
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 29 September 2021
Sec. Gut Endocrinology
Volume 12 - 2021 | https://doi.org/10.3389/fendo.2021.726368
This article is part of the Research TopicProglucagon-Derived PeptidesView all 25 articles
 Farzad Asadi1,2
Farzad Asadi1,2 Savita Dhanvantari1,2,3,4*
Savita Dhanvantari1,2,3,4*Patients with diabetes mellitus exhibit hyperglucagonemia, or excess glucagon secretion, which may be the underlying cause of the hyperglycemia of diabetes. Defective alpha cell secretory responses to glucose and paracrine effectors in both Type 1 and Type 2 diabetes may drive the development of hyperglucagonemia. Therefore, uncovering the mechanisms that regulate glucagon secretion from the pancreatic alpha cell is critical for developing improved treatments for diabetes. In this review, we focus on aspects of alpha cell biology for possible mechanisms for alpha cell dysfunction in diabetes: proglucagon processing, intrinsic and paracrine control of glucagon secretion, secretory granule dynamics, and alterations in intracellular trafficking. We explore possible clues gleaned from these studies in how inhibition of glucagon secretion can be targeted as a treatment for diabetes mellitus.
Glucagon is a 29-amino acid peptide hormone produced by the alpha (α) cells of the pancreatic islet. It is known as the primary glucose counter-regulatory hormone, as its main physiological function is to maintain euglycemia by its actions on the liver to promote glycogenolysis and gluconeogenesis. This glucose counterregulation is impaired in both Type 1 (T1D) and Type 2 (T2D) diabetes (1). Intensive insulin therapy in T1D replaces the beta cell deficiency and corrects fasting and postprandial hyperglycemia, but at increased risks of hypoglycemia (2). These findings suggest that insulin therapy alone may not be adequate for optimal glycemic control. In fact, as stated by the bihormonal hypothesis, diabetes progression may be dependent on both excessive glucagon secretion and insulin deficiency (3), and the glucagonocentric hypothesis ascribes even more importance to alpha cell dysfunction in diabetic hyperglycemia (4). Support for the latter hypothesis seemed to be provided by studies showing that glucagon receptor knockout mice are resistant to the effects of beta cell deficiency induced by streptozotocin treatment (5, 6), providing evidence that blocking glucagon action can provide glycemic control even in the relative absence of insulin. However, more recent studies have shown that blocking the glucagon receptor in absolute insulin deficiency does not prevent hyperglycemia (7, 8), indicating that residual insulin signalling is required in order for glucagon receptor antagonism to be effective.
Nonetheless, the idea that blocking glucagon action could be an additional therapy for diabetes has sparked interest in the development of potential pharmacological interventions that target the hepatic glucagon receptor. Blocking glucagon action can be achieved through: i) glucagon receptor antagonists, in particular small molecule antagonists, which can allosterically or competitively inhibit glucagon action (9–11); ii) glucagon receptor neutralizing antibodies (12); and iii) antisense oligonucleotides against the glucagon receptor (13). However, blockade of the glucagon receptor may induce α-cell hyperplasia and exacerbate hyperaminoacidemia (14–16) through impairments in a liver-alpha cell axis [reviewed in (17)], and increased risk of hyperlipidemia (18). Therefore, long-term use of glucagon receptor blockers may result in harmful metabolic sequelae.
An alternate and possibly safer strategy may be to directly target the intracellular mechanisms that govern the secretion of glucagon from the alpha cell. In this review, we discuss aspects of alpha cell biology that may provide such targets: proglucagon processing, sorting, exocytosis and intracellular trafficking, as well as mechanisms of intrinsic and intra-islet regulation of glucagon secretion.
The alpha cell secretory pathway begins with the synthesis of proglucagon in the endoplasmic reticulum. It is then transported through the Golgi to the trans-Golgi network (TGN). Budding immature secretory granules from the TGN contain proglucagon, its processing enzymes and many other proteins (19). But how does proglucagon find its way to the site of granule budding? Based on one hypothesis, proglucagon may contain a sorting signal within its structure that directs it to a specific sorting receptor on the membrane of the TGN.
Investigations into the mechanisms of the sorting of prohormones have revealed the existence of intrinsic sorting signals that can take the form of amphipathic loops, such as for POMC (20) or proinsulin (21), or amphipathic alpha helices, as is the case for the N-terminal region of prosomatostatin, and in the C-terminal regions of PC1/3, PC2, PC5/6a, and CPE (22–24). These signals may interact directly with membrane lipids, in particular with lipid raft regions (25) or with sorting receptors within the TGN, to be sorted into secretory granules. To this end, it has been proposed that membrane-bound form of the processing enzyme carboxypeptidase E (CPE) may be a prohormone sorting receptor (21, 26–28). It was shown that ablation of CPE disrupted the regulated secretion of proopiomelanocortin (POMC), proenkephalin and proinsulin in related cell lines and the CPEfat mouse model, in which CPE is degraded within the pituitary. Additionally, CPE may interact other resident granule proteins, secretogranin III and chromogranin A, to facilitate the sorting of POMC (29) and neuropeptides (30).
Another possible mechanism of prohormone sorting to granules may be simply through selective retention while constitutively secreted proteins are removed from the nascent immature granule. Evidence for this mechanism lies in the fact that the protein composition of immature secretory granules is altered during the process of granule maturation (31). In this context, proinsulin and the enzymes involved in the post-translational processing to mature insulin are retained within the beta cell secretory granule, while other proteins designated for constitutive secretion are removed (31).
By considering all of these findings, it is likely that both receptor-mediated and retention mechanisms operate in the sorting of prohormones into secretory granules. In this scenario, prohormones could be sorted into secretory granules by means of sorting signals, followed by retention within the granule as maturation of secretory granules takes place. The maturation process involves alterations in the components and composition of the secretory granule, by removal of constitutively-secreted proteins, acidification of the granule milieu, and exclusion of water to condense the intragranular environment.
The cellular events underlying the sorting of proglucagon to secretory granules have not been fully elucidated, and studying this mechanism is complicated by the multi-step processing of proglucagon. The processing of proglucagon in the alpha cell is largely governed by the prohormone convertase (PC) family of enzymes. Proglucagon processing begins early in the secretory pathway (TGN or immature secretory granule) with cleavage at K70R71, which yields glicentin and major proglucagon fragment (MPGF) (Figure 1A) (32). This site is accessible to a number of processing enzymes, and in the alpha cell, is likely cleaved by furin or PC1/3 in the absence of PC2 (33). Subsequent cleavage of glicentin by PC2 at K31R32 results in the production of mature glucagon. This cleavage event likely occurs within the mature secretory granule since the enzymatic activity of PC2 is optimal at the acidic pH and millimolar calcium concentrations within secretory granules (33–36). Thus, the sorting of proglucagon into the secretory granule is vital for the generation of active glucagon, and storage within granules assures a robust secretory response in response to physiological need.
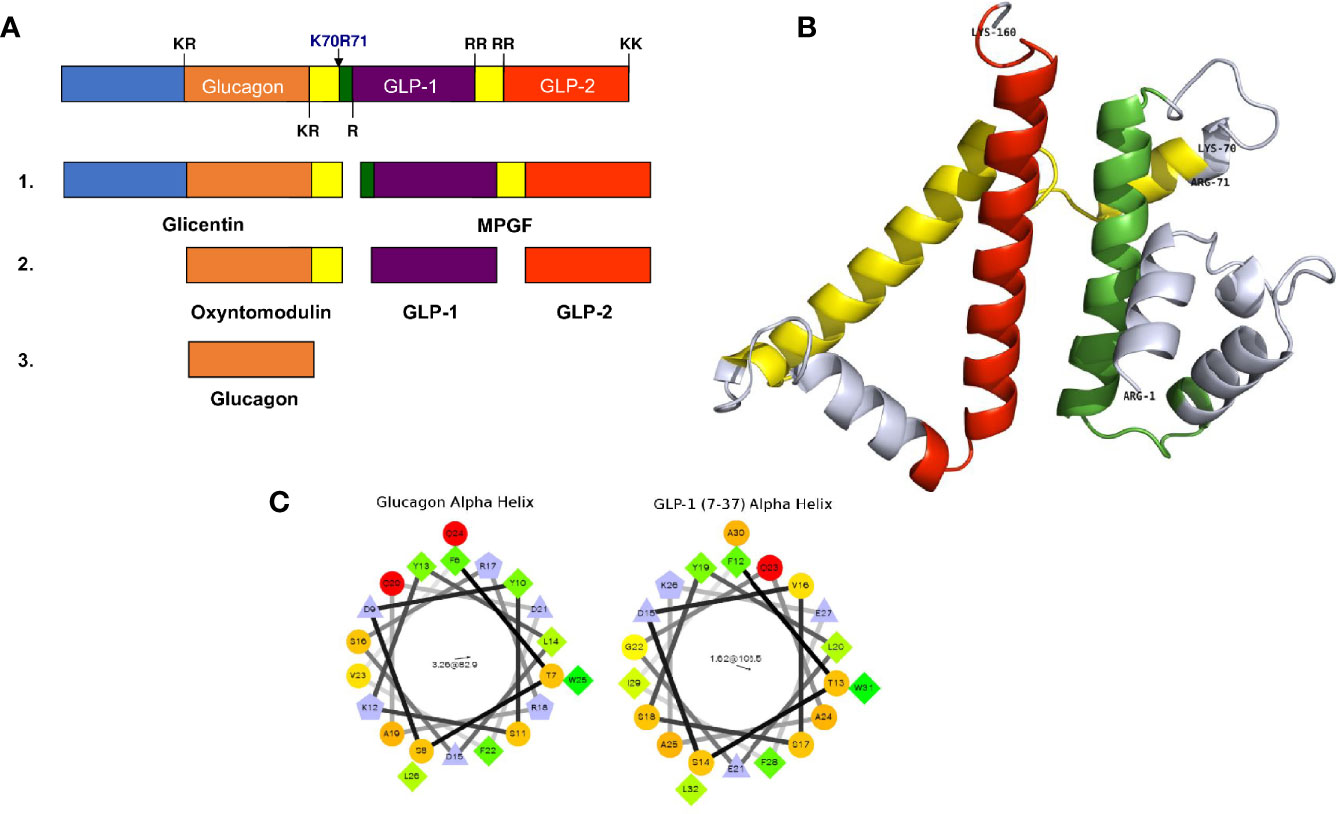
Figure 1 Proglucagon processing and sorting signals. (A) A schematic representation of proglucagon showing the major prohormone processing sites that yield the peptides glicentin, oxyntomodulin, glucagon, GLP-1 and GLP-2. (B) Computational modelling of the structure of proglucagon showing the alpha helical structures of glucagon (green), GLP-1 (yellow) and GLP-2 (red) (from reference 28) © 2013 Society for Endocrinology. (C) Helical wheel projections of the alpha helices contained within glucagon and GLP-1 (7–37) that function as sorting signals to direct proglucagon to the regulated secretory pathway.
Is there evidence for sorting signals and a sorting receptor for proglucagon? Using the alpha cell line αTC1-6, it was shown that siRNA-mediated knockdown of CPE increased constitutive secretion of glucagon; however, the processing of proglucagon to glucagon remained unchanged, indicating that CPE may have an effect on secretion, but not intracellular sorting (28). The search for sorting signals provided more clarity on the mechanisms of proglucagon sorting. Computational modelling of proglucagon indicates a largely disordered structure comprising alpha helices, some of which correspond to glucagon, GLP-1 and GLP-2 (Figure 1B). Using Fc-tagged proglucagon-derived peptides that could be detected by immunoprecipitation and immunofluorescence microscopy, it was shown that two dipolar α-helices containing hydrophobic patches with three charged residues within the sequences play roles as sorting signals. One amphipathic α-helix was located within the amino acid sequence of glucagon (SDYSKYLDSRRAQDFVQWLMN), and one within GLP-1 (SDVSSYLEGQAAKEFLAWLVK) (32) (Figure 1C). Interestingly, Fc-glicentin was sorted to secretory granules, but Fc-MPGF was not, suggesting that the sorting signal within GLP-1 is masked when contained within the MPGF sequence. These results indicate that the sorting of proglucagon into secretory granules occurs prior to the initial processing event, such that processing occurs exclusively within the granule. Another possibility is that processing to glicentin and MPGF occurs first, with the prediction that glicentin is sorted into granules and processed to glucagon, while MPGF is not sorted, or very inefficiently sorted into granules. Given the emerging evidence of significant amounts of GLP-1 being produced in alpha cells in healthy human islets (37), this model would predict that proglucagon is first sorted into granules, then processed by both PC2 and PC1/3 to yield glucagon and GLP-1, respectively. This proglucagon processing profile changes in diabetes; in human and rodent islets, there is a significant increase in the processing of proglucagon to GLP-1. This alteration in the processing profile was first shown in rats treated with STZ; both PC1/3 and PC2 mRNA levels increased, concomitant with increases in glucagon and GLP-1 secretion (38). These results have since been corroborated in several studies showing significant elevations in PC1/3 expression and activity and GLP-1 production and secretion in αTC1-6 cells (39) and InR1G9 cells (40) grown in media containing high glucose concentrations, well as in islets from rodent models of diabetes. A recent study showed that up to 40% of alpha cells in human islets obtained from healthy donors contain both GLP-1 and glucagon and this proportion increases to 60% in islets from donors with Type 2 diabetes (37). The expression of PC1/3 and the production of GLP-1 and glucagon are increased in response to the diabetes-related pro-inflammatory cytokine, IL-6, in isolated human and mouse islets (41), and in the db/db (42), and ob/ob (43) mouse models of obesity-associated diabetes. Alpha TC1 cells engineered to be depleted in PC2 overexpress PC1/3, and subsequently produce sufficient amounts of GLP-1 to overcome STZ-induced hyperglycemia in mice (44, 45). In contrast to these findings, islets from the NOD mouse model of type 1 diabetes mouse showed a small but significant increase in GLP-1 and PC1/3 in alpha cells (42), but no change in the amount of secreted intra-islet GLP-1.
