cell Biol (2004) 24(24):10941–53. doi: 10.1128/MCB.24.24.10941-10953.2004
42. Ro SH, Semple IA, Park H, Park H, Park HW, Kim M, et al. Sestrin2 Promotes Unc-51-Like Kinase 1 Mediated Phosphorylation of P62/Sequestosome-1. FEBS J (2014) 281(17):3816–27. doi: 10.1111/febs.12905
43. Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, et al. Sestrins Activate Nrf2 by Promoting P62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. Cell Metab (2013) 17(1):73–84. doi: 10.1016/j.cmet.2012.12.002
44. Hetz C, Saxena S. ER Stress and the Unfolded Protein Response in Neurodegeneration. Nat Rev Neurol (2017) 13(8):477–91. doi: 10.1038/nrneurol.2017.99
45. Pluquet O, Pourtier A, Abbadie C. The Unfolded Protein Response and Cellular Senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am J Physiol Cell Physiol (2015) 308(6):C415–25. doi: 10.1152/ajpcell.00334.2014
46. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of Endoplasmic Reticulum Stress-Induced Apoptosis. EMBO Rep (2006) 7(9):880–5. doi: 10.1038/sj.embor.7400779
47. Saveljeva S, Cleary P, Mnich K, Ayo A, Pakos-Zebrucka K, Patterson JB, et al. Endoplasmic Reticulum Stress-Mediated Induction of SESTRIN 2 Potentiates Cell Survival. Oncotarget (2016) 7(11):12254–66. doi: 10.18632/oncotarget.7601
48. Kim HJ, Joe Y, Kim SK, Park SU, Park J, Chen Y, et al. Carbon Monoxide Protects Against Hepatic Steatosis in Mice by Inducing Sestrin-2 via the PERK-Eif2α-ATF4 Pathway. Free Radic Biol Med (2017) 110:81–91. doi: 10.1016/j.freeradbiomed.2017.05.026
49. Ding B, Parmigiani A, Divakaruni AS, Archer K, Murphy AN, Budanov AV. Sestrin2 is Induced by Glucose Starvation via the Unfolded Protein Response and Protects Cells From non-Canonical Necroptotic Cell Death. Sci Rep (2016) 6:22538. doi: 10.1038/srep22538
50. Piché ME, Tchernof A, Després JP. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ Res (2020) 126(11):1477–500. doi: 10.1161/CIRCRESAHA.120.316101
51. Polyzos SA, Kountouras J, Mantzoros CS. Obesity and Nonalcoholic Fatty Liver Disease: From Pathophysiology to Therapeutics. Metabolism (2019) 92:82–97. doi: 10.1016/j.metabol.2018.11.014
52. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-Activated Protein Kinase: Ancient Energy Gauge Provides Clues to Modern Understanding of Metabolism. Cell Metab (2005) 1(1):15–25. doi: 10.1016/j.cmet.2004.12.003
53. Caballero B. Humans Against Obesity: Who Will Win? Adv Nutr (2019) 10(suppl_1):S4–s9. doi: 10.1093/advances/nmy055
54. Mossmann D, Park S, Hall MN. mTOR Signalling and Cellular Metabolism are Mutual Determinants in Cancer. Nat Rev Cancer (2018) 18(12):744–57. doi: 10.1038/s41568-018-0074-8
55. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
56. Zoncu R, Efeyan A, Sabatini DM. mTOR: From Growth Signal Integration to Cancer, Diabetes and Ageing. Nat Rev Mol Cell Biol (2011) 12(1):21–35. doi: 10.1038/nrm3025
57. Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, et al. Maintenance of Metabolic Homeostasis by Sestrin2 and Sestrin3. Cell Metab (2012) 16(3):311–21. doi: 10.1016/j.cmet.2012.08.004
58. Kimball SR, Gordon BS, Moyer JE, Dennis MD, Jefferson LS. Leucine Induced Dephosphorylation of Sestrin2 Promotes Mtorc1 Activation. Cell Signal (2016) 28(8):896–906. doi: 10.1016/j.cellsig.2016.03.008
59. Yoon MS. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients (2017) 9(11):1176. doi: 10.3390/nu9111176
60. Lamming DW, Sabatini DM. A Central Role for mTOR in Lipid Homeostasis. Cell Metab (2013) 18(4):465–9. doi: 10.1016/j.cmet.2013.08.002
61. Zhu Z, Yang C, Iyaswamy A, Krishnamoorthi S, Sreenivasmurthy SG, Liu J, et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int J Mol Sci (2019) 20(3):728. doi: 10.3390/ijms20030728
