
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 02 September 2021
Sec. Cellular Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.742215
This article is part of the Research TopicHypoxia, Oxidative Stress, and Endocrine CancersView all 8 articles
 Andria Rakotomalala1,2Alexandre Escande3,4,5Alessandro Furlan1,2
Andria Rakotomalala1,2Alexandre Escande3,4,5Alessandro Furlan1,2 Samuel Meignan1,2*Eric Lartigau3,4,5
Samuel Meignan1,2*Eric Lartigau3,4,5Radiotherapy is an important component of cancer treatment, with approximately 50% of all cancer patients receiving radiation therapy during their course of illness. Nevertheless, solid tumors frequently exhibit hypoxic areas, which can hinder therapies efficacy, especially radiotherapy one. Indeed, hypoxia impacts the six parameters governing the radiotherapy response, called the « six Rs of radiation biology » (for Radiosensitivity, Repair, Repopulation, Redistribution, Reoxygenation, and Reactivation of anti-tumor immune response), by inducing pleiotropic cellular adaptions, such as cell metabolism rewiring, epigenetic landscape remodeling, and cell death weakening, with significant clinical repercussions. In this review, according to the six Rs, we detail how hypoxia, and associated mechanisms and pathways, impact the radiotherapy response of solid tumors and the resulting clinical implications. We finally illustrate it in hypoxic endocrine cancers through a focus on anaplastic thyroid carcinomas.
Because of its high cytotoxic potential in solid tumors, radiation therapy is a standard of care in many solid tumors (1, 2). The mechanism of action relies on ROS production, notably through water radiolysis, and DNA damages induction, especially double strand breaks (DSBs), leading to cell death. Aiming at killing cancer cells while preserving healthy cells, radiotherapy is mainly delivered through fractionated schemes. The success of fractionated radiotherapy depends on multiple sub-cellular, cellular, and microenvironmental parameters, together referred to as the “5Rs of radiation therapy”: Repair (of irradiation-induced DNA damages), Redistribution (of cells within the cell cycle), Repopulation (of tumor cells after radiation), Reoxygenation (of the surviving cells) and, more recently, intrinsic Radiosensitivity (3). Moreover, it is now accepted that immune response could play a critical role in radiotherapy response, leading to the emergence of a 6th R: Reactivation (of the anti-tumor immune response) (4) (Figure 1).
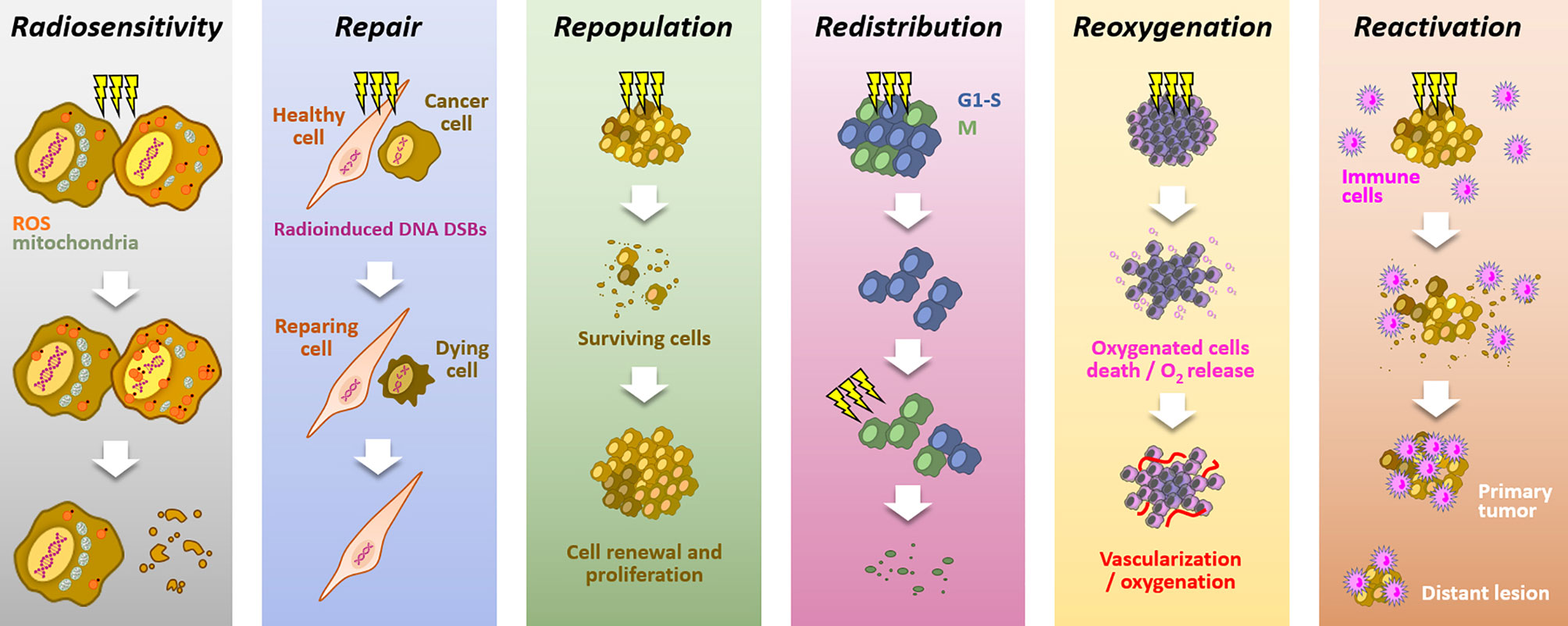
Figure 1 The six “Rs” dictating the response to radiotherapy. Radiotherapy response depends on six parameters: Radiosensitivity, refers to the cell-intrinsic mechanisms (e.g., metabolic adaption, ROS detoxification), explaining differences in cell responses to irradiations; Repair, refers to the cell capacity to survive by repairing radio-induced damages (particularly DSBs), in theory, more characteristic of healthy cells; Repopulation, refers to the tumor cells capacity to grow following a radiotherapy fraction; Redistribution, refers to the progression of cancer cells from radioresistant cell cycle phases (i.e., G1/S) toward more radiosensitive phases (i.e., G2/M), between radiotherapy fractions; Reoxygenation, refers to the oxygen level recovery following irradiation, due to well-oxygenated cells death and tumor vascularization; Reactivation, refers to the triggering of a systemic anti-tumor immune response following irradiation-induced immunogenic cell deaths.
However, numerous tumors fail to be controlled with radiotherapy, representing a significant cause of disease progression and mortality in cancer. There is a growing consensus that tumor heterogeneity is one of the principal explanations for treatment failure (5). In particular, oxygenation level is generally reduced and heterogenous within solid tumors, compared to the oxygenation found in associated healthy tissues (6, 7). In this sense, hypoxic regions are considered to be present in about 50% of solid tumors, and hypoxia is one of the most studied causes of radioresistance (8, 9). Indeed, hypoxia correlates with a poor prognosis after radiotherapy in various cancer types (10–12). Experimental evidence confirms the crucial impact of the low oxygen level (hypoxia) on the efficiency of cell irradiation.
Hypoxia is a consequence of the high tumor cell proliferation rate and the abnormal structure of the tumor vasculature (13). Depending on the distance of the blood vessels and their transient collapses, hypoxia can be chronic, meaning diffusion-limited, or acute, meaning transient and perfusion-limited. Basically, within most solid tumors, oxygen level fluctuates between physioxia (about 8% O2, i.e., 60 mmHg), hypoxia (about 1% O2, i.e., 7.5 mmHg), and anoxia (0% oxygen) (14). The unquestionable master regulators of the transcriptional response to hypoxia are the HIF transcription factors (Hypoxia Inducible Factor), especially HIF-1. The regulation of HIF-1 is very dynamic to adapt quickly to the oxygen concentration. HIF-1 is composed of two subunits, HIF-1α and HIF-1β. Under physioxia, HIF-1α is hydroxylated on 402 and/or 564 proline residues by oxygen-dependent prolyl hydroxylase domain proteins (PHD1-3). This hydroxylated form will interact with the tumor suppressor protein Von Hippel-Lindau (pVHL), which recruits an E3-ubiquitine-ligase complex tagging HIF-1α by polyubiquitination for proteasomal degradation. Thus, in the presence of oxygen, the HIF-1α subunit is continuously produced and degraded, which regulates HIF-1 activity. Under hypoxia, oxygen-dependent PHDs activity is attenuated, HIF-1α is thus stabilized and accumulates to finally translocate to the nucleus where it dimerizes with the constitutively expressed HIF-1β subunit. Thus formed, the HIF-1 heterodimer will then recruit transcriptional coactivators (p300/CBP) and bind to hypoxia response elements (HREs) present in various gene promoters to initiate a transcriptional program leading to cell adaptation to hypoxia, notably through induction of metabolic switch, oxygen transport increase, and angiogenesis. In the same family, HIF-2 presents a similar structure. As HIF-1, HIF-2 is a heterodimer of the HIF-2α subunit combined with the HIF-1β one. The oxygen-dependent alpha-subunit HIF-2α is similarly regulated to HIF-1α (Figure 2). HIF-1 and HIF-2 modulate their own set of target genes and appear differentially regulated according to the hypoxia level and duration (15). By their role in hypoxia adaption, both HIF factors have a pleiotropic impact on the cell response to irradiation. Supporting this, in head and neck squamous cell carcinomas, HIF-1α and HIF-2α expressions positively correlate with a higher rate of local failure after chemoradiotherapy treatment (16, 17). Of course, hypoxia can also impact cellular behaviors through HIF-independent mechanisms and pathways. For example, severe hypoxia can induce endoplasmic reticulum stress and thus activate UPR (Unfolded Protein Response) pathways.
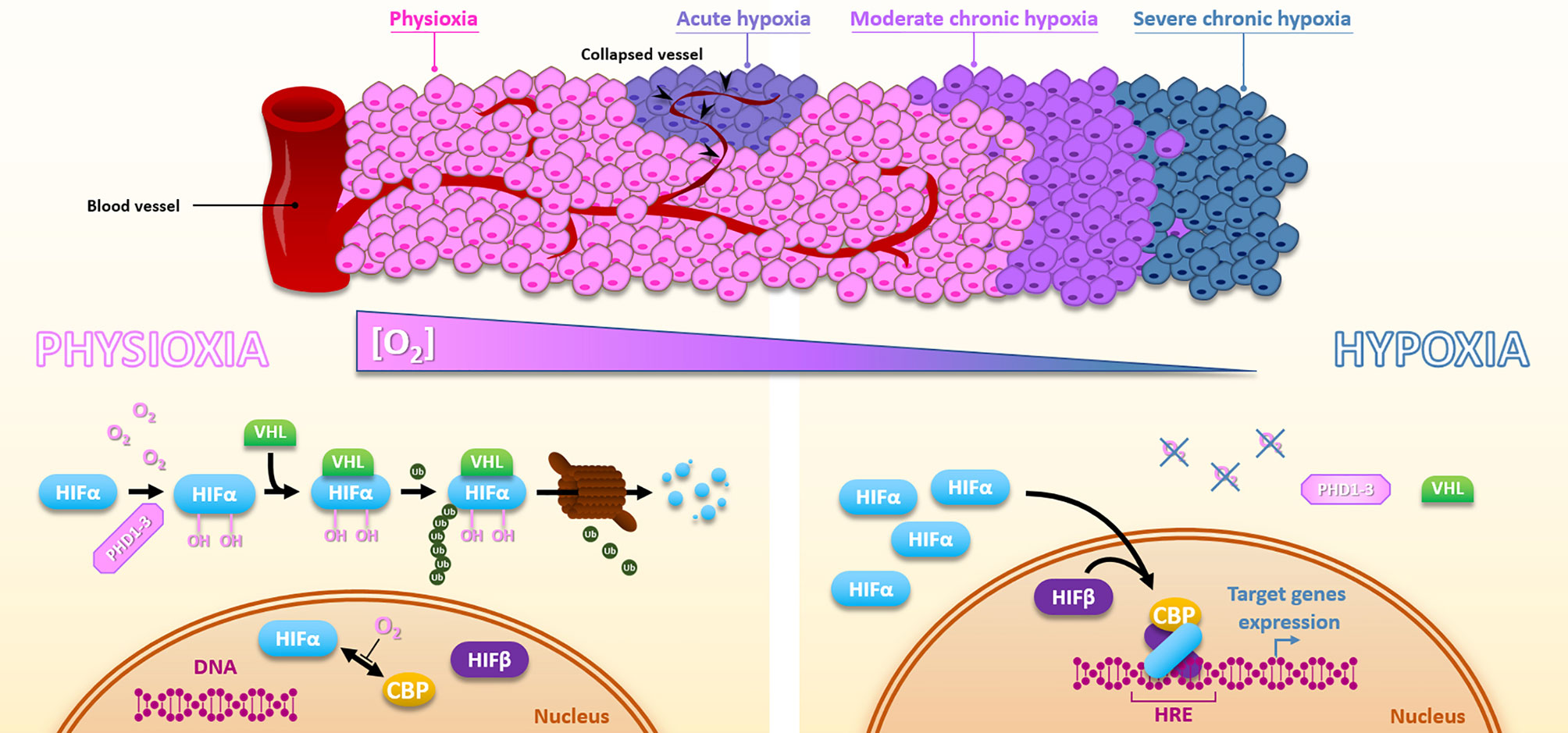
Figure 2 Hypoxia and HIFs regulation. Due to the distance to the blood vessels, cancer cells are well oxygenated or under moderate to severe chronic hypoxia. In addition, vessels collapse can cause sporadic acute hypoxic areas. Under physioxia, HIFα is hydroxylated by PHDs, poly-ubiquitinylated via VHL, and then degraded by the proteasomal way. Oxygen also inhibits HIFα-CBP interaction. Inversely, hypoxia inactivates PHDs, HIFα is subsequently stabilized, translocates into the nucleus, and forms a complex with HIFβ and CBP, which activates HREs-containing genes expression.
This review proposes to outline how low oxygen levels and hypoxia-associated tumor cell adaptions affect the six Rs of radiation therapy (here photon radiation therapy, the most widely used) and thereby its efficiency in solid tumors treatment.
“Radiosensitivity” defines the intrinsic sensitivity of tumor cells to radiation therapy. This property appears very heterogeneous within solid tumors and is impacted at two levels by hypoxia.
First, the molecular oxygen level directly impacts cell radiosensitivity via the “oxygen effect.” Actually, radiation-induced DNA damages are less likely to lead to cell killing under hypoxia (7). To explain that, the “oxygen fixation hypothesis” is based on the fact that oxygen reacts with radiation-induced DNA radicals to stabilize them. In a hypoxia or anoxia context, compounds containing sulfhydryl groups spontaneously reverse radiation-induced DNA radicals by reduction (18). In vitro, the “oxygen fixation hypothesis” is supported by the radioresistance induced by an oxygen depletion only during irradiation (19, 20). Another hypothesis formulated by Richardson and Harper suggests a central role for radiation impacts on mitochondria to explain the “oxygen effect.” In fact, radiations increase cell mitochondrial ROS content and disturb mitochondrial metabolism, which is more prejudicial for aerobic cells than hypoxic cells (21). The oxygen tension thus appears to be a determinant for the intrinsic tumor cell “Radiosensitivity,” and the “oxygen effect” may partly explain the radioresistant phenotype of hypoxic tumor areas.
