 Fengchao Lang
Fengchao Lang Abhishek Jha
Abhishek Jha Leah Meuter2
Leah Meuter2 Karel Pacak
Karel Pacak Chunzhang Yang
Chunzhang Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 20 September 2021
Sec. Cancer Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.731096
This article is part of the Research Topic Pheochromocytoma and Paraganglioma: Prognosis, Treatment and Molecular Mechanisms View all 7 articles
Carotid body paragangliomas (PGLs) are rare neuroendocrine tumors that develop within the adventitia of the medial aspect of the carotid bifurcation. Carotid body PGLs comprise about 65% of head and neck paragangliomas, however, their genetic background remains elusive. In the present study, we report one case of carotid body PGL with a somatic mutation in the gene encoding isocitrate dehydrogenase 2 (IDH2). The missense mutation in IDH2 resulted in R172G amino acid substitution, which exhibits neomorphic activity and production of D-2-hydroxyglutarate.
Paragangliomas (PGLs) are neuroendocrine tumors originating from the chromaffin cells in the neural crest. PGLs can arise near the carotid artery, along nerve pathways in the head and neck. Several studies recently revealed that PGLs are heterogeneous and can be categorized into disease Clusters I and II, based on their molecular subtypes (1, 2). Cluster I PGLs carry genetic abnormalities in metabolic pathways, such as loss-of-function mutations in the genes that encode the succinate dehydrogenase (SDHx) and Von Hippel-Lindau (VHL) tumor suppressor protein (3). Our recent studies also found that gain-of-function mutations in the oxygen-sensing domain in hypoxia inducible factor 2 (EPAS1) can also be related to the development of Cluster I tumors (4). Functional studies further indicate that Cluster I PGLs share many common molecular signatures that are relevant to disease pathogenesis, including a pseudohypoxia phenotype, metabolic reprogramming, and epigenetic shifts. Cluster II PGLs carry mutations in the kinase signaling pathways, such as genes that encode NF1, RET, and MAX (5).
Isocitrate dehydrogenases (IDHs) are metabolic enzymes that are involved in Krebs cycle metabolism. IDH1 is a cytoplasmic enzyme that converts isocitrate into α-ketoglutarate (α-KG) in an NADP+-dependent manner. IDH2 is a mitochondrial enzyme that catalyzes a similar reaction with IDH1. IDH3 is a mitochondrial enzyme that catalyzes irreversible oxidative decarboxylation of isocitrate through an NAD+ dependent manner (6, 7). Several pioneering studies revealed that missense mutations in IDH1/2 are frequently identified in several types of human malignancies, including lower grade glioma, acute myeloid leukemia, cholangiocarcinoma, and chondrosarcoma (8–11). Cancer-associated IDH mutations result in amino acid substitutions at the catalytic center of the enzyme, which lead to neomorphic activity (change-of-function). The mutant IDHs consume α-KG and NADPH and produce D-2-hydroxyglutarate (D-2-HG) and NADP+ (12, 13). Many recent findings have demonstrated that D-2-HG is an oncometabolite, which promotes oncogenesis through metabolic reprogramming, epigenetic shifts, and activation of oncogenic pathways (14, 15). Notably, the glutaminolytic and reductive carboxylation are remarkably impacted by cancer-associated IDH mutations, which results in a distinctive metabolic signature among solid cancers (16). The presence of an IDH mutation in PGL is of significant interest. Zhang et al. reported one case of an IDH heterozygous mutation accompanied by an ATRX mutation in one PGL case (17). The mutation of IDH2 was firstly described in a case of head and neck paraganglioma (18). In the present study, we provided detail histological analysis and 18F-FDOPA positron emission tomography (PET) scan about this case. The presence of IDH2-R172G mutation was confirmed by an antibody targeting the mutant enzymes. The R172G mutation results in the production of D-2-HG.
The tumor specimen and the patient were previously reported (18). A right carotid body tumor was surgically resected for histology, genetics, and biochemistry analysis.
