 Mohd Shazli Draman1,2
Mohd Shazli Draman1,2 Lei Zhang
Lei Zhang Colin Dayan
Colin Dayan Marian Ludgate
Marian Ludgate- 1Thyroid Research Group, Cardiff University School of Medicine, Cardiff, United Kingdom
- 2KPJ Healthcare University College, Nilai, Malaysia
Graves’ orbitopathy (GO) is a complex and poorly understood disease in which extensive remodeling of orbital tissue is dominated by adipogenesis and hyaluronan production. The resulting proptosis is disfiguring and underpins the majority of GO signs and symptoms. While there is strong evidence for the thyrotropin receptor (TSHR) being a thyroid/orbit shared autoantigen, the insulin-like growth factor 1 receptor (IGF1R) is also likely to play a key role in the disease. The pathogenesis of GO has been investigated extensively in the last decade with further understanding of some aspects of the disease. This is mainly derived by using in vitro and ex vivo analysis of the orbital tissues. Here, we have summarized the features of GO pathogenesis involving target autoantigens and their signaling pathways.
Introduction
Graves’ orbitopathy (GO) or thyroid eye disease is the most common overt thyroidal manifestation of Graves’ disease (GD) with substantial morbidity and socioeconomic impact (1–4). Extensive orbital tissue remodelling in GO is mainly shown as adipose tissue expansion and tissue edema via increased adipogenesis and hyaluronan production, respectively. These pathogenetic processes produce disfiguring proptosis and underpin all GO signs and symptoms. There is a close clinical and temporal association between GD and GO suggesting an autoimmune response to common antigen/s in the orbit and thyroid gland. The thyrotropin receptor (TSHR) is expressed in orbital adipose tissue (OAT) (5–8) and virtually all patients with hyperthyroid GO have thyroid stimulating antibodies (TSAB). Therefore, the TSHR is the most logical candidate (9), which is further supported by the existence of TSHR-induced GO in an animal model (10). The incidence of GO is estimated to be 16/100,000 in females and 2.9/100,000 in males (11). On the other hand, the prevalence estimate is about 10/10,000 (12). A recent meta-analysis reported that current GD patients have a milder phenotype than in the past; as a consequence, a smaller proportion display GO symptoms (13). As with other autoimmune conditions there is female preponderance towards the condition with 6:1 female to male ratio, although in GO the ratio is less skewed than in GD. In addition, most patients with GO have reduced quality of life (QOL) (14) and suffer long-term psychological distress due to the disfiguring appearance of the proptosis, also known as exophthalmos (15). Available treatments for GO are unsatisfactory and more research is needed to address the pathophysiology of the disease which may lead to early pre-clinical diagnosis promoting preventative/early interventions. This in turn will improve long-term morbidity and socioeconomic impact.
Adipogenesis
Adipogenesis is a process in which preadipocytes differentiate into mature adipocytes to form adipose tissues. Our current understanding of adipogenesis has been largely derived by using the murine 3T3L1 cell line. This cell line can spontaneously differentiate into adipocytes when maintained in a high concentration of fetal calf serum for several weeks but the process can be accelerated by employing adipogenic cocktails including insulin, steroid and 3- isobutyl-1-methylxanthine (IBMX) (16). Further components of the differentiation cocktails may also include proliferation-activated receptor gamma (PPARγ) agonists such as pioglitazone and indomethacin (17). Insulin, in common with insulin-like growth factor-1 (IGF-1) activates PI3 kinase (18) and MAP (mitogen-activated protein kinase) (19) pathways. Phosphorylation of protein kinase B (PKB/Akt) in turn phosphorylates forkhead box protein O1 (FOXO1) causing it to exit from the nucleus leading to increased transcription of adipogenic genes (20). Steroids induce the expression of the early adipogenic gene, CCAAT enhancer binding protein delta (C/EBP-δ). This transcription factor contributes to an increase in PPAR-γ expression and production of prostacyclin leading to elevated intracellular cAMP. IBMX is a nonselective phosphodiesterase inhibitor whose presence further elevates levels of intracellular cAMP and protein kinase A (PKA). IBMX is thus required for transcriptional activation of the master regulator of adipogenesis, PPARγ.
Adipogenesis contributes to OAT expansion because a fibroblast has an approximate diameter of 30 microns, whereas the diameter of a mature adipocyte is approximately 150 microns, i.e. 5 times larger. The increased adipogenesis has been demonstrated by using in vitro cultures of human fibroblasts and analysis of ex vivo samples from patients with GO (21). By using both in vitro lineage specific differentiation protocols and flow cytometry, studies have indicated that orbital fibroblasts (OF) possess mesenchymal stem cell (MSC) properties including positivity for Thy-1 (CD90) which is a marker of MSC (22–25). In the orbit, Thy-1 negative OF can be induced to differentiate when cultured in appropriate adipogenic medium whereas Thy-1 positive cells are more likely to undergo differentiation to myofibroblasts and cause fibrosis (23, 24). The orbital fibroblast is also able to undergo neurogenesis, myogenesis, osteogenesis and chondrogenesis in vitro, indicating their pluripotency (22, 25).
Extra-Cellular Matrix
Several extracellular matrix (ECM) components are overproduced in GO including collagens and glycosaminoglycans (GAGs). The excess ECM accumulation in OAT and extraocular muscle (EOM) lead to oedema with consequent proptosis and diplopia respectively (26). The main GAG produced in GO is hyaluronic acid, which is generated by three synthase enzymes (HAS1, HAS2 and HAS3) and broken down by hyaluronidases. Activation of cAMP-protein kinase A signaling via the TSHR, increases cAMP response element binding protein (CREB) at CREB binding sites in the promoters of HAS1 and HAS2 genes, thereby enhancing hyaluronan production (27).
TSHR Intracellular Pathways
Several studies, including from our group, have shown that activation of the TSHR in OF leads to an increase in hyaluronan production and adipogenesis (20, 28). TSHR expression has been shown to increase during adipogenesis (5). We demonstrated that ‘neutral’ TSHR antibodies were capable of binding but had no effect on traditional TSHR signaling pathways (described below) (29). Indeed, TSHR signaling may be far more complex than initially thought (30). Little is known about the effects of TSHR activation at various stages during differentiation. The downstream cascade triggered by TSHR will depend on the types and abundance of guanine-nucleotide binding proteins (G proteins) available in the cell (31). G protein coupled receptors (GPCR) can exist as monomers or oligomers. Oligomerization is the term used to describe dimeric, tetrameric, or higher-order complexes between GPCR monomers. The activation of different GPCR complexes will have major influence on subsequent G protein signaling pathways. The evidence that TSHR may exist in an oligomeric state was initially provided by studies using antibodies (32) and more recently by fluorescence resonance energy transfer (FRET) technology (33). Interestingly, the presence of dimerization influences TSHR behavior. Unstimulated TSHRs have been shown to form oligomers that return to the monomer state with TSH (34). TSHR autoantibodies with stimulating properties are (TSAB) proposed to favor formation of TSHR dimers, whilst TSHR blocking antibodies, are unable to bring about this conformational change. After TSH binding, a constitutively oligomeric TSHR dissociates into active monomers (or dimers when TSAB bind). Subsequently the monomers or dimers are recruited to the lipid rafts and interact with G proteins, thereby initiating the signaling cascade. In the case of TSH, the signal is rapid and brief because of faster movement of monomers into the lipid rafts, in contrast to the slow motion of the dimers. Multivalent blocking TSHR antibodies may cross-link the oligomers, thus preventing them from dissociating and impeding their entry into lipid rafts (35). In cells with low levels of TSHR expression, homo-heterodimer formation is less likely. This may change during adipogenesis, as TSHR expression increases, and may lead to activation of different signaling cascades from that predominating in orbital fibroblasts.
TSHR is known to activate mainly the guanine-nucleotide protein alpha stimulation (Gs)-cAMP pathway. In addition, TSHR may activate several other G protein subtypes, as detailed below (36, 37), non G protein pathways such as β-arrestin-1 (38) and other signalling pathways (39, 40). When TSH binds to its receptor, GTP replaces GDP in the heterotrimeric G protein, which dissociates into Gsα and Gβγ subunits with the former activating all isoforms of adenylate cyclase (41). This enzyme increases levels of cAMP in the cell and activates PKA, also known as cAMP-dependent protein kinase. The activated PKA phosphorylates multiple downstream target proteins one of which is cAMP responsive element binding protein (CREB). CREB then binds to its receptors on the promoter region of the DNA exerting various gene transcription processes including expression of thyroglobulin (TG), thyroid peroxidase (TPO), sodium iodide symporter (NIS), the thyroid transcription factors TTF1/NKx2.1, TTF2/FoxE1, and PAX (42, 43). Every intermediary in the pathway described above may additionally interact with different molecules belonging to other pathways.
In human thyrocytes and rat FRTL-5, guanidine binding protein alpha a/alpha 11 (Gαq/α11) coupling has been shown to stimulate Protein kinase C (PKC) pathways by generating phospholipase C (PLCβ). The PKC pathways has been associated with hyaluronan generation in GO (44). Activation of PKC pathways requires supraphysiological TSH concentrations although not all research agrees with this finding (45). PLC catalyses hydrolysis of phosphatidylinositol in cell membranes yielding di-acyl-glycerol (DAG) and inositol tri phosphate (IP3) as second messengers. DAG directly stimulates PKC. IP3 increases cytosolic Ca+2 levels which act through a number of effectors including PKC itself (46) and Nuclear Factor of Activated T-cells (NFAT) transcription factor protein. NFAT plays an important role in cytokine gene transcription regulation (47). Calcium via calmodulin –a calcium sensor protein - activates the serine/threonine phosphatase calcineurin (inhibited by cyclosporin and FK506). This in turn rapidly dephosphorylates NFAT proteins, resulting in a conformational change that exposes a nuclear localization signal leading to NFAT nuclear import (48). TSHR may also couple to guanine nucleotide binding protein alpha inhibition (Gαi1), which inhibits adenylyl cyclase and decreases cAMP levels. The accompanying Gβγ dimers may induce multitudes of other pathways, including adenylyl cyclase, PI3K/Akt (PKB)-FOXO and PLC cascades (49–51). Others have reported that TSHR activation of OF signals via p70s6 kinase (52). The finding may explain our lack of success when using gain-of-function mutants of the TSHR, which signal predominantly via Gsα, to stimulate adipogenesis (28) and concurs with the study from van Ziejl et al. who investigated TSH/TSAB induced hyaluronan production (53). It contrasts with the studies of Neumann and colleagues, who report increased M22-mediated cAMP, even at baseline. However, these authors maintain their OF in a semi-adipogenic medium which likely increases TSHR expression (54).
Our previous work has demonstrated that adipogenesis and HA production, are linked in the orbit. HA accumulation increases in the orbit during adipogenesis but not in other fat depots (55). In this study, adipogenesis in orbital preadipocytes was accompanied by HA accumulation and significantly increased HAS2 transcripts (but not HAS 1 and 3). In contrast, adipogenic differentiation in subcutaneous preadipocyte-fibroblasts significantly decreased secreted HA and HAS2 transcript levels. IGF-I alone did not increase HAS2 levels, but inhibition of PKB/Akt increased orbital HAS2 transcripts but not subcutaneous preadipocytes. Furthermore, our study suggested that mTORC1 negative feedback in IGF1–PI3K–Akt signalling is absent in OF but present in subcutaneous adipose tissue (55). The difference might be explained by the fact that human OF originate from neural crest, while subcutaneous adipose tissue is of mesodermal origin. In addition, our most recent studies demonstrated a depot specific fatty acid-uptake driven adipogenesis with unique gene signatures in OAT. These result in hyperplastic-type expansion of adipocytes in GO (56, 57). Taken together, these findings suggest a very distinctive mechanism underlying the orbital adipogenesis process.
Insulin Like Growth Factor -1 Receptor Signalling
While there is strong evidence supporting the role of TSHR in GO, IGF1R is also likely to play a key role in the disease progress. The IGF1R was first proposed by Weightman and colleagues who demonstrated high affinity IGF1 binding sites in OF (58). More recently extensive work from Terry Smith and his colleagues has confirmed this finding and further showed that TSHR and IGF1R co-localize to orbital cell membranes (59). The same group has further reported a wide range of IGF1R mediated effects in OF including increases in proliferation, GAG production and cytokine production (60, 61). Our own study demonstrated that activation of TSHR and IGR1R has additive effect on HAS2 transcripts/HA production (62). Krieger et al. found that M22 stimulation of HA secretion by OF involves cross talk between IGF-1R and TSHR. The relationship relies on TSHR activation per se rather than direct activation of IGF-1R which leads to synergistic stimulation of HA secretion (63). TSH induced ERK phosphorylation can be blocked by an IGF-1R-blocking monoclonal antibody suggesting that IGF-1R might mediate some TSH-provoked signalling. Further studies have highlighted the importance of down stream factors of IGF1–PI3K signalling and revealed that FOXOs, may mediate both TSHR and IGF1R signalling pathways in GO (64). The notion is further supported by recent successful trial of teprotumumab- monoclonal antibody which blocks IGF1R - in reducing proptosis in patients with GO (65, 66). Whilst effective medical treatment for GO is welcome, some concerns have been raised about these trials including the lack of orbital imaging and the fact that despite QOL scores being improved in the teprotumumab group, all patients scores remained low (67). Furthermore, the activation of Fibroblast Growth factor (FGF) and its receptor has been shown to increase the expression of Insulin like growth factor-2 (IGF-2) in mesenchymal stem cells via IGF-2 and IGF1-R (68). The FGF signalling pathway has also been shown to play a role in OAT expansion in GO (69). Our most recent study used RNA-seq analysis to demonstrate that FGFs, FGFR2, IGF-2 and IGF1-R were highly expressed in OAT compared with white adipose tissue, supporting the aforementioned successful trial of IGF1R inhibition in GO (56).
TSHR Variants
To add to the complexity of the molecular events associated with GO, several TSHR variants have been described which lack the transmembrane domain. If the variants are expressed as protein, they would yield soluble receptor products which could serve as TSH/TRAB binding proteins or even as autoantigens. Early northern blot analysis of thyroid tissue identified the expected full-length transcript plus 2 additional transcripts at 1.3 and 1.6 kb (70); the transcripts were also detected in OF (71). Of interest, the exon 1-8 variant is similar in structure to the TSHR A subunit which is generated following cleavage of the full-length receptor (72, 73). Furthermore, induced murine models of GD and GO are more effective when immunizing with the A subunit than with the complete TSHR (74, 75). We have reported that the 1.3 variant is expressed as a protein and can affect TSHR activation (76). Thus, these variants could have impact on the pathogenesis of GO by inducing further production of TSAB or protect against GO by ‘neutralizing’ TSAB, respectively.
Discussion
TSHR and IGF1 signaling are important in orbital tissues (summarized in Figure 1) but more complex than generally thought. Although these signals are mainly activated through G protein signalling pathways, other cascades may also be involved. As our understanding expands, additional extracellular or intracellular factors, which regulate signaling, may be identified. The abundance of the receptors may also dictate which pathways are activated. The recent success of TSHR extracellular domain crystallization is likely to catapult these areas of research and may lead to further alternative treatment strategies for GO (77).
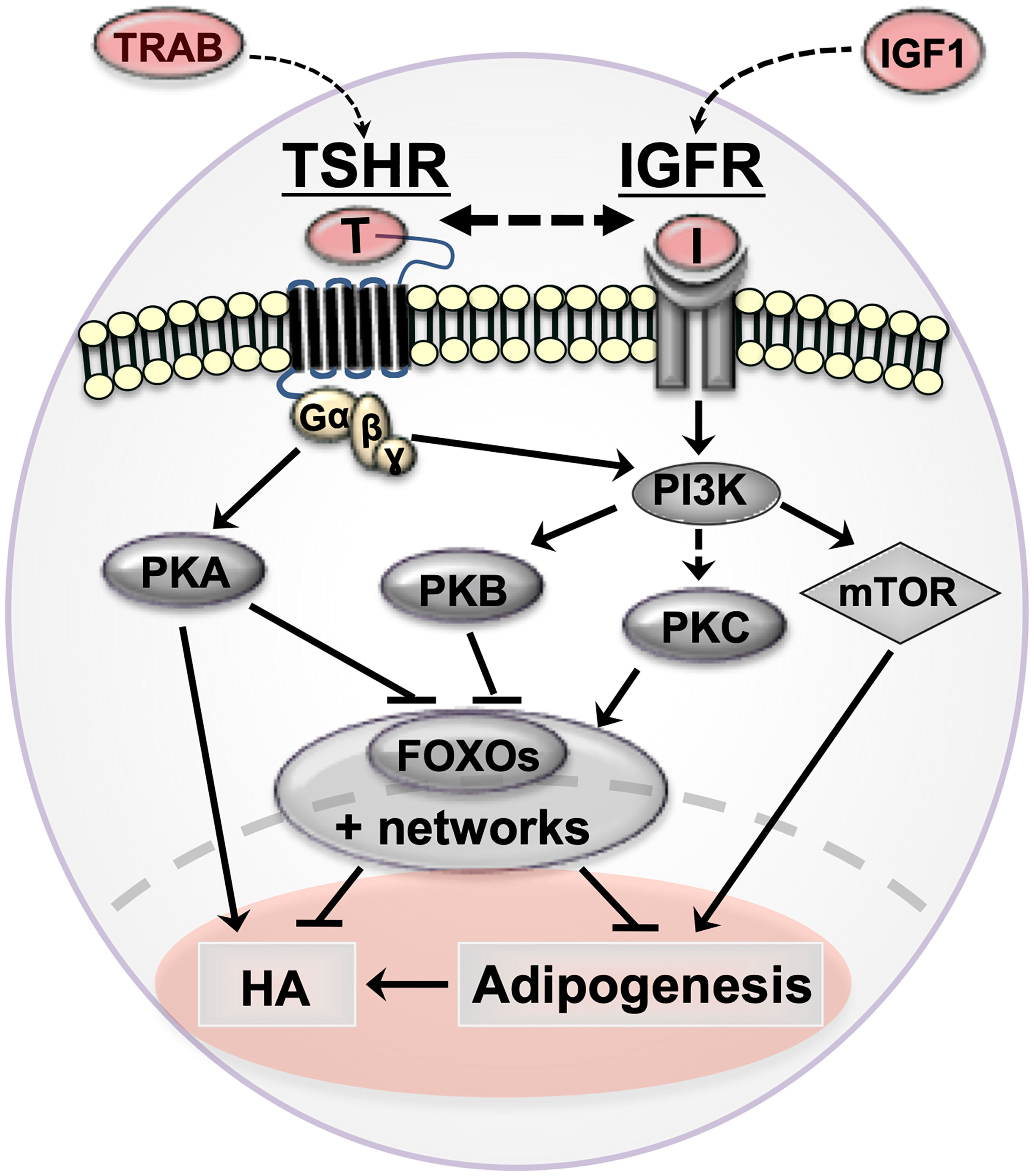
Figure 1 Cartoon summarizing orbital fibroblast signaling cascades in Graves’ orbitopathy (GO) and how they affect pathogenetic mechanisms (adipogenesis and hyaluronan production). TSHR/TRAB and IGFR/IGF are shown in red with arrows indicating the possible crosstalk between the pathways in GO. thyrotropin receptor (TSHR, serpentine structure); TSHR auto-antibodies (TRAB); Insulin-like growth factor 1 receptor (IGF-1R) and IGF1; protein kinase A (PKA); protein kinase B (PKB/Akt); protein kinase C (PKC); phosphoinositide 3-kinase (PI3K); forkhead box protein O (FOXO); hyaluronan production (HA).
As discussed above, a human monoclonal anti-IGF-1R-blocking antibody, Teprotumumab has been approved by FDA for treatment of patients with GO specifically in reducing proptosis and has recently been reported to be highly effective in active GO (65). The potential for treatments based on TSHR antagonism, which have been demonstrated to be effective in vitro, is keenly anticipated either with blocking antibodies or small molecule antagonists which in theory could inhibit both TSHR and IGF-1R related and/or unrelated pathways (78). The beneficial effects on GD and GO following administration of a monoclonal TSHR blocking antibody (TBAB) in a patient with thyroid cancer has recently been described (79). Furthermore, manipulating the two pathways concomitantly may provide even more effective treatment for GO and merits investigation.
Author Contributions
MD and LZ wrote the manuscript with input from ML. CD and ML reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
GO, Graves’ orbitopathy; TSAB, thyroid stimulating antibodies; TSHR, thyrotropin receptor; IGF1, insulin like growth factor 1; IBMX, 3-isobutyl-1-methylxanthine; PPARγ, peroxisome proliferator-activated receptors gamma; PI3 kinase Phosphoinositide-3- kinase; MAP, mitogen-activated protein kinase; FOXO, Forkhead Box O1; C/EBP-δ, CCAAT-enhancer-binding proteins Delta; cAMP, Cyclic adenosine monophosphate; PKA, protein kinase A; GCRP, G protein coupled receptors; FRET, fluorescence resonance energy transfer; CREB, cAMP responsive element binding protein; TG, thyroglobulin; TPO, thyroid peroxidase; NIS, sodium iodide symporter (NIS), TTF, thyroid transcription factors; PKC, Protein kinase C; PLC phospholipase C; DAG, di-acyl-glycerol; NFAT, Nuclear factor of activated T-cells; HA, hyaluronic acid; GAG, glycosaminoglycan; SNP, single nucleotide polymorphism.
References
1. Reddy SV, Jain A, Yadav SB, Sharma K, Bhatia E. Prevalence of Graves' Ophthalmopathy in Patients With Graves' Disease Presenting to a Referral Centre in North India. Indian J Med Res (2014) 139(1):99–104.
2. Tellez M, Cooper J, Edmonds C. Graves' Ophthalmopathy in Relation to Cigarette Smoking and Ethnic Origin. Clin Endocrinol (1992) 36(3):291–4. doi: 10.1111/j.1365-2265.1992.tb01445.x
3. Wiersinga WM, Bartalena L. Epidemiology and Prevention of Graves' Ophthalmopathy. Thyroid (2002) 12(10):855–60. doi: 10.1089/105072502761016476
4. Tanda ML, Piantanida E, Liparulo L, Veronesi G, Lai A, Sassi L, et al. Prevalence and Natural History of Graves' Orbitopathy in a Large Series of Patients With Newly Diagnosed Graves' Hyperthyroidism Seen at a Single Center. J Clin Endocrinol Metab (2013) 98(4):1443–9. doi: 10.1210/jc.2012-3873
5. Crisp M, Starkey KJ, Lane C, Ham J, Ludgate M. Adipogenesis in Thyroid Eye Disease. Invest Ophthalmol Visual Sci (2000) 41(11):3249–55.
6. Kumar S, Coenen MJ, Scherer PE, Bahn RS. Evidence for Enhanced Adipogenesis in the Orbits of Patients With Graves' Ophthalmopathy. J Clin Endocrinol Metab (2004) 89(2):930–5. doi: 10.1210/jc.2003-031427
7. Bahn RS, Dutton CM, Natt N, Joba W, Spitzweg C, Heufelder AE. Thyrotropin Receptor Expression in Graves' Orbital Adipose/Connective Tissues: Potential Autoantigen in Graves' Ophthalmopathy. J Clin Endocrinol Metab (1998) 83(3):998–1002. doi: 10.1210/jc.83.3.998
8. Roselli-Rehfuss L, Robbins LS, Cone RD. Thyrotropin Receptor Messenger Ribonucleic Acid Is Expressed in Most Brown and White Adipose Tissues in the Guinea Pig. Endocrinology (1992) 130(4):1857–61. doi: 10.1210/endo.130.4.1547715
9. Khoo DH, Ho SC, Seah LL, Fong KS, Tai ES, Chee SP, et al. The Combination of Absent Thyroid Peroxidase Antibodies and High Thyroid-Stimulating Immunoglobulin Levels in Graves' Disease Identifies a Group at Markedly Increased Risk of Ophthalmopathy. Thyroid (1999) 9(12):1175–80. doi: 10.1089/thy.1999.9.1175
10. Banga JP, Moshkelgosha S, Berchner-Pfannschmidt U, Eckstein A. Modeling Graves' Orbitopathy in Experimental Graves' Disease. Horm Metab Res (2015) 47(10):797–803. doi: 10.1055/s-0035-1555956
11. Lazarus JH. Epidemiology of Graves' Orbitopathy (GO) and Relationship With Thyroid Disease. Best Pract Res Clin Endocrinol Metab (2012) 26(3):273–9. doi: 10.1016/j.beem.2011.10.005
12. Perros P, Hegedus L, Bartalena L, Marcocci C, Kahaly GJ, Baldeschi L, et al. Graves' Orbitopathy as a Rare Disease in Europe: A European Group on Graves' Orbitopathy (EUGOGO) Position Statement. Orphanet J Rare Dis (2017) 12(1):72. doi: 10.1186/s13023-017-0625-1
13. Ippolito S, Cusini C, Lasalvia P, Gianfagna F, Veronesi G, Gallo D, et al. Change in Newly Diagnosed Graves' Disease Phenotype Between the Twentieth and the Twenty-First Centuries: Meta-Analysis and Meta-Regression. J Endocrinol Invest (2021) 44(8):1707–18. doi: 10.1007/s40618-020-01479-z
14. Wiersinga WM. Quality of Life in Graves' Ophthalmopathy. Best Pract Res Clin Endocrinol Metab (2012) 26(3):359–70. doi: 10.1016/j.beem.2011.11.001
15. Coulter I, Frewin S, Krassas GE, Perros P. Psychological Implications of Graves' Orbitopathy. Eur J Endocrinol Eur Fed Endocrine Societies (2007) 157(2):127–31. doi: 10.1530/EJE-07-0205
16. Rosen ED, Spiegelman BM. Molecular Regulation of Adipogenesis. Annu Rev Cell Dev Biol (2000) 16:145–71. doi: 10.1146/annurev.cellbio.16.1.145
17. Tontonoz P, Hu E, Spiegelman BM. Stimulation of Adipogenesis in Fibroblasts by PPAR Gamma 2, a Lipid-Activated Transcription Factor. Cell (1994) 79(7):1147–56. doi: 10.1016/0092-8674(94)90006-x
18. Xu J, Liao K. Protein Kinase B/AKT 1 Plays a Pivotal Role in Insulin-Like Growth Factor-1 Receptor Signaling Induced 3T3-L1 Adipocyte Differentiation. J Biol Chem (2004) 279(34):35914–22. doi: 10.1074/jbc.M402297200
19. Qiu Z, Wei Y, Chen N, Jiang M, Wu J, Liao K. DNA Synthesis and Mitotic Clonal Expansion Is Not a Required Step for 3T3-L1 Preadipocyte Differentiation Into Adipocytes. J Biol Chem (2001) 276(15):11988–95. doi: 10.1074/jbc.M011729200
20. Zhang L, Paddon C, Lewis MD, Grennan-Jones F, Ludgate M. Gsalpha Signalling Suppresses PPARgamma2 Generation and Inhibits 3T3L1 Adipogenesis. J Endocrinol (2009) 202(2):207–15. doi: 10.1677/JOE-09-0099
21. Starkey KJ, Janezic A, Jones G, Jordan N, Baker G, Ludgate M. Adipose Thyrotrophin Receptor Expression Is Elevated in Graves' and Thyroid Eye Diseases Ex Vivo and Indicates Adipogenesis in Progress In Vivo. J Mol Endocrinol (2003) 30(3):369–80. doi: 10.1677/jme.0.0300369
22. Brandau S, Bruderek K, Hestermann K, Gortz GE, Horstmann M, Mattheis S, et al. Orbital Fibroblasts From Graves' Orbitopathy Patients Share Functional and Immunophenotypic Properties With Mesenchymal Stem/Stromal Cells. Invest Ophthalmol Visual Sci (2015) 56(11):6549–57. doi: 10.1167/iovs.15-16610
23. Smith TJ, Koumas L, Gagnon A, Bell A, Sempowski GD, Phipps RP, et al. Orbital Fibroblast Heterogeneity May Determine the Clinical Presentation of Thyroid-Associated Ophthalmopathy. J Clin Endocrinol Metab (2002) 87(1):385–92. doi: 10.1210/jcem.87.1.8164
24. Sorisky A, Pardasani D, Gagnon A, Smith TJ. Evidence of Adipocyte Differentiation in Human Orbital Fibroblasts in Primary Culture. J Clin Endocrinol Metab (1996) 81(9):3428–31. doi: 10.1210/jcem.81.9.8784110
25. Kozdon K, Fitchett C, Rose GE, Ezra DG, Bailly M. Mesenchymal Stem Cell-Like Properties of Orbital Fibroblasts in Graves' Orbitopathy. Invest Ophthalmol Visual Sci (2015) 56(10):5743–50. doi: 10.1167/iovs.15-16580
26. Laurberg P, Berman DC, Pedersen IB, Andersen S, Carle A. Double Vision Is a Major Manifestation in Moderate to Severe Graves' Orbitopathy, But It Correlates Negatively With Inflammatory Signs and Proptosis. J Clin Endocrinol Metab (2015) 100(5):2098–105. doi: 10.1210/jc.2014-4557
27. Zhang L, Bowen T, Grennan-Jones F, Paddon C, Giles P, Webber J, et al. Thyrotropin Receptor Activation Increases Hyaluronan Production in Preadipocyte Fibroblasts: Contributory Role in Hyaluronan Accumulation in Thyroid Dysfunction. J Biol Chem (2009) 284(39):26447–55. doi: 10.1074/jbc.M109.003616
28. Zhang L, Baker G, Janus D, Paddon CA, Fuhrer D, Ludgate M. Biological Effects of Thyrotropin Receptor Activation on Human Orbital Preadipocytes. Invest Ophthalmol Visual Sci (2006) 47(12):5197–203. doi: 10.1167/iovs.06-0596
29. Metcalfe R, Jordan N, Watson P, Gullu S, Wiltshire M, Crisp M, et al. Demonstration of Immunoglobulin G, A, and E Autoantibodies to the Human Thyrotropin Receptor Using Flow Cytometry. J Clin Endocrinol Metab (2002) 87(4):1754–61. doi: 10.1210/jcem.87.4.8411
30. Latif R, Morshed SA, Zaidi M, Davies TF. The Thyroid-Stimulating Hormone Receptor: Impact of Thyroid-Stimulating Hormone and Thyroid-Stimulating Hormone Receptor Antibodies on Multimerization, Cleavage, and Signaling. Endocrinol Metab Clinics North Am (2009) 38(2):319–41. doi: 10.1016/j.ecl.2009.01.006
31. Wess J. Molecular Basis of Receptor/G-Protein-Coupling Selectivity. Pharmacol Ther (1998) 80(3):231–64. doi: 10.1016/S0163-7258(98)00030-8
32. Graves PN, Vlase H, Bobovnikova Y, Davies TF. Multimeric Complex Formation by the Thyrotropin Receptor in Solubilized Thyroid Membranes. Endocrinology (1996) 137(9):3915–20. doi: 10.1210/endo.137.9.8756566
33. Latif R, Graves P, Davies TF. Oligomerization of the Human Thyrotropin Receptor: Fluorescent Protein-Tagged hTSHR Reveals Post-Translational Complexes. J Biol Chem (2001) 276(48):45217–24. doi: 10.1074/jbc.M103727200
34. Latif R, Graves P, Davies TF. Ligand-Dependent Inhibition of Oligomerization at the Human Thyrotropin Receptor. J Biol Chem (2002) 277(47):45059–67. doi: 10.1074/jbc.M206693200
35. Moffett S, Brown DA, Linder ME. Lipid-Dependent Targeting of G Proteins Into Rafts. J Biol Chem (2000) 275(3):2191–8. doi: 10.1074/jbc.275.3.2191
36. Allgeier A, Offermanns S, Van Sande J, Spicher K, Schultz G, Dumont JE. The Human Thyrotropin Receptor Activates G-Proteins Gs and Gq/11. J Biol Chem (1994) 269(19):13733–5.
37. Laugwitz KL, Allgeier A, Offermanns S, Spicher K, Van Sande J, Dumont JE, et al. The Human Thyrotropin Receptor: A Heptahelical Receptor Capable of Stimulating Members of All Four G Protein Families. Proc Natl Acad Sci USA (1996) 93(1):116–20. doi: 10.1073/pnas.93.1.116
38. Boutin A, Eliseeva E, Gershengorn MC, Neumann S. Beta-Arrestin-1 Mediates Thyrotropin-Enhanced Osteoblast Differentiation. FASEB J (2014) 28(8):3446–55. doi: 10.1096/fj.14-251124
39. Büch TRH, Biebermann H, Kalwa H, Pinkenburg O, Hager D, Barth H, et al. G13-Dependent Activation of MAPK by Thyrotropin. J Biol Chem (2008) 283(29):20330–41. doi: 10.1074/jbc.M800211200
40. Laurent E, Mockel J, Van Sande J, Graff I, Dumont JE. Dual Activation by Thyrotropin of the Phospholipase C and Cyclic AMP Cascades in Human Thyroid. Mol Cell Endocrinol (1987) 52(3):273–8. doi: 10.1016/0303-7207(87)90055-4
41. Raspe E, Dumont JE. Robert Feulgen Lecture 1991. Control and Role of Major Signalling Cascades of the Thyrocyte. Prog Histochem Cytochem (1992) 26(1-4):1–29. doi: 10.1016/s0079-6336(11)80074-4
42. Postiglione MP, Parlato R, Rodriguez-Mallon A, Rosica A, Mithbaokar P, Maresca M, et al. Role of the Thyroid-Stimulating Hormone Receptor Signaling in Development and Differentiation of the Thyroid Gland. Proc Natl Acad Sci USA (2002) 99(24):15462–7. doi: 10.1073/pnas.242328999
43. Vassart G, Dumont JE. The Thyrotropin Receptor and the Regulation of Thyrocyte Function and Growth. Endocrine Rev (1992) 13(3):596–611. doi: 10.1210/edrv-13-3-596
44. Wang HS, Tung WH, Tang KT, Wong YK, Huang GJ, Wu JC, et al. TGF-Beta Induced Hyaluronan Synthesis in Orbital Fibroblasts Involves Protein Kinase C betaII Activation In Vitro. J Cell Biochem (2005) 95(2):256–67. doi: 10.1002/jcb.20405
45. D'Arcangelo D, Silletta MG, Di Francesco AL, Bonfitto N, Di Cerbo A, Falasca M, et al. Physiological Concentrations of Thyrotropin Increase Cytosolic Calcium Levels in Primary Cultures of Human Thyroid Cells. J Clin Endocrinol Metab (1995) 80(4):1136–43. doi: 10.1210/jcem.80.4.7714082
46. Jhon DY, Lee HH, Park D, Lee CW, Lee KH, Yoo OJ, et al. Cloning, Sequencing, Purification, and Gq-Dependent Activation of Phospholipase C-Beta 3. J Biol Chem (1993) 268(9):6654–61. doi: 10.1016/S0021-9258(18)53300-7
47. Macian F. NFAT Proteins: Key Regulators of T-Cell Development and Function. Nat Rev Immunol (2005) 5(6):472–84. doi: 10.1038/nri1632
48. Luo C, Shaw KT, Raghavan A, Aramburu J, Garcia-Cozar F, Perrino BA, et al. Interaction of Calcineurin With a Domain of the Transcription Factor NFAT1 That Controls Nuclear Import. Proc Natl Acad Sci USA (1996) 93(17):8907–12. doi: 10.1073/pnas.93.17.8907
49. Sunahara RK, Dessauer CW, Gilman AG. Complexity and Diversity of Mammalian Adenylyl Cyclases. Annu Rev Pharmacol Toxicol (1996) 36:461–80. doi: 10.1146/annurev.pa.36.040196.002333
50. Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-Kinases: A Conserved Family of Signal Transducers. Trends Biochem Sci (1997) 22(7):267–72. doi: 10.1016/S0968-0004(97)01061-X
51. Rhee SG, Bae YS. Regulation of Phosphoinositide-Specific Phospholipase C Isozymes. J Biol Chem (1997) 272(24):15045–8. doi: 10.1074/jbc.272.24.15045
52. Bell A, Gagnon A, Grunder L, Parikh SJ, Smith TJ, Sorisky A. Functional TSH Receptor in Human Abdominal Preadipocytes and Orbital Fibroblasts. Am J Physiol Cell Physiol (2000) 279(2):C335–40. doi: 10.1152/ajpcell.2000.279.2.C335
53. van Zeijl CJ, Fliers E, van Koppen CJ, Surovtseva OV, de Gooyer ME, Mourits MP, et al. Thyrotropin Receptor-Stimulating Graves' Disease Immunoglobulins Induce Hyaluronan Synthesis by Differentiated Orbital Fibroblasts From Patients With Graves' Ophthalmopathy Not Only via Cyclic Adenosine Monophosphate Signaling Pathways. Thyroid (2011) 21(2):169–76. doi: 10.1089/thy.2010.0123
54. Neumann S, Pope A, Geras-Raaka E, Raaka BM, Bahn RS, Gershengorn MC. A Drug-Like Antagonist Inhibits Thyrotropin Receptor–Mediated Stimulation of cAMP Production in Graves' Orbital Fibroblasts. Thyroid (2012) 22:839–43. doi: 10.1089/thy.2011.0520
55. Zhang L, Grennan-Jones F, Lane C, Rees DA, Dayan CM, Ludgate M. Adipose Tissue Depot-Specific Differences in the Regulation of Hyaluronan Production of Relevance to Graves' Orbitopathy. J Clin Endocrinol Metab (2012) 97(2):653–62. doi: 10.1210/jc.2011-1299
56. Zhang L, Evans A, von Ruhland C, Draman MS, Edkins S, Vincent AE, et al. Distinctive Features of Orbital Adipose Tissue (OAT) in Graves' Orbitopathy. Int J Mol Sci (2020) 21(23):9145–62. doi: 10.3390/ijms21239145
57. Zhang L, Rai P, Miwa S, Draman MS, Rees DA, Haridas AS, et al. The Role of Mitochondria Linked Fatty-Acid Uptake-Driven Adipogenesis in Graves' Orbitopathy. Endocrinology (2021) 162(12):bqab188. doi: 10.1210/endocr/bqab188
58. Weightman DR, Perros P, Sherif IH, Kendall-Taylor P. Autoantibodies to IGF-1 Binding Sites in Thyroid Associated Ophthalmopathy. Autoimmunity (1993) 16(4):251–7. doi: 10.3109/08916939309014643
59. Tsui S, Naik V, Hoa N, Hwang CJ, Afifiyan NF, Sinha Hikim A, et al. Evidence for an Association Between Thyroid-Stimulating Hormone and Insulin-Like Growth Factor 1 Receptors: A Tale of Two Antigens Implicated in Graves' Disease. J Immunol (Baltimore Md 1950) (2008) 181(6):4397–405. doi: 10.4049/jimmunol.181.6.4397
60. Smith TJ, Janssen JA. Building the Case for Insulin-Like Growth Factor Receptor-I Involvement in Thyroid-Associated Ophthalmopathy. Front Endocrinol (Lausanne) (2016) 7:167. doi: 10.3389/fendo.2016.00167
61. Smith TJ, Janssen J. Insulin-Like Growth Factor-I Receptor and Thyroid-Associated Ophthalmopathy. Endocrine Rev (2019) 40(1):236–67. doi: 10.1210/er.2018-00066
62. Zhang L, Grennan-Jones F, Draman MS, Lane C, Morris D, Dayan CM, et al. Possible Targets for Nonimmunosuppressive Therapy of Graves' Orbitopathy. J Clin Endocrinol Metab (2014) 99(7):E1183–90. doi: 10.1210/jc.2013-4182
63. Krieger CC, Neumann S, Place RF, Marcus-Samuels B, Gershengorn MC. Bidirectional TSH and IGF-1 Receptor Cross Talk Mediates Stimulation of Hyaluronan Secretion by Graves' Disease Immunoglobins. J Clin Endocrinol Metab (2015) 100(3):1071–7. doi: 10.1210/jc.2014-3566
64. Zhang L, Ji QH, Ruge F, Lane C, Morris D, Tee AR, et al. Reversal of Pathological Features of Graves' Orbitopathy by Activation of Forkhead Transcription Factors, FOXOs. J Clin Endocrinol Metab (2016) 101(1):114–22. doi: 10.1210/jc.2015-2932
65. Kahaly GJ, Douglas RS, Holt RJ, Sile S, Smith TJ. Teprotumumab for Patients With Active Thyroid Eye Disease: A Pooled Data Analysis, Subgroup Analyses, and Off-Treatment Follow-Up Results From Two Randomised, Double-Masked, Placebo-Controlled, Multicentre Trials. Lancet Diabetes Endocrinol (2021) 9(6):360–72. doi: 10.1016/S2213-8587(21)00056-5
66. Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, Tang RA, et al. Teprotumumab for Thyroid-Associated Ophthalmopathy. N Engl J Med (2017) 376(18):1748–61. doi: 10.1056/NEJMoa1614949
67. Piantanida E, Bartalena L. Teprotumumab: A New Avenue for the Management of Moderate-to-Severe and Active Graves' Orbitopathy? J Endocrinol Invest (2017) 40(8):885–7. doi: 10.1007/s40618-017-0717-8
68. Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, Werbowetski-Ogilvie T, et al. IGF and FGF Cooperatively Establish the Regulatory Stem Cell Niche of Pluripotent Human Cells. Vitro Nat (2007) 448(7157):1015–21. doi: 10.1038/nature06027
69. Schrijver B, Kooiman MA, Kasteleijn E, van Holten-Neelen C, Virakul S, Paridaens D, et al. Basic Fibroblast Growth Factor Induces Adipogenesis in Orbital Fibroblasts: Implications for the Pathogenesis of Graves' Orbitopathy. Thyroid (2019) 29(3):395–404. doi: 10.1089/thy.2018.0544
70. Graves PN, Tomer Y, Davies TF. Cloning and Sequencing of a 1.3 KB Variant of Human Thyrotropin Receptor mRNA Lacking the Transmembrane Domain. Biochem Biophys Res Commun (1992) 187(2):1135–43. doi: 10.1016/0006-291x(92)91315-h
71. Paschke R, Metcalfe A, Alcalde L, Vassart G, Weetman A, Ludgate M. Presence of Nonfunctional Thyrotropin Receptor Variant Transcripts in Retroocular and Other Tissues. J Clin Endocrinol Metab (1994) 79(5):1234–8. doi: 10.1210/jcem.79.5.7962314
72. Quellari M, Desroches A, Beau I, Beaudeux E, Misrahi M. Role of Cleavage and Shedding in Human Thyrotropin Receptor Function and Trafficking. Eur J Biochem (2003) 270(17):3486–97. doi: 10.1046/j.1432-1033.2003.03718.x
73. Rapoport B, McLachlan SM. The Thyrotropin Receptor in Graves' Disease. Thyroid (2007) 17(10):911–22. doi: 10.1089/thy.2007.0170
74. Zhao SX, Tsui S, Cheung A, Douglas RS, Smith TJ, Banga JP. Orbital Fibrosis in a Mouse Model of Graves' Disease Induced by Genetic Immunization of Thyrotropin Receptor cDNA. J Endocrinol (2011) 210(3):369–77. doi: 10.1530/joe-11-0162
75. Moshkelgosha S, So PW, Deasy N, Diaz-Cano S, Banga JP. Cutting Edge: Retrobulbar Inflammation, Adipogenesis, and Acute Orbital Congestion in a Preclinical Female Mouse Model of Graves' Orbitopathy Induced by Thyrotropin Receptor Plasmid-In Vivo Electroporation. Endocrinology (2013) 154(9):3008–15. doi: 10.1210/en.2013-1576
76. Draman MS, Grennan-Jones F, Taylor P, Muller I, Evans S, Haridas A, et al. Expression of Endogenous Putative TSH Binding Protein in Orbit. Curr Issues Mol Biol (2021) 43:1794–804. doi: 10.3390/cimb43030126
77. Miller-Gallacher J, Sanders P, Young S, Sullivan A, Baker S, Reddington SC, et al. Crystal Structure of a Ligand-Free Stable TSH Receptor Leucine-Rich Repeat Domain. J Mol Endocrinol (2019) 62(3):117–28. doi: 10.1530/JME-18-0213
78. Neumann S, Krieger CC, Gershengorn MC. Targeting TSH and IGF-1 Receptors to Treat Thyroid Eye Disease. Eur Thyroid J (2020) 9(Suppl 1):59–65. doi: 10.1159/000511538
Keywords: thyroid eye disease, adipogenesis, hyaluronan, TSAB, TSHR, IGF1R
Citation: Draman MS, Zhang L, Dayan C and Ludgate M (2021) Orbital Signaling in Graves’ Orbitopathy. Front. Endocrinol. 12:739994. doi: 10.3389/fendo.2021.739994
Received: 12 July 2021; Accepted: 21 October 2021;
Published: 09 November 2021.
Edited by:
Rauf Latif, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Husnia Marrif, Marrif Biotech, CanadaEliana Angela Piantanida, University of Insubria, Italy
Copyright © 2021 Draman, Zhang, Dayan and Ludgate. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zhang, emhhbmdMMTRAY2YuYWMudWs=
†Senior Author: Marian Ludgate