 Michael S. Kuefner
Michael S. Kuefner- Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden
The phospholipases A2 (PLA2) superfamily encompasses enzymes commonly found in mammalian tissues and snake venom. Many of these enzymes have unique tissue distribution, function, and substrate specificity suggesting distinct biological roles. In the past, much of the research on secretory PLA2s has analyzed their roles in inflammation, anti-bacterial actions, and atherosclerosis. In recent studies utilizing a variety of mouse models, pancreatic islets, and clinical trials, a role for many of these enzymes in the control of metabolism and insulin action has been revealed. In this review, this research, and the unique contributions of the PLA2 enzymes in insulin resistance and metabolism.
Introduction
Metabolic syndrome constitutes an array of pathophysiologies, including obesity, glucose intolerance, and dyslipidemia. The most common feature between these pathophysiologies is insulin resistance, a condition in which cells fail to respond normally to insulin. One factor associated with insulin resistance is triacylglycerol accumulation in muscle, liver, and fat (1, 2). While triacylglycerols are a marker for insulin resistance (3), they may not be causal. Instead, cellular lipids with signaling roles (diacylglycerols, fatty acids, phospholipids, etc.) may fill this role, as accumulation of a variety of lipid species can cause insulin resistance (4–6).
One group of enzymes that produce these lipids are secretory phospholipases A2 (sPLA2s), a protein family found in mammalian tissues and snake venom which hydrolyze glycerophospholipid sn-2 ester bonds, generating a non-esterified free fatty acid and a lysophospholipid. Secretory phospholipases A2s are low molecular weight enzymes (~14 kDa), most of which require millimolar amounts of Ca2+ to function. Twelve sPLA2 isoforms have been identified thus far, of which ten are catalytically active (IB, IIA, IIC, IID, IIE, IIF, III, V, X, XIIA), and two are dormant (XIIB and otoconin-90) (7, 8). These enzymes have varied expression patterns and substrate preferences, signifying diversity in their biological roles. Over the past couple decades of research, the majority of studies concerning sPLA2s dealt with their roles in cardiovascular disease, inflammation, antimicrobial actions, and membrane remodeling. However, a discussion on how sPLA2s may regulate or impact glucose metabolism, insulin signaling, and metabolism is lacking. Recent research has identified that at least 7 of these sPLA2s modulate glucose metabolism, presumably by generation of fatty acids or lipoproteins that influence lipid metabolism and mobilization, alterations in fatty acid oxidation, or other mechanisms involved in insulin signaling and obesity (Table 1).
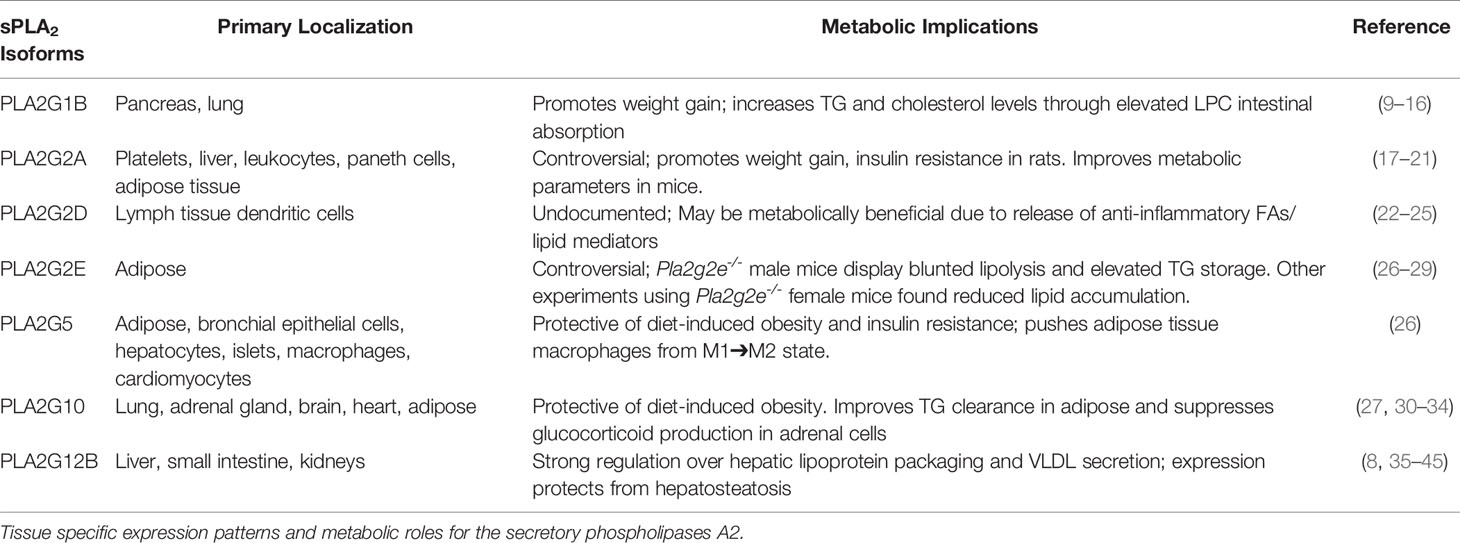
Table 1 Metabolic roles of sPLA2 isoforms.
PLA2G1B
The metabolic role of PLA2G1B has been clarified with Pla2g1b knockout (Pla2g1b-/-) mice or pancreatic acinar cell-specific PLA2G1B overexpression. PLA2G1B is mainly expressed in pancreatic acinar cells and the lungs, and only displays its enzymatic activity after feeding, as this causes the enzyme’s release into the pancreatic fluid and subsequent secretion into the intestinal lumen where it is proteolytically cleaved from an inactive state to its active form (46). Activated PLA2G1B contributes to lipid metabolism and absorption of lysophospholipids, particularly lysophosphatidylcholine (LPC). Pla2g1b-/- mice on a C57BL/6 background fed a hypercaloric diet (58.5% fat, 25% sucrose, 16.5% protein) for either 3 or 10 weeks are resistant to diet-induced obesity (9). Pla2g1b-/- mice showed a 37% reduction in plasma triglyceride (TG) levels primarily due to a decrease in hepatic VLDL production and an increase in TG-rich lipoprotein clearance. Pla2g1b-/- mice also displayed a 61% reduction in plasma cholesterol following 10 weeks on the hypercaloric diet compared to age-matched wild-type controls. Notably, Pla2g1b-/- Ldlr-/- mice fed the same hypercaloric diet for 10 weeks displayed a similar phenotype, including reductions in fasting glucose, insulin, and plasma lipids (10). Similarly, wild-type mice consuming a high-fat, high-carbohydrate diet supplemented with the general sPLA2 inhibitor, methyl indoxam, showed a reduction in body weight after 10 weeks (11). This decrease in body weight was accompanied by enhanced glucose tolerance and suppression of post-prandial plasma lysophospholipid levels. Transgenic mice over-expressing the human PLA2G1B in pancreatic acinar cells gained more weight when given the hypercaloric high-fat/high-carb diet, and these mice also had reduced glucose tolerance and insulin resistance (12).