Besides, hypoxia promotes additional pleiotropic cellular adaptions through hypoxia-associated signaling pathways, which can prime cancer cells for radiotherapy resistance. These include cell metabolism rewiring, ROS detoxification, autophagy, and resistance to cell death (Figure 3).
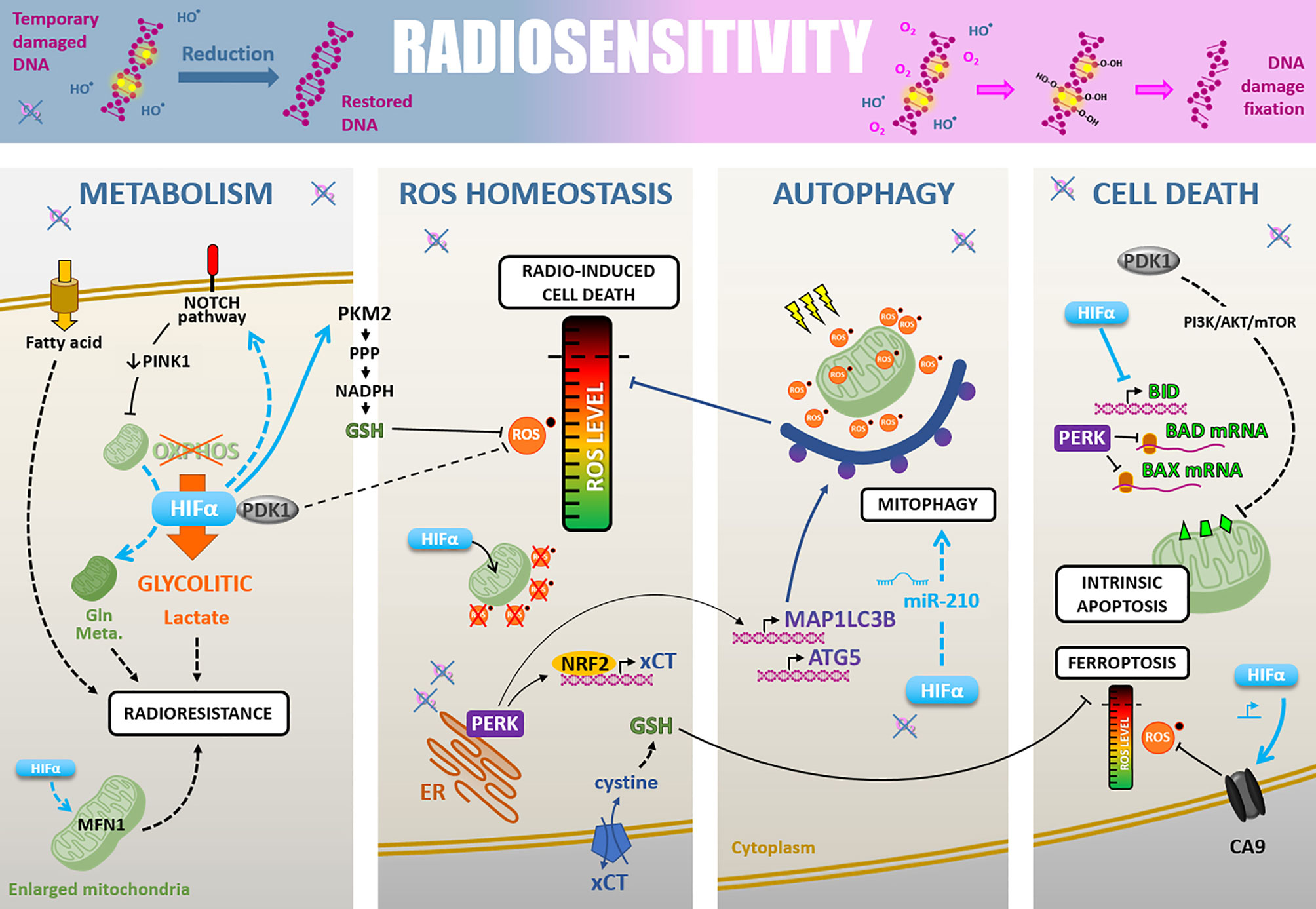
Figure 3 Radiosensitivity. Cell-intrinsic radiosensitivity can be linked to the fixation of radiation-induced DNA damages in the presence of oxygen, whereas a hypoxic environment favors the reduction of DNA radicals and return to a restored DNA (upper part). Hypoxia also impacts radiosensitivity through different cellular adaptions. i) The cell metabolism modifications, mainly PDK1-mediated glycolytic switch leading to lactate production, but also fatty acid uptake, NOTCH-dependent mitochondrial glutamine metabolism increase, and mitochondria fusion. ii) The ROS homeostasis is maintained under the critical level by PDK1 increase, HIFα-mediated GSH regeneration, HIFα translocation to mitochondria leading to their ROS production decrease, and PERK-mediated xCT expression promoting GSH synthesis. iii) Induction of HIFα- and PERK-mediated autophagy of ROS-productive damaged mitochondria. iiii) The cell death regulation through a HIFα-, PDK1- and PERK-mediated inhibition of antiapoptosis modulators BID, BAD, and BAX, and a HIFα-induced CA9 expression decreasing ROS level and inhibiting ferroptosis.
Hypoxia modifies cancer cell metabolism (22). Yet, cell metabolism changes strongly correlate with radioresistance in various cancer models (23). The consensus is that hypoxia drives a metabolic switch from an OXPHOS metabolism toward a glycolytic metabolism (24). This glycolytic flux leads to an increased lactate production which correlates with a particularly poor prognosis in HNSCC and uterine cervix cancers treated by radiotherapy (25, 26). Moreover, the lactate concentration positively correlates with tumor hypoxia and relative resistance to a fractionated radiation therapy regimen (30 fractions within six weeks) in HNSCC xenograft models (27, 28). Following that, under hypoxic conditions, HIF-1α knock-down diminishes the tumor lactate level, increases basal and maximal oxygen consumption rate (OCR), and sensitizes HNSCC xenografts to high-dose single-fraction radiotherapy (29).
At the molecular level, various factors involved in hypoxia-induced metabolic modulations confer radioresistant properties to cancer cells. Among them, pyruvate dehydrogenase kinase 1 (PDK1), a HIF-1 target gene, inactivates the pyruvate dehydrogenase (PDH) responsible for pyruvate to acetyl-CoA conversion to supply tricarboxylic acid cycle (TCA) (30). So, PDK1 inhibits OXPHOS metabolism and forces cells to rely on glycolytic metabolism. In HNSCC, PDK1 expression appears to have a critical role in maintaining glycolytic metabolism under hypoxia through a HIF-1α dependent way and correlates with a dismal prognosis (31). Following the example of PDK1, another mechanism involved in the OXPHOS down-regulation and tumor aggressiveness under hypoxia (1% O2) is the modification of mitochondrial mass and morphology by PINK1 down-regulation through HIF-1-mediated NOTCH signaling activation in hepatocellular carcinoma cells, a cancer type highly resistant to radiotherapy (32).
Although hypoxia favors glycolytic metabolism over mitochondrial OXPHOS, mitochondria still keep a role in cell death and radiation therapy resistance under hypoxia (33). Chronic hypoxia (72h, 1% O2) actually modifies mitochondria morphology through a mitochondria fusion excess mediated by a HIF-1α, Mitofusin I, BNIP3, and BNIP3L dependent way in several cancer cell types (i.e., colon, lung, cervix, renal cancer cells). These functional enlarged mitochondria confer a cell death resistance and could, by the way, prime hypoxic cancer cells to radioresistance (34, 35). Moreover, hypoxia can enhance mitochondrial glutamine metabolism of NSCLCC through a HIF-1α dependent manner and glutamine metabolism targeting can induce radiosensitization (36, 37).
Also, monounsaturated fatty acids are essential cellular compounds whose generation depends on oxygen. In lung adenocarcinoma cells, chronic severe hypoxia induces an adaptive up-regulation of SKG1 (human serum and glucocorticoid-inducible kinase) to promote unsaturated fatty acid uptake and thus shunt their oxygen-dependent generation. Matschke and colleagues showed that this hypoxia-linked adaption confers radioresistance in vitro in this model (38).
Metabolism and ROS homeostasis are closely associated, and hypoxia significantly impacts ROS detoxification through metabolism rewiring and other ways. For example, PDK1, which expression increases under hypoxia, affects cell metabolism and thus maintains a low basal ROS level in hypoxic cells (30). As radiotherapy efficacy relies on ROS production, hypoxia can favor tumor cell-intrinsic radioresistance by promoting ROS detoxification processes.
In addition, to sustain a glycolytic metabolism, hypoxia can support glycolysis branched pathways such as pentose phosphate pathway (PPP), which participate in cellular redox homeostasis (39, 40). Mechanistically, hypoxia (1% O2) induces PKM2 (Pyruvate Kinase M2) isoform expression in breast cancer cells through HIF-1α recruitment on PKM2 gene by KDM8 (41). The particularly weak activity of the PKM2 isoform slows the glycolytic flux down and favors glucose-6-phosphate accumulation to fuel PPP. During PPP, the 6-phosphogluconate dehydrogenase reduces NADP+ to NADPH which participates in reduced glutathione (GSH) regeneration, a cellular antioxidant highly involved in cellular ROS detoxification. So, by promoting PPP, hypoxia increases the cellular antioxidant potential and could counteract radiation therapy efficacy. Furthermore, severe cyclic hypoxia can increase GSH level through an up-regulation of two mitochondrial metabolite carriers, SLC25A1 and SLC25A10, in various cancer cell types (i.e., glioblastoma, lung, and prostate cancer). These factors participate in NADPH cellular pool maintenance and sustain mitochondrial metabolism under hypoxia, notably fatty acids uptake, which has been linked to the radioresistance phenomenon (38). Consequently, severe cyclic hypoxia is associated with reducing basal mitochondrial ROS content and a high ROS detoxification potential, conferring resistance to radiotherapy (42, 43).
Beyond their canonical role of transcription factors, HIFs also protect cells from oxidative stress more directly. Indeed, under hypoxia or during oxidative stress, mitochondrial translocation of HIF-1α reduces mitochondrial ROS production and protects cancer cells from cell death (44), and possibly from irradiation-induced mitochondrial ROS production. Following the example of HIF-1α, HIF-2α can also prevent ROS overload and protect renal carcinoma cells from irradiation-induced cell killing (45).
In addition to the HIF-dependent response, severe hypoxia leads to endoplasmic reticulum (ER) stress and induces unfolded protein response (UPR). The PERK (PRKR-like ER kinase) axis of the UPR is particularly involved in cell survival during severe hypoxia or anoxia. In a subcutaneous murine glioma model, the tumor hypoxic fraction resists a high single dose of radiation therapy by activating the PERK/eIF2α/GADD34c axis, when HIF-1α is rather involved in post-irradiation re-progression. This PERK axis drives glutamate-cystine antiporter xCT expression, promoting GSH synthesis and cell radioresistance (46). In this sense, it has been shown that PERK phosphorylates the oxidative homeostasis master regulator NRF2 and promotes its nuclear translocation driving the expression of antioxidant factors, notably xCT antiporter (47–49). Küper and colleagues have recently shown that NRF2 stabilizes HIF-1α by direct interaction and plays a critical role in pancreatic and lung cancer cell radioresistance under hypoxia (1% O2) in vitro (50).
In several cancer models, hypoxia exposure, as well as radiation treatment, lead to autophagy initiation (51). Autophagy preferentially occurs in hypoxic tumor regions and is observed along the entire gradient of hypoxia (52). Although autophagy can participate in radiation sensitivity in certain contexts, it is largely described as a cytoprotective mechanism, protecting the cell from extensive damages, notably after irradiation, by recycling damaged cellular components (e.g., mitochondria). Thus, hypoxia-induced autophagy can constitute a radioresistance mechanism in several cancer models, as demonstrated in osteosarcoma cells, a highly hypoxic and radioresistant cancer type (53, 54). Moreover, targeting specific autophagy-related genes (e.g., ATG3) can sensitize pharyngeal, breast, and rectum resistant cancer cell lines to single-dose and fractionated irradiations in vitro (55).
At the molecular level, during severe hypoxia (<0.02% O2), the PERK axis of UPR sustains autophagy by inducing MAP1LC3B (microtubule-associated protein 1 light chain 3β) and ATG5 (autophagy-related gene 5) expression in vitro and in vivo. This PERK-induced autophagy is a hypoxia-mediated adaption that diminishes cancer cell radiosensitivity, in vitro and in vivo (52). To a smaller extent, it has also been reported that the “hypoxamir” miR-210 can participate in the initiation of hypoxia-induced autophagy and promotes colon cancer cells radioresistance (56).
Finally, hypoxia-induced autophagy can highly reduce irradiation-linked oxidative stress and counteracts radiation therapy efficacy in various cancer cell models (e.g., osteosarcoma and NSCLC cells) (53, 57).
Radiation therapy relies on DNA damages and ROS overload induction to trigger cell death by various modalities (58). Among them, the cell death by mitotic catastrophe and the apoptosis processes are notably controlled by the BCL-2 family proteins, containing the pro-apoptotic members BID, BAD, and BAX (59). Yet, hypoxia (1-2% O2) and anoxia (<0.1% O2) correlate with the down-regulation of these proteins in colorectal cancer cells in vitro. In addition, these pro-apoptotic factors are widely absent from tumors hypoxic areas in vivo. Mechanistically, hypoxia-mediated BID down-regulation depends on its transcriptional repression by HIF-1α (60). Also, PDK1 (a HIF-1α target gene) drives PI3K/AKT/mTOR pathway activation, decreases BAX expression, and renders hepatocellular carcinoma cells resistant to irradiations (61). Likewise, the decrease of BAD and BAX protein levels can also be due to reducing their translation rate, possibly linked to the PERK activation observed under severe hypoxia (60). So, hypoxia can confer a radioresistant phenotype by down-regulate certain pro-apoptotic factors, which have been recently shown as essential for irradiation-induced apoptosis (58).
Besides apoptosis, recent studies suggest that other kinds of cell death are highly involved in radiation therapy efficacy. It’s notably the case for ferroptosis, an iron and ROS-dependent cell death (62), from which cancer cells are protected under hypoxia (63). Indeed, it has been shown that carbonic anhydrase 9 (CA9), a well-known target of HIFs factors, can participate in mesothelioma cells ferroptosis resistance by regulating ROS, lipid peroxidation, and mitochondrial Fe2+ levels under hypoxia (64). Also, by promoting ROS detoxification, notably through NRF2-mediated xCT expression (an anti-ferroptosis factor) and decreasing mitochondrial ROS production, hypoxia can counteract radiation-induced ferroptosis (65).
Since low oxygen level leads to selecting cells that can overcome and adapt to this stress, cyclic hypoxia promotes apoptosis-resistant cell expansion and further diminishes global radiation-induced cell death within the tumor. For example, cyclic hypoxia selects lung cancer cells resistant to the intrinsic apoptosis pathway. These selected cells harbor a radioresistant phenotype linked to a defective conformational change of BAX after irradiation (66). Also, a recent study defines hypoxia molecular hallmarks across 19 tumor types. It shows that hypoxic stress highly impacts the clonal evolution of tumors and drives an enrichment for specific molecular alterations (67). Among these hypoxic-enriched molecular alterations, TP53 loss of function is frequently observed and directly associated with an apoptotic potential loss. Of note, TP53 mutations drive the radioresistance phenomenon, notably in some pediatric high-grade gliomas characterized by their radioresistance (68).