62. Herzig S, Shaw RJ. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat Rev Mol Cell Biol (2018) 19(2):121–35. doi: 10.1038/nrm.2017.95
63. Han J, Wang Y. Mtorc1 Signaling in Hepatic Lipid Metabolism. Protein Cell (2018) 9(2):145–51. doi: 10.1007/s13238-017-0409-3
64. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol Cell (2008) 30(2):214–26. doi: 10.1016/j.molcel.2008.03.003
65. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice. Cell Metab (2011) 13(4):376–88. doi: 10.1016/j.cmet.2011.03.009
66. Budanov AV, Karin M. P53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell (2008) 134(3):451–60. doi: 10.1016/j.cell.2008.06.028
67. Sanli T, Linher-Melville K, Tsakiridis T, Singh G. Sestrin2 Modulates AMPK Subunit Expression and its Response to Ionizing Radiation in Breast Cancer Cells. PLoS One (2012) 7(2):e32035. doi: 10.1371/journal.pone.0032035
68. Peng M, Yin N, Li MO. Sestrins Function as Guanine Nucleotide Dissociation Inhibitors for Rag GTPases to Control Mtorc1 Signaling. Cell (2014) 159(1):122–33. doi: 10.1016/j.cell.2014.08.038
69. Liu X, Niu Y, Yuan H, Huang J, Fu L. AMPK Binds to Sestrins and Mediates the Effect of Exercise to Increase Insulin-Sensitivity Through Autophagy. Metabolism (2015) 64(6):658–65. doi: 10.1016/j.metabol.2015.01.015
70. Caron A, Richard D, Laplante M. The Roles of mTOR Complexes in Lipid Metabolism. Annu Rev Nutr (2015) 35:321–48. doi: 10.1146/annurev-nutr-071714-034355
71. Han X, Ding C, Zhang G, Pan R, Liu Y, Huang N, et al. Liraglutide Ameliorates Obesity-Related Nonalcoholic Fatty Liver Disease by Regulating Sestrin2-Mediated Nrf2/HO-1 Pathway. Biochem Biophys Res Commun (2020) 525(4):895–901. doi: 10.1016/j.bbrc.2020.03.032
72. Fang Z, Kim HG, Huang M, Chowdhury K, Li MO, Liangpunsakul S, et al. Sestrin Proteins Protect Against Lipotoxicity-Induced Oxidative Stress in the Liver via Suppression of C-Jun N-Terminal Kinases. Cell Mol Gastroenterol Hepatol (2021) 12(3):921–42. doi: 10.1016/j.jcmgh.2021.04.015
73. Quan N, Wang L, Chen X, Luckett C, Cates C, Rousselle T, et al. Sestrin2 Prevents Age-Related Intolerance to Post Myocardial Infarction via AMPK/PGC-1α Pathway. J Mol Cell Cardiol (2018) 115:170–8. doi: 10.1016/j.yjmcc.2018.01.005
74. Morrison A, Chen L, Wang J, Zhang M, Yang H, Ma Y, et al. Sestrin2 Promotes LKB1-Mediated AMPK Activation in the Ischemic Heart. FASEB J (2015) 29(2):408–17. doi: 10.1096/fj.14-258814
75. Quan N, Sun W, Wang L, Chen X, Bogan JS, Zhou X, et al. Sestrin2 Prevents Age-Related Intolerance to Ischemia and Reperfusion Injury by Modulating Substrate Metabolism. FASEB J (2017) 31(9):4153–67. doi: 10.1096/fj.201700063R
76. Ren D, Quan N, Fedorova J, Zhang J, He Z, Li J. Sestrin2 Modulates Cardiac Inflammatory Response Through Maintaining Redox Homeostasis During Ischemia and Reperfusion. Redox Biol (2020) 34:101556. doi: 10.1016/j.redox.2020.101556
77. Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of Peroxiredoxins by P53-Regulated Sestrins, Homologs of Bacterial AhpD. Science (2004) 304(5670):596–600. doi: 10.1126/science.1095569
78. Liu Y, Li M, Sun M, Zhang Y, Li X, Sun W, et al. Sestrin2 is an Endogenous Antioxidant That Improves Contractile Function in the Heart During Exposure to Ischemia and Reperfusion Stress. Free Radic Biol Med (2021) 165:385–94. doi: 10.1016/j.freeradbiomed.2021.01.048
79. Sun X, Han F, Lu Q, Li X, Ren D, Zhang J, et al. Empagliflozin Ameliorates Obesity-Related Cardiac Dysfunction by Regulating Sestrin2-Mediated AMPK-mTOR Signaling and Redox Homeostasis in High-Fat Diet-Induced Obese Mice. Diabetes (2020) 69(6):1292–305.
80. Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, et al. The Sestrins Interact With GATOR2 to Negatively Regulate the Amino-Acid-Sensing Pathway Upstream of Mtorc1. Cell Rep (2014) 9(1):1–8. doi: 10.1016/j.celrep.2014.09.014
81. Kim JS, Ro SH, Kim M, Park HW, Semple IA, Park H, et al. Sestrin2 Inhibits Mtorc1 Through Modulation of GATOR Complexes. Sci Rep (2015) 5:9502. doi: 10.1038/srep09502
82. Dong XC. The Potential of Sestrins as Therapeutic Targets for Diabetes. Expert Opin Ther Targets (2015) 19(8):1011–5. doi: 10.1517/14728222.2015.1044976
83. Goyal R, Jialal I. “Diabetes Mellitus Type 2”. In: StatPearls. Treasure Island (FL: StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC (2021).
84. Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, et al. Impaired Glucose Transport as a Cause of Decreased Insulin-Stimulated Muscle Glycogen Synthesis in Type 2 Diabetes. N Engl J Med (1999) 341(4):240–6. doi: 10.1056/NEJM199907223410404
85. Freeman AM, Pennings N. ”Insulin Resistance. In: StatPearls”. Treasure Island (FL: StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC (2021).
86. Kowalsky AH, Namkoong S, MettEtal E, Park HW, Kazyken D, Fingar DC, et al. The GATOR2-Mtorc2 Axis Mediates Sestrin2-Induced AKT Ser/Thr Kinase Activation. J Biol Chem (2020) 295(7):1769–80. doi: 10.1074/jbc.RA119.010857
87. Sundararajan S, Jayachandran I, Balasubramanyam M, Mohan V, Venkatesan B, Manickam N. Sestrin2 Regulates Monocyte Activation Through AMPK-mTOR Nexus Under High-Glucose and Dyslipidemic Conditions. J Cell Biochem (2018) 120(5):8201–13. doi: 10.1002/jcb.28102
88. Chen M, Xi Y, Chen K, Xiao P, Li S, Sun X, et al. Upregulation Sestrin2 Protects Against Hydrogen Peroxide-Induced Oxidative Damage Bovine Mammary Epithelial Cells via a Keap1-Nrf2/ARE Pathway. J Cell Physiol (2021) 236(1):392–404. doi: 10.1002/jcp.29867
89. Zick Y. Uncoupling Insulin Signalling by Serine/Threonine Phosphorylation: A Molecular Basis for Insulin Resistance. Biochem Soc Trans (2004) 32(Pt 5):812–6. doi: 10.1042/BST0320812
90. Tavares MR, Pavan IC, Amaral CL, Meneguello L, Luchessi AD, Simabuco FM. The S6K Protein Family in Health and Disease. Life Sci (2015) 131:1–10. doi: 10.1016/j.lfs.2015.03.001
91. Wullschleger S, Loewith R, Hall MN. TOR Signaling in Growth and Metabolism. Cell (2006) 124(3):471–84. doi: 10.1016/j.cell.2006.01.016
92. Hardie DG, Ross FA, Hawley SA. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat Rev Mol Cell Biol (2012) 13(4):251–62. doi: 10.1038/nrm3311
93. Tao R, Xiong X, Liangpunsakul S, Dong XC. Sestrin 3 Protein Enhances Hepatic Insulin Sensitivity by Direct Activation of the Mtorc2-Akt Signaling. Diabetes (2015) 64(4):1211–23. doi: 10.2337/db14-0539
94. Lee JH, Cho US, Karin M. Sestrin Regulation of TORC1: Is Sestrin a Leucine Sensor? Sci Signal (2016) 9(431):re5. doi: 10.1126/scisignal.aaf2885
95. Rinella ME. Nonalcoholic Fatty Liver Disease: A Systematic Review. JAMA (2015) 313(22):2263–73. doi: 10.1001/jama.2015.5370
96. Pierantonelli I, Svegliati-Baroni G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation (2019) 103(1):e1–e13. doi: 10.1097/TP.0000000000002480
97. Ashraf NU, Sheikh TA. Endoplasmic Reticulum Stress and Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Free Radic Res (2015) 49(12):1405–18. doi: 10.3109/10715762.2015.1078461
98. Sahini N, Borlak J. Recent Insights Into the Molecular Pathophysiology of Lipid Droplet Formation in Hepatocytes. Prog Lipid Res (2014) 54:86–112. doi: 10.1016/j.plipres.2014.02.002
