 Sugyeong Jo
Sugyeong Jo Sungsoon Fang
Sungsoon Fang- 1Department of Medical Science, Brain Korea 21 Project for Medical Science, Yonsei University College of Medicine, Seoul, South Korea
- 2Severance Biomedical Science Institute, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, South Korea
Increased incidence of type I and type II diabetes has been prevailed worldwide. Though the pathogenesis of molecular mechanisms remains still unclear, there are solid evidence that disturbed immune homeostasis leads to pancreatic β cell failure. Currently, autoimmunity and uncontrolled inflammatory signaling pathways have been considered the major factors in the pathogenesis of diabetes. Many components of immune system have been reported to implicate pancreatic β cell failure, including helper T cells, cytotoxic T cells, regulatory T cells and gut microbiota. Immune modulation of those components using small molecules and antibodies, and fecal microbiota transplantation are undergoing in many clinical trials for the treatment of type I and type II diabetes. In this review we will discuss the basis of molecular pathogenesis focusing on the disturbed immune homeostasis in type I and type II diabetes, leading to pancreatic β cell destruction. Finally, we will introduce current therapeutic strategies and clinical trials by modulation of immune system for the treatment of type I and type II diabetes patients.
Introduction
Diabetes is a complex disorder caused by multiple genetic and environmental factors. For example, sex differences in genetic and environmental factors such as sex hormones, sex chromosomes and sex-specific epigenetic modification are associated with the development of diabetes (1–4). Although there are numerous risk factors for diabetes, dysfunction of pancreatic β cells is a common feature of both type 1 diabetes (T1D) and type 2 diabetes (T2D). Pancreatic β cells play crucial roles in the regulation of glucose homeostasis. They have molecular sensors to recognize a rise in blood glucose and to produce insulin for the maintenance of blood glucose levels. While destruction of pancreatic β cells leads to development of T1D, T2D occurs when β cells fail to secrete sufficient insulin to compensate for insulin resistance. Numerous studies have shown that multiple molecular mechanisms, including autoimmune, inflammation and metabolic stress are risk factors for the development of β cell failure. Autoimmune-mediated β cell dysfunction is due to β cell autoantigens and immune cell infiltration of the pancreatic islets (insulitis) (5). The autoantibodies have been identified in the sera of T1D patients and these autoantibodies serve as biomarkers for the prediction of T1D (6). Besides, a spontaneous animal model of T1D, non-obese diabetic (NOD) mice has shown the presence of autoreactive T cells and antigen-presenting cells (APCs) in islets, implying that autoreactive T cells recognize β cell autoantigen to infiltrate islets and contribute to β cell destruction for the development of T1D (7). In addition to T1D, islet inflammation also contributes to β cell dysfunction in T2D. Infiltration of macrophages and increased expression of pro-inflammatory cytokines have been observed in islets of rodent models including high-fat diet (HFD)-fed mice, db/db mice and GK rat (8, 9). Glucotoxicity and lipotoxicity have been shown to induce β cell death in T2D. Chronic exposure of β cells to high glucose altered glucose-stimulated insulin secretion (GSIS) (10). Although short-term exposure to saturated free fatty acids (FFAs) is able to stimulate insulin secretion, prolonged exposure to saturated FFAs dramatically suppresses GSIS by increasing oxidative stress in β cells (11).
Recent studies have reported that the gut microbiota is associated with both T1D and T2D (12–14). The microbiota plays a critical role to regulate energy metabolism by fermentation of carbohydrates and production of metabolites such as bile acids, short-chain fatty acids (SCFAs) including acetate, propionate, and butyrate. The metabolites induced by gut microbiota are involved in glucose homeostasis. For example, SCFAs stimulate secretion of glucagon-like peptide 1 (GLP-1), an incretin to potentiate insulin secretion for glucose homeostasis (15). Thus, alteration of gut microbiota may contribute to disturbed incretin-mediated insulin signaling in diabetes patients (16–18). And, it has been reported that children with high genetic risks for developing T1D have distinct features of microbiome compared with a healthy control group (19). Though the underlying molecular mechanisms are still unclear, the alteration of gut microbiota in early life has been shown to correlate with the presence of autoantibodies and autoimmunity in pancreatic islets, suggesting that gut microbiota plays a pivotal role to modulate immune responses in islets for the development of T1D (20). Likewise, numerous studies using metagenomics and 16S rRNA-based high-throughput sequencing have reported compositional and functional changes of gut microbiota in T2D patients (17, 21, 22). Cohort studies from Europe and China commonly demonstrated a decrease of butyrate-producing gut bacteria in T2D patients compared to the normal group (21–23). The reduction of butyrate-producing gut bacteria contributed to both T1D and T2D development, implying that gut microbiota alteration is crucial risk factor for the pathogenesis of diabetes.
Herein, we will provide an overview of the pathogenesis and therapeutic strategies of diabetes in terms of immune modulation using gut microbiota. Given that immune modulation strongly contributes to the destruction of pancreatic β cells for the development of diabetes, the crosstalk between the gut microbiota and immune system would be promising therapeutic strategy for the treatment of diabetes.
The Pathogenesis of Diabetes
Autoimmunity in Type I Diabetes
It has been widely accepted that T1D is an organ-specific autoimmune disease. Autoantibodies in pancreatic islet serve as diagnosis markers to predict the pathogenesis of T1D in both NOD mice and T1D human patients (24, 25). These autoantibodies recognize specific self-antigens include insulin, glutamic acid decarboxylase (GAD), zinc transporter (ZnT8), and insulinoma-antigen 2 (IA-2) in pancreatic islets. Though the molecular mechanisms of how autoantigens are processed remain still unclear, endoplasmic reticulum (ER) stress-mediated misfolded proteins in pancreatic β cells may contribute to the process of these self-antigens (26–28). Antigen-presenting cells (APCs) such as macrophages and dendritic cells (DCs) have been shown to infiltrate in β cells and present autoantigens to naïve CD4+ T cell for T cell activation. After recognizing autoantigens, naïve CD4+ T cell differentiate into autoreactive CD4+ T cells, leading to promote B cells to produce autoantibodies and activate CD8+ T cells to differentiate into cytotoxic T cell (5). These autoreactive CD4+ T cells and CD8+ T cells are key drivers of autoimmune reaction to destroy pancreatic β cells.
Recently, it has been reported that polymorphism of Human leukocyte antigens (HLA) class II encoding DQ and DR is a genetic determinant of T1D (29). In NOD mice, a peptide derived from insulin was reported to serve as one of the autoantigens. This peptide was able to bind major histocompatibility complex (MHC) class II molecule H2-Ag7, resulting in activation of CD4+ T cells (30). HLA DR4-restricted CD4+ T cells from T1D patients are also able to respond to pre-proinsulin (PPI)-derived epitope (31). Once activated by self-antigens, the activated CD4+ T cells then secrete interleukin-2 (IL-2) to provide ‘help’ CD8+ T cell activation.
In addition to CD4+ T cells, cytotoxic CD8+ T cells are predominant lymphocytes infiltrating the pancreatic islets, resulting in β cell destruction. HLA class I has been shown to be overexpressed in pancreatic islets of early T1D patients, resulting in infiltration of CD8+ T cells and insulitis (32). Thus, MHC class I-deficient NOD. β2M−/− mice decreased infiltration of CD8+ T cells into pancreatic islets, leading to ameliorate pathogenesis of T1D (33).
Recently, it has been reported that β cell peptides including insulin B chain, GAD and Islet-specific glucose-6-phosphatase catalytic subunit–related protein (IGRP) are prone to bind HLA-A2 in T1D patients and autoreactive CD8+ T cell response to many HLA-A*0201–restricted β cell peptides (34). Once activated, autoreactive CD8+ T cells release cytotoxic granules such as perforin, granzyme B and proinflammatory cytokines, including IFN-γ leading to destruction of pancreatic β cells (35). These proinflammatory cytokines increase the expression levels of MHC class I and chemokine CXCL10 which promote T cell infiltration in human β cells (32).
On the other hand, FOXP3+ regulatory T cells (Tregs) are known as immunosuppressive cells. Treg directly suppress the proliferation and activation of effector T cells or dendritic cells and secrete anti-inflammatory cytokines such as IL-10 (36). However, the function of FOXP3+ Treg is altered in the onset of T1D and the dysfunction of Treg may contribute to the pathogenesis of T1D (37, 38).
Inflammatory Signaling in Type II Diabetes
Type 2 diabetes (T2D) is a multifactorial disease that resulted from the combination of both genetic and environmental factors. This disease is associated with hyperglycemia and is characterized by insulin resistance and dysfunction or death of β-cells, leading to insufficient insulin secretion for glycemic homeostasis.
Insulin resistance is highly associated with diet-induced obesity (39, 40). In the condition of obesity, dysfunction of adipose tissue which is characterized by enlarged adipocytes and abnormal secretion of adipokines and inflammatory cytokines promotes the progression of insulin resistance (41, 42). Adipocyte hypertrophy generally increase the expression of pro-inflammatory cytokines including tumor-necrosis factor-α (TNF-α), IL-6, and IL-1β in the human patients with insulin resistance. Furthermore, the adipocytes release macrophage chemoattractant protein-1 (MCP-1) to promote macrophage infiltration in adipose tissue (43). The infiltration of macrophages with secretion of proinflammatory cytokines contribute to insulin resistance, leading to increase of lipolysis and plasma FFA in the T2D patients (44).
Similar with adipose tissues, dysfunction of pancreatic β cells in T2D is involved in islet inflammation through increased proinflammatory cytokines and infiltration of immune cells. The islet inflammation has been observed in several T2D mouse model including Psammomys obesus rat, HFD-fed mice, db/db mouse and GK rat (45). Chronic hyperglycemia induces the production of IL-1β in pancreatic β cells which is implicated in insulin resistance as well as β cell dysfunction (46). Treatment of IL-1 receptor antagonist to HFD-fed mice resulted in protection from apoptotic β cell death and improved GSIS secretion by blocking IL-1β signaling (47). IL-1 receptor antagonisms inhibited macrophage infiltration into β cell, leading to improved hyperglycemia in GK rats (48). In addition, exposure of isolated human islets to high glucose levels resulted in an increase of IL-1β secretion to activate NF-κB, a key regulator of inflammation, and Fas (CD95) signaling to induce dysfunctional β cell (49). These findings suggest that glucotoxicity implicates an islet inflammatory process in T2D.