To sum up, the low oxygenation both reduces radiotherapy efficacy via the physico-chemical “oxygen effect” and via hypoxia-induced biological adaptions. While hypoxia downplays the OXPHOS metabolism, the mitochondria are not inert and conserve a crucial role in hypoxia-mediated radioresistance phenomenon. The metabolic remodeling towards pathways known to enhance antioxidant protections, accompanied by increased autophagy, result in an exacerbate ability to maintain ROS homeostasis even after ionizing radiation. Last but not least, hypoxia drives and selects resistance to various radiation-induced cell death. Altogether, hypoxia undeniably enhances cancer cell’s intrinsic radioresistance and participates in radiotherapy treatment failure. In this frame, a phase I clinical trial (NCT01163487) evaluates tumor hypoxia by PET-scan in recurrent head and neck cancers and the possibility to use dichloroacetate (DCA), a drug targeting PDK1, in these tumors. Furthermore, a phase II clinical trial (NCT02432417) in glioblastoma assesses the efficiency of combining the standard chemoradiotherapy regimen with an autophagy inhibition by chloroquine. In this study, the hypoxia within the tumor is monitored by PET-scan imaging before and after the chloroquine treatment. A challenge for the effectiveness of autophagy-inhibition combined with radiotherapy remains to dichotomize contexts in which autophagy is cytoprotective rather than nonprotective, cytostatic, or cytotoxic.
Radiation therapy causes direct and indirect (via ROS) DNA damages. The “Repair” defines the deferential capability of normal versus tumor cells to rely on DNA damage response (DDR) pathways after a radiotherapy fraction. Among radiation-induced DNA damages, double-strand breaks (DSB) are those responsible for the higher part of radiotherapy-induced cell death (69).
In eukaryotic cells, two major pathways drive DSBs repair; the non-homologous end joining (NHEJ) in most of the cases and the high-fidelity homologous recombination repair (HRR) (70). An increase in those DSBs repair mechanisms’ efficiency correlates with cancer cell resistance to radiation therapy (71, 72). Several studies have shown that hypoxia impacts those DSBs repair mechanisms (7, 73, 74). Nevertheless, hypoxia globally has a versatile role on cells’ DSBs repair capacity in vitro, either promoting or reducing it, depending on cellular model, hypoxia level, duration, and alternation with reoxygenation. For example, Kumareswaran and colleagues showed that continuous and very severe hypoxia (0.2% O2), as well as anoxia, cause a defective irradiation-induced DSBs repair in non-cancerous human fibroblasts (19). On the contrary, Wozny et al. recently showed that continuous but less pronounced hypoxia (1% O2) leads to an increased NHEJ initiation and correlates with radioresistance in HNSCC cells (75).
Such discrepancies may find some explanations with various molecular mechanisms differentially impacted by hypoxia in different cellular contexts (Figure 4). On the one hand, it is frequently reported that hypoxia favors the initiation of DSBs repair mechanisms by increasing ATM (ataxia-telangiectasia mutated protein) and DNA-PKcs (DNA-dependent protein kinase catalytic subunit) expression and phosphorylation on ser1981 and ser2056, respectively. This notably occurs through MAPK, Scr, and AMPK signaling pathways (20, 76–78). Once activated, ATM and DNA-PKcs catalyze the phosphorylation of several substrates, notably histone H2AX at serine 139 (ɣH2AX), which signaled DSBs before effectors processing. Thus, hypoxia can prime cancer cells for DSBs repair initiation (79). In this sense, it has been recently demonstrated that hypoxia (1% O2) is associated with a higher ɣH2AX foci decay rate and enhanced RAD51 recruitment in irradiated HNSCC cells, suggesting a higher efficacy of HRR under hypoxia in this model (75, 80).
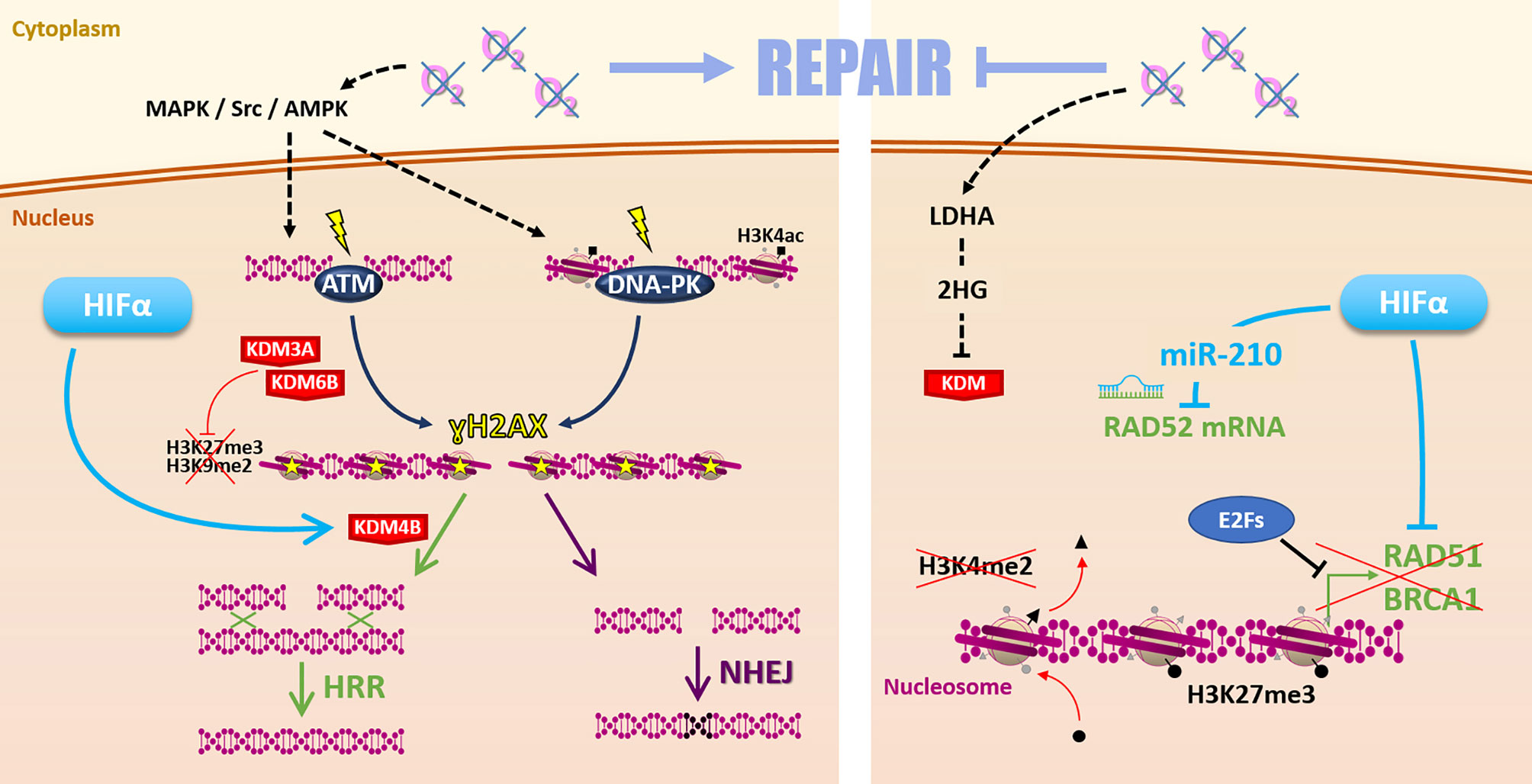
Figure 4 Repair. Hypoxia can favor radiation-induced DNA damages repair (left side) by MAPK/Src/AMPK-dependent ATM and DNA-PK activation. Hypoxia-induced modulation of KDM3A, KDM6B, and KDM4B expression changes the epigenetic landscape favoring DSBs repair by HRR or NHEJ. However, low oxygen levels can also affect the Repair (right side) notably via 2-hydroxyglutarate-dependent KDM inhibition. Moreover, HIFα and E2Fs inhibit HRR-related genes RAD51, BRCA1 expression. Hypoxia-induced epigenetic changes (as loss of H3K4me2 and gain of H3K27me3 marks) inhibit these genes’ expression. HIFα can also inhibit RAD52 mRNA through the miR-210.
Contrarily, on the other hand, hypoxia reduces the expression of several key HRR effectors such as NBN, MRE11, RAD51, and BRCA1 in different cancer cell models (20, 81, 82). Mechanistically, hypoxia-induced down-regulation of RAD51 and BRCA1 can be mediated by E2Fs factors (83, 84). Also, the reduced BRCA1 and RAD51 expressions in VHL-mutated cancers highlight the involvement of HIFs in HRR factors down-regulation and radiation-induced DSBs persistence (85). A mechanism of HRR genes down-regulation is driven by the HIFs-dependent miR-210, which targets RAD52 mRNA (86). Altogether, the hypothesis that hypoxia down-regulates HRR to favor fast but error-prone NHEJ mechanism to repair radiation-induced DSBs emerged in some models (84, 87).
Concerning NHEJ pathway effectors, oxygen level impacts remain elusive and appear highly dependent on hypoxia conditions (7, 73). For example, in prostate and breast cancer cells maintained under severe (0.2% O2) and moderate (3% O2) hypoxia, respectively, both HRR-related genes (e.g., RAD51 and BRCA1) and NHEJ-related genes (e.g., Ku70, LIG4, and XRCC4) are down-regulated (88, 89). On the contrary, in breast cancer, NSCLCC, and colon cancer cells, very severe hypoxia (0.01%O2) has no impact on NHEJ activity (82).
In addition, the deep relationship between hypoxia and epigenetic also impacts the “Repair” (90). Indeed, hypoxia-induced epigenetic modulations can directly impact the DDR orchestration, which relies on chromatin remodeling (91, 92). For example, hypoxia-enhanced acetylation at lysine 14 of histone 3 (H3K14ac) favors DNA-PK complex formation on DNA (79). This potentially primes hypoxic cells for post-irradiation DSBs repair initiation. It has also been shown that hypoxia can modify the epigenetic landscape (global loss of H3K27me3 and H3K9me2) by modulating epigenetic enzyme expressions, such as KDM3A and KDM6B and finally promotes DDR activity and radioresistance in HNSCC (80). Moreover, via HIFs, hypoxia can also increase KDM4B expression participating in HRR orchestration (93–95). Nevertheless, these KDM enzymes are α-ketoglutarate-dependent enzymes and are inhibited by L-2-hydroxyglutarate (2HG), a metabolite notably produced under hypoxia by lactate dehydrogenase A (LDHA) (96, 97). Thus, in that case, hypoxia-associated metabolism can also disturb HRR activity but by radiosensitizing cells (93). In this sense, hypoxia can also promote transcriptional silencing of the HRR-related genes BRCA1 and RAD51 through the induction of LSD1-mediated demethylation of the histone 3 lysine 4 (H3K4) and EZH2-mediated trimethylation of the histone 3 lysine 27 (H3K27) (98, 99).
Finally, hypoxia impacts “Repair” through various multimodal mechanisms leading to apparently antagonist cellular consequences. Globally, hypoxia appears to favor DSBs repair initiation and epigenetic orchestration but down-regulate HRR effectors. The consequent impact is highly dependent on the cancer model and hypoxia type. In this context, it seems complicated to develop a generalizable “Repair”-based therapeutic strategies to overcome hypoxic cells radioresistance. Still, a recent study demonstrated that, contrary to photon radiation therapy, hypoxia (1% O2) doesn’t enhance the repair of carbon ion irradiation-induced DSBs, suggesting that the choice of irradiation particles can overcome hypoxia-mediated repair enhancement in HNSCC (75). In this frame, a phase II clinical trial evaluates the benefit of an increased radiation dose and the use of carbon ion radiotherapy for hypoxic HNSCCs based on F-MISO-PET hypoxia-imaging (NCT03865277). Further studies in other cancer models and with different hypoxia parameters are needed to corroborate these promising findings.
The “Repopulation” defines the renewal and proliferation of surviving cancer cells following irradiation. This phenomenon represents one of the main reasons for the failure of conventionally fractionated radiation therapy.
It is now widely accepted that cancer stem cells (CSCs) are involved in cancer progression and post-radiotherapy tumor recurrence through their self-renewal properties (100). Also, CSCs are frequently associated with hypoxic niches which favor stemness maintenance (101, 102). Hypoxia, particularly chronic hypoxia, promotes the self-renewal capacity of persistent CSCs (103). For example, hypoxia maintains CD133-positive glioblastoma cells in an undifferentiated state and enhances their self-renewal activity through a HIF-1α dependent way (104). In addition, in these cells, HIF-2α overexpression under hypoxia also plays an essential role in maintaining self-renewal capacities in vitro and in vivo (105).
Mechanistically, hypoxia favors CSC self-renewal by stemness-associated signaling activation. In breast cancer cells, HIF-2α stabilization under chronic hypoxia actives the WNT/β-catenin signaling pathway (106). Hypoxia can also induce AKT phosphorylation and thus participate in CSCs self-renewal by the AKT/WNT/β-catenin axis (107–109). Furthermore, the WNT/β-catenin signaling pathway drives post-radiotherapy progression in xenograft models of prostate cancer cells overexpressing HIF-1α (110). Another stemness-associated pathway potentially responsible for hypoxia-enhanced repopulation is the NOTCH pathway. NOTCH signaling pathway appears highly responsible for HIF-mediated maintenance of self-renewal properties in glioblastoma and breast cancer cells (106, 111). As WNT/β-catenin, the NOTCH signaling pathway is preferentially activated under chronic hypoxia in a HIF-2α dependent way (106), even if HIF-1α can also activate the NOTCH pathway by stabilizing NICD through direct interaction (111). STAT3 activation is a third pathway by which hypoxia promotes CSCs expansion. Through a HIF-1α-dependent way, hypoxia modulates glioma cells secretome, which induces STAT3 phosphorylation and subsequently promotes CSC self-renewal capacity in vitro and in vivo (112). Following these studies, HIFs factors appear highly involved in glioblastoma stem cells’ self-renewal properties that could explain the post-radiotherapy progression characteristic of this tumor type.
Hypoxia also plays a role in repopulation by regulating cyclin expressions, notably via HIFs factors, thus promoting radiation-surviving cancer cell proliferation. HIF-2α can drive cyclin D1 and cyclin D2 expression, notably through c-MYC activation for the latter, and promote cancer cell proliferation (113, 114). Following the example of HIF-2α, HIF-1α drives NOCTH1-mediated cell proliferation through cyclin D1 expression in melanoma cells under severe hypoxia (0.5%) (115). Also, hypoxia can impact cyclin expressions via the control of epigenetic enzymes. Indeed, Kim and collaborators have shown that, under hypoxia (1% O2), KDM4B induces cyclin A1 expression and favors gastric cancer cell proliferation following irradiation (95).