If both glucagon and GLP-1 are produced in a proportion of alpha cells, and are both sorted to secretory granules, the question arises: are they sorted to distinct granule populations, and released under different glucose conditions? These questions may have been answered in a very recent islet granule peptidomics study showing that both human and mouse islets produce 300-1000 times more glucagon than active GLP-1 (46), indicating that the processing of proglucagon to active GLP-1 in alpha cells is very inefficient. Also in this study, analysis of secreted proglucagon-derived peptides showed that both glucagon and active GLP-1 were released in parallel in response to either low (1 mM), medium (6 mM) or high (16.7mM) glucose concentrations. Therefore, both glucagon and GLP-1 are likely stored in the same granules and secreted under the same conditions, with glucagon being the dominant peptide, and perhaps serving as the intra-islet GLP-1R agonist (47).
The control of hyperglucagonemia obviously targets glucagon secretion. But what mechanism(s) are potentially druggable? Inhibition of glucagon secretion by glucose from alpha cells is a long-standing puzzle in islet biology. Unlike insulin secretion from beta cells which is primarily driven by prevailing glucose levels, there is no one single factor that governs glucagon secretion from the alpha cell. Intrinsic glucose sensing, intra-islet paracrine secretion and factors from the alpha cell itself all interact to generate a complex network that regulates glucagon secretion.
In order to examine the direct effects of glucose on glucagon secretion in the absence of paracrine inputs, isolated mouse pancreatic alpha cells, clonal hamster In-R1-G9 cells (48, 49), clonal mouse αTC1-6 and -9 cells (39, 50, 51) and dispersed alpha cells from human islets (52) have been used. All of these preparations show a bimodal response to increasing glucose concentrations. In the range from 1 to ~7 mM, glucagon secretion is suppressed in a dose-dependent manner, and above 7 mM, glucagon secretion increases (Figure 2A). This secretion profile suggests intrinsic mechanisms alone can operate in regulating glucagon secretion below 7 mM glucose, and that these mechanisms may be ineffective at higher glucose concentrations. However, such conclusions must be interpreted with caution, as single dispersed alpha cells are in a highly abnormal environment, and alpha cell lines are not representative of the normal alpha cell phenotype, as discussed in more detail below.
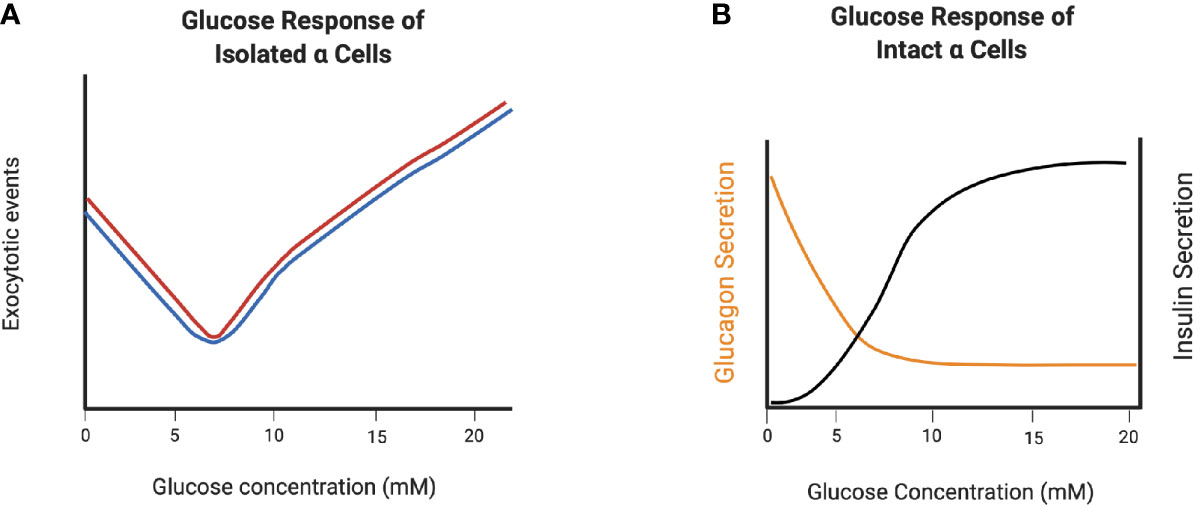
Figure 2 Glucagon secretion from dispersed alpha cells and alpha cells in intact islets demonstrate the role of paracrine regulation at high glucose concentrations. (A) V-shape curve of glucagon exocytosis in response to glucose in dispersed non-diabetic (black) and T2D (red) human α-cells. (B) Glucagon secretion from intact islets in response to glucose. Created with BioRender.com.
The alpha cell secretory response to both glucose is likely more accurately captured in isolated, intact mouse and human islets, where the paracrine regulatory environment and cell-cell contacts are intact. Similar to dispersed alpha cells, increasing the glucose concentration from 1 to 7 mM dose dependently decreases glucagon secretion from mouse alpha cells (53) and human alpha cells (52) within intact islets, and remains low as glucose levels increase beyond 7 mM, a concentration at which insulin secretion is stimulated (Figure 2B). Therefore, paracrine inputs are significant factors in the inhibition of glucagon secretion as glucose concentrations increase above euglycemia.
One mechanism underlying the intrinsic response to glucose is the direct effect on alpha cell electrical activity. At low (1 mM) glucose concentrations, alpha cells in intact mouse and human islets exhibit low KATP activity and are electrically active (54–56) and as glucose concentrations increase, KATP activity is inhibited. This is an apparent physiological paradox, since further closure of KATP channels should more strongly depolarize the plasma membrane and result in opening of voltage-dependent Ca2+ channels (VDCCs), as in beta cells. In alpha cells, glucagon exocytosis is coupled to the opening of P/Q-type VDCCs (55). A recent review by Zhang et al. (57) nicely explains this apparent paradox: membrane depolarization evoked by KATP inhibition reduces the amplitude of action potentials from Na+ channels. It is this reduction in the amplitude of action potentials that result in the closure of the voltage-dependent P/Q-type Ca2+ channels, leading to inhibition of glucagon granule exocytosis. Therefore, the intrinsic regulation of glucagon secretion by glucose may be explained primarily by the unique electrical properties of the alpha cell, and secondarily by glucose metabolism.
It has been also proposed that another mechanism for the direct effect of glucose on glucagon secretion is the regulation of intracellular Ca2+ flux through store-operated current (SOC) (58, 59). Under low glucose conditions, Ca2+ depletion of the ER causes the translocation of stromal interaction molecule 1 (STIM1) from the ER to the subplasmalemmal junctions, leading to clustering with Orai1, resulting in SOC activation. In contrast, exposure of alpha cells to high glucose conditions and subsequent high ATP levels activates the sarcoendoplasmic reticulum calcium transport ATPase (SERCA) pump, increasing Ca2+ sequestration from the cytoplasm into the ER. Filling the store with Ca2+ inhibits the translocation of STIM1, turns off CRAC channel subunit, reduces VDCC activity and suppresses glucagon secretion (59, 60). However, this hypothesis implicated L-type VDCCs in glucagon exocytosis, which have been shown to mediate intracellular Ca2+ oscillations, but not granule exocytosis, in alpha cells (54).
There is evidence that glucagon secretion becomes uncoupled from intracellular Ca2+ levels under high glucose conditions (61), suggesting the presence of alternate or additional intracellular signalling pathways in the regulation of glucagon secretion. In particular, cAMP signalling may play a key role in the alpha cell secretory response to insulin and somatostatin (62). There is one report that cAMP may also mediate intrinsic glucose sensing within the alpha cell. Using genetically encoded fluorescent cAMP biosensors, it was shown that high glucose suppressed subplasmalemmal cAMP levels in isolated mouse and human islets (63). This effect was not abolished in Ca2+-free media, nor in the presence of insulin receptor or SSTR2 antagonists. Conversely, sustained high cAMP levels abolished the suppression of glucagon secretion by high glucose concentrations. There may specific isoforms of adenylyl cyclase and phosphodiesterases that are associated with cAMP-regulated glucagon secretion/hyperglycemia [reviewed in (64)], and exactly how glucose interacts with these components of cAMP signalling remains to be determined.
Lastly, intrinsic glucose sensing by the alpha cell may also be mediated by the nutrient sensors AMP-activated protein kinase (AMPK) and its downstream target, mammalian target of rapamycin complex 1 (mTORC1). In a series of studies that manipulated alpha cell expression of AMPK itself (65) and its upstream effectors PASK (66) and LKB1 (67), it was shown that components of this nutrient-sensing pathway can mediate the low glucose-induced secretion of glucagon. One of these proteins, PASK, is down-regulated in T2D human islets, thus indicating that components of the AMPK pathway may be potential targets for controlling hyperglucagonemia. Using innovative mouse models that selectively targeted activators and inhibitors of mTORC1, it was shown that loss of mTORC1 activity resulted in a loss of the glucose counter-regulatory response and reduction in response to alpha cell secretagogues (68). Interestingly, depletion of the mTORC1 inhibitor TSC2 in alpha cells resulted in a mouse model of hyperglucagonemia and glucagon resistance (69), which will be an excellent resource for studies on mechanisms of hyperglucagonemia.
Therefore, the mechanisms underlying the intrinsic response to glucose may provide potential targets for the control of abnormally up-regulated glucagon secretion in diabetes.
The beta cell secretory granule contains a number of agents that act directly or indirectly on the alpha cell to inhibit glucagon secretion, and also generally modulate mechanisms of alpha cell biology, such as proliferation. Insulin, the primary cargo, is a potent suppressor of glucagon secretion and operates through several mechanisms. Upon binding to its receptor on the alpha cell, signaling through PI3 kinase is activated, reducing KATP-channel activity, causing plasma membrane hyperpolarization and reduced activity of the P/Q type Ca2+ channel (70). Mice lacking the insulin receptor on alpha cells (αIRKO) exhibit hyperglycemia and hyperglucagonemia, indicating that insulin receptor signalling is required for an appropriate alpha cell secretory response to glucose (71). Alpha cell insulin resistance may underlie the abnormal up-regulation of glucagon secretion Type 2 diabetes (72). Additionally, these results also indicate that insulin alone is not sufficient to regulate glycemia in the face of hyperglucagonemia.
Along with insulin, gamma amino butyric acid (GABA) is also released from the beta cell and is a potent suppressor of glucagon secretion from alpha cells (73, 74). Activating the GABAA receptor in alpha cells results in Cl- influx into the cells which hyperpolarizes the membrane and reduces glucagon secretion (75). As well, there is coordination between insulin and GABAA receptor activity, as insulin action leads to the translocation of GABAA receptor to the cell membrane (76), thus augmenting the inhibitory effects of GABA. In addition, GABA also inhibits mTOR activity to suppress alpha cell proliferation. In type 1 diabetes, the destruction of beta cells leads to a reduction in the amount of secreted GABA, resulting in the activation of mTOR and alpha cell proliferation (77). In addition to effects on alpha cell proliferation, some studies have suggested that pharmacologic activation of GABAA receptor by artemisinins or GABA may alter alpha cell identity and trans-differentiate adult alpha cells to beta-like cells (78–80), and have led to clinical trials investigating GABA receptor agonists as protection against the development of diabetes. However, there is still some debate on this topic, as transdifferentiation could not be induced either in isolated mouse islets in which both insulin and glucagon were tagged with fluorescent reporters (81) or in an alpha cell-specific lineage tracing model (82). In any case, the reported immunomodulatory effects of GABA, together with either GLP-1 (83) or the SGLT2 inhibitor empagliflozin (84) also protect newly formed beta cells in the inflammatory environment of T1D, and thus also indirectly restore normal regulation of alpha cell mass and glucagon secretion.