Genomic DNA was extracted from tumor tissue and patient blood using DNeasy Blood & Tissue kit (QIAGEN). Library preparation and exome enrichment were conducted using SureSelect V7 kit (Agilent). The sequencing was performed on a NovaSeq S4-300 chip. The resulting FASTQ data was aligned to the human reference genome for variant annotation.
Sanger sequencing was performed as previously described (19). Genomic DNA was amplified through polymerase chain reaction (PCR) using Herculase II Fusion DNA polymerase (Agilent). The amplicon was purified using Monarch PCR & DNA Cleanup Kit (New England Biolabs, Inc.) and sent for Sanger sequencing (Eurofin). The primers used for PCR and sequencing were: F: 5’-CAG AGA CAA GAG GAT GGC TAG G -3’ and R: 5’-TGT AAA ACG ACG GCC AGT GTC TGG CTG TGT TGT TGC TTG-3’. Sequence analysis was performed using Geneious Pro software (version 11.1.4).
Immunohistochemistry was performed as previously described. Tumor tissues were fixed and sectioned to 10-micron slices. The slices were labeled with primary antibodies and probed with Vectastain ABC kit (Vector laboratories). The slices were then exposed to 3,3′-Diaminobenzidine and mounted in Permount mounting medium (Fisher Chemical). The samples were then visualized by light microscopy (Olympus BX-43). The primary antibody used in this study was anti-IDH1/2 Mutant (R132/R172), clone MsMab-1 (1:200, MilliPore).
Mutagenesis was conducted as previously described (20). The pCMV6-IDH2 wild type plasmid (Origene) was used as a template. The coding sequence of IDH2 was transferred to pLenti-C-myc-DDK-IRES-puro plasmid (Origene). The mutagenesis reaction was performed using a Quikchange Mutagenesis Lightning kit (Agilent). The sequence of the mutant plasmid was confirmed by sequencing the entire coding region of IDH2. The plasmid was then used for lentivirus packaging. The hpheo-1 cells were kindly provided by Dr. Hans Ghayee (University of Florida). The hpheo-1 cells were infected with lentivirus with IDH2 coding sequence and selected by puromycin. The mutagenesis primers used were: 5’- CCA TGG GCG TGC CCG CCA ATG GTG ATG -3’; 5’- CAT CAC CAT TGG CGG GCA CGC CCA TGG-3’.
The tissue specimens or cell pellets were lysed with Radioimmunoprecipitation assay buffer (RIPA buffer, Thermo Fisher) and quantified using the DC Protein Assay (Bio-Rad). Protein samples were resolved on 4-12% Bis-tris gel (Thermo Fisher) and transferred to PVDF membrane (MilliPore). The membranes were labeled with primary antibodies and visualized through chemiluminescence kit (Bio-Rad). The primary antibodies used in this study include anti-IDH1/2 Mutant (R132/R172), clone MsMab-1 (1:2,000, MilliPore); Flag (1:5,000, Origene); and β-actin (1:5,000, Cell Signaling Technology).