To conclude, hypoxia widely impacts the “Repopulation” through HIFs master regulators (Figure 5). In several cancer models, chronic hypoxia and HIF-2α appear critical, either by favoring CSCs self-renewal via WNT and NOTCH signaling pathways or by modulating cyclin expressions through c-MYC activation for example. In this frame, the ɣ-secretase/NOTCH signaling pathway inhibitor RO4929097 has been tested in association with radiotherapy through several phase I/II clinical trials, notably for gliomas or breast cancers brain metastases (NCT01119599, NCT01217411). Nevertheless, these studies were prematurely terminated, and the clinical interest in inhibiting the NOTCH pathway in hypoxic tumors remains to be assessed.
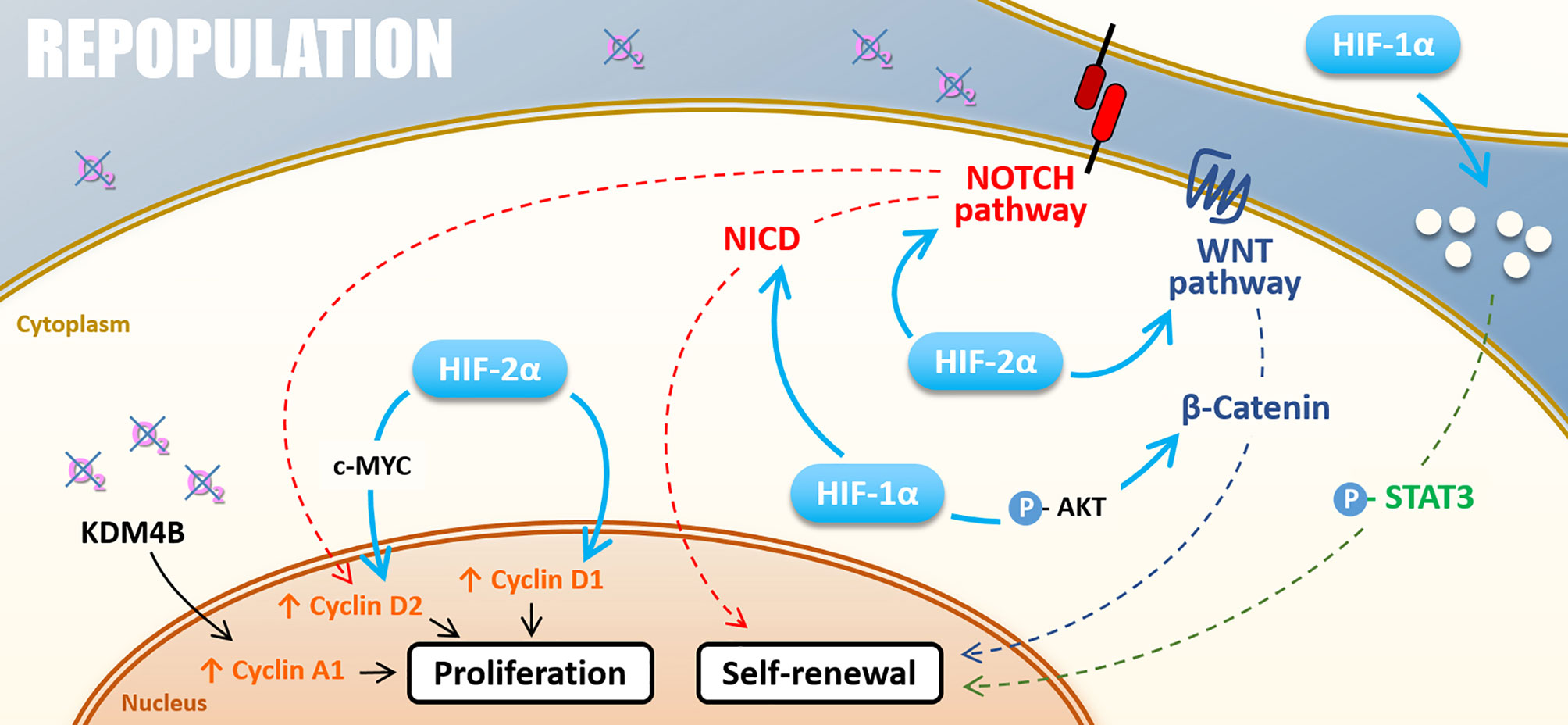
Figure 5 Repopulation. Under hypoxia, cell proliferation can be stimulated by KDM4B-dependant Cyclin A1 up-regulation and Cyclin D2 up-regulation driven by HIF-2α through c-MYC or thought NOTCH pathway under hypoxia. HIF-2α also directly induces Cyclin D1 expression. The self-renewal is positively regulated by NOTCH and WNT/β-catenin pathways activated by both HIF-1α and HIF-2α. HIF-1α also activates the STAT3 pathway and cell renewal through the modification of cell secretome.
Cancer cells exhibit differential radiosensitivity depending on the cell cycle phase. Because cells in mitosis already pass through the last G2/M cell cycle checkpoint, they are the most sensitive to a mitotic catastrophe following radiations. Those in the G1 and S-phases are, on the contrary, more radioresistant (116). “Redistribution” defines the fact that these cells that survive the first fraction of radiation will progress into a more sensitive phase of the cell cycle and will be more sensitive to the following fraction. In this context, hypoxia, in particular acute hypoxia, via HIF-1α stabilization, counteracts radiation therapy efficiency by favoring cell-cycle arrest in less sensible phases (i.e., G0/G1 and S-phases) (117).
Indeed, HIF-1α modulates many cell cycle regulation processes such as CHK1 (checkpoint kinase 1) activation and cyclin-dependent kinase inhibitor (CKI) expression like p21CIP1 (110). CHK1 and p21CIP1 are involved in cell cycle arrest in G1 or S-phase, by inhibiting CDC25A (cell division cycle 25A) activating-phosphatase or direct inhibition of the cyclin E/CDK2 complex, respectively. In this sense, in prostate cancer cells overexpressing HIF-1α, irradiation leads to a re-assortment in favor of G0/G1 and S-phase, in accordance with CHK1 phosphorylation and p21 up-regulation. This correlates with a radioresistant phenotype in vitro and in vivo (110). Mechanistically, under hypoxia (1% O2), HIF-1α displaces MYC from CDKN1A (p21CIP1) promoter and induces p21CIP1 up-regulation (118). Hypoxia also induces p27KIP1 and p57KIP2 up-regulation, two other CKIs targeting G1/S transition (119–121). In several cancer cell types (i.e., colon, cervical and hepatocellular cancer cells), another hypoxia-induced mechanism that leads to G0/G1 and S-phase accumulation is the HIF-1α dependent down-regulation of CDC25A protein level (122). In addition to these mechanisms, it can also be assumed that hypoxia-induced ATM activation (described in the “Repair” section), which activates CHK2 and thus inhibits CDC25A, participates to cell cycle re-assortment in less radiosensitive phases during hypoxia (123).
In addition to their implication in “Repopulation” through their renewal properties, CSCs present in hypoxic niches can as well impact “Redistribution.” Indeed, CSCs can harbor a quiescent behavior under hypoxia. In glioblastoma, CSCs of the peri-necrotic zone, expressing HIF-1α, harbor a quiescent phenotype highlighted by the suppression of the RNA polymerase II phosphorylation at serine 2 (RNApII-S2P), a marker of transcriptionally less active quiescent cells (124). Moreover, in breast cancer cells, cyclic hypoxia selects slow-cycling CSCs, which accumulate in G0/G1 phase and are rarer in G2/M phases (125). At the molecular level, Ju and colleagues have recently shown that hypoxia (1% O2) induces CSN8 (COP9 signalosome 8) expression, which correlates with several quiescence markers expression (i.e., NR2F1, DEC2, and p27KIP1), a decrease of MYC expression, and a lower level of ki67 in colorectal cancer cells (126). Thus, CSN8 appears to be involved in a hypoxia-induced quiescent state and could, in this context, impact cell response to irradiation.
To sum up, hypoxia can counteract the “Redistribution” after radiation therapy by favoring cell cycle arrest within most radioresistant cell cycle phases (i.e., G0/G1 and S-phase). Actually, HIF-1α promotes CKI expressions (such as p21CIP1) and CDC25A repression leading to cell cycle arrest in G0/G1 and S phases (Figure 6). In this sense, hypoxia can also maintain CSCs in a quiescent state. Nevertheless, determine in which context hypoxia counteracts the “Redistribution” (via cell cycle arrest) rather than favors the “Repopulation” (via cell expansion) after radiotherapy remains an open question.
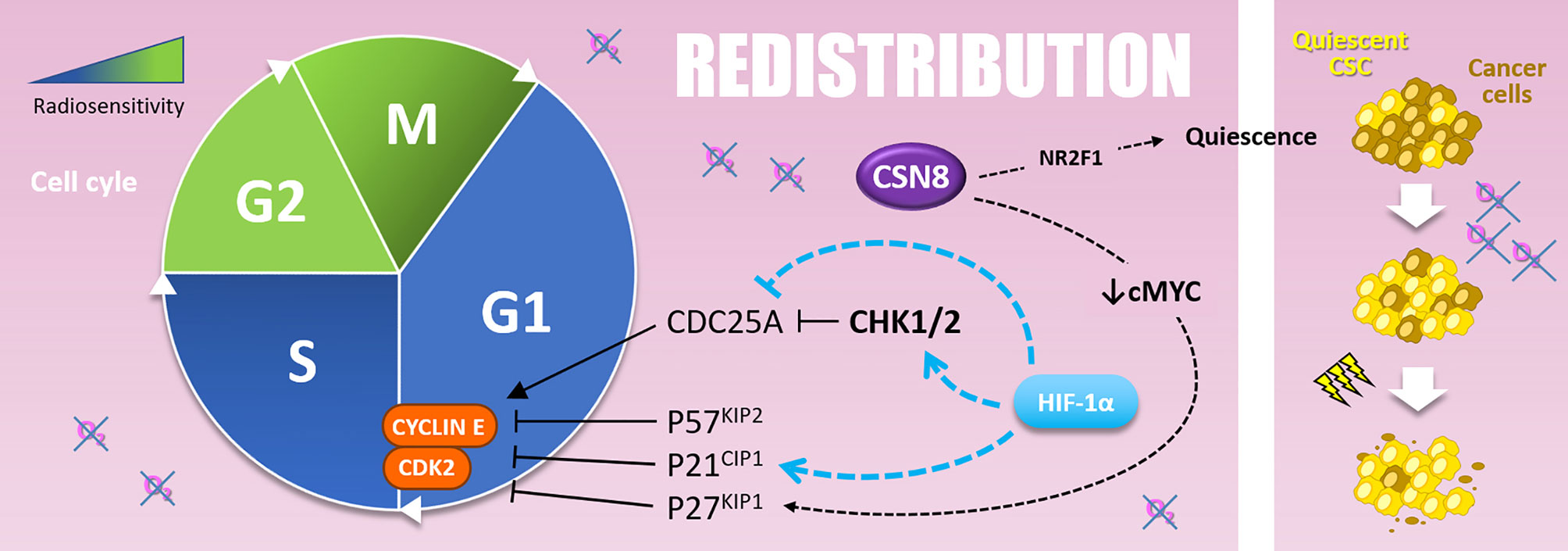
Figure 6 Redistribution. Cells are more resistant to radiotherapy in the G1 and, above all, in the S phase of the cell cycle. The G1-S transition is notably driven by the Cyclin E and CDK2, inhibited by P57KIP2, P21CIP1, and P27KIP1. Hypoxia upregulates P57KIP2 and P27KIP1 as well as P21CIP1 in a HIF-1α dependent manner. The overexpression of CSN8 correlates with cMYC decrease and P27KIP1 expression. It also correlates with NR2F1 quiescence marker expression. Quiescent CSC, selected by hypoxic stress, are more resistant to irradiation and participate in radioresistance (right side).
Hypoxic cells are up to three times more radioresistant than well-oxygenated ones. In this context, “Reoxygenation” defines the fact that, between radiotherapy fractions, well-oxygenated cells death leads to oxygen release, reduction of oxygen demand, and tumor bulk shrinkage allowing better oxygen diffusion and angiogenesis. Thus, the “Reoxygenation” turns back initially refractory hypoxic areas to a more radiosensitive state. Nevertheless, it is essential to note that irradiation and the subsequent reoxygenation induce oxidative stress that paradoxically stabilizes HIF-1 (127) (Figure 7).
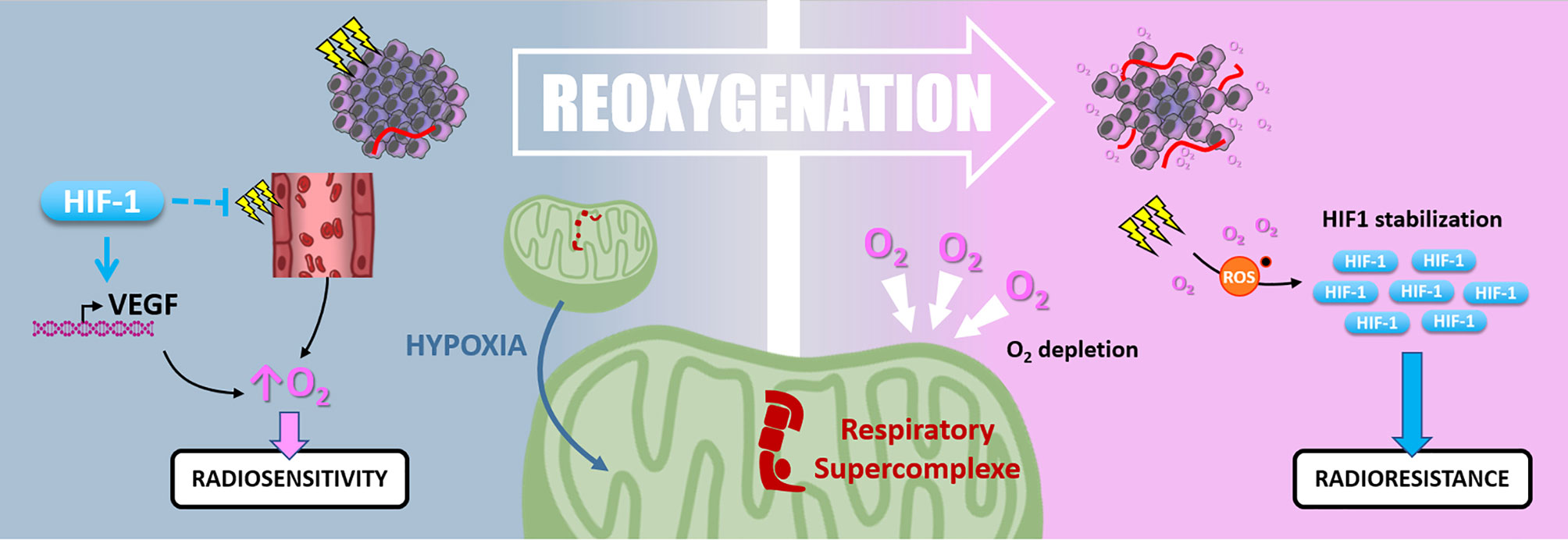
Figure 7 Reoxygenation. Radiotherapy induces tissue reoxygenation through O2 release and angiogenesis. Hypoxia also favors reoxygenation since HIF1 protects endothelial cells from irradiation and promotes VEGF expression. Nevertheless, hypoxia induces mitochondrial respiratory supercomplexes formation, which could persist after reoxygenation and cause high O2 consumption and depletion. Oxidative stress due to irradiation and reoxygenation stabilizes HIF-1, which can maintain radioresistance.