Given the strong evidence supporting PLA2G1B inhibition as an avenue for improving metabolic health, a discussion on the mode of action is warranted. Absorption of LPC into the portal blood, plasma, and livers was reduced in Pla2g1b-/- mice fasted for 12 hours followed by a glucose-lipid mixed meal (13). These data suggest that phospholipid digestion in the intestinal lumen and absorption of the digested lysophospholipid product through the portal blood is caused directly by PLA2G1B enzymatic activity following a meal (13). While PLA2G1B is the major enzyme for phospholipid hydrolysis within the intestinal lumen, other lipolytic enzymes may compensate in its absence to preserve lipid and cholesterol absorption (14). In regards to the enhanced glucose tolerance in Pla2g1b-/- mice, there is evidence that LPC alone has an adverse effect on hyperglycemia as shown with a glucose tolerance test (GTT) (13). Furthermore, Pla2g1b-/- mice have elevated fatty acid oxidation which can be directly suppressed by LPC, suggesting PLA2G1B enzymatic products reduce fatty acid oxidation (15).
Taken together, studies on the metabolic impact of PLA2G1B inhibition indicate it has metabolic effects in response to feeding. Recent experiments provide evidence that the benefits of PLA2G1B inhibition mimic those seen in response to bariatric surgery in mice, including prevention of dyslipidemia, and protection and remission from diet-induced Type 2 diabetes (16).
PLA2G2A
PLA2G2A is induced by several cytokines and second messengers including interleukins 1 and 6 (IL-1 and IL-6), tumor necrosis factor (TNFα), lipopolysaccharides (LPS), and cyclic AMP, suggesting a pro-inflammatory role (47–49). The Pla2g2a knockout (Pla2g2a-/-) BALB/c mice have less joint inflammation than their wild-type counterparts under inflammatory conditions (50). With the focus of PLA2G2A studies being on its role in inflammation and atherosclerosis, studies on the actions of PLA2G2A in metabolism are quite limited. One consistent observation of the metabolic studies involving PLA2G2A is that its expression is up-regulated in response to a high-fat diet (17–19). In male Wistar rats, Pla2g2a expression was elevated 6-fold after 16 weeks on a high-carbohydrate high-fat (HCHF) diet (17). At 8 weeks of age, these rats were orally administered the PLA2G2A inhibitor KH064, which drastically reduced weight gain, fat mass, and prevented an increase of adipocyte crown formation and macrophage infiltration seen in the wild-type rats (17). Inhibition of Pla2g2a by KH064 was also accompanied by an increase in lipolytic gene expression, attributed to an increase in hormone-sensitive lipase (HSL) phosphorylation. Lastly, treatment with KH064 improved glucose tolerance and insulin sensitivity as assessed by GTTs and ITTs (17).
The metabolic phenotype of male C57BL/6 mice genetically engineered to overexpress the human PLA2G2A gene has also been assessed (51). PLA2G2A overexpression improved glucose clearance and insulin sensitivity in GTTs and ITTs, thereby alleviating obesogenic symptoms in response to HFD (18, 20). C57BL/6 mice normally do not express the murine Pla2g2a due to a frameshift mutation in exon 3 (52). Overexpression of human PLA2G2A protected mice from weight gain on a high-fat diet compared to wild-type C57BL/6 mice after 10 weeks. PLA2G2A expression also enhanced oxygen consumption (VO2) and energy expenditure. The expression of thermogenic genes in brown adipose tissue (BAT), including uncoupling protein 1 (UCP1), peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), and Sirtuin-1 (SIRT1) was elevated (18, 20). PLA2G2A-expressing primary adipocytes from epididymal and inguinal white adipose tissue (WAT), and interscapular BAT showed elevated abundance of several proteins involved in adaptive thermogenesis compared to C57BL/6 wild-type adipocytes (20). To accompany this phenotype, mice expressing PLA2G2A also had reduced 6-hour fasting blood glucose levels and an increase in glucose transporter type 4 (GLUT4) in BAT suggesting that PLA2G2A enhances BAT glucose utilization (20).
PLA2G2A contributes to the inflammatory response, but the enzyme’s role in obesity and metabolism is still unclear. Contributing to this, current investigations of PLA2G2As metabolic role used different designs. The human PLA2G2A gene was expressed in mice, whereas the rat Pla2g2a enzyme was inhibited pharmacologically (17, 51). Overexpression of PLA2G2A in mice may alter the expression of other secretory phospholipases in a variety of tissues, which could influence metabolism. Similarly, the activity of various sPLA2 isozymes has not been examined in response to PLA2G2A inhibition by KH064. Moreover, food intake following KH064 administration was not reported in this study, and the impact of KH064 on intestinal lipid absorption was not assessed.
PLA2G2E
The expression of PLA2G2E is elevated in the white adipose tissue (WAT) and BAT of female C57BL/6 mice fed a HFD (26). Conversely, female Pla2g2e-/- mice gain less weight on a HFD over 18 weeks, with marked reductions in fat mass, hepatic lipid deposition, plasma aspartate aminotransferase (AST), and alanine aminotransferase (ALT) levels. Thus, the level of PLA2G2E may affect adiposity and liver metabolism. PLA2G2E preferentially hydrolyzes PE and PS on very low-density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL), although with weak enzymatic activity compared to other sPLA2s (27, 28). Mass spectrometry of the lipoprotein particles from Pla2g2e-/- mice revealed a reduction in phospholipids (PL), triglyceride (TG), and cholesterol accumulation in VLDL, LDL, and HDL (26). These data suggest that Pla2g2e may promote obesity through elevated hepatic lipogenesis and VLDL assembly in the liver. Anionic phospholipids decrease the affinity for ApoE to bind the LDL receptor (LDL-r), which could impact lipoprotein particle clearance (53). Finally, whether lysophosphatidylserine (LPS) or lysophosphatidylethanolamine (LPE) impacts metabolism has yet to be determined.