Stock solutions of D- or L-2-hydroxyglutaric acid (D- or L-2-HG) disodium salt (10 mM), and the internal standards (IS) D or L-2-HG -13C5 disodium salt (5 mM) were prepared in water and stored at -50°C until use. Calibration standard of D- and L-2-HG were prepared by serial dilutions of the stock with 80% methanol. The working IS mixture (1 µM each) was prepared by diluting the stock with 50% methanol. The TSPC (N-(p-toluenesulfonyl)-L-phenylalanyl chloride) derivatization reagent was prepared in acetonitrile (1 mg/mL). Tissues were homogenized in 80% methanol (15-75 mg tissue/mL solvent) using the Bead Rupter (Omni International). The supernatant was diluted with 80% methanol if needed to ensure the 2-HG level was within the linear range of the calibration curve. Samples were analyzed in triplicate. TSPC derivatization was carried out according to Cheng et al. (21) with slight modification. In a 0.5 mL micro-centrifuge tube, 30 µL of sample (or standard) were mixed with 10 µL of IS and then vacuum dried. Two µL of pyridine and 100 µL of TSPC were added to the dried samples. After 30 min, the reaction mixture was vacuum dried and reconstituted with 30 µL of 50% acetonitrile for LC/MS/MS. LC was performed with a Prominence 20AD system (Shimadzu). The injection volume was 2 µL. Separation was achieved at 35°C with a 2.1 x 100 mm, 2.7 µm Cortecs C18 column (Waters). Mobile phase A was 5 mM ammonium acetate in water and mobile phase B was methanol. The flow rate was 300 µL/min and peaks were eluted with a 5-min gradient from 25%-40%B. MS/MS was performed with a TSQ Quantiva triple quadrupole mass spectrometer (Thermo Fisher) operating at selected reaction monitoring mode with negative electrospray ionization. The TSPC derivatized peaks were detected using the following m/z precursor > product ions: 2-HG (448 > 318); 2-HG -13C5 (453 > 318). D- and L-2-HG in the homogenates were determined by a 9-level calibration curve (0.05-25 µM) with linear regression (1/x weighting) using the Thermo Xcalibur software.
Statistical analysis was performed with GraphPad Prism software (Version 6.01). A student t-test was applied for statistical comparisons. All statistical tests were two-tailed. Results are shown as Mean ± SEM, and P < 0.05 was considered statistically significant.