Other beta cell secreted factors that have direct effects on the alpha cell include serotonin, adenosine, and Zn2+. Direct effects of serotonin are mediated by activation of the serotonin receptor, 5-HT1FR, on α-cells, which reduces intracellular cAMP to suppress glucagon secretion (85, 86). In patients with long-standing T2D, the proportion of alpha cells expressing 5-HT1FR is decreased, suggesting that reduced serotonin action on alpha cells may play a role in hyperglucagonemia of diabetes. In STZ-treated mice, administration of the 5-HT1FR agonist LY344864 alleviated hyperglucagonemia and hyperglycemia. However, insulin-induced hypoglycemia was worsened, suggesting that the effects of serotonin are glucose-independent (85). Therefore, while alpha cell HT1FR may be a potential target for the treatment of hyperglucagonemia, it may not be an ideal target.
The effects of adenosine are mediated by the adenosine A1 receptor (Adora1), in which activation is coupled to opening of KATP channels, hyperpolarization of the cell membrane and prevention of granule exocytosis. In NOD mice, autoantibody-positive people and people with long-term T1D, alpha cells gradually lose Adora1 expression, suggesting that the hyperglucagonemia of diabetes is associated with a loss of adenosine action (87).
The Zn2+ transporter, ZnT8, is encoded by Slc30A8, and variants of this gene are associated with an increased risk of T2D and impaired glucose tolerance. ZnT8 is located in the secretory granule membrane of both α-and β-cells. There is a direct relationship between expression of the proglucagon gene and Slc30A8 in α-cells (88). However, there are conflicting findings about the effects of Zn2+ on glucagon secretion (88). There are reports that, in isolated mouse islets and alpha cells, Zn2+ administration decreases glucagon secretion (89), or has no effect (90). In contrast, treatment of isolated human islets with similar concentrations of Zn2+ enhanced glucagon secretion (55). The reason for these discordant results is not clear; it may be that exogenously administered Zn2+ or Zn2+ secreted from the beta cell does not have a direct role in glucagon secretion in normal physiology. Interestingly, in mice lacking Slc30A8/ZnT8 specifically in alpha cells, there is a heightened secretory response to 1 mM glucose (91), and overexpression of ZnT8 specifically in alpha cells restricted glucagon secretion in response to 1 mM glucose (92), suggesting autocrine regulation by Zn2+.
Somatostatin is a well-known tonic inhibitor of glucagon secretion. Somatostatin binds to the SSTR2 receptor subtype on alpha cells (93), which is coupled to the inhibitory Gi subunit, resulting in decreased production of cAMP as a mechanism for the suppression of glucagon secretion (62). Another mechanism may be more closely coupled to Ca2+ flux and inhibition of glucagon granule exocytosis by de-priming of the alpha cell secretory granules located near L-type Ca2+ channels (94). Notably, secretion of somatostatin and inhibition of glucagon secretion both occur at 3 mM glucose, indicating that the alpha cell response to low glucose may be fine-tuned by somatostatin (95). In rat pancreatic preparations perfused with an SSTR2 antagonist, the suppression of glucagon secretion by 3.5-5 mM glucose was lost (96), suggesting that somatostatin may be required for the fine control of glucagon secretion at euglycemia. However, in isolated human islets, blockade of SSTR2 did not affect suppression of glucagon secretion at 6 mM glucose (55), perhaps reflecting species-specific differences or differences in the models (perfused pancreas vs static islet culture).
There is also evidence that somatostatin is required for the high glucose-induced suppression of glucagon secretion: in perifused islets from mice lacking the prosomatostatin gene (SST-/- mice) (97), glucagon secretion was elevated at 10 and 20 mM glucose. Interestingly, insulin secretion was also elevated, indicating that both insulin and somatostatin are required for the suppression of glucagon secretion at high glucose concentrations. In intact human islets, high glucose (10 mM) inhibition of glucagon exocytosis was lost after administration of the SSTR2 antagonist CYN154806 (52). In diabetes, circulating and pancreatic somatostatin, together with SST mRNA, are elevated. However, expression of SSTR2 on alpha cells is decreased in T2D due to increased receptor internalization (52), indicating alpha cell somatostatin resistance. Together with alpha cell insulin resistance, this could be another mechanism in the hyperglucagonemia of diabetes. Alternatively, somatostatin resistance may be a dominant and direct mechanism of hyperglucagonemia, as eliminating the insulin receptor on delta cells completely abolishes the glucagonostatic effect of insulin, indicating an indirect glucagonostatic effect for insulin (98).
The emerging role of somatostatin in the regulation of alpha cell function and glucagon secretion has been further highlighted by one study in which mice were engineered for optogenetic activation of beta cells to study the paracrine regulation of alpha cells (99). By this approach, opto-activation of beta cells both suppressed alpha cell electrical activity and stimulated action potentials in delta cells mediated by gap junction currents. The suppressive effect of beta cell activation was lost in the presence of the SSTR2 antagonist CYN 154806 (99), indicating that somatostatin secretion stimulated by beta cell electrical activity is critical for the suppression of glucagon secretion. Subsequent modelling predicted that a reduction in gap junction connections between beta and delta cells, perhaps caused by disruptions in islet architecture in T2D (100), may contribute to the hyperglucagonemia of diabetes. Thus these findings highlight a central role for delta cells in the context of intra-islet regulation of glucagon secretion, and may have implications for designing drugs for the treatment of hyperglucagonemia of diabetes.
The alpha cell itself displays plasticity during the progression of diabetes. In addition to the mechanisms above that describe changes in responses to glucose and paracrine effectors, there are alterations within the alpha cell, including proglucagon processing and secretion of proglucagon-derived peptides, and remodelling of the secretory granules themselves in terms of exocytotic behavior and contents, and alterations in intracellular trafficking pathways.
Secreted glucagon from alpha cells can stimulate its secretion through an autocrine effect. It has been shown that glucagon stimulates glucagon secretion from the rat and mouse isolated alpha cells in an autocrine manner through glucagon receptor-stimulated cAMP signaling (101). In αTC1-9 cells and mouse islets, exogenous glucagon administration, as well as secreted glucagon stimulated by 1 mM glucose, increased glucagon secretion and proglucagon gene transcription through the PKA-cAMP-CREB signalling pathway in a glucagon receptor-dependent manner (102). The apparent interplay between glucagon and its receptor on the alpha cell appears to be of a positive feedback loop, controlled by the pulsatile nature of glucagon secretion.
In addition to glucagon, a novel proglucagon-derived peptide, proglucagon 1-61 (PG 1-61) comprised of GRPP and glucagon, was identified as a major molecular form of glucagon in plasma from human patients with hyperglucagonemia-associated conditions: Type 2 diabetes and renal dysfunction, morbid obesity or gastric bypass surgery, and only after oral ingestion of macronutrients (103). This N-terminally extended form of immunoreactive glucagon was not found in healthy controls, leading the authors to speculate that PG 1-61, and molecular heterogeneity of glucagon in general, could be a biomarker for alpha cell dysfunction. Administration of PG 1-61 decreased glucagon secretion in healthy rats, diverging from the positive feedback observed with glucagon administration. Interestingly, this effect was not observed in diabetic rats, suggesting an impairment in this distinct feedback loop in the alpha cell.
The interplay between glucagon, insulin and somatostatin in the regulation of glucagon secretion at various levels of glucose is illustrated in Figure 3. In diabetes, beta cell deficiency, together with alpha cell insulin and somatostatin resistance, all contribute to alpha cell dysfunction and a loss of the regulation of glucagon secretion, resulting in hyperglucagonemia.
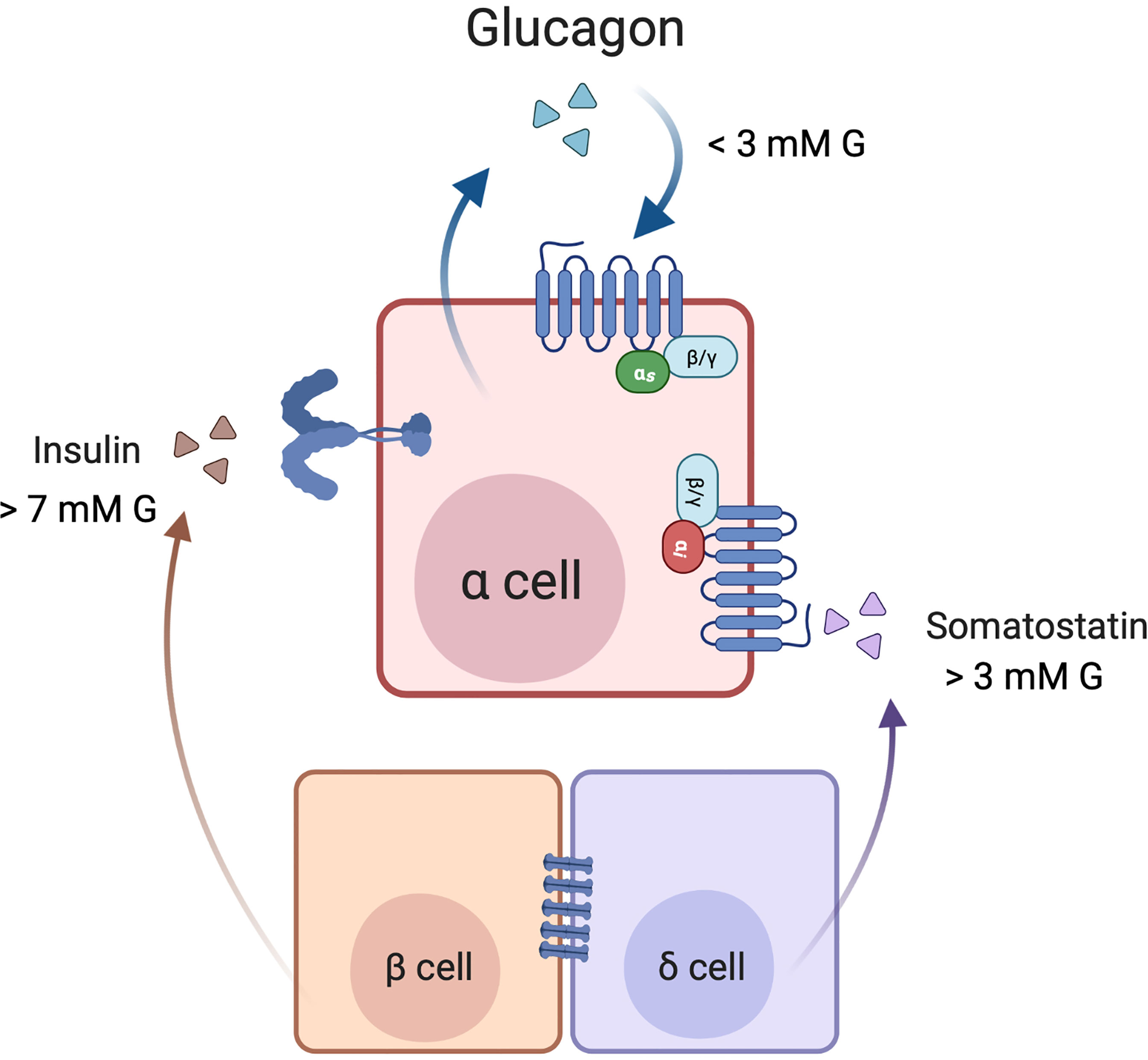
Figure 3 Cross-talk among α, β, and δ-cells in the paracrine regulation of glucagon secretion. Under low glucose (1-3 mM) conditions, secreted glucagon may act in an autocrine feed-forward loop. In high glucose (> 7 mM) conditions, glucose metabolism in β cells leads to insulin release, and insulin inhibits glucagon secretion by signalling through its receptor on the alpha cell. Additionally, electrical coupling of the beta and delta cells through gap junctions contributes to somatostatin release. Somatostatin may also be secreted independently of insulin when glucose concentrations are > 3 mM. Somatostatin binds to SST receptor 2 (SSTR2) on the α cell membrane, where signalling through Gi inhibits glucagon secretion. Created with BioRender.com.