99. Martinez-Lopez N, Singh R. Autophagy and Lipid Droplets in the Liver. Annu Rev Nutr (2015) 35:215–37. doi: 10.1146/annurev-nutr-071813-105336
100. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation (2019) 139(10):e56–e528. doi: 10.1161/CIR.0000000000000659
101. Buja LM. Myocardial Ischemia and Reperfusion Injury. Cardiovasc Pathol (2005) 14(4):170–5. doi: 10.1016/j.carpath.2005.03.006
102. Yellon DM, Hausenloy DJ. Myocardial Reperfusion Injury. N Engl J Med (2007) 357(11):1121–35. doi: 10.1056/NEJMra071667
103. Cadenas S. ROS and Redox Signaling in Myocardial Ischemia-Reperfusion Injury and Cardioprotection. Free Radic Biol Med (2018) 117:76–89. doi: 10.1016/j.freeradbiomed.2018.01.024
104. Son Y, Kim S, Chung HT, Pae HO. Reactive Oxygen Species in the Activation of MAP Kinases. Methods Enzymol (2013) 528:27–48. doi: 10.1016/B978-0-12-405881-1.00002-1
105. Fan P, Xie XH, Chen CH, Peng X, Zhang P, Yang C, et al. Molecular Regulation Mechanisms and Interactions Between Reactive Oxygen Species and Mitophagy. DNA Cell Biol (2019) 38(1):10–22. doi: 10.1089/dna.2018.4348
106. Lambeth JD. NOX Enzymes and the Biology of Reactive Oxygen. Nat Rev Immunol (2004) 4(3):181–9. doi: 10.1038/nri1312
107. Russell RR 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-Activated Protein Kinase Mediates Ischemic Glucose Uptake and Prevents Postischemic Cardiac Dysfunction, Apoptosis, and Injury. J Clin Invest (2004) 114(4):495–503. doi: 10.1172/JCI19297
108. Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, et al. Macrophage Migration Inhibitory Factor Stimulates AMP-Activated Protein Kinase in the Ischaemic Heart. Nature (2008) 451(7178):578–82. doi: 10.1038/nature06504
109. Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, et al. Impaired Macrophage Migration Inhibitory Factor-AMP-Activated Protein Kinase Activation and Ischemic Recovery in the Senescent Heart. Circulation (2010) 122(3):282–92. doi: 10.1161/CIRCULATIONAHA.110.953208
110. Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and Activation of Heart PFK-2 by AMPK has a Role in the Stimulation of Glycolysis During Ischaemia. Curr Biol (2000) 10(20):1247–55. doi: 10.1016/S0960-9822(00)00742-9
111. Hue L, Rider MH. The AMP-Activated Protein Kinase: More Than an Energy Sensor. Essays Biochem (2007) 43:121–37. doi: 10.1042/bse0430121
112. Seo K, Ki SH, Shin SM. Sestrin2-AMPK Activation Protects Mitochondrial Function Against Glucose Deprivation-Induced Cytotoxicity. Cell Signal (2015) 27(7):1533–43. doi: 10.1016/j.cellsig.2015.03.003
113. Mohany KM, Al Rugaie O. Association of Serum Sestrin 2 and Betatrophin With Serum Neutrophil Gelatinase Associated Lipocalin Levels in Type 2 Diabetic Patients With Diabetic Nephropathy. J Diabetes Metab Disord (2020) 19(1):249–56. doi: 10.1007/s40200-020-00498-0
Keywords: Sestrin2, AMPK, mTOR, metabolism, metabolic diseases
Citation: Gong L, Wang Z, Wang Z and Zhang Z (2021) Sestrin2 as a Potential Target for Regulating Metabolic-Related Diseases. Front. Endocrinol. 12:751020. doi: 10.3389/fendo.2021.751020
Received: 31 July 2021; Accepted: 11 October 2021;
Published: 03 November 2021.
Edited by:
Xiaodong Sun, Affiliated Hospital of Weifang Medical University, ChinaReviewed by:
Zhen Wang, University of Mississippi Medical Center School of Dentistry, United StatesCopyright © 2021 Gong, Wang, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiguo Zhang, emhhbmd6aGlnQGpsdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.