In contrast, another study using male Pla2g2e-/- mice discovered the enzyme regulates lipolysis in adipocytes, likely through enhanced ERK1/2 signaling (29). Pla2g2e-/- mice had increased epididymal fat compared to C57BL/6 wild-type mice and accumulated more TG in the SVF isolated from adipose tissue. Over-expression of Pla2g2e in OP9 stem cells or treatment of 3T3-L1 cells with Pla2g2e protein reduced lipid accumulation and increased release of free glycerol, indicative of elevated lipolysis (29). These Pla2g2e-/- animals had reduced ERK1/2 signaling and HSL, the intracellular lipase responsible for hydrolyzing TG to FFAs. Finally, treatment of adipocytes with mouse Pla2g2e protein induced ERK1/2 signaling, demonstrating that Pla2g2e regulates adipocyte lipolysis through ERK/HSL signaling (29).
Currently, the understanding of the role of PLA2G2E in obesity and metabolism is limited to the two studies described above which report contrasting phenotypes with Pla2g2e-/- mice. A notable difference between these studies is the use of females versus males, and the contrasting results suggest sex-specific differences in Pla2g2e activity and its impact on obesity may be involved.
PLA2G5
PLA2G5 is mainly expressed in WAT and protects from diet-induced obesity. PLA2G5 expression is elevated in response to HFD feeding in female C57BL/6 mice (26). When placed on a HFD, Pla2g5-/- mice gained a large amount of weight, mainly from increased visceral fat mass (26). In GTTs and ITTs, the Pla2g5-/- mice had impaired glucose tolerance and insulin resistance. Furthermore, there was a striking induction of plasma ALT levels and hepatic fat deposition, indicating exacerbated hepatosteatosis (26). PLA2G5 preferentially hydrolyzes phosphatidylcholine (PC) in low-density lipoprotein (LDL) (54). In the Pla2g5-/- mice phospholipid, cholesterol, and TG levels were considerably higher in LDL (26). When transgenic mice overexpressing PLA2G5 in adipocytes were put on HFD, they showed better insulin sensitivity and a decreased expression of pro-inflammatory genes in WAT (26). These data suggest PLA2G5-mediated hydrolysis of PC and other phospholipids may reduce local adipose tissue inflammation which appears to have a beneficial impact on whole-body insulin sensitivity.
PLA2G5 also modulates bone marrow-derived macrophage (BMDM) polarization. BMDM treated with palmitic acid (PA) or lipopolysaccharides induces the inflammatory response. Addition of recombinant PLA2G5 enzyme augments the expression of M2 markers Arg1 and Cd206 in the BMDM, suggesting that PLA2G5 has anti-inflammatory effects (26). The capacity for PLA2G5 to push macrophage polarization from an M1 to M2 state broadens its impact as the metabolic benefits of M2 macrophages are well documented (55–57). Genetic deletion of Th2 or M2 inducers increases the risk for metabolic disorders (56), and M2 macrophage infusion into obese mice has proven to be effective in treating obesity and improving insulin sensitivity (57). In humans, M2 macrophages are more prevalent in adipose tissue from lean individuals (55). While additional work needs to be done to elucidate the effects of PLA2G5 on metabolism, these actions appear to be partly mediated by the fatty acid and/or PC-released induction of M2 macrophage polarization in adipose tissue.
The role of PLA2G5 in glucose-stimulated insulin secretion (GSIS) is complex. GSIS is decreased in isolated pancreatic islets from PLA2G5 knockout mice and in pancreatic MIN6 cells following siRNA-mediated PLA2G5 knockdown (58). Additionally, PLA2G5 overexpression in MIN6 cells enhanced GSIS and increased AA release into the media with no change in prostaglandin E2 (PGE2) abundance (58). In contrast to the studies with isolated islets, the GSIS of Pla2g5-/- mice was increased compared to WT mice (58). The elevated GSIS was attributed to increased pancreatic islet size and number of proliferating cells in the pancreatic β-islets of the Pla2g5 KO mice. The in vivo data from this study suggests a reduction in the release of AA, a fatty acid contained in over 30% of glycerolipids in rodent islets (59), is beneficial for insulin secretion and β-cell proliferation. Amino acids generally induce GSIS, while inhibition of the release of AA inhibits GSIS (60). Paradoxically, a major metabolite of AA is PGE2, a well-known inhibitor of GSIS (61–63). The role of AA metabolites in GSIS will be discussed further in the next section regarding Pla2g10. The data suggest that PLA2G5 regulates insulin secretion and β-cell proliferation, which may be dependent on the amount of AA released versus the amount of AA used for PGE2 production.
PLA2G10
Studies with transgenic mice provided evidence that PLA2G10 mediates adipogenesis and has thereby led to the hypothesis that it protects from diet-induced obesity (30, 31). PLA2G10 binds to zwitterionic phospholipids such as PC with high affinity, releasing AA and LPC. PLA2G10 is expressed in a variety of tissues including the lungs, adrenal glands, pancreas, brain, heart, and adipose tissue (27, 32–34). Pla2g10-/- mice on a C57BL/6 background gain more weight and adipose mass over 40 weeks compared to chow-fed wild-type mice (30). The effects of Pla2g10 deletion are directly on WAT, as there were no changes in food intake or respiration. Pre-adipocytes prepared from WAT of Pla2g10-/- mice accumulated more TG when induced to differentiate ex vivo, suggesting that PLA2G10 has a direct effect in adipose tissue to reduce lipid accumulation. Similarly, when the OP9 cell line was modified to overexpress PLA2G10, TG accumulation was reduced following differentiation (30). In addition, the cells had reduced expression of multiple lipogenic genes including sterol regulatory element-binding protein 1c (SREBP-1c), stearoyl-CoA desaturase-1 (SCD-1), and fatty acid synthase (FAS) (31, 64–66). Interestingly, the reduction in lipogenic gene expression arose from the ability of PLA2G10 to generate lipid products that suppress liver X receptor (LXR) activity.
PLA2G10 is expressed in adrenal cells and has a regulatory role in adrenal corticosteroid production. Overexpression of PLA2G10 in C57BL/6 mice resulted in a 30-40% reduction in corticosteroid production, and this effect was reversed by methyl indoxam administration (31). The elevated glucocorticoid level did not arise from elevated ACTH or ACTH responsiveness. PLA2G10 overexpression dramatically reduced expression of the LXR-target gene steroidogenic acute regulatory protein (StAR), a nuclear-encoded mitochondrial protein that mediates the rate-limiting step of steroid synthesis (67). As in the adipocytes, Pla2g10 generated a ligand that reduced LXR activity.
Pla2G10 is also expressed in the pancreatic beta cells and suppresses GSIS (68). Like Pla2g5, Pla2g10 generates AA. However, this pool of AA is converted to prostaglandin E2, which binds to the EP3 receptor. EP3 elevates cAMP leading to decreased insulin secretion. Why Pla2g5 and Pla2g10 have opposite effects on GSIS is unclear. However, Pla2g10 has to be proteolytically activated, and this may give it access to a different pool of phospholipids. Finally, older Pla2g10 KO mice appear to be protected from age-related glucose intolerance.