The glucose-dependent insulinotropic actions of intestinal GLP-1 on the beta cell are well known. GLP-1 also suppresses glucagon secretion in both healthy people and people with type 2 diabetes (104), and poorly-controlled type 1 diabetes (105). The emerging evidence of GLP-1 being produced and secreted by the pancreatic alpha cell has led to a debate on which source of GLP-1 suppresses glucagon secretion from pancreatic alpha cells. To investigate this question, Chambers et al. (106) generated a Gcg knockout mouse and then by reactivation of Gcg in L-cells or alpha cells, showed that islet-generated GLP-1 was primarily responsible for glucose homeostasis by promoting glucose-stimulated insulin secretion and suppressing glucagon secretion. The gut-derived GLP-1 binds to its receptor on local afferent vagal nerve terminals, which ultimately signals for satiety, delaying gastric emptying and suppression of hepatic glucose release (106, 107). However, this model may not translate well to human islets due to differences in islet architecture, and in light of the recent findings that glucagon is the dominant peptide hormone secreted from human alpha cells (46).
The search for a GLP-1 receptor on alpha cells has been hampered by a lack of a reliable GLP-1 receptor antibody (108, 109). GLP-1 appears to mildly reduce action potentials in the alpha cell membrane at 1 mM glucose in isolated mouse alpha cells, and this effect is blocked by the GLP-1R antagonist exendin (9-39) (110), therefore suggesting the presence of GLP-1R, perhaps at a very low density, on a small proportion of alpha cells. The development of near infra-red and fluorescent analogues of GLP-1R ligands has enabled both in vivo (111, 112) and high-resolution tissue imaging (113, 114) of GLP-1R with high specificity, sensitivity, and reproducibility. Coupled with super-resolution microscopy, the fluorescent analog LUXendin645 revealed that 5% of alpha cells in murine islets expressed GLP-1R (114). Given the already small proportion of alpha cells in the mouse islet, the contribution of direct alpha cell action to the glucagonostatic effect of GLP-1 is likely very small.
Islet GLP-1 may also exert its effects through receptors on delta cells (115), resulting in stimulation of somatostatin secretion and inhibition of glucagon secretion via SSTR2 on alpha cells (116, 117). This paracrine effect could not be detected in isolated normal human islets (110); nonetheless, this mechanism may be clinically relevant in the treatment of T2D, as experiments in human islets showed that the GLP-1R agonist liraglutide enhanced somatostatin secretion to reduce hyperglucagonemia induced by the SGLT2 inhibitor dapagliflozin (118).
As drugs targeted to the control of glucagon secretion are now being developed for the treatment of hyperglucagonemia, a deeper understanding of the dynamics of the alpha cell secretory granule is critical for identifying effective targets. However, the study of glucagon granule trafficking and exocytosis presents several technological challenges. Commonly used cell lines such as InR1-G9, αTC1-6 and αTC1-9, while useful for preliminary studies on trafficking and secretion, as a rule do not exhibit robust secretory responses to glucose or other secretagogues. The αTC1-6 cell line in particular differs from primary alpha cells in their complement of transcriptional, epigenetic and metabolic factors (119, 120) which may explain the blunted secretory response to glucose. Dispersed primary alpha cells may offer a slightly better alternative, but as discussed above, both cell lines and dispersed primary alpha cells exhibit aberrant glucagon exocytosis patterns at high glucose levels, likely due to the absence of paracrine inputs and juxtamembrane contacts. The greatest advances in gleaning the mechanisms of glucagon granule exocytosis have been made using patch-clamp approaches in isolated rodent or human islets. In such preparations, alpha cells can identified by their unique electrophysiological signature under low glucose conditions (121) or, in the case of mouse islets, by genetically-encoded fluorescence reporters such as YFP (122, 123) or tdTomato (124).
After proglucagon processing and granule maturation, glucagon is stored in the alpha cell secretory granule until a stimulus triggers exocytosis. As in beta cells, there may be different functional pools of secretory granules: a reserve pool and a readily releasable pool that is primed and situated at the sites of exocytosis. Quantitative ultrastructural analysis of murine islets has shown that, in the presence of 1mM glucose, the mouse α-cell contains ~4400 secretory granules, of which ~140 are in close proximity to the plasma membrane, or primed (125). This means that the reserve pool is large and can resupply the readily releasable pool to maintain euglycemia over extended periods of time. In the presence of 16.7 mM glucose, the numbers of the docked secretory granules increase to ~310, due to both decreased secretion and increased docking in response to increased glucose metabolism (125). Kinetic modelling of the exocytotic behaviour of alpha cell secretory granules under low glucose conditions predicts the relationship between intracellular Ca2+ levels and granule dynamics and mobilization, by which sequential Ca2+ binding events promote granule fusion with the plasma membrane (126).
Following docking, secretory granules are primed through the action of the SNARE protein complex. This complex contains two subsets of proteins; i) the t-SNAREs syntaxin 1A and SNAP-25, located in the plasma membrane; and ii) the v-SNAREs VAMP2 and synaptotagmin VII, which are located in the granule membrane (127). The Ca2+ dependence of glucagon granule exocytosis is due to the actions of synaptotagmin VII (128), a major Ca2+-responsive component. Under low glucose conditions, SNAP-25 and syntaxin 1A are translocated to the plasma membrane. SNAP-25 itself may play a role in the transportation of granules from the releasable pool to the readily releasable pool, and then mediates their fusion with plasma membrane via interaction with syntaxin 1A (125, 126). Following Ca2+ influx and binding, the C2 domain of synaptotagmin VII binds to syntaxin 1A and forms the SNARE complex to prime the secretory granules for exocytosis (125) in a mechanism similar to that in beta cells (129, 130).
Alpha cell membrane SNAREs are located in close proximity to some of the ion channels that govern alpha cell membrane depolarization and Ca2+ entry, possibly at the site of cholesterol-rich lipid microdomains (131). Modelling data predicts that membrane microdomains of P/Q-type Ca2+ channels are closely coupled to glucagon exocytosis (132), suggesting that granules in the readily releasable pool cluster at specific sites within the membrane. Live imaging of exocytosis using a proglucagon-luciferase reporter showed spatial clustering of glucagon secretion sites in αTC1-6 cells (133). Future studies may reveal some interesting dynamics with SNARE proteins that may fine-tune the alpha cell secretory response to glucose and paracrine inputs.
Could disruption of these molecular mechanisms contribute to the hyperglucagonemia of diabetes? In dispersed alpha cells from patients with T2D, voltage-dependent Ca2+ currents were normal; nonetheless, secretory granules remained in the reserve pool for a longer period of time compared to non-diabetic alpha cells, and exocytosis was actually impaired (52). However, neither membrane potential nor exocytosis was responsive to insulin or (to a greater extent) somatostatin, in contrast to normal alpha cells in which both were significantly reduced. Therefore, in T2D, hyperglucagonemia may result from insulin and somatostatin resistance at the level of the readily releasable pool of granules. In alpha cells of patients with T1D, expression levels of genes encoding SNARE proteins, ion channels and cAMP signalling molecules were disrupted (134), which could explain the impaired glucose counter-regulatory response and the inappropriately elevated levels of postprandial glucagon in T1D. Combining patch-clamp electrophysiological measurements with single-cell RNA sequencing (patch-seq) in human islets has given high-resolution insight into mechanisms underlying impairments in alpha cell function in diabetes (135) at the level of granule exocytosis. Further characterization of the link between electrophysiological signatures and the genes regulating the dynamics of granule exocytosis will reveal new mechanisms of alpha cell dysfunction in diabetes.
Identifying new pathways or networks that control glucagon granule biogenesis and trafficking may identify novel targets for the control of hyperglucagonemia in addition to yielding a greater understanding of alpha cell biology in both health and disease. There is an emerging hypothesis that glucagon secretion can be controlled by trafficking through the endosomal-lysosomal pathway, similar to insulin (136), and below, we highlight some recent studies that suggest glucagon may regulated through such an alternate trafficking pathway.
Brefeldin A-inhibited guanine nucleotide exchange protein 3 (BIG3) is a member of the Arf-GEF family of proteins, and was initially found in a database search and found to inhibit insulin granule biogenesis and insulin secretion (137). A subsequent study found that it had a similar role in regulating glucagon granule production and exocytosis (138). Although these studies inferred action of BIG3 through whole-body knockouts and not cell-specific or conditional knockouts, some more recent studies have shown that BIG3 is localized to LAMP1+ lysosomes in hypothalamic neurons (139), thus implicating BIG3 in lysosomal trafficking, at least in neurons. Whether BIG3 can mediate glucagon trafficking through lysosomes remains to be investigated.
The composition and cargo of the alpha cell secretory granule may also hold some determinants of glucagon secretion. While it is known that granule contents and composition are modified during normal granule maturation, a more complete picture of granule remodeling and heterogeneity in the context of intracellular trafficking networks in normal physiology and in diabetes is required. In an effort to identify networks of secretory granule proteins that interact with glucagon and regulate its trafficking and secretion, proteomic analysis was conducted on αTC1-6 cell secretory granule lysates immunoprecipitated with tagged glucagon (51). This qualitative study demonstrated the plasticity in the network of proteins interacting with glucagon in response to insulin or GABA under high (25 mM) or low (5.5 mM) glucose. Stathmin-2, a member of the family of neuronal phosphoproteins that associates with the secretory pathway in neurons, was identified as a candidate protein for the regulation of glucagon secretion and subsequently shown to modulate glucagon secretion through the lysosomal pathway (140) and may be down-regulated in diabetes in humans (141) and in mice (142). Therefore, disruptions in the routing of glucagon through the lysosomal pathway may contribute to the hyperglucagonemia of diabetes (Figure 4).
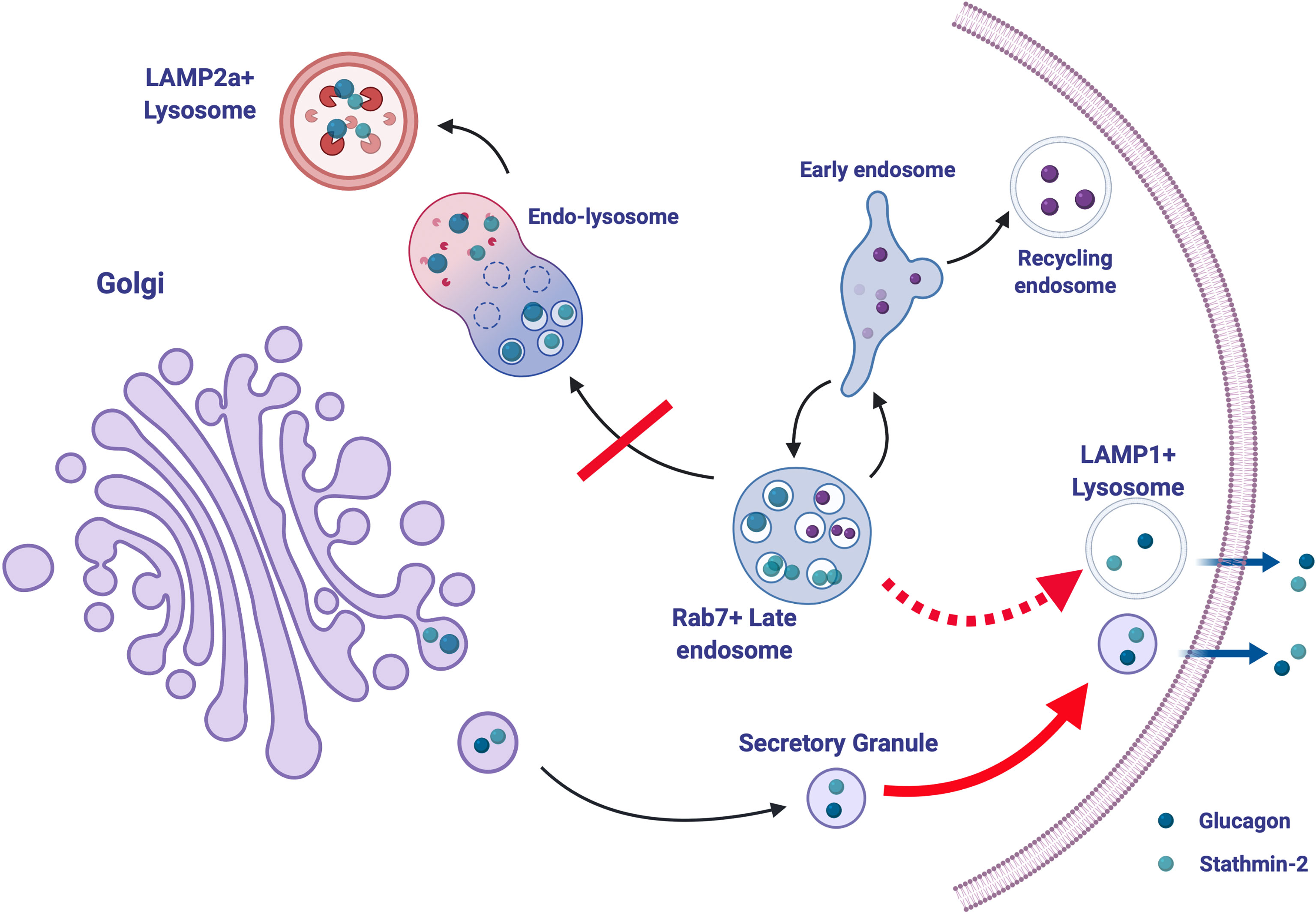
Figure 4 Stathmin-2-mediated lysosomal trafficking modulates glucagon secretion. Glucagon (dark blue) and stathmin-2 (light blue) are normally sorted to secretory granules from the Golgi in alpha cells. Stathmin-2 overexpression diverts glucagon-containing secretory granules to lysosomes (black arrows), thus reducing glucagon secretion. In the relative absence of stathmin-2, this regulatory pathway is disrupted; transport of glucagon and stathmin-2 to the LAMP2+ lysosome is inhibited and instead is re-routed to lysosomes (possibly LAMP1+) adjacent to the plasma membrane, thus and allowing for excess glucagon secretion (dotted red arrow). Additionally, secretion from secretory granules is also enhanced (solid red arrow). Created with BioRender.com.