Likewise, lipotoxicity also has physiological impacts on the function of β cells. FFAs as well as TAG and cholesterol levels have been shown to be elevated in HFD-fed mice (47). Increase of plasma FFAs contributes to insulin resistance and impaired insulin secretion by inducing ER stress and oxidative stress. Moreover, excessive level of FFAs in skeletal muscle serves as toxic lipids such as ceramide and diacylglyceride, leading to incomplete fatty acid oxidation (50). These toxic FFA metabolites and incomplete fatty acid oxidation contribute to ER stress, oxidative stress and the generation of reactive oxygen species (ROS). Metabolic products of FFAs activate pro-inflammatory signaling pathways such as PKC and JNK leading to impaired insulin signaling. Saturated FFA also can activate toll-like receptor (TLR) signaling in β cells, followed by immune responses (51). FFA-induced TLR-mediated signaling has been shown to promote infiltration of macrophages through the secretion of chemokines such as CCL2 and CXCL1 in β cells (51). TLR4-mediated signaling also induced inflammation through increased production of pro-inflammatory cytokines such as IL-1β and IL-6. Thus, insulin resistance-mediated hyperglycemia and lipotoxicity induce inflammatory signaling to disturb pancreatic β cell homeostasis, leading to β cell failure in T2D patients (Figure 1).
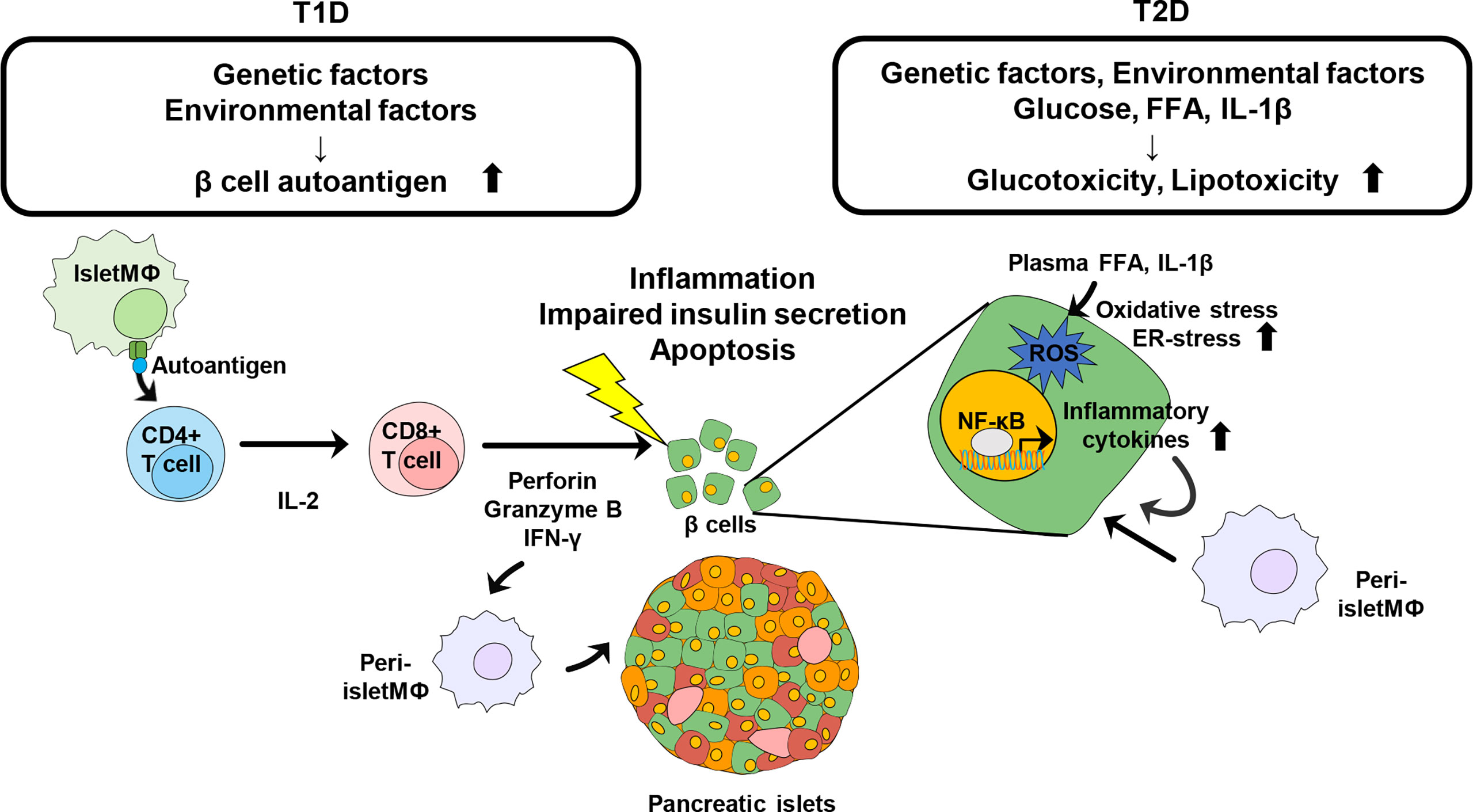
Figure 1 Schematic view of Immune modulation contributing to the destruction of pancreatic β cells in diabetes. Genetic and environmental factors contribute to generation of islet autoantigens. Intra islet macrophage or DC recognize the autoantigen and present it to naïve CD4+ T cells. Activated CD4+ T cells further activate CD8+ T cells to directly damage β cells and further induce the infiltration of other immune cells, leading to progression of T1D. Obesity is a risk factor of pathogenesis of T2D. Increased glucose, plasma FFA, and IL-1β promote oxidative stress and ER-stress in pancreatic β cells to induce insulin resistance as well as β cell destruction. Inflammatory cytokines recruit other immune cells into pancreatic islets and β cells and trigger further inflammation.
Immune Modulation by Microbiota
Recently, several studies have reported compositional changes of gut microbiota in both T1D and T2D and the alteration may contribute to the development of diabetes (52–54). The gut microbiota interacts with the host immune system via multiple mechanisms involved in TLR-mediated signaling and microbial products such as SCFAs (55, 56). In a rodent study, knock out of Myd88, an adaptor protein for multiple TLRs, has been shown to protect NOD mice from the development of T1D in specific pathogen free (SPF) condition. However, these Myd88-deficient NOD mice (NOD.Myd88−/−) in germ free condition developed T1D, implying that gut microbiota plays a pivotal role in pathogenesis of T1D in NOD mice (57). These findings propose that Myd88-dependent TLR signaling is crucial to T1D development and microbiota is required for the protective effects in the absence of TLR signaling in Myd88-deficient NOD mice.
Gut microbiota-mediated TLR signaling pathways also regulate the development of T2D. Lipopolysaccharides (LPS), a component of Gram-negative bacteria are known to promote inflammation by induction of pro-inflammatory cytokines (58). In a rodent study, it has been observed that HFD can change the composition of gut microbiota and increase plasma LPS concentration, resulting in low grade chronic inflammation (59). Microbiota-induced LPS binds to CD14/TLR4 complex and activates pro-inflammatory pathways leading to insulin resistance and β cell dysfunction (60, 61). In addition, HFD feeding reduced the expression of genes related to intestinal tight junction proteins and increases intestinal permeability, leading to impaired gut epithelial barrier (62). This impaired gut barrier also increases plasma LPS levels leading to LPS-induced inflammation and insulin resistance (63, 64). Thus, the gut microbiota-mediated LPS signaling pathways can modulate pro- or anti-diabetogenic pathways through multiple inflammatory signaling pathways.
The gut microbiota produces SCFAs by fermentation of nondigestible carbohydrates. The SCFAs including acetate, propionate and butyrate have an influence on immune systems in both T1D and T2D. Acetate can prevent T1D by reducing the population of autoreactive T cells in the pancreatic lymph node (65). The concentration of IL-21, an inflammatory cytokine contributing to T1D pathogenesis was also reduced in acetate-fed NOD mice. In addition to acetate, butyrate can also increase IL-10RA expression and potentiate integrity of intestinal epithelial cell (IEC) barrier through IL-10 signaling, leading to reduction of gut permeability (12, 66). Butyrate has also been reported to promote the generation of regulatory T cells which may be involved in autoimmunity suppression in T1D and lactate can be converted to butyrate by intestinal microbiota (67, 68). Thus, a low abundance of lactate- and butyrate-producing bacteria was observed within the gut microbiota of children with islet autoantibodies (20). As consistent, rodent study reported that the concentration of acetate and butyrate in germ free NOD.Myd88g−/− is much lower than in SPF ones (65). Altogether, these results clearly proposed that microbiota modulates autoimmunity through metabolites, such as acetate and butyrate in T1D.
SCFAs also play pivotal roles during pathogenesis of T2D. SCFA binds to FFAR2 or FFAR3, G protein-coupled receptors which are expressed in gut, pancreas, white adipose tissue, and immune cells (69). SCFA-mediated FFAR2 and/or FFAR3 signaling stimulate the secretion of glucagon-like peptide-1 (GLP-1), a well-known incretin from the intestinal L cells, to potentiate insulin secretion from pancreatic β cells. Thus, SCFA-mediated GLP-1 secretion leads to decrease the incidence of T2D by ameliorating glucose homeostasis. In addition to SCFAs, the secretion of GLP-1 can be stimulated by other gut microbial metabolic products, such as hydrogen sulfide(H2S), a gas metabolite (70).
Gut microbiota regulates host bile acid metabolism through farnesoid x receptor (FXR) and TGR5 receptor. In general, primary bile acids are synthesized in the liver from cholesterol by CYP7A1, rate-limiting enzyme in the bile acid biosynthetic pathway, followed by conjugation to glycine and taurine for the secretion into intestinal lumen upon food intake (71). In the intestinal tract, gut microbiota converts primary bile acids to secondary bile acids. While primary bile acids prefer to bind FXR suppresses the hepatic synthesis of bile acids, secondary bile acids tend to bind TGR5 receptor and stimulates GLP-1 secretion from intestinal L-cells (72) (Figure 2).
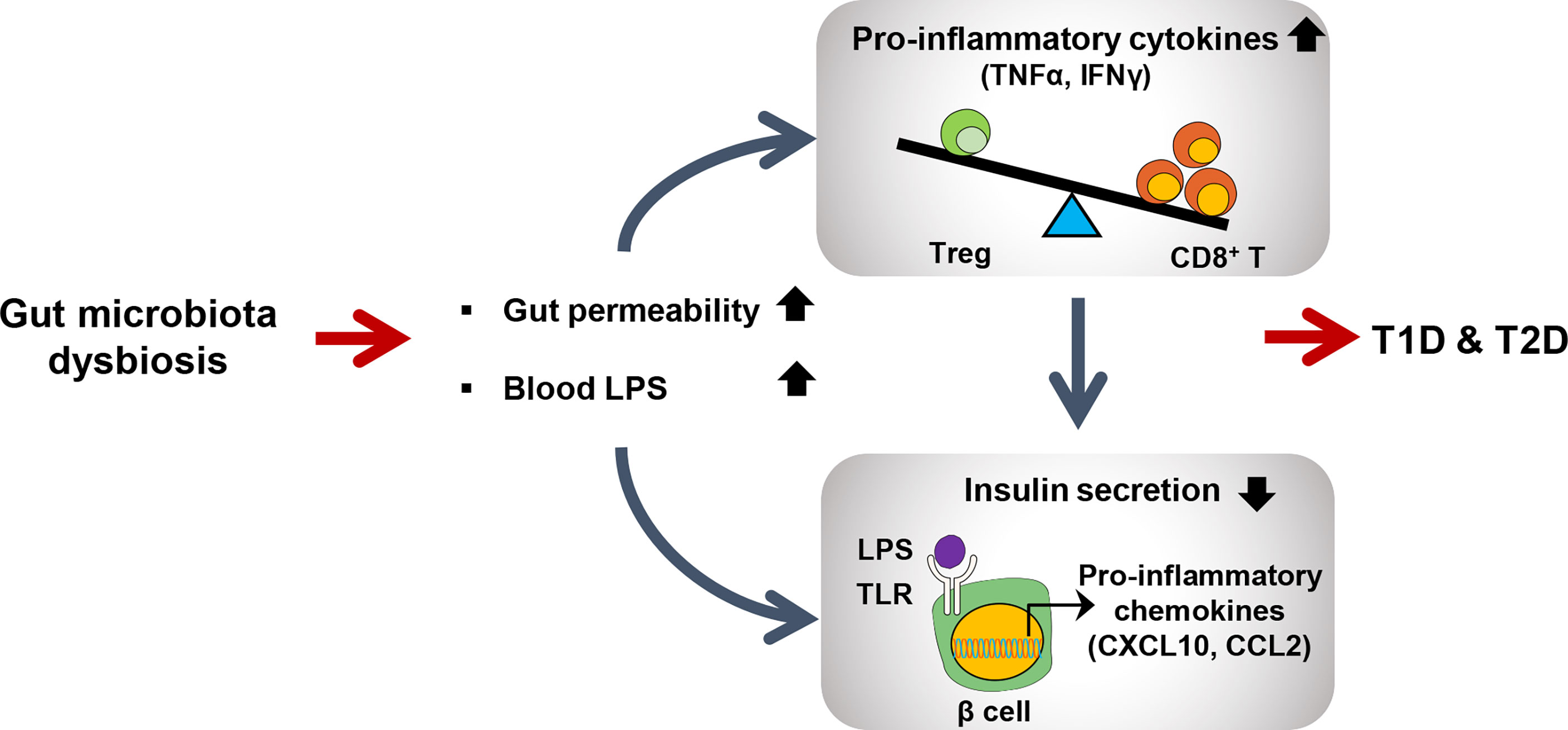
Figure 2 Schematic view of gut microbiota dysbiosis contributing to both type 1 and type2 diabetes. Dysbiosis leads to increased intestinal permeability and LPS level in blood. Elevated LPS level activates CD8+ T cells to produce proinflammatory cytokines to further activate immune signaling pathways in many peripheral tissues including pancreatic beta cells. LPS-induced TLR signaling in pancreatic beta cells promotes inflammation to reduce insulin secretion.
Therapeutic Strategies Using Immune Modulation in T1D
Anti-CD3 Antibody
CD4+ T and CD8+ effector T cells are key factors of pancreatic β cell destruction and overt T1D development. Therefore, targeting T cells has been considered the most effective approach to the progression of T1D. Anti-CD3 monoclonal antibody (mAb) is a one of promising therapeutic approaches targeting T cells. Although the exact mechanism of anti-CD3 mAb is not clear, it has been considered to deplete autoreactive T cells and preserve regulatory T cell activity (73). In NOD mouse, treatment of anti-CD3 mAb induced long-lasting remission to ameliorate diabetic pathogenesis including restoration of normoglycemia (74). Thus, numerous human clinical studies using humanized anti-CD3 mAb has been reported for the treatment of pancreatic β cell destruction (74). In 1986, muromonab-CD3(Orthoclone OKT3), the first antibody recognizing CD3 molecules on human T cells, was approved in the US, but it is currently not available cause its side effects such as cytokine storm. Therefore, two humanized anti-CD3 antibodies engineered to improve the side effect of OKT3 called teplizumab and otelixizumab were developed. These antibodies retained binding regions of OKT3 have been investigated in numerous clinical studies for the prevention of T1D and resulted in preserved β cells (75–78).
Anti-CD20 Antibody
B cells act as APC and have a pathogenic role in T1D. Therefore, approaches targeting B cells also have been taken to alter T1D progression. Anti-CD20 or anti-CD22 therapy showed prevention or reversal of T1D in NOD by depleting B (79, 80). Especially, treatment of rituximab, anti-CD20 mAb, transiently depleted B cells and delayed the rate of C peptide decline in T1D compared with a placebo group, although these effects were diminished after 2 years (81).
Anti-Thymocyte Globulin (ATG)
Anti-thymocyte globulin (ATG) is another approach to target T cells for the prevention of pancreatic β cell destruction. ATG is rabbit-derived IgG against human T cells and thymocytes. ATG has been reported to deplete T cells through apoptosis and expand regulatory T cells (79, 82). Indeed, in a clinical trial, low-dose ATG has been shown to reduce glycated hemoglobin (Hb1Ac), a clinical marker of glycemic control, up to 2 years compared to placebo (80).
Fusion Proteins: Alefacept and Abatacept
Fusion proteins, alefacept and abatacept also can deplete T cells and preserve β cell function. Alefacept consists of LFA-3 molecules and Fc portion of IgG and binds CD2 of effector memory T cell (83). Administration of alefacept led to increase of Treg/Teff ratio compared with placebo treatment (84). Abatacept is a CTLA4-IgG fusion protein that inhibits APC function to prevent T cell activation by blocking CD80 and CD86 on APC (85). In a clinical trial, the group of abatacept maintained lower HbA1c level compared with placebo after 3 years from T1D diagnosis (86). In addition, decreased level of follicular helper T cells involved in proliferation and isotype switching of B cells, was found in abatacept-treated T1d patients (87). These findings suggest that targeting T cells can modulate autoimmunity to prevent pancreatic β cell failure, although most of the above clinical trials failed to reach the endpoints in T1D.
Autoantigen-Specific Therapies
β cell autoantigen-specific therapies is promising strategies to suppress autoimmune responses by selectively suppressing autoreactive T cell activation or expanding Treg (88, 89). One of these approaches has been applied with a peptide vaccine. For example, vaccination with insulin mimotope, an antigen-mimicking peptide, resulted in the prevention of T1D in NOD by converting naïve T cells into FOXP3+ Treg (90). Other novel insulin mimotopes also promoted induction of insulin-specific FOXP3+ Treg in humanized mice (91).
Furthermore, APC-based therapies with DCs and immunosuppressive macrophages and have attracted clinical interests in T1D. Administration of tolerogenic DC loaded with encoding autoantigen can suppress autoreactive T cells. T1D patients treated tolerogenic DCs which can induce proinsulin-specific Treg maintained C peptide level for 6 months (92, 93). Adaptive transfer of immunosuppressive macrophages prevented T1D in NOD through anti-inflammatory responses (94).
Administration of cytokines such as IL-2 also prevents autoimmunity in T1D. IL-2 is essential not only to the activation of T cells but also the expansion of FOXP3+ Treg. It has been reported that FOXP3+ Treg preferentially responds to low-dose of IL-2 versus T cells (95). In NOD studies, low-dose of IL-2 increased Treg population to prevent the progression of diabetes (95). Furthermore, administration of low dose of aldesleukin, a recombinant IL-2, increased the population of Treg without drug-relative adverse effects in T1D patients versus placebo (96). Currently, a phase 2 clinical trial evaluating the effect of ultra-low dose aldesleukin on the preservation of β cell function is ongoing (NCT03782636) (96) (Table 1).
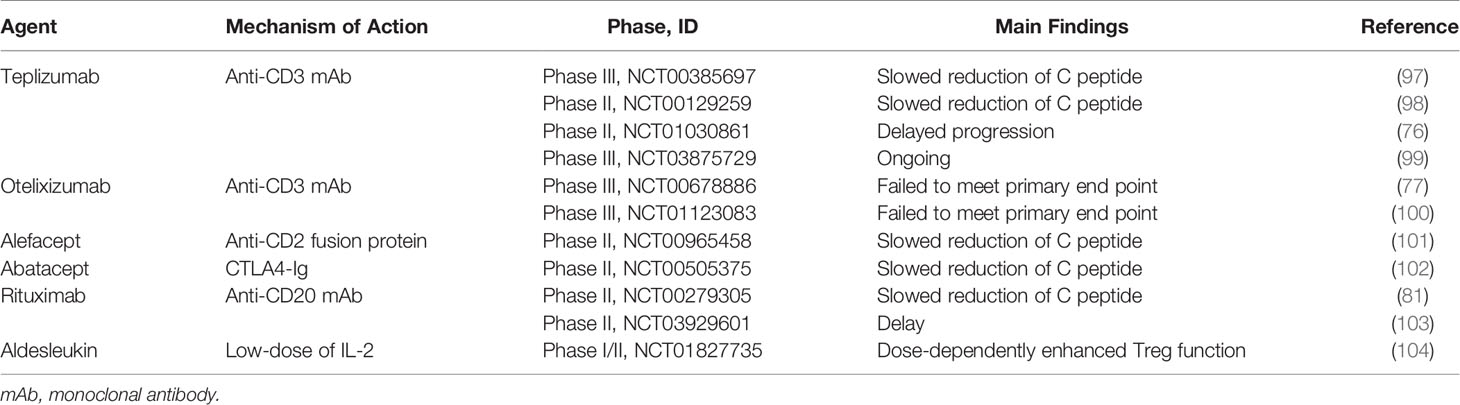
Table 1 Summary of major clinical trials for anti-inflammatory therapies on T1D.
Anti-Inflammatory Therapeutic Strategies for T2D
T2D is a chronic metabolic disorder associated with obesity. Abnormal fat accumulation in adipose tissue and non-adipose organs including liver and pancreatic β cells promotes inflammatory responses such as the production of pro-inflammatory cytokines and chemokines, resulting in insulin resistance and β cell dysfunction. Therefore, anti-inflammatory approaches have been considered as a therapeutic strategy for the prevention of T2D.
Metformin
Metformin is one of the most common drugs for the treatment of T2D. Metformin reduces blood glucose levels by suppressing hepatic glucose production and improving insulin sensitivity. In addition, metformin has a pivotal role in anti-inflammatory responses through inhibition of NF-κB signaling (105). Although the mechanisms of metformin remain unclear, it has been widely accepted that metformin acts via activation of AMP-activated protein kinase (AMPK). AMPK plays a potent anti-inflammatory effect by inhibiting NF-κB and regulating redox balance (106). Treatment of metformin increases phosphorylation of AMPK, enhances insulin sensitivity, and decreased ceramide and DAG related to saturated FFA in HFD-fed mice (107). Another study also demonstrated that metformin treatment increased Treg population and decreased Th17 by inducing FGF21 production in HFD-fed mice (108).
GLP-1 Receptor Agonist & DPP4i
GLP-1 receptor agonists (GLP-1 RAs) and dipeptidyl peptidase 4 inhibitors (DPP-4is) have been widely prescribed for the T2D patients in clinic. GLP-1 RA such as liraglutide and exendin-4 binds to GLP-1 receptor in pancreatic β cell to stimulate Ca2+ influx and insulin secretion (109). Furthermore, GLP-1 RA has anti-inflammatory effects by suppressing expression of proinflammatory cytokines and chemokines. Exendin-4 treatment to human islet cells suppressed the expression of inflammatory genes and protected β cells from cytokine-induced apoptosis (110, 111). In addition, GLP-1 RA reduced inflammatory macrophage activation and inflammatory cytokines such as IL-1β and IL-6 in T2D patients (112).
DPP-4is are anti-hyperglycemic drugs that increase insulin secretion and reduce glucose level by preventing the activity of DPP-4 degrading incretins such as GLP-1. These drugs also have anti-inflammatory potential. DPP-4i including sitagliptin suppresses nod-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome in human macrophages via inhibition PKC pathway and reduced inflammatory cytokines including TNF-α, IL-6, IL-1β in WAT (113, 114). In human studies, sitagliptin treatment reduced levels of Hb1Ac and inflammatory cytokines in T2D patients (115).
IL-1 Receptor Antagonist
Hyperglycemia increases the production of IL-1 which drives dysfunction of β cells. Therefore, blocking IL-1 signaling pathway can reduce islet inflammation and delay T2D development. In rodent studies, IL-1 receptor antagonist (IL-1Ra) reduced hyperglycemia and enhanced function of β cells by reducing expression of TNF-α, MCP-1 and IL-6 in liver and islet (47, 48). In a clinical trial, administration of anakinra, IL-1R blockade, increased insulin secretion and improved glycemia in T2D patients versus placebo (116). Canakinumab, an anti-IL-1β antibody, also reduced plasma IL-6 level, leading to improvement of glycemia in T2D patients (117) (Table 2).
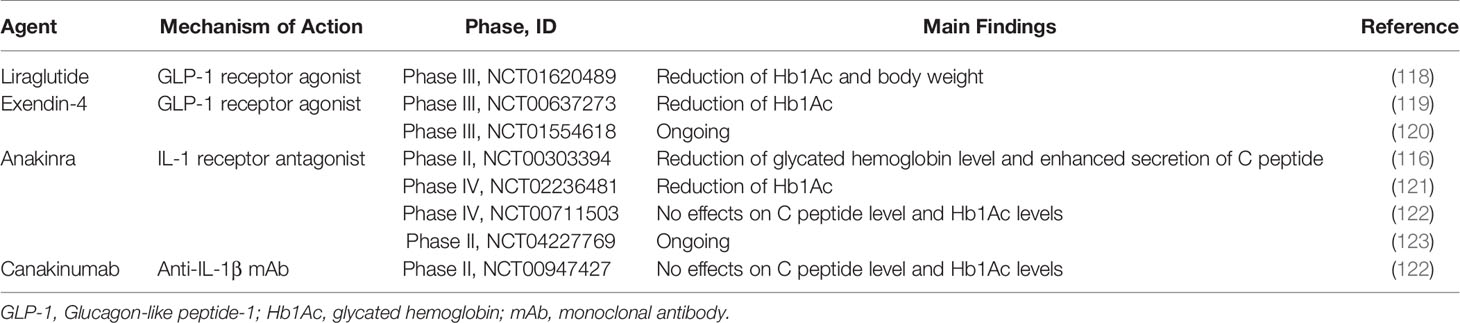
Table 2 Summary of major clinical trials for anti-inflammatory therapies on T2D.
Gut Microbiota as Novel Therapeutic Approach for Diabetes
Recently, several studies have demonstrated that gut microbiota has important roles in the development of diabetes. Therefore, targeting gut microbiota has been considered as therapeutic approaches for diabetes.
Probiotics
Probiotics are live microorganisms regarded as beneficial to host gut microbiota homeostasis. Several studies demonstrated that adequate consumption of probiotics can improve immune responses, insulin resistance, and insulin secretion by modulation of gut microbiota (124–126). For example, Lactobacillus johnsonii N62 support maintain IEC barrier by modifying intestinal microbiota and Lactobacillus rhamnosus GG is known to attenuate LPS-induced inflammation by reducing TRL4 expression (127–129). Lactobacillus reuteri also has been reported that enhance intestinal barrier function and have anti-inflammatory effects by suppressing T cell response or inducing Treg (130). In NOD mouse model, administration of a probiotic combination including Lactobacillus acidophilus and Bifidobacterium lactis inhibited T1D development by reducing gut permeability, CD8+ T cells polarization and increasing Treg population (131). In humans, numerous clinical studies using probiotics for T1D patients are ongoing. Probiotics has been also considered for the prevention of T2D. Treatment of 14 probiotics decreased Hb1Ac level and increased C peptide level in db/db mice by increasing SCFA-producing bacteria and reducing levels of Escherichia and inflammatory cytokines (132). This compositional change of gut microbiota contributes to improving intestinal permeability and β cell function by increasing GLP-1 secretion. In clinical studies, administration of Lactobacillus acidophilus or Bifidobacterium bifidum also enhanced glycemic control and reduced inflammatory cytokines and oxidative stress in T2D patients versus placebo (133, 134).
Fecal Microbiota Transplantation
Recently, fecal microbiota transplantation (FMT) has considered as a therapeutic strategy for diabetes. Oral transfer of fecal bacteria from NOD.Myd88−/− altered gut microbial composition of diabetes-prone NOD, leading to a delay in the progression of T1D (135). In a randomized controlled trial, the transfer of fecal bacteria from healthy donors improved β cell function in T1D patients by reducing CXCR3+ T cells (136). Furthermore, a FMT treatment to HFD and streptozotocin-induced T2D mice model improved insulin resistance and destruction of β cells by a decrease of inflammatory cytokines (137). In humans, FMT from lean donors to obese recipients for 6 weeks increased levels of butyrate-producing intestinal microbiota to improve insulin sensitivity (138). This result implies that FMT treatment also promising therapeutic strategy for the prevention of T2D (Table 3).
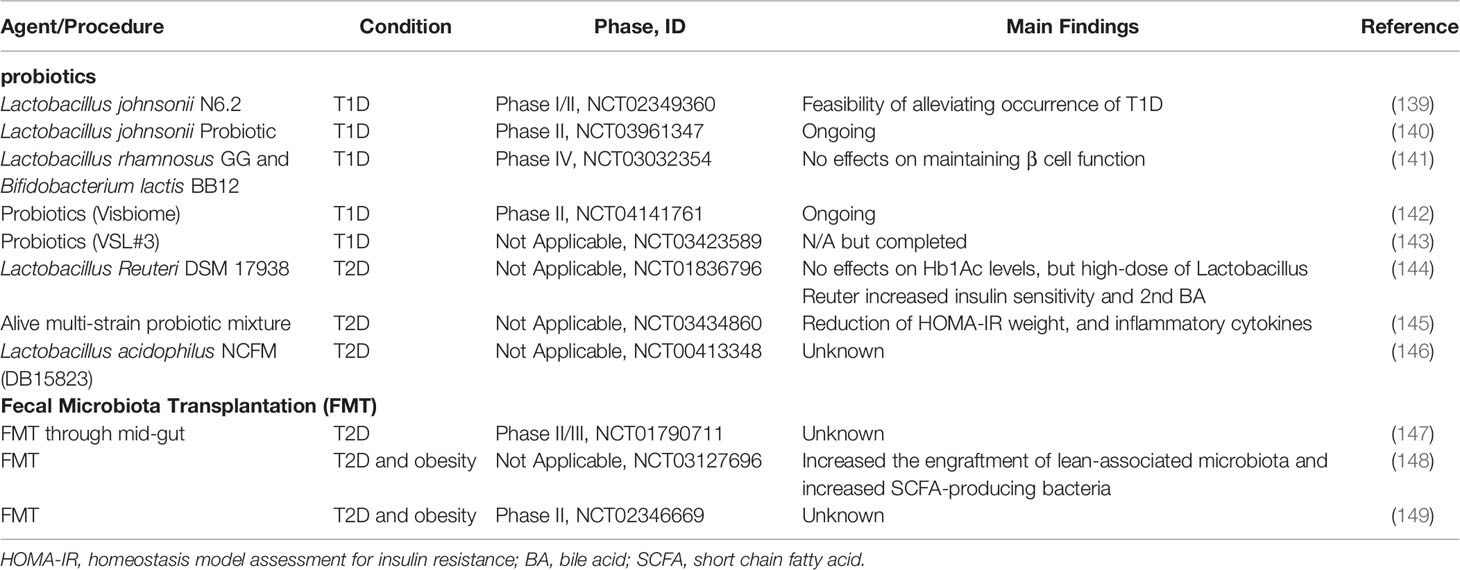
Table 3 Summary of current clinical trials evaluating the efficacy of probiotics or FMT on diabetes.
Conclusions
Diabetes is a multifactorial disease caused by genetic factors as well as environmental factors that affect abnormal immune modulation, leading to the dysfunction and destruction of pancreatic β cells. Although the mechanisms stimulating the change of the immune responses are diverse, and are not fully understood yet, targeting effector CD4+ T cells and CD8+ T cells, key mediators of β cell damage, has been the focus of immunotherapeutic approaches to prevent diabetes. Indeed, anti-CD3 mAb therapy such as teplizumab showed promising results including the preservation of β cell function in clinical studies. However, most of initial phase III trials using anti-CD3 mAbs in patients with T1D were failed because they did not meet their primary endpoint. Moreover, several adverse effects were revealed including rash or Epstein-Barr virus (EBV) reactivation. Therefore, novel therapeutic strategies are required to prevent adverse effects for the treatment of T1D.
The aberrant immune modulation via obesity is also crucial for the development of T2D. Although the immunomodulatory mechanisms of T2D progression remain unclear, it has been believed that obesity-mediated metabolic stresses such as ER-stress and oxidative stress contributed to T2D development by induction of pro-inflammatory responses. Anti-inflammatory agents such as metformin as well as GLP-1 RA and DPP-4i are widely used to treat T2D patients. Blocking IL-1 signaling is also one of the immunotherapeutic strategies for the treatment of T2D.
Currently, the compositional change of gut microbiota has been associated with T1D and T2D. Although the roles of gut microbiota are still not fully understood, several studies have demonstrated that the presence or absence of specific microbiota can contribute to immune modulation, leading to the development of diabetes by abnormally regulating host metabolisms. Interestingly, FMT has attracted attention as a therapeutic strategy for diabetes. Several studies have reported that FMT can improve β cell destruction and delay the progression of T1D in mouse models or humans. Altogether, numerous studies by immune modulation are ongoing for the prevention and treatment for diabetes patients. And novel therapeutic strategies to minimize adverse effects are still required for the development of immune modulator to prevent T1D and T2D.
Author Contributions
SF drafted and revised the manuscript. SJ wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Bio and Medical Technology Development Program (NRF-2017M3A9F3046538), National Research Foundation (NRF-2021R1A2C2009749), Ministry of Health and Welfare (HR18C0012) and faculty research grant from the Yonsei University College of Medicine (6-2018-0098) to SF.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Östman J, Lönnberg G, Arnqvist H, Blohme G, Bolinder J, Schnell AE, et al. Gender Differences and Temporal Variation in the Incidence of Type 1 Diabetes: Results of 8012 Cases in the Nationwide Diabetes Incidence Study in Sweden 1983–2002. J Internal Med (2008) 263(4):386–94. doi: 10.1111/j.1365-2796.2007.01896.x
2. Oh J-Y, Barrett-Connor E, Wedick NM, Wingard DL. Endogenous Sex Hormones and the Development of Type 2 Diabetes in Older Men and Women: The Rancho Bernardo Study. Diabetes Care (2002) 25(1):55–60. doi: 10.2337/diacare.25.1.55
3. Chen X, McClusky R, Chen J, Beaven SW, Tontonoz P, Arnold AP, et al. The Number of X Chromosomes Causes Sex Differences in Adiposity in Mice. PloS Genet (2012) 8(5):e1002709. doi: 10.1371/journal.pgen.1002709
4. Hall E, Volkov P, Dayeh T, Esguerra JLS, Salö S, Eliasson L, et al. Sex Differences in the Genome-Wide DNA Methylation Pattern and Impact on Gene Expression, microRNA Levels and Insulin Secretion in Human Pancreatic Islets. Genome Biol (2014) 15(12):1–22. doi: 10.1186/s13059-014-0522-z
5. Yoon J-W, Jun H-S. Autoimmune Destruction of Pancreatic β Cells. Am J Ther (2005) 12(6):580–91. doi: 10.1097/01.mjt.0000178767.67857.63
6. Pihoker C, Gilliam LK, Hampe CS, Lernmark Å. Autoantibodies in Diabetes. Diabetes (2005) 54(suppl 2):S52–61. doi: 10.2337/diabetes.54.suppl_2.S52
7. Delovitch TL, Singh B. The Nonobese Diabetic Mouse as a Model of Autoimmune Diabetes: Immune Dysregulation Gets the NOD. Immunity (1997) 7(6):727–38. doi: 10.1016/S1074-7613(00)80392-1
8. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, et al. Increased Number of Islet-Associated Macrophages in Type 2 Diabetes. Diabetes (2007) 56(9):2356–70. doi: 10.2337/db06-1650
9. Ehses JA, Ellingsgaard H, Böni-Schnetzler M, Donath MY. Pancreatic Islet Inflammation in Type 2 Diabetes: From α and β Cell Compensation to Dysfunction. Arch Physiol Biochem (2009) 115(4):240–7. doi: 10.1080/13813450903025879
10. Khaldi M, Guiot Y, Gilon P, Henquin J-C, Jonas J-C. Increased Glucose Sensitivity of Both Triggering and Amplifying Pathways of Insulin Secretion in Rat Islets Cultured for 1 Wk in High Glucose. Am J Physiology-Endocrinology Metab (2004) 287(2):E207–E17. doi: 10.1152/ajpendo.00426.2003
11. Oprescu AI, Bikopoulos G, Naassan A, Allister EM, Tang C, Park E, et al. Free Fatty Acid–Induced Reduction in Glucose-Stimulated Insulin Secretion: Evidence for a Role of Oxidative Stress In Vitro and In Vivo. Diabetes (2007) 56(12):2927–37. doi: 10.2337/db07-0075
12. Vaarala O, Atkinson MA, Neu J. The “Perfect Storm” for Type 1 Diabetes: The Complex Interplay Between Intestinal Microbiota, Gut Permeability, and Mucosal Immunity. Diabetes (2008) 57(10):2555–62. doi: 10.2337/db08-0331
13. Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The Gut Microbiota as an Environmental Factor That Regulates Fat Storage. Proc Natl Acad Sci (2004) 101(44):15718–23. doi: 10.1073/pnas.0407076101
14. Zhang X, Shen D, Fang Z, Jie Z, Qiu X, Zhang C, et al. Human Gut Microbiota Changes Reveal the Progression of Glucose Intolerance. PloS One (2013) 8(8):e71108. doi: 10.1371/journal.pone.0071108
15. Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, et al. Short-Chain Fatty Acids Stimulate Glucagon-Like Peptide-1 Secretion via the G-Protein–Coupled Receptor FFAR2. Diabetes (2012) 61(2):364–71. doi: 10.2337/db11-1019
16. Leiva-Gea I, Sánchez-Alcoholado L, Martín-Tejedor B, Castellano-Castillo D, Moreno-Indias I, Urda-Cardona A, et al. Gut Microbiota Differs in Composition and Functionality Between Children With Type 1 Diabetes and MODY2 and Healthy Control Subjects: A Case-Control Study. Diabetes Care (2018) 41(11):2385–95. doi: 10.2337/dc18-0253
17. Larsen N, Vogensen FK, Van Den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut Microbiota in Human Adults With Type 2 Diabetes Differs From Non-Diabetic Adults. PloS One (2010) 5(2):e9085. doi: 10.1371/journal.pone.0009085
18. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut Microbiota in Children With Type 1 Diabetes Differs From That in Healthy Children: A Case-Control Study. BMC Med (2013) 11(1):1–12. doi: 10.1186/1741-7015-11-46
19. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The Human Gut Microbiome in Early-Onset Type 1 Diabetes From the TEDDY Study. Nature (2018) 562(7728):589–94. doi: 10.1038/s41586-018-0620-2
20. De Goffau MC, Luopajärvi K, Knip M, Ilonen J, Ruohtula T, Härkönen T, et al. Fecal Microbiota Composition Differs Between Children With β-Cell Autoimmunity and Those Without. Diabetes (2013) 62(4):1238–44. doi: 10.2337/db12-0526
21. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A Metagenome-Wide Association Study of Gut Microbiota in Type 2 Diabetes. Nature (2012) 490(7418):55–60. doi: 10.1038/nature11450
22. Li Q, Chang Y, Zhang K, Chen H, Tao S, Zhang Z. Implication of the Gut Microbiome Composition of Type 2 Diabetic Patients From Northern China. Sci Rep (2020) 10(1):1–8. doi: 10.1038/s41598-020-62224-3
23. Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut Metagenome in European Women With Normal, Impaired and Diabetic Glucose Control. Nature (2013) 498(7452):99–103. doi: 10.1038/nature12198
24. Koo BK, Chae S, Kim KM, Kang MJ, Kim EG, Kwak SH, et al. Identification of Novel Autoantibodies in Type 1 Diabetic Patients Using a High-Density Protein Microarray. Diabetes (2014) 63(9):3022–32. doi: 10.2337/db13-1566
25. Yu L, Robles DT, Abiru N, Kaur P, Rewers M, Kelemen K, et al. Early Expression of Antiinsulin Autoantibodies of Humans and the NOD Mouse: Evidence for Early Determination of Subsequent Diabetes. Proc Natl Acad Sci (2000) 97(4):1701–6. doi: 10.1073/pnas.040556697
26. Clark AL, Urano F. Endoplasmic Reticulum Stress in Beta Cells and Autoimmune Diabetes. Curr Opin Immunol (2016) 43:60–6. doi: 10.1016/j.coi.2016.09.006
27. Todd DJ, Lee A-H, Glimcher LH. The Endoplasmic Reticulum Stress Response in Immunity and Autoimmunity. Nat Rev Immunol (2008) 8(9):663–74. doi: 10.1038/nri2359
28. Phelps EA, Cianciaruso C, Michael IP, Pasquier M, Kanaani J, Nano R, et al. Aberrant Accumulation of the Diabetes Autoantigen GAD65 in Golgi Membranes in Conditions of ER Stress and Autoimmunity. Diabetes (2016) 65(9):2686–99. doi: 10.2337/db16-0180
29. Hu X, Deutsch AJ, Lenz TL, Onengut-Gumuscu S, Han B, Chen W-M, et al. Additive and Interaction Effects at Three Amino Acid Positions in HLA-DQ and HLA-DR Molecules Drive Type 1 Diabetes Risk. Nat Genet (2015) 47(8):898. doi: 10.1038/ng.3353
30. Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, et al. Prime Role for an Insulin Epitope in the Development of Type 1 Diabetes in NOD Mice. Nature (2005) 435(7039):220–3. doi: 10.1038/nature03523
31. Congia M, Patel S, Cope AP, De Virgiliis S, Sønderstrup G. T Cell Epitopes of Insulin Defined in HLA-DR4 Transgenic Mice Are Derived From Preproinsulin and Proinsulin. Proc Natl Acad Sci (1998) 95(7):3833–8. doi: 10.1073/pnas.95.7.3833
32. Marroqui L, Dos Santos RS, de Brachène AC, Marselli L, Marchetti P, Eizirik DL. Interferon-α Mediates Human Beta Cell HLA Class I Overexpression, Endoplasmic Reticulum Stress and Apoptosis, Three Hallmarks of Early Human Type 1 Diabetes. Diabetologia (2017) 60(4):656–67. doi: 10.1007/s00125-016-4201-3
33. De Jersey J, Snelgrove SL, Palmer SE, Teteris SA, Mullbacher A, Miller JF, et al. β Cells Cannot Directly Prime Diabetogenic CD8 T Cells in Nonobese Diabetic Mice. Proc Natl Acad Sci (2007) 104(4):1295–300. doi: 10.1073/pnas.0610057104
34. Ouyang Q, Standifer NE, Qin H, Gottlieb P, Verchere CB, Nepom GT, et al. Recognition of HLA Class I–Restricted β-Cell Epitopes in Type 1 Diabetes. Diabetes (2006) 55(11):3068–74. doi: 10.2337/db06-0065
35. Pinkse GG, Tysma OH, Bergen CA, Kester MG, Ossendorp F, van Veelen PA, et al. Autoreactive CD8 T Cells Associated With β Cell Destruction in Type 1 Diabetes. Proc Natl Acad Sci (2005) 102(51):18425–30. doi: 10.1073/pnas.0508621102
36. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T Cell-Derived Interleukin-10 Limits Inflammation at Environmental Interfaces. Immunity (2008) 28(4):546–58. doi: 10.1016/j.immuni.2008.02.017
37. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective Suppressor Function in CD4+ CD25+ T-Cells From Patients With Type 1 Diabetes. Diabetes (2005) 54(1):92–9. doi: 10.2337/diabetes.54.1.92
38. Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The Effector T Cells of Diabetic Subjects Are Resistant to Regulation via CD4+ FOXP3+ Regulatory T Cells. J Immunol (2008) 181(10):7350–5. doi: 10.4049/jimmunol.181.10.7350
39. Kahn BB, Flier JS. Obesity and Insulin Resistance. J Clin Invest (2000) 106(4):473–81. doi: 10.1172/JCI10842
40. Winzell MS, Ahrén B. The High-Fat Diet–Fed Mouse: A Model for Studying Mechanisms and Treatment of Impaired Glucose Tolerance and Type 2 Diabetes. Diabetes (2004) 53(suppl 3):S215–S9. doi: 10.2337/diabetes.53.suppl_3.S215
41. Goossens GH. The Role of Adipose Tissue Dysfunction in the Pathogenesis of Obesity-Related Insulin Resistance. Physiol Behav (2008) 94(2):206–18. doi: 10.1016/j.physbeh.2007.10.010
42. Lagathu C, Yvan-Charvet L, Bastard J-P, Maachi M, Quignard-Boulange A, Capeau J, et al. Long-Term Treatment With Interleukin-1β Induces Insulin Resistance in Murine and Human Adipocytes. Diabetologia (2006) 49(9):2162–73. doi: 10.1007/s00125-006-0335-z
43. Sartipy P, Loskutoff DJ. Monocyte Chemoattractant Protein 1 in Obesity and Insulin Resistance. Proc Natl Acad Sci (2003) 100(12):7265–70. doi: 10.1073/pnas.1133870100
44. Cawthorn WP, Sethi JK. TNF-α and Adipocyte Biology. FEBS Lett (2008) 582(1):117–31. doi: 10.1016/j.febslet.2007.11.051
45. Böni-Schnetzler M, Meier DT eds. Islet Inflammation in Type 2 Diabetes. Springer: Seminars in Immunopathology (2019).
46. Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. Glucose-Induced β Cell Production of IL-1β Contributes to Glucotoxicity in Human Pancreatic Islets. J Clin Invest (2002) 110(6):851–60. doi: 10.1172/JCI200215318
47. Sauter NS, Schulthess FT, Galasso R, Castellani LW, Maedler K. The Antiinflammatory Cytokine Interleukin-1 Receptor Antagonist Protects From High-Fat Diet-Induced Hyperglycemia. Endocrinology (2008) 149(5):2208–18. doi: 10.1210/en.2007-1059
48. Ehses J, Lacraz G, Giroix M-H, Schmidlin F, Coulaud J, Kassis N, et al. IL-1 Antagonism Reduces Hyperglycemia and Tissue Inflammation in the Type 2 Diabetic GK Rat. Proc Natl Acad Sci (2009) 106(33):13998–4003. doi: 10.1073/pnas.0810087106
49. Ortis F, Cardozo AK, Crispim D, Störling J, Mandrup-Poulsen T, Eizirik D. Cytokine-Induced Proapoptotic Gene Expression in Insulin-Producing Cells Is Related to Rapid, Sustained, and Nonoscillatory Nuclear Factor-κb Activation. Mol Endocrinol (2006) 20(8):1867–79. doi: 10.1210/me.2005-0268
50. Tumova J, Andel M, Trnka J. Excess of Free Fatty Acids as a Cause of Metabolic Dysfunction in Skeletal Muscle. Physiol Res (2016) 65(2):193. doi: 10.33549/physiolres.932993
51. Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, et al. Saturated Fatty Acid and TLR Signaling Link β Cell Dysfunction and Islet Inflammation. Cell Metab (2012) 15(4):518–33. doi: 10.1016/j.cmet.2012.01.023
52. Brugman S, Klatter F, Visser J, Wildeboer-Veloo A, Harmsen H, Rozing J, et al. Antibiotic Treatment Partially Protects Against Type 1 Diabetes in the Bio-Breeding Diabetes-Prone Rat. Is the Gut Flora Involved in the Development of Type 1 Diabetes? Diabetologia (2006) 49(9):2105–8. doi: 10.1007/s00125-006-0334-0
53. Patterson E, Marques TM, O’Sullivan O, Fitzgerald P, Fitzgerald GF, Cotter PD, et al. Streptozotocin-Induced Type-1-Diabetes Disease Onset in Sprague–Dawley Rats Is Associated With an Altered Intestinal Microbiota Composition and Decreased Diversity. Microbiology (2015) 161(1):182–93. doi: 10.1099/mic.0.082610-0
54. Navab-Moghadam F, Sedighi M, Khamseh ME, Alaei-Shahmiri F, Talebi M, Razavi S, et al. The Association of Type II Diabetes With Gut Microbiota Composition. Microbial pathogenesis (2017) 110:630–6. doi: 10.1016/j.micpath.2017.07.034
55. Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota Regulates Type 1 Diabetes Through Toll-Like Receptors. Proc Natl Acad Sci (2015) 112(32):9973–7. doi: 10.1073/pnas.1508740112
56. Puddu A, Sanguineti R, Montecucco F, Viviani GL. Evidence for the Gut Microbiota Short-Chain Fatty Acids as Key Pathophysiological Molecules Improving Diabetes. Mediators Inflammation (2014) 2014. doi: 10.1155/2014/162021
57. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate Immunity and Intestinal Microbiota in the Development of Type 1 Diabetes. Nature (2008) 455(7216):1109–13. doi: 10.1038/nature07336
58. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes (2007) 56(7):1761–72. doi: 10.2337/db06-1491
59. Cani PD, Osto M, Geurts L, Everard A. Involvement of Gut Microbiota in the Development of Low-Grade Inflammation and Type 2 Diabetes Associated With Obesity. Gut Microbes (2012) 3(4):279–88. doi: 10.4161/gmic.19625
60. Kim JJ, Sears DD. TLR4 and Insulin Resistance. Gastroenterology Res Pract (2010) 2010. doi: 10.1155/2010/212563
61. Garay-Malpartida HM, Mourão RF, Mantovani M, Santos IA, Sogayar MC, Goldberg AC. Toll-Like Receptor 4 (TLR4) Expression in Human and Murine Pancreatic Beta-Cells Affects Cell Viability and Insulin Homeostasis. BMC Immunol (2011) 12(1):1–8. doi: 10.1186/1471-2172-12-18
62. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet–Induced Obesity and Diabetes in Mice. Diabetes (2008) 57(6):1470–81. doi: 10.2337/db07-1403
63. Jayashree B, Bibin Y, Prabhu D, Shanthirani C, Gokulakrishnan K, Lakshmi B, et al. Increased Circulatory Levels of Lipopolysaccharide (LPS) and Zonulin Signify Novel Biomarkers of Proinflammation in Patients With Type 2 Diabetes. Mol Cell Biochem (2014) 388(1):203–10. doi: 10.1007/s11010-013-1911-4
64. Miranda MCG, Oliveira RP, Torres L, Aguiar SLF, Pinheiro-Rosa N, Lemos L, et al. Frontline Science: Abnormalities in the Gut Mucosa of Non-Obese Diabetic Mice Precede the Onset of Type 1 Diabetes. J leukocyte Biol (2019) 106(3):513–29. doi: 10.1002/JLB.3HI0119-024RR
65. Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut Microbial Metabolites Limit the Frequency of Autoimmune T Cells and Protect Against Type 1 Diabetes. Nat Immunol (2017) 18(5):552–62. doi: 10.1038/ni.3713
66. Zheng L, Kelly CJ, Battista KD, Schaefer R, Lanis JM, Alexeev EE, et al. Microbial-Derived Butyrate Promotes Epithelial Barrier Function Through IL-10 Receptor–Dependent Repression of Claudin-2. J Immunol (2017) 199(8):2976–84. doi: 10.4049/jimmunol.1700105
67. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, Deroos P, et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
68. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The Orphan G Protein-Coupled Receptors GPR41 and GPR43 are Activated by Propionate and Other Short Chain Carboxylic Acids. J Biol Chem (2003) 278(13):11312–9. doi: 10.1074/jbc.M211609200
69. Ichimura A, Hasegawa S, Kasubuchi M, Kimura I. Free Fatty Acid Receptors as Therapeutic Targets for the Treatment of Diabetes. Front Pharmacol (2014) 5:236. doi: 10.3389/fphar.2014.00236
70. Pichette J, Fynn-Sackey N, Gagnon J. Hydrogen Sulfide and Sulfate Prebiotic Stimulates the Secretion of GLP-1 and Improves Glycemia in Male Mice. Endocrinology (2017) 158(10):3416–25. doi: 10.1210/en.2017-00391
71. Staels B, Fonseca VA. Bile Acids and Metabolic Regulation: Mechanisms and Clinical Responses to Bile Acid Sequestration. Diabetes Care (2009) 32(suppl 2):S237–S45. doi: 10.2337/dc09-S355
72. Kim H, Fang S. Crosstalk Between FXR and TGR5 Controls Glucagon-Like Peptide 1 Secretion to Maintain Glycemic Homeostasis. Lab Anim Res (2018) 34(4):140–6. doi: 10.5625/lar.2018.34.4.140
73. Penaranda C, Tang Q, Bluestone JA. Anti-CD3 Therapy Promotes Tolerance by Selectively Depleting Pathogenic Cells While Preserving Regulatory T Cells. J Immunol (2011) 187(4):2015–22. doi: 10.4049/jimmunol.1100713
74. Chatenoud L, Thervet E, Primo J, Bach J-F. Anti-CD3 Antibody Induces Long-Term Remission of Overt Autoimmunity in Nonobese Diabetic Mice. Proc Natl Acad Sci (1994) 91(1):123–7. doi: 10.1073/pnas.91.1.123
75. Kung P, Goldstein G, Reinherz EL, Schlossman SF. Monoclonal Antibodies Defining Distinctive Human T Cell Surface Antigens. Science (1979) 206(4416):347–9. doi: 10.1126/science.314668
76. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. New Engl J Med (2019) 381(7):603–13. doi: 10.1056/NEJMoa1902226
77. Aronson R, Gottlieb PA, Christiansen JS, Donner TW, Bosi E, Bode BW, et al. Low-Dose Otelixizumab Anti-CD3 Monoclonal Antibody DEFEND-1 Study: Results of the Randomized Phase III Study in Recent-Onset Human Type 1 Diabetes. Diabetes Care (2014) 37(10):2746–54. doi: 10.2337/dc13-0327
78. Kuhn C, Weiner HL. Therapeutic Anti-CD3 Monoclonal Antibodies: From Bench to Bedside. Immunotherapy (2016) 8(8):889–906. doi: 10.2217/imt-2016-0049
79. Mohty M. Mechanisms of Action of Antithymocyte Globulin: T-Cell Depletion and Beyond. Leukemia (2007) 21(7):1387–94. doi: 10.1038/sj.leu.2404683
80. Haller MJ, Long SA, Blanchfield JL, Schatz DA, Skyler JS, Krischer JP, et al. Low-Dose Anti-Thymocyte Globulin Preserves C-Peptide, Reduces HbA1c, and Increases Regulatory to Conventional T-Cell Ratios in New-Onset Type 1 Diabetes: Two-Year Clinical Trial Data. Diabetes (2019) 68(6):1267–76. doi: 10.2337/db19-0057
81. Pescovitz MD, Greenbaum CJ, Bundy B, Becker DJ, Gitelman SE, Goland R, et al. B-Lymphocyte Depletion With Rituximab and β-Cell Function: Two-Year Results. Diabetes Care (2014) 37(2):453–9. doi: 10.2337/dc13-0626
82. Lopez M, Clarkson MR, Albin M, Sayegh MH, Najafian N. A Novel Mechanism of Action for Anti-Thymocyte Globulin: Induction of CD4+ CD25+ Foxp3+ Regulatory T Cells. J Am Soc Nephrol (2006) 17(10):2844–53. doi: 10.1681/ASN.2006050422
83. da Silva AJ, Brickelmaier M, Majeau GR, Li Z, Su L, Hsu Y-M, et al. Alefacept, an Immunomodulatory Recombinant LFA-3/IgG1 Fusion Protein, Induces CD16 Signaling and CD2/CD16-Dependent Apoptosis of CD2+ Cells. J Immunol (2002) 168(9):4462–71. doi: 10.4049/jimmunol.168.9.4462
84. Rigby MR, Harris KM, Pinckney A, DiMeglio LA, Rendell MS, Felner EI, et al. Alefacept Provides Sustained Clinical and Immunological Effects in New-Onset Type 1 Diabetes Patients. J Clin Invest (2016) 125(8):3285–96. doi: 10.1172/JCI81722
85. Moreland L, Bate G, Kirkpatrick P. Abatacept. Nat Rev Drug Discovery (2006) 5(3):185–6. doi: 10.1038/nrd1989
86. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Costimulation Modulation With Abatacept in Patients With Recent-Onset Type 1 Diabetes: Follow-Up 1 Year After Cessation of Treatment. Diabetes Care (2014) 37(4):1069–75. doi: 10.2337/dc13-0604
87. Edner NM, Heuts F, Thomas N, Wang CJ, Petersone L, Kenefeck R, et al. Follicular Helper T Cell Profiles Predict Response to Costimulation Blockade in Type 1 Diabetes. Nat Immunol (2020) 21(10):1244–55. doi: 10.1038/s41590-020-0744-z
88. Serra P, Santamaria P. Antigen-Specific Therapeutic Approaches for Autoimmunity. Nat Biotechnol (2019) 37(3):238–51. doi: 10.1038/s41587-019-0015-4
89. Smith EL, Peakman M. Peptide Immunotherapy for Type 1 Diabetes—Clinical Advances. Front Immunol (2018) 9:392. doi: 10.3389/fimmu.2018.00392
90. Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of Type 1 Diabetes in Mice by Tolerogenic Vaccination With a Strong Agonist Insulin Mimetope. J Exp Med (2011) 208(7):1501–10. doi: 10.1084/jem.20110574
91. Serr I, Fürst RW, Achenbach P, Scherm MG, Gökmen F, Haupt F, et al. Type 1 Diabetes Vaccine Candidates Promote Human Foxp3+ Treg Induction in Humanized Mice. Nat Commun (2016) 7(1):1–18. doi: 10.1038/ncomms10991
92. Eggenhuizen PJ, Ng BH, Ooi JD. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int J Mol Sci (2020) 21(19):7015. doi: 10.3390/ijms21197015
93. Nikolic T, Zwaginga JJ, Uitbeijerse BS, Woittiez NJ, de Koning EJ, Aanstoot H-J, et al. Safety and Feasibility of Intradermal Injection With Tolerogenic Dendritic Cells Pulsed With Proinsulin Peptide—for Type 1 Diabetes. Lancet Diabetes Endocrinol (2020) 8(6):470–2. doi: 10.1016/S2213-8587(20)30104-2
94. Parsa R, Andresen P, Gillett A, Mia S, Zhang X-M, Mayans S, et al. Adoptive Transfer of Immunomodulatory M2 Macrophages Prevents Type 1 Diabetes in NOD Mice. Diabetes (2012) 61(11):2881–92. doi: 10.2337/db11-1635
95. Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, et al. IL-2 Reverses Established Type 1 Diabetes in NOD Mice by a Local Effect on Pancreatic Regulatory T Cells. J Exp Med (2010) 207(9):1871–8. doi: 10.1084/jem.20100209
96. Marcovecchio ML, Wicker LS, Dunger DB, Dutton SJ, Kopijasz S, Scudder C, et al. Interleukin-2 Therapy of Autoimmunity in Diabetes (ITAD): A Phase 2, Multicentre, Double-Blind, Randomized, Placebo-Controlled Trial. Wellcome Open Res (2020) 5. doi: 10.12688/wellcomeopenres.15697.1
97. Hagopian W, Ferry RJ, Sherry N, Carlin D, Bonvini E, Johnson S, et al. Teplizumab Preserves C-Peptide in Recent-Onset Type 1 Diabetes: Two-Year Results From the Randomized, Placebo-Controlled Protégé Trial. Diabetes (2013) 62(11):3901–8. doi: 10.2337/db13-0236
98. Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W, et al. Teplizumab (Anti-CD3 mAb) Treatment Preserves C-Peptide Responses in Patients With New-Onset Type 1 Diabetes in a Randomized Controlled Trial: Metabolic and Immunologic Features at Baseline Identify a Subgroup of Responders. Diabetes (2013) 62(11):3766–74. doi: 10.2337/db13-0345
99. Clinicaltrials.Gov. Recent-Onset Type 1 Diabetes Trial Evaluating Efficacy and Safety of Teplizumab (2019). Available at: https://ClinicalTrials.gov/show/NCT03875729 (Accessed 2021 May 27).
100. Clinicaltrials.Gov. Trial of Otelixizumab for Adolescents and Adults With Newly Diagnosed Type 1 Diabetes Mellitus (Autoimmune): DEFEND-2 (2010). Available at: https://ClinicalTrials.gov/show/NCT01123083 (Accessed 2021 May 27).
101. Rigby MR, DiMeglio LA, Rendell MS, Felner EI, Dostou JM, Gitelman SE, et al. Targeting of Memory T Cells With Alefacept in New-Onset Type 1 Diabetes (T1DAL Study): 12 Month Results of a Randomised, Double-Blind, Placebo-Controlled Phase 2 Trial. Lancet Diabetes Endocrinol (2013) 1(4):284–94. doi: 10.1016/S2213-8587(13)70111-6
102. Cabrera SM, Engle S, Kaldunski M, Jia S, Geoffrey R, Simpson P, et al. Innate Immune Activity as a Predictor of Persistent Insulin Secretion and Association With Responsiveness to CTLA4-Ig Treatment in Recent-Onset Type 1 Diabetes. Diabetologia (2018) 61(11):2356–70. doi: 10.1007/s00125-018-4708-x
103. Clinicaltrials.Gov. Rituximab and Abatacept for Prevention or Reversal of Type 1 Diabetes (2020). Available at: https://ClinicalTrials.gov/show/NCT03929601 (Accessed 2021 May 27).
104. Todd JA, Evangelou M, Cutler AJ, Pekalski ML, Walker NM, Stevens HE, et al. Regulatory T Cell Responses in Participants With Type 1 Diabetes After a Single Dose of Interleukin-2: A non-Randomised, Open Label, Adaptive Dose-Finding Trial. PloS Med (2016) 13(10):e1002139. doi: 10.1371/journal.pmed.1002139
105. Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, et al. Metformin Inhibits Proinflammatory Responses and Nuclear Factor-κb in Human Vascular Wall Cells. Arteriosclerosis thrombosis Vasc Biol (2006) 26(3):611–7. doi: 10.1161/01.ATV.0000201938.78044.75
106. Wang S, Song P, Zou M-H. AMP-Activated Protein Kinase, Stress Responses and Cardiovascular Diseases. Clin Sci (2012) 122(12):555–73. doi: 10.1042/CS20110625
107. Zabielski P, Hady HR, Chacinska M, Roszczyc K, Gorski J, Blachnio-Zabielska AU. The Effect of High Fat Diet and Metformin Treatment on Liver Lipids Accumulation and Their Impact on Insulin Action. Sci Rep (2018) 8(1):1–11. doi: 10.1038/s41598-018-25397-6
108. Kim EK, Lee SH, Jhun JY, Byun JK, Jeong JH, Lee S-Y, et al. Metformin Prevents Fatty Liver and Improves Balance of White/Brown Adipose in an Obesity Mouse Model by Inducing FGF21. Mediators Inflammation (2016) 2016. doi: 10.1155/2016/5813030
109. Lee Y-S, Jun H-S. Anti-Inflammatory Effects of GLP-1-Based Therapies Beyond Glucose Control. Mediators Inflammation (2016) 2016. doi: 10.1155/2016/3094642
110. Velmurugan K, Balamurugan A, Loganathan G, Ahmad A, Hering BJ, Pugazhenthi S. Antiapoptotic Actions of Exendin-4 Against Hypoxia and Cytokines are Augmented by CREB. Endocrinology (2012) 153(3):1116–28. doi: 10.1210/en.2011-1895
111. Li L, El-Kholy W, Rhodes C, Brubaker P. Glucagon-Like Peptide-1 Protects Beta Cells From Cytokine-Induced Apoptosis and Necrosis: Role of Protein Kinase B. Diabetologia (2005) 48(7):1339–49. doi: 10.1007/s00125-005-1787-2
112. Hogan AE, Gaoatswe G, Lynch L, Corrigan MA, Woods C, O’Connell J, et al. Glucagon-Like Peptide 1 Analogue Therapy Directly Modulates Innate Immune-Mediated Inflammation in Individuals With Type 2 Diabetes Mellitus. Diabetologia (2014) 57(4):781–4. doi: 10.1007/s00125-013-3145-0
113. Birnbaum Y, Bajaj M, Qian J, Ye Y. Dipeptidyl Peptidase-4 Inhibition by Saxagliptin Prevents Inflammation and Renal Injury by Targeting the Nlrp3/ASC Inflammasome. BMJ Open Diabetes Res Care (2016) 4(1):e000227. doi: 10.1136/bmjdrc-2016-000227
114. Zhuge F, Ni Y, Nagashimada M, Nagata N, Xu L, Mukaida N, et al. DPP-4 Inhibition by Linagliptin Attenuates Obesity-Related Inflammation and Insulin Resistance by Regulating M1/M2 Macrophage Polarization. Diabetes (2016) 65(10):2966–79. doi: 10.2337/db16-0317
115. Satoh-Asahara N, Sasaki Y, Wada H, Tochiya M, Iguchi A, Nakagawachi R, et al. A Dipeptidyl Peptidase-4 Inhibitor, Sitagliptin, Exerts Anti-Inflammatory Effects in Type 2 Diabetic Patients. Metabolism (2013) 62(3):347–51. doi: 10.1016/j.metabol.2012.09.004
116. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1–Receptor Antagonist in Type 2 Diabetes Mellitus. New Engl J Med (2007) 356(15):1517–26. doi: 10.1056/NEJMoa065213
117. Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J, et al. Effects of Interleukin-1β Inhibition With Canakinumab on Hemoglobin A1c, Lipids, C-Reactive Protein, Interleukin-6, and Fibrinogen: A Phase IIb Randomized, Placebo-Controlled Trial. Circulation (2012) 126(23):2739–48. doi: 10.1161/CIRCULATIONAHA.112.122556
118. Zobel EH, von Scholten BJ, Goldman B, Persson F, Hansen TW, Rossing P. Pleiotropic Effects of Liraglutide in Patients With Type 2 Diabetes and Moderate Renal Impairment: Individual Effects of Treatment. Diabetes Obes Metab (2019) 21(5):1261–5. doi: 10.1111/dom.13638
119. Wysham C, Bergenstal R, Malloy J, Yan P, Walsh B, Malone J, et al. DURATION-2: Efficacy and Safety of Switching From Maximum Daily Sitagliptin or Pioglitazone to Once-Weekly Exenatide. Diabetic Med (2011) 28(6):705–14. doi: 10.1111/j.1464-5491.2011.03301.x
120. Clinicaltrials.Gov. Safety and Efficacy Study of Exenatide Once Weekly in Adolescents With Type 2 Diabetes (2011). Available at: https://ClinicalTrials.gov/show/NCT01554618 (Accessed 2021 May 27).
121. Ruscitti P, Masedu F, Alvaro S, Airò P, Battafarano N, Cantarini L, et al. Anti-Interleukin-1 Treatment in Patients With Rheumatoid Arthritis and Type 2 Diabetes (TRACK): A Multicentre, Open-Label, Randomised Controlled Trial. PloS Med (2019) 16(9):e1002901. doi: 10.1371/journal.pmed.1002901
122. Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Interleukin-1 Antagonism in Type 1 Diabetes of Recent Onset: Two Multicentre, Randomised, Double-Blind, Placebo-Controlled Trials. Lancet (2013) 381(9881):1905–15. doi: 10.1016/S0140-6736(13)60023-9
123. Clinicaltrials.Gov. IL-1-Receptor Antagonist During Cephalic Phase of Insulin Secretion in Health and Type 2 Diabetes (2020). Available at: https://ClinicalTrials.gov/show/NCT04227769 (Accessed 2021 May 27).
124. Naito E, Yoshida Y, Makino K, Kounoshi Y, Kunihiro S, Takahashi R, et al. Beneficial Effect of Oral Administration of Lactobacillus Casei Strain Shirota on Insulin Resistance in Diet-Induced Obesity Mice. J Appl Microbiol (2011) 110(3):650–7. doi: 10.1111/j.1365-2672.2010.04922.x
125. Andreasen AS, Larsen N, Pedersen-Skovsgaard T, Berg RM, Møller K, Svendsen KD, et al. Effects of Lactobacillus Acidophilus NCFM on Insulin Sensitivity and the Systemic Inflammatory Response in Human Subjects. Br J Nutr (2010) 104(12):1831–8. doi: 10.1017/S0007114510002874
126. Niibo M, Shirouchi B, Umegatani M, Morita Y, Ogawa A, Sakai F, et al. Probiotic Lactobacillus Gasseri SBT2055 Improves Insulin Secretion in a Diabetic Rat Model. J dairy Sci (2019) 102(2):997–1006. doi: 10.3168/jds.2018-15203
127. Valladares R, Sankar D, Li N, Williams E, Lai K-K, Abdelgeliel AS, et al. Lactobacillus Johnsonii N6. 2 Mitigates the Development of Type 1 Diabetes in BB-DP Rats. PloS One (2010) 5(5):e10507. doi: 10.1371/journal.pone.0010507
128. Li N, Russell WM, Douglas-Escobar M, Hauser N, Lopez M, Neu J. Live and Heat-Killed Lactobacillus Rhamnosus GG: Effects on Proinflammatory and Anti-Inflammatory Cytokines/Chemokines in Gastrostomy-Fed Infant Rats. Pediatr Res (2009) 66(2):203–7. doi: 10.1203/PDR.0b013e3181aabd4f
129. Li Y, Yang S, Lun J, Gao J, Gao X, Gong Z, et al. Inhibitory Effects of the Lactobacillus Rhamnosus GG Effector Protein HM0539 on Inflammatory Response Through the TLR4/MyD88/NF-Кb Axis. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.551449
130. Mu Q, Tavella VJ, Luo XM. Role of Lactobacillus Reuteri in Human Health and Diseases. Front Microbiol (2018) 9:757. doi: 10.3389/fmicb.2018.00757
131. Kim TK, Lee J-C, Im S-H, Lee M-S. Amelioration of Autoimmune Diabetes of NOD Mice by Immunomodulating Probiotics. Front Immunol (2020) 11:1832. doi: 10.3389/fimmu.2020.01832
132. Wang Y, Dilidaxi D, Wu Y, Sailike J, Sun X. Nabi X-H. Composite Probiotics Alleviate Type 2 Diabetes by Regulating Intestinal Microbiota and Inducing GLP-1 Secretion in Db/Db Mice. Biomedicine Pharmacotherapy (2020) 125:109914. doi: 10.1016/j.biopha.2020.109914
133. Tonucci LB, Dos Santos KMO, de Oliveira LL, Ribeiro SMR, Martino HSD. Clinical Application of Probiotics in Type 2 Diabetes Mellitus: A Randomized, Double-Blind, Placebo-Controlled Study. Clin Nutr (2017) 36(1):85–92. doi: 10.1016/j.clnu.2015.11.011
134. Ejtahed HS, Mohtadi-Nia J, Homayouni-Rad A, Niafar M, Asghari-Jafarabadi M, Mofid V. Probiotic Yogurt Improves Antioxidant Status in Type 2 Diabetic Patients. Nutrition (2012) 28(5):539–43. doi: 10.1016/j.nut.2011.08.013
135. Peng J, Narasimhan S, Marchesi JR, Benson A, Wong FS, Wen L. Long Term Effect of Gut Microbiota Transfer on Diabetes Development. J Autoimmun (2014) 53:85–94. doi: 10.1016/j.jaut.2014.03.005
136. De Groot P, Nikolic T, Pellegrini S, Sordi V, Imangaliyev S, Rampanelli E, et al. Faecal Microbiota Transplantation Halts Progression of Human New-Onset Type 1 Diabetes in a Randomised Controlled Trial. Gut (2021) 70(1):92–105. doi: 10.1136/gutjnl-2020-322630
137. Wang H, Lu Y, Yan Y, Tian S, Zheng D, Leng D, et al. Promising Treatment for Type 2 Diabetes: Fecal Microbiota Transplantation Reverses Insulin Resistance and Impaired Islets. Front Cell infection Microbiol (2020) 9:455. doi: 10.3389/fcimb.2019.00455
138. Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, et al. Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome. Gastroenterology (2012) 143(4):913–6.e7. doi: 10.1053/j.gastro.2012.06.031
139. Marcial GE, Ford AL, Haller MJ, Gezan SA, Harrison NA, Cai D, et al. Lactobacillus Johnsonii N6. 2 Modulates the Host Immune Responses: A Double-Blind, Randomized Trial in Healthy Adults. Front Immunol (2017) 8:655. doi: 10.3389/fimmu.2017.00655
140. Clinicaltrials.Gov. Lactobacillus Johnsonii Supplementation in Adults With T1D (2020). Available at: https://ClinicalTrials.gov/show/NCT03961347 (Accessed 2021 May 27).
141. Groele L, Szajewska H, Szalecki M, Świderska J, Wysocka-Mincewicz M, Ochocińska A, et al. Lack of Effect of Lactobacillus Rhamnosus GG and Bifidobacterium Lactis Bb12 on Beta-Cell Function in Children With Newly Diagnosed Type 1 Diabetes: A Randomised Controlled Trial. BMJ Open Diabetes Res Care (2021) 9(1):e001523. doi: 10.1136/bmjdrc-2020-001523
142. Clinicaltrials.Gov. Probiotics in Newly Diagnosed T1D (2019). Available at: https://ClinicalTrials.gov/show/NCT04141761 (Accessed 2021 May 27).
143. Clinicaltrials.Gov. Modulation of Type 1 Diabetes Susceptibility Through the Use of Probiotics (2018). Available at: https://ClinicalTrials.gov/show/NCT03423589 (Accessed 2021 May 27).
144. Mobini R, Tremaroli V, Ståhlman M, Karlsson F, Levin M, Ljungberg M, et al. Metabolic Effects of L Actobacillus Reuteri DSM 17938 in People With Type 2 Diabetes: A Randomized Controlled Trial. Diabetes Obes Metab (2017) 19(4):579–89. doi: 10.1111/dom.12861
145. Kobyliak N, Falalyeyeva T, Mykhalchyshyn G, Kyriienko D, Komissarenko I. Effect of Alive Probiotic on Insulin Resistance in Type 2 Diabetes Patients: Randomized Clinical Trial. Diabetes Metab Syndrome: Clin Res Rev (2018) 12(5):617–24. doi: 10.1016/j.dsx.2018.04.015
146. Tao Y-W, Gu Y-L, Mao X-Q, Zhang L, Pei Y-F. Effects of Probiotics on Type II Diabetes Mellitus: A Meta-Analysis. J Trans Med (2020) 18(1):1–11. doi: 10.1186/s12967-020-02274-3
147. Clinicaltrials.Gov. Fecal Microbiota Transplantation on Type 2 Diabetes Mellitus (2012). Available at: https://ClinicalTrials.gov/show/NCT01790711 (Accessed 2021 May 27).
148. Ng SC, Xu Z, Mak JWY, Yang K, Liu Q, Zuo T, et al. Microbiota Engraftment After Faecal Microbiota Transplantation in Obese Subjects With Type 2 Diabetes: A 24-Week, Double-Blind, Randomised Controlled Trial. Gut (2021). doi: 10.1136/gutjnl-2020-323617
149. Clinicaltrials.Gov. Fecal Microbiota Transplantation for Diabetes Mellitus Type II in Obese Patients (2016). Available at: https://ClinicalTrials.gov/show/NCT02346669 (Accessed 2021 May 27).
Keywords: diabetes, pancreatic β cell, immune modulation, autoimmunity, inflammation, gut microbiota
Citation: Jo S and Fang S (2021) Therapeutic Strategies for Diabetes: Immune Modulation in Pancreatic β Cells. Front. Endocrinol. 12:716692. doi: 10.3389/fendo.2021.716692
Received: 29 May 2021; Accepted: 30 July 2021;
Published: 17 August 2021.
Edited by:
Zong Wei, Mayo Clinic Arizona, United StatesReviewed by:
Eiji Yoshihara, Lundquist Institute for Biomedical Innovation, United StatesMarcia Hiriart, Universidad Nacional Autonoma de Mexico, Mexico
Ji Min Lee, Kangwon National University, South Korea
Copyright © 2021 Jo and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sungsoon Fang, c2ZhbmdAeXVocy5hYw==