The current data on PLA2G10 indicate it impacts multiple aspects of metabolism and hormonal action. PLA2G10 expression reduces weight gain and overall adiposity in mice. Concerning hormone actions, Pla2g10 decreases corticosteroid production in adrenal cells (31). Excessive use or production of glucocorticoids will induce insulin resistance, weight gain, and adiposity while also exacerbating Type 2 diabetes (69–72). However, Pla2g10 reduces GSIS, giving this phospholipase a complex contribution to the overall metabolic state.
PLA2G12B
PLA2G12B is the only phospholipase implicated in metabolism showing no catalytic activity due to a point mutation in its active site, and thus it is hypothesized to act as a ligand for receptors that are currently unidentified (8). Using Pla2g12b-/- mice fed an ad libitum chow diet, knockout of the Pla2g12b gene increased TG, cholesterol, and free fatty acids in the liver, resulting in severe hepatosteatosis (35). Hepatocyte nuclear factor-4alpha (HNF-4α) and its co-activator PGC-1α induce Pla2g12b expression (35, 36), resulting in the induction of genes involved in lipoprotein packaging (microsomal triglyceride transfer protein, MTP) and VLDL secretion (37, 38). Moreover, liver-specific HNF-4α-/- mice are phenotypically similar to Pla2g12b-/- mice, as both lines have reduced serum TG and cholesterol levels and display severe hepatosteatosis (35, 39). These observations suggest that PLA2G12B is one gene involved in the control of lipid metabolism downstream of HNF-4α. Infection of mice with an adenovirus encoding Pla2g12b into Pla2g12b-/- mice improves hepatic VLDL secretion and restores the decline in serum TGs (35). These data indicate that PLA2G12B is under the regulation of HNF-4α and plays a metabolic role by regulating lipoprotein packaging and VLDL secretion in the liver. More recent data has shown that estrogen-related receptor γ (ERRγ) transcriptionally regulates PLA2G12B through binding to its promoter region to regulate hepatic VLDL-TG secretion (73). In Pla2g12b-/- mice, ERRγ fails to regulate VLDL-TG secretion and large hepatic lipid droplets result. Importantly, ERRγ is implicated in a wide range of physiologic roles including metabolic homeostasis, especially hepatic glucose metabolism (40). The findings associating PLA2G12B with ERRγ may be one avenue by which ERRγ mediates hepatic glucose production.
Conclusion
The recent studies discussed in this review show that sPLA2s can influence metabolic diseases such as Type 2 diabetes and obesity, at least partially through alterations in lipid production and mobilization (Figure 1); and while controversy exists regarding whether elevated lipids directly cause insulin resistance, pre-clinical and clinical data indicate an association between elevated lipids and lipoproteins with insulin resistance (74–77). Furthermore, one issue in analyzing the metabolic impact of sPLA2s is many of the isoforms effect intertwined pathologies such as atherosclerosis, heart disease, and cancer (41, 55, 57, 64–67, 69–72). For this reason, future studies on sPLA2s should consider their roles based on tissue localization, as their distinct functions may be altered based on the tissue being analyzed. Furthermore, the expression and compensation of other sPLA2 isoforms in transgenic animal models is another factor that might result in large phenotypic changes, and thus should also be observed to advance what we know about sPLA2s in metabolic diseases.
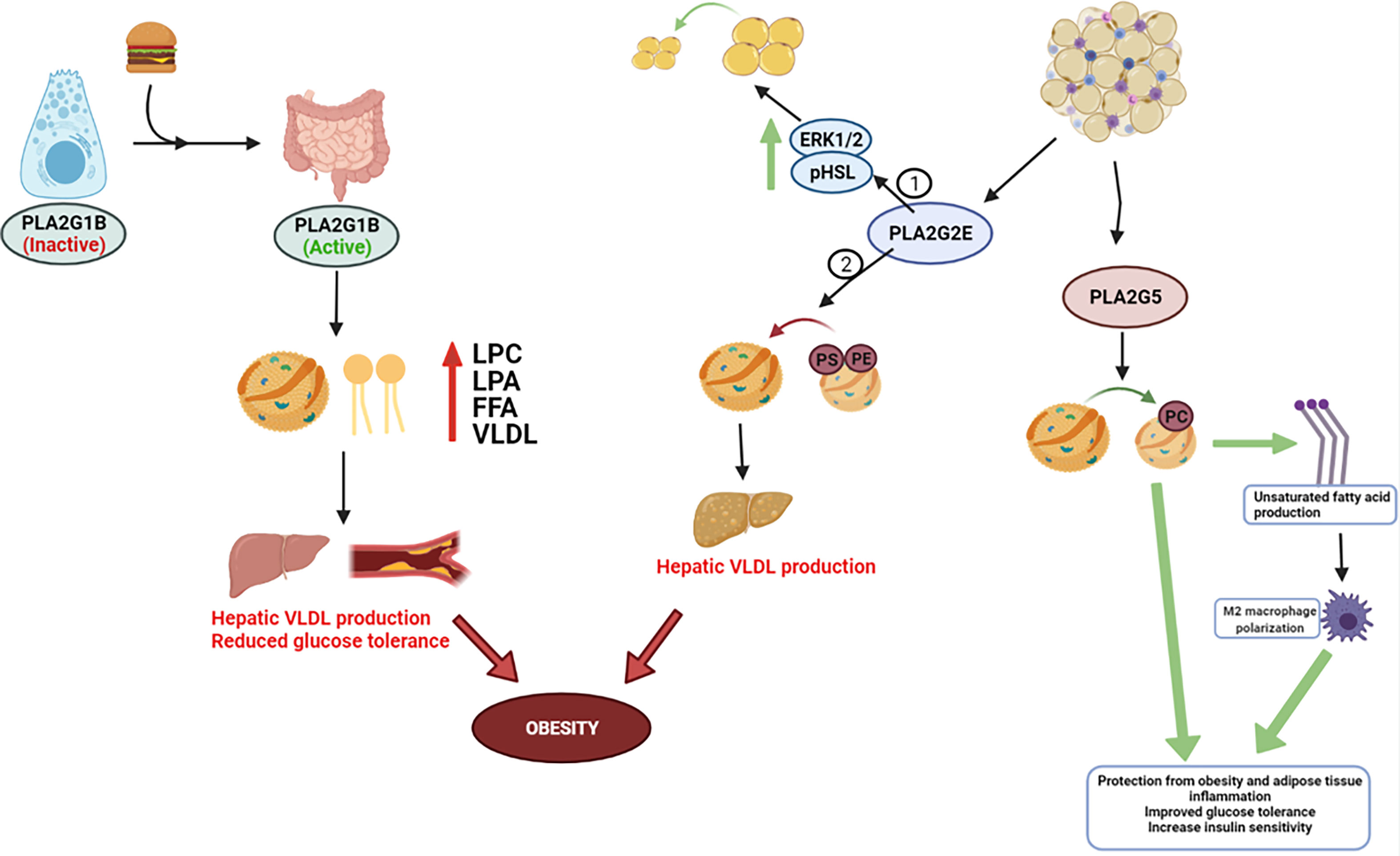
Figure 1 Metabolic role for PLA2s. PLA2G1B is released by pancreatic acinar cells into the pancreatic juice following a meal and then secreted into the intestinal lumen, where it eventually becomes activated. Phospholipid digestion by PLA2G1B results in elevated lysophosphatidylcholine (LPC), lysophosphatidic acid (LPA), and free fatty acids absorption in the portal blood, plasma, and liver, and increases hepatic very-low density lipoprotein (VLDL) production. Elevated enzymatic activity in the intestinal lumen can progress obesity, reduce glucose tolerance, and exacerbate insulin resistance – most notably due to elevated release/absorption of LPC and overarching dyslipidemia. PLA2G2E and PLA2G5 are expressed in WAT. 2 studies have contradicting results in PLA2G2Es metabolic role – 1 study discovered PLA2G2E increases ERK1/2 signaling and HSL phosphorylation to increase lipolysis, whereas Study 2 found PLA2G2E may promote obesity through hepatic lipogenesis and VLDL production. PLA2G5 preferentially hydrolyzes PC-rich phospholipids and promotes M2 macrophage polarization, which appears to have a beneficial impact on LDL lipid normalization and whole-body insulin sensitivity.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