Glucagon trafficking and exocytosis may also be controlled through nutrient-driven pathways. The nutrient sensor O-GlcNAc transferase (OGT) catalyses the O-glycosylation of several proteins including those involved in the conventional secretory pathway (143) and autophagosome-lysosome fusion (144). In mice lacking OGT specifically in alpha cells, glucagon secretion, cell content and alpha cell mass are reduced (145). Possible mechanisms include lack of O-glycosylation of FOXA1 and FOXA2, which regulate genes encoding proteins involved in proglucagon processing and glucagon secretion (146). Whether other trafficking proteins are affected, and how alpha cell function is affected in diabetes in these mice, is not yet known.
So what are the implications of glucagon trafficking through the lysosomal pathway in diabetes? Lysosomal trafficking and autophagy in the beta cell may be a possible mechanism of insulin secretory defects in diabetes, with a recent study providing evidence for impairment of lysosomal function in human T1D (147). How does lysosomal function contribute to defects in alpha cell function? It is tempting to hypothesize that impairments in lysosomal biogenesis and trafficking result in both reduced insulin secretion in the beta cell and unregulated glucagon secretion from the alpha cell. Further investigation into the altered dynamics of glucagon trafficking in the alpha cell in diabetes may reveal key roles for the lysosome in the regulation of glucagon secretion, thus identifying a potential new target for the treatment of hyperglucagonemia.
Finally, some excellent single-cell transcriptomics and epigenomics databases are being generated that reveal the dynamics of intracellular trafficking networks at the transcriptional level in human pancreatic alpha cells in both health and diabetes (148–150). The mapping of T2D-associated genetic variants with RNA-seq of human islets (151) may reveal risk factors associated with defects in alpha cell function A novel immunocompromised mouse model in which glucagon-encoding codons were deleted while preserving both GLP-1 and GLP-2 will provide an innovative and much-needed resource for the study of the regulation of glucagon secretion from human islets in vivo (152). In this study, transplantation of islets from people with T2D resulted in hyperglucagonemia with apparent alpha cell insulin resistance, revealing intrinsic alpha cell defects in T2D. Moreover, defects in alpha cell function were more apparent than in isolated islets, thus emphasizing the utility of such an in vivo system to investigate the molecular mechanisms of glucagon secretion in human islets, and the testing of possible treatments for hyperglucagonemia.
While the development of glucagon receptor antagonists and other inhibitors of glucagon action has provided some possibilities for the treatment of hyperglucagonemia, there are significant side effects that result from impaired hepatic metabolism and potentially uncontrolled alpha cell proliferation. The advantage to developing such drugs, however, lie in the fact that the glucagon receptor is an easily available target. In contrast, targeting glucagon secretion as a means to treat hyperglucagonemia may alleviate concerns about effects on the liver and alpha cell mass; however, there are potentially many more targets within the alpha cell secretory pathway, and many of those may not be easily accessible for drug treatment. The ongoing discovery of novel proteins and networks that regulate the secretion of glucagon will shed further light on alpha cell biology in health and disease while also searching for improved means to control hyperglucagonemia and hyperglycemia of diabetes.
SD and FA co-wrote the manuscript. FA was SD’s PhD student and this review was largely adapted from FA’s thesis. All authors contributed to the article and approved the submitted version.
This work was funded by a Natural Sciences and Engineering Research Council Discovery Grant to SD.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stanley S, Moheet A, Seaquist ER. Central Mechanisms of Glucose Sensing and Counterregulation in Defense of Hypoglycemia. Endocr Rev (2019) 40:768–88. doi: 10.1210/er.2018-00226
2. DCCT Research Group. Hypoglycemia in the Diabetes Control and Complications Trial. Diabetes (1997) 46:271–86. doi: 10.2337/diab.46.2.271
3. Unger R, Orci L. The Essential Role of Glucagon in the Pathogenesis of Diabetes Mellitus. Lancet (1975) 1:14–6. doi: 10.1016/S0140-6736(75)92375-2
4. Unger RH, Cherrington AD. Glucagonocentric Restructuring of Diabetes: A Pathophysiologic and Therapeutic Makeover. J Clin Invest (2012) 122:4–12. doi: 10.1172/JCI60016
5. Lee Y, Wang M-Y, Du XQ, Charron MJ, Unger RH. Glucagon Receptor Knockout Prevents Insulin-Deficient Type 1 Diabetes in Mice. Diabetes (2011) 60:391–7. doi: 10.2337/db10-0426
6. Conarello SL, Jiang G, Mu J, Li Z, Woods J, Zycband E, et al. Glucagon Receptor Knockout Mice are Resistant to Diet-Induced Obesity and Streptozotocin-Mediated Beta Cell Loss and Hyperglycaemia. Diabetologia (2006) 50:142–50. doi: 10.1007/s00125-006-0481-3
7. Neumann UH, Ho JSS, Mojibian M, Covey SD, Charron MJ, Kieffer TJ. Glucagon Receptor Gene Deletion in Insulin Knockout Mice Modestly Reduces Blood Glucose and Ketones But Does Not Promote Survival. Mol Metab (2016) 5:731–6. doi: 10.1016/j.molmet.2016.05.014
8. Damond N, Thorel F, Moyers JS, Charron MJ, Vuguin PM, Powers AC, et al. Blockade of Glucagon Signaling Prevents or Reverses Diabetes Onset Only If Residual β-Cells Persist. Elife (2016) 5:1–18. doi: 10.7554/eLife.13828
9. Kazda CM, Ding Y, Kelly RP, Garhyan P, Shi C, Lim CN, et al. Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients With Type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diabetes Care (2016) 39:1241–9. doi: 10.2337/dc15-1643
10. Yang B, Gelfanov VM, Perez-Tilve D, DuBois B, Rohlfs R, Levy J, et al. Optimization of Truncated Glucagon Peptides to Achieve Selective, High Potency, Full Antagonists. J Med Chem (2021) 64:4697–708. doi: 10.1021/acs.jmedchem.0c02069
11. Lee CY, Choi H, Park EY, Nguyen TTL, Maeng HJ, Mee Lee K, et al. Synthesis and Anti-Diabetic Activity of Novel Biphenylsulfonamides as Glucagon Receptor Antagonists. Chem Biol Drug Des (2021) 00:1–18. doi: 10.1111/cbdd.13928
12. Okamoto H, Cavino K, Na E, Krumm E, Kim SY, Cheng X, et al. Glucagon Receptor Inhibition Normalizes Blood Glucose in Severe Insulin-Resistant Mice. Proc Natl Acad Sci (2017) 114:2753–8. doi: 10.1073/pnas.1621069114
13. Liang Y, Osborne MC, Monia BP, Bhanot S, Gaarde WA, Reed C, et al. Reduction in Glucagon Receptor Expression by an Antisense Oligonucleotide Ameliorates Diabetic Syndrome in Db/Db Mice. Diabetes (2004) 53:410–7. doi: 10.2337/diabetes.53.2.410
14. Kim J, Okamoto H, Huang ZJ, Anguiano G, Chen S, Liu Q, et al. Amino Acid Transporter Slc38a5 Controls Glucagon Receptor Inhibition-Induced Pancreatic α Cell Hyperplasia in Mice. Cell Metab (2017) 25:1348–61.e8. doi: 10.1016/j.cmet.2017.05.006
15. Wei R, Gu L, Yang J, Yang K, Liu J, Le Y, et al. Antagonistic Glucagon Receptor Antibody Promotes α-Cell Proliferation and Increases β-Cell Mass in Diabetic Mice. iScience (2019) 16:326–39. doi: 10.1016/j.isci.2019.05.030
16. Galsgaard KD, Winther-Sørensen M, Ørskov C, Kissow H, Poulsen SS, Vilstrup H, et al. Disruption of Glucagon Receptor Signaling Causes Hyperaminoacidemia Exposing a Possible Liver-Alpha-Cell Axis. Am J Physiol Metab (2018) 314:E93–E103. doi: 10.1152/ajpendo.00198.2017
17. Wewer Albrechtsen NJ, Pedersen J, Galsgaard KD, Winther-Sørensen M, Suppli MP, Janah L, et al. The Liver–α-Cell Axis and Type 2 Diabetes. Endocr Rev (2019) 40:1353–66. doi: 10.1210/er.2018-00251
18. Guan H-P, Yang X, Lu K, Wang S-P, Castro-Perez JM, Previs S, et al. Glucagon Receptor Antagonism Induces Increased Cholesterol Absorption. J Lipid Res (2015) 56:2183–95. doi: 10.1194/jlr.M060897
19. Tooze S. Biogenesis of Secretory Granules in the Trans-Golgi Network of Neuroendocrine and Endocrine Cells. Biochim Biophys Acta (1998) 1404:231–44. doi: 10.1016/s0167-4889(98)00059-7
20. Cool DR, Fenger M, Snell CR, Loh YP. Identification of the Sorting Signal Motif Within Pro-Opiomelanocortin for the Regulated Secretory Pathway. J Biol Chem (1995) 270:8723–9. doi: 10.1074/jbc.270.15.8723
21. Dhanvantari S, Shen F, Adams T, Snell CR, Zhang C, Mackin RB, et al. Disruption of a Receptor-Mediated Mechanism for Intracellular Sorting of Proinsulin in Familial Hyperproinsulinemia. Mol Endocrinol (2003) 17:1856–67. doi: 10.1210/me.2002-0380
22. Zhang C-F, Dhanvantari S, Lou H, Loh YP. Sorting of Carboxypeptidase E to the Regulated Secretory Pathway Requires Interaction of its Transmembrane Domain With Lipid Rafts. Biochem J (2003) 369:453–60. doi: 10.1042/bj20020827
23. Dikeakos JD, Mercure C, Lacombe M-J, Seidah NG, Reudelhuber TL. PC1/3, PC2 and PC5/6A Are Targeted to Dense Core Secretory Granules by a Common Mechanism. FEBS J (2007) 274:4094–102. doi: 10.1111/j.1742-4658.2007.05937.x
24. Dikeakos JD, Di Lello P, Lacombe M-J, Ghirlando R, Legault P, Reudelhuber TL, et al. Functional and Structural Characterization of a Dense Core Secretory Granule Sorting Domain From the PC1/3 Protease. Proc Natl Acad Sci USA (2009) 106:7408–13. doi: 10.1073/pnas.0809576106
25. Dhanvantari S, Loh YP. Lipid Raft Association of Carboxypeptidase E Is Necessary for Its Function as a Regulated Secretory Pathway Sorting Receptor. J Biol Chem (2000) 275:29887–93. doi: 10.1074/jbc.M005364200