At first, hypoxia appears to favor the tumor reoxygenation as VEGF is a target gene of HIF factors, and HIF-1 protects tumor endothelial cells from cell death following irradiation. According to these observations, hypoxia supports the oxygen level recovery inside the tumor and thus could enhance radiotherapy response subsequently.
Harada and colleagues have shown that hypoxic tumor areas are drastically reoxygenated 24 hours following irradiation. Surprisingly, this reoxygenation leads to an increase of HIF-1 activity through a glucose-dependent AKT/mTOR signaling activation (128). Moreover, Moeller and colleagues described that in vivo, HIF-1 pharmacological inhibition at the time of the reoxygenation post-irradiation sensitizes tumor cells (127). So, even after oxygen level recovering, HIF-1α-driven radioresistance mechanisms can be maintained. In addition, the high oxygen demand of some cancer cells can reduce the global oxygen availability inside the tumor and maintain a hypoxic environment. In this frame, in pancreatic ductal adenocarcinomas (PDAC) cells, an adaption mechanism to survive nearly anoxic conditions is the formation of mitochondrial respiratory supercomplexes. In this context, PDAC cells conserve oxidative metabolism and maintain their oxygen consumption despite a very low oxygen level (129). It can be assumed that this kind of cell, containing respiratory supercomplexes, disturbs radiotherapy-induced reoxygenation by maintaining oxygen depletion. Interestingly, in a recent study by Taylor and colleagues, the extent of tumor reoxygenation appears heterogeneous after five fractions of radiotherapy in murine orthotopic pancreatic PDX (patient-derived xenograft) containing hypoxic areas. This study showed that the level of reoxygenation dictates tumor growth inhibition following radiation therapy (130).
Finally, the precise role of “Reoxygenation” on radiotherapy response appears ambiguous. On the one hand, the consensus is that the oxygen level increase radiosensitizes tumor cells via the “oxygen effect”. Nevertheless, on the other hand, reoxygenation induces radioresistance through hypoxia-related signaling (i.e., HIF-1 stabilization mediated by oxidative stress). Further studies are needed to determine whether initial tumor hypoxia affects the reoxygenation extent following radiotherapy, particularly fractionated protocols. Since the 60’s, the “Reoxygenation” concept gives rise to strategies aiming to increase tumor oxygenation during irradiation, such as using hyperbaric oxygen or administration of O2 at atmospheric pressure. Some of them were able to improve the local control of the tumor but appeared too difficult to set up in a clinical routine (131). More recently, several pre-clinical studies established a rationale for combining radiotherapy with HIF-1 inhibitor (132, 133), leading, for example, to a phase I clinical trial using PX-478 in advanced solid tumors (NCT00522652).
Radiotherapy has long been considered immunosuppressive through lymphocytes killing. Conversely, there is growing evidence assuming that irradiation efficacy may also rely on the induction of an anti-tumor immune response, depending on radiotherapy protocol, leading to a 6th R rising: the “Reactivation” of anti-tumor immune response (4). Supporting this idea, ablative (high dose) radiation therapy efficacy on melanoma xenograft is significantly reduced in immunodeficient nude mice than in immunocompetent models. Indeed, ablative radiotherapy increases tumor antigens presentation by dendritic cells (DCs), stimulating effector T-cells priming and expansion (134). Furthermore, high single-dose irradiation triggers DCs, macrophages, and primed T-cells infiltration in the irradiated tumor (135). As mentioned above, ionizing radiations lead to various types of cell death, including immunogenic cell deaths (e.g., necrosis, necroptosis), depending on the fraction dose. For example, a high dose, notably during ablative radiotherapy, can induce necrosis (136). Also, ionizing radiations can cause necroptosis, notably in endocrine cancers (e.g., anaplastic thyroid and adrenocortical cancers) (137). Moreover, radiation-induced DNA damages leading to the mitotic catastrophe can end in necrosis or senescence. Then senescent cells could, in some cases, undergo necrosis (58). Altogether, these radiation-induced immunogenic cell deaths can trigger an anti-tumor immune response following radiation therapy (138). Mechanistically, immunogenic cancer cell deaths release tumor-associated antigens. These tumor-associated antigens are processed by antigen-presenting cells, which stimulates effector T-cells and triggers a systemic anti-tumor immune response. This is in line with a clinical observation called the “abscopal effect,” which consists of an immune-dependent regression of distant lesions after irradiation of the primary tumor (139, 140). So, innate and adaptive immune response players are involved in this radiotherapy-driven anti-tumor response. Yet, hypoxia modulates several immune player functions (141, 142) and participates in establishing an immunosuppressive microenvironment (Figure 8).
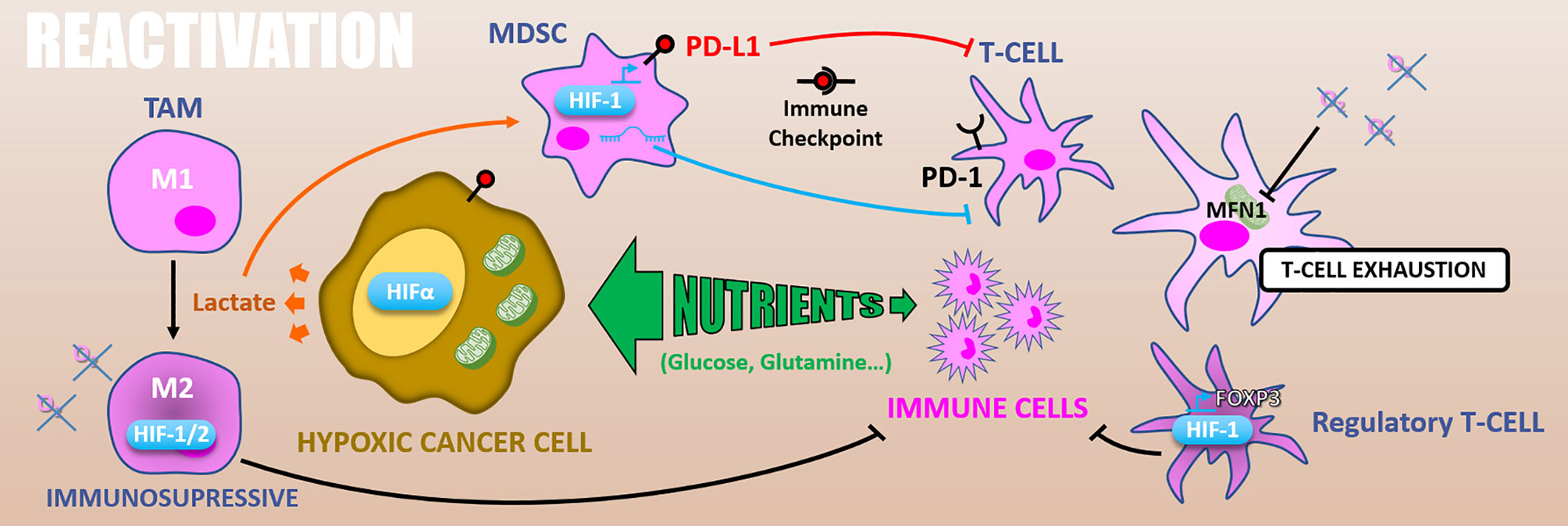
Figure 8 Reactivation. Because of exacerbated metabolism, hypoxic cancer cells consume nutrients from the microenvironment and starve immune cells. In addition, glycolytic metabolism produces lactate which polarizes TAMs in M2 immuno-suppressive and activates MDSCs inhibiting T-cells. HIFs expression in TAMs increases their immunosuppressive activity. Also, in MDSCs HIF-1 promotes PD-L1 expression that inhibits T-cell through PD-1/PD-L1 immune checkpoint. HIF-1α differentiates T-cells into immunosuppressive regulatory T-cells via FOXP3 expression. Finally, hypoxia can cause T-cells exhaustion by reducing their energetic metabolism through MFN1 down-regulation.
Besides, some nutrients (notably glucose and glutamine) essential for the anti-tumor function of immune cells (e.g., T-cells, macrophages) are depleted in the tumor microenvironment because of consumption by cancer cells (143). This phenomenon by which cancer cells starve immune ones is called “metabolic competition” and appears amplified by hypoxia-related cancer cell metabolic remodeling.
In addition, glycolytic metabolism of hypoxic tumors results in lactate accumulation in the tumor microenvironment, leading to tumor-associated macrophages (TAMs) polarization toward an M2 immuno-suppressive phenotype (144) and can attenuate the radiotherapy-driven immune response. Moreover, the HIF-1α dependent lactate production by pancreatic ductal adenocarcinoma (PDAC) cells induces the activation of myeloid-derived suppressor cells (MDSCs) that reduces effector T-cells expansion and anti-tumor activity. This lactate-driven MDSCs activation is thus a major contributor to PDAC radioresistance (145). Supporting this work, the combination of hypoxia-inducible pro-drug with immune checkpoint inhibitors in prostate adenocarcinoma murine models highly reduces MDSCs density and functions inside the tumor. It thus restores T-cell infiltration and their anti-tumor activity (146).
Hypoxia also has a direct impact on the immune effector’s metabolism. As mentioned above, effector T-cells, notably T CD8+ lymphocytes, are required for optimal radiation therapy response. However, in nasopharyngeal carcinoma tissues, hypoxia causes T-cells exhaustion by reducing their energetic metabolism through an MFN1 (Mitofusin 1) down-regulation via a miR-24 dependent repression of the MYC transcription factor (147). HIF factors have as well an impact on immune effector cells. For example, HIF-1α stabilization and miR-210 expression are involved in hypoxia-enhanced MDSCs immunosuppressive functions (145, 148, 149). Also, in murine transgenic breast cancer models, it has been shown that TAMs exert an HIF-1α dependent immunosuppressive effect triggering a hypoxia-induced suppression of T-cell functions. Indeed, a specific knock-out of HIF-1α in macrophages decreases tumor aggressiveness (150). Similarly, HIF-2α suppression in TAMs reduces the tumor aggressiveness in a murine colon carcinoma model, indicating a HIF-2α role in hypoxia-induced immunosuppression (151). HIF-1α also favors CD4+ T-cells differentiation into immunosuppressive regulatory T-cells by promoting FOXP3 transcription factor expression (152).
Another way by which hypoxia can counteract immune reactivation following ionizing radiations is by increasing immune checkpoints proteins expression. In this sense, PD-L1 (Programmed death 1 – ligand 1) is an HIF-1α target gene and is upregulated under hypoxia (0.1% O2) in MDSCs, in dendritic cells, as well as in cancer cells (153). Complementarily, hypoxia drives PD-1 (Programmed death 1) expression at the surface of T-cells and thus favors the PD-1/PD-L1 axis (147). Through CD86 upregulation in dendritic cells, hypoxia also promotes another well-known immune checkpoint, the CTLA4 (Cytotoxic T-lymphocyte-associated protein 4)/CD86 axis (154).
To sum up, radiation therapy induces immunogenic cancer cell deaths and can trigger a systemic anti-tumor immune response. However, hypoxia inside solid tumors counteracts this mechanism of action by promoting an immuno-suppression due to its impacts on cell metabolism and through HIFs factors, both in cancer and immune cells. As described in this section, hypoxia drives immune checkpoints proteins expression, encouraging the combination of radiotherapy with immune checkpoint inhibitors for patients with hypoxic tumors (155). Adding a hypoxia-inducible pro-drug to this treatment combination could durably restore anti-tumor immune response, and maximize radiotherapy response (146, 156).
As most solid tumors, endocrine cancers frequently exhibit hypoxic areas. Thyroid cancer is the most common endocrine malignancy. Anaplastic thyroid carcinomas (ATCs) account for 2-3% of thyroid gland neoplasms and are particularly aggressive tumors harboring a 3 to 4 months median survival (157). In this context, even though ATCs are often described as a radioresistant disease, adjuvant radiotherapy appears to increase the overall survival of patients with non-metastatic ATCs (158). Also, radiotherapy could offer a local control in advanced ATCs or for inoperable patients (159). Indeed, multimodal therapy with radiotherapy and chemotherapy (doxorubicin or cisplatin) is associated with the most extended survival and is considered a standard of care for these patients.
In ATCs, cancer cell metabolism relies on the reverse Warburg effect (i.e., lactate uptake to fuel OXPHOS metabolism), frequently related to a hypoxic state (160, 161). In their study, Nakajo and collaborators concluded that glucose metabolism but also hypoxic conditions might be associated with progression in patients with metastatic thyroid cancer following radioactive iodine treatment (iodine-131) (162). Based on their metabolic profile, particularly their exacerbated glycolytic flux, a clinical trial assesses the benefit of using 18F-FDG-PET-guided radiotherapy in refractory thyroid cancers (NCT03191643). Most significantly, ATCs are necrotic tumors, supporting their hypoxic property (163). This hypoxic environment has been described to increase thyroid CSC-enriched side population, which are deeply involved in the “Repopulation” and also in the “Redistribution” parameters (164). Regarding “repopulation”, some authors have described a better overall survival depending on the schedule fractionation used, particularly in the case of twice-daily treatment (165). Furthermore, another modality, neutron radiation therapy, could also be explored as its effectiveness may be less sensitive to hypoxia (166). In the clinic, neutrons provides better survival for thyroid cancer in small patient series (167). However, these studies need to be confirmed, given the large discrepancies about this topic in the literature.While HIF-1α is not detectable in healthy thyroids, ATCs exhibit high levels of nuclear HIF-1α and consistently overexpress the HIF target gene CA9 (168). Of note, as previously mentioned in the “Radiosensitivity” section, the CA9 is involved in cellular ROS homeostasis and can participate in irradiation-induced ferroptosis resistance. Additionally, HIF-1α silencing has been described to promote thyroid cancer cells apoptosis (169). Thereafter, Kim and colleagues showed that pharmacological targeting of HIF-1α can be a promising approach for thyroid cancers treatment (170). Based on this in vitro rationale, further studies are still needed to assess if HIFs inhibition could improve radiotherapy efficacy and thus represent a promising approach in the clinic.
Altogether, there is currently a huge amount of evidence on the critical role of hypoxia in radioresistance, via a strong modulation of the 6Rs of radiotherapy. Preclinical and clinical studies are ongoing, which should now further help us in defining the potential of hypoxia-related strategies aiming to improve the efficacy of radiation therapy against cancers.
