 Ibrahim Mohammed
Ibrahim Mohammed Morley D. Hollenberg
Morley D. Hollenberg Hong Ding
Hong Ding Chris R. Triggle
Chris R. Triggle- 1Department of Medical Education, Weill Cornell Medicine-Qatar, Al-Rayyan, Qatar
- 2Inflammation Research Network and Snyder Institute for Chronic Diseases, Department of Physiology & Pharmacology, University of Calgary Cumming School of Medicine, Calgary, AB, Canada
- 3Department of Medicine, University of Calgary Cumming School of Medicine, Calgary, AB, Canada
- 4Departments of Medical Education and Pharmacology, Weill Cornell Medicine-Qatar, Al-Rayyan, Qatar
The numerous beneficial health outcomes associated with the use of metformin to treat patients with type 2 diabetes (T2DM), together with data from pre-clinical studies in animals including the nematode, C. elegans, and mice have prompted investigations into whether metformin has therapeutic utility as an anti-aging drug that may also extend lifespan. Indeed, clinical trials, including the MILES (Metformin In Longevity Study) and TAME (Targeting Aging with Metformin), have been designed to assess the potential benefits of metformin as an anti-aging drug. Preliminary analysis of results from MILES indicate that metformin may induce anti-aging transcriptional changes; however it remains controversial as to whether metformin is protective in those subjects free of disease. Furthermore, despite clinical use for over 60 years as an anti-diabetic drug, the cellular mechanisms by which metformin exerts either its actions remain unclear. In this review, we have critically evaluated the literature that has investigated the effects of metformin on aging, healthspan and lifespan in humans as well as other species. In preparing this review, particular attention has been placed on the strength and reproducibility of data and quality of the study protocols with respect to the pharmacokinetic and pharmacodynamic properties of metformin. We conclude that despite data in support of anti-aging benefits, the evidence that metformin increases lifespan remains controversial. However, via its ability to reduce early mortality associated with various diseases, including diabetes, cardiovascular disease, cognitive decline and cancer, metformin can improve healthspan thereby extending the period of life spent in good health. Based on the available evidence we conclude that the beneficial effects of metformin on aging and healthspan are primarily indirect via its effects on cellular metabolism and result from its anti-hyperglycemic action, enhancing insulin sensitivity, reduction of oxidative stress and protective effects on the endothelium and vascular function.
Introduction
Metformin is a synthetic biguanide, orally effective and insulin sensitizing anti-diabetic drug, which for most patients is the first line anti-hyperglycemic for the treatment of type 2 diabetes mellitus (T2DM) (1). Metformin was first synthesized in 1922 and its development was based on knowledge from folk medicine that the active, but toxic, constituent from Galega officinalis (French lilac) that could treat ‘sweet urine’ was the guanidine, galegine (2, 3). Metformin was introduced to treat T2DM in France in 1958 and now after over 60 years is used on a daily basis by over 150 million people (4). For the majority of people metformin is not only an inexpensive drug as it is off patent but also a safe drug with the most prominent side effects being gastrointestinal-related that occur in about 20-30% of patients and can include abdominal pain, bloating, diarrhea, nausea and vomiting (5, 6). The GI-side effects are dose-dependent and do not usually limit its use; however, in approximately 5% of subjects these side effects can result in the need to switch to an alternative drug (5–9). The GI side effects can be linked to the accumulation of metformin in gut enterocytes and enhanced glucose utilization resulting in lactate generation, as well as metformin-induced changes in the microbiota (6, 10). More serious side effects include lactic acidosis, which is, however, very rare and is reduced by avoiding its use in patients with severe renal and liver dysfunction (9, 11–15). Several studies, but not all, have reported that the chronic use of metformin results in malabsorption issues and vitamin B12 deficiency, affecting between 6 and 30% of users. Thus, monitoring B12 levels will be essential if the use of metformin is expanded to become the panacea for all reasons and all diseases (16–20). On the positive side, metformin use leads to weight reduction in many patients, reduces HbA1c and its use is not associated with hypoglycemia. In addition, data from the Diabetes Prevention Program (DPP) indicates that metformin is effective in preventing the development of diabetes in patients with pre-diabetes and its use following a 10-year follow up associated with an average 2.1-kg weight loss (7). However, metformin has not been approved as a weight loss drug in part because its effects are highly variable and in women with PCOS despite weight loss visceral fat may not be reduced (21).
According to the WHO, diabetes and cardiovascular disease contribute approximately 22 million deaths annually (22). Given that metformin is so widely used, there is an opportunity to determine whether metformin does have anti-aging properties. This goal raises the question as to what really defines an anti-aging drug? We will use the definition that an “anti-aging action refers to the prevention, and treatment of age-related diseases” in other words “positively affects healthspan, the period of life spent in good health and free of disabling diseases, or healthy lifespan”. We acknowledge that the use of the term ‘healthspan’ is considered by some to be controversial as it cannot readily be quantified and therefore is subjective; however, the term is extensively used. Therefore, to be consistent, we retain its use, accepting the limitations that the term simply reflects a reduction in the risk of disabling disease. That reduction in itself could increase lifespan (23). Healthspan is thus distinct from extending life expectancy, or lifespan (longevity), which can be readily quantified, and is defined as “the length of time an organism lives, or total lifespan”.
The use of metformin in the setting of T2DM increased dramatically with the results from the United Kingdom Prospective Diabetes Study (UKPDS), a prospective, 20-year randomized, multicenter of patients with T2DM. The study, published in 1998, reported the cardiovascular (CV) benefits of the use of metformin for diabetes (24). It has long been assumed that the anti-diabetic benefits of metformin are due to its hepatic-mediated actions in human subjects and that other clinical benefits are secondary to its effects on glucose and lipid metabolism. We now know there are extra-hepatic sites of action of metformin with the gut being a major contributor to its clinical benefits as an anti-hyperglycemic drug via modulating glucagon-like peptide 1 (GLP-1) levels by a duodenal AMPK-mediated pathway, as well as effects resulting from modulating the microbiota (25–29). The benefits of metformin in reducing microvascular disease have been supported by a 10-year follow up of the original UKPDS report (30). Similarly, Johnson et al. accessed the Saskatchewan Health Administrative databases for a retrospective study and also reported reduced cardiovascular morbidity and mortality benefits of metformin monotherapy versus sulfonylurea monotherapy (31). Additional supportive pre-clinical and clinical evidence has been provided by Nesti and Natali (32). Collectively these data indicate that metformin can reduce both mortality and disease-accelerated aging of the cardiovascular system. Based on evidence from both clinical and pre-clinical studies metformin, independent of its anti-hyperglycemic actions, directly protects the endothelium (33–35). An accumulation of sometimes controversial data has opened the debate as to whether metformin’s favorable effects extend beyond its anti-hyperglycemic actions and include a role as an anti-aging drug that enhances healthspan and extends lifespan thus raising the hope that it could be the “Fountain of Youth” (26, 27, 29, 36–42).
In addition to its benefits as an anti-hyperglycemic agent, metformin also lowers the atherosclerotic burden in non-diabetic persons at risk of developing T2DM (1, 7). Interest in the potential clinical benefits of metformin has been extended to include repurposing for polycystic ovary syndrome (PCOS), preeclampsia, cancer, rheumatoid arthritis, malaria, and antibiotic and antiviral actions (43–48). Based on this ever-expanding catalogue of the benefits of metformin, the title of a review article refers to metformin as: “Metformin, the aspirin of the 21st century—” (27).
In this review we summarize the published evidence that argues for, or against, an anti-aging effect of metformin. Furthermore we describe the putative anti-aging pathways that metformin activates and discuss with reference to ongoing clinical trials whether the use of metformin is translated into positive effects on both healthspan and lifespan. An important issue when evaluating the potential benefits of a drug in the treatment of any given medical condition is the strength and reproducibility of the evidence and a particular concern when translating data from pre-clinical studies of a drug to therapeutic efficacy (49–51).
The Putative Relationship Between Metformin and Aging
Based on a systematic review of 53 studies, Campbell et al. concluded that independent of its therapeutic efficacy as an anti-diabetic drug, the use of metformin results in a reduction of all-cause mortality associated with diseases that accelerate aging, including cancer and cardiovascular disease (37).
The genes and cell signaling pathways related to the cell cycle, DNA repair cell death, mitochondria, immunity, nutrient signaling and the growth hormone Insulin Growth Factor-1 (IGF-1) mediated via the PI3K/AKT/mTOR (phosphoinositide 3-kinase/AKT (protein kinase B)/mammalian target for rapamycin) pathway have received extensive investigations as targets for anti-aging strategies (36, 52, 53). The potential for biguanides to be used as geroproetctors was described as early as 1980 (54). Metformin via its insulin sensitizing actions reduces insulin and thereby should also normalize IGF-1 levels (55, 56). The effects of metformin on IGF-1 are linked to metformin as an activator of AMP-activated kinase (AMPK) and inhibition of signaling through the mTOR pathway. mTOR is a highly conserved serine-threonine kinase that has important roles in the regulation of cell metabolism, including nutrient signaling and growth mediated by IGF-1 (57, 58). Signaling via mTOR is linked to accelerated aging and dysregulation of mTOR signaling is also linked to the progression of cancer, inflammatory and neurological diseases, as well as T2DM (59). AMPK is a key regulator of many cellular pathways that are linked to both healthspan and lifespan, including the benefits of calorie restriction. Thus, as an activator of AMP metformin has come under the microscope as a potential anti-aging drug and its potential role as an anti-aging drug promoted (36, 41, 60). Worthy of note is that the sensitivity to AMPK declines with age thus promoting the argument that activators of AMPK, such as metformin, could delay aging (61).
The lifespan and healthspan of invertebrates are extended by mutations in pathways such as the DAF-2, a gene that encodes for the IGF-1 pathway in the nematode Caenorhabditis elegans (C. elegans), or the mTOR signaling pathways that are linked to accelerating the aging process (60, 62–65). The addition of metformin to the diet can delay aging and increase life span in both C. elegans and rodents (66–68) (see Table 1). In C. elegans the beneficial effects of metformin on lifespan are, at least in part, argued to be mediated through changing the microbiome and modifying microbial folate and methionine metabolism and potentially due to an anti-oxidant action via transcription factors, SKN-1/Nrf2, a requirement for AMPK and the upstream serine-threonine kinase, liver kinase B1 (LKB1) (67, 71, 86). However, in these studies with C. elegans, very high concentrations of metformin, from 10 to 150 mM, were used (67, 86), and are the equivalent to a dose in humans of approximately 5 kg/day. Such doses in humans would be fatal and one would expect the same in C. elegans where toxicity is offset by the effects of metformin on the microbiome and inhibition of bacterial folate metabolism. Indeed, in the absence of bacteria, metformin shortened lifespan implying that it is the anti-bacterial action of metformin that explains the enhancement of lifespan in C. elegans (67). Furthermore, in old C. elegans all concentrations of metformin, 10, 25 and 50 mM, proved toxic and reduced lifespan (72). The enhanced toxicity to metformin in the older worms was linked to reduced mitochondria abundance and the reduced ability to generate ATP – see Table 1 for details (72).

Table 1 Summaries of studies cited in the review with a focus on the effects of metformin on lifespan.
The same pathways studied in C. elegans have also been investigated in mammals, where lifespan and healthspan were prolonged through modifying the ‘aging’ pathways or through drugs, including metformin (60, 87). As for C. elegans, metformin use in humans will change the microbiome. This action is an important determinant for the anti-hyperglycemic therapeutic effects, the gastrointestinal side effects and possibly the anti-aging effects of metformin (88, 89). Metformin has a bioavailability of approximately 50% with the unabsorbed drug exiting in the stool and following a dose of up to 2550-mg/day; the concentration of metformin in the gut could reach mM levels. In addition, and also via gut-mediated action, metformin enhances the release of glucagon-like peptide-1 (GLP-1). It is the release of GLP-1 that contributes substantially to the antihyperglycemic effects of metformin (26, 90, 91). Additional support for the importance of gut-mediated mechanisms for metformin’s actions is the knowledge that IV administered metformin is ineffective in lowering hyperglycemia and was demonstrated using a hyperglycemic clamp technique to determine the relationship between plasma metformin concentration and hepatic glucose production and disposal in T2DM patients (92).
The recognition of the role of both the gut microbiome and the release of GLP-1 as important contributors to the anti-diabetic effects of metformin has been a significant advance in understanding how metformin mediates its therapeutic actions including the contribution to the variable weight loss that is associated with metformin (93). An additional factor in weight loss is the novel cytokine, Growth Differentiation Factor 15 (GDF15), which is a member of the transforming growth factor β superfamily and also known as macrophage inhibitory cytokine-1 (MIC-1. Secretion of GDF15 has been linked to a number of physiological and pathophysiological functions and expression levels are elevated in response to stress stimuli and associated with stress pathway transcription factors, CHOP (C/EBP homologous Protein) and ATF4 (activating transcription factor 4) (94–96). As a result of an analysis of data from the ORIGIN trial (Outcome Reduction with Initial Glargine Intervention), GDF15 was identified as a biomarker linked to positive CV outcomes and also elevated in those patients receiving metformin (80, 81). The association between metformin and GDF15 was strengthened by data showing that metformin prevented weight gain in high fat fed mice, but not in mice lacking GDF15, or lacking its receptor, glial cell-derived neurotrophic factor family receptor alpha-like (GFRAL), or following treatment with a GRAL antagonist; however, GDF15 was not required for the antihyperglycemic effects of metformin (83). A number of studies have reported a positive correlation between the use of metformin and enhanced levels of GDF15 (97). Of additional interest and based on a proteomic analysis of the plasma from 240 healthy, disease-free, humans in the age range 22-93 years-old, GDF15 was identified as the protein most positively correlated with chronological age and also known to reduce appetite via an action in the hind-brain (82, 95). Collectively, these data suggest a link between the putative anti-aging effects of metformin, weight loss and CV benefits that are distinct from the effects of metformin on glucose homeostasis. GDF15 also signals through the epidermal growth factor (EGF) family of receptors and the PI3K, and AKT signaling pathways raising the question whether an elevation of GDF15 is necessarily protective (94, 98). Elevated levels of GDF15 have been linked to tumor growth and poor prognosis in cancer patients raising the question as to whether GDF15 serves as a tumor suppressor or promoter and a target for the treatment of cancer (84, 85, 99). See Table 1.
AMP-Activated Protein Kinase
AMPK functions as an energy sensor that coordinates multiple protective and energy-conserving signaling pathways including the pathways activated by caloric restriction and referred to as the fuel gauge of the cell by serving as an energy sensor (100, 101) (Figure 1). AMPK is activated through metabolic stress and acts as a cellular regulator of lipid and glucose metabolism (102, 103). Hepatic gluconeogenesis is inhibited by AMPK activation, which also enhances insulin sensitivity, muscle glucose absorption, and fatty acid oxidation (103, 104). Additionally, metformin inhibits the inflammatory response through nuclear factor κB (NFκB) inhibition via pathways involving AMPK (104, 105) (Table 2 and Figure 1). An increase in the activity of AMPK would also explain the protective effects of metformin on endothelial function via the activation of endothelial nitric oxide synthase (eNOS) thereby countering the negative effects of a diabetic milieu on cardiovascular function (119). AMPK inhibits signaling via mTOR and through this action could contribute to the reduced incidence of some cancers that has been associated with the use of metformin: see Table 2. Furthermore, reduced sensitivity to AMPK activation with age or disease could result in reduced healthspan and lifespan (61). Although these data support the argument that metformin enhances both healthspan and lifespan, caution is needed because of the high concentrations of metformin, well above therapeutic levels in humans that have been used in some of the pre-clinical in vitro studies.
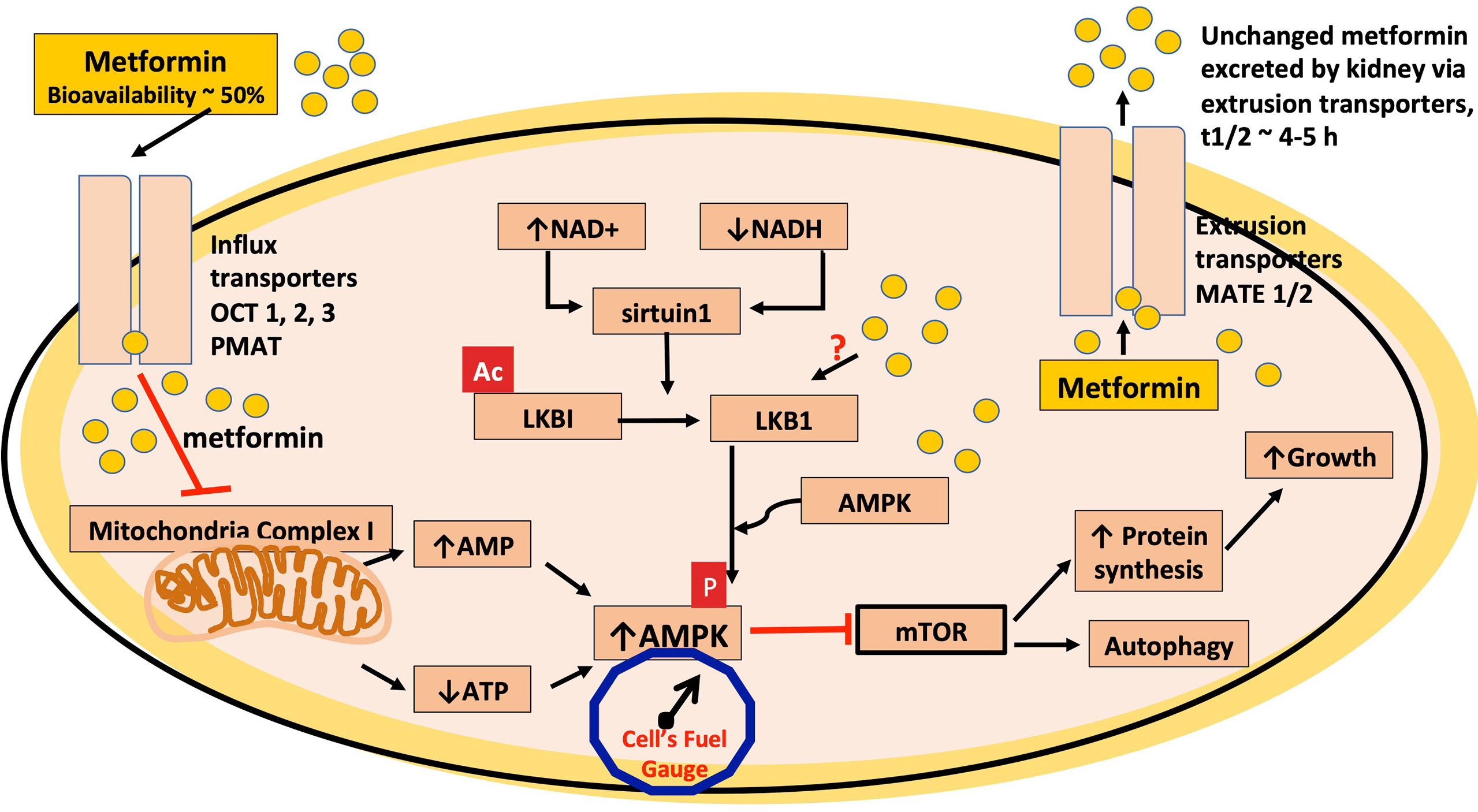
Figure 1 Metformin inhibits mitochondrial complex 1. In this schematic, metformin is transported into the cell via the organic cation transporters, OCT 1, 2 and 3 and the plasma membrane monoamine transporter (PMAT). The transport of metformin out of the cell is mediated through the multidrug and toxin extrusion (MATE1/2) transporters. Metformin is thought to mediate most of its cellular effects via activation of AMPK and, in this schematic, metformin inhibits the electron transport chain of mitochondrial complex 1, which leads to a reduction in ATP levels, increasing the AMP/ATP ratio, thus increasing AMPK activation and also reduces the generation of reactive oxygen species (ROS). AMPK activation leads to an inhibition of the mTOR pathway, which would contribute to the antitumor effects of metformin. Metformin also has been shown to activate AMPK via the serine-threonine liver kinase B1 (LKB1) where phosphorylation (p) (activation) of AMPK occurs. The protein product of SIRT1, sirtuin1, is an upstream deacetylase, which activates LKB1 via deacetylation as indicated in the figure by loss of ac, at times of cellular stress and decreased cellular energy, when NAD+/NADH ratio is high and is also a putative site of action for metformin.