26. Cool DR, Normant E, Shen F, Chen H, Pannell L, Zhang Y, et al. Carboxypeptidase E Is a Regulated Secretory Pathway Sorting Receptor: Genetic Obliteration Leads to Endocrine Disorders in Cpe Fat Mice. Cell (1997) 88:73–83. doi: 10.1016/S0092-8674(00)81860-7
27. Irminger JC, Verchere CB, Meyer K, Halban PA. Proinsulin Targeting to the Regulated Pathway Is Not Impaired in Carboxypeptidase E-Deficient Cpe(fat)/Cpe(fat) Mice. J Biol Chem (1997) 272:27532–4. doi: 10.1074/jbc.272.44.27532
28. McGirr R, Guizzetti L, Dhanvantari S. The Sorting of Proglucagon to Secretory Granules is Mediated by Carboxypeptidase E and Intrinsic Sorting Signals. J Endocrinol (2013) 217:229–40. doi: 10.1530/JOE-12-0468
29. Hosaka M, Watanabe T, Sakai Y, Kato T, Takeuchi T. Interaction Between Secretogranin III and Carboxypeptidase E Facilitates Prohormone Sorting Within Secretory Granules. J Cell Sci (2005) 118:4785–95. doi: 10.1242/jcs.02608
30. Plá V, Paco S, Ghezali G, Ciria V, Pozas E, Ferrer I, et al. Secretory Sorting Receptors Carboxypeptidase E and Secretogranin III in Amyloid β-Associated Neural Degeneration in Alzheimer’s Disease. Brain Pathol (2013) 23:274–84. doi: 10.1111/j.1750-3639.2012.00644.x
31. Arvan P, Halban PA. Sorting Ourselves Out: Seeking Consensus on Trafficking in the Beta-Cell. Traffic (2004) 5:53–61. doi: 10.1111/j.1600-0854.2004.00152.x
32. Guizzetti L, McGirr R, Dhanvantari S. Two Dipolar α-Helices Within Hormone-Encoding Regions of Proglucagon are Sorting Signals to the Regulated Secretory Pathway. J Biol Chem (2014) 289:14968–80. doi: 10.1074/jbc.M114.563684
33. Dey A, Lipkind GM, Rouillé Y, Norrbom C, Stein J, Zhang C, et al. Significance of Prohormone Convertase 2, PC2, Mediated Initial Cleavage at the Proglucagon Interdomain Site, Lys70-Arg71, to Generate Glucagon. Endocrinology (2005) 146:713–27. doi: 10.1210/en.2004-1118
34. Rouille Y, Westermark G, Martin SK, Steiner DF. Proglucagon is Processed to Glucagon by Prohormone Convertase PC2 in Alpha TC1-6 Cells. Proc Natl Acad Sci (1994) 91:3242–6. doi: 10.1073/pnas.91.8.3242
35. Dhanvantari S, Seidah NG, Brubaker PL. Role of Prohormone Convertases in the Tissue-Specific Processing of Proglucagon. Mol Endocrinol (1996) 10:342–55. doi: 10.1210/me.10.4.342
36. Furuta M, Zhou A, Webb G, Carroll R, Ravazzola M, Orci L, et al. Severe Defect in Proglucagon Processing in Islet Alpha-Cells of Prohormone Convertase 2 Null Mice. J Biol Chem (2001) 276:27197–202. doi: 10.1074/jbc.M103362200
37. Campbell SA, Golec DP, Hubert M, Johnson J, Salamon N, Barr A, et al. Human Islets Contain a Subpopulation of Glucagon-Like Peptide-1 Secreting α Cells That is Increased in Type 2 Diabetes. Mol Metab (2020) 39:101014. doi: 10.1016/j.molmet.2020.101014
38. Nie Y, Nakashima M, Brubaker PL, Li QL, Perfetti R, Jansen E, et al. Regulation of Pancreatic PC1 and PC2 Associated With Increased Glucagon-Like Peptide 1 in Diabetic Rats. J Clin Invest (2000) 105:955–65. doi: 10.1172/JCI7456
39. McGirr R, Ejbick CE, Carter DE, Andrews JD, Nie Y, Friedman TC, et al. Glucose Dependence of the Regulated Secretory Pathway in αtc1-6 Cells. Endocrinology (2005) 146:4514–23. doi: 10.1210/en.2005-0402
40. Liu P, Song J, Liu H, Yan F, He T, Wang L, et al. Insulin Regulates Glucagon-Like Peptide-1 Secretion by Pancreatic Alpha Cells. Endocrine (2018) 62:394–403. doi: 10.1007/s12020-018-1684-3
41. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 Enhances Insulin Secretion by Increasing Glucagon-Like Peptide-1 Secretion From L Cells and Alpha Cells. Nat Med (2011) 17:1481–9. doi: 10.1038/nm.2513
42. O’Malley TJ, Fava GE, Zhang Y, Fonseca VA, Wu H. Progressive Change of Intra-Islet GLP-1 Production During Diabetes Development. Diabetes Metab Res Rev (2014) 30:661–8. doi: 10.1002/dmrr.2534
43. Kilimnik G, Kim A, Steiner DF, Friedman TC, Hara M. Intraislet Production of GLP-1 by Activation of Prohormone Convertase 1/3 in Pancreatic α-Cells in Mouse Models of -Cell Regeneration. Islets (2010) 2:149–55. doi: 10.4161/isl.2.3.11396
44. Wideman RD, Gray SL, Covey SD, Webb GC, Kieffer TJ. Transplantation of PC1/3-Expressing Alpha-Cells Improves Glucose Handling and Cold Tolerance in Leptin-Resistant Mice. Mol Ther (2009) 17:191–8. doi: 10.1038/mt.2008.219
45. Wideman RD, Covey SD, Webb GC, Drucker DJ, Kieffer TJ. A Switch From Prohormone Convertase (PC)-2 to PC1/3 Expression in Transplanted Alpha-Cells Is Accompanied by Differential Processing of Proglucagon and Improved Glucose Homeostasis in Mice. Diabetes (2007) 56:2744–52. doi: 10.2337/db07-0563.AUC
46. Galvin SG, Kay RG, Foreman R, Larraufie P, Meek CL, Biggs E, et al. The Human and Mouse Islet Peptidome: Effects of Obesity and Type 2 Diabetes, and Assessment of Intraislet Production of Glucagon-Like Peptide-1. J Proteome Res (2021) x:acs.jproteome.1c00463. doi: 10.1021/acs.jproteome.1c00463
47. Runge S, Wulff BS, Madsen K, Bräuner-Osborne H, Knudsen LB. Different Domains of the Glucagon and Glucagon-Like Peptide-1 Receptors Provide the Critical Determinants of Ligand Selectivity. Br J Pharmacol (2003) 138:787–94. doi: 10.1038/sj.bjp.0705120
48. Salehi A, Vieira E, Gylfe E. Paradoxical Stimulation of Glucagon Secretion by High Glucose Concentrations. Diabetes (2006) 55:2318–23. doi: 10.2337/db06-0080
49. Gylfe E. Glucose Control of Glucagon Secretion—‘there’s a Brand-New Gimmick Every Year’. Ups J Med Sci (2016) 121:120–32. doi: 10.3109/03009734.2016.1154905
50. Whalley NM, Pritchard LE, Smith DM. White a. Processing of Proglucagon to GLP-1 in Pancreatic α-Cells: Is This a Paracrine Mechanism Enabling GLP-1 to Act on β-Cells? J Endocrinol (2011) 211:99–106. doi: 10.1530/JOE-11-0094
51. Asadi F, Dhanvantari S. Plasticity in the Glucagon Interactome Reveals Novel Proteins That Regulate Glucagon Secretion in α-TC1-6 Cells. Front Endocrinol (Lausanne) (2019) 10:792. doi: 10.3389/fendo.2018.00792
52. Omar-Hmeadi M, Lund PE, Gandasi NR, Tengholm A, Barg S. Paracrine Control of α-Cell Glucagon Exocytosis is Compromised in Human Type-2 Diabetes. Nat Commun (2020) 11:1–11. doi: 10.1038/s41467-020-15717-8
53. Le Marchand SJ, Piston DW. Glucose Suppression of Glucagon Secretion: Metabolic and Calcium Responses From Alpha-Cells in Intact Mouse Pancreatic Islets. J Biol Chem (2010) 285:14389–98. doi: 10.1074/jbc.M109.069195
54. Quoix N, Cheng-xue R, Mattart L, Zeinoun Z, Guiot Y, Beauvois M, et al. Glucose and Pharmacological Modulators of ATP-Sensitive K+ Channels Control [Ca2+]c by Different Mechanisms in Isolated Mouse {Alpha}-Cells. Diabetes (2009) 58:412–21. doi: 10.2337/db07-1298.N.Q
55. Ramracheya R, Ward C, Shigeto M, Walker JN, Amisten S, Zhang Q, et al. Membrane Potential-Dependent Inactivation of Voltage-Gated Ion Channels in α-Cells Inhibits Glucagon Secretion From Human Islets. Diabetes (2010) 59:2198–208. doi: 10.2337/db09-1505
56. Zhang Q, Ramracheya R, Lahmann C, Tarasov A, Bengtsson M, Braha O, et al. Role of KATP Channels in Glucose-Regulated Glucagon Secretion and Impaired Counterregulation in Type 2 Diabetes. Cell Metab (2013) 18:871–82. doi: 10.1016/j.cmet.2013.10.014
57. Zhang Q, Dou H, Rorsman P. ‘Resistance is Futile?’ – Paradoxical Inhibitory Effects of K ATP Channel Closure in Glucagon-Secreting α-Cells. J Physiol (2020) 598:4765–80. doi: 10.1113/JP279775
58. Liu Y-J, Vieira E, Gylfe E. A Store-Operated Mechanism Determines the Activity of the Electrically Excitable Glucagon-Secreting Pancreatic α-Cell. Cell Calcium (2004) 35:357–65. doi: 10.1016/j.ceca.2003.10.002
59. Tian G, Tepikin AV, Tengholm A, Gylfe E. cAMP Induces Stromal Interaction Molecule 1 (STIM1) Puncta But Neither Orai1 Protein Clustering Nor Store-Operated Ca2+ Entry (SOCE) in Islet Cells. J Biol Chem (2012) 287:9862–72. doi: 10.1074/jbc.M111.292854
60. Watts M, Sherman A. Modeling the Pancreatic α-Cell: Dual Mechanisms of Glucose Suppression of Glucagon Secretion. Biophys J (2014) 106:741–51. doi: 10.1016/j.bpj.2013.11.4504
61. Le Marchand SJ, Piston DW. Glucose Decouples Intracellular Ca2+ Activity From Glucagon Secretion in Mouse Pancreatic Islet Alpha-Cells. PloS One (2012) 7:e47084. doi: 10.1371/journal.pone.0047084
62. Elliott AD, Ustione A, Piston DW. Somatostatin and Insulin Mediate Glucose-Inhibited Glucagon Secretion in the Pancreatic α-Cell by Lowering cAMP. Am J Physiol Endocrinol Metab (2015) 308:E130–43. doi: 10.1152/ajpendo.00344.2014
63. Yu Q, Shuai H, Ahooghalandari P, Gylfe E, Tengholm A. Glucose Controls Glucagon Secretion by Directly Modulating cAMP in Alpha Cells. Diabetologia (2019) 62:1212–24. doi: 10.1007/s00125-019-4857-6
64. Hughes JW, Ustione A, Lavagnino Z, Piston DW. Regulation of Islet Glucagon Secretion: Beyond Calcium. Diabetes Obes Metab (2018) 20:127–36. doi: 10.1111/dom.13381
65. Leclerc I, Sun G, Morris C, Fernandez-Millan E, Nyirenda M, Rutter GA. AMP-Activated Protein Kinase Regulates Glucagon Secretion From Mouse Pancreatic Alpha Cells. Diabetologia (2011) 54:125–34. doi: 10.1007/s00125-010-1929-z
66. Da Silva Xavier G, Farhan H, Kim H, Caxaria S, Johnson P, Hughes S, et al. Per-Arnt-Sim (PAS) Domain-Containing Protein Kinase is Downregulated in Human Islets in Type 2 Diabetes and Regulates Glucagon Secretion. Diabetologia (2011) 54:819–27. doi: 10.1007/s00125-010-2010-7
67. Sun G, da Silva Xavier G, Gorman T, Priest C, Solomou A, Hodson DJ, et al. LKB1 and Ampkα1 are Required in Pancreatic Alpha Cells for the Normal Regulation of Glucagon Secretion and Responses to Hypoglycemia. Mol Metab (2015) 4:277–86. doi: 10.1016/j.molmet.2015.01.006
68. Bozadjieva N, Blandino-Rosano M, Chase J, Dai XQ, Cummings K, Gimeno J, et al. Loss of Mtorc1 Signaling Alters Pancreatic α Cell Mass and Impairs Glucagon Secretion. J Clin Invest (2017) 127:4379–93. doi: 10.1172/JCI90004
69. Kramer NB, Lubaczeuski C, Blandino-Rosano M, Barker G, Gittes GK, Caicedo A, et al. Glucagon Resistance and Decreased Susceptibility to Diabetes in a Model of Chronic Hyperglucagonemia. Diabetes (2021) 70:477–91. doi: 10.2337/db20-0440
70. Gromada J, Franklin I, Wollheim CB. Alpha-Cells of the Endocrine Pancreas: 35 Years of Research But the Enigma Remains. Endocr Rev (2007) 28:84–116. doi: 10.1210/er.2006-0007
71. Kawamori D, Kulkarni RN. Insulin Modulation of Glucagon Secretion: The Role of Insulin and Other Factors in the Regulation of Glucagon Secretion. Islets (2009) 1:276–9. doi: 10.4161/isl.1.3.9967
72. Tsuchiyama N, Takamura T, Ando H, Sakurai M, Shimizu A, Kato KI, et al. Possible Role of α-Cell Insulin Resistance in Exaggerated Glucagon Responses to Arginine in Type 2 Diabetes. Diabetes Care (2007) 30:2583–7. doi: 10.2337/dc07-0066
73. Wendt A, Birnir B, Buschard K, Gromada J, Salehi A, Sewing S, et al. Glucose Inhibition of Glucagon Secretion From Rat α-Cells Is Mediated by GABA Released From Neighboring β-Cells. Diabetes (2004) 53:1038–45. doi: 10.2337/diabetes.53.4.1038
74. Li C, Liu C, Nissim I, Chen J, Chen P, Doliba N, et al. Regulation of Glucagon Secretion in Normal and Diabetic Human Islets by ??-Hydroxybutyrate and Glycine. J Biol Chem (2013) 288:3938–51. doi: 10.1074/jbc.M112.385682
75. Rorsman P, Berggren PO, Bokvist K, Ericson H, Möhler H, Ostenson CG SP. Glucose-Inhibition of Glucagon Secretion Involves Activation of GABAA-Receptor Chloride Channels. Nature (1989) 341:233–6. doi: 10.1038/341233a0
76. Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, et al. Intra-Islet Insulin Suppresses Glucagon Release via GABA-GABAA Receptor System. Cell Metab (2006) 3:47–58. doi: 10.1016/j.cmet.2005.11.015
77. Feng AL, Xiang Y, Gui L, Kaltsidis G, Feng Q, Lu W. Paracrine GABA and Insulin Regulate Pancreatic Alpha Cell Proliferation in a Mouse Model of Type 1 Diabetes. Diabetologia (2017) 60:1033–42. doi: 10.1007/s00125-017-4239-x
78. Jin Li J, Casteels T, Frogne T, Ingvorsen C, Honore C, Courtney M, et al. Artemisinins Target GABAA Receptor Signaling and Impair α Cell Identity. Cell (2017) 168:86–100.e15. doi: 10.1016/j.cell.2016.11.010
79. Weir GC, Bonner-Weir S. GABA Signaling Stimulates β Cell Regeneration in Diabetic Mice. Cell (2017) 168:7–9. doi: 10.1016/j.cell.2016.12.006
80. Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, et al. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-Like Cell Neogenesis. Cell (2017) 168:73–85.e11. doi: 10.1016/j.cell.2016.11.002
81. van der Meulen T, Lee S, Noordeloos E, Donaldson CJ, Adams MW, Noguchi GM, et al. Artemether Does Not Turn α Cells Into β Cells. Cell Metab (2018) 27:218–225.e4. doi: 10.1016/j.cmet.2017.10.002
82. Ackermann AM, Moss NG, Kaestner KH. GABA and Artesunate Do Not Induce Pancreatic α-to-β Cell Transdifferentiation In Vivo. Cell Metab (2018) 28:787–792.e3. doi: 10.1016/j.cmet.2018.07.002
83. Son DO, Liu W, Li X, Prud’homme GJ, Wang Q. Combined Effect of GABA and Glucagon-Like Peptide-1 Receptor Agonist on Cytokine-Induced Apoptosis in Pancreatic β-Cell Line and Isolated Human Islets. J Diabetes (2019) 11:563–72. doi: 10.1111/1753-0407.12881
84. Daems C, Welsch S, Boughaleb H, Vanderroost J, Robert A, Sokal E, et al. Early Treatment With Empagliflozin and GABA Improves β -Cell Mass and Glucose Tolerance in Streptozotocin-Treated Mice. J Diabetes Res (2019) 2019:1–14. doi: 10.1155/2019/2813489
85. Woalder. Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion From Alpha Cells. Cell Rep (2016) 17:3281–91. doi: 10.1016/j.celrep.2016.11.072
86. Bennet H, Balhuizen A, Medina A, Dekker Nitert M, Ottosson Laakso E, Essén S, et al. Altered Serotonin (5-HT) 1D and 2A Receptor Expression May Contribute to Defective Insulin and Glucagon Secretion in Human Type 2 Diabetes. Peptides (2015) 71:113–20. doi: 10.1016/j.peptides.2015.07.008
87. Yip L, Taylor C, Whiting CC, Fathman CG. Diminished Adenosine A1 Receptor Expression in Pancreatic a-Cells May Contribute to the Patholog Y of Type 1 Diabetes. Diabetes (2013) 62:4208–19. doi: 10.2337/db13-0614
88. Ishihara H, Wollheim CB. Is Zinc an Intra-Islet Regulator of Glucagon Secretion? Diabetol Int (2016) 7:106–10. doi: 10.1007/s13340-016-0259-x
89. Franklin I, Gromada J, Gjinovci A, Theander S. WCB. Beta Cell Secretory Products Activate Alpha Cell ATP-Dependent Potassium Channels to Inhibit Glucagon Release. Diabetes (2005) 54:1808–15. doi: 10.2337/diabetes.54.6.1808
90. Ravier MA, Rutter GA. Glucose or Insulin, But Not Zinc Ions, Inhibit Glucagon Secretion From Mouse Pancreatic [Alpha]-Cells. Diabetes (2005) 54:1789–97. doi: 10.2337/diabetes.54.6.1789
91. Solomou A, Philippe E, Chabosseau P, Migrenne-li S, Gaitan J, Lang J, et al. Over-Expression of Slc30a8 / ZnT8 Selectively in the Mouse α Cell Impairs Glucagon Release and Responses to Hypoglycemia. Nutr Metab (Lond) (2016) 13:46. doi: 10.1186/s12986-016-0104-z
92. Solomou A, Meur G, Bellomo E, Hodson DJ, Tomas A, Li SM, et al. The Zinc Transporter Slc30a8/ZnT8 is Required in a Subpopulation of Pancreatic α-Cells for Hypoglycemia-Induced Glucagon Secretion. J Biol Chem (2015) 290:21432–42. doi: 10.1074/jbc.M115.645291
93. Strowski MZ, Parmar RM, Blake AD, Schaeffer JM. Somatostatin Inhibits Insulin and Glucagon Secretion via Two Receptor Subtypes : An In Vitro Study of Pancreatic Islets From Somatostatin Receptor 2 Knockout Mice. Endocrinology (2000) 141:111–7. doi: 10.1210/endo.141.1.7263
94. Gromada J, Hoy M, Bushcard K, Salehi A, Rorsman P. Somatostatin Inhibits Exocytosis in Rat Pancreatic a-Cells by Gi2-Dependent Activation of Calcineurin and Depriming of Secretory Granules. J Physiol (2001) 535:519–32. doi: 10.1111/j.1469-7793.2001.00519.x
95. Rutter GA. Regulating Glucagon Secretion: Somatostatin in the Spotlight. Diabetes (2009) 58:299–301. doi: 10.2337/db08-1534
96. Xu SFS, Andersen DB, Izarzugaza JMG, Kuhre RE, Holst JJ. In the Rat Pancreas, Somatostatin Tonically Inhibits Glucagon Secretion and Is Required for Glucose-Induced Inhibition of Glucagon Secretion. Acta Physiol (2020) 229:1–15. doi: 10.1111/apha.13464
97. Hauge-Evans AC, King AJ, Carmignac D, Richardson CC, Robinson ICAF, Low MJ, et al. Somatostatin Secreted by Islet -Cells Fulfills Multiple Roles as a Paracrine Regulator of Islet Function. Diabetes (2009) 58:403–11. doi: 10.2337/db08-0792
98. Vergari E, Knudsen JG, Ramracheya R, Salehi A, Zhang Q, Adam J, et al. Insulin Inhibits Glucagon Release by SGLT2-Induced Stimulation of Somatostatin Secretion. Nat Commun (2019) 10:139. doi: 10.1038/s41467-018-08193-8
99. Briant LJB, Reinbothe TM, Spiliotis I, Miranda C, Rodriguez B, Rorsman P. Δ-Cells and β-Cells Are Electrically Coupled and Regulate α-Cell Activity Via Somatostatin. J Physiol (2018) 596:197–215. doi: 10.1113/JP274581
100. Kilimnik G, Zhao B, Jo J, Periwal V, Witkowski P, Misawa R, et al. Altered Islet Composition and Disproportionate Loss of Large Islets in Patients With Type 2 Diabetes. PloS One (2011) 6:e27445. doi: 10.1371/journal.pone.0027445
101. Ma X, Zhang Y, Gromada J, Sewing S, Berggren P-O, Buschard K, et al. Glucagon Stimulates Exocytosis in Mouse and Rat Pancreatic α-Cells by Binding to Glucagon Receptors. Mol Endocrinol (2005) 19:198–212. doi: 10.1210/me.2004-0059
102. Leibiger B, Moede T, Muhandiramlage TP, Kaiser D, Vaca Sanchez P, Leibiger IB, et al. Glucagon Regulates its Own Synthesis by Autocrine Signaling. Proc Natl Acad Sci USA (2012) 109:20925–30. doi: 10.1073/pnas.1212870110
103. Wewer Albrechtsen NJ, Kuhre RE, Hornburg D, Jensen CZ, Hornum M, Dirksen C, et al. Circulating Glucagon 1-61 Regulates Blood Glucose by Increasing Insulin Secretion and Hepatic Glucose Production. Cell Rep (2017) 21:1452–60. doi: 10.1016/j.celrep.2017.10.034
104. Hare KJ, Knop FK, Asmar M, Madsbad S, Deacon CF, Holst JJ, et al. Preserved Inhibitory Potency of GLP-1 on Glucagon Secretion in Type 2 Diabetes Mellitus. J Clin Endocrinol Metab (2009) 94:4679–87. doi: 10.1210/jc.2009-0921
105. Garg M, Ghanim H, Kuhadiya N, Green K, Hejna J, Abuaysheh S, et al. Liraglutide Acutely Suppresses Glucagon, Lipolysis and Ketogenesis in Type 1 Diabetes. Diabetes Obes Metab (2017) 19:1306–11. doi: 10.1111/dom.12944
106. Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim K-S, et al. The Role of Pancreatic Preproglucagon in Glucose Homeostasis in Mice. Cell Metab (2017) 25:927–34.e3. doi: 10.1016/j.cmet.2017.02.008
107. Habener JF, Stanojevic V. Pancreas and Not Gut Mediates the GLP-1-Induced Glucoincretin Effect. Cell Metab (2017) 25:757–8. doi: 10.1016/j.cmet.2017.03.020
108. Fava GE, Dong EW, Wu H. Intra-Islet Glucagon-Like Peptide-1. J Diabetes Complicat (2017) 30:1651–8. doi: 10.1016/j.jdiacomp.2016.05.016.INTRA-ISLET
109. Holst J, Christensen M, Lund A, De Heer J, Svendsen B, Kielgast U, et al. Regulation of Glucagon Secretion by Incretins. Diabetes Obes Metab (2011) 13:89–94. doi: 10.1111/j.1463-1326.2011.01452.x
110. Ramracheya R, Chapman C, Chibalina M, Dou H, Miranda C, González A, et al. GLP-1 Suppresses Glucagon Secretion in Human Pancreatic Alpha-Cells by Inhibition of P/Q-Type Ca 2+ Channels. Physiol Rep (2018) 6:1–17. doi: 10.14814/phy2.13852
111. Boss M, Bos D, Frielink C, Sandker G, Ekim S, Marciniak C, et al. Targeted Optical Imaging of the Glucagon-Like Peptide 1 Receptor Using Exendin-4irdye800cw. J Nucl Med (2020) 61:1066–71. doi: 10.2967/JNUMED.119.234542
112. Roberts S, Khera E, Choi C, Navaratna T, Grimm J, Thurber GM, et al. Optoacoustic Imaging of Glucagon-Like Peptide 1 Receptor With a Near-Infrared Exendin-4 Analog. J Nucl Med (2020) 62:839–48. doi: 10.2967/jnumed.120.252262 jnumed.120.252262.
113. Azad BB, Rota V. Design, Synthesis and In Vitro Characterization of Glucagon-Like Peptide-1 Derivatives for Pancreatic Beta Cell Imaging by SPECT. Bioorg Med Chem (2010) 18:1265–72. doi: 10.1016/j.bmc.2009.12.032
114. Ast J, Arvaniti A, Fine NHF, Nasteska D, Ashford FB, Stamataki Z, et al. Super-Resolution Microscopy Compatible Fluorescent Probes Reveal Endogenous Glucagon-Like Peptide-1 Receptor Distribution and Dynamics. Nat Commun (2020) 11:467. doi: 10.1038/s41467-020-14309-w
115. Waser B, Blank A, Karamitopoulou E, Perren A, Reubi JC. Glucagon-Like-Peptide-1 Receptor Expression in Normal and Diseased Human Thyroid and Pancreas. Mod Pathol (2015) 28:391–402. doi: 10.1038/modpathol.2014.113
116. de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon-Like Peptide-1, But Not Glucose-Dependent Insulinotropic Peptide, Inhibits Glucagon Secretion via Somatostatin (Receptor Subtype 2) in the Perfused Rat Pancreas. Diabetologia (2008) 51:2263–70. doi: 10.1007/s00125-008-1149-y
117. Ørgaard A, Holst JJ. The Role of Somatostatin in GLP-1-Induced Inhibition of Glucagon Secretion in Mice. Diabetologia (2017) 60:1731–9. doi: 10.1007/s00125-017-4315-2
118. Saponaro C, Gmyr V, Thévenet J, Moerman E, Delalleau N, Pasquetti G, et al. The GLP1R Agonist Liraglutide Reduces Hyperglucagonemia Induced by the SGLT2 Inhibitor Dapagliflozin via Somatostatin Release. Cell Rep (2019) 28:1447–54.e4. doi: 10.1016/j.celrep.2019.07.009
119. Lawlor N, Youn A, Kursawe R, Ucar D, Stitzel ML. Alpha TC1 and Beta-TC-6 Genomic Profiling Uncovers Both Shared and Distinct Transcriptional Regulatory Features With Their Primary Islet Counterparts. Sci Rep (2017) 7:1–14. doi: 10.1038/s41598-017-12335-1
120. Stamenkovic JA, Andersson LE, Adriaenssens AE, Bagge A, Sharoyko VV, Gribble F, et al. Inhibition of the Malate-Aspartate Shuttle in Mouse Pancreatic Islets Abolishes Glucagon Secretion Without Affecting Insulin Secretion. Biochem J (2015) 468:49–63. doi: 10.1042/BJ20140697
121. Briant LJB, Zhang Q, Vergari E, Kellard JA, Rodriguez B, Ashcroft FM, et al. Functional Identification of Islet Cell Types by Electrophysiological Fingerprinting. J R Soc Interface (2017) 14:20160999. doi: 10.1098/rsif.2016.0999
122. Ackermann AM, Zhang J, Heller A, Briker A, Kaestner KH. High-Fidelity Glucagon-CreER Mouse Line Generated by CRISPR-Cas9 Assisted Gene Targeting. Mol Metab (2016) 6:236–44. doi: 10.1016/j.molmet.2017.01.003
123. Quoix N, Cheng-Xue R, Guiot Y, Herrera PL, Henquin JC, Gilon P. The GluCre-ROSA26EYFP Mouse: A New Model for Easy Identification of Living Pancreatic Alpha-Cells. FEBS Lett (2007) 581:4235–40. doi: 10.1016/j.febslet.2007.07.068
124. Shiota C, Prasadan K, Guo P, Fusco J, Xiao X, Gittes GK. GcgCreERT2knockin Mice as a Tool for Genetic Manipulation in Pancreatic Alpha Cells. Diabetologia (2017) 60:2399–408. doi: 10.1007/s00125-017-4425-x
125. Andersson SA, Pedersen MG, Vikman J, Eliasson L. Glucose-Dependent Docking and SNARE Protein-Mediated Exocytosis in Mouse Pancreatic Alpha-Cell. Pflugers Arch Eur J Physiol (2011) 462:443–54. doi: 10.1007/s00424-011-0979-5
126. González-Vélez V, Dupont G, Gil A, González A, Quesada I. Model for Glucagon Secretion by Pancreatic α-Cells. PloS One (2012) 7:e32282. doi: 10.1371/journal.pone.0032282
127. Gerber SH, Sudhof TC. Molecular Determinants of Regulated Exocytosis. Diabetes (2002) 51:3–11. doi: 10.2337/diabetes.51.2007.S3
128. Gustavsson N, Wei S, Hoang DN, Lao Y, Zhang Q, Radda GK, et al. Synaptotagmin-7 Is a Principal Ca 2+ Sensor for Ca 2+ -Induced Glucagon Exocytosis in Pancreas. J Physiol (2009) 587:1169–78. doi: 10.1113/jphysiol.2008.168005
129. Jewell JL, Oh E, Thurmond DC. Exocytosis Mechanisms Underlying Insulin Release and Glucose Uptake : Conserved Roles for Munc18c and Syntaxin 4. Am J Physiol Regul Integr Comp Physiol (2010) 298:R517–31. doi: 10.1152/ajpregu.00597.2009
130. Gandasi NR, Yin P, Riz M, Chibalina MV, Cortese G, Lund P, et al. Ca 2 + Channel Clustering With Insulin-Containing Granules Is Disturbed in Type 2 Diabetes. J Clin Invest (2017) 127:2353–64. doi: 10.1172/JCI88491
131. Xia F, Leung YM, Gaisano G, Gao X, Chen Y, Fox JEM, et al. Targeting of Voltage-Gated K+ and Ca2+ Channels and Soluble N-Ethylmaleimide-Sensitive Factor Attachment Protein Receptor Proteins to Cholesterol-Rich Lipid Rafts in Pancreatic α-Cells: Effects on Glucagon Stimulus-Secretion Coupling. Endocrinology (2007) 148:2157–67. doi: 10.1210/en.2006-1296
132. Montefusco F, Pedersen MG. Mathematical Modelling of Local Calcium and Regulated Exocytosis During Inhibition and Stimulation of Glucagon Secretion From Pancreatic Alpha-Cells. J Physiol (2015) 593:4519–30. doi: 10.1113/JP270777
133. Yokawa S, Suzuki T, Inouye S, Inoh Y, Suzuki R, Kanamori T, et al. Visualization of Glucagon Secretion From Pancreatic α Cells by Bioluminescence Video Microscopy: Identification of Secretion Sites in the Intercellular Contact Regions. Biochem Biophys Res Commun (2017) 485:725–30. doi: 10.1016/j.bbrc.2017.02.114
134. Brissova M, Haliyur R, Saunders D, Shrestha S, Dai C, Blodgett DM, et al. α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep (2018) 22:2667–76. doi: 10.1016/j.celrep.2018.02.032
135. Camunas-Soler J, Dai XQ, Hang Y, Bautista A, Lyon J, Suzuki K, et al. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab (2020) 31:1017–31.e4. doi: 10.1016/j.cmet.2020.04.005
136. Zhou Y, Liu Z, Zhang S, Zhuang R, Liu H, Liu X, et al. RILP Restricts Insulin Secretion Through Mediating Lysosomal Degradation of Proinsulin. Diabetes (2020) 69:67–82. doi: 10.2337/db19-0086
137. Li H, Wei S, Cheng K, Gounko NV, Ericksen RE, Xu A, et al. BIG3 Inhibits Insulin Granule Biogenesis and Insulin Secretion. EMBO Rep (2014) 15:714–22. doi: 10.1002/embr.201338181
138. Li H, Liu T, Lim J, Gounko NV, Hong W, Han W. Increased Biogenesis of Glucagon-Containing Secretory Granules and Glucagon Secretion in BIG3-Knockout Mice. Mol Metab (2015) 4:246–52. doi: 10.1016/j.molmet.2015.01.001
139. Liu T, Li H, Hong W, Han W. Brefeldin A-Inhibited Guanine Nucleotide Exchange Protein 3 is Localized in Lysosomes and Regulates GABA Signaling in Hippocampal Neurons. J Neurochem (2016) 139:748–56. doi: 10.1111/jnc.13859
140. Asadi F, Dhanvantari S. Stathmin-2 Mediates Glucagon Secretion From Pancreatic α-Cells. Front Endocrinol (Lausanne) (2020) 11:29. doi: 10.3389/fendo.2020.00029
141. Bramswig NC, Everett LJ, Schug J, Dorrell C, Liu C, Luo Y, et al. Epigenomic Plasticity Enables Human Pancreatic α to β Cell Reprogramming. J Clin Invest (2013) 123:1275–84. doi: 10.1172/JCI66514
142. Asadi F, Dhanvantari S. Misrouting of Glucagon and Stathmin-2 Towards Lysosomal System of α-Cells in Glucagon Hypersecretion of Diabetes. bioRxiv (2021) 0–1. doi: 10.1101/2021.04.08.439083
143. Cho HJ. Mook-Jung I. O-GlcNAcylation Regulates Endoplasmic Reticulum Exit Sites Through Sec31A Modification in Conventional Secretory Pathway. FASEB J (2018) 32:4641–57. doi: 10.1096/fj.201701523R
144. Zhang X, Wang L, Lak B, Li J, Jokitalo E, Wang Y. GRASP55 Senses Glucose Deprivation Through O-GlcNAcylation to Promote Autophagosome-Lysosome Fusion. Dev Cell (2018) 45:245–61.e6. doi: 10.1016/j.devcel.2018.03.023
145. Essawy A, Jo S, Beetch M, Lockridge A, Gustafson E. Alejandro EU. O-Linked N-Acetylglucosamine Transferase (OGT) Regulates Pancreatic α-Cell Function in Mice. J Biol Chem (2021) 296:100297. doi: 10.1016/j.jbc.2021.100297
146. Masson MH, Poisson C, Guérardel A, Mamin A, Philippe J, Gosmain Y. Foxa1 and Foxa2 Regulate β-Cell Differentiation, Glucagon Biosynthesis, and Secretion. Endocrinology (2014) 155:3781–92. doi: 10.1210/en.2013-1843
147. Muralidharan C, Conteh AM, Marasco MR, Crowder JJ, Kuipers J, de Boer P, et al. Pancreatic Beta Cell Autophagy is Impaired in Type 1 Diabetes. Diabetologia (2021) 64:865–77. doi: 10.1007/s00125-021-05387-6
148. Chiou J, Zeng C, Cheng Z, Han JY, Schlichting M, Miller M, et al. Single-Cell Chromatin Accessibility Identifies Pancreatic Islet Cell Type– and State-Specific Regulatory Programs of Diabetes Risk. Nat Genet (2021) 53:455–66. doi: 10.1038/s41588-021-00823-0
149. Rai V, Quang DX, Erdos MR, Cusanovich DA, Daza RM, Narisu N, et al. Single-Cell ATAC-Seq in Human Pancreatic Islets and Deep Learning Upscaling of Rare Cells Reveals Cell-Specific Type 2 Diabetes Regulatory Signatures. Mol Metab (2020) 32:109–21. doi: 10.1016/j.molmet.2019.12.006
150. Dai X, Camunas-soler J, Briant LJB, Santos T, Aliya F, Nie A, et al. Heterogenous Impairment of a α-Cell Function in Type 2 Diabetes is Linked to Cell Maturation State. bioRxiv (2021). doi: 10.1101/2021.04.08.435504
151. Viñuela A, Varshney A, van de Bunt M, Prasad RB, Asplund O, Bennett A, et al. Genetic Variant Effects on Gene Expression in Human Pancreatic Islets and Their Implications for T2D. Nat Commun (2020) 11:4912. doi: 10.1038/s41467-020-18581-8
Keywords: glucagon, secretion, trafficking, alpha cell, hyperglucagonemia, proglucagon processing, secretory granule
Citation: Asadi F and Dhanvantari S (2021) Pathways of Glucagon Secretion and Trafficking in the Pancreatic Alpha Cell: Novel Pathways, Proteins, and Targets for Hyperglucagonemia. Front. Endocrinol. 12:726368. doi: 10.3389/fendo.2021.726368
Received: 16 June 2021; Accepted: 13 September 2021;
Published: 29 September 2021.
Edited by:
Andrei I. Tarasov, Ulster University, United KingdomReviewed by:
Quan Zhang, University of Oxford, United KingdomCopyright © 2021 Asadi and Dhanvantari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Savita Dhanvantari, c2RoYW52YW5AbGF3c29uaW1hZ2luZy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.