I would like to thank the Wenner-Gren Foundation of Sweden and their post-doctoral fellowship program for the support on this work (UDP2020-0101).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
I would like to thank Juleen R. Zierath, Professor and Integrative Physiology research group leader at the Karolinska Institutet in Stockholm, Sweden, and Edwards A. Park, Professor in the Pharmacology Department at the University of Tennessee Health Science Center for their constant support and continued discussion about this review. I would also like to express my sincere gratitude to the Wenner-Gren Foundation in Sweden for their continued financial support of my work at the Karolinska Institute.
References
1. Zammit VA, Waterman IJ, Topping D, McKay G. Insulin Stimulation of Hepatic Triacylglycerol Secretion and the Etiology of Insulin Resistance. J Nutr (2001) 131(8):2074–7. doi: 10.1093/jn/131.8.2074
2. Ma M, Liu H, Yu J, He S, Li P, Ma C, et al. Triglyceride Is Independently Correlated With Insulin Resistance and Islet Beta Cell Function: A Study in Population With Different Glucose and Lipid Metabolism States. Lipids Health Dis (2020) 19(1):121. doi: 10.1186/s12944-020-01303-w
3. Lee EY, Yang HK, Lee J, Kang B, Yang Y, Lee S-H, et al. Triglyceride Glucose Index, A Marker of Insulin Resistance, is Associated With Coronary Artery Stenosis in Asymptomatic Subjects With Type 2 Diabetes. Lipids Health Dis (2016) 15(1):155.
4. Lipina C, Hundal HS. Sphingolipids: Agents Provocateurs in the Pathogenesis of Insulin Resistance. Diabetologia (2011) 54(7):1596–607. doi: 10.1186/s12944-016-0324-2
5. Jornayvaz FR, Shulman GI. Diacylglycerol Activation of Protein Kinase Cϵ and Hepatic Insulin Resistance. Cell Metab (2012) 15(5):574–84. doi: 10.1016/j.cmet.2012.03.005
6. D’Souza K, Paramel GV, Kienesberger PC. Lysophosphatidic Acid Signaling in Obesity and Insulin Resistance. Nutrients (2018) 10(4):399. doi: 10.3390/nu10040399
7. Murakami M, Sato H, Miki Y, Yamamoto K, Taketomi Y. A New Era of Secreted Phospholipase a₂. J Lipid Res (2015) 56(7):1248–61. doi: 10.1194/jlr.R058123
8. Rouault M, Bollinger JG, Lazdunski M, Gelb MH, Lambeau G. Novel Mammalian Group XII Secreted Phospholipase A2 Lacking Enzymatic Activity. Biochemistry (2003) 42(39):11494–503. doi: 10.1021/bi0349930
9. Hollie NI, Hui DY. Group 1B Phospholipase A2 Deficiency Protects Against Diet-Induced Hyperlipidemia in Mice. J Lipid Res (2011) 52(11):2005–11. doi: 10.1194/jlr.M019463
10. Hollie NI, Konaniah ES, Goodin C, Hui DY. Group 1b Phospholipase A2 Inactivation Suppresses Atherosclerosis and Metabolic Diseases in LDL Receptor-Deficient Mice. Atherosclerosis (2014) 234(2):377–80. doi: 10.1016/j.atherosclerosis.2014.03.027
11. Hui DY, Cope MJ, Labonte ED, Chang HT, Shao J, Goka E, et al. The Phospholipase A(2) Inhibitor Methyl Indoxam Suppresses Diet-Induced Obesity and Glucose Intolerance in Mice. Br J Pharmacol (2009) 157(7):1263–9. doi: 10.1111/j.1476-5381.2009.00308.x
12. Cash JG, Kuhel DG, Goodin C, Hui DY. Pancreatic Acinar Cell-Specific Over-Expression of Group 1B Phospholipase A(2) Exacerbates Diet-Induced Obesity and Insulin Resistance in Mice. Int J Obes (2011) 35(6):877–81. doi: 10.1038/ijo.2010.215
13. Labonté ED, Kirby RJ, Schildmeyer NM, Cannon AM, Huggins KW, Hui DY. Group 1b Phospholipase A(2)–Mediated Lysophospholipid Absorption Directly Contributes to Postprandial Hyperglycemia. Diabetes (2006) 55(4):935–41. doi: 10.2337/diabetes.55.04.06.db05-1286
14. Richmond BL, Boileau AC, Zheng S, Huggins KW, Granholm NA, Tso P, et al. Compensatory Phospholipid Digestion Is Required for Cholesterol Absorption in Pancreatic Phospholipase A(2)-Deficient Mice. Gastroenterology (2001) 120(5):1193–202. doi: 10.1053/gast.2001.23254
15. Labonté ED, Pfluger PT, Cash JG, Kuhel DG, Roja JC, Magness DP, et al. Postprandial Lysophospholipid Suppresses Hepatic Fatty Acid Oxidation: The Molecular Link Between Group 1B Phospholipase A(2) and Diet-Induced Obesity. FASEB J (2010) 24(7):2516–24. doi: 10.1096/fj.09-144436
16. Cash JG, Konaniah E, Hegde N, Kuhel DG, Watanabe M, Romick-Rosendale L, et al. Therapeutic Reduction of Lysophospholipids in the Digestive Tract Recapitulates the Metabolic Benefits of Bariatric Surgery and Promotes Diabetes Remission. Mol Metab (2018). 16:55–64. doi: 10.1016/j.molmet.2018.07.009
17. Iyer A, Lim J, Poudyal H, Reid RC, Suen JY, Webster J, et al. An Inhibitor of Phospholipase A(2) Group IIA Modulates Adipocyte Signaling and Protects Against Diet-Induced Metabolic Syndrome in Rats. Diabetes (2012) 61(9):2320–9. doi: 10.2337/db11-1179
18. Kuefner MS, Pham K, Redd JR, Stephenson EJ, Harvey I, Deng X, et al. Secretory Phospholipase A2 Group IIA Modulates Insulin Sensitivity and Metabolism. J Lipid Res (2017) 58(9):1822–33. doi: 10.1194/jlr.M076141