AR, AE, AF, SM, and EL wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Buckley AM, Lynam-Lennon N, O’Neill H, O’Sullivan J. Targeting Hallmarks of Cancer to Enhance Radiosensitivity in Gastrointestinal Cancers. Nat Rev Gastroenterol Hepatol (2020) 17:298–313. doi: 10.1038/s41575-019-0247-2
2. Schaue D, McBride WH. Opportunities and Challenges of Radiotherapy for Treating Cancer. Nat Rev Clin Oncol (2015) 12:527–40. doi: 10.1038/nrclinonc.2015.120
3. Steel GG, McMillan TJ, Peacock JH. The 5Rs of Radiobiology. Int J Radiat Biol (1989) 56:1045–8. doi: 10.1080/09553008914552491
4. Boustani G, Laurent A, Mirjolet. The 6th R of Radiobiology: Reactivation of Anti-Tumor Immune Response. Cancers (2019) 11:860. doi: 10.3390/cancers11060860
5. Alfonso JCL, Berk L. Modeling the Effect of Intratumoral Heterogeneity of Radiosensitivity on Tumor Response Over the Course of Fractionated Radiation Therapy. Radiat Oncol (2019) 14:88. doi: 10.1186/s13014-019-1288-y
6. Muz B, de la Puente P, Azab F, Azab AK. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. HP (2015) 3:83–92. doi: 10.2147/HP.S93413
7. Begg K, Tavassoli M. Inside the Hypoxic Tumour: Reprogramming of the DDR and Radioresistance. Cell Death Discovery (2020) 6:77. doi: 10.1038/s41420-020-00311-0
8. Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Imaging Hypoxia to Improve Radiotherapy Outcome. Nat Rev Clin Oncol (2012) 9:674–87. doi: 10.1038/nrclinonc.2012.171
9. Horsman MR, Overgaard J. The Impact of Hypoxia and Its Modification of the Outcome of Radiotherapy. J Radiat Res (2016) 57:i90–8. doi: 10.1093/jrr/rrw007
10. Brizel DM, Sibley GS, Prosnitz LR, Scher RL, Dewhirst MW. Tumor Hypoxia Adversely Affects the Prognosis of Carcinoma of the Head and Neck. Int J Radiat Oncol Biol Phys (1997) 38:285–9. doi: 10.1016/S0360-3016(97)00101-6
11. Fyles AW, Milosevic M, Wong R, Kavanagh M-C, Pintilie M, Sun A, et al. Oxygenation Predicts Radiation Response and Survival in Patients With Cervix Cancer. Radiotherapy Oncol (1998) 48:149–56. doi: 10.1016/S0167-8140(98)00044-9
12. Milosevic M, Warde P, Ménard C, Chung P, Toi A, Ishkanian A, et al. Tumor Hypoxia Predicts Biochemical Failure Following Radiotherapy for Clinically Localized Prostate Cancer. Clin Cancer Res (2012) 18:2108–14. doi: 10.1158/1078-0432.CCR-11-2711
13. Rey S, Schito L, Koritzinsky M, Wouters BG. Molecular Targeting of Hypoxia in Radiotherapy. Adv Drug Deliv (2017) 109:45–62. doi: 10.1016/j.addr.2016.10.002
14. Barker HE, Paget JTE, Khan AA, Harrington KJ. The Tumour Microenvironment After Radiotherapy: Mechanisms of Resistance and Recurrence. Nat Rev Cancer (2015) 15:409–25. doi: 10.1038/nrc3958
15. Koh MY, Powis G. Passing the Baton: The HIF Switch. Trends Biochem Sci (2012) 37:364–72. doi: 10.1016/j.tibs.2012.06.004
16. Aebersold DM, Burri P, Beer KT, Laissue J, Djonov V, Greiner RH, et al. Expression of Hypoxia-Inducible Factor-1: A Novel Predictive and Prognostic Parameter in the Radiotherapy of Oropharyngeal Cancer. Cancer Res (2001) 61(7):2911–6.
17. Koukourakis MI, Simopoulos C, Gatter KC, Phil D, Harris AL, Phil D. Hypoxia-Inducible Factor (HIF1A and HIF2A), Angiogenesis, and Chemoradiotherapy Outcome of Squamous Cell Head-and-Neck Cancer. Int J Radiat Oncol Biol Phys (2002) 53(5):1192–202. doi: 10.1016/s0360-3016(02)02848-1
18. Wang H, Jiang H, Van De Gucht M, De Ridder M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers (2019) 11:112. doi: 10.3390/cancers11010112
19. Kumareswaran R, Ludkovski O, Meng A, Sykes J, Pintilie M, Bristow RG. Chronic Hypoxia Compromises Repair of DNA Double-Strand Breaks to Drive Genetic Instability. J Cell Sci (2012) 125:189–99. doi: 10.1242/jcs.092262
20. Hauth F, Toulany M, Zips D, Menegakis A. Cell-Line Dependent Effects of Hypoxia Prior to Irradiation in Squamous Cell Carcinoma Lines. Clin Trans Radiat Oncol (2017) 5:12–9. doi: 10.1016/j.ctro.2017.06.001
21. Richardson RB, Harper M-E. Mitochondrial Stress Controls the Radiosensitivity of the Oxygen Effect: Implications for Radiotherapy. Oncotarget (2016) 7:21469–83. doi: 10.18632/oncotarget.7412
22. Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-Modified Cancer Cell Metabolism. Front Cell Dev Biol (2019) 7:4. doi: 10.3389/fcell.2019.00004
23. Tang L, Wei F, Wu Y, He Y, Shi L, Xiong F, et al. Role of Metabolism in Cancer Cell Radioresistance and Radiosensitization Methods. J Exp Clin Cancer Res (2018) 37:87. doi: 10.1186/s13046-018-0758-7
24. Denko NC. Hypoxia, HIF1 and Glucose Metabolism in the Solid Tumour. Nat Rev Cancer (2008) 8:705–13. doi: 10.1038/nrc2468
25. Brizel DM, Walenta S, Mueller-Klieser W. Elevated Tumor Lactate Concentrations Predict for an Increased Risk of Metastases in Head-and-Neck Cancer. Int J Radiat Oncol Biol Phys (2001) 51:5. doi: 10.1016/S0360-3016(01)01630-3
26. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, et al. High Lactate Levels Predict Likelihood of Metastases, Tumor Recurrence, and Restricted Patient Survival in Human Cervical Cancers. Cancer Res (2000) 60:916–21.
27. Sattler UGA, Meyer SS, Quennet V, Hoerner C, Knoerzer H, Fabian C, et al. Glycolytic Metabolism and Tumour Response to Fractionated Irradiation. Radiotherapy Oncol (2010) 94:102–9. doi: 10.1016/j.radonc.2009.11.007
28. Quennet V, Yaromina A, Zips D, Rosner A, Walenta S, Baumann M, et al. Tumor Lactate Content Predicts for Response to Fractionated Irradiation of Human Squamous Cell Carcinomas in Nude Mice. Radiotherapy Oncol (2006) 81:130–5. doi: 10.1016/j.radonc.2006.08.012
29. Leung E, Cairns RA, Chaudary N, Vellanki RN, Kalliomaki T, Moriyama EH, et al. Metabolic Targeting of HIF-Dependent Glycolysis Reduces Lactate, Increases Oxygen Consumption and Enhances Response to High-Dose Single-Fraction Radiotherapy in Hypoxic Solid Tumors. BMC Cancer (2017) 17:418. doi: 10.1186/s12885-017-3402-6
30. Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
31. Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL. PDK-1 Regulates Lactate Production in Hypoxia and Is Associated With Poor Prognosis in Head and Neck Squamous Cancer. Br J Cancer (2008) 98:1975–84. doi: 10.1038/sj.bjc.6604356
32. Kung-Chun Chiu D, Pui-Wah Tse A, Law C-T, Ming-Jing Xu I, Lee D, Chen M, et al. Hypoxia Regulates the Mitochondrial Activity of Hepatocellular Carcinoma Cells Through HIF/HEY1/PINK1 Pathway. Cell Death Dis (2019) 10:934. doi: 10.1038/s41419-019-2155-3
33. McCann E, O’Sullivan J, Marcone S. Targeting Cancer-Cell Mitochondria and Metabolism to Improve Radiotherapy Response. Trans Oncol (2021) 14:100905. doi: 10.1016/j.tranon.2020.100905
34. Chiche J, Rouleau M, Gounon P, Brahimi-Horn MC, Pouysségur J, Mazure NM. Hypoxic Enlarged Mitochondria Protect Cancer Cells From Apoptotic Stimuli. J Cell Physiol (2009) 222(3):648–57. doi: 10.1002/jcp.21984
35. Grosso S, Doyen J, Parks SK, Bertero T, Paye A, Cardinaud B, et al. MiR-210 Promotes a Hypoxic Phenotype and Increases Radioresistance in Human Lung Cancer Cell Lines. Cell Death Dis (2013) 4:e544–4. doi: 10.1038/cddis.2013.71
36. Jiang Z-F, Wang M, Xu J-L, Ning Y-J. Hypoxia Promotes Mitochondrial Glutamine Metabolism Through HIF1α-GDH Pathway in Human Lung Cancer Cells. Biochem Biophys Res Commun (2017) 483:32–8. doi: 10.1016/j.bbrc.2017.01.015
37. Meijer TWH, Peeters WJM, Dubois LJ, van Gisbergen MW, Biemans R, Venhuizen J-H, et al. Targeting Glucose and Glutamine Metabolism Combined With Radiation Therapy in non-Small Cell Lung Cancer. Lung Cancer (2018) 126:32–40. doi: 10.1016/j.lungcan.2018.10.016
38. Matschke J, Wiebeck E, Hurst S, Rudner J, Jendrossek V. Role of SGK1 for Fatty Acid Uptake, Cell Survival and Radioresistance of NCI-H460 Lung Cancer Cells Exposed to Acute or Chronic Cycling Severe Hypoxia. Radiat Oncol (2016) 11:75. doi: 10.1186/s13014-016-0647-1
39. Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. IJMS (2019) 20:238. doi: 10.3390/ijms20020238
40. Sattler UGA, Mueller-Klieser W. The Anti-Oxidant Capacity of Tumour Glycolysis. Int J Radiat Biol (2009) 85:963–71. doi: 10.3109/09553000903258889
41. Wang H-J, Hsieh Y-J, Cheng W-C, Lin C-P, Lin Y-S, Yang S-F, et al. JMJD5 Regulates PKM2 Nuclear Translocation and Reprograms HIF-1 -Mediated Glucose Metabolism. Proc Natl Acad Sci (2014) 111:279–84. doi: 10.1073/pnas.1311249111
42. Hlouschek J, Hansel C, Jendrossek V, Matschke J. The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells With Tolerance to Cycling Severe Hypoxia. Front Oncol (2018) 8:170. doi: 10.3389/fonc.2018.00170
43. Hlouschek J, Ritter V, Wirsdörfer F, Klein D, Jendrossek V, Matschke J. Targeting SLC25A10 Alleviates Improved Antioxidant Capacity and Associated Radioresistance of Cancer Cells Induced by Chronic-Cycling Hypoxia. Cancer Lett (2018) 439:24–38. doi: 10.1016/j.canlet.2018.09.002
44. Li H-S, Zhou Y-N, Li L, Li S-F, Long D, Chen X-L, et al. HIF-1α Protects Against Oxidative Stress by Directly Targeting Mitochondria. Redox Biol (2019) 25:101109. doi: 10.1016/j.redox.2019.101109
45. Bertout JA, Majmundar AJ, Gordan JD, Lam JC, Ditsworth D, Keith B, et al. HIF2 Inhibition Promotes P53 Pathway Activity, Tumor Cell Death, and Radiation Responses. Proc Natl Acad Sci (2009) 106:14391–6. doi: 10.1073/pnas.0907357106
46. Rouschop KM, Dubois LJ, Keulers TG, van den Beucken T, Lambin P, Bussink J, et al. PERK/eIF2 Signaling Protects Therapy Resistant Hypoxic Cells Through Induction of Glutathione Synthesis and Protection Against ROS. Proc Natl Acad Sci (2013) 110:4622–7. doi: 10.1073/pnas.1210633110
47. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol Cell Biol (2003) 23:7198–209. doi: 10.1128/MCB.23.20.7198-7209.2003
48. Habib E, Linher-Melville K, Lin H-X, Singh G. Expression of xCT and Activity of System Xc– Are Regulated by NRF2 in Human Breast Cancer Cells in Response to Oxidative Stress. Redox Biol (2015) 5:33–42. doi: 10.1016/j.redox.2015.03.003
49. Salaroglio IC, Panada E, Moiso E, Buondonno I, Provero P, Rubinstein M, et al. PERK Induces Resistance to Cell Death Elicited by Endoplasmic Reticulum Stress and Chemotherapy. Mol Cancer (2017) 16:91. doi: 10.1186/s12943-017-0657-0
50. Küper A, Baumann J, Göpelt K, Baumann M, Sänger C, Metzen E, et al. Overcoming Hypoxia-Induced Resistance of Pancreatic and Lung Tumor Cells by Disrupting the PERK-NRF2-HIF-Axis. Cell Death Dis (2021) 12:82. doi: 10.1038/s41419-020-03319-7
51. Patel NH, Sohal SS, Manjili MH, Harrell JC, Gewirtz DA. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat Res (2020) 194:103. doi: 10.1667/RADE-20-00009
52. Rouschop KMA, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The Unfolded Protein Response Protects Human Tumor Cells During Hypoxia Through Regulation of the Autophagy Genes MAP1LC3B and ATG5. J Clin Invest (2010) 120:127–41. doi: 10.1172/JCI40027
53. Feng H, Wang J, Chen W, Shan B, Guo Y, Xu J, et al. Hypoxia-Induced Autophagy as an Additional Mechanism in Human Osteosarcoma Radioresistance. J Bone Oncol (2016) 5:67–73. doi: 10.1016/j.jbo.2016.03.001
54. Zou Y, Hu G, Zhao X, Lu T, Zhu F, Yu S, et al. Hypoxia-Induced Autophagy Contributes to Radioresistance via C-Jun-Mediated Beclin1 Expression in Lung Cancer Cells. J Huazhong Univ Sci Technol [Med Sci] (2014) 34:761–7. doi: 10.1007/s11596-014-1349-2
55. Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked Autophagy Sensitizes Resistant Carcinoma Cells to Radiation Therapy. Cancer Res (2008) 68:1485–94. doi: 10.1158/0008-5472.CAN-07-0562
56. Sun Y, Xing X, Liu Q, Wang Z, Xin Y, Zhang P, et al. Hypoxia-Induced Autophagy Reduces Radiosensitivity by the HIF-1α/miR-210/Bcl-2 Pathway in Colon Cancer Cells. Int J Oncol (2015) 46:750–6. doi: 10.3892/ijo.2014.2745
57. Chen X, Wang P, Guo F, Wang X, Wang J, Xu J, et al. Autophagy Enhanced the Radioresistance of non-Small Cell Lung Cancer by Regulating ROS Level Under Hypoxia Condition. Int J Radiat Biol (2017) 93:764–70. doi: 10.1080/09553002.2017.1325025
58. Adjemian S, Oltean T, Martens S, Wiernicki B, Goossens V, Vanden Berghe T, et al. Ionizing Radiation Results in a Mixture of Cellular Outcomes Including Mitotic Catastrophe, Senescence, Methuosis, and Iron-Dependent Cell Death. Cell Death Dis (2020) 11:1003. doi: 10.1038/s41419-020-03209-y
59. Castedo M, Perfettini J-L, Roumier T, Andreau K, Medema R, Kroemer G. Cell Death by Mitotic Catastrophe: A Molecular Definition. Oncogene (2004) 23:2825–37. doi: 10.1038/sj.onc.1207528