The patient was a 57-year-old female presenting with a history of multinodular goiter and benign thyroid nodules through fine needle aspiration (FNA). The patient also has a family history of thyroid cancer. The patient presented approximately 2.5 x 1.5 cm mass at bifurcation of right common carotid artery with minimal vascularity. Whole-body CT and neck MRI revealed a 2.7 x 2.0 cm mass in the right neck suggestive of carotid body PGL (Figures 1A, B). The mass was also positive on 18F-fluorodopa (18F-FDOPA) PET/CT scan (Figure 1C). There was no evidence of any other pheochromocytomas (PHEOs) or PGLs on the aforementioned scans. A 2.8 x 1.8 x 1.1 cm tumor mass was surgically resected, and the operative findings and histopathology confirmed right carotid body PGL. Immunohistochemical studies also showed positivity for chromogranin A and synaptophysin. Six years after surgery, based on follow-up imaging with whole-body 18F-FDOPA (Figure 1D) and 68Ga-DOTATATE PET/CT (Figure 1E), as well as whole-body CT and neck MRI, there was no evidence of any recurrent or new PGL.
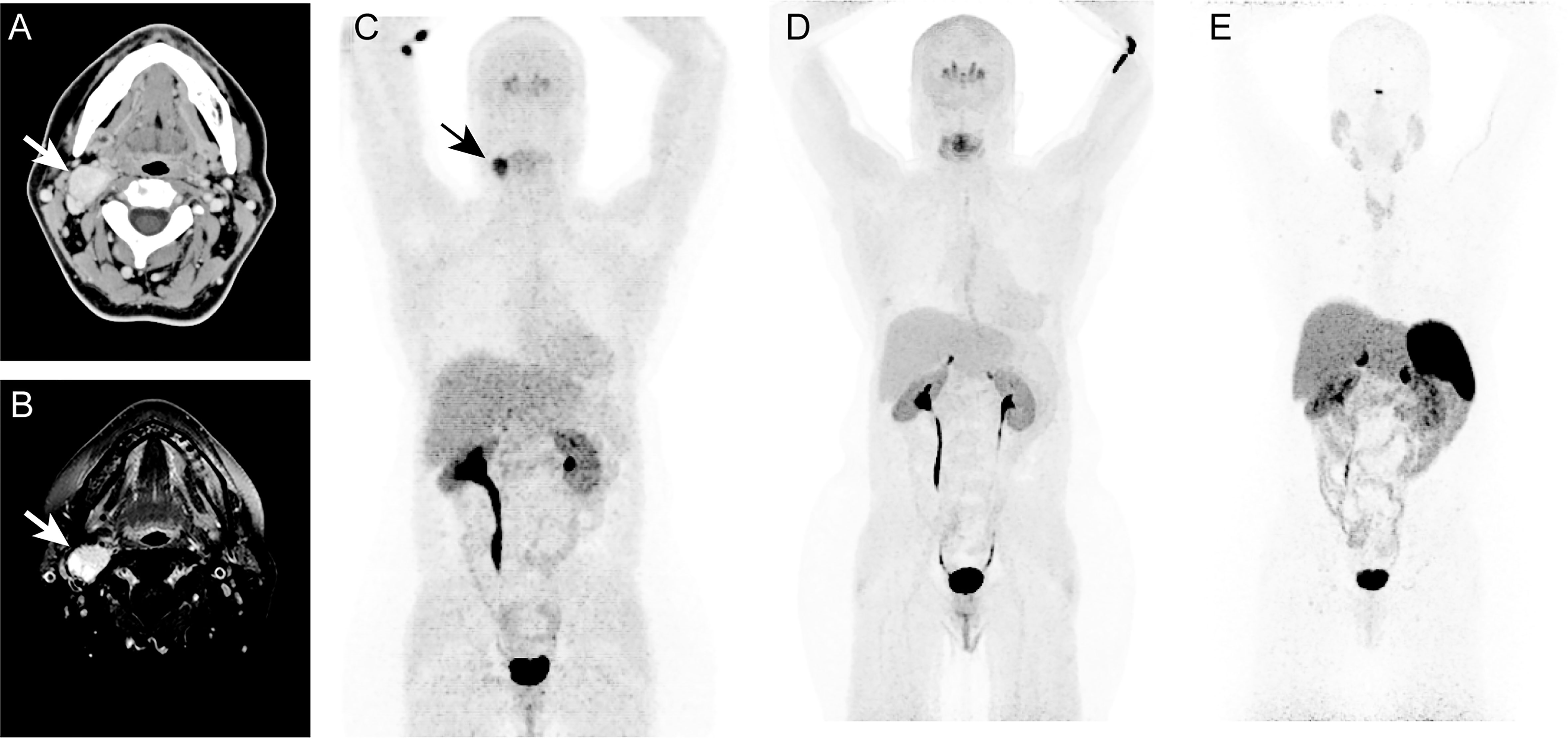
Figure 1 In this figure of a 47-year-old-woman, axial images (A, B) of contrast enhanced computed tomography (CT, A) and T1 weighted, fat suppressed magnetic resonance imaging (B) demonstrates an intensely enhancing 2.7 x 2.0 cm mass (arrows; A, B) at the bifurcation of right common carotid artery causing splaying of right internal and external carotid arteries. The anterior maximum intensity projection (MIP, C–E) of 18F-fluorodopa (18F-FDOPA) positron emission tomography (PET) demonstrates an uptake in the carotid region (arrow, C). This mass was identified as a right carotid body paraganglioma. Six years later, follow-up functional PET/CT imaging with 18F-FDOPA (D) and 68Ga-DOTATATE (E) shows no evidence of pheochromocytoma/paragangliomas (PHEOs/PGLs). Similarly, whole-body CT (not shown here) imaging and neck MRI (not shown here) were negative for PHEOs/PGLs.
This patient was enrolled under the protocol Diagnosis, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma (ClinicalTrials.gov Identifier: NCT00004847), which was approved by the Eunice Kennedy Shriver National Institutes of Child Health and Human Development and Institutional Review Board. Written informed consent was obtained from the patient.