19. Sharma P, Levesque T, Boilard E, Park EA. Thyroid Hormone Status Regulates the Expression of Secretory Phospholipases. Biochem Biophys Res Commun (2014) 444(1):56–62. doi: 10.1016/j.bbrc.2014.01.003
20. Kuefner MS, Deng X, Stephenson EJ, Pham K, Park EA. Secretory Phospholipase A2 Group IIA Enhances the Metabolic Rate and Increases Glucose Utilization in Response to Thyroid Hormone. FASEB J (2019) 33(1):738–49. doi: 10.1096/fj.201800711R
21. Monroy-Munoz IE, Angeles-Martinez J, Posadas-Sanchez R, Villarreal-Molina T, Alvarez-Leon E, Flores-Dominguez C, et al. PLA2G2A Polymorphisms Are Associated With Metabolic Syndrome and Type 2 Diabetes Mellitus. Results Genet Atherosclerotic Dis Mexican Study Immunobiol (2017) 222(10):967–72. doi: 10.1016/j.imbio.2016.08.014
22. Miki Y, Yamamoto K, Taketomi Y, Sato H, Shimo K, Kobayashi T, et al. Lymphoid Tissue Phospholipase A(2) Group IID Resolves Contact Hypersensitivity by Driving Antiinflammatory Lipid Mediators. J Exp Med (2013) 210(6):1217–34. doi: 10.1084/jem.20121887
23. Valentin E, Koduri RS, Scimeca JC, Carle G, Gelb MH, Lazdunski M, et al. Cloning and Recombinant Expression of a Novel Mouse-Secreted Phospholipase A2. J Biol Chem (1999) 274(27):19152–60. doi: 10.1074/jbc.274.27.19152
24. Ishizaki J, Suzuki N, Higashino K, Yokota Y, Ono T, Kawamoto K, et al. Cloning and Characterization of Novel Mouse and Human Secretory Phospholipase A(2)s. J Biol Chem (1999) 274(35):24973–9. doi: 10.1074/jbc.274.35.24973
25. Shakhov AN, Rubtsov AV, Lyakhov IG, Tumanov AV, Nedospasov SA. SPLASH (PLA2IID), A Novel Member of Phospholipase A2 Family, Is Associated With Lymphotoxin Deficiency. Genes Immun (2000) 1(3):191–9. doi: 10.1038/sj.gene.6363659
26. Sato H, Taketomi Y, Ushida A, Isogai Y, Kojima T, Hirabayashi T, et al. The Adipocyte-Inducible Secreted Phospholipases PLA2G5 and PLA2G2E Play Distinct Roles in Obesity. Cell Metab (2014) 20(1):119–32. doi: 10.1016/j.cmet.2014.05.002
27. Suzuki N, Ishizaki J, Yokota Y, Higashino K-i, Ono T, Ikeda M, et al. Structures, Enzymatic Properties, and Expression of Novel Human and Mouse Secretory Phospholipase A2s. J Biol Chem (2000) 275(8):5785–93. doi: 10.1074/jbc.275.8.5785
28. Yamamoto K, Miki Y, Sato H, Nishito Y, Gelb MH, Taketomi Y, et al. Expression and Function of Group IIE Phospholipase A(2) in Mouse Skin. J Biol Chem (2016) 291(30):15602–13. doi: 10.1074/jbc.M116.734657
29. Zhi H, Qu L, Wu F, Chen L, Tao J. Group IIE Secretory Phospholipase A2 Regulates Lipolysis in Adipocytes. Obesity (2015) 23(4):760–8. doi: 10.1002/oby.21015
30. Li X, Shridas P, Forrest K, Bailey W, Webb NR. Group X Secretory Phospholipase A(2) Negatively Regulates Adipogenesis in Murine Models. FASEB J (2010) 24(11):4313–24. doi: 10.1096/fj.10-154716
31. Shridas P, Bailey WM, Boyanovsky BB, Oslund RC, Gelb MH, Webb NR. Group X Secretory Phospholipase A2 Regulates the Expression of Steroidogenic Acute Regulatory Protein (StAR) in Mouse Adrenal Glands. J Biol Chem (2010) 285(26):20031–9. doi: 10.1074/jbc.M109.090423
32. Cupillard L, Koumanov K, Mattéi M-G, Lazdunski M, Lambeau G. Cloning, Chromosomal Mapping, and Expression of a Novel Human Secretory Phospholipase A2. J Biol Chem (1997) 272(25):15745–52. doi: 10.1074/jbc.272.25.15745
33. Morioka Y, Saiga A, Yokota Y, Suzuki N, Ikeda M, Ono T, et al. Mouse Group X Secretory Phospholipase A2 Induces a Potent Release of Arachidonic Acid From Spleen Cells and Acts as a Ligand for the Phospholipase A2 Receptor. Arch Biochem Biophysics (2000) 381(1):31–42. doi: 10.1006/abbi.2000.1977
34. Sato H, Isogai Y, Masuda S, Taketomi Y, Miki Y, Kamei D, et al. Physiological Roles of Group X-Secreted Phospholipase A2 in Reproduction, Gastrointestinal Phospholipid Digestion, and Neuronal Function. J Biol Chem (2011) 286(13):11632–48. doi: 10.1074/jbc.M110.206755
35. Guan M, Qu L, Tan W, Chen L, Wong C-W. Hepatocyte Nuclear Factor-4 Alpha Regulates Liver Triglyceride Metabolism in Part Through Secreted Phospholipase A2 GXIIB. Hepatology (2011) 53(2):458–66. doi: 10.1002/hep.24066
36. Aljakna A, Choi S, Savage H, Hageman Blair R, Gu T, Svenson KL, et al. Pla2g12b and Hpn Are Genes Identified by Mouse ENU Mutagenesis That Affect HDL Cholesterol. PloS One (2012) 7(8):e43139. doi: 10.1371/journal.pone.0043139
37. Sheena V, Hertz R, Nousbeck J, Berman I, Magenheim J, Bar-Tana J. Transcriptional Regulation of Human Microsomal Triglyceride Transfer Protein by Hepatocyte Nuclear Factor-4α. J Lipid Res (2005) 46(2):328–41. doi: 10.1194/jlr.M400371-JLR200
38. Yin L, Ma H, Ge X, Edwards PA, Zhang Y. Hepatic Hnf4α Is Essential for Maintaining Triglyceride and Cholesterol Homeostasis. Arteriosclerosis Thrombosis Vasc Biol (2011) 31(2):328–36. doi: 10.1161/ATVBAHA.110.217828