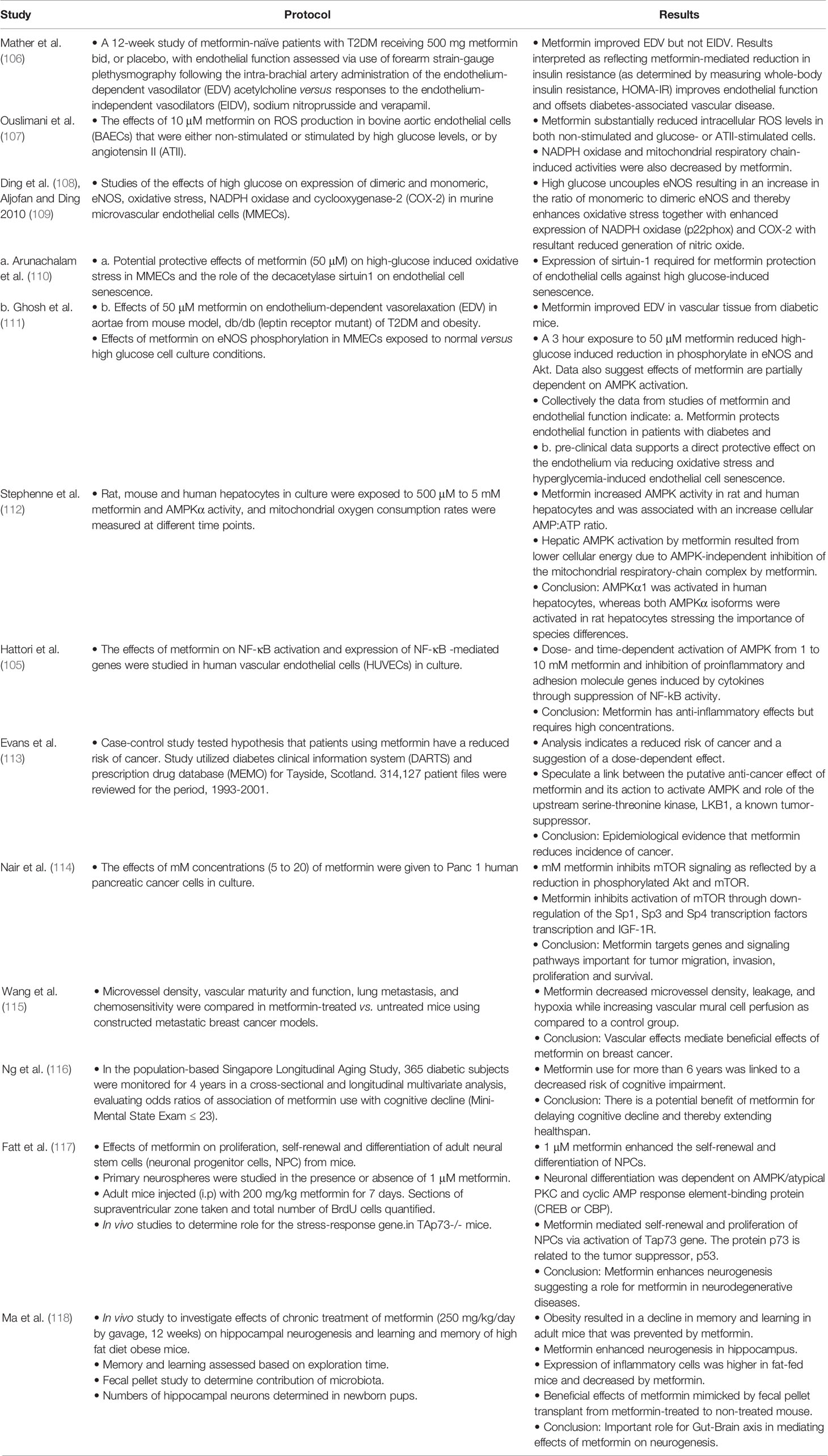
Table 2 Summaries of studies that focus on cellular actions of metformin to activate AMPK, reduce generation of reactive oxygen species, improve vascular function, mediate anti-inflammatory effects and the potential to treat cancer and neurodegenerative diseases.
AMPK is regulated by two upstream serine threonine AMPK kinases: LKB1, and Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) and is also positively regulated by the nicotinamide adenine dinucleotide (NAD+) deacetylase, sirtuin-1. Sirtuin-1 is the protein product of the putative anti-aging gene SIRT1, which targets lysine residues in proteins, including histones and the tumor-suppressors, LKB1 and p53 (120, 121). In endothelial cells, metformin has been shown to enhance phosphorylation and increase the activity of LKB1 (122). Senescence is a major contributor to aging and the development of cardiovascular disease (123), and sirtuin-1 expression is required for metformin to protect endothelial cells against hyperglycemia-induced senescence (110). Furthermore, in silico analysis suggests that metformin could directly activate SIRT1 (124).
Of significance and supporting a potential anti-cancer action of metformin is that LKB1 is a tumor suppressor and that mutations in LKB1 are observed in numerous cancers that result in the reduction of the inhibitory effect of the LKB1/AMPK pathway on the pro-proliferative signaling via mTOR (125, 126). Collectively these data provide a link between metformin, AMPK, LKB1, sirtuin-1, and cellular mechanisms that could enhance both healthspan and lifespan by reducing both cellular senescence and the activation of pro-proliferative pathways.
AMPK as a Target for Metformin
Considerable attention has been placed on the liver and skeletal muscle as the primary sites of action of metformin resulting from the activation of AMPK secondary to an inhibitory action on mitochondrial complex 1. This inhibition would in turn decrease the ATP/AMP ratio activating AMPK (127–129). A small increase in the level of AMP enhances AMPK via three mechanisms: (i) allosteric activation by AMP binding to the Υ subunit (ii) promotion of phosphorylation of Thr-172, and (iii) inhibition of dephosphorylation of Thr-172 (130). Furthermore, an increase in AMP also decreases adenylate cyclase activation and thereby lowers glucagon release (129). This mechanism is an attractive hypothesis to explain how metformin mediates a multitude of cellular effects via AMPK (100). Evidence that metformin targets mitochondrial complex 1 is supported by data from several studies (112). The role of complex 1 inhibition in mediating the effects of metformin is summarized in Figure 1, and much of the published literature assumes this inhibition is indeed the mechanism whereby the drug mediates its effects via AMPK, and the reduction of the generation of ROS from complex 1 (131). However, several other mechanisms to explain the cellular actions of metformin have also been advanced [see (39, 132)].
As pointed out by Fontaine, a problem associated with accepting the argument that the therapeutic effects of metformin result from inhibition of complex 1, as depicted in Figure 1, are the high concentrations used in the cell culture studies described by El Mir et al. and Owen et al. (127, 128, 133). For instance, activation of AMPK by metformin has been observed at much lower metformin concentrations in comparable cell culture protocols using metformin concentrations that are within the upper limits of its therapeutic plasma levels (4, 134). In addition, studies have also reported AMPK/LKB1 independent effects of metformin on gluconeogenesis (135). On the other hand and based on thermodynamic considerations, Owen et al. (128) argue that with a mitochondrial membrane potential of – 180mV metformin could accumulate 1000 fold thus potentially providing a concentration within the mitochondria that would inhibit complex 1 [see also Vial et al. (136)]. Partial support for this hypothesis has been provided by Chien et al. who used a cell culture protocol with human embryonic kidney (HEK) cells that were overexpressed with the organic cation transporter 1 (OCT1) and exposed for 60 minutes to 5 μM 14C-metformin (137). Chien et al. found that higher concentrations of metformin were found in the HEK cells with overexpressed OCT1 versus control HEK cells (268+/-11 vs. 26.4+/-11 μM) (137). On the basis of this enhanced uptake, Chien et al. argue that metformin can be trapped in intracellular organelles such as microsomes and mitochondria (137). In a similar protocol, the in vitro and in vivo effects of metformin on mitochondria function were investigated in vitro in primary murine hepatocytes and Hepa 1-6 cells (murine hepatoma cells derived from the BW7756 tumor) and an in vivo protocol with high-fat diet (HFD) treated mice (138). In the in vitro studies, Hepa1-6 cells were exposed to either 75 or ‘supra-pharmacological’ 1000 μM metformin; however, even following exposure to 1000 μM, no inhibitory effects were seen on the activities of complex I-IV, and the subcellular distribution of metformin indicated that metformin remained primarily in the cytosol with levels in mitochondria less than 70 μM (139). The in vivo arm of the study involved treatment of the HFD mice, including AMPKα1/2 knockout mice, with 50-mg/kg/day metformin in the drinking water and then assaying hepatocytes in vitro with the results indicating that complex 1 activity, mitochondria density and mitochondrial DNA were all increased via an AMPK-dependent mechanism (138). It is also worthy of note that the IC50 for metformin to inhibit mitochondrial complex 1 is 19–66 mM (140, 141).
Collectively these data cast doubt as to the contribution of inhibition of complex 1 as the primary target and explanation for the therapeutic effects of metformin both as an anti-diabetic and as an anti-aging drug. This conclusion is also supported in reviews (133, 142), and by clinical data from T2DM patients treated with metformin that show normal mitochondrial complex 1 respiration in skeletal muscle biopsies (143). As discussed below, it is important to consider the pharmacokinetic properties of metformin when extrapolating data from in vitro studies to potential benefits when used clinically.
Metformin and Mitochondrial Function
Mitochondria play a critical role in oxidative metabolism and a link has been made in patients between obesity, insulin resistance, and defective mitochondria oxidative processes that results in a buildup of toxic intermediate metabolites (144). Several studies have also provided evidence that mitochondrial function is impaired in diabetes and thus the additional insult of metformin-induced dysfunction would be detrimental to patients with T2DM (145, 146), as was seen with the effects of metformin on old (AD10) C. elegans as previously discussed (72). Complex 1 activity in mitochondria from skeletal muscle biopsies was lower in T2DM and obese subjects compared to diabetic lean subjects (147). Although these data do not entirely negate the argument that metformin mediates its anti-hyperglycemic actions via an inhibitory action on mitochondria complex 1, the evidence strongly suggests that further inhibition of function would worsen, and contribute to, the toxic effects of metformin, such as lactic acidosis, that are, albeit very rare (11). As suggested by Wang et al., metformin may play an important role in the ‘quality-control’ regulation of mitochondria and shift the cellular dynamics to a healthy population via mitophagy and eliminate damaged mitochondria (138) [see also (148)]. AMPK has also been implicated in the regulation of mitochondrial biogenesis, thus providing another link between metformin, AMPK, and improved mitochondrial function (149, 150). Mitochondrial function declines with age due either to ROS and/or an accumulation of mutations in mitochondrial DNA (151). If metformin can offset the decline in mitochondrial function, as some studies suggest, then this effect could contribute to enhancing both healthspan and lifespan (138, 149, 150). Interestingly, both metformin and the anti-oxidant, resveratrol, inhibit ROS-associated mitochondria fission (152). Numerous studies have also reported that resveratrol has anti-aging effects in several species with its effects, as also described for metformin, linked to the deacetylase, sirtuin-1 (153). That said, the concentrations of resveratrol present in the diet, e.g. in wines, are far below the amounts that might have any effect on aging. Collectively, these data suggest an important role for metformin in the regulation of mitochondria function that could link to beneficial effects on both healthspan and lifespan.
Pharmacokinetic Properties of Metformin
In order to extrapolate data from in vitro studies to in vivo therapeutic effectiveness, it is important to review the pharmacokinetic properties of metformin (34, 133, 154–156). When prescribed in the usual oral dosage range of 250 to ~2550 mg/day for the treatment of T2DM and in the absence of reduced renal function, it is unlikely that peak plasma levels of metformin would exceed 15-20 μM, with trough levels of between 1-5 μM, and a plasma half-life of approximately 3 to 5 hours. Metformin is a strongly basic hydrophilic drug with a pKa of approximately 11 is not metabolized and is excreted principally by the kidney. The expression levels of the saturable organic cation transporters will determine the rate of absorption of metformin from the gut and the subsequent cellular distribution and excretion of metformin. There are three distinct classes of organic cation-selective transporters that can be utilized by metformin: Organic Cation Transporter (OCT) 1, 2 and 3 (SLC22A1, A2, A3); Plasma membrane Monoamine Transporter (PMAT; SLC29A4); and Multidrug And Toxin Extrusion protein (MATE) 1 and 2 (SLC47A1, A2), to enter and leave cells (Figure 1) (157). A high expression levels of influx transporters in cells might allow a much higher accumulation of metformin resulting in selective toxicity and cell death as has been postulated to explain the anti-cancer effects of the drug (158). However, with a plasma half-life of approximately 3 to 5 hours, and subject to the schedule of dosing, it is unlikely, that other than in gut enterocytes, a significant cellular accumulation of metformin occurs when the drug is used for the treatment of T2DM (34, 155, 156, 159, 160).
It is important that results from in vitro studies using high micromolar and millimolar concentrations of metformin, often for long-exposure times, are looked at critically as such data may not be readily translated to effects when used in humans. A useful comparison of the doses of metformin used therapeutically versus pre-clinical studies was made by Badrick and Renehan, indicating that for pre-clinical studies done in vivo, the doses ranged from x2 to x45 therapeutic doses and that for studies done in vitro, concentrations ranged from x25 to x1000 higher than those observed in patients (161).
Justification for the use of high concentrations of metformin in in vitro studies has been provided. Thus, Onken and Driscoll (71) reported that the anti-aging actions of metformin in C. elegans were only seen with 50 mM metformin and not lower concentrations and stated: “C. elegans has a highly protective cuticle and intestinal lining that generally limit drug uptake such that it is not unusual for polar drugs to be applied at a concentration 1000 fold higher than their predicted affinity for the target; physiological levels of drug in animals are anticipated to be much lower.” [(71); see Table 1]. Although this rationale may be true for C. elegans, the absorption and distribution of metformin in mammals is also dependent on organic cation transporters (OCTs) and therefore a similar argument should be applied to other species unless it can be shown that C. elegans has a very low expression level of influx transporters and high expression of extrusion transporters thus justifying the use of high mM doses. Similarly, others have defended drug levels as matching plasma levels in humans and not acknowledged the short half-life of metformin; a drug that is not metabolized, for which plasma levels rapidly drop as metformin is extruded from cellular sites and excreted by the kidney.
Metformin and Mortality in Diabetic Patients
Data from multiple studies indicate that patients with T2DM who are prescribed metformin have improved survival rates when compared to non-diabetic controls. In a study using retrospective observational data from the UK Clinical Practice Research Datalink, T2DM patients on metformin or sulfonylurea monotherapy were compared to non-diabetic control groups that were age and sex matched (69) (see Table 1). The data show that matched non-diabetics had 15% lower median survival times versus diabetics on metformin monotherapy and that those diabetics on sulfonylurea monotherapy had a 38% lower survival time compared to the metformin group (37, 69). Collectively, these data imply a clear mortality benefit associated with the use of metformin. However, the lower survival rate of those in the sulfonylurea-treated group might reflect a negative effect of sulfonylurea drugs on mortality as has been reported in subsequent studies (162). On the other hand, a recent systematic review concluded that metformin significantly lowered all-cause mortality and cardiovascular events in patients with T2DM and mild/moderate chronic kidney disease (163). Similarly, a meta-analysis with the objective of determining the cardiovascular benefits of metformin in combination with newer anti-diabetic drugs including incretins (GLP-1 agonists), dipeptidyl peptidase-4 (DPP-4) inhibitors, and sodium-glucose co-transporter 2 (SGLT2) inhibitors revealed both positive and neutral effects of adding metformin (164, 165). Nonetheless, in the absence of appropriately designed placebo-driven randomized control studies (RCTs) that include comparison with GLP-1 agonists, DPP-4 and SGLT2 inhibitors, doubt persists if, in fact, metformin does reduce the cardiovascular risk associated with diabetes (166).
Metformin and Endothelial Function and Cardiovascular Disease
The endothelium plays a critical role in the regulation of cardiovascular function and not least as a source of the important signaling molecule, nitric oxide (NO). Furchgott and Zawadski were the first to report that an intact and undamaged endothelium was essential in order for acetylcholine to mediate a vasodilator response (167). We now refer to acetylcholine as an endothelium-dependent vasodilator, and we also recognize that endothelial dysfunction, which can be defined as a reduced vasodilator response to acetylcholine, is an early prognostic indicator for the onset of cardiovascular disease (168–171). Robert Furchgott, together with Louis Ignarro and Ferid Murad, shared the 1998 Nobel Prize for Physiology and Medicine “for their discoveries concerning nitric oxide as a signalling molecule in the cardiovascular system”. Remarkably it was the much earlier microscopy work of Dr. Rudolf Altschul in Saskatoon Canada that recognized the link between hypercholesterolemia and the pathological changes in the endothelium. Based on his comparisons of blood vessels of patients who died from cardiovascular disease versus other causes Dr. Altschul stated in the preface to his 1954 book:
“Because the great majority of heart diseases is caused by coronary thrombosis, which is a lesion of the vessels supplying the heart proper, we may as well cancel the first word in “cardiovascular” and conclude that, in North America most people who die a natural death succumb to a vascular disease. Blood vessels, however, are primarily endothelial tubes with secondary, accessorial walls, and, therefore, it may be postulated that the endothelium has a great importance in our life and that its failure will cause the death of many of us.
It has been said that one is as old as one’s arteries. In view of the supreme importance of the endothelium in arterial function I should like to modify, or rather simplify this statement by saying that one is as old as one’s endothelium” (172).
The obvious question is: “Does metformin exert a direct anti-aging effect on the endothelium?”. Based on critical reviews of the literature, the answer appears to be “yes”, although the clinical data could be interpreted as secondary to metformin’s actions to enhance insulin sensitivity and lower blood glucose levels (34, 106, 173). There is an extensive literature based on data from pre-clinical and clinical studies that indicates metformin protects endothelial function from the effects of diabetes (33). Positive clinical data have been provided by Mather et al. who used forearm strain-gauge plethysmography to assess the effects of metformin on forearm blood flow following the intra-brachial artery administration of acetylcholine versus responses to the endothelium-independent vasodilators, sodium nitroprusside and verapamil, in patients with T2DM (106). The patients, prior to the start of the 12-week trial, were metformin naïve and either received metformin (500 mg, twice each day) or placebo (106). Mather et al. reported that compared to placebo, metformin improved endothelium-dependent, but not endothelium-independent vasodilation thus highlighting the primary dysfunction in blood flow was not the vascular smooth muscle response but was secondary to endothelial dysfunction (106). A larger randomized placebo-controlled study with 390 patients receiving daily doses of between 850 and 2550 mg metformin focused on changes in biomarkers of endothelial dysfunction and patients were followed for 52 months (174). The results complement the plethysmography data reported by Mather et al. (106) with reduced levels of biomarkers of endothelial dysfunction including von Willebrand factor, soluble vascular adhesion molecule-1, tissue type plasminogen activator (t-PA), plasminogen activator inhibitor-1 (PAI-1), but not soluble E-selectin (sE-selectin), or urinary albumin (174). Data from a metabolomics analysis of T2DM patients treated with metformin has indicated that levels of the amino acid citrulline were lower than controls with comparable data from tissues from the db/db mouse model of T2DM and obesity. The metabolomics data can links the lowering of citrulline to the increase in NO generation from arginine and improved eNOS activity, thus providing support to the literature that metformin protects the cardiovascular system via promoting the generation of NO (175).
As previously discussed, metformin directly protects the endothelium from hyperglycemia-induced dysfunction and premature senescence and implicates a crucial role for the SIRT1-derived deacetylase, sirtuin-1 (110, 111). Sirtuin-1 plays an essential role in the regulation of angiogenesis, a protective role against oxidative stress and cardiovascular disease and higher expression levels of sirtuin-1 are associated with reducing disease (176–178). Sirtuin-1 also has been reported to promote endothelial cell proliferation and suppress senescence of porcine primary aortic endothelial cells via a signaling mechanism requiring the expression of the serine-threonine kinase, LKB1, the upstream regulator of AMPK (179). Sirtuin-1 plays a positive role in regulating endothelium-dependent vasodilation via the deacetylation of lysines 496 and 506 on endothelial nitric oxide synthase (eNOS) (180). The positive effects of metformin to offset hyperglycemia-induced endothelial dysfunction reflect an increase in the generation of NO (110, 111). In both studies, Arunachalam et al. (110) and Ghosh et al. (111) used 50 μM metformin and justified this concentration on the basis that estimates of metformin levels in the portal circulation following oral absorption are between 40 and 70 μM (4, 181). Of significance is that much lower concentrations of metformin, between 1 and 10 μM, have also been shown to protect endothelium-dependent vasodilation in murine aortae (182). In their study, Triggle et al. demonstrated a critical role for the orphan nuclear receptor, Nr4a1, in metformin’s endothelial-protective effects as the benefits of metformin were absent in orphan-nuclear-receptor Nr4a1-null murine aorta tissues (182). Furthermore, at the low micromolar concentrations used in this study, neither mitochondrial complex-I nor complex-III oxygen consumption rates were inhibited, but were when 500-μM metformin was used. Collectively, these data suggest that a novel mechanism of action is responsible for mediating the protective effects of metformin in the vasculature (Figure 2).
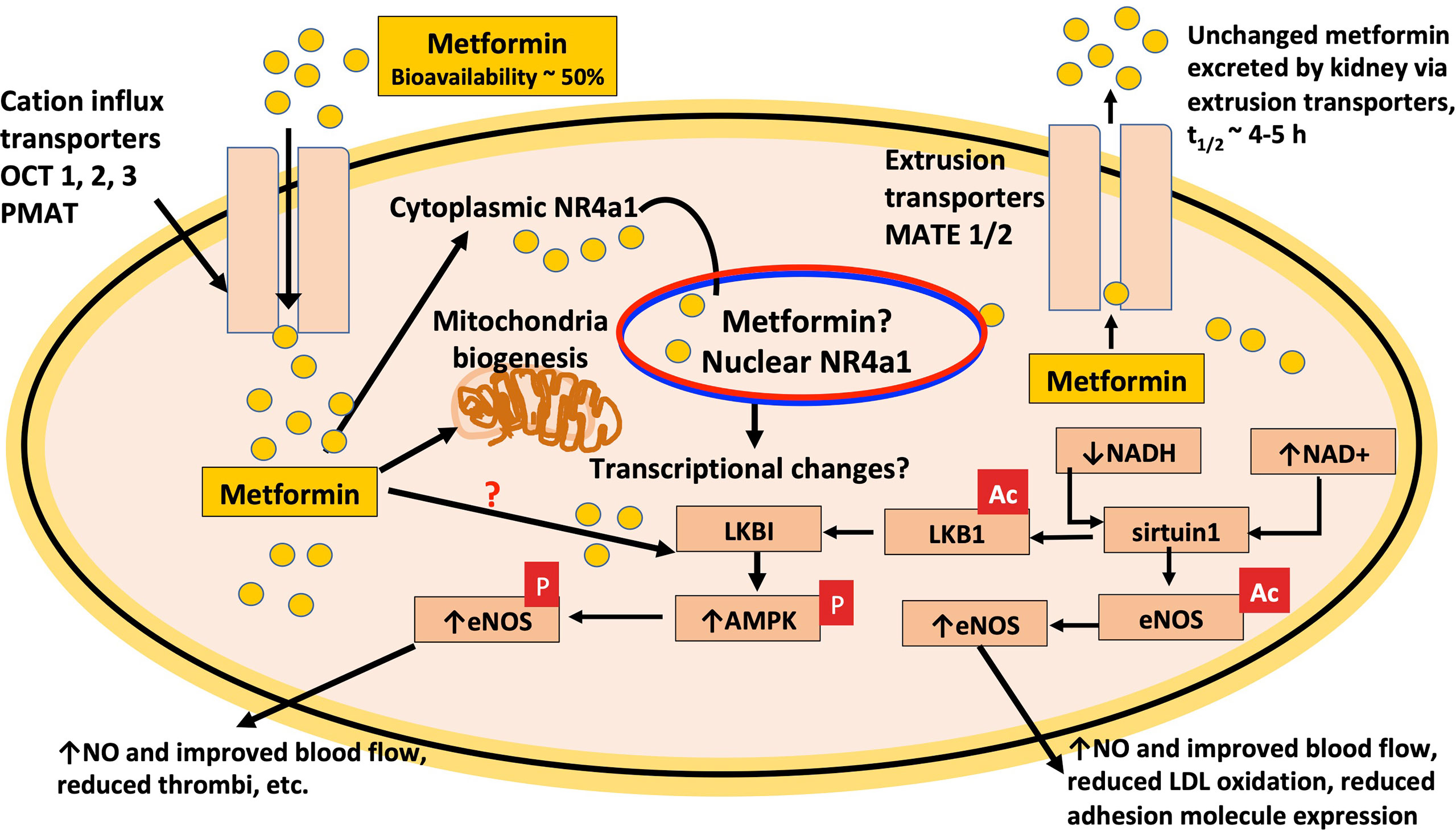
Figure 2 Metformin protects endothelial function. Pre-clinical and clinical data indicates that metformin has direct effects to protect the endothelium from diabetes-induced dysfunction and treatment results in improved function of endothelial nitric oxide synthase (eNOS), the generation of nitric oxide (NO) and improved blood flow that facilitates glucose disposal. Based on in vitro data, the effects of metformin are dependent on the expression of the NAD+ dependent deacetylase, sirtuin1, which targets lysine residues on eNOS as reflected in the figure by removal of ac, the activation of AMPK, and the nuclear orphan receptor, NR4A1. The latter has important links to the regulation of metabolism. OCT3 transporter expression in nuclear membrane facilitates metformin transport into the nucleus (183).
To unravel the precise signaling pathway linking metformin to the NR4A nuclear receptor family requires further studies, but it is worthy of note that the orphan receptor, Nur77, regulates LKB1 localization and is also important for the regulation of glucose uptake into C212 mouse muscle cells (184, 185). Additional evidence that metformin protects mitochondrial function has been provided by Wang et al., reported that treatment of streptozotocin (STZ)-induced diabetic mice with metformin (300 mg/kg/day) in their drinking water for 4 weeks suppressed mitochondrial fission through inhibition of dynamin-related protein (Drp1) via an AMPK-dependent process and enhanced mitochondrial fusion (186). Wang et al. have reported that metformin protects complex 1 activity in a cell culture protocol in hepatocytes but enhances mitochondria fission, thereby promoting healthy mitochondrial function via mitophagy (138).
Metformin and other drugs used for T2DM, like the SGLT2 inhibitors, may also reduce hyperglycemia-induced elevated endothelial ROS, independent of negative effects on complex 1, such as via inhibition of NADPH oxidase or blocking the entry of glucose into the endothelium (32, 107–109, 187, 188) (Table 2). Thus, based on both pre-clinical and clinical data, we can conclude that metformin elicits important protective effects on vascular function that help offset the advance of vascular-related diseases and thereby improves healthspan.
Metformin and Hyperglycemic Memory
Hyperglycemic memory was first described in humans as the resistance to preventing the development of diabetic retinopathy despite achieving good glycemic control in patients (189). Hyperglycemic memory contributes to the pathophysiology of diabetes despite the initiation of intensive glycemic control (190, 191). Data from studies of tissues from streptozotocin-induced diabetic rats and also of endothelial cells in culture indicate that glucose-elevated fibronectin expression is not reversed when normal glycaemia is restored (192).
Metformin has been shown to reverse hyperglycemic memory. In bovine retinal capillary endothelial cells (BRECs) and retinas from diabetic rats, hyperglycemia-induced elevated levels of NF-κB, and Bax, a pro-apoptotic gene, were sustained even after returning to normoglycemia (193). BRECs where SIRT1 was knocked down with siRNA knockdown demonstrated an increased sensitivity to hyperglycemic stress, whereas SIRT1 overexpression, or exposure to metformin, inhibited the increase of mitochondrial ROS by upregulation of LKB1, and suppressed the expression of NF-B and the apoptosis regulator protein, Bax (193). Several other studies using a variety of different cell types have reported that metformin inhibits NF-κB activation, decreases the production of inflammatory cytokines and the genes that code for the inflammatory response thus supporting the healthspan benefits of metformin (105, 194–196).
Exercise, Metformin, and Healthspan
Exercise activates AMPK, which in turn enhances glucose uptake into muscle and improves insulin sensitivity thus helping to offset the negative effects of obesity, diabetes and cardiovascular disease and thereby reducing morbidity and improving healthspan (197). It is important to note that exercise has been shown to have positive effects on the endothelium and, via improved eNOS function, enhances endothelium-dependent vasodilation. Metformin has also been reported to offset the effects of aging in the elderly and in cardiovascular disease (194, 198–200). On the other hand data from the Diabetes Prevention Program study of 3234 pre-diabetic subjects over an average of 2.8 years who were randomized to receive placebo, metformin (850 mg bid), or lifestyle intervention, indicated that although both metformin and lifestyle changes were effective in reducing the risk of developing diabetes, lifestyle intervention was more effective (7).
Since both exercise and metformin can improve glycemic control and since both mediate their effects via the activation of AMPK, this suggests that there should be at least an additive effect when metformin use is combined with exercise. Unfortunately, based on a prospective, double-blinded, randomized, controlled study additive effects of benefits were not observed (201). In this study, men and women with pre-diabetes followed an exercise protocol for 12 weeks with no drug, versus metformin alone (2000 mg/day), versus a combination, or, exercise plus placebo. The results indicated that although both metformin and exercise improved skeletal muscle insulin sensitivity by 55 and 90% respectively the combination resulted in only a 30% enhancement. The results were similar for effects on systolic BP and C-Reactive Protein (CRP) that were reduced by 7 to 8% versus 20-25% respectively (201). In addition, metformin blunted the exercise-induced increase in VO2peak (201). The authors suggest that the negative effect of metformin on exercise results from metformin lowering ROS levels, thus reducing the effects of ROS to activate AMPK. The data suggest that exercise, and not metformin, is the “ideal drug” (201).
Additional doubts about the benefits of combining metformin and exercise come from two studies with older adults. Konopka et al. reported that metformin (2000, or 1500 mg/day for those experiencing GI upset) attenuated exercise induced increases in whole-body insulin sensitivity and also reduced exercise-induced increases in mitochondrial respiration in patients with family history or risk factors for T2DM (202). In the double-blinded Metformin to Augment Strength Training Effective Response in Seniors (MASTERS) trial (203), metformin, despite an increase in AMPK signaling, blunted the exercise-induced hypertrophic response in skeletal muscle in healthy men and women over the age of 65 who took part in a supervised progressive resistance exercise training program over 14 weeks following a 2 week metformin treatment (1700 mg/day, or placebo) (203). Comparable conclusions were reached based on the data from the Look AHEAD randomized intensive lifestyle intervention trial that metformin provided minimal additional benefit (204). Exercise is universally endorsed as the ‘Gold Standard’ for improving cardio-respiratory health and analysis of the benefits of exercise for 26 different chronic diseases reflects positively on “Exercise as Medicine” (205). But the negative effects of metformin on exercise-induced benefits raise concerns about the use of metformin for anything other than for approved diseases such as T2DM.
Calorie Restriction and Nutrient Signaling Pathways
Several genetic mutations, such as in the C. elegans DAF-16 transcription signaling pathway, involving nutrient-sensing pathways have been described and linked to a role in extending lifespan. The data infer the mutations cause a physiological state similar to that experienced during periods of reduced calorie intake (206). Caloric restriction of 10-15% without malnutrition has been suggested to have a role in increasing lifespan in humans (70) (see Table 1). Furthermore, caloric restriction has been found to extend the life span of several organisms including S. cerevisiae (yeast), C. elegans, fish, rodents, and rhesus monkeys (206, 207). Caloric restriction reduces the generation of growth hormone, insulin, IGF1, and other growth factors, all of which have been shown to hasten aging and increase mortality in a number of species (see Figure 3) (206).
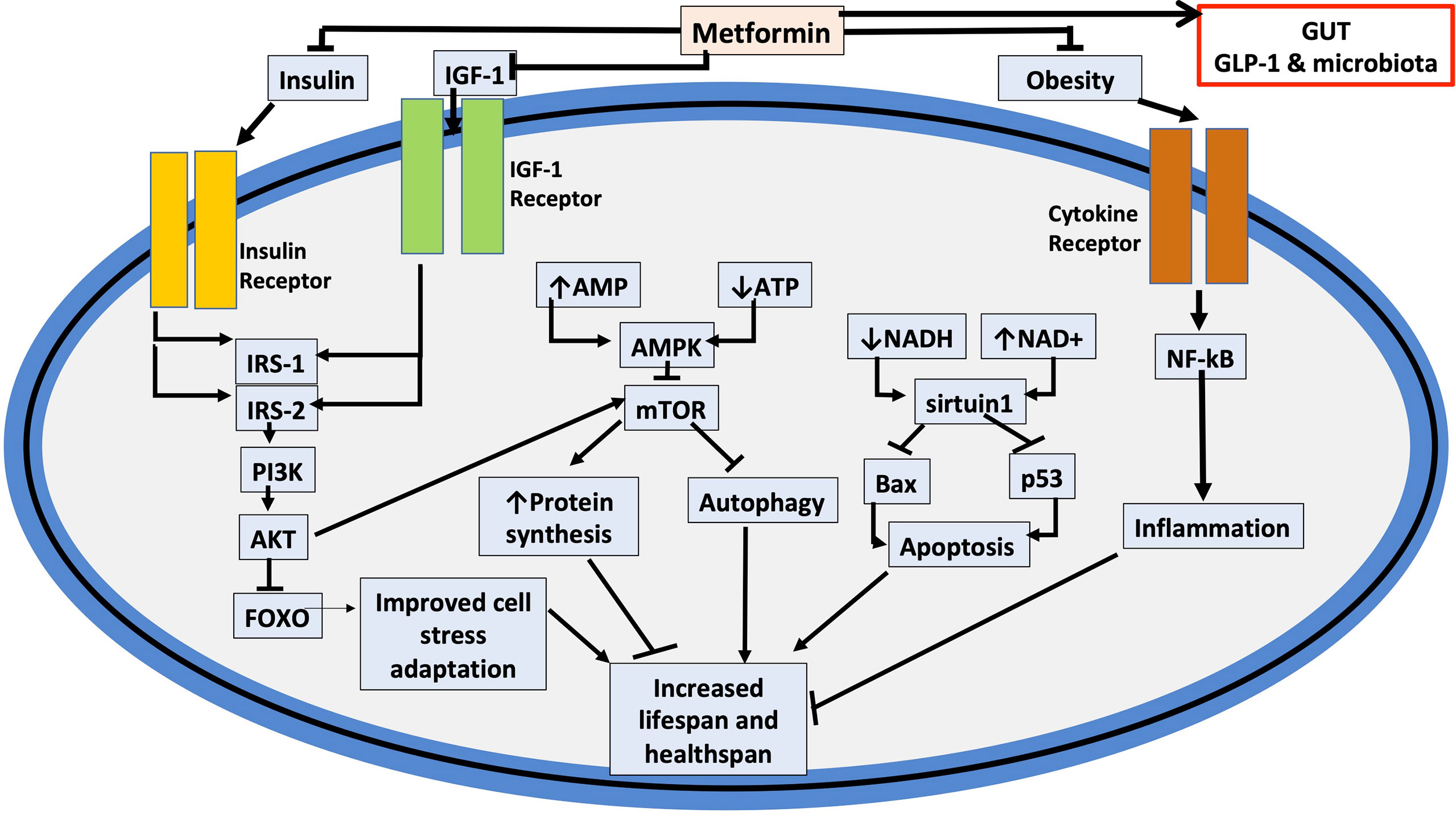
Figure 3 Potential cellular targets for metformin that affect healthspan and lifespan. The figure depicts how metformin may affect cell aging and indicates a potential action in the gut where, prior to absorption, merformin modulates the microbiome as well as enhances release of glucagon-like factor 1 (GLP-1). Important links are also indicated to the insulin (IRS: Insulin Receptor Substrate) and insulin-like growth factor-1 (IGF-1) signaling pathways as well as to tumor suppressors including p53, and inflammation and cytokine signaling. (PI3K: Phosphatidylinositol 3-kinase); (AKT: protein kinase B); (FOXO: Forkhead Box O3); (SIRT1: NAD-dependent deacetylase sirtuin-1); (Bax: Bcl-2-associated X protein). As a result of metformin moderating the cellular signaling pathways mediated by insulin, IGF-1, and cytokines, both, healthspan and lifespan are increased. Metformin also, inhibits the inflammatory pathway and increases AMPK activation, which inhibits mTOR, a primary target for cell aging modulation. Inflammation, apoptosis, autophagy, cell survival, and protein synthesis are all affected by these mechanisms and are all linked to accelerated aging.
A longitudinal study suggested that long-term calorie restriction of 30% significantly reduces age-related deaths in adult rhesus monkeys and a 50% lower incidence of cancer and cardiovascular disease as compared to control animals (208). Apart from lifespan extension, caloric restriction also reduces the risk factors for major diseases including diabetes and cardiovascular disease in rodents (209, 210). Studies by the Calorie Restriction Society, a group of people who chose to limit their calorie consumption with the intention of extending their life span, included adult men and women (mean BMI, 19.6; mean age, 51 years; age range, 35-82 years) whose diet of nutrient-dense foods, consisted of approximately 1800 kcal/day for an average of 6.5 years and consumed 30% fewer calories than age and sex-matched adults on a standard Western diet (210–212). Those on the calorie-restricted diet demonstrated a number of metabolic improvements including body fat, lower blood pressure, and improved insulin sensitivity, and lipid profile (210–212). Meta-analysis has also demonstrated that dietary restrictions decreased levels of circulating IGF-1 in humans (213); however, although much of the evidence indicates that lower levels of IGF-1 benefit enhanced lifespan, IGF-1 does play important roles in homeostasis and not just during childhood thus raising the concern of potential negative effects of excessive lowering of the growth hormone.
Collectively, these observations support the benefits of calorie restriction and heighten the interest in pharmacologic agents, such as metformin, as calorie restriction mimetics; however, there are questions and limitations to address that include [see (214, 215)]: For instance: 1. What level of calorie restriction is required and is acceptable for optimal benefit? 2. How to avoid the effects of severe calorie restriction that can result in malnutrition and adversely affect health particularly in those with low BMI? (214, 216). These questions are important as not all studies have demonstrated a clear relationship between BMI, being overweight, obese and mortality although the risk of CVD is elevated (217). Dietary intervention studies such as with the Women’s Health Initiative RCT have also generated contradictory results with respect to CVD risk (218).
The cellular processes that mediate the benefits of calorie restriction on lifespan in mammalian species remain controversial and although it is tempting to assume an important role for AMPK as a key nutrient sensor, there are a number of caveats that limit a positive correlation in mammalian species versus more convincing evidence in lower eukaryotic organisms, such as C. elegans (219). For instance, as discussed in the next section, despite activating AMPK, metformin has not reproducibly been shown to enhance lifespan in rodents and notably less, or not effective in older animals, including C. elegans (66, 72, 74, 75, 77, 78). In addition, applying data obtained from studies with C. elegans and rodents to intervention studies in a diverse human population raises obvious limitations.
Calorie Restriction Mimetics
The National Institute on Aging Interventions Testing Program has investigated the effectiveness of a variety of pharmacologic agents, including aspirin, metformin, nordihydroguaiaretic acid (NDGA), and rapamycin to determine whether they prolong lifespan in mice (220). Of significance is that metformin, like rapamycin, is known to inhibit mTOR signaling, and the inhibition of the mTOR signaling pathway with rapamycin has been shown to extend lifespan in C. elegans, S. cerevisiae, and Drosophila melanogaster (fruit fly) (221–225). The lifespan-extending effects of metformin have been investigated by Martin-Montalvo et al. in male mice, and indicate that long-term treatment with 0.1 percent metformin w/w supplemented in the diet and beginning in middle age, increased healthspan and lifespan (76) (see Table 1). A higher dose of 1% metformin w/w, on the other hand, was toxic and decreased the average lifespan of mice by 14.4% (76). The effects of metformin were reported to be similar to those of calorie restriction, including improved insulin sensitivity and lower cholesterol levels (76). However, Strong et al. were unable to reproduce the positive lifespan data with the 0.1% metformin protocol in mice, although in combination with rapamycin (14 ppm) lifespan was extended (77). Strong et al. provided the explanation that the insulin-sensitizing effect of metformin offsets the negative effects of rapamycin on glucose homeostasis (77). Smith et al. have demonstrated that calorie restriction, but not metformin (in the diet 300 mg/kg/day), extended lifespan in male Fisher-344 rats, leading to the conclusion that metformin is not a calorie restriction mimetic (CRM) (78) (see Table 1 for a summary).
Collectively, these results demonstrated that the mTOR pathway has a role in lifespan extension in mammals, but raise the question as to why, with the exception of the study by Martin-Montalvo et al., metformin does not extend lifespan in mammals whereas rapamycin does (76). Blagosklonny argued that the metabolic side effects of rapamycin are a consequence of it acting as a CRM and are required to mediate its positive effects on lifespan (226). If we accept this argument and also accept that metformin, despite inhibiting mTOR, is not a CRM (77, 78) then metformin would not be expected to extend lifespan. One caveat to consider for all ageing studies using rodent models relates to the marked genomic differences between rodents and humans in terms of the response to inflammatory disease (227). No doubt, both the innate and adaptive immune responses to inflammation play a key role in the ageing process, and differences in these responses between rodents and humans merit attention. In this regard, metformin may play an important role, due to its potential impact on the innate immune response and the generation of ROS caused by inflammatory cytokines.
Metformin and Autophagy
Autophagy is a process necessary for the removal of damaged proteins and organelles and plays an important role in the regulation of cell aging, providing a supply of nutrients to maintain cellular function during starvation, and inhibition of autophagy mimics accelerated aging (52, 228, 229). Furthermore, calorie restriction is a strong inducer of autophagy and increases the lifespan in C. elegans (230).
Xie et al. investigated the role of chronic AMPK activation by metformin in restoring cardiomyocyte autophagy in OVE26 diabetic mice, a model for type one diabetes (231). Isolated hearts, from the diabetic mice showed a substantial reduction in AMPK function and cardiomyocyte autophagy as well as mitochondria aggregations dispersed between poorly organized myofibrils and increased apoptosis that was reversed following chronic treatment with metformin (231). Song et al. have reported a link between SIRT1, AMPK, and metformin-induced autophagy thereby supporting a synergistic relationship between the deacetylase, sirtuin-1, and metformin-mediated effects on aging (232).
In mice, overexpression of Atg5, the protein product of the essential gene for the autophagosome, boosts autophagy and, more importantly, induces anti-aging phenotypes including enhanced insulin sensitivity and motor control (233). Additionally, embryonic fibroblasts cultured from Atg5 transgenic mice are less affected by oxidative stress-induced cell death, a tolerance reversible by an autophagy inhibitor (233). Furthermore, siRNA targeting Atg5 blocks metformin’s activation of autophagy flux and cell death in adenocarcinoma cells in culture, albeit based on the use of high concentrations (1 to 4 mM and a 12-hour exposure) of metformin (234).
Collectively, data from these studies suggest a link between metformin, autophagy, and extension of lifespan. However, under conditions when tumor microvascular endothelial cells in culture are exposed to glucose starvation, metformin inhibits autophagy via inhibition of the mTOR pathway and a partially AMPK-independent mechanism in (235). The effects of metformin to inhibit autophagy were only seen following a 48-hour incubation with 2 mM metformin, and lower concentrations that are in the therapeutic range, including 50 μM, were ineffective (235). Thus, again the question is raised as to whether effects reported from in vitro studies with mM concentrations of metformin can be translated to a therapeutic effect in humans.
Metformin and a Decreased Incidence of Cancer
Diabetes has been associated with an enhanced risk for the development of various cancers (236). A retrospective study published in 2005 reported that patients with diabetes who had been treated with metformin for T2DM had a lower risk of cancer and highlighted the possible link between metformin and the serine-threonine tumor suppressor, LKB1, as a mechanism for the reduced risk (113) (see Table 2). Similarly, the link between metformin and the activation of AMPK has been emphasized as the basis for the anti-proliferative effects of metformin (43).
Extensive support for a protective effect of metformin against cancer has been provided by numerous, but not all studies (237, 238). For instance, no association has been shown between the use of metformin and a lower incidence of bladder cancer and concerns have also been expressed how data from observational studies are analyzed (239, 240). The National Cancer Institute (https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/metformin-hydrochloride) lists a number of on-going clinical trials involving metformin for the following: Her2 positive breast cancer, head and neck squamous cell cancer, endometrial and ovarian cancers, multiple myeloma, lymphocytic leukemia, and thyroid cancer. Examples include a Phase II study, NCT02028221, designed to determine whether metformin reduces obesity-associated breast cancer risk and due to be completed in mid-2021. A Phase III trial, NCT01101438, “Metformin Versus Placebo on Recurrence and Survival in Early Stage Breast Cancer” is due to be completed in early 2022.
A logical target whereby metformin could mediate its putative antiproliferative effects in cancer is via inhibition of mTOR and the serine-threonine kinase, ribosomal S6K (pS6K), either via activation of AMPK or via an AMPK-independent pathway (241, 242). It has also been argued that the inhibition of mitochondrial complex 1 is an important contributor to the cytotoxic effects of metformin and has been observed in cancer cells and supported by data showing reduced inhibition of tumor growth in cancer cells expressing a metformin-resistant yeast complex 1, NDI1 (136, 243).
The metabolic changes that occur as a result of diabetes (hyperinsulinemia, hyperglycemia, and dyslipidemia) potentiate signaling pathways and may increase the oncogenic nature of breast tissue through accelerating cell growth and migration, angiogenesis, increasing metastasis, and decreasing the response to chemotherapy (158, 244–247). These metabolic pathways, rather than a direct anti-proliferative action, may be the target as Metformin decreases hepatic gluconeogenesis, improves insulin sensitivity, reduces insulin and blood glucose levels, and these effects, which also will reduce tumor growth, rather than a direct anti-proliferative action may be the primary target of metformin (55, 248).
As previously stated when used to treat diabetes peak plasma concentrations of metformin are usually less than 20 μM (155, 160). However, many in vitro studies have used mM concentrations of metformin to demonstrate an anti-proliferative action in tumor cells in culture [see (114, 115); Table 2]. Chandel et al. argue that higher concentrations of metformin are necessary in in vitro cell culture protocols because the abundance of growth factors and nutrients, such as glucose, reduces the sensitivity to the inhibitory effects of metformin thus reflecting the importance of glucose and the Warburg effect for cancer cell growth as well as the importance of glycemic control in diabetes (249). Studies of the effects of glucose concentration on the anti-proliferative effects of metformin on breast cancer cell growth in different human cancer lines In vitro studies reveal that triple negative breast cancer cells (TNBC) are particularly sensitive to the pro-proliferative effects of glucose, and TNBC cells are more sensitive to metformin at lower levels of glucose (250). Zordoky et al. reported that metformin significantly inhibited growth in cells cultured in normoglycemic conditions, and only for the cells grown in normoglycemic conditions did metformin induce significant AMPK activation (251). Samuel et al. have also demonstrated using a cell culture protocol that higher levels of glucose reduce the ability of metformin to inhibit cancer cell proliferation (247). In contrast to the data from in vitro studies, a retrospective analysis of patients with T2DM and TNBC by Bayraktar et al. indicated that treatment with adjuvant metformin was not associated with a significantly improved survival and concluding the need for data from prospective Phase III randomized studies (252).
An argument to explain the selective action of metformin in some cancers but not all is that there is a differential expression of the influx and efflux transporters in tumor cells that allows for the intracellular accumulation of metformin in the cancer cell and resultant selective toxicity (158). Cai et al. who compared metformin uptake levels and inhibiting activity on cancer cell growth in a human breast cancer cell line (BT-20) deficient in OCTs and a BT-20 cell line overexpressing organic cation transporter 3 (OCT3), OCT3-BH20 cells: OCT3 is also a predominant transporter in human breast neoplasms (241). [14C]-Metformin uptake was 13 times higher in OCT3-BT20 cells than in BT-20 cells, and associated with increased AMPK phosphorylation and decreased pS6K phosphorylation in OCT3-BT20 cells and these results were substantiated in mice with OCT3 overexpressing tumors (241). Comparable data has also been reported from a study with a high-fat diet rat model of breast cancer showing a positive association with the expression of OCT2 and accumulation of [D6]-metformin isotope (253). In contrast and although LnCaP, a prostate cancer line, proved to be particularly sensitive to metformin and in a concentration range that was within that expected clinically and correlated with a high expression of mRNA for OCT3 and low expression of MATE2 for the other cell lines, very high concentrations, up to 10mM, of metformin were required to see significant inhibition, and a strong significant correlation between inhibition of proliferation and MATE2 expression was not seen (254). A limitation of the studies investigating expression levels of the cation transporters is that in the absence of adequate specific transporter antibodies quantification of transporter protein was not possible, and correlations were based entirely on mRNA data. Collectively, these data suggest that expression levels of the uptake and extrusion transporters are not necessarily the limiting factors determining the anti-proliferative effects of metformin and that genetically determined variations in signaling pathways are variably important as are nutrient levels in the tumor microenvironment (249, 254).
In conclusion, since beneficial effects of metformin are not seen in all cancers further studies are required to determine whether the anti-cancer actions are direct or are secondary to the positive effects of metformin on healthspan that are apparent in obese patients such as improved glucose homeostasis, enhanced insulin sensitivity and reduced signaling through the IGF-1–mTOR pathway (114, 242), Table 2.
The Connection Between Metformin and Improved Neurological Function
Diabetes-associated hyperglycemia, hyperinsulinemia, elevated oxidative stress, vascular disease, and inflammation are all linked to cognitive decline and, as reflected by a meta-analysis, it was concluded that metformin reduces cognitive decline and dementia in T2DM subjects (255).
In the Singapore Longitudinal Aging Study 2,365 subjects with diabetes (age ≥ 55) were monitored for 4 years – see Table 2 (116). In a cross-sectional and longitudinal multivariate analysis, odds ratios of association of the chronic use of metformin with cognitive disability (Mini-Mental State Exam ≤ 23) were evaluated demonstrating a substantial inverse relationship with cognitive impairment (116). In a large observational study, 67,731 participants, who had no evidence of dementia, were non-diabetic, and aged ≥ 65 were followed from January 2004 to December 2009, to observe the onset of T2DM and compare the risk of the incidence of dementia associated with the use of anti-diabetic drugs and reported that metformin reduced the risk of developing dementia (256). In another clinical trial, 58 participants who had both depression and T2DM received either metformin or placebo for 24 weeks concluded that metformin improved cognitive performance (257).
A molecular basis for the effects of metformin on cognitive function is suggested by data using adult murine neural stem cells in culture that shows metformin in the concentration range 500 nM to 1 μM enhanced proliferation and self renewal dependent on the transcription factor, Tap73, and enhanced neuronal differentiation via the AMPK-atypical protein kinase C (aPKC)-CREB-binding protein (CBP) pathway [(117): Table 2]. Data from a study with high-fat diet mice has also linked the learning and memory restorative benefits of metformin in the diet (250 mg/kg/day) to the microbiota with positive data from a fecal transplantation protocol [(118): Table 2]. The potential to use metformin to treat Alzheimer’s is also provided by data indicating that metformin targets monoacylglycerol lipase, an enzyme responsible for generating pro-inflammatory eicosanoids from 2-arachidonoyl glycerol (258). An overlooked hypothesis is that the ability of metformin to enhance CNS vascular function, as it does in the periphery (106), may also contribute to improved CNS function.
Clinical Trials to Assess Effects of Metformin on Aging, Healthspan, and Lifespan
Designed as a crossover study, the Metformin in Longevity Study (MILES) is a double-blinded study where the subjects act as their own placebo control group [(79) Table 1]. MILES (https://clinicaltrials.gov/ct2/show/NCT02432287) commenced October 2014 and was conducted on 14 elderly participants with impaired glucose tolerance to determine whether metformin (1700 mg/day) can cause physiological and transcriptomic changes in muscle and adipose tissues after 6 weeks of treatment and also to determine which pathways are affected by metformin and outline possible molecular intermediates involved in metformin’s mechanism of action (79). Data from the MILES trial indicate that metformin modified multiple pathways associated with aging including metabolic pathways, collagen trimerization and extracellular matrix (ECM) remodeling, adipose tissue and fatty acid metabolism, mitochondria, and the MutS genes, MSH2 and MSH3, which play a role in DNA mismatch repair, a process that declines with age (79).
Dysregulation of adipose tissue metabolism has been previously linked to an age‐related process of ECM deposition and the effects of metformin on fatty acid metabolism have been previously shown to mimic those of other interventions that increase lifespan in model organisms (36, 259, 260). The results of the MILES study underscore metformin’s targeting of multiple mechanisms of aging. However, it is important to note a concluding sentence from Kulkarni et al.: “These findings remain to be validated in other tissues and study designs and do not yet allow us to identify the primary site of action of metformin, which then may trigger the observed changes in gene expression” (79).
Targeting Aging with Metformin (TAME) trial is a double-blinded, placebo-controlled, multicenter trial that is planned to involve 14 research centers in the USA, and subject to funding and approval, will enroll 3000 ethnically diverse, non-diabetic subjects aged 65–80 (60, 261). The objectives of TAME are: 1. Clinical outcomes as measured by the appearance of new age-related chronic diseases; 2. Functional outcomes such as changes in mobility as measured by gait speed over 10 meters [also see (262)], as well as measures of cognitive impairment; 3. Biomarkers of aging such as for inflammation and senescence (261). The study plan for TAME is that patients will be given a daily dose of metformin (1500 mg) for 6 years, with an estimated follow-up period of more than 3.5 years (261). It is argued that TAME trial outcomes will give more insight on whether metformin decreases the risk of developing age-dependent diseases, excluding diabetes, in non-diabetic individuals, and potentially provide a tool to target aging itself and not related diseases individually (60, 261). However, as expressed both in this review and by others there are concerns about the age –dependent effects of metformin and in older organisms, including C.elegans, rodents and humans, the effects of metformin are variable and may be detrimental [see (202)]. It is also worthy of citing a concluding statement and caution by Pyrkov et al. from their dataset analysis of more than 500,000 people from Russia, UK and USA to assess biological age and the potential for extending lifespan: “The proximity of the critical point revealed in this work indicates that the apparent human lifespan limit is not likely to be improved by therapies aimed against specific chronic diseases or frailty syndrome.” (263).
A number of other clinical trials are underway that address some of the concerns noted in this review: NCT04264897 (Does Insulin Sensitivity Impact the Potential of Metformin to Slow Aging) due to be completed in April 2024, which is a randomized, placebo driven, double-blinded Phase 3 study with 148 participants and the objective to compare the effects of metformin on insulin sensitivity and mitochondria function in patients who are insulin-sensitive versus those who are insulin resistant. NCT02570672 (‘Metformin for Preventing Frailty in High-risk Older Adults’) is a Phase 2 study involving 120 subjects aged 65 to 90 years old with pre-diabetes and treated with 1000 mg metformin bid, versus placebo, and due for completion in October 2024. Frailty will be determined using a standardized assessment (264). A Phase 1 study already underway, Role of Metformin on Muscle Health of Older Adults ‘(NCT03107884), and due for completion in April 2022 is designed to determine whether metformin treatment of elderly subjects offsets the negative effects of bed rest on lipid accumulation, inflammation, insulin resistance, and muscle loss. Collectively the results from these studies will be valuable in better assessing the benefits of metformin on healthspan and also fine-tuning future larger studies such as TAME.
Conclusions
Based on a 60-year history of use as an anti-diabetic drug for the treatment of T2DM, metformin is accepted as a comparatively safe drug. Metformin is no longer protected by patents and thus is comparatively inexpensive. Collectively, these attributes together with an extensive literature supportive of benefits in the settings of diabetes, obesity, cardiovascular disease and, arguably, cancer and dementia could justify its wider use as a prophylactic to offset the effects of aging and enhance healthspan and lifespan. In this review we have also highlighted and critiqued some of the key clinical and laboratory-based studies that provide data supportive of the hypothesis that metformin, independent of its anti-hyperglycemic actions, has benefits that in principle can slow cellular aging and enhance healthspan and lifespan. Metformin, via its direct protective effects on vascular function, may slow the aging process via improved blood flow and provide protection against age-related cognitive decline. However, not all of the data is supportive and metformin, as shown in C. elegans and mice, may be less effective, or ineffective, in older humans. We have also stressed that, based on the pharmacokinetic properties of metformin, caution is needed before extrapolating from in vitro cell-based studies done with comparatively high metformin concentrations to clinical effectiveness with plasma concentrations in the range of 20 micromolar or lower. This issue is a particular concern with the in vitro studies reporting an anti-proliferative effect of metformin and promoting its use as an adjunct for the treatment of various cancers as, in most of these studies, the concentration/dose of metformin that have been used would most likely prove toxic in patients. Furthermore, a dependence on the use of metformin as a prophylactic to delay aging could serve to decrease the incentive to pursue the proven benefits of lifestyle changes such as improved diet and exercise. Moreover, the long-term chronic use of metformin would require attention to the potential occurrence of vitamin B12 deficiency. On this basis, we conclude that metformin should not be seen as a ‘quick fix’ panacea for aging at the expense of non-pharmacologic interventions such as diet, exercise, and related lifestyle changes. Indeed, the use of metformin may negate some of the positive effects of exercise and lifestyle and less favorable effects in older subjects as was also emphasized by the Diabetes Prevention Program (265, 266). On the more positive side, we do accept that the use of metformin in the treatment of patients with T2DM is associated with a positive benefit on healthspan. By lowering plasma glucose levels and body weight, metformin improves the metabolic profile of the patient and thereby reduces the severity and risk of other diseases associated with diabetes such as cardiovascular, cancer, and also neurodegenerative diseases (267). The importance of the gut-brain axis in mediating the therapeutic effects of metformin is also emphasized, as are the potential beneficial effects of metformin to protect against neurodegenerative disorders. Finally, although the evidence for lifespan expansion in mammalian species is not conclusive, a full analysis and follow-up of clinical trials, including MILES and TAME, may provide more definitive answers as to whether metformin should be promoted beyond its use to treat T2DM, as a drug that enhances both healthspan and lifespan. Of particular importance is the need for evidence from prospective studies of the effects of metformin on subjects of different age groups, free of chronic diseases, which will help determine if metformin has benefits beyond those of reducing pre-existing disease burden.
Author Contributions
IM and CT participated in the manuscript conceptualization and writing the first draft. HD, MH, IM, and CT contributed equally to literature and manuscript review and revisions of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The publication of this article was funded by the Department of Medical Education, Weill Cornell Medicine.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rojas LBA, Gomes MB. Metformin: An Old But Still the Best Treatment for Type 2 Diabetes. Diabetol Metab Syndr (2013) 5:6. doi: 10.1186/1758-5996-5-6
3. Bailey C, Day C. Metformin: Its Botanical Background. Pract Diabetes Int (2004) 21:115–7. doi: 10.1002/pdi.606
4. He L, Wondisford FE. Metformin Action: Concentration Matters. Cell Metab (2015) 21:159–62. doi: 10.1016/j.cmet.2015.01.003
5. Dujic T, Zhou K, Donnelly LA, Tavendale R, Palmer CNA, Pearson ER. Association of Organic Cation Transporter 1 With Intolerance to Metformin in Type 2 Diabetes: A GoDARTS Study. Diabetes (2015) 64:1786–93. doi: 10.2337/db14-1388
6. McCreight LJ, Bailey CJ, Pearson ER. Metformin and the Gastrointestinal Tract. Diabetologia (2016) 59:426–35. doi: 10.1007/s00125-015-3844-9
7. The Diabetes Prevention Program Research Group. Long-Term Safety, Tolerability, and Weight Loss Associated With Metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care (2012) 35:731–7. doi: 10.2337/dc11-1299
8. Inzucchi SE, Lipska KJ, Mayo H, Bailey CJ, McGuire DK. Metformin in Patients With Type 2 Diabetes and Kidney Disease: A Systematic Review. JAMA (2014) 312:2668. doi: 10.1001/jama.2014.15298
9. Kalantar-Zadeh K, Kovesdy CP. Should Restrictions Be Relaxed for Metformin Use in Chronic Kidney Disease? No, We Should Never Again Compromise Safety! Diabetes Care (2016) 39:1281–6. doi: 10.2337/dc15-2327
10. Bailey CJ, Mynett KJ, Page T. Importance of the Intestine as a Site of Metformin-Stimulated Glucose Utilization. Br J Pharmacol (1994) 112:671–5. doi: 10.1111/j.1476-5381.1994.tb13128.x
11. Rifkin SI, McFarren C, Juyyadi R, Weinstein SS. Prolonged Hemodialysis for Severe Metformin Intoxication. Renal Failure (2011) 33:459–61. doi: 10.3109/0886022X.2011.568132
12. Duong JK, Furlong TJ, Roberts DM, Graham GG, Greenfield JR, Williams KM, et al. The Role of Metformin in Metformin-Associated Lactic Acidosis (MALA): Case Series and Formulation of a Model of Pathogenesis. Drug Saf (2013) 36:733–46. doi: 10.1007/s40264-013-0038-6
13. Flory JH, Hennessy S. Metformin Use Reduction in Mild to Moderate Renal Impairment: Possible Inappropriate Curbing of Use Based on Food and Drug Administration Contraindications. JAMA Intern Med (2015) 175:458–9. doi: 10.1001/jamainternmed.2014.6936
14. Bakris GL, Molitch ME. Should Restrictions Be Relaxed for Metformin Use in Chronic Kidney Disease? Yes, They Should Be Relaxed! What’s the Fuss? Diabetes Care (2016) 39:1287–91. doi: 10.2337/dc15-2534
15. Huang W, Castelino RL, Peterson GM. Lactate Levels With Chronic Metformin Use: A Narrative Review. Clin Drug Investig (2017) 37:991–1007. doi: 10.1007/s40261-017-0564-6
16. de Jager J, Kooy A, Lehert P, Wulffelé MG, van der Kolk J, Bets D, et al. Long Term Treatment With Metformin in Patients With Type 2 Diabetes and Risk of Vitamin B-12 Deficiency: Randomised Placebo Controlled Trial. BMJ (2010) 340:c2181. doi: 10.1136/bmj.c2181
17. Kos E, Liszek MJ, Emanuele MA. Effect of Metformin Therapy on Vitamin D and Vitamin B(1)(2) Levels in Patients With Type 2 Diabetes Mellitus. Endocr Pract (2012) 18:179–84. doi: 10.4158/EP11009.OR
18. Aroda VR, Edelstein SL, Goldberg RB, Knowler WC, Marcovina SM, Orchard TJ, et al. Long-Term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab (2016) 101:1754–61. doi: 10.1210/jc.2015-3754
19. Elhadd T, Ponirakis G, Dabbous Z, Siddique M, Chinnaiyan S, Malik RA. Metformin Use Is Not Associated With B12 Deficiency or Neuropathy in Patients With Type 2 Diabetes Mellitus in Qatar. Front Endocrinol (2018) 9:248. doi: 10.3389/fendo.2018.00248
20. Reinstatler L, Qi YP, Williamson RS. Association of Biochemical B(1)(2) Deficiency With Metformin Therapy and Vitamin B(1)(2) Supplements: The National Health and Nutrition Examination Survey, 1999-2006. Diabetes Care (2012) 35:327–33. doi: 10.2337/dc11-1582
21. Lord J, Thomas R, Fox B, Acharya U, Wilkin T. The Effect of Metformin on Fat Distribution and the Metabolic Syndrome in Women With Polycystic Ovary Syndrome–A Randomised, Double-Blind, Placebo-Controlled Trial. BJOG (2006) 113:817–24. doi: 10.1111/j.1471-0528.2006.00966.x
22. World Health Organization. Cardiovascular diseases. Available at: https://www.who.int/westernpacific/health-topics/cardiovascular-diseases (Accessed May 10, 2021).
23. Kaeberlein M. How Healthy Is the Healthspan Concept? GeroScience (2018) 40:361–4. doi: 10.1007/s11357-018-0036-9
24. UKPDS 34: UK Prospective Diabetes Study (UKPDS) Group. Effect of Intensive Blood-Glucose Control With Metformin on Complications in Overweight Patients With Type 2 Diabetes (UKPDS 34. Lancet (1998) 352:854–65. doi: 10.1016/S0140-6736(98)07037-8
25. Duca FA, Côté CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM, et al. Metformin Activates a Duodenal Ampk-Dependent Pathway to Lower Hepatic Glucose Production in Rats. Nat Med (2015) 21:506–11. doi: 10.1038/nm.3787
26. Buse JB, DeFronzo RA, Rosenstock J, Kim T, Burns C, Skare S, et al. The Primary Glucose-Lowering Effect of Metformin Resides in the Gut, Not the Circulation: Results From Short-Term Pharmacokinetic and 12-Week Dose-Ranging Studies. Diabetes Care (2016) 39:198–205. doi: 10.2337/dc15-0488
27. Romero R, Erez O, Huttemann M, Maymon E, Panaitescu B, Conde-Agudelo A, et al. Metformin, the Aspirin of the 21st Century: Its Role in Gestational Diabetes Mellitus, Prevention of Preeclampsia and Cancer, and the Promotion of Longevity. Am J Obstet Gynecol (2017) 217:382–02. doi: 10.1016/j.ajog.2017.06.003
28. Glossmann HH, Lutz OMD. Metformin and Aging: A Review. Gerontol nd (2019) 65(6):581–90. doi: 10.1159/000502257
29. Vallianou NG, Stratigou T, Tsagarakis S. Metformin and Gut Microbiota: Their Interactions and Their Impact on Diabetes. Hormones (Athens) (2019) 18:141–4. doi: 10.1007/s42000-019-00093-w
30. Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HAW. 10-Year Follow-Up of Intensive Glucose Control in Type 2 Diabetes. N Engl J Med (2008) 359:1577–89. doi: 10.1056/NEJMoa0806470
31. Johnson J, Simpson S, Toth E, Majumdar S. Reduced Cardiovascular Morbidity and Mortality Associated With Metformin Use in Subjects With Type 2 Diabetes. Diabetes Med (2005) 22:497–502. doi: 10.1111/j.1464-5491.2005.01448.x
32. Nesti L, Natali A. Metformin Effects on the Heart and the Cardiovascular System: A Review of Experimental and Clinical Data. Nutr Metab Cardiovasc Dis (2017) 27:657–69. doi: 10.1016/j.numecd.2017.04.009
33. Nafisa A, Gray SG, Cao Y, Wang T, Xu S, Wattoo FH, et al. Endothelial Function and Dysfunction: Impact of Metformin. Pharmacol Ther (2018) 192:150–62. doi: 10.1016/j.pharmthera.2018.07.007
34. Triggle CR, Ding H, Marei I, Anderson TJ, Hollenberg MD. Why the Endothelium? The Endothelium as a Target to Reduce Diabetes-Related Associated Vascular Disease. Can J Physiol Pharmacol (2020) 98:415–30. doi: 10.1139/cjpp-2019-0677
35. Kalyani RR. Glucose-Lowering Drugs to Reduce Cardiovascular Risk in Type 2 Diabetes. N Engl J Med (2021) 384:1248–60. doi: 10.1056/NEJMcp2000280
36. Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The Critical Role of Metabolic Pathways in Aging. Diabetes (2012) 61:1315–22. doi: 10.2337/db11-1300
37. Campbell JM, Bellman SM, Stephenson MD, Lisy K. Metformin Reduces All-Cause Mortality and Diseases of Ageing Independent of Its Effect on Diabetes Control: A Systematic Review and Meta-Analysis. Ageing Res Rev (2017) 40:31–44. doi: 10.1016/j.arr.2017.08.003
38. Podhorecka M, Ibanez B, Dmoszyńska A. Metformin – Its Potential Anti-Cancer and Anti-Aging Effects. Postepy Hig Med Dosw (Online) (2017) 2;71(0):170–5. doi: 10.5604/01.3001.0010.3801
39. Glossmann HH, Lutz OMD. Pharmacology of Metformin – An Update. Eur J Pharmacol (2019) 865:172782. doi: 10.1016/j.ejphar.2019.172782
40. Glossmann HH, Lutz O. Is the Classical Oral Antidiabetic Metformin the Novel Miracle Drug to Combat Aging? Biochem (Lond) (2021) 43:48–56. doi: 10.1042/bio_2021_118
41. Kulkarni AS, Gubbi S, Barzilai N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab (2020) 32:15–30. doi: 10.1016/j.cmet.2020.04.001
42. Soukas AA, Hao H, Wu L. Metformin as Anti-Aging Therapy: Is It for Everyone? Trends Endocrinol Metab (2019) 30:745–55. doi: 10.1016/j.tem.2019.07.015
43. Anwar MA, Kheir WA, Eid S, Fares J, Liu X, Eid AH, et al. Colorectal and Prostate Cancer Risk in Diabetes: Metformin, an Actor Behind the Scene. J Cancer (2014) 5:736–44. doi: 10.7150/jca.9726
44. Vera IM, Grilo Ruivo MT, Lemos Rocha LF, Marques S, Bhatia SN, Mota MM, et al. Targeting Liver Stage Malaria With Metformin. JCI Insight (2019) 4:e127441. doi: 10.1172/jci.insight.127441
45. Malhotra A, Hepokoski M, McCowen KC, Y-J Shyy J. ACE2, Metformin, and COVID-19. IScience (2020) 23:101425. doi: 10.1016/j.isci.2020.101425
46. Matsuoka Y, Morimoto S, Fujishiro M, Hayakawa K, Kataoka Y, Suzuki S, et al. Metformin Repositioning in Rheumatoid Arthritis. Clin Exp Rheumatol (2020) 39:763–8.
47. Masadeh MM, Alzoubi KH, Masadeh MM, Aburashed ZO. Metformin as a Potential Adjuvant Antimicrobial Agent Against Multidrug Resistant Bacteria. Clin Pharmacol: Adv Appl (2021) 13:83–90. doi: 10.2147/CPAA.S297903
48. Samuel SM, Varghese E, Busselberg D. Therapeutic Potential of Metformin in COVID-19: Reasoning for Its Protective Role. Trends Microbiol (2021) S0966-842X(21):00063–9. doi: 10.1016/j.tim.2021.03.004
49. Begley CG, Ioannidis JPA. Reproducibility in Science: Improving the Standard for Basic and Preclinical Research. Circ Res (2015) 116:116–26. doi: 10.1161/CIRCRESAHA.114.303819
50. Baker M. 1,500 Scientists Lift the Lid on Reproducibility. Nature (2016) 533:452–4. doi: 10.1038/533452a
51. Nichols JD, Oli MK, Kendall WL, Boomer GS. Opinion: A Better Approach for Dealing With Reproducibility and Replicability in Science. Proc Natl Acad Sci USA (2021) 118:e2100769118. doi: 10.1073/pnas.2100769118
52. de Cabo R, Carmona-Gutierrez D, Bernier M, Hall MN, Madeo F. The Search for Antiaging Interventions: From Elixirs to Fasting Regimens. Cell (2014) 157:1515–26. doi: 10.1016/j.cell.2014.05.031
53. Kowalczyk A, Partha R, Clark NL, Chikina M. Pan-Mammalian Analysis of Molecular Constraints Underlying Extended Lifespan. ELife (2020) 9:e51089. doi: 10.7554/eLife.51089
54. Anisimov VN. Effect of Buformin and Diphenylhydantoin on the Life Span, Estrous Function and Spontaneous Tumor Incidence in Rats. Voprosy Onkologii (1980) 26:42–8.
55. Dowling RJO, Niraula S, Chang MC, Done SJ, Ennis M, McCready DR, et al. Changes in Insulin Receptor Signaling Underlie Neoadjuvant Metformin Administration in Breast Cancer: A Prospective Window of Opportunity Neoadjuvant Study. Breast Cancer Res: BCR (2015) 17:32. doi: 10.1186/s13058-015-0540-0
56. Yang X, Kord-Varkaneh H, Talaei S, Clark CCT, Zanghelini F, Tan SC, et al. The Influence of Metformin on IGF-1 Levels in Humans: A Systematic Review and Meta-Analysis. Pharmacol Res (2020) 151:104588. doi: 10.1016/j.phrs.2019.104588
57. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
58. Johnson SC. Nutrient Sensing, Signaling and Ageing: The Role of IGF-1 and mTOR in Ageing and Age-Related Disease. In: Harris JR, Korolchuk VI, editors. Biochemistry and Cell Biology of Ageing: Part I Biomedical Science, vol. 90. Singapore: Springer Singapore (2018). p. 49–97. doi: 10.1007/978-981-13-2835-0_3
59. Dazert E, Hall MN. mTOR Signaling in Disease. Curr Opin Cell Biol (2011) 23:744–55. doi: 10.1016/j.ceb.2011.09.003
60. Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a Tool to Target Aging. Cell Metab (2016) 23:1060–5. doi: 10.1016/j.cmet.2016.05.011
61. Salminen A, Kaarniranta K. AMP-Activated Protein Kinase (AMPK) Controls the Aging Process via an Integrated Signaling Network. Ageing Res Rev (2012) 11:230–41. doi: 10.1016/j.arr.2011.12.005
62. Friedman DB, Johnson TE. Three Mutants That Extend Both Mean and Maximum Life Span of the Nematode, Caenorhabditis Elegans, Define the Age-1 Gene. J Gerontol (1988) 43:B102–9. doi: 10.1093/geronj/43.4.B102
63. Bartke A, Wright JC, Mattison JA, Ingram DK, Miller RA, Roth GS. Extending the Lifespan of Long-Lived Mice. Nature (2001) 414:412. doi: 10.1038/35106646
64. Blagosklonny MV. TOR-Driven Aging: Speeding Car Without Brakes. Cell Cycle (2009) 8:4055–9. doi: 10.4161/cc.8.24.10310
65. Vitale G, Pellegrino G, Vollery M, Hofland LJ. ROLE of IGF-1 System in the Modulation of Longevity: Controversies and New Insights From a Centenarians’ Perspective. Front Endocrinol (2019) 10:27. doi: 10.3389/fendo.2019.00027
66. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, et al. Metformin Slows Down Aging and Extends Life Span of Female SHR Mice. Cell Cycle (2008) 7:2769–73. doi: 10.4161/cc.7.17.6625
67. Cabreiro F, Au C, Leung K-Y, Vergara-Irigaray N, Cochemé HM, Noori T, et al. Metformin Retards Aging in C. Elegans by Altering Microbial Folate and Methionine Metabolism. Cell (2013) 153:228–39. doi: 10.1016/j.cell.2013.02.035
68. De Haes W, Frooninckx L, Van Assche R, Smolders A, Depuydt G, Billen J, et al. Metformin Promotes Lifespan Through Mitohormesis via the Peroxiredoxin PRDX-2. Proc Natl Acad Sci USA (2014) 111:E2501–9. doi: 10.1073/pnas.1321776111
69. Bannister CA, Holden SE, Jenkins-Jones S, Morgan CL, Halcox JP, Schernthaner G, et al. Can People With Type 2 Diabetes Live Longer Than Those Without? A Comparison of Mortality in People Initiated With Metformin or Sulphonylurea Monotherapy and Matched, Non-Diabetic Controls. Diabetes Obes Metab (2014) 16:1165–73. doi: 10.1111/dom.12354
70. Willcox BJ, Willcox DC. Caloric Restriction, Caloric Restriction Mimetics, and Healthy Aging in Okinawa: Controversies and Clinical Implications. Curr Opin Clin Nutr Metab Care (2014) 17:51–8. doi: 10.1097/MCO.0000000000000019
71. Onken B, Driscoll M. Metformin Induces a Dietary Restriction-Like State and the Oxidative Stress Response to Extend C. elegans Healthspan via AMPK, LKB1 and SKN-1. PloS One (2010) 5:8758. doi: 10.1371/journal.pone.0008758
72. Espada L, Dakhovnik A, Chaudhari P, Martirosyan A, Miek L, Poliezhaieva T, et al. Loss of Metabolic Plasticity Underlies Metformin Toxicity in Aged Caenorhabditis Elegans. Nat Metab (2020) 2:1316–31. doi: 10.1038/s42255-020-00307-1
73. Anisimov VN, Semenchenko AV, Yashin AI. Insulin and Longevity: Antidiabetic Biguanides as Geroprotectors. Biogerontology (2003) 4:297–307. doi: 10.1023/A:1026299318315
74. Anisimov VN, Berstein LM, Popovich IG, Zabezhinski MA, Egormin PA, Piskunova TS, et al. If Started Early in Life, Metformin Treatment Increases Life Span and Postpones Tumors in Female SHR Mice. Aging (2011) 3:148–57. doi: 10.18632/aging.100273
75. Alfaras I, Mitchell SJ, Mora H, Lugo DR, Warren A, Navas-Enamorado I, et al. Health Benefits of Late-Onset Metformin Treatment Every Other Week in Mice. NPJ Aging Mech Dis (2017) 3:16. doi: 10.1038/s41514-017-0018-7
76. Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, et al. Metformin Improves Healthspan and Lifespan in Mice. Nat Commun (2013) 4:2192. doi: 10.1038/ncomms3192
77. Strong R, Miller RA, Antebi A, Astle CM, Bogue M, Denzel MS, et al. Longer Lifespan in Male Mice Treated With a Weakly Estrogenic Agonist, an Antioxidant, an α-Glucosidase Inhibitor or a Nrf2-Inducer. Aging Cell (2016) 15:872–84. doi: 10.1111/acel.12496
78. Smith DL, Elam CF, Mattison JA, Lane MA, Roth GS, Ingram DK. Metformin Supplementation and Life Span in Fischer-344 Rats. J Gerontol A Biol Sci Med Sci (2010) 65:468–74. doi: 10.1093/gerona/glq033
79. Kulkarni AS, Brutsaert EF, Anghel V, Zhang K, Bloomgarden N, Pollak M, et al. Metformin Regulates Metabolic and Nonmetabolic Pathways in Skeletal Muscle and Subcutaneous Adipose Tissues of Older Adults. Aging Cell (2018) 17(2):e12723. doi: 10.1111/acel.12723
80. Gerstein HC, Paré G, McQueen MJ, Haenel H, Lee SF, Pogue J, et al. Identifying Novel Biomarkers for Cardiovascular Events or Death in People With Dysglycemia. Circulation (2015) 132:2297–304. doi: 10.1161/CIRCULATIONAHA.115.015744
81. Gerstein HC, Pare G, Hess S, Ford RJ, Sjaarda J, Raman K, et al. Growth Differentiation Factor 15 as a Novel Biomarker for Metformin. Diabetes Care (2017) 40:280–3. doi: 10.2337/dc16-1682
82. Tanaka T, Biancotto A, Moaddel R, Moore AZ, Gonzalez-Freire M, Aon MA, et al. Plasma Proteomic Signature of Age in Healthy Humans. Aging Cell (2018) 17:e12799. doi: 10.1111/acel.12799
83. Coll AP, Chen M, Taskar P, Rimmington D, Patel S, Tadross JA, et al. GDF15 Mediates the Effects of Metformin on Body Weight and Energy Balance. Nature (2020) 578:444–8. doi: 10.1038/s41586-019-1911-y
84. Modi A, Dwivedi S, Roy D, Khokhar M, Purohit P, Vishnoi J, et al. Growth Differentiation Factor 15 and Its Role in Carcinogenesis: An Update. Growth Factors (2019) 37:190–207. doi: 10.1080/08977194.2019.1685988
85. Wischhusen J, Melero I, Fridman WH. Growth/Differentiation Factor-15 (GDF-15): From Biomarker to Novel Targetable Immune Checkpoint. Front Immunol (2020) 11:951. doi: 10.3389/fimmu.2020.00951
86. Wu L, Zhou B, Oshiro-Rapley N, Li M, Paulo JA, Webster CM, et al. An Ancient, Unified Mechanism for Metformin Growth Inhibition in C. Elegans and Cancer. Cell (2016) 167:1705–18. doi: 10.1016/j.cell.2016.11.055
87. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-Induced Insulin Resistance Is Mediated by mTORC2 Loss and Uncoupled From Longevity. Science (2012) 335:1638–43. doi: 10.1126/science.1215135
88. MetaHIT consortium, Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, et al. Disentangling Type 2 Diabetes and Metformin Treatment Signatures in the Human Gut Microbiota. Nature (2015) 528:262–6. doi: 10.1038/nature15766
89. Bryrup T, Thomsen CW, Kern T, Allin KH, Brandslund I, Jørgensen NR, et al. Metformin-Induced Changes of the Gut Microbiota in Healthy Young Men: Results of a Non-Blinded, One-Armed Intervention Study. Diabetologia (2019) 62:1024–35. doi: 10.1007/s00125-019-4848-7
90. Wu T, Horowitz M, Rayner CK. New Insights Into the Anti-Diabetic Actions of Metformin: From the Liver to the Gut. Expert Rev Gastroenterol Hepatol (2017) 11:157–66. doi: 10.1080/17474124.2017.1273769
91. Bahne E, Sun EWL, Young RL, Hansen M, Sonne DP, Hansen JS, et al. Metformin-Induced Glucagon-Like Peptide-1 Secretion Contributes to the Actions of Metformin in Type 2 Diabetes. JCI Insight (2018) 3:e93936. doi: 10.1172/jci.insight.93936
92. Sum C-F, Webster JM, Johnson AB, Catalano C, Cooper BG, Taylor R. The Effect of Intravenous Metformin on Glucose Metabolism During Hyperglycaemia in Type 2 Diabetes. Diabetic Med (1992) 9:61–5. doi: 10.1111/j.1464-5491.1992.tb01716.x
93. Shah M, Vella A. Effects of GLP-1 on Appetite and Weight. Rev Endocrine Metab Disord (2014) 15:181–7. doi: 10.1007/s11154-014-9289-5
94. Mimeault M, Batra SK. Divergent Molecular Mechanisms Underlying the Pleiotropic Functions of Macrophage Inhibitory Cytokine-1 in Cancer. J Cell Physiol (2010) 224:626–35. doi: 10.1002/jcp.22196
95. Hsu J-Y, Crawley S, Chen M, Ayupova DA, Lindhout DA, Higbee J, et al. Non-Homeostatic Body Weight Regulation Through a Brainstem-Restricted Receptor for GDF15. Nature (2017) 550:255–9. doi: 10.1038/nature24042
96. Day EA, Ford RJ, Smith BK, Mohammadi-Shemirani P, Morrow MR, Gutgesell RM, et al. Metformin-Induced Increases in GDF15 Are Important for Suppressing Appetite and Promoting Weight Loss. Nat Metab (2019) 1:1202–8. doi: 10.1038/s42255-019-0146-4
97. Ouyang J, Isnard S, Lin J, Fombuena B, Peng X, Chen Y, et al. GDF-15 as a Weight Watcher for Diabetic and Non-Diabetic People Treated With Metformin. Front Endocrinol (2020) 11:581839. doi: 10.3389/fendo.2020.581839
98. Adela R, Banerjee SK. GDF-15 as a Target and Biomarker for Diabetes and Cardiovascular Diseases: A Translational Prospective. J Diabetes Res (2015) 2015:1–14. doi: 10.1155/2015/490842
99. Welsh JB, Sapinoso LM, Kern SG, Brown DA, Liu T, Bauskin AR, et al. Large-Scale Delineation of Secreted Protein Biomarkers Overexpressed in Cancer Tissue and Serum. Proc Natl Acad Sci (2003) 100:3410–5. doi: 10.1073/pnas.0530278100
100. Hardie DG, Carling D. The AMP-Activated Protein Kinase–Fuel Gauge of the Mammalian Cell? Eur J Biochem (1997) 1;246(2):259–73. doi: 10.1111/j.1432-1033.1997.00259.x
101. Hardie DG, Hawley SA, Scott JW. AMP-Activated Protein Kinase – Development of the Energy Sensor Concept. J Physiol (2006) 574:7–15. doi: 10.1113/jphysiol.2006.108944
102. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J Clin Invest (2001) 108:1167–74. doi: 10.1172/JCI13505
103. Myers RW, Guan H-P, Ehrhart J, Petrov A, Prahalada S, Tozzo E, et al. Systemic Pan-AMPK Activator MK-8722 Improves Glucose Homeostasis But Induces Cardiac Hypertrophy. Science (2017) 357:507–11. doi: 10.1126/science.aah5582
104. Saisho Y. Metformin and Inflammation: Its Potential Beyond Glucose-Lowering Effect. Endocr Metab Immune Disord Drug Targets (2015) 15:196–205. doi: 10.2174/1871530315666150316124019
105. Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin Inhibits Cytokine-Induced Nuclear Factor kappaB Activation via AMP-Activated Protein Kinase Activation in Vascular Endothelial Cells. Hypertension (2006) 47:1183–8. doi: 10.1161/01.HYP.0000221429.94591.72
106. Mather KJ, Verma S, Anderson TJ. Improved Endothelial Function With Metformin in Type 2 Diabetes Mellitus. J Am Coll Cardiol (2001) 37:1344–50. doi: 10.1016/s0735-1097(01)01129-9
107. Ouslimani N, Peynet J, Bonnefont-Rousselot D, Thérond P, Legrand A, Beaudeux J-L. Metformin Decreases Intracellular Production of Reactive Oxygen Species in Aortic Endothelial Cells. Metabolism (2005) 54:829–34. doi: 10.1016/j.metabol.2005.01.029
108. Ding H, Aljofan M, Triggle CR. Oxidative Stress and Increased eNOS and NADPH Oxidase Expression in Mouse Microvessel Endothelial Cells. J Cell Physiol (2007) 212:682–9. doi: 10.1002/jcp.21063
109. Aljofan M, Ding H. High Glucose Increases Expression of Cyclooxygenase-2, Increases Oxidative Stress and Decreases the Generation of Nitric Oxide in Mouse Microvessel Endothelial Cells. J Cell Physiol (2010) 222:669–75. doi: 10.1002/jcp.21986
110. Arunachalam G, Samuel SM, Marei I, Ding H, Triggle CR. Metformin Modulates Hyperglycaemia-Induced Endothelial Senescence and Apoptosis Through SIRT1. Br J Pharmacol (2014) 171:523–35. doi: 10.1111/bph.12496
111. Ghosh S, Lakshmanan AP, Hwang MJ, Kubba H, Mushannen A, Triggle CR, et al. Metformin Improves Endothelial Function in Aortic Tissue and Microvascular Endothelial Cells Subjected to Diabetic Hyperglycaemic Conditions. Biochem Pharmacol (2015) 98:412–21. doi: 10.1016/j.bcp.2015.10.008
112. Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, et al. Metformin Activates AMP-Activated Protein Kinase in Primary Human Hepatocytes by Decreasing Cellular Energy Status. Diabetologia (2011) 54:3101–10. doi: 10.1007/s00125-011-2311-5
113. Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and Reduced Risk of Cancer in Diabetic Patients. BMJ (2005) 330:1304–5. doi: 10.1136/bmj.38415.708634.F7
114. Nair V, Sreevalsan S, Basha R, Abdelrahim M, Abudayyeh A, Rodrigues Hoffman A, et al. Mechanism of Metformin-Dependent Inhibition of Mammalian Target of Rapamycin (mTOR) and Ras Activity in Pancreatic Cancer: Role of Specificity Protein (Sp) Transcription Factors. J Biol Chem (2014) 289:27692–701. doi: 10.1074/jbc.M114.592576
115. Wang J-C, Li G-Y, Wang B, Han S-X, Sun X, Jiang Y-N, et al. Metformin Inhibits Metastatic Breast Cancer Progression and Improves Chemosensitivity by Inducing Vessel Normalization via PDGF-B Downregulation. J Exp Clin Cancer Res (2019) 38:235. doi: 10.1186/s13046-019-1211-2
116. Ng TP, Feng L, Yap KB, Lee TS, Tan CH, Winblad B. Long-Term Metformin Usage and Cognitive Function Among Older Adults With Diabetes. J Alzheimers Dis (2014) 41:61–8. doi: 10.3233/JAD-131901
117. Fatt M, Hsu K, He L. Metformin Acts on Two Different Molecular Pathways to Enhance Adult Neural Precursor Proliferation/Self-Renewal and Differentiation. Stem Cell Rep (2015) 5:988–95. doi: 10.1016/j.stemcr.2015.10.014
118. Ma X, Xiao W, Li H, Pang P, Xue F, Wan L, et al. Metformin Restores Hippocampal Neurogenesis and Learning and Memory via Regulating Gut Microbiota in the Obese Mouse Model. Brain Behav Immun (2021) 95:68–83. doi: 10.1016/j.bbi.2021.02.011
119. Fisslthaler B, Fleming I. Activation and Signaling by the AMP-Activated Protein Kinase in Endothelial Cells. Circ Res (2009) 105:114–27. doi: 10.1161/CIRCRESAHA.109.201590
120. Grabowska W, Sikora E, Bielak-Zmijewska A. Sirtuins, a Promising Target in Slowing Down the Ageing Process. Biogerontology (2017) 18:447–76. doi: 10.1007/s10522-017-9685-9
121. Kupis W, Pałyga J, Tomal E, Niewiadomska E. The Role of Sirtuins in Cellular Homeostasis. J Physiol Biochem (2016) 72:371–80. doi: 10.1007/s13105-016-0492-6
122. Xie Z, Dong Y, Scholz R, Neumann D, Zou MH. Phosphorylation of LKB1 at Serine 428 by Protein C-Zeta Is Required for Metformin-Enhanced Activation of the AMP-Activated Protein Kinase in Endothelial Cells. Circulation (2008) 117:952–62. doi: 10.1161/CIRCULATIONAHA.107.744490
123. Katsuumi G, Shimizu I, Yoshida Y, Minamino T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front Cardiovasc Med (2018) 5:18. doi: 10.3389/fcvm.2018.00018
124. Cuyàs E, Verdura S, Llorach-Parés L, Fernández-Arroyo S, Joven J, Martin-Castillo B, et al. Metformin Is a Direct SIRT1-Activating Compound: Computational Modeling and Experimental Validation. Front Endocrinol (2018) 9:657. doi: 10.3389/fendo.2018.00657
125. Faubert B, Vincent EE, TGriss T, Samborska B, Izreig S, Svensson RU, et al. Loss of the Tumor Suppressor LKB1 Promotes Metabolic Reprogramming of Cancer Cells via HIF-1α. PNAS (2014) 111:2554–9. doi: 10.1073/pnas.1312570111
126. Momcilovic M, Shackelford DB. Targeting LKB1 in Cancer – Exposing and Exploiting Vulnerabilities. Br J Cancer (2015) 113:574–84. doi: 10.1038/bjc.2015.261
127. El-Mir M-Y, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J Biol Chem (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
128. Owen MR, Doran E, Halestrap AP. Evidence That Metformin Exerts Its Anti-Diabetic Effects Through Inhibition of Complex 1 of the Mitochondrial Respiratory Chain. Biochem J (2000) 348 Pt 3:607–14. doi: 10.1042/bj3480607
129. Hardie DG. AMPK: A Target for Drugs and Natural With Effects on Both Diabetes and Cancer. Diabetes (2013) 62:10 2337 13–0368. doi: 10.2337/db13-0368
130. Hardie DG, Salt IP SAH, Davies SP. AMP-Activated Protein Kinase: An Ultrasensitive System for Monitoring Cellular Energy Charge. Biochem J (1999) 338:717–22. doi: 10.1042/bj3380717
131. El-Mir M-Y, Detaille D, R-Villanueva G, Delgado-Esteban M, Guigas B, Attia S, et al. Neuroprotective Role of Antidiabetic Drug Metformin Against Apoptotic Cell Death in Primary Cortical Neurons. J Mol Neurosci (2008) 34:77–87. doi: 10.1007/s12031-007-9002-1
132. Viollet B, Guigas B, Garcia NS, Leclerc J, Foretz M, Andreelli F. Cellular and Molecular Mechanisms of Metformin: An Overview. Clin Sci (2012) 122:253–70. doi: 10.1042/CS20110386
133. Fontaine E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front Endocrinol (Lausanne) (2018) 9:753. doi: 10.3389/fendo.2018.00753
134. Meng S, Cao J, He Q, Xiong L, Chang E, Radovick S, et al. Metformin Activates AMP-Activated Protein Kinase by Promoting Formation of the αβγ Heterotrimeric Complex. J Biol Chem (2015) 290:3793–802. doi: 10.1074/jbc.M114.604421
135. Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin Inhibits Hepatic Gluconeogenesis in Mice Independently of the LKB1/AMPK Pathway via a Decrease in Hepatic Energy State. J Clin Invest (2010) 120:2355–69. doi: 10.1172/JCI40671
136. Vial G, Detaille D, Guigas B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front Endocrinol (2019) 10:294. doi: 10.3389/fendo.2019.00294
137. Chien HC, Zur AA, Maurer TS, Yee SW, Tolsma J, Jasper P, et al. Rapid Method to Determine Intracellular Drug Concentrations in Cellular Uptake Assays: Application to Metformin in Organic Cation Transporter 1-Transfected Human Embryonic Kidney 293 Cells. Drug Metab Dispos (2016) 44:356–64. doi: 10.1124/dmd.115.066647
138. Wang Y, An H, Liu T, Qin C, Sesaki H, Guo S, et al. Metformin Improves Mitochondrial Respiratory Activity Through Activation of AMPK. Cell Rep (2019) 29:1511–23. doi: 10.1016/j.celrep.2019.09.070
139. Wilcock C, Wyre ND, Bailey CJ. Subcellular Distribution of Metformin in Rat Liver. J Pharm Pharmacol (1991) 43:442–4. doi: 10.1111/j.2042-7158.1991.tb03507.x
140. Dykens JA, Jamieson J, Marroquin L, Nadanaciva S, Billis PA, Will Y. Biguanide-Induced Mitochondrial Dysfunction Yields Increased Lactate Production and Cytotoxicity of Aerobically-Poised HepG2 Cells and Human Hepatocytes In Vitro. Toxicol Appl Pharmacol (2008) 233:203–10. doi: 10.1016/j.taap.2008.08.013
141. Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of Metformin and Other Biguanides on Oxidative Phosphorylation in Mitochondria. Biochem J (2014) 462:475–87. doi: 10.1042/BJ20140620
142. Panfoli I, Puddu A, Bertola N, Ravera S, Maggi D. The Hormetic Effect of Metformin: “Less Is More”? Int J Mol Sci (2021) 22:6297. doi: 10.3390/ijms22126297
143. Larsen S, Rabøl R, Hansen CN, Madsbad S, Helge JW, Dela F. Metformin-Treated Patients With Type 2 Diabetes Have Normal Mitochondrial Complex I Respiration. Diabetologia (2012) 55:443–9. doi: 10.1007/s00125-011-2340-0
144. Sergi D, Naumovski N, Heilbronn LK, Abeywardena M, O’Callaghan N, Lionetti L, et al. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Mechanisms to the Impact of Diet. Front Physiol (2019) 10:532. doi: 10.3389/fphys.2019.00532
145. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of Mitochondria in Human Skeletal Muscle in Type 2 Diabetes. Diabetes (2002) 51:2944–50. doi: 10.2337/diabetes.51.10.2944
146. Sivitz WI, Yorek MA. Mitochondrial Dysfunction in Diabetes: From Molecular Mechanisms to Functional Significance and Therapeutic Opportunities. Antioxid Redox Signaling (2010) 12:537–77. doi: 10.1089/ars.2009.2531
147. Ritov VB, Menshikova EV, Azuma K, Wood R, Toledo FGS, Goodpaster BH, et al. Deficiency of Electron Transport Chain in Human Skeletal Muscle Mitochondria in Type 2 Diabetes Mellitus and Obesity. Am J Physiol Endocrinol Metab (2010) 298:E49–58. doi: 10.1152/ajpendo.00317.2009
148. Liesa M, Shirihai OS. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab (2013) 17:491–506. doi: 10.1016/j.cmet.2013.03.002
149. Kluge MA, Fetterman JL, Vita JA. Mitochondria and Endothelial Function. Circ Res (2013) 112:1171–88. doi: 10.1161/CIRCRESAHA.111.300233
150. Li C, Reif MM, Craige SM, Kant S, Keaney JF. Endothelial AMPK Activation Induces Mitochondrial Biogenesis and Stress Adaptation via eNOS-Dependent Mtorc1 Signaling. Nitric Oxide (2016) 55–56:45–53. doi: 10.1016/j.niox.2016.03.003
151. Bratic A, Larsson N-G. The Role of Mitochondria in Aging. J Clin Invest (2013) 123:951–7. doi: 10.1172/JCI64125
152. Li A, Zhang S, Li J, Liu K, Huang F, Liu B. Metformin and Resveratrol Inhibit Drp1-Mediated Mitochondrial Fission and Prevent ER Stress-Associated NLRP3 Inflammasome Activation in the Adipose Tissue of Diabetic Mice. Molec Cell Endo (2016) 434:36–47. doi: 10.1016/j.mce.2016.06.008
153. Morselli E, Maiuri M, Markaki M. Caloric Restriction and Resveratrol Promote Longevity Through the Sirtuin-1-Dependent Induction of Autophagy. Cell Death Dis (2010) 1:e10. doi: 10.1038/cddis.2009.8
154. Pentikäinen PJ, Neuvonen PJ, Penttilä A. Pharmacokinetics of Metformin After Intravenous and Oral Administration to Man. Eur J Clin Pharmacol (1979) 16:195–202. doi: 10.1007/BF00562061
155. Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, et al. Clinical Pharmacokinetics of Metformin. Clin Pharmacokinet (2011) 50:81–98. doi: 10.2165/11534750-000000000-00000
156. Kinaan M, Ding H, Triggle CR. Metformin: An Old Drug for the Treatment of Diabetes But a New Drug for the Protection of the Endothelium. Med Princ Pract (2015) 24:401–15. doi: 10.1159/000381643
157. Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin Pathways: Pharmacokinetics and Pharmacodynamics. Pharmacogenet Genomics (2012) 22(11):820–7. doi: 10.1097/FPC.0b013e3283559b22
158. Samuel SM, Varghese E, Kubatka P, Triggle CR, Büsselberg D. Metformin: The Answer to Cancer in a Flower? Current Knowledge and Future Prospects of Metformin as an Anti-Cancer Agent in Breast Cancer. Biomolecules (2019) 9:846. doi: 10.3390/biom9120846
159. Bailey CJ, Wilcock C, Scarpello JHB. Metformin and the Intestine. Diabetologia (2008) 51:1552–3. doi: 10.1007/s00125-008-1053-5
160. Christensen MMH, Højlund K, Hother-Nielsen O, Stage TB, Damkier P, Beck-Nielsen H, et al. Steady-State Pharmacokinetics of Metformin Is Independent of the OCT1 Genotype in Healthy Volunteers. Eur J Clin Pharmacol (2015) 71:691–7. doi: 10.1007/s00228-015-1853-8
161. Badrick E, Renehan AG. Diabetes and Cancer: 5 Years Into the Recent Controversy. Eur J Cancer (2014) 50:2119–25. doi: 10.1016/j.ejca.2014.04.032
162. Simpson SH. Dose-Response Relation Between Sulfonylurea Drugs and Mortality in Type 2 Diabetes Mellitus: A Population-Based Cohort Study. Can Med Assoc J (2006) 174:169–74. doi: 10.1503/cmaj.050748
163. Hu Y, Lei M, Ke G, Huang X, Peng X, Zhong L, et al. Metformin Use and Risk of All-Cause Mortality and Cardiovascular Events in Patients With Chronic Kidney Disease—A Systematic Review and Meta-Analysis. Front Endocrinol (2020) 11:559446. doi: 10.3389/fendo.2020.559446
164. Gebrie D, Getnet D, Manyazewal T. Cardiovascular Safety and Efficacy of Metformin-SGLT2i Versus Metformin-Sulfonylureas in Type 2 Diabetes: Systematic Review and Meta-Analysis of Randomized Controlled Trials. Sci Rep (2021) 11:137. doi: 10.1038/s41598-020-80603-8
165. Zaccardi F, Kloecker DE, Buse JB, Mathieu C, Khunti K, Davies MJ. Use of Metformin and Cardiovascular Effects of New Classes of Glucose-Lowering Agents: A Meta-Analysis of Cardiovascular Outcome Trials in Type 2 Diabetes. Diabetes Care (2021) 44:32–4. doi: 10.2337/dc20-2080
166. Griffin SJ, Leaver JK, Irving GJ. Impact of Metformin on Cardiovascular Disease: A Meta-Analysis of Randomised Trials Among People With Type 2 Diabetes. Diabetologia (2017) 60:1620–9. doi: 10.1007/s00125-017-4337-9
167. Furchgott RF, Zawadzki JV. The Obligatory Role of Endothelial Cells in the Relaxation of Arterial Smooth Muscle by Acetylcholine. Nature (1980) 288:373–6. doi: 10.1038/b288373a0
168. Leung W-H, Lau C-P, Wong C-K. Beneficial Effect of Cholesterol-Lowering on Coronary Endothelium-Dependent Relaxation in Hypercholsterolaemic Patients. Lancet (1993) 341:1496–500. doi: 10.1016/0140-6736(93)90634-S
169. Anderson TJ, Uehata A, Gerhard MD, Meredith IT, Knab S, Delagrange D, et al. Close Relation of Endothelial Function in the Human Coronary and Peripheral Circulations. J Am Coll Cardiol (1995) 26:1235–4. doi: 10.1016/0735-1097(95)00327-4
170. Landmesser U, Hornig B, Drexler H. Endothelial Function: A Critical Determinant in Atherosclerosis? Circulation (2004) 109(21 Suppl 1):II27–33. doi: 10.1161/01.CIR.0000129501.88485.1f
171. Lerman A, Zeiher AM. Endothelial Function: Cardiac Events. Circulation (2005) 111:363–8. doi: 10.1161/01.CIR.0000153339.27064.14
172. Altschul A. Endothelium. Its Development, Morphology, Function and Pathology. New York: The MacMillan Company (1954).
173. Salvatore T, Pafundi PC, Galiero R, Rinaldi L, Caturano A, Vetrano E, et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines (2020) 9:3. doi: 10.3390/biomedicines9010003
174. de Jager J, Kooy A, Schalkwijk C, van der Kolk J, Lehert P, Bets D, et al. Long-Term Effects of Metformin on Endothelial Function in Type 2 Diabetes: A Randomized Controlled Trial. J Internal Med (2014) 275:59–70. doi: 10.1111/joim.12128
175. Adam J, Brandmaier S, Leonhardt J, Scheerer MF, Mohney RP, Xu T, et al. Metformin Effect on Nontargeted Metabolite Profiles in Patients With Type 2 Diabetes and in Multiple Murine Tissues. Diabetes (2016) 65:3776–85. doi: 10.2337/db16-0512
176. Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, et al. Sirt1 Regulates Aging and Resistance to Oxidative Stress in the Heart. Circ Res (2007) 100(10):1512–21. doi: 10.1161/01.RES.0000267723.65696.4a
177. Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, et al. SIRT1 Controls Endothelial Angiogenic Functions During Vascular Growth. Genes Dev (2007) 21:2644–58. doi: 10.1101/gad.435107
178. Elibol B, Kilic U. High Levels of SIRT1 Expression as a Protective Mechanism Against Disease-Related Conditions. Front Endocrinol (2018) 9:614. doi: 10.3389/fendo.2018.00614
179. Zu Y, Liu L, Lee MY, Xu C, Liang Y, Man RY, et al. SIRT1 Promotes Proliferation and Prevent Senescenc Through Targeting LKB1 in Primary Porcine Aortic Endothelial Cells. Circ Res (2010) 106:1384–93. doi: 10.1161/CIRCRESAHA.109.215483
180. Mattagajasingh I, Kim C-S, Naqvi A, Yamamori T, Hoffman TA, Jung S-B, et al. SIRT1 Promotes Endothelium-Dependent Vascular Relaxation by Activating Endothelial Nitric Oxide Synthase. Proc Natl Acad Sci (2007) 104:14855–60. doi: 10.1073/pnas.0704329104
181. Wilcock C, Bailey CJ. Accumulation of Metformin by Tissues of the Normal and Diabetic Mouse. Xenobiotica (1994) 24:49–57. doi: 10.3109/00498259409043220
182. Triggle CR, Venu VK, Saifeddine M, Alston LA, Motahhary M, Ding H, et al. Prevention of Hyperglycaemia-Related Oxidative-Stress-Induced Endothelial Dysfunction by Metformin: Novel Involvement of Orphan Nuclear Receptor, NR4a1/Nur77. BritJPharmacol (2019) 176:2993–4. doi: 10.1111/bph.14681
183. Gasser PJ, Hurley MM, Chan J, Pickel VM. Organic Cation Transporter 3 (OCT3) Is Localized to Intracellular and Surface Membranes in Select Glial and Neuronal Cells Within the Basolateral Amygdaloid Complex of Both Rats and Mice. Brain Struct Funct (2017) 222:1913–28. doi: 10.1007/s00429-016-1315-9
184. Zhan YY, Chen Y, Zhang Q, Zhuang JJ, Tian M, Chen HZ, et al. The OrphanNuclear Receptor Nur77 Regulates LKB1 Localization and Activates AMPK. Nat Chem Biol (2012) 8:897–904. doi: 10.1038/nchembio.1069
185. Mohankumar K, Lee J, Wu CS, Sun Y, Safe S. Bis-Indole–Derived NR4A1 Ligands and Metformin Exhibit NR4A1-Dependent Glucose Metabolism and Uptake in C2C12 Cells. Endocrinology (2018) 159:1950–63. doi: 10.1210/en.2017-03049
186. Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, et al. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes (2017) 66:193–205. doi: 10.2337/db16-0915
187. Gallo A, Ceolotto G, Pinton P, Iori E, Murphy E, Rutter GA, et al. Metformin Prevents Glucose-Induced Protein Kinase C-Beta2 Activation in Human Umbilical Vein Endothelial Cells Through an Antioxidant Mechanism. Diabetes (2005) 54:1123–31. doi: 10.2337/diabetes.54.4.1123
188. El-Daly M, Pulakazhi Venu VK, Saifeddine M, Mihara K, Kang S, Fedak PWM, et al. Hyperglycaemic Impairment of PAR2-Mediated Vasodilation: Prevention by Inhibition of Aortic Endothelial Sodium-Glucose-Co-Transporter-2 and Minimizing Oxidative Stress. Vasc Pharmacol (2018) 109:56–71. doi: 10.1016/j.vph.2018.06.006
189. Engerman RL, Kern TS. Progression of Incipient Diabetic Retinopathy During Good Glycemic Control. Diabetes (1987) 36:808–12. doi: 10.2337/diab.36.7.808
190. Paneni F, Volpe M, Luscher TF, Cosentino F. SIRT1, p66Shc, and Set7/9 in Vascular Hyperglycemic Memory: Bringing All the Strands Together. Diabetes (2013) 62:1800–7. doi: 10.2337/db12-1648
191. Paneni F, Costantino S, Volpe M, Lüscher TF, Cosentino F. Epigenetic Signatures and Vascular Risk in Type 2 Diabetes: A Clinical Perspective. Atherosclerosis (2013) 230:191–7. doi: 10.1016/j.atherosclerosis.2013.07.003
192. Roy S, Sala R, Cagliero E, Lorenzi M. Overexpression of Fibronectin Induced by Diabetes or High Glucose: Phenomenon With a Memory. Proc Natl Acad Sci U.S.A. (1990) 87:404–8. doi: 10.1073/pnas.87.1.404
193. Zheng Z, Chen H, Li J, Li T, Zheng B, Zheng Y, et al. Sirtuin 1-Mediated Cellular Metabolic Memory of High Glucose via the LKB1/AMPK/ROS Pathway and Therapeutic Effects of Metformin. Diabetes (2012) 61:217–28. doi: 10.2337/db11-0416
194. Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu Rev Pathol (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
195. Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, et al. Metformin Inhibits the Senescence-Associated Secretory Phenotype by Interfering With IKK/NF-κB Activation. Aging Cell (2013) 12:489–98. doi: 10.1111/acel.12075
196. Hirsch HA, Iliopoulos D, Struhl K. Metformin Inhibits the Inflammatory Response Associated With Cellular Transformation and Cancer Stem Cell Growth. Proc Natl Acad Sci (2013) 110:972–7. doi: 10.1073/pnas.1221055110
197. O’Neill HM. AMPK and Exercise: Glucose Uptake and Insulin Sensitivity. Diabetes Metab J (2013) 37:1. doi: 10.4093/dmj.2013.37.1.1
198. Golbidi S, Laher I. Exercise and Aging Endothelium. J Diabetes Res (2013) 2016:789607. doi: 10.1155/2013/78960
199. Sallam N, Laher I. Exercise Modulates Oxidative Stress and Inflammation in Aging and Cardiovascular Diseases. Oxid Med Cell Longev (2016) 2016:7239639. doi: 10.1155/2016/7239639
200. Liu H-W, Chang S-J. Moderate Exercise Suppresses NF-κB Signaling and Activates the SIRT1-AMPK-PGC1α Axis to Attenuate Muscle Loss in Diabetic db/db Mice. Front Physiol (2018) 9:636. doi: 10.3389/fphys.2018.00636
201. Malin SK, Braun B. Impact of Metformin on Exercise-Induced Metabolic Adaptations to Lower Type 2 Diabetes Risk. Exercise Sport Sci Rev (2016) 44:4–11. doi: 10.1249/JES.0000000000000070
202. Konopka AR, Laurin JL, Schoenberg HM, Reid JJ, Castor WM, Wolff CA, et al. Metformin Inhibits Mitochondrial Adaptations to Aerobic Exercise Training in Older Adults. Aging Cell (2019) 18:e12880. doi: 10.1111/acel.12880
203. Walton RG, Dungan CM, Long DE, Tuggle SC, Kosmac K, Peck BD, et al. Metformin Blunts Muscle Hypertrophy in Response to Progressive Resistance Exercise Training in Older Adults: A Randomized, Double-Blind, Placebo-Controlled, Multicenter Trial: The MASTERS Trial. Aging Cell (2019) 18:e13039. doi: 10.1111/acel.13039
204. Terada T, Boulé NG. Does Metformin Therapy Influence the Effects of Intensive Lifestyle Intervention? Exploring the Interaction Between First Line Therapies in the Look AHEAD Trial. Metabolism (2019) 94:39–46. doi: 10.1016/j.metabol.2019.01.004
205. Pedersen BK, Saltin B. Exercise as Medicine - Evidence for Prescribing Exercise as Therapy in 26 Different Chronic Diseases. Scand J Med Sci Sports (2015) 25:1–72. doi: 10.1111/sms.12581
206. Fontana L, Partridge L, Longo VD. Extending Healthy Life Span–From Yeast to Humans. Science (2010) 328:321–6. doi: 10.1126/science.1172539
207. Anderson RM, Shanmuganayagam D, Weindruch R. Caloric Restriction and Aging: Studies in Mice and Monkeys. Toxicol Pathol (2009) 37:47–51. doi: 10.1177/0192623308329476
208. Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, et al. Caloric Restriction Delays Disease Onset and Mortality in Rhesus Monkeys. Science (2009) 325:201–4. doi: 10.1126/science.1173635
209. Guo Z, Mitchell-Raymundo F, Yang H, Ikeno Y, Nelson J, Diaz V, et al. Dietary Restriction Reduces Atherosclerosis and Oxidative Stress in the Aorta of Apolipoprotein E-Deficient Mice. Mech Ageing Dev (2002) 123:1121–31. doi: 10.1016/s0047-6374(02)00008-8
210. Fontana L, Klein S, Holloszy JO, Premachandra BN. Effect of Long-Term Calorie Restriction With Adequate Protein and Micronutrients on Thyroid Hormones. J Clin Endocrinol Metab (2006) 91:3232–5. doi: 10.1210/jc.2006-0328
211. Fontana L, Meyer TE, Klein S, Holloszy JO. Long-Term Calorie Restriction Is Highly Effective in Reducing the Risk for Atherosclerosis in Humans. Proc Natl Acad Sci USA (2004) 101:6659–63. doi: 10.1073/pnas.0308291101
212. Meyer TE, Kovács SJ, Ehsani AA, Klein S, Holloszy JO, Fontana L. Long-Term Caloric Restriction Ameliorates the Decline in Diastolic Function in Humans. J Am Coll Cardiol (2006) 47:398–402. doi: 10.1016/j.jacc.2005.08.069
213. Lettieri-Barbato D, Giovannetti E, Aquilano K. Effects of Dietary Restriction on Adipose Mass and Biomarkers of Healthy Aging in Human. Aging (2016) 8:3341–55. doi: 10.18632/aging.101122
214. Fontana L, Klein S. Aging, Adiposity, and Calorie Restriction. JAMA (2007) 297:986–94. doi: 10.1001/jama.297.9.986
215. Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C, et al. Interventions to Slow Aging in Humans: Are We Ready? Aging Cell (2015) 14:497–510. doi: 10.1111/acel.12338
216. Flegal KM. Excess Deaths Associated With Underweight, Overweight, and Obesity. JAMA (2005) 293:1861. doi: 10.1001/jama.293.15.1861
217. Khan SS, Ning H, Wilkins JT, Allen N, Carnethon M, Berry JD, et al. Association of Body Mass Index With Lifetime Risk of Cardiovascular Disease and Compression of Morbidity. JAMA Cardiol (2018) 3:280. doi: 10.1001/jamacardio.2018.0022
218. Howard BV, Van Horn L, Hsia J, Manson JE, Stefanick ML, Wassertheil-Smoller S, et al. Low-Fat Dietary Pattern and Risk of Cardiovascular Disease: The Women’s Health Initiative Randomized Controlled Dietary Modification Trial. JAMA (2006) 295:655. doi: 10.1001/jama.295.6.655
219. Cantó C, Auwerx J. Calorie Restriction: Is AMPK a Key Sensor and Effector? Physiology (2011) 26:214–24. doi: 10.1152/physiol.00010.2011
220. Strong R, Miller RA, Astle CM, Floyd RA, Flurkey K, Hensley KL, et al. Nordihydroguaiaretic Acid and Aspirin Increase Lifespan of Genetically Heterogeneous Male Mice. Aging Cell (2008) 7:641–50. doi: 10.1111/j.1474-9726.2008.00414.x
221. Powers RW. Extension of Chronological Life Span in Yeast by Decreased TOR Pathway Signaling. Genes Dev (2006) 20:174–84. doi: 10.1101/gad.1381406
222. Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, et al. Mechanisms of Life Span Extension by Rapamycin in the Fruit Fly Drosophila Melanogaster. Cell Metab (2010) 11:35–46. doi: 10.1016/j.cmet.2009.11.010
223. Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, et al. TOR Signaling and Rapamycin Influence Longevity by Regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab (2012) 15:713–24. doi: 10.1016/j.cmet.2012.04.007
224. Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR Is a Key Modulator of Ageing and Age-Related Disease. Nature (2013) 493:338–45. doi: 10.1038/nature11861
225. Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, et al. Metformin Inhibits Hepatic Mtorc1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab (2017) 25:463–71. doi: 10.1016/j.cmet.2016.12.009
226. Blagosklonny MV. Rapamycin-Induced Glucose Intolerance: Hunger or Starvation Diabetes. Cell Cycle (2011) 10:4217–24. doi: 10.4161/cc.10.24.18595
227. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic Responses in Mouse Models Poorly Mimic Human Inflammatory Diseases. Proc Natl Acad Sci (2013) 110:3507–12. doi: 10.1073/pnas.1222878110
228. Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell (2008) 132:27–42. doi: 10.1016/j.cell.2007.12.018
229. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and Aging. Cell (2011) 146:682–95. doi: 10.1016/j.cell.2011.07.030
230. Jia K, Levine B. Autophagy Is Required for Dietary Restriction-Mediated Life Span Extension in C. elegans. Autophagy (2007) 3:597–9. doi: 10.4161/auto.4989
231. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of Cardiac Functions by Chronic Metformin Treatment Is Associated With Enhanced Cardiac Autophagy in Diabetic OVE26 Mice. Diabetes (2011) 60:1770–8. doi: 10.2337/db10-0351
232. Song YM, Lee Y, Kim J-W, Ham D-S, Kang E-S, Cha BS, et al. Metformin Alleviates Hepatosteatosis by Restoring SIRT1-Mediated Autophagy Induction via an AMP-Activated Protein Kinase-Independent Pathway. Autophagy (2015) 11:46–59. doi: 10.4161/15548627.2014.984271
233. Pyo J-O, Yoo S-M, Ahn H-H, Nah J, Hong S-H, Kam T-I, et al. Overexpression of Atg5 in Mice Activates Autophagy and Extends Lifespan. Nat Commun (2013) 4:2300. doi: 10.1038/ncomms3300
234. Nazim UMD, Moon J-H, Lee J-H, Lee Y-J, Seol J-W, Eo S-K, et al. Activation of Autophagy Flux by Metformin Downregulates Cellular FLICE-Like Inhibitory Protein and Enhances TRAIL- Induced Apoptosis. Oncotarget (2016) 7:23468–81. doi: 10.18632/oncotarget.8048
235. Samuel SM, Ghosh S, Majeed Y, Arunachalam G, Emara MM, Ding H, et al. Metformin Represses Glucose Starvation Induced Autophagic Response in Microvascular Endothelial Cells and Promotes Cell Death. Biochem Pharmacol (2017) 132:118–32. doi: 10.1016/j.bcp.2017.03.001
236. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and Cancer: A Consensus Report. Diabetes Care (2010) 33:1674–85. doi: 10.2337/dc10-0666
237. Gandini S, Puntoni M, Heckman-Stoddard BM, Dunn BK, Ford L, DeCensi A, et al. Metformin and Cancer Risk and Mortality: A Systematic Review and Meta-Analysis Taking Into Account Biases and Confounders. Cancer Prev Res (2014) 7:867–85. doi: 10.1158/1940-6207.CAPR-13-0424
238. Vancura A, Bu P, Bhagwat M, Zeng J, Vancurova I. Metformin as an Anticancer Agent. Trends Pharmacol Sci (2018) 39:867–78. doi: 10.1016/j.tips.2018.07.006
239. Mamtani R, Pfanzelter N, Haynes K, Finkelman BS, Wang X, Keefe SM, et al. Incidence of Bladder Cancer in Patients With Type 2 Diabetes Treated With Metformin or Sulfonylureas. Diabetes Care (2014) 37:1910–7. doi: 10.2337/dc13-1489
240. Suissa S, Azoulay L. Metformin and Cancer: Mounting Evidence Against an Association: Figure 1. Diabetes Care (2014) 37:1786–8. doi: 10.2337/dc14-0500
241. Cai H, Zhang Y, Han TK, Everett RS, Thakker DR. Cation-Selective Transporters Are Critical to the AMPK-Mediated Antiproliferative Effects of Metformin in Human Breast Cancer Cells. Int J Cancer (2016) 138:2281–92. doi: 10.1002/ijc.29965
242. Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B, et al. Metformin, Independent of AMPK, Inhibits mTORC1 in a Rag GTPase-Dependent Manner. Cell Metab (2010) 11:390–401. doi: 10.1016/j.cmet.2010.03.014
243. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. Elife (Cambridge (2014) 3:2242. doi: 10.7554/eLife.02242
244. Pelton K, Coticchia CM, Curatolo AS, Schaffner CP, Zurakowski D, Solomon KR, et al. Hypercholesterolemia Induces Angiogenesis and Accelerates Growth of Breast Tumors In Vivo. Am J Pathol (2014) 184:2099–110. doi: 10.1016/j.ajpath.2014.03.006
245. Samuel SM, Varghese E, Varghese S, Büsselberg D. Challenges and Perspectives in the Treatment of Diabetes Associated Breast Cancer. Cancer Treat Rev (2018) 70:98–111. doi: 10.1016/j.ctrv.2018.08.004
246. Ferroni P, Riondino S, Buonomo O, Palmirotta R, Guadagni F, Roselli M. Type 2 Diabetes and Breast Cancer: The Interplay Between Impaired Glucose Metabolism and Oxidant Stress. Oxid Med Cell Longev (2015) 2015:183928. doi: 10.1155/2015/183928
247. Mathews Samuel S, Satheesh NJ, Ghosh S, Büsselberg D, Majeed Y, Ding H, et al. Treatment With a Combination of Metformin and 2-Deoxyglucose Upregulates Thrombospondin-1 in Microvascular Endothelial Cells: Implications in Anti-Angiogenic Cancer Therapy. Cancers (2019) 11:1737. doi: 10.3390/cancers11111737
248. Lei Y, Yi Y, Liu Y, Liu X, Keller ET, Qian C-N, et al. Metformin Targets Multiple Signaling Pathways in Cancer. Chin J Cancer (2017) 36:17. doi: 10.1186/s40880-017-0184-9
249. Chandel NS, Avizonis D, Reczek CR, Weinberg SE, Menz S, Neuhaus R, et al. Are Metformin Doses Used in Murine Cancer Models Clinically Relevant? Cell Metab (2016) 23:569–70. doi: 10.1016/j.cmet.2016.03.010
250. Wahdan-Alaswad R, Fan Z, Edgerton SM, Liu B, Deng X-S, Arnadottir SS, et al. Glucose Promotes Breast Cancer Aggression and Reduces Metformin Efficacy. Cell Cycle (2013) 12:3759–69. doi: 10.4161/cc.26641
251. Zordoky BNM, Bark D, Soltys CL, Sung MM, Dyck JRB. The Anti-Proliferative Effect of Metformin in Triple-Negative MDA-MB-231 Breast Cancer Cells Is Highly Dependent on Glucose Concentration: Implications for Cancer Therapy and Prevention. Biochim Biophys Acta (2014) 1840:1943–57. doi: 10.1016/j.bbagen.2014.01.023
252. Bayraktar S, Hernadez-Aya LF, Lei X, Meric-Bernstam F, Litton JK, Hsu L, et al. Effect of Metformin on Survival Outcomes in Diabetic Patients With Triple Receptor-Negative Breast Cancer. Cancer (2012) 118:1202–11. doi: 10.1002/cncr.26439
253. Checkley LA, Rudolph MC, Wellberg EA, Giles ED, Wahdan-Alaswad RS, Houck JA, et al. Metformin Accumulation Correlates With Organic Cation Transporter 2 Protein Expression and Predicts Mammary Tumor Regression In Vivo. Cancer Prev Res (Phila) (2017) 10:198–207. doi: 10.1158/1940-6207.CAPR-16-0211-T
254. Chowdhury S, Yung E, Pintilie M, Muaddi H, Chaib S, Yeung M, et al. MATE2 Expression Is Associated With Cancer Cell Response to Metformin. PloS One (2016) 11:e0165214. doi: 10.1371/journal.pone.0165214
255. Campbell JM, Stephenson MD, de Courten B, Chapman I, Bellman SM, Aromataris E. Metformin Use Associated With Reduced Risk of Dementia in Patients With Diabetes: A Systematic Review and Meta-Analysis. J Alzheimer’s Dis (2018) 65:1225–36. doi: 10.3233/JAD-180263
256. Cheng C, Lin C-H, Tsai Y-W, Tsai C-J, Chou P-H, Lan T-H. Type 2 Diabetes and Antidiabetic Medications in Relation to Dementia Diagnosis. J Gerontol A Biol Sci Med Sci (2014) 69:1299–305. doi: 10.1093/gerona/glu073
257. Guo M, Mi J, Jiang Q-M, Xu J-M, Tang Y-Y, Tian G, et al. Metformin may Produce Antidepressant Effects Through Improvement of Cognitive Function Among Depressed Patients With Diabetes Mellitus. Clin Exp Pharmacol Physiol (2014) 41:650–6. doi: 10.1111/1440-1681.12265
258. Syal C, Kosaraju J, Hamilton L, Aumont A, Chu A, Sarma SN, et al. Dysregulated Expression of Monoacylglycerol Lipase Is a Marker for Anti-Diabetic Drug Metformin-Targeted Therapy to Correct Impaired Neurogenesis and Spatial Memory in Alzheimer’s Disease. Theranostics (2020) 10:6337–60. doi: 10.7150/thno.44962
259. Sun K, Kusminski CM, Scherer PE. Adipose Tissue Remodeling and Obesity. J Clin Invest (2011) 121:2094–101. doi: 10.1172/JCI45887
260. Luo T, Nocon A, Fry J, Sherban A, Rui X, Jiang B, et al. AMPK Activation by Metformin Suppresses Abnormal Extracellular Matrix Remodeling in Adipose Tissue and Ameliorates Insulin Resistance in Obesity. Diabetes (2016) 65:2295–310. doi: 10.2337/db15-1122
261. Justice JN, Niedernhofer L, Robbins PD, Aroda VR, Espeland MA, Kritchevsky SB, et al. Development of Clinical Trials to Extend Healthy Lifespan. Cardiovasc Endocrinol Metab (2018) 7:80–3. doi: 10.1097/XCE.0000000000000159
262. Studenski S. Gait Speed and Survival in Older Adults. JAMA (2011) 305:50. doi: 10.1001/jama.2010.1923
263. Pyrkov TV, Avchaciov K, Tarkhov AE, Menshikov LI, Gudkov AV, Fedichev PO. Longitudinal Analysis of Blood Markers Reveals Progressive Loss of Resilience and Predicts Human Lifespan Limit. Nat Commun (2021) 12:2765. doi: 10.1038/s41467-021-23014-1
264. Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J, et al. Frailty in Older Adults: Evidence for a Phenotype. Journals Gerontol Ser A: Biol Sci Med Sci (2001) 56:M146–57. doi: 10.1093/gerona/56.3.M146
265. The Diabetes Prevention Program Research Group. [Prepared by Jill Crandall, David Schade, Yong Ma, Wilfred Y. Fujimoto, Elizabeth Barrett-Connor, Sarah Fowler, Sam Dagogo-Jack, Reubin Andres]. The Influence of Age on the Effects of Lifestyle Modification and Metformin in Prevention of Diabetes. J Gerontol Ser A: Biol Sci Med Sci (2006) 61:1075–81. doi: 10.1093/gerona/61.10.1075
266. Diabetes Prevention Program Research Group. Reduction in the Incidence of Type 2 Diabetes With Lifestyle Intervention or Metformin. N Engl J Med (2002) 346:393–403. doi: 10.1056/NEJMoa012512
Keywords: metformin, aging, healthspan and lifespan, AMP-Kinase, calorie restriction mimetic, diabetes, cardiovascular and neurodegenerative diseases, cancer
Citation: Mohammed I, Hollenberg MD, Ding H and Triggle CR (2021) A Critical Review of the Evidence That Metformin Is a Putative Anti-Aging Drug That Enhances Healthspan and Extends Lifespan. Front. Endocrinol. 12:718942. doi: 10.3389/fendo.2021.718942
Received: 10 June 2021; Accepted: 15 July 2021;
Published: 05 August 2021.
Edited by:
James Harper, Sam Houston State University, United StatesReviewed by:
Adiv Johnson, Independent researcher, Tucson, AZ, United StatesAdam Konopka, University of Wisconsin-Madison, United States
Andrzej Bartke, Southern Illinois University School of Medicine, United States
Copyright © 2021 Mohammed, Hollenberg, Ding and Triggle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chris R. Triggle, Y2h0MjAxMUBxYXRhci1tZWQuY29ybmVsbC5lZHU=; Ibrahim Mohammed, aWJtMjAwMUBxYXRhci1tZWQuY29ybmVsbC5lZHU=