Histopathological examination confirmed the typical PGL morphology (Figure 2A). The tumor tissue from the patient exhibited strong immunopositivity for the IDH2 mutant enzyme (Figure 2B). The IDH2 mutant immunopositivity was not seen in PGLs with other genetic backgrounds, such as NF2, SDHx or VHL. Further, we examined the expression of IDH2 mutant through immunoblotting. The IDH2 mutant was expressed in the protein lysate from the patient, but not in PGLs with other genetic backgrounds as described above (Figure 2C). The specificity of the IDH2 mutant antibody was confirmed by testing the ectopic expression of the IDH2 R172G variant in hPheo1 cell (Figure 2C).
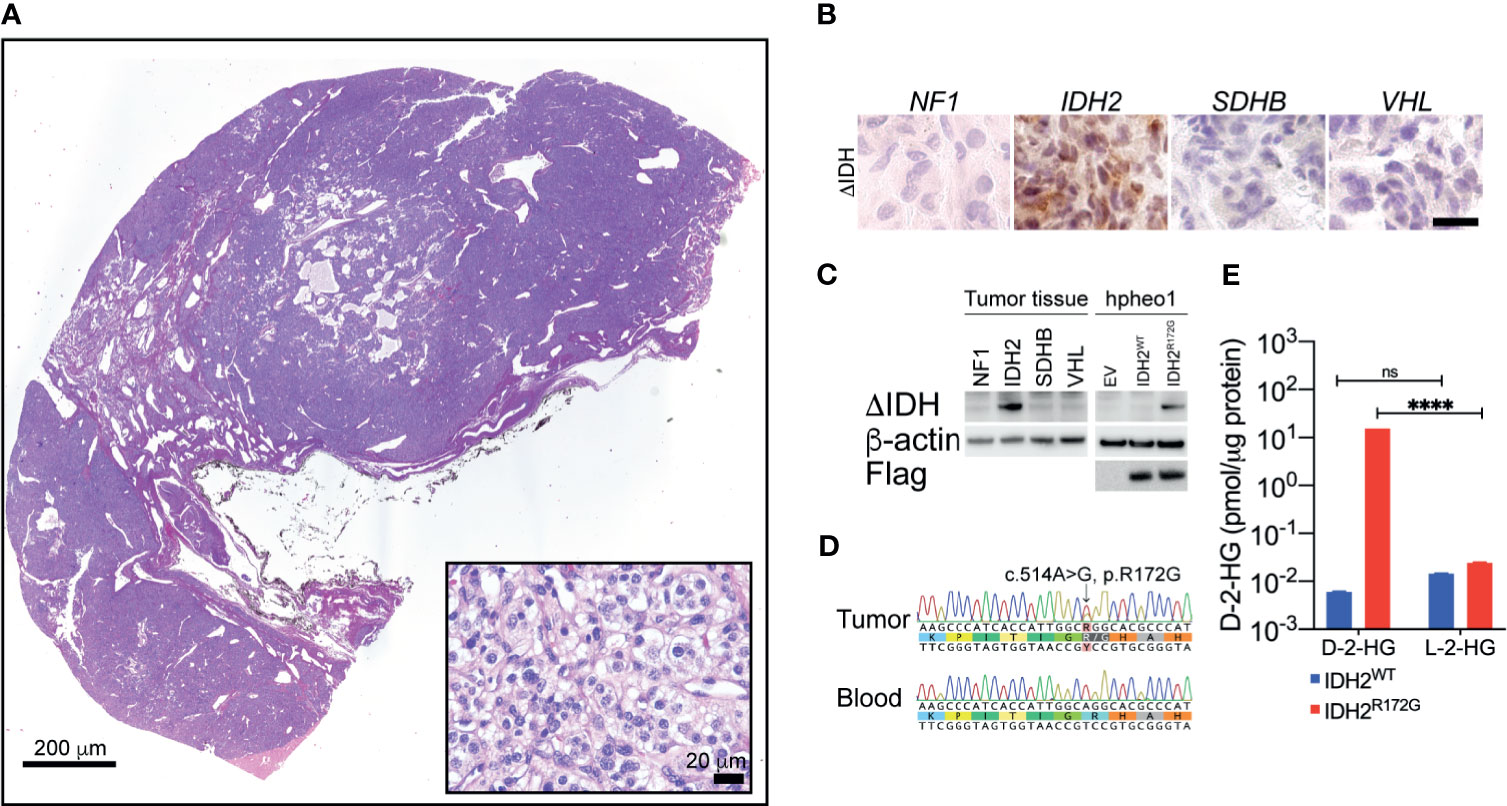
Figure 2 Hematoxylin and eosin staining revealed typical morphology of neuroendocrine tumors (A). Immunohistochemistry showed strong IDH2 mutant expression in the tissue specimen from the index patient (B). Immunoblotting showed the expression of the IDH2 mutant enzyme in cancer tissue. Ectopic expression of IDH2 was used as a positive control (C). Sanger sequencing showed a somatic mutation of the IDH2 gene in cancer tissue. The variant is not seen in blood DNA (D). Mass spec analysis revealed the presence of high-level D-2-HG in cancer tissue. The level of L-2-HG was found not significantly changed. A non-IDH-mutated tumor specimen was used as a control (E). ****p < 0.0001; ns, no significance.
Whole exome sequencing was performed based on DNA specimens from carotid body PGL and patient blood. A somatic change in IDH2 was identified at the location of arginine 172 residue. A follow-up Sanger sequencing confirmed the presence of IDH2 c.514A>G variant, which leads to R172G amino acid substitution (Figure 2D).
To confirm the catalytic function of the IDH2 mutant in cancer tissue, we performed the mass spec measurement on D-2-HG in frozen cancer tissue. We identified 15.39 pmol/µg protein, which was significantly higher than in non-IDH-mutant cancer tissue (0.0059 pmol/µg protein, Figure 2E, ****p<0.0001). The level of L-2-HG was found not significantly changed between the tested samples (Figure 2E, IDH2WT: 0.01436 pmol/µg protein, IDH2R172G: 0.2407 pmol/µg protein).