39. Hayhurst GP, Lee Y-H, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte Nuclear Factor 4α (Nuclear Receptor 2a1) Is Essential for Maintenance of Hepatic Gene Expression and Lipid Homeostasis. Mol Cell Biol (2001) 21(4):1393–403. doi: 10.1128/MCB.21.4.1393-1403.2001
40. Kim D-K, Gang G-T, Ryu D, Koh M, Kim Y-N, Kim SS, et al. Inverse Agonist of Nuclear Receptor Errγ Mediates Antidiabetic Effect Through Inhibition of Hepatic Gluconeogenesis. Diabetes (2013) 62(9):3093–102. doi: 10.2337/db12-0946
41. Murakami M TY, Sato H, Yamamoto K. Secreted Phospholipase A2 Revisited. J Biol Chem (2011) 150:233–55. doi: 10.1093/jb/mvr088
42. Ahn S-H, Shah YM, Inoue J, Morimura K, Kim I, Yim S, et al. Hepatocyte Nuclear Factor 4α in the Intestinal Epithelial Cells Protects Against Inflammatory Bowel Disease. Inflammatory Bowel Dis (2008) 14(7):908–20. doi: 10.1002/ibd.20413
43. Liu Q, Yang M, Fu X, Liu R, Sun C, Pan H, et al. Activation of Farnesoid X Receptor Promotes Triglycerides Lowering by Suppressing Phospholipase A2 G12B Expression. Mol Cell Endocrinol (2016) 436:93–101. doi: 10.1016/j.mce.2016.07.027
44. Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, et al. Intestinal FXR Agonism Promotes Adipose Tissue Browning and Reduces Obesity and Insulin Resistance. Nat Med (2015) 21:159. doi: 10.1038/nm.3760
45. Trabelsi MS, Daoudi M, Prawitt J, Ducastel S, Touche V, Sayin SI, et al. Farnesoid X Receptor Inhibits Glucagon-Like Peptide-1 Production by Enteroendocrine L Cells. Nat Commun (2015) 6:7629. doi: 10.1038/ncomms8629
46. Hui DY. Group 1B Phospholipase A2 in Metabolic and Inflammatory Disease Modulation. Biochim Biophys Acta (BBA) Mol Cell Biol Lipids (2018)6:784–8. doi: 10.1016/j.bbalip.2018.07.001
47. Antonio V, Brouillet A, Janvier B, Monne C, Bereziat G, Andreani M, et al. Transcriptional Regulation of the Rat Type IIA Phospholipase A2 Gene by cAMP and Interleukin-1β in Vascular Smooth Muscle Cells: Interplay of the CCAAT/enhancer Binding Protein (C/EBP), Nuclear Factor-κb and Ets Transcription Factors. Biochem J (2002) 368(2):415–24. doi: 10.1042/bj20020658
48. Crowl RM, Stoller TJ, Conroy RR, Stoner CR. Induction of Phospholipase A2 Gene Expression in Human Hepatoma Cells by Mediators of the Acute Phase Response. J Biol Chem (1991) 266(4):2647–51. doi: 10.1016/S0021-9258(18)52293-6
49. Massaad C, Paradon M, Jacques C, Salvat C, Bereziat G, Berenbaum F, et al. Induction of Secreted Type IIA Phospholipase A2 Gene Transcription by Interleukin-1β: ROLE OF C/EBP FACTORS. J Biol Chem (2000) 275(30):22686–94. doi: 10.1074/jbc.M001250200
50. Boilard E, Lai Y, Larabee K, Balestrieri B, Ghomashchi F, Fujioka D, et al. A Novel Anti-Inflammatory Role for Secretory Phospholipase A2 in Immune Complex-Mediated Arthritis. EMBO Mol Med (2010) 2(5):172–87. doi: 10.1002/emmm.201000072
51. Kuefner MS, Pham K, Redd JR, Stephenson EJ, Harvey I, Deng X, et al. Secretory Phospholipase A2 Group IIA (PLA2G2A) Modulates Insulin Sensitivity and Metabolism. J Lipid Res (2017). 9:1822–33. doi: 10.1194/jlr.M076141
52. Kennedy BP, Payette P, Mudgett J, Vadas P, Pruzanski W, Kwan M, et al. A Natural Disruption of the Secretory Group II Phospholipase A2 Gene in Inbred Mouse Strains. J Biol Chem (1995) 270(38):22378–85. doi: 10.1074/jbc.270.38.22378
53. Yamamoto T, Ryan RO. Anionic Phospholipids Inhibit Apolipoprotein E–low-Density Lipoprotein Receptor Interactions. Biochem Biophys Res Commun (2007) 354(3):820–4. doi: 10.1016/j.bbrc.2007.01.066
54. Sato H, Kato R, Isogai Y, Saka G, Ohtsuki M, Taketomi Y, et al. Analyses of Group III Secreted Phospholipase A2 Transgenic Mice Reveal Potential Participation of This Enzyme in Plasma Lipoprotein Modification, Macrophage Foam Cell Formation, and Atherosclerosis. J Biol Chem (2008) 283(48):33483–97. doi: 10.1074/jbc.M804628200
55. Hill AA, Bolus WR, Hasty AH. A Decade of Progress in Adipose Tissue Macrophage Biology. Immunol. Rev (2014) 262(1):134–52. doi: 10.1111/imr.12216
56. Odegaard JI, Chawla A. The Immune System as a Sensor of the Metabolic State. Immunity (2013) 38(4):644–54. doi: 10.1016/j.immuni.2013.04.001
57. Zhang Q, Hao H, Xie Z, Cheng Y, Yin Y, Xie M, et al. M2 Macrophages Infusion Ameliorates Obesity and Insulin Resistance by Remodeling Inflammatory/Macrophages’ Homeostasis in Obese Mice. Mol Cell Endocrinol (2017) 443:63–71. doi: 10.1016/j.mce.2017.01.005
58. Shridas P, Noffsinger VP, Trumbauer AC, Webb NR. The Dual Role of Group V Secretory Phospholipase A2 in Pancreatic Beta-Cells. Endocrine (2017) 58(1):47–58. doi: 10.1007/s12020-017-1379-1
59. Ramanadham S, Bohrer A, Mueller M, Jett P, Gross RW, Turk J. Mass Spectrometric Identification and Quantitation of Arachidonate-Containing Phospholipids in Pancreatic Islets: Prominence of Plasmenylethanolamine Molecular Species. Biochemistry (1993) 32(20):5339–51. doi: 10.1021/bi00071a009