60. Erler JT, Cawthorne CJ, Williams KJ, Koritzinsky M, Wouters BG, Wilson C, et al. Hypoxia-Mediated Down-Regulation of Bid and Bax in Tumors Occurs via Hypoxia-Inducible Factor 1-Dependent and -Independent Mechanisms and Contributes to Drug Resistance. Mol Cell Biol (2004) 24:2875–89. doi: 10.1128/MCB.24.7.2875-2889.2004
61. Bamodu OA, Chang H-L, Ong J-R, Lee W-H, Yeh C-T, Tsai J-T. Elevated PDK1 Expression Drives PI3K/AKT/MTOR Signaling Promotes Radiation-Resistant and Dedifferentiated Phenotype of Hepatocellular Carcinoma. Cells (2020) 9:746. doi: 10.3390/cells9030746
62. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The Role of Ferroptosis in Ionizing Radiation-Induced Cell Death and Tumor Suppression. Cell Res (2020) 30:146–62. doi: 10.1038/s41422-019-0263-3
63. Fuhrmann DC, Mondorf A, Beifuß J, Jung M, Brüne B. Hypoxia Inhibits Ferritinophagy, Increases Mitochondrial Ferritin, and Protects From Ferroptosis. Redox Biol (2020) 36:101670. doi: 10.1016/j.redox.2020.101670
64. Li Z, Jiang L, Chew SH, Hirayama T, Sekido Y, Toyokuni S. Carbonic Anhydrase 9 Confers Resistance to Ferroptosis/Apoptosis in Malignant Mesothelioma Under Hypoxia. Redox Biol (2019) 26:101297. doi: 10.1016/j.redox.2019.101297
65. Fan Z, Wirth A-K, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 Pathway Promotes Cell Proliferation and Diminishes Ferroptosis. Oncogenesis (2017) 6:e371. doi: 10.1038/oncsis.2017.65
66. Weinmann M, Jendrossek V, Güner D, Goecke B, Belka C. Cyclic Exposure to Hypoxia and Reoxygenation Selects for Tumor Cells With Defects in Mitochondrial Apoptotic Pathways. FASEB J (2004) 18(15):1906–8. doi: 10.1096/fj.04-1918fje
67. Bhandari V, Hoey C, Liu LY, Lalonde E, Ray J, Livingstone J, et al. Molecular Landmarks of Tumor Hypoxia Across Cancer Types. Nat Genet (2019) 51:308–18. doi: 10.1038/s41588-018-0318-2
68. Werbrouck C, Barret E, Kergrohen T, Mondini M, Beccaria K, Boddaert N, et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin Cancer Res (2019) 25(22):6788–800. doi: 10.1158/1078-0432.CCR-19-0126
69. Frankenberg-Schwager M. Review of Repair Kinetics for DNA Damage Induced in Eukaryotic Cells In Vitro by Ionizing Radiation. Radiotherapy Oncol (1989) 14:307–20. doi: 10.1016/0167-8140(89)90143-6
70. Mladenov E, Magin S, Soni A, Iliakis G. DNA Double-Strand Break Repair as Determinant of Cellular Radiosensitivity to Killing and Target in Radiation Therapy. Front Oncol (2013) 3:113. doi: 10.3389/fonc.2013.00113
71. Ho V, Chung L, Singh A, Lea V, Abubakar A, Lim SH, et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) Complex in Rectal Cancer Correlates With Poor Response to Neoadjuvant Radiotherapy and Prognosis. BMC Cancer (2018) 18:869. doi: 10.1186/s12885-018-4776-9
72. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature (2006) 444:756–60. doi: 10.1038/nature05236
73. Kaplan AR, Glazer PM. Impact of Hypoxia on DNA Repair and Genome Integrity. Mutagenesis (2020) 35:61–8. doi: 10.1093/mutage/gez019
74. Chang WH, Lai AG. Transcriptional Landscape of DNA Repair Genes Underpins a Pan-Cancer Prognostic Signature Associated With Cell Cycle Dysregulation and Tumor Hypoxia. DNA Repair (2019) 78:142–53. doi: 10.1016/j.dnarep.2019.04.008
75. Wozny A-S, Alphonse G, Cassard A, Malésys C, Louati S, Beuve M, et al. Impact of Hypoxia on the Double-Strand Break Repair After Photon and Carbon Ion Irradiation of Radioresistant HNSCC Cells. Sci Rep (2020) 10:21357. doi: 10.1038/s41598-020-78354-7
76. Hashimoto T, Murata Y, Urushihara Y, Shiga S, Takeda K, Hosoi Y. Severe Hypoxia Increases Expression of ATM and DNA-PKcs and it Increases Their Activities Through Src and AMPK Signaling Pathways. Biochem Biophys Res Commun (2018) 505:13–9. doi: 10.1016/j.bbrc.2018.09.068
77. Marampon F, Gravina GL, Zani BM, Popov VM, Fratticci A, Cerasani M, et al. Hypoxia Sustains Glioblastoma Radioresistance Through ERKs/DNA-PKcs/HIF-1α Functional Interplay. Int J Oncol (2014) 44:2121–31. doi: 10.3892/ijo.2014.2358
78. Zhou J, Wu K, Gao D, Zhu G, Wu D, Wang X, et al. Reciprocal Regulation of Hypoxia-Inducible Factor 2α and GLI1 Expression Associated With the Radioresistance of Renal Cell Carcinoma. Int J Radiat Oncol Biol Phys (2014) 90:942–51. doi: 10.1016/j.ijrobp.2014.06.065
79. Bouquet F, Ousset M, Biard D, Fallone F, Dauvillier S, Frit P, et al. A DNA-Dependent Stress Response Involving DNA-PK Occurs in Hypoxic Cells and Contributes to Cellular Adaptation to Hypoxia. J Cell Sci (2011) 124:1943–51. doi: 10.1242/jcs.078030
80. Macedo-Silva C, Miranda-Gonçalves V, Lameirinhas A, Lencart J, Pereira A, Lobo J, et al. JmjC-KDMs KDM3A and KDM6B Modulate Radioresistance Under Hypoxic Conditions in Esophageal Squamous Cell Carcinoma. Cell Death Dis (2020) 11:1068. doi: 10.1038/s41419-020-03279-y
81. Cowman S, Fan YN, Pizer B, Sée V. Decrease of Nibrin Expression in Chronic Hypoxia Is Associated With Hypoxia-Induced Chemoresistance in Some Brain Tumour Cells. BMC Cancer (2019) 19:300. doi: 10.1186/s12885-019-5476-9
82. Bindra RS, Schaffer PJ, Meng A, Woo J, Lizardi P, Hedley DW, et al. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol Cell Biol (2004) 24(19):8504–18. doi: 10.1128/MCB.24.19.8504-8518.2004
83. Bindra RS, Glazer PM. Repression of RAD51 Gene Expression by E2F4/p130 Complexes in Hypoxia. Oncogene (2007) 26:2048–57. doi: 10.1038/sj.onc.1210001
84. Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, et al. Hypoxia-Induced Down-Regulation of BRCA1 Expression by E2Fs. Cancer Res (2005) 65:11597–604. doi: 10.1158/0008-5472.CAN-05-2119
85. Scanlon SE, Hegan DC, Sulkowski PL, Glazer PM. Suppression of Homology-Dependent DNA Double-Strand Break Repair Induces PARP Inhibitor Sensitivity in VHL -Deficient Human Renal Cell Carcinoma. Oncotarget (2018) 9:4647–60. doi: 10.18632/oncotarget.23470
86. Crosby ME, Kulshreshtha R, Ivan M, Glazer PM. MicroRNA Regulation of DNA Repair Gene Expression in Hypoxic Stress. Cancer Res (2009) 69:1221–9. doi: 10.1158/0008-5472.CAN-08-2516
87. Scanlon SE, Glazer PM. Multifaceted Control of DNA Repair Pathways by the Hypoxic Tumor Microenvironment. DNA Repair (2015) 32:180–9. doi: 10.1016/j.dnarep.2015.04.030
88. Meng AX, Jalali F, Cuddihy A, Chan N, Bindra RS, Glazer PM, et al. Hypoxia Down-Regulates DNA Double Strand Break Repair Gene Expression in Prostate Cancer Cells. Radiotherapy Oncol (2005) 76:168–76. doi: 10.1016/j.radonc.2005.06.025
89. Fanale D, Bazan V, Caruso S, Castiglia M, Bronte G, Rolfo C, et al. Hypoxia and Human Genome Stability: Downregulation of BRCA2 Expression in Breast Cancer Cell Lines. BioMed Res Int (2013) 2013:1–8. doi: 10.1155/2013/746858
90. Cabrera-Licona A, Pérez-Añorve IX, Flores-Fortis M, Moral-Hernández O, del, González-de la Rosa CH, Suárez-Sánchez R, et al. Deciphering the Epigenetic Network in Cancer Radioresistance. Radiotherapy Oncol (2021) 159:48–59. doi: 10.1016/j.radonc.2021.03.012
91. Camuzi D, de Amorim Í, Ribeiro Pinto L, Oliveira Trivilin L, Mencalha A, Soares Lima S. Regulation Is in the Air: The Relationship Between Hypoxia and Epigenetics in Cancer. Cells (2019) 8:300. doi: 10.3390/cells8040300
92. Karakaidos P, Karagiannis D, Rampias T. Resolving DNA Damage: Epigenetic Regulation of DNA Repair. Molecules (2020) 25:2496. doi: 10.3390/molecules25112496
93. Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate Produced by Neomorphic IDH Mutations Suppresses Homologous Recombination and Induces PARP Inhibitor Sensitivity. Sci Transl Med (2017) 9:eaal2463. doi: 10.1126/scitranslmed.aal2463
94. Hwang SY, Heo K, Kim JS, Im JW, Lee SM, Cho M, et al. Emodin Attenuates Radioresistance Induced by Hypoxia in HepG2 Cells via the Enhancement of PARP1 Cleavage and Inhibition of JMJD2B. Oncol Rep (2015) 33:1691–8. doi: 10.3892/or.2015.3744
95. Kim J-G, Yi JM, Park S-J, Kim J-S, Son TG, Yang K, et al. Histone Demethylase JMJD2B-Mediated Cell Proliferation Regulated by Hypoxia and Radiation in Gastric Cancer Cell. Biochim Biophys Acta (BBA) - Gene Regul Mech (2012) 1819:1200–7. doi: 10.1016/j.bbagrm.2012.10.001
96. Intlekofer AM, Dematteo RG, Venneti S, Finley LWS, Lu C, Judkins AR, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab (2015) 22:304–11. doi: 10.1016/j.cmet.2015.06.023
97. Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-Mediated Increases in L -2-Hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metab (2015) 22:291–303. doi: 10.1016/j.cmet.2015.06.021
98. Chang C-J, Yang J-Y, Xia W, Chen C-T, Xie X, Chao C-H, et al. EZH2 Promotes Expansion of Breast Tumor Initiating Cells Through Activation of RAF1-β-Catenin Signaling. Cancer Cell (2011) 19:86–100. doi: 10.1016/j.ccr.2010.10.035
99. Lu Y, Chu A, Turker MS, Glazer PM. Hypoxia-Induced Epigenetic Regulation and Silencing of the BRCA1 Promoter. Mol Cell Biol (2011) 31:3339–50. doi: 10.1128/MCB.01121-10
100. Liu Y, Yang M, Luo J, Zhou H. Radiotherapy Targeting Cancer Stem Cells “Awakens” Them to Induce Tumour Relapse and Metastasis in Oral Cancer. Int J Oral Sci (2020) 12:19. doi: 10.1038/s41368-020-00087-0
101. Emami Nejad A, Najafgholian S, Rostami A, Sistani A, Shojaeifar S, Esparvarinha M, et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int (2021) 21:62. doi: 10.1186/s12935-020-01719-5
102. Najafi M, Farhood B, Mortezaee K, Kharazinejad E, Majidpoor J, Ahadi R. Hypoxia in Solid Tumors: A Key Promoter of Cancer Stem Cell (CSC) Resistance. J Cancer Res Clin Oncol (2020) 146:19–31. doi: 10.1007/s00432-019-03080-1
103. Phillips TM, McBride WH, Pajonk F. The Response of CD24 –/Low/CD44 + Breast Cancer–Initiating Cells to Radiation. JNCI: J Natl Cancer Institute (2006) 98:1777–85. doi: 10.1093/jnci/djj495
104. Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia Promotes Expansion of the CD133-Positive Glioma Stem Cells Through Activation of HIF-1α. Oncogene (2009) 28:3949–59. doi: 10.1038/onc.2009.252
105. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell (2009) 15:501–13. doi: 10.1016/j.ccr.2009.03.018
106. Yan Y, Liu F, Han L, Zhao L, Chen J, Olopade OI, et al. HIF-2α Promotes Conversion to a Stem Cell Phenotype and Induces Chemoresistance in Breast Cancer Cells by Activating Wnt and Notch Pathways. J Exp Clin Cancer Res (2018) 37:256. doi: 10.1186/s13046-018-0925-x
107. Liu L, Zhu X-D, Wang W-Q, Shen Y, Qin Y, Ren Z-G, et al. Activation of β-Catenin by Hypoxia in Hepatocellular Carcinoma Contributes to Enhanced Metastatic Potential and Poor Prognosis. Clin Cancer Res (2010) 16:2740–50. doi: 10.1158/1078-0432.CCR-09-2610
108. Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, et al. Regulation of Mammary Stem/Progenitor Cells by PTEN/Akt/β-Catenin Signaling. PloS Biol (2009) 7:e1000121. doi: 10.1371/journal.pbio.1000121
109. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic Agents Increase Breast Cancer Stem Cells via the Generation of Tumor Hypoxia. Proc Natl Acad Sci (2012) 109:2784–9. doi: 10.1073/pnas.1018866109
110. Luo Y, Li M, Zuo X, Basourakos S, Zhang J, Zhao J, et al. β−Catenin Nuclear Translocation Induced by HIF−1α Overexpression Leads to the Radioresistance of Prostate Cancer. Int J Oncol (2018) 52(6):1827–40. doi: 10.3892/ijo.2018.4368
111. Qiang L, Wu T, Zhang H-W, Lu N, Hu R, Wang Y-J, et al. HIF-1α Is Critical for Hypoxia-Mediated Maintenance of Glioblastoma Stem Cells by Activating Notch Signaling Pathway. Cell Death Differ (2012) 19:284–94. doi: 10.1038/cdd.2011.95
112. Almiron Bonnin DA, Havrda MC, Lee MC, Liu H, Zhang Z, Nguyen LN, et al. Secretion-Mediated STAT3 Activation Promotes Self-Renewal of Glioma Stem-Like Cells During Hypoxia. Oncogene (2018) 37:1107–18. doi: 10.1038/onc.2017.404
113. Baba M, Hirai S, Yamada-Okabe H, Hamada K, Tabuchi H, Kobayashi K, et al. Loss of Von Hippel-Lindau Protein Causes Cell Density Dependent Deregulation of CyclinD1 Expression Through Hypoxia-Inducible Factor. Oncogene (2003) 22:2728–38. doi: 10.1038/sj.onc.1206373
114. Gordan JD, Bertout JA, Hu C-J, Diehl JA, Simon MC. HIF-2α Promotes Hypoxic Cell Proliferation by Enhancing C-Myc Transcriptional Activity. Cancer Cell (2007) 11:335–47. doi: 10.1016/j.ccr.2007.02.006
115. Bedogni B, Warneke JA, Nickoloff BJ, Giaccia AJ, Powell MB. Notch1 Is an Effector of Akt and Hypoxia in Melanoma Development. J Clin Invest (2008) 118:3660–70. doi: 10.1172/JCI36157
116. Pawlik TM, Keyomarsi K. Role of Cell Cycle in Mediating Sensitivity to Radiotherapy. Int J Radiat Oncol Biol Phys (2004) 59:928–42. doi: 10.1016/j.ijrobp.2004.03.005
117. Kato Y, Yashiro M, Fuyuhiro Y, Kashiwagi S, Matsuoka J, Hirakawa T, et al. Effects of Acute and Chronic Hypoxia on the Radiosensitivity of Gastric and Esophageal Cancer Cells. Anticancer Res (2011) 31(10):3369–75.
118. Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1α Induces Cell Cycle Arrest by Functionally Counteracting Myc. EMBO J (2004) 23:1949–56. doi: 10.1038/sj.emboj.7600196
119. Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV. Hypoxia Inhibits G1/S Transition Through Regulation of P27 Expression. J Biol Chem (2001) 276:7919–26. doi: 10.1074/jbc.M010189200
120. Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in Cartilage: HIF-1α Is Essential for Chondrocyte Growth Arrest and Survival. Genes Dev (2001) 15(21):2865–76. doi: 10.1101/gad.934301
121. Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-Inducible Factor 1α Is Essential for Cell Cycle Arrest During Hypoxia. Mol Cell Biol (2003) 23(1):359–69. doi: 10.1128/MCB.23.1.359-369.2003
122. Hammer S, To KKW, Yoo Y-G, Koshiji M, Huang LE. Hypoxic Suppression of the Cell Cycle Gene CDC25A in Tumor Cells. Cell Cycle (2007) 6:1919–26. doi: 10.4161/cc.6.15.4515
123. Gibson SL, Bindra RS, Glazer PM. Hypoxia-Induced Phosphorylation of Chk2 in an Ataxia Telangiectasia Mutated–Dependent Manner. Cancer Res (2005) 65:10734–41. doi: 10.1158/0008-5472.CAN-05-1160
124. Ishii A, Kimura T, Sadahiro H, Kawano H, Takubo K, Suzuki M, et al. Histological Characterization of the Tumorigenic “Peri-Necrotic Niche” Harboring Quiescent Stem-Like Tumor Cells in Glioblastoma. PloS One (2016) 11:e0147366. doi: 10.1371/journal.pone.0147366
125. Carcereri de Prati A, Butturini E, Rigo A, Oppici E, Rossin M, Boriero D, et al. Metastatic Breast Cancer Cells Enter Into Dormant State and Express Cancer Stem Cells Phenotype Under Chronic Hypoxia: Tumor Breast Dormancy in Hypoxic Microenvironment. J Cell Biochem (2017) 118:3237–48. doi: 10.1002/jcb.25972
126. Ju S, Wang F, Wang Y, Ju S. CSN8 Is a Key Regulator in Hypoxia-Induced Epithelial–Mesenchymal Transition and Dormancy of Colorectal Cancer Cells. Mol Cancer (2020) 19:168. doi: 10.1186/s12943-020-01285-4
127. Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation Activates HIF-1 to Regulate Vascular Radiosensitivity in Tumors: Role of Reoxygenation, Free Radicals, and Stress Granules. Cancer Cell (2004) 5(5):429–41. doi: 10.1016/s1535-6108(04)00115-1
128. Harada H, Itasaka S, Kizaka-Kondoh S, Shibuya K, Morinibu A, Shinomiya K, et al. The Akt/mTOR Pathway Assures the Synthesis of HIF-1α Protein in a Glucose- and Reoxygenation-Dependent Manner in Irradiated Tumors. J Biol Chem (2009) 284:5332–42. doi: 10.1074/jbc.M806653200
129. Hollinshead KER, Parker SJ, Eapen VV, Encarnacion-Rosado J, Sohn A, Oncu T, et al. Respiratory Supercomplexes Promote Mitochondrial Efficiency and Growth in Severely Hypoxic Pancreatic Cancer. Cell Rep (2020) 33:108231. doi: 10.1016/j.celrep.2020.108231
130. Taylor E, Zhou J, Lindsay P, Foltz W, Cheung M, Siddiqui I, et al. Quantifying Reoxygenation in Pancreatic Cancer During Stereotactic Body Radiotherapy. Sci Rep (2020) 10:1638. doi: 10.1038/s41598-019-57364-0
131. Rockwell S, Dobrucki I, Kim E, Marrison S, Vu V. Hypoxia and Radiation Therapy: Past History, Ongoing Research, and Future Promise. CMM (2009) 9:442–58. doi: 10.2174/156652409788167087
132. Harada H, Itasaka S, Zhu Y, Zeng L, Xie X, Morinibu A, et al. Treatment Regimen Determines Whether an HIF-1 Inhibitor Enhances or Inhibits the Effect of Radiation Therapy. Br J Cancer (2009) 100:747–57. doi: 10.1038/sj.bjc.6604939
133. Kabakov AE, Yakimova AO. Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing. Cancers (2021) 13:1102. doi: 10.3390/cancers13051102
134. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic Effects of Ablative Radiation on Local Tumor Require CD8+ T Cells: Changing Strategies for Cancer Treatment. Blood (2009) 114:589–95. doi: 10.1182/blood-2009-02-206870
135. Lugade AA, Moran JP, Gerber SA, Rose C, Frelinger JG, Lord EM. Local Radiation Therapy of B16 Melanoma Tumors Increases the Generation of Tumor Antigen-Specific Effector Cells That Traffic to the Tumor. J Immunol (2005) 174(12):7516–23. doi: 10.4049/jimmunol.174.12.7516
136. Lauber K. Dying Cell Clearance and Its Impact on the Outcome of Tumor Radiotherapy. Front Oncol (2012) 2:116. doi: 10.3389/fonc.2012.00116
137. Nehs MA. Necroptosis Is a Novel Mechanism of Radiation-Induced Cell Death in Anaplastic Thyroid and Adrenocortical Cancers. Surgery (2011) 150(6):1032–9. doi: 10.1016/j.surg.2011.09.012
138. Pasparakis M, Vandenabeele P. Necroptosis and Its Role in Inflammation. Nature (2015) 517(7534):311–20. doi: 10.1038/nature14191
139. Garelli E, Rittmeyer A, Putora PM, Glatzer M, Dressel R, Andreas S. Abscopal Effect in Lung Cancer: Three Case Reports and a Concise Review. Immunotherapy (2019) 11:1445–61. doi: 10.2217/imt-2019-0105
140. Dagoglu N, Karaman S, Caglar HB, Oral EN. Abscopal Effect of Radiotherapy in the Immunotherapy Era: Systematic Review of Reported Cases. Cureus (2019) 11(2):e4103. doi: 10.7759/cureus.4103
141. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The Hypoxic Tumour Microenvironment. Oncogenesis (2018) 7:10. doi: 10.1038/s41389-017-0011-9
142. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: A Key Player in Antitumor Immune Response. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol-Cell Physiol (2015) 309:C569–79. doi: 10.1152/ajpcell.00207.2015
143. Riera-Domingo C, Audigé A, Granja S, Cheng W-C, Ho P-C, Baltazar F, et al. Immunity, Hypoxia, and Metabolism–The Ménage À Trois of Cancer: Implications for Immunotherapy. Physiol Rev (2020) 100:1–102. doi: 10.1152/physrev.00018.2019
144. Colegio OR. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
145. Yang X, Lu Y, Hang J, Zhang J, Zhang T, Huo Y, et al. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol Res (2020) 8:1440–51. doi: 10.1158/2326-6066.CIR-20-0111
146. Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted Hypoxia Reduction Restores T Cell Infiltration and Sensitizes Prostate Cancer to Immunotherapy. J Clin Invest (2018) 128:5137–49. doi: 10.1172/JCI96268
147. Liu Y-N, Yang J-F, Huang D-J, Ni H-H, Zhang C-X, Zhang L, et al. Hypoxia Induces Mitochondrial Defect That Promotes T Cell Exhaustion in Tumor Microenvironment Through MYC-Regulated Pathways. Front Immunol (2020) 11:1906. doi: 10.3389/fimmu.2020.01906
148. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn J-I, Cheng P, et al. HIF-1α Regulates Function and Differentiation of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. J Exp Med (2010) 207:2439–53. doi: 10.1084/jem.20100587
149. Noman MZ, Janji B, Hu S, Wu JC, Martelli F, Bronte V, et al. Tumor-Promoting Effects of Myeloid-Derived Suppressor Cells Are Potentiated by Hypoxia-Induced Expression of miR-210. Cancer Res (2015) 75:3771–87. doi: 10.1158/0008-5472.CAN-15-0405
150. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage Expression of Hypoxia-Inducible Factor-1α Suppresses T-Cell Function and Promotes Tumor Progression. Cancer Res (2010) 70:7465–75. doi: 10.1158/0008-5472.CAN-10-1439
151. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan L-J, et al. Hypoxia-Inducible Factor 2α Regulates Macrophage Function in Mouse Models of Acute and Tumor Inflammation. J Clin Invest (2010) 120:2699–714. doi: 10.1172/JCI39506
152. Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J. Hypoxia Controls CD4+CD25+ Regulatory T-Cell Homeostasis via Hypoxia-Inducible Factor-1α. Eur J Immunol (2008) 38:2412–8. doi: 10.1002/eji.200838318
153. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 Is a Novel Direct Target of HIF-1α, and Its Blockade Under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J Exp Med (2014) 211:781–90. doi: 10.1084/jem.20131916
154. Köhler T, Reizis B, Johnson RS, Weighardt H, Förster I. Influence of Hypoxia-Inducible Factor 1α on Dendritic Cell Differentiation and Migration: Immunomodulation. Eur J Immunol (2012) 42:1226–36. doi: 10.1002/eji.201142053
155. Eckert F, Zwirner K, Boeke S, Thorwarth D, Zips D, Huber SM. Rationale for Combining Radiotherapy and Immune Checkpoint Inhibition for Patients With Hypoxic Tumors. Front Immunol (2019) 10:407. doi: 10.3389/fimmu.2019.00407
156. Bailleul Q, Navarin P, Arcicasa M, Bal-Mahieu C, Carcaboso AM, Le Bourhis X, et al. Evofosfamide Is Effective Against Pediatric Aggressive Glioma Cell Lines in Hypoxic Conditions and Potentiates the Effect of Cytotoxic Chemotherapy and Ionizing Radiations. Cancers (2021) 13:1804. doi: 10.3390/cancers13081804
157. Lin B, Ma H, Ma M, Zhang Z, Sun Z, Hsieh I, et al. The Incidence and Survival Analysis for Anaplastic Thyroid Cancer: A SEER Database Analysis. Am J Transl Res (2019) 11(9):5888–96.
158. Saeed NA, Kelly JR, Deshpande HA, Bhatia AK, Burtness BA, Judson BL, et al. Adjuvant External Beam Radiotherapy for Surgically Resected, Nonmetastatic Anaplastic Thyroid Cancer. Head Neck (2020) 42:1031–44. doi: 10.1002/hed.26086
159. Augustin T, Oliinyk D, Rauch J, Koehler VF, Spitzweg C, Belka C, et al. Radiation to the Primary Tumor in Metastatic Anaplastic Thyroid Cancer. In Vivo (2021) 35:461–5. doi: 10.21873/invivo.12279
160. Johnson JM, Lai SY, Cotzia P, Cognetti D, Luginbuhl A, Pribitkin EA, et al. Mitochondrial Metabolism as a Treatment Target in Anaplastic Thyroid Cancer. Semin Oncol (2015) 42:915–22. doi: 10.1053/j.seminoncol.2015.09.025
161. Gill K S, Tassone P. Thyroid Cancer Metabolism: A Review. Thyroid Disord Ther (2016) 05:200. doi: 10.4172/2167-7948.1000200
162. Nakajo M, Jinguji M, Tani A, Kajiya Y, Nandate T, Kitazano I, et al. [18f]-FDG-PET/CT and [18F]-FAZA-PET/CT Hypoxia Imaging of Metastatic Thyroid Cancer: Association With Short-Term Progression After Radioiodine Therapy. Mol Imaging Biol (2020) 22:1609–20. doi: 10.1007/s11307-020-01516-6
163. Lee JW, Yoon DY, Choi CS, Chang SK, Yun EJ, Seo YL, et al. Anaplastic Thyroid Carcinoma: Computed Tomographic Differentiation From Other Thyroid Masses. Acta Radiol (2008) 49:321–7. doi: 10.1080/02841850701813120
164. Mahkamova K, Latar N, Aspinall S, Meeson A. Hypoxia Increases Thyroid Cancer Stem Cell-Enriched Side Population. World J Surg (2018) 42:350–7. doi: 10.1007/s00268-017-4331-x
165. Jacobsen A-B, Grøholt KK, Lorntzsen B, Osnes TA, Falk RS, Sigstad E. Anaplastic Thyroid Cancer and Hyperfractionated Accelerated Radiotherapy (HART) With and Without Surgery. Eur Arch Otorhinolaryngol (2017) 274:4203–9. doi: 10.1007/s00405-017-4764-8
166. Warenius HM, White R, Peacock JH, Hanson J, Britten RA, Murray D. The Influence of Hypoxia on the Relative Sensitivity of Human Tumor Cells to 62.5 MeV (P→Be) Fast Neutrons and 4 MeV Photons. Radiat Res (2000) 154:54–63. doi: 10.1667/00337587(2000)154[0054:TIOHOT]2.0.CO;2
167. Chapman TR, Laramore GE, Bowen SR, Orio PF. Neutron Radiation Therapy for Advanced Thyroid Cancers. Adv Radiat Oncol (2016) 1:148–56. doi: 10.1016/j.adro.2016.05.001
168. Burrows N, Resch J, Cowen RL, von Wasielewski R, Hoang-Vu C, West CM, et al. Expression of Hypoxia-Inducible Factor 1α in Thyroid Carcinomas. Endocrine-Related Cancer (2010) 17:61–72. doi: 10.1677/ERC-08-0251
169. Ding Z-Y, Huang Y-J, Tang J-D, Li G, Jiang P-Q, Wu H-T. Silencing of Hypoxia-Inducible Factor-1α Promotes Thyroid Cancer Cell Apoptosis and Inhibits Invasion by Downregulating WWP2, WWP9, VEGF and VEGFR2. Exp Ther Med (2016) 12:3735–41. doi: 10.3892/etm.2016.3826
Keywords: radiotherapy, hypoxia, radioresistance, HIF, thyroid cancer
Citation: Rakotomalala A, Escande A, Furlan A, Meignan S and Lartigau E (2021) Hypoxia in Solid Tumors: How Low Oxygenation Impacts the “Six Rs” of Radiotherapy. Front. Endocrinol. 12:742215. doi: 10.3389/fendo.2021.742215
Received: 15 July 2021; Accepted: 13 August 2021;
Published: 02 September 2021.
Edited by:
Ralf Jockers, Université de Paris, FranceReviewed by:
Rosalba Senese, University of Campania Luigi Vanvitelli, ItalyCopyright © 2021 Rakotomalala, Escande, Furlan, Meignan and Lartigau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samuel Meignan, cy1tZWlnbmFuQG8tbGFtYnJldC5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.