In the present study, we described the presence of a somatic IDH2 mutation in a carotid body PGL (Figure 1). Sequencing analysis revealed a heterozygous IDH1 R172G mutation in tumor tissue, whereas this mutation was absent in germline DNA (Figure 2D). Further, we confirmed the presence of an IDH2 mutant enzyme through immunohistochemistry and immunoblotting (Figures 2B, C). The IDH2 mutation exhibited neomorphic activity, as mass spec analysis revealed substantially elevated D-2-HG levels in tumor tissue (Figure 2E).
Mutations of IDH are highly prevalent genetic abnormalities in human cancers (22). Cancer-associated IDH mutations occur in the catalytic center, resulting in changes in the catalytic function, production of D-2-HG, epigenetic reprogramming, and tumorigenesis (23, 24). The identification of an IDH2 mutation in a carotid body PGL not only broadens the understanding of these mutations in human cancers (particularly PGL), but it also highlights the role of metabolic deficiency in tumorigenesis of these tumors, implying possible therapeutic regimen by targeting the distinctive metabolic signature. Several small molecule inhibitors against IDH mutant enzymes are currently under investigation through clinical studies, which may be helpful for patients with IDH-mutated PGLs.
Due to the critical role of IDH mutation in several types human malignancies, pharmacological grade inhibitors have been developed to suppress the neomorphic activity of IDH mutant enzymes (25, 26). Several IDH mutant inhibitors are currently examined through clinical studies, such as AG-120 (e.g., NCT02073994 and NCT02074839), AG-221 (e.g., NCT02273739 and NCT03744390), AG-881 (e.g., NCT02481154 and NCT04164901), and BAY1436032 (e.g., NCT02746081 and NCT04603001). The discovery of an IDH mutation in a neuroendocrine tumor suggests the possible application of these inhibitors as potential therapeutic regimens for this type of malignancy. Follow up studies are highly encouraged to explore the prevalence of IDH mutations in neuroendocrine tumors. In-depth cancer biology studies will also be helpful to reveal the role of IDH mutations in the pathogenesis of neuroendocrine tumors.