60. Ramanadham S, Hsu FF, Zhang S, Jin C, Bohrer A, Song H, et al. Apoptosis of Insulin-Secreting Cells Induced by Endoplasmic Reticulum Stress Is Amplified by Overexpression of Group VIA Calcium-Independent Phospholipase A2 (Ipla2 Beta) and Suppressed by Inhibition of Ipla2 Beta. Biochemistry (2004) 43(4):918–30. doi: 10.1021/bi035536m
61. Robertson RP, Tsai P, Little SA, Zhang HJ, Walseth TF. Receptor-Mediated Adenylate Cyclase-Coupled Mechanism for PGE2 Inhibition of Insulin Secretion in HIT Cells. Diabetes (1987) 36(9):1047–53. doi: 10.2337/diab.36.9.1047
62. Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in Health and Disease. J Lipid Res (2009) 50 Suppl:S423–8. doi: 10.1194/jlr.R800094-JLR200
63. Yajima H, Komatsu M, Schermerhorn T, Aizawa T, Kaneko T, Nagai M, et al. cAMP Enhances Insulin Secretion by an Action on the ATP-Sensitive K+ Channel-Independent Pathway of Glucose Signaling in Rat Pancreatic Islets. Diabetes (1999) 48(5):1006–12. doi: 10.2337/diabetes.48.5.1006
64. Chu K, Miyazaki M, Man WC, Ntambi JM. Stearoyl-Coenzyme A Desaturase 1 Deficiency Protects Against Hypertriglyceridemia and Increases Plasma High-Density Lipoprotein Cholesterol Induced by Liver X Receptor Activation. Mol Cell Biol (2006) 26(18):6786–98. doi: 10.1128/MCB.00077-06
65. Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, et al. Direct and Indirect Mechanisms for Regulation of Fatty Acid Synthase Gene Expression by Liver X Receptors. J Biol Chem (2002) 277(13):11019–25. doi: 10.1074/jbc.M111041200
66. Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of Mouse Sterol Regulatory Element-Binding Protein-1c Gene (SREBP-1c) by Oxysterol Receptors, LXRalpha and LXRbeta. Genes Dev (2000) 14(22):2819–30. doi: 10.1101/gad.844900
67. Stocco DM. StAR Protein and the Regulation of Steroid Hormone Biosynthesis. Annu Rev Physiol (2001) 63:193–213. doi: 10.1146/annurev.physiol.63.1.193
68. Shridas P, Zahoor L, Forrest KJ, Layne JD, Webb NR. Group X Secretory Phospholipase A2 Regulates Insulin Secretion Through a Cyclooxygenase-2-Dependent Mechanism. J Biol Chem (2014) 289(40):27410–7. doi: 10.1074/jbc.M114.591735
69. Asensio C, Muzzin P, Rohner-Jeanrenaud F. Role of Glucocorticoids in the Physiopathology of Excessive Fat Deposition and Insulin Resistance. Int J Obes Related Metab Disord: J Int Assoc Study Obes (2004) 28 Suppl 4:S45–52. doi: 10.1038/sj.ijo.0802856
70. Pivonello R, De Leo M, Vitale P, Cozzolino A, Simeoli C, De Martino MC, et al. Pathophysiology of Diabetes Mellitus in Cushing’s Syndrome. Neuroendocrinology (2010) 92 Suppl 1:77–81. doi: 10.1159/000314319
71. Stojanovska L, Rosella G, Proietto J. Evolution of Dexamethasone-Induced Insulin Resistance in Rats. Am J Physiol (1990) 258(5 Pt 1):E748–56. doi: 10.1152/ajpendo.1990.258.5.E748
72. Zakrzewska KE, Cusin I, Stricker-Krongrad A, Boss O, Ricquier D, Jeanrenaud B, et al. Induction of Obesity and Hyperleptinemia by Central Glucocorticoid Infusion in the Rat. Diabetes (1999) 48(2):365–70. doi: 10.2337/diabetes.48.2.365
73. Chen L, Wu M, Zhang S, Tan W, Guan M, Feng L, et al. Estrogen-Related Receptor γ Regulates Hepatic Triglyceride Metabolism Through Phospholipase A2 G12B. FASEB J (2019) 33(7):7942–52. doi: 10.1096/fj.201802704R
74. He Q, Gao Z, Yin J, Zhang J, Yun Z, Ye J. Regulation of HIF-1{Alpha} Activity in Adipose Tissue by Obesity-Associated Factors: Adipogenesis, Insulin, and Hypoxia. Am J Physiol Endocrinol Metab (2011) 300(5):E877–85. doi: 10.1152/ajpendo.00626.2010
75. Mykkänen L, Haffner SM, Rainwater DL, Karhapää P, Miettinen H, Laakso M. Relationship of LDL Size to Insulin Sensitivity in Normoglycemic Men. Arteriosclerosis Thrombosis Vasc Biol (1997) 17(7):1447–53. doi: 10.1161/01.ATV.17.7.1447
76. Petersen KS, Bowen KJ, Tindall AM, Sullivan VK, Johnston EA, Fleming JA, et al. The Effect of Inflammation and Insulin Resistance on Lipid and Lipoprotein Responsiveness to Dietary Intervention. Curr Developments Nutr (2020) 4(11). 11. doi: 10.1093/cdn/nzaa160
Keywords: phospholipase A2, metabolism and obesity, Type 2 diabetes, insulin resistance, glucose homeostasis, lipid metabolism
Citation: Kuefner MS (2021) Secretory Phospholipase A2s in Insulin Resistance and Metabolism. Front. Endocrinol. 12:732726. doi: 10.3389/fendo.2021.732726
Received: 29 June 2021; Accepted: 09 August 2021;
Published: 27 August 2021.
Edited by:
Jeff M. P. Holly, University of Bristol, United KingdomReviewed by:
Guillermo Romero, University of Pittsburgh, United StatesCarsten Schmitz-Peiffer, Garvan Institute of Medical Research, Australia
Copyright © 2021 Kuefner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael S. Kuefner, TWljaGFlbC5rdWVmbmVyQGtpLnNl