The datasets presented in this article are not readily available because they contain patient information and confidential personally identifiable information (PII). Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Eunice Kennedy Shriver National Institutes of Child Health and Human Development and Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
CY and KP designed the study. FL and AJ performed the experiment and analysis. FL and CY wrote the manuscript. AJ, LM, and KP revised the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the Intramural Research Program of the National Cancer Institute (NCI).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We appreciate Drs. King Chan and Thorkell Andresson for their help in mass spec quantification on metabolites.
1. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell (2017) 31:181–93. doi: 10.1016/j.ccell.2017.01.001
2. Galan SR, Kann PH. Genetics and Molecular Pathogenesis of Pheochromocytoma and Paraganglioma. Clin Endocrinol (Oxf) (2013) 78:165–75. doi: 10.1111/cen.12071
3. Pillai S, Gopalan V, Smith RA, Lam AK. Updates on the Genetics and the Clinical Impacts on Phaeochromocytoma and Paraganglioma in the New Era. Crit Rev Oncol Hematol (2016) 100:190–208. doi: 10.1016/j.critrevonc.2016.01.022
4. Yang C, Sun MG, Matro J, Huynh TT, Rahimpour S, Prchal JT, et al. Novel HIF2A Mutations Disrupt Oxygen Sensing, Leading to Polycythemia, Paragangliomas, and Somatostatinomas. Blood (2013) 121:2563–6. doi: 10.1182/blood-2012-10-460972
5. Crona J, Taieb D, Pacak K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr Rev (2017) 38:489–515. doi: 10.1210/er.2017-00062
6. Huh TL, Kim YO, Oh IU, Song BJ, Inazawa J. Assignment of the Human Mitochondrial NAD+ -Specific Isocitrate Dehydrogenase Alpha Subunit (IDH3A) Gene to 15q25.1–>Q25.2by in Situ Hybridization. Genomics (1996) 32:295–6. doi: 10.1006/geno.1996.0120
7. Ugur B, Bao H, Stawarski M, Duraine LR, Zuo Z, Lin YQ, et al. The Krebs Cycle Enzyme Isocitrate Dehydrogenase 3a Couples Mitochondrial Metabolism to Synaptic Transmission. Cell Rep (2017) 21:3794–806. doi: 10.1016/j.celrep.2017.12.005
8. Cairns RA, Mak TW. Oncogenic Isocitrate Dehydrogenase Mutations: Mechanisms, Models, and Clinical Opportunities. Cancer Discovery (2013) 3:730–41. doi: 10.1158/2159-8290.CD-13-0083
9. Kipp BR, Voss JS, Kerr SE, Barr Fritcher EG, Graham RP, Zhang L, et al. Isocitrate Dehydrogenase 1 and 2 Mutations in Cholangiocarcinoma. Hum Pathol (2012) 43:1552–8. doi: 10.1016/j.humpath.2011.12.007
10. Waitkus MS, Diplas BH, Yan H. Isocitrate Dehydrogenase Mutations in Gliomas. Neuro Oncol (2016) 18:16–26. doi: 10.1093/neuonc/nov136
11. Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate Dehydrogenase Mutations in Myeloid Malignancies. Leukemia (2017) 31:272–81. doi: 10.1038/leu.2016.275
12. Han S, Liu Y, Cai SJ, Qian M, Ding J, Larion M, et al. IDH Mutation in Glioma: Molecular Mechanisms and Potential Therapeutic Targets. Br J Cancer (2020) 122:1580–9. doi: 10.1038/s41416-020-0814-x
13. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature (2009) 462:739–44. doi: 10.1038/nature08617
14. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of Alpha-Ketoglutarate-Dependent Dioxygenases. Cancer Cell (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
15. Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator Dysfunction and Oncogene Activation in IDH Mutant Gliomas. Nature (2016) 529:110–4. doi: 10.1038/nature16490
16. Parker SJ, Metallo CM. Metabolic Consequences of Oncogenic IDH Mutations. Pharmacol Ther (2015) 152:54–62. doi: 10.1016/j.pharmthera.2015.05.003
17. Zhang J, Jiang J, Luo Y, Li X, Lu Z, Liu Y, et al. Molecular Evaluation of a Sporadic Paraganglioma With Concurrent IDH1 and ATRX Mutations. Endocrine (2018) 61:216–23. doi: 10.1007/s12020-018-1617-1
18. Richter S, Gieldon L, Pang Y, Peitzsch M, Huynh T, Leton R, et al. Metabolome-Guided Genomics to Identify Pathogenic Variants in Isocitrate Dehydrogenase, Fumarate Hydratase, and Succinate Dehydrogenase Genes in Pheochromocytoma and Paraganglioma. Genet Med (2019) 21:705–17. doi: 10.1038/s41436-018-0106-5
19. Yang C, Hong CS, Prchal JT, Balint MT, Pacak K, Zhuang Z. Somatic Mosaicism of EPAS1 Mutations in the Syndrome of Paraganglioma and Somatostatinoma Associated With Polycythemia. Hum Genome Var (2015) 2:15053. doi: 10.1038/hgv.2015.53
20. Yang C, Wang H, Zhu D, Hong CS, Dmitriev P, Zhang C, et al. Mutant Glucocerebrosidase in Gaucher Disease Recruits Hsp27 to the Hsp90 Chaperone Complex for Proteasomal Degradation. Proc Natl Acad Sci USA (2015) 112:1137–42. doi: 10.1073/pnas.1424288112
21. Cheng QY, Xiong J, Huang W, Ma Q, Ci W, Feng YQ, et al. Sensitive Determination of Onco-Metabolites of D- and L-2-Hydroxyglutarate Enantiomers by Chiral Derivatization Combined With Liquid Chromatography/Mass Spectrometry Analysis. Sci Rep (2015) 5:15217. doi: 10.1038/srep15217
22. Liu Y, Lang F, Chou FJ, Zaghloul KA, Yang C. Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications. Biomedicines (2020) 8:294. doi: 10.3390/biomedicines8090294
23. Clark O, Yen K, Mellinghoff IK. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clin Cancer Res (2016) 22:1837–42. doi: 10.1158/1078-0432.CCR-13-1333
24. Dang L, Su SM. Isocitrate Dehydrogenase Mutation and (R)-2-Hydroxyglutarate: From Basic Discovery to Therapeutics Development. Annu Rev Biochem (2017) 86:305–31. doi: 10.1146/annurev-biochem-061516-044732
25. Dang L, Yen K, Attar EC. IDH Mutations in Cancer and Progress Toward Development of Targeted Therapeutics. Ann Oncol (2016) 27:599–608. doi: 10.1093/annonc/mdw013
Keywords: paraganglioma, carotid body tumor, PET/CT, isocitrate dehydrogenase 2, IDH2, D-2-hydroxyglutarate
Citation: Lang F, Jha A, Meuter L, Pacak K and Yang C (2021) Identification of Isocitrate Dehydrogenase 2 (IDH2) Mutation in Carotid Body Paraganglioma. Front. Endocrinol. 12:731096. doi: 10.3389/fendo.2021.731096
Received: 26 June 2021; Accepted: 30 July 2021;
Published: 20 September 2021.
Edited by:
Wen Zhou, Case Western Reserve University, United StatesCopyright © 2021 Lang, Jha, Meuter, Pacak and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunzhang Yang, eWFuZ2MyQG5paC5nb